

Abstract
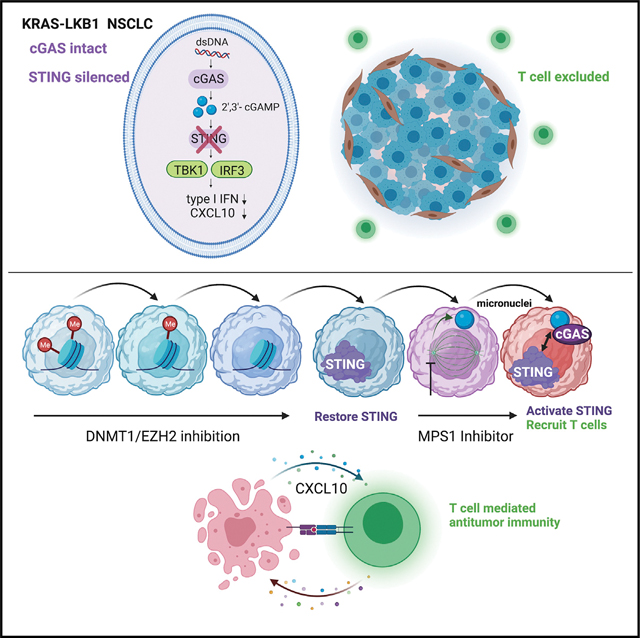
KRAS-LKB1 (KL) mutant lung cancers silence STING owing to intrinsic mitochondrial dysfunction, resulting in T cell exclusion and resistance to programmed cell death (ligand) 1 (PD-[L]1) blockade. Here we discover that KL cells also minimize intracellular accumulation of 2′3′-cyclic GMP-AMP (2′3′-cGAMP) to further avoid downstream STING and STAT1 activation. An unbiased screen to co-opt this vulnerability reveals that transient MPS1 inhibition (MPS1i) potently re-engages this pathway in KL cells via micronuclei generation. This effect is markedly amplified by epigenetic de-repression of STING and only requires pulse MPS1i treatment, creating a therapeutic window compared with non-dividing cells. A single course of decitabine treatment followed by pulse MPS1i therapy restores T cell infiltration in vivo, enhances anti-PD-1 efficacy, and results in a durable response without evidence of significant toxicity.
In brief
KRAS-LKB1 (KL) lung cancers epigenetically silence STING and resist PD-1 blockade. Here, Kitajima et al. discover that MPS1 inhibition strongly reactivates KL cGAS-STING signaling following epigenetic STING de-repression. Microfluidic and animal models demonstrate potent T/NK cell recruitment by this combination, reversing anti-PD-1 resistance and revealing a strategy translatable to the clinic.
Graphical Abstract
INTRODUCTION
Immune checkpoint blockade (ICB) exhibits significant therapeutic efficacy in many cancers, including non-small cell lung cancer (NSCLC). Recent extensive efforts have identified biological markers that are predictive of favorable response, including higher programmed cell death ligand 1 (PD-L1) expression, the degree of tumor mutation burden, and the number of tumor-infiltrating lymphocytes residing in the tumor microenvironment (TME) (Keenan et al., 2019). Conversely, certain somatic mutations, including those that impair interferon sensing or major histocompatibility (MHC) class I display, allow cancer cells to evade cancer immunosurveillance and promote resistance to ICB (Keenan et al., 2019; Spranger and Gajewski, 2018).
Aberrant cytoplasmic DNA accumulation after the invasion of microbial pathogens or cytosolic leakage of self-DNA are detected by cyclic guanosine monophosphate (GMP)-AMP synthase (cGAS), which produces the second messenger 2′3′-cyclic GMP-AMP (2′3′-cGAMP) that directly activates its downstream target, stimulator of interferon genes (STING) (Li and Chen, 2018). STING then translocates from the endoplasmic reticulum (ER) toward the ER-Golgi intermediate compartment (ERGIC), and subsequently activates TANK-binding kinase 1 (TBK1) to phosphorylate and activate IRF3 (Hopfner and Hornung, 2020; Zhang et al., 2020). Since IRF3-induced cytokines, including type I interferon and CXCL10, play a central role in enhancing antigen presentation and cytotoxic T cell recruitment into tumors, intact STING signaling is increasingly recognized as a key determinant of therapeutic antitumor immunity (Kwon and Bakhoum, 2020).
The physiological function of the STING pathway has been well-studied in immune cells such as antigen-presenting cells (Deng et al., 2014; Woo et al., 2014). However, it is becoming increasingly clear that the activity of the cancer cell-intrinsic STING pathway defines their immunogenicity and the efficacy of ICB (Falahat et al., 2019, 2021; Guan et al., 2021; Lu et al., 2021; Mahadevan et al., 2021). Recently, we reported that STING expression is epigenetically silenced in KRAS-LKB1 (KL)-mutated NSCLC cells through EZH2-mediated histone H3K27 methylation, as well as DNMT1-induced 5-methylcytosine accumulation around its promoter (Kitajima et al., 2019). Mechanistically, KL cells exhibit cytoplasmic accumulation of mtDNA owing to an autophagic defect, which selects for STING silencing to protect cells from STAT1-induced cytotoxicity. Loss of tumor cell STING signaling in KL cells impairs infiltration of cytotoxic T cells into the TME (Campisi et al., 2020), and is associated with resistance to anti-PD-1 therapy in clinic (Koyama et al., 2016; Rizvi et al., 2018; Skoulidis et al., 2018). These findings suggest that re-engagement of STING activity might represent a promising strategy to restore immunogenicity of KL tumors. In fact, STING over-expression in KRAS-mutated NSCLC cells with lentiviral vectors induced STAT1-dependent cell death along with elevated secretion of STING downstream cytokines specifically in KL cells, in contrast with LKB1 wild-type (WT) cells (Kitajima et al., 2019).
The importance of the STING pathway as a pharmacologic target has accelerated the development of synthetic STING agonists such as ADU-S100 and MK-1454, which are based on the structure of natural STING ligand cyclic di-nucleotides (CDN) (Kwon and Bakhoum, 2020). However, stimulation of cancer cell-intrinsic STING signaling by these molecules is limited by low cell membrane permeability owing to the intrinsic negative charges and hydrophilicity, while immune cells express import channels such as SLC19A1 and SLC46A2 (Cordova et al., 2021; Luteijn et al., 2019; Ritchie et al., 2019). Furthermore, exogenous STING agonist injection can promote T cell cytotoxicity and death (Gulen et al., 2017).
An alternative strategy to activate cancer cell-intrinsic STING signaling is to promote endogenous 2′3′-cGAMP production via cGAS activation. Recently, many studies have revealed that genotoxic therapies, including radiation therapy and treatment with targeted or cytotoxic DNA-damaging agents, can trigger the activation of the STING pathway through aberrant accumulation of cytoplasmic DNA in a cGAS-dependent manner (Reislander et al., 2020). For example, treatment with poly ADP-ribose polymerase (PARP) inhibitors such as olaparib causes genomic instability leading to cytosolic leakage of self-DNA specifically in homologous recombination-deficient cancer cells associated with the BRCA1/2 mutation (Ding et al., 2018; Pantelidou et al., 2019; Reislander et al., 2019). In addition, treatment with certain chemotherapy reagents such as cisplatin and paclitaxel induces cancer cell-intrinsic STING signaling via accumulation of DNA damage (Grabosch et al., 2019; Zierhut et al., 2019). However, most KL cells are BRCA1/2-proficient, and there is no a priori evidence that this or any other previously described approach is optimal to re-engage STING signaling in KL cells.
Although cGAS expression is often suppressed in certain types of cancer cells such as melanoma (Konno et al., 2018), most KL cell lines still express cGAS (Kitajima et al., 2019), suggesting that inducing cytoplasmic DNA accumulation in KL cells could be an effective approach to impair their viability and to increase their immunogenicity. We therefore conducted a systematic set of studies to examine the unique sensitivity of KL cells to intracellular accumulation of 2′3′-cGAMP, and then conducted a focused screen to identify existing clinical stage drugs that could co-opt this vulnerability, especially in combination with epigenetic therapies that force them to express STING.
RESULTS
LKB1 inactivation sensitizes KRAS mutant NSCLC cells to 2′3′-cGAMP
We first used treatment with 2′3′-cGAMP or ADU-S100 to examine the sensitivity of KRAS-Tp53 (KP) mutated or KL NSCLC cell lines to exogenous CDN exposure. We previously reported that a subset of KL cell lines maintains low levels of STING protein expression (STINGLow), whereas others exhibit undetectable STING levels owing to concurrent high DNMT1 expression (STINGAbsent) (Kitajima et al., 2019). STINGLow KL cells secreted CXCL10 in response to either 2′3′-cGAMP or ADU-S100 in a dose-dependent manner, especially H1944, H2122, and H647 cells that retain higher STING levels compared with H1355 cells (Figure S1A); in contrast, STINGAbsent cell lines failed to respond to exogenous CDNs even at high doses, consistent with their lack of STING expression (Figure 1A). Notably, KP cell lines (H2009, H358, H441, and H1792 cells) exhibited a generally weaker response to STING agonists despite their higher STING expression, with two of four KP cell lines such as H2009 and H1792 lacking response, even at a high dose (Figures 1A and S1A). KL cell response to extracellular CDNs was still modest as compared with endothelial cells (human umbilical cord vein endothelial cells) or monocytic THP-1 cells, which also have different membrane permeabilities (Figure S1B) (Cordova et al., 2021; Luteijn et al., 2019; Ritchie et al., 2019). However, these findings suggested that STING-positive KL cell lines might be particularly sensitive to the intracellular accumulation of 2′3′-cGAMP.
Figure 1. KL cells exhibit low tolerability to accumulation of intracellular 2′3′-cGAMP.
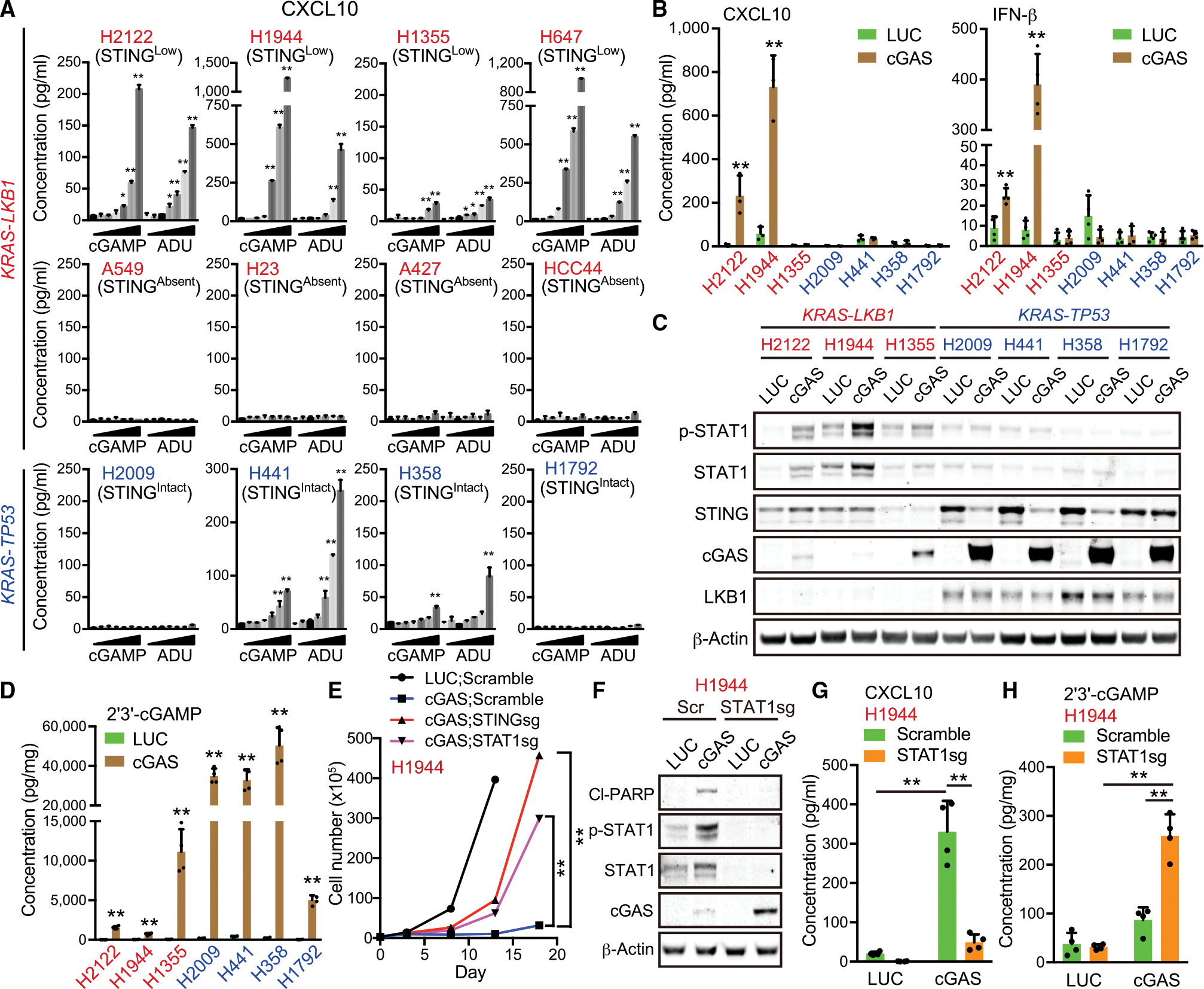
(A) Enzyme-linked immunosorbent assay (ELISA) of human CXCL10 levels in conditioned medium (CM) derived from KL (red) or KP (blue) NSCLC cells treated with or without 3.125, 6.25, 12.5, 25, 50, or 100 μM 2′3′-cGAMP or ADU-S100 for 24 h (n = 3). H2122, H1355, H23, and HCC44 KL cell lines have a p53 mutation.
(B) ELISA of human CXCL10 or IFN-β levels in CM derived from KL or KP cells transduced with the indicated vectors (n = 3).
(C) Immunoblot (IB) of the indicated proteins in KL or KP cells transduced with the indicated vectors.
(D) ELISA of intracellular 2′3′-cGAMP levels in KL (red) or KP (blue) cells transduced with the indicated vectors (n = 4). © Total cell number of H1944 cells transduced with the indicated vectors at each measuring point (day 0, day 3, day 8, day 13, or day 18).
(F–H) IB of the indicated proteins (F), or ELISA of human CXCL10 in CM (G) or intracellular 2′3′-cGAMP levels (H) in H1944 cells transduced with the indicated vectors (n = 4).
All quantitative data are represented as mean ± SD p values were calculated by unpaired two-tailed Student’s t test (B, D, and E), or one-way analysis of variance followed by Dunnet’s post hoc test (A), or two-way ANOVA(G and H) followed by Sidak’s post hoc test (G and H), *p < 0.05, **p < 0.01. See also Figure S1.
We, therefore, directly compared sensitivity to endogenous intracellular 2′3′-cGAMP by forced over-expression of cGAS across multiple STINGLow KL versus KP cancer cell lines. H2122 and H1944 cGAS-expressing cells produced significantly higher levels of both CXCL10 and interferon (IFN)-β and failed to downregulate STING levels as a counter-regulatory response, in contrast to KP cells (Figures 1B and 1C). These same KL cell lines also uniquely activated STAT1 after cGAS transduction, in contrast with KP cells, despite achieving much lower levels of cGAS over-expression (Figures 1C and S1C) and generating significantly lower levels of 2′3′-cGAMP as compared with KP cell lines (Figure 1D). These data further suggested hypersensitivity of the KL cellular state to intracellular 2′3′-cGAMP. Indeed, H1944 KL cells could instead tolerate over-expression of a catalytically inactive mutant cGAS (K414R) (Dai et al., 2019); conversely, LKB1 depletion hindered WT cGAS accumulation in KP cells (Figures S1D–S1G). Together, these data reveal that LKB1 inactivation is not only associated with STING silencing, but also cGAS intolerance and 2′3′-cGAMP hypersensitivity.
We previously determined that STAT1 activation contributes to KL intolerance of STING signaling (Kitajima et al., 2019). We, therefore, also assessed its role in mediating this upstream intolerance to cGAS expression and 2′3′-cGAMP generation. Indeed, the inhibition of STAT1, as well as STING, similarly attenuated cGAS-induced growth suppression, induction of apoptosis, and CXCL10 production in multiple STINGLow KL cell lines, including H1944, H2122, and H1355 cells (Figures 1E–1G, S1H, and S1I). Furthermore, STAT1-depleted STINGLow KL cells exhibited higher exogenous cGAS expression and intracellular 2′3′-cGAMP accumulation after cGAS over-expression (Figures 1F, 1H, and S1J). Thus, KL cells limit intracellular 2′3′-cGAMP accumulation, at least in part to avoid the downstream cytotoxicity associated with activation of STAT1.
Identification of MPS1 inhibition as a potent inducer of endogenous 2′3′-cGAMP production in KL cells
Owing to their particular hypersensitivity, we used H1944 cells as a model system in which to conduct an unbiased screen of cytotoxic chemotherapies or targeted DNA-damaging agents for their ability to upregulate endogenous 2′3′-cGAMP production after pulse treatment, including cGAS-deficient H2122 cells as a counter-screen (Figures 2A and 2B). Indeed, we validated selective induction of 2′3′-cGAMP by the double-stranded DNA (dsDNA) mimic poly(dA:dT) in H1944 cells, but not H2122 cells (Figure 2C). We focused in particular on DNA-damaging agents already used in the clinic or in clinical trials; we examined a variety of chemotherapy drugs, including cisplatin, docetaxel, etoposide, vinorelbine, pemetrexed, and methotrexate, and molecularly targeted drugs against DNA replication and repair pathways, including olaparib (PARP inhibitor), barasertib (Aurora B inhibitor), MK5108 (Aurora A inhibitor), MK1775 (WEE1 inhibitor), ceralasertib (ATR inhibitor), prexasertib (CHK1 inhibitor), CFI-402257 (MPS1 inhibitor), and volasertib (PLK1 inhibitor). Additionally, we included hydroxyurea or nocodazole as controls to induce S-phase or M-phase cell-cycle arrest. We identified the median inhibition concentration (IC50) for each compound for cell viability in H1944 cells and used this concentration for the screen (Figures S2A and Table S1). We confirmed that each agent induced cellular cytotoxicity and/or DNA damage response at this concentration, as measured by the induction of propidium iodide positive cells, cleaved PARP fragments, or histone γH2A.X expression (Figures S2B and S2C).
Figure 2. Screening of DNA-damaging agents to extract the drugs activating the STING pathway in KL cells.
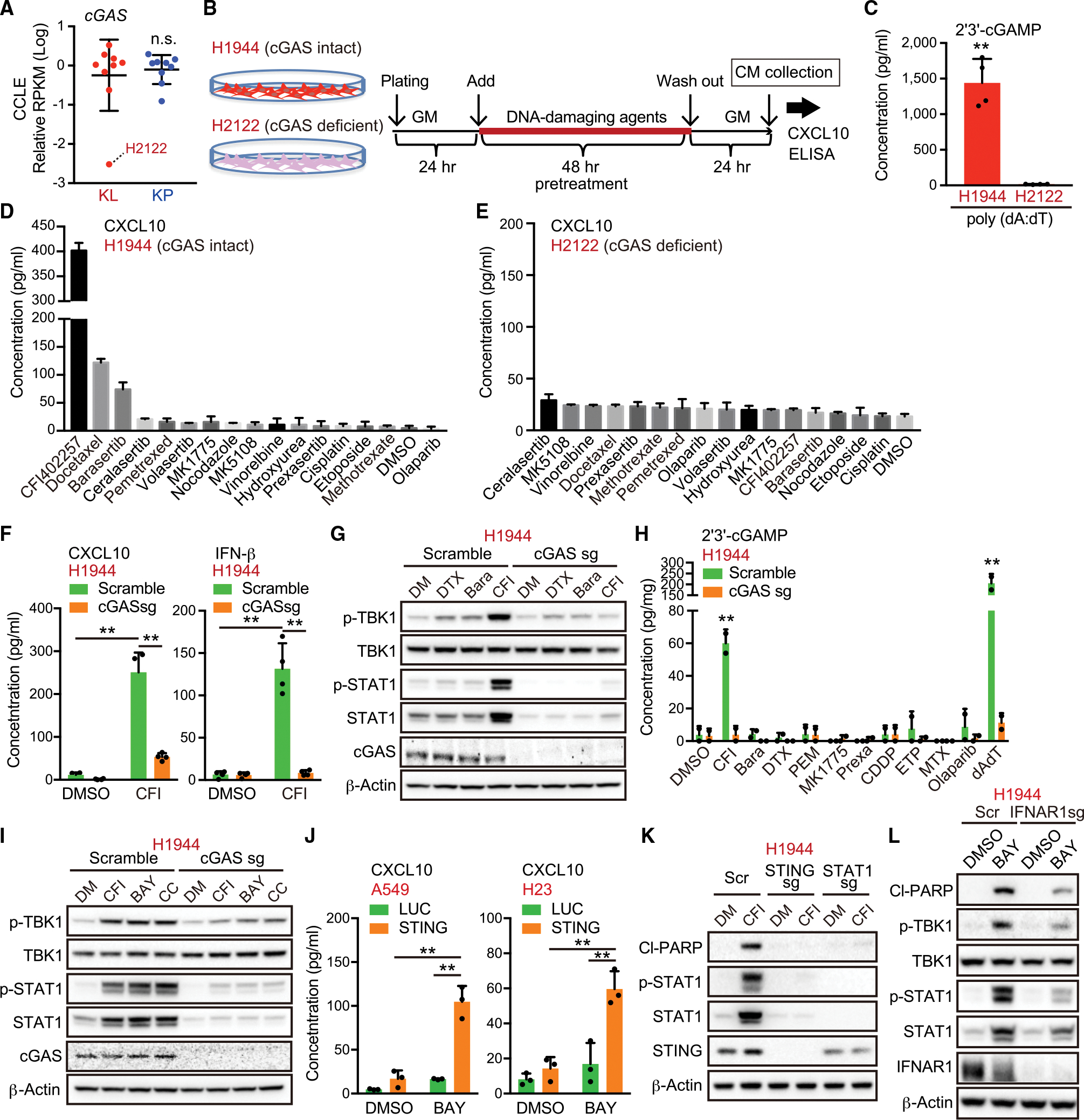
(A) Relative RPKM values of cGAS in KL and KP cells from CCLE.
(B) Schedule of drug treatment for the screening. GM, growth medium; CM, conditioned medium.
(C) Intracellular 2′3′-cGAMP levels in H2122 or H1944 cells treated with 0.5 μg/mL poly (dA:dT) (n = 4).
(D and E) Enzyme-linked immunosorbent assay (ELISA) of human CXCL10 in CM derived from H1944 (D) or H2122 (E) cells treated with the indicated DNA-damaging agents in accordance with the schedule for the screening (n = 2).
(F) ELISA of human CXCL10 or IFN-β levels in CM derived from H1944 cells transduced with the indicated vectors, treated with 200 nM CFI-402257 (n = 4).
(G and H) Immunoblot (IB) of the indicated proteins (G), or intracellular 2′3′-cGAMP levels (H), in H1944 cells transduced with the indicated vectors, and treated with the indicated DNA-damaging agents (n = 2). Bara, barasertib; CDDP, cisplatin; CFI, CFI-402257; DTX, docetaxel; ETP, etoposide Pr exa, prexasertib; MTX, methotrexate; PEM, pemetrexed.
(I, K, and L) IB of the indicated proteins in H1944 cells transduced with the indicated vectors, and treated with 200 nM CFI-402257, 100 nM BAY-1217389, or 250 nM CC-671.
(J) ELISA of human CXCL10 levels in CM derived from H1944 cells transduced with the indicated vectors, and treated with 100 nM BAY-1217389 (n = 3).
All quantitative data are represented as mean ± standard deviation; p values were calculated by unpaired two-tailed Student t test (A, C, and H), or two-way analysis of variance followed by Sidak’s post hoc test (F, J), **p < 0.01. See also Figure S2 and Table S1.
We identified three compounds with the ability to induce significant CXCL10 secretion in H1944 cells after a 48-h pulse treatment, all of which were anti-mitotics: docetaxel, barasertib, and especially CFI-402257, a selective inhibitor of the spindle assembly checkpoint (SAC) kinase monopolar kinase (MPS1; also known as TTK protein kinase) (Mason et al., 2017) (Figure 2D). The degree of CXCL10 induction did not correlate with the amount of cytotoxicity or DNA damage response induced by each agent, suggesting that the mechanism was not related to the degree of DNA damage induced (Figures S2B and S2C). Furthermore, the ability of each agent to induce CXCL10 in H2122 cells was abrogated, suggestive of cGAS dependence (Figure 2E). Indeed, we confirmed that CXCL10 and IFN-β secretion in H1944 cells after pulse treatment with CFI-402257 was markedly attenuated by cGAS depletion (Figure 2F), as was STAT1 activation (Figure 2G). CFI-402257 was also the only agent that yielded detectable levels of intracellular 2′3′-cGAMP, which was similarly ablated by cGAS depletion (Figure 2H). Taken together, as compared with other DNA-damaging agents that activate cGAS-STING in other contexts (Ding et al., 2018; Grabosch et al., 2019; Pantelidou et al., 2019; Reislander et al., 2019; Zierhut et al., 2019), these data revealed pulse MPS1 inhibition as the most potent inducer of intracellular 2′3′-cGAMP and downstream STING pathway activation in KL cells.
To rule out a potential off-target effect of CFI-402257 treatment, we also examined the effects of two additional MPS1 inhibitors, BAY-1217389 and CC-671, which induced similar cGAS-dependent activation of STING signaling in H1944 cells (Figure 2I); all of these findings were reproducible in an additional STINGLow KL cell line (Figures S2D and S2E). In addition, genetic depletion of MPS1 using small interfering RNA significantly upregulated STING downstream signaling and CXCL10 secretion more potently than Aurora B depletion, in a cGAS-dependent manner (Figures S2F and S2G). In contrast, the reconstitution of STING in STINGabsent KL cells such as A549 and H23 cells also sensitized them to MPS1 inhibition, leading to CXCL10 secretion and STAT1 activation (Figures 2J and S2H). We further confirmed that MPS1 inhibitor-induced cellular cytotoxicity and growth arrest in KL cells required the activation of STING signaling after a 48-h pulse treatment and, to a lesser degree, during continuous 96-h SAC inhibition, which is also capable of arresting KP cells (Figures 2K and S2I–S2K). Notably, IFN-α and -β receptor subunit 1 (IFNAR1) depletion also attenuated cell death and growth arrest, and CXCL10 induction by pulse MPS1 inhibitor treatment in KL cells (Figures S2L and S2M). Phosphorylation of TBK1 and IFN-β secretion, which are upstream of IFNAR1, were also attenuated by IFNAR1 depletion (Figures 2L, S2M, and S2N). These data implied that the additional formation of a positive feedback loop between STING signaling and type l IFN contributed to their hypersensitivity to pulse MPS1 inhibition, potentially independently of its direct anti-mitotic effects.
MPS1 inhibition activates the STING pathway via micronuclei formation in KL cells
MPS1 is a critical regulator of the SAC and MPS1 inhibition in other contexts is known to facilitate massive chromosome missegregation (London and Biggins, 2014). We, therefore, hypothesized that these effects could be related to formation of micronuclei, known activators of cGAS-STING signaling generated by mitotic slippage and subsequent progression into G1 (Mackenzie et al., 2017). Indeed, treatment of H1944 cells with CFI-402257 using the same 48-h pulse treatment schedule (Figure 2B) generated abundant micronuclei, as compared with other drugs, including docetaxel or barasertib (Figures 3A and 3B). Furthermore, the induction of CXCL10 messenger RNA (mRNA) expression in H1944 cells directly correlated with the number of micronuclei induced by these DNA-damaging agents, with CFI-402257 representing the clear outlier (Figure 3C). Additionally, Lamin B2 over-expression, which is sufficient to inhibit micronuclei disruption (Hatch et al., 2013) and could protect against cGAS recognition, attenuated CXCL10 secretion induced by MPS1 inhibitor pulse treatment (Figures S3A and S3B).
Figure 3. MPS1 inhibition induces micronuclei formation and subsequent STING activation in KL cells.
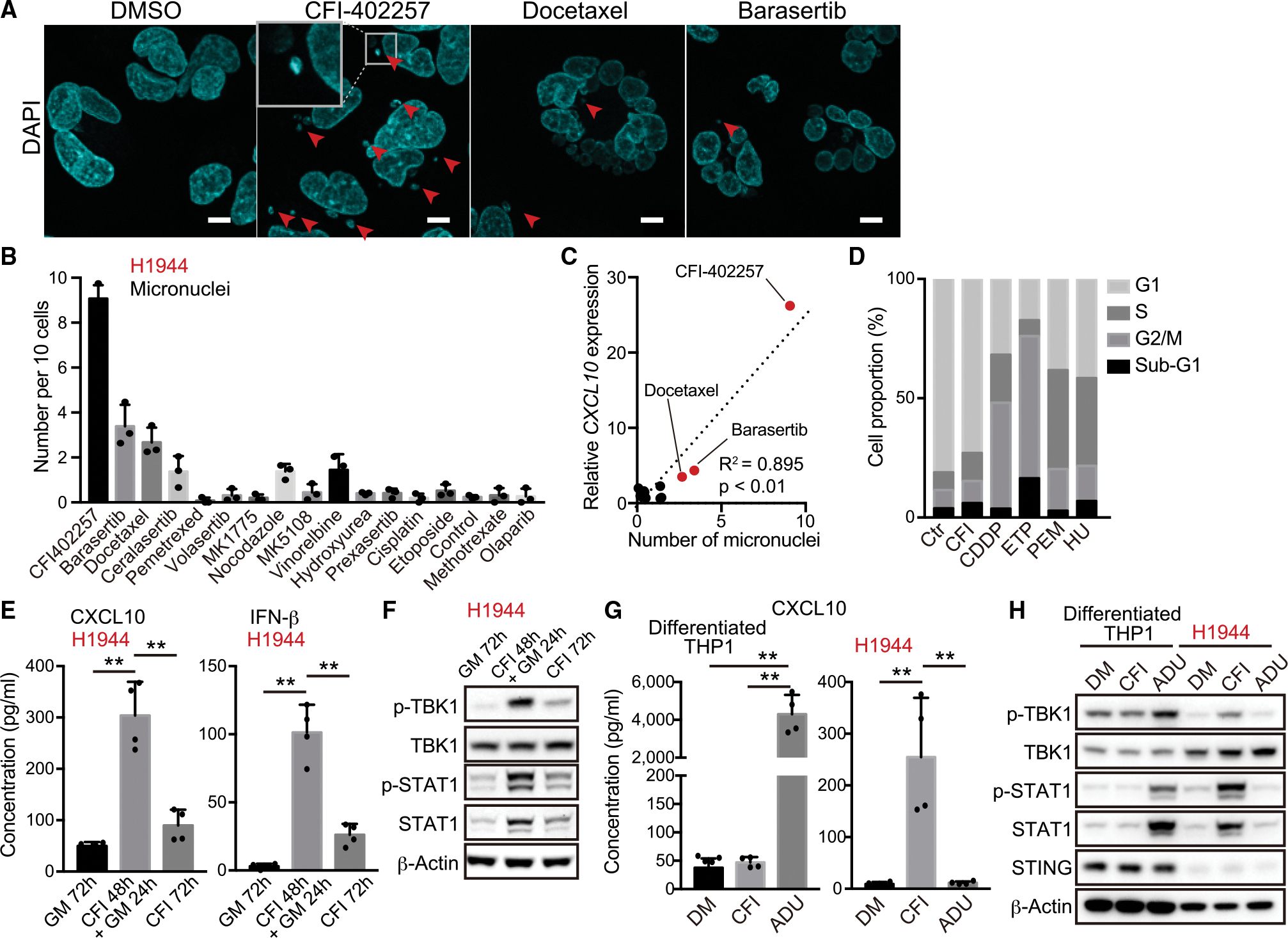
(A) Representative confocal microscope images of DAPI staining in H1944 cells treated with 200 nM CFI-402257, 5 nM docetaxel, or 200 nM barasertib. Arrows indicate micronuclei. Inset highlights micronucleus. Scale bars, 10 μm.
(B) Number of micronuclei in H1944 cells treated with the indicated DNA-damaging agents (n = 3).
(C) Relative messenger RNA (mRNA) expression of CXCL10 (y axis) versus number of micronucleus (x axis) in H1944 cells treated with the indicated DNA-damaging agents. R2 values and p values for the correlation (Pearson’s r correlation) are shown.
(D) Quantification of cell cycle analysis through propidium iodide staining for the cells after treatment with 200 nM CFI-402257 (CFI), 2.5 μM cisplatin (CDDP), 5 μM etoposide (ETP), 500 nM pemetrexed (PEM), or 50 μM hydroxyurea (HU) for 48 h.
(E and F) Enzyme-linked immunosorbent assay (ELISA) of human CXCL10 or IFN-β levels in CM (E), or immunoblot (IB) of the indicated proteins (F) in H1944 cells treated with 200 nM CFI-402257 (n = 4). GM, growth medium.
(G and H) ELISA of human CXCL10 in CM (G), or IB of the indicated proteins (H) in H1944 or THP1 cells treated with 200 nM CFI-402257, or 10 μM ADU-S100 for 24 h (n = 4). THP1 cells were differentiated to macrophages in the presence of 25 nM phorbol 12-myristate13-acetate (PMA) for 48 h.
All quantitative data are represented as mean ± standard deviation; p values were calculated by one-way analysis of variance followed by Tukey’s post hoc test (E and G), **p < 0.01. See also Figure S3.
Since treatment with docetaxel or barasertib at the IC50 used generated micronuclei and partially activated CXCL10 production in a cGAS-dependent manner (Figures S3C and S3D), we assessed whether lowering their concentrations could recapitulate the effects of CFI-402257 treatment after pulse treatment. However, CXCL10 induction was not upregulated at lower concentrations of these drugs and, in fact, tended to decrease in a dose-dependent manner (Figure S3E). These data are consistent with the concept that drugs that induce a potent mitotic arrest do not efficiently create micronuclei, since micronuclei arise when a broken chromosome improperly segregates during mitosis, with subsequent re-entry into G1 phase (Harding et al., 2017). Indeed, in contrast with other DNA-damaging agents such as cisplatin or etoposide, which strongly induced G2/M arrest, or pemetrexed or hydroxyurea which induced S-phase arrest, release from CFI-402257 treatment resulted in comparable cell cycle status with control cells, revealing the ability to progress past mitosis (Figure 3D). Also consistent with this idea, cGAS-STING-induced CXCL10 and IFN-β secretion and activation of STAT1 was substantially weaker during continuous exposure to CFI-402257 over 72 h, as compared with the pulse 48-h treatment and 24-h release (Figures 3E and 3F). Additionally, in WT-LKB1 reconstituted H1944 cells, which restores STING expression but also impairs cell growth owing to its tumor suppressive function, ADU-S100 sensitivity was enhanced while MPS1 inhibition (MPS1i) impact was dampened, and this required intact LKB1 kinase activity (Figures S3F–S3L).
Based on these observations, we hypothesized that MPS1 inhibition might also induce micronuclei more efficiently in proliferating cancer cells as compared with non-genomically altered, non-proliferative cells in the TME, such as immune cells and endothelial cells. Indeed, treatment with CFI-402257 did not significantly activate the STING pathway in terminally differentiated macrophage-like THP1 cells following phorbol 12-myristate13-acetate treatment, in contrast with their high sensitivity to 10 μM ADU-S100 treatment (Figures 3G and 3H). Conversely, treatment with CFI-402257 more efficiently activated STING signaling in H1944 cells compared with treatment with 10 μM ADU-S100, a concentration that had a negligible impact (Figures 1A, 3G, and 3H). Collectively, these data reveal a unique property of MPS1 inhibition in activating cGAS-STING signaling in proliferating KL cells as compared with other cells in the TME.
We also explored the differential impact of pulse MPS1i across a broader spectrum of lung cancer cell lines. We detected cGAS by immunofluorescence staining in multiple KL vs KP cell lines and successfully labeled micronuclei generated following pulse MPS1i treatment, revealing similar numbers regardless of KL or KP status (Figures S3–S3O). Importantly, in consonance with the preferential sensitivity of STINGLow KL cells to cGAS-STING activation, we only observed substantial CXCL10 induction in H1944 and H647 cells. We also examined the impact of pulsed MPS1i in KRAS WT cells with mutant or intact LKB1 status. While STING expression still tracked with LKB1, a strong sensitivity to exogenous 2′3′cGAMP or MPS1i was not observed (Figures S3P–S3R), potentially consistent with a recent report demonstrating a distinct immune biology of human KL tumors (Ricciuti et al., 2022). Finally, we tested the hypothesis that the defective autophagy present in KL cells could contribute to their particular sensitivity to micronuclei (Zhao et al., 2021) and found that ATG5 KO increased sensitivity of multiple KP lines to pulse MPS1i (Figures S3S and S3T). Thus, pulse MPS1 inhibition potently generates micronuclei, which preferentially activates cGAS-STING signaling in KL cells.
Combination treatment with MPS1 and epigenetic inhibitors drives potent STING activation in KL cells
Since KL cells epigenetically silence STING, we hypothesized that unleashing STING expression might robustly sensitize them to MPS1 inhibition, including cell lines with baseline STING absence. We first explored pretreatment with the EZH2 inhibitor GSK126, which is able to de-repress STING in STINGLow KL cells lines H1944 and H1355 cells (Kitajima et al., 2019) (Figures S4A and S4B). Pretreatment of H1944 or H1355 cells with GSK126 markedly enhanced CXCL10 and IFN-β secretion induced by pulse MPS1i treatment, which remained dependent on intact cGAS, STING, or STAT1 (Figures 4A and S4C). Furthermore, combination therapy with GSK126 followed by MPS1i treatment synergized to induce potent TBK1 and STAT1 activation, as well as PARP cleavage, which was similarly blocked by the deletion of cGAS, STING, or STAT1 (Figures 4B and S4D). GSK126 combination therapy not only upregulated STING levels, but also increased STING colocalization with the ERGIC, indicative of translocation to its active state where it complexes with TBK1 to induce downstream signaling (Figures 4C and 4D).
Figure 4. Combination treatment with MPS1 and epigenetic inhibitors cooperatively activate the STING pathway.
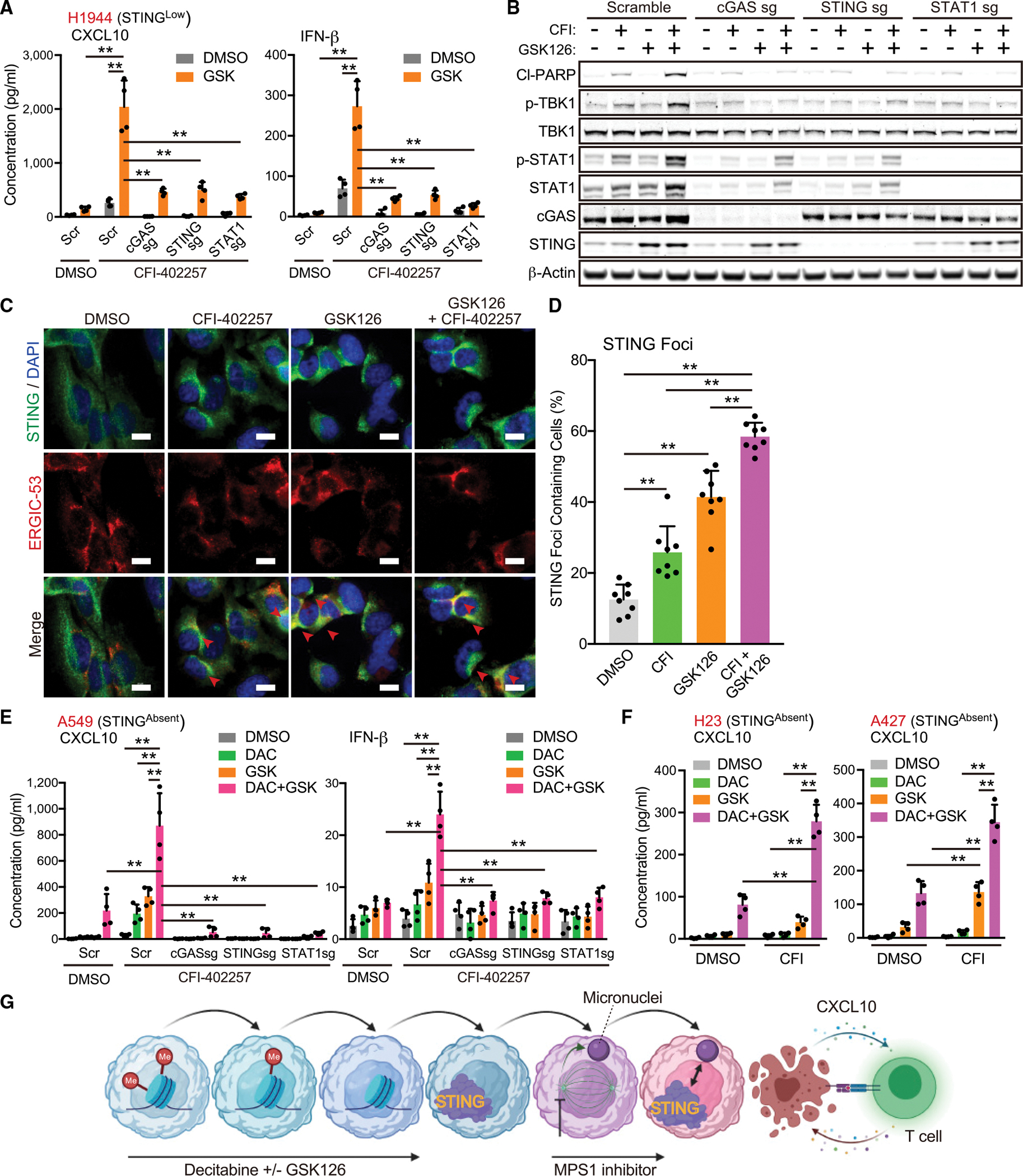
(A and B) Enzyme-linked immunosorbent assay (ELISA) of human CXCL10 or IFN-β levels in CM (A), or immunoblot (IB) of the indicated proteins (B) in H1944 transduced with the indicated vectors, and treated with the indicated drugs (5 μM GSK, and/or 200 nM CFI) in accordance with pretreatment schedule (Figure S4A) (n = 4).
(C and D) Fluorescent images (C) and quantification of STING foci containing cells (arrows) (D) in H1944 cells treated with the indicated drugs (5 μM GSK and/or 200 nM CFI) (n = 8). Scale bars, 10 μm.
(E and F) ELISA of human CXCL10 or IFN-β levels in CM derived from A549, H23, or A427 transduced with the indicated vectors, and treated with the indicated drugs (100 nM DAC, 5 μM GSK, and/or 200 nM CFI) (n = 4).
(G) Schematic of the concept of sequential combination therapy with epigenetic inhibitors and MPS1 inhibitor. Schematic is created with BioRender.com.
All quantitative data are represented as mean ± standard deviation; p values were calculated by one-way (D) followed by Tukey’s post hoc test, or two-way (A, E, and F) analysis of variance followed by Sidak’s post hoc test, **p < 0.01. See also Figure S4.
In contrast with STINGLow H1944 cells, STING downstream signaling was not upregulated after MPS1i treatment in STINGAbsent KL cells (Figure S3K). We previously reported that superimposed DNA hypermethylation of the promoter region of STING is a key epigenetic modification to abolish STING expression in STINGAbsent KL cells (Kitajima et al., 2019). We, therefore, examined the consequences of MPS1i following treatment of these cell lines with the DNMT inhibitor decitabine (DAC) alone or in combination with GSK126 to restore STING expression. Remarkably, A549 cells, which were almost completely resistant to CFI-402257 treatment alone, exhibited potent induction of CXCL10 and IFN-β secretion following DAC with or without GSK126 treatment (Figure 4E). We also confirmed that this effect was directly related to the restoration of cGAS-STING signaling, since deletion of cGAS, STING, or STAT1 completely suppressed secretion of CXCL10 and IFN-β induced by these treatments (Figure 4E). We further confirmed that DAC and GSK126 treatment primed response to MPS1i across multiple additional STINGAbsent KL cell lines (Figure 4F). Taken together, these data reveal that de-repressing STING by targeting its epigenetic silencing markedly enhances the impact of MPS1i in KL cells, co-opting their de-regulated cell cycle progression and potently restoring CXCL10 production and type I IFN signaling (Figure 4G).
MPS1 inhibition upregulates HLA expression in KL cells and enhances immune cell chemotaxis
We previously showed that the re-activation of cGAS-STING signaling and CXCL10 in KL cells promotes T cell extravasation from the vasculature and intra-tumoral T cell recruitment (Campisi et al., 2020; Kitajima et al., 2019). Furthermore, recent work has implicated the downregulation of MHC class I, also a STAT1 target, in KL cell immune evasion (Deng et al., 2021). To explore the potential impact of MPS1i-induced STING activation on antitumor immunity, we examined surface expression of MHC class I molecules HLA-A/B/C in H1944 cells. As compared with exogenous ADU-S100 treatment, intracellular 2′3′-cGAMP induction via pulse MPS1i significantly induced the expression of MHC class I (Figure 5A). We also confirmed STING and type l IFN dependence of this phenomenon, since STING or IFNAR1 depletion abrogated the impact of MPS1i on MHC class I induction (Figures 5A and S5A). KL cells are also known to maintain low levels of PD-L1 expression (Koyama et al., 2016; Skoulidis et al., 2018). Similarly, we observed that pulse MPS1i also induced PD-L1 expression on the cell surface, which was suppressed by STING depletion (Figure 5B). Next, we examined whether the restoration of STING expression in A549 STINGAbsent KL cells might also promote the MPS1 inhibitor-mediated induction of MHC class I. Consistent with its impact on STING upregulation (Figure S4B), we observed that pretreatment with DAC with or without GSK126 upregulated both MHC class I and PD-L1 expression in response to CFI-402257 treatment, with combined DAC and GSK126 pretreatment priming the most potent effect (Figures 5C, 5D, and S5B).Taken together, these data reveal that the MPS1 inhibitor-induced activation of STING can restore MHC class I expression, as well as PD-L1, on both STINGLow and STINGAbsent KL cells after epigenetic priming.
Figure 5. MPS1 inhibition upregulates HLAs expression and immune infiltration into peri-tumor region.
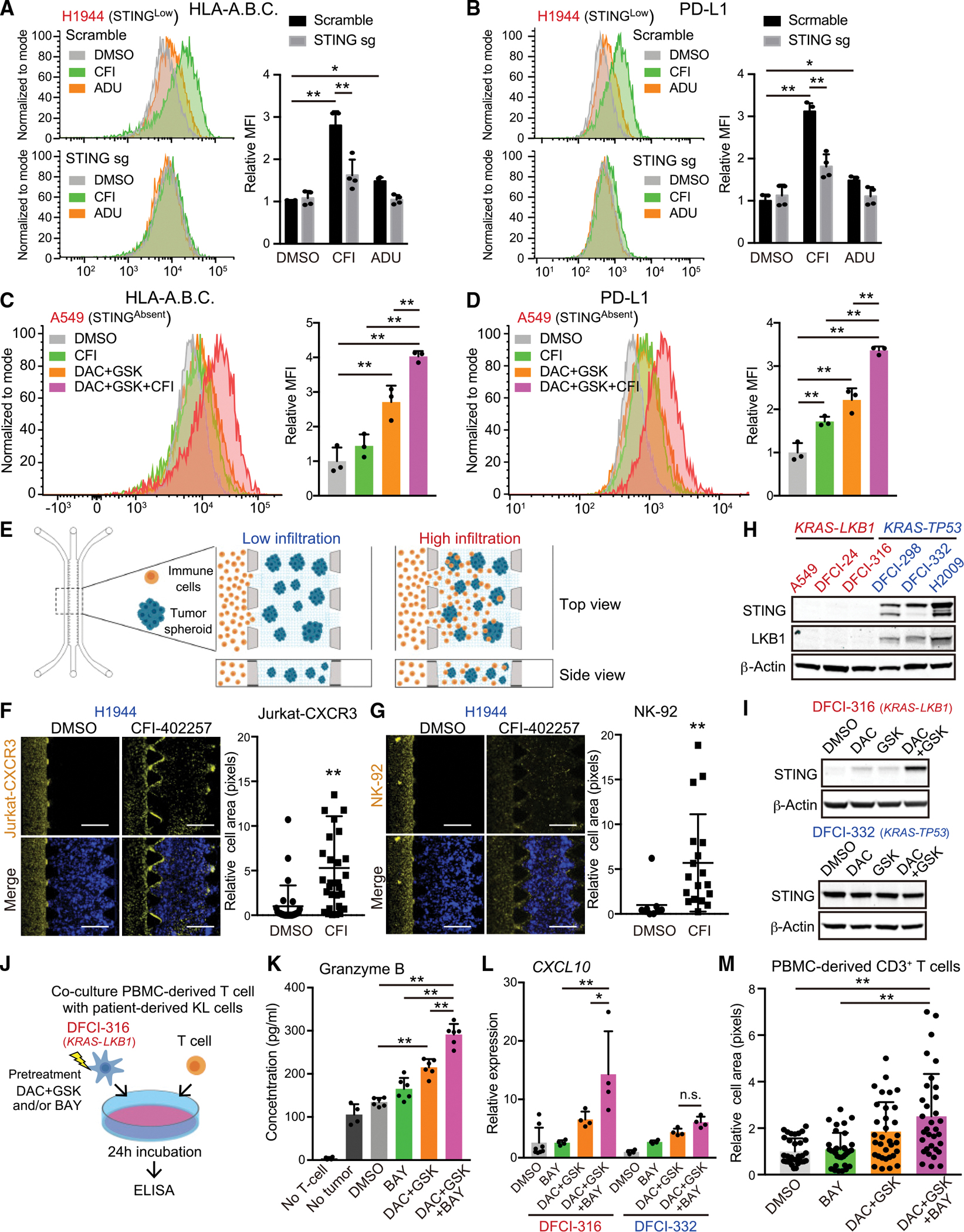
(A and B) HLA-A.B.C (A) or PD-L1 (B) expression on the cell surface in H1944 cells transduced with the indicated vectors, and treated with the indicated drugs (200 nM CFI, or 25 μM ADU) Data are representative of four independent experiments. Mean fluorescence intensity (MFI) was quantified by FlowJo (right) (n = 4).
(C and D) HLA-A.B.C (C) or PD-L1 (D) expression on the cell surface in A549 cells treated with the indicated drugs (100 nM DAC, 5 μM GSK, and/or 200 nM CFI). Data are representative of three independent experiments. Mean fluorescence intensity (MFI) was quantified by FlowJo (right) (n = 3).
(E) Schematic of immune cell migration assay using a 3D microfiuidic device with tumor spheroids embedded in a central collagen-filled channel and with immune cells co-cultured in a side channel.
(F and G) Representative images of Jurkat-CXCR3 (F) or NK-92 (G) cells migration. Immune cells infiltration into peri-tumor region is quantified by ImageJ (n = 18). Values were normalized to DMSO control. Scale bars, 500 μm.
(H and I) IB of the indicated proteins in patient-derived KL or KP cells (H), and DFCI-316 or DFCI-332 cells treated with 100nM DAC, 5 μM GSK126, and/or 100nM BAY-1217389 in accordance with pretreatment schedule as shown in Figure S4A (I).
(J) Schematic of co-culture PBMC-derived T-cells with patient-derived KL cells pretreated with 100 nM DAC, 5 μM GSK126, and/or 100 nM BAY-1217389.
(K) Enzyme-linked immunosorbent assay (ELISA) of human granzyme B in CM derived from DFCI-316 cells co-cultured with PBMC-derived T-cells (n = 6).
(L and M) ELISA of human CXCL10 in CM derived from DFCI-316 or DFCI-332 cells treated with 100 nM DAC, 5 μM GSK126, and/or 100 nM BAY-1217389 (L) (n = 4), and the ratio of infiltration of PBMC-derived T-cells into peri-tumor region using immune cell migration assay (see STAR Methods) (M) (n = 33).
All quantitative data are represented as mean ± standard deviation; p values were calculated by the unpaired two-tailed Student t test (F and G), or one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test (C, D, K, and M) or two-way ANOVA followed by Sidak’s post hoc test (A, B, and L), *p < 0.05, **p < 0.01. See also Figure S5.
We next investigated immune cell chemotaxis following MPS1 inhibition using a previously described three-dimensional (3D) microfluidic system (Figure 5E) (Kitajima et al., 2019; Ritter et al., 2020). We treated H1944 KL cells with CFI-402257 or DMSO control in two-dimensional culture, formed tumor spheroids using an ultra-low attachment plate, and then embedded them in collagen into the central channel of a 3D microfluidic device. As expected, CXCR3-reconstituted Jurkat T cell (Figure S5C) migration was only observed in the presence of H1944 KL cells that were pretreated with CFI-402257 (Figure 5F), consistent with their enhanced CXCL10 production (Figure 2D). In addition, we also used NK-92 cells, which endogenously express CXCR3 (Figure S5D), for this immune cell migration assay. Similarly, we observed that treatment with CFI-402257 also accelerated the migration of NK-92 cells from the side channel toward tumor cell spheroids (Figure 5G). In addition, consistent with restoration of STING expression and subsequent activation of STING downstream, we observed that pretreatment with DAC with or without GSK126 enhanced the migration of both Jurkat-CXCR3 and NK-92 cells induced by MPS1i treatment in A549 STINGAbsent KL cells (Figures S5E and S5F).
Next, we examined whether MPS1i treatment upregulates antigen presentation and immune cell recruitment in models more closely obtained from patients. We examined multiple Dana-Farber patient-derived xenograft (PDX) cell lines and confirmed low to absent STING expression in KL cells similar to A549 STINGAbsent KL cells, which was restored with DAC plus GSK126 treatment (Figures 5H and 5I). Conversely, patient-derived KP cells retained STING expression, and the treatment of epigenetic inhibitors did not upregulate STING expression (Figures 5H and 5I). Indeed, the pretreatment of patient-derived KL cells with epigenetic and pulse MPS inhibition, and then co-culture with allogeneic T cells derived from peripheral blood mononuclear cells (PBMCs), reveals significantly enhanced granzyme B induction particularly, revealing increased immunogenicity (Figures 5J and 5K). Moreover, this same pretreatment promoted CXCL10 secretion and migration of PBMC-derived CD3+ T cells into PDX KL spheroids in 3D culture (Figures 5L and 5M). Taken together, these results reveal that epigenetic and pulse MPS1 inhibitor treatment in KL cells can improve antigen presentation and T cell and natural killer (NK) cell recruitment.
Sequential DNMT/MPS1 inhibitor pulse treatment induces durable therapeutic efficacy in a syngeneic murine KL model in vivo
Since re-activation of STING signaling by MPS inhibition in KL cells might thus promote antitumor immunity as well as intrinsic cell death, we next established a syngeneic murine KL model to examine therapeutic consequences in vivo. As the process of immune editing in naturally arising human lung cancers versus engineered mouse tumors is vastly different, we first characterized cell lines derived from multiple murine Kras mutant lung cancer models (CMT167, 393P, Lacun3, and LLC1) to select a model that most closely recapitulates the biological features of human KL lung cancers. Among these cell lines, and in contrast to those derived from poorly immunogenic genetically engineered mouse models (Koyama et al., 2016), we found that 393P cells naturally upregulate DNMT1 to suppress STING and that LKB1 inactivation further abrogated Sting expression in this model, inverting cytokine secretion to promote IL-6 release (Figures 6A–6C and S6A). Indeed, the pretreatment of 393P-KL cells with DAC not only restored Sting protein levels, but also uniquely promoted high CXCL10 expression and MPS1i induced STING signaling (Figures 6D, 6E, S6B, and S6C). Furthermore, whereas parental 393P-K cells were immunogenic and responsive to PD-1 blockade, 393P-KL cells exhibited relative anti-PD-1 resistance (Figure S6D), and increased infiltration of CD11b+ Ly-6G+ granulocytes in the TME, all consistent with their baseline IL-6 upregulation and modeling of in vivo KL immunobiology (Figure S6E) (Koyama et al., 2016; Skoulidis et al., 2018).
Figure 6. Sequential combination therapy with MPS1 and DNMT inhibitor enhances intra-tumoral T cell infiltration in syngeneic murine KL model.
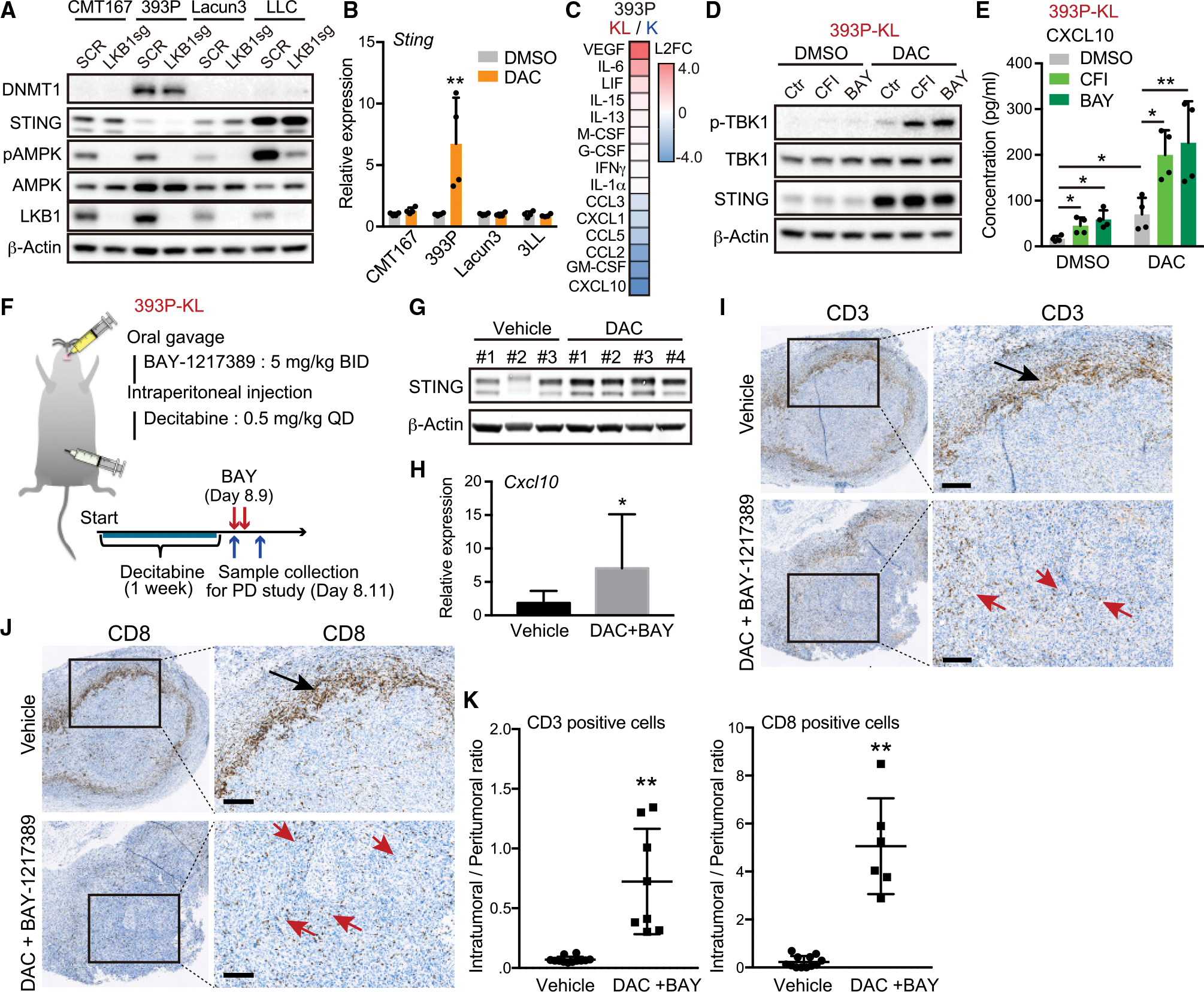
(A) Immunoblot (IB) of the indicated proteins in murine lung cancer cells transduced with the indicated vectors.
(B) Quantitative RT-PCR of Sting in murine lung cancer cells treated with 100 nM DAC for 5 days (n = 4).
(C) Heatmap of cytokine profiles in CM derived from 393P-K or 393P-KL cells. Scores = log2 fold change (393P-KL/393P-K). Cytokines indicating log2 fold change (L2FC) > 0.2 or L2FC < −0.2 are shown in the heatmap.
(D and E) IB of the indicated proteins (D), or Enzyme-linked immunosorbent assay (ELISA) of mouse CXCL10 levels in CM derived from 393P-KL cells (E) treated with the indicated drugs (100 nM DAC, and/or 200 nM CFI or 100 nM BAY) in accordance with pretreatment schedule (n = 4).
(F) Schematic of pharmacodynamics study with MPS1 and DNMT inhibitor in syngeneic murine KL model.
(G and H) IB of the indicated proteins (G), or quantitative RT-PCR of Cxcl10 (H) in tumor tissues derived from mice treated with the indicated drugs (each group, n = 4).
(I–K) Representative CD3 (I) or CD8 (J) IHC images and quantitative analysis (K) from 393P-KL tumors treated with vehicle or combination of DAC and BAY-1217389. Arrows highlight peri-tumoral localization (black) and intra-tumoral localization (red) of CD3+ or CD8+ T cells. QuPath (see STAR Methods) was used to quantify CD3+ or CD8+ T cell infiltration (n = 6). Scale bar, 200 μM.
All quantitative data are represented as mean ± standard deviation; p values were calculated by an unpaired two-tailed Student t test (B, H, and K), or two-way analysis of variance followed by Sidak’s post hoc test (E). *p < 0.05, **p < 0.01. See also Figure S6.
Next, using this syngeneic 393P-KL model, we examined a pulse schedule of combination therapy with DAC and MPS1 inhibition, using the clinically relevant compound BAY-1217389 because of its well-established in vivo dosing (Maia et al., 2018; Wengner et al., 2016). Initially, in a pharmacodynamic study, 393P-KL tumors in the syngeneic 129S2/SvPasCrl mice were treated daily with 0.5 mg/kg DAC for 7 days and tumors were harvested 24 h after the last dose to check for restoration of STING in vivo (Figure 6F). As observed in our in vitro study, we confirmed by immunoblot that STING expression was increased after DAC treatment versus vehicle control (Figure 6G). Next, we assessed the impact of subsequent pulse treatment with twice daily 5 mg/kg BAY-1217389 for 2 days. DAC treatment followed by BAY-1217389 significantly increased Cxcl10 expression by mRNA in bulk tumors (Figures 6F and 6H). Examination of vehicle- or combination-treated tumors by IHC for total CD3+ and CD8+ T cells revealed pronounced T cell exclusion of control tumors, in further agreement with their STING silencing and the phenotype of KL human tumors (Figures 6I–6K and S6F–S6H) (Kitajima et al., 2019; Skoulidis et al., 2018). As expected, the restoration of STING and CXCL10 expression after DAC/MPS1i treatment promoted redistribution of CD3+ and specifically CD8+ T cells from the tumor periphery to the tumor interior, suggesting that this combination could potentially restore tumor immunogenicity in vivo (Figures 6I–6K and S6F–S6H).
To test this, we performed an efficacy study using the same short-term pulse regimen of the sequential therapy with DAC/MPS1i in 393P-KL syngeneic model. This treatment induced potent inhibition of tumor growth (Figures 7A and 7B). To directly assess the role of CD8+ T cell redistribution in mediating this therapeutic effect, we tested the effects of CD8+ depletion using a CD8-depleting antibody (Figure 7A). Consistent with a key role for CD8+ T cell immunity, the impact of sequential therapy of DAC and BAY-1217389 was significantly attenuated by CD8+ T cell depletion (Figures 7B, S7A, and S7B). Additionally, to further assess the role of an intact immune system, we used immune-deficient NSG mice. The 393P-KL tumors grew robustly in NSG mice and sequential treatment with DAC + MPS1i resulted in only partial tumor growth inhibition with aggressive rebound within 8 days after stopping treatment (Figure S7C). Next, to directly investigate the dependency of this response on activation of tumor cell STING, we established STING-depleted 393P-KL cells and then evaluated the efficacy of pulse combination therapy (Figure S7D). As expected, STING-depleted 393P-KL showed impaired CXCL10 response after MPS1i treatment in vitro (Figure S7E), which translated to inactivity of DAC/MPS1i treatment in vivo (Figure 7C).
Figure 7. Sequential combination therapy shows durable therapeutic effect in syngeneic murine KL model.
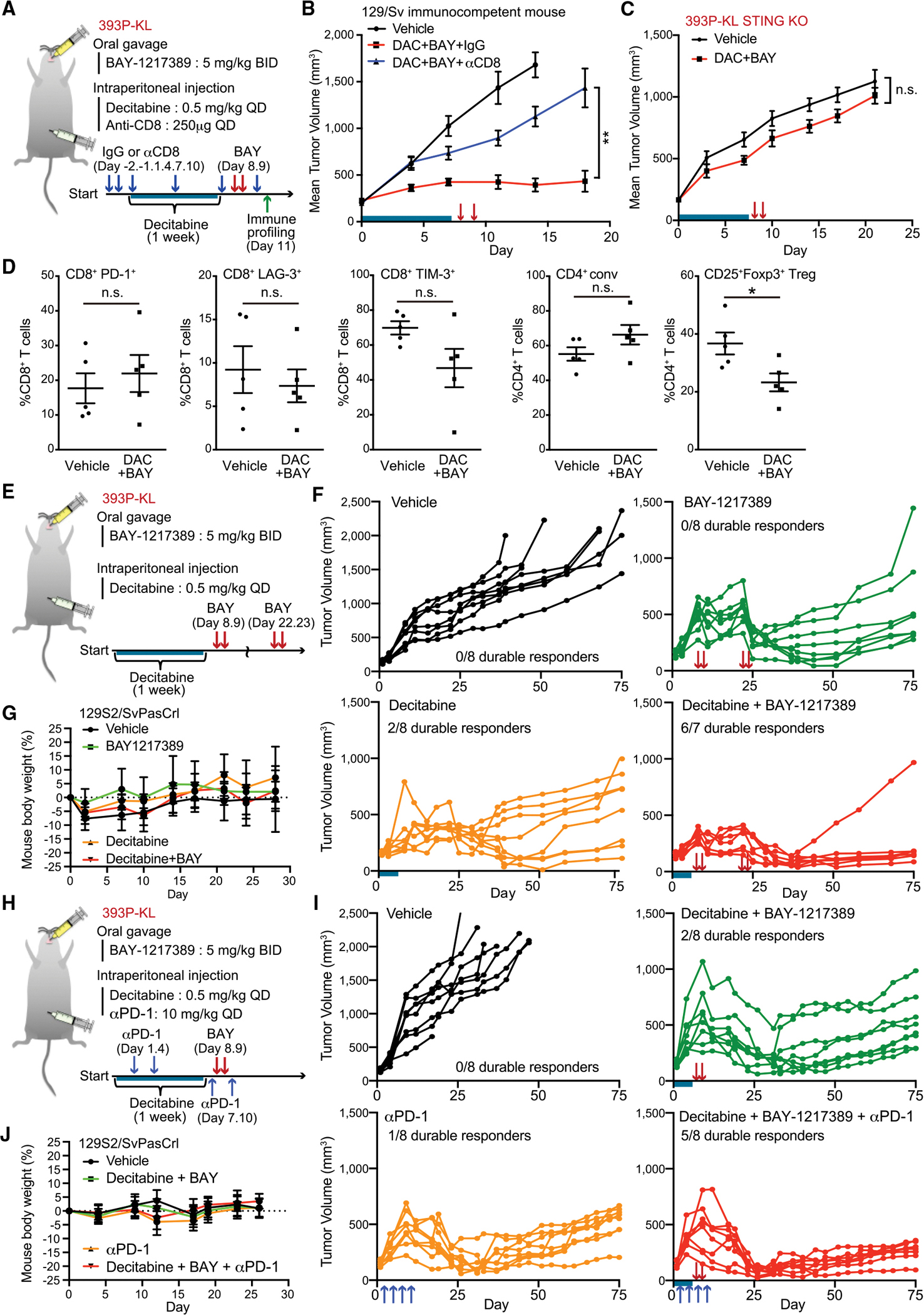
(A) Schematic of short-term efficacy study, CD8+ T cell depletion study, and immune profiling with MPS1 and DNMT inhibitor in syngeneic murine KL model.
(B) Mean tumor volume of 393P-KL cells after subcutaneous inoculation into syngeneic 129S2/SvPasCrl mice treated with anti-CD8 neutralization antibody. Mice were treated with anti-CD8 antibody, and/or DAC and BAY-1217389 in accordance with the schedule shown in Figure 7A (n = 8). Blue bar, DAC treatment; red arrows, BAY-1217389 treatment.
(C) Mean tumor volume of STING KO 393P-KL cells after subcutaneous inoculation into syngeneic 129S2/SvPasCrl mice. Mice were treated with DAC from day 1 to day 7 and BAY-1217389 on day 8, 9 (n = 8). Blue bar; DAC treatment. Red arrows, BAY-1217389 treatment.
(D) Flow cytometric analysis of immune cell populations in tumor tissue treated with or without DAC and BAY-1217389 (n = 5). Tumor tissue were collected and analyzed after 48 h from second BAY-1217389 treatment. n.s., not significant.
(E and H) Schematic of long-term efficacy study with MPS1 inhibitor, DNMT inhibitor and/or anti-PD1 antibody in syngeneic murine KL model.
(F and G) Tumor volume of 393P-KL cells (F) and mouse body weight (G) after subcutaneous inoculation into syngeneic 129S2/SvPasCrl mice followed by BAY-1217389 on day 8, 9, 21, and 22 (as shown in red arrows) and/or DAC from day1 to day7 (as shown in blue bar) (n = 8).
(I and J) Tumor volume of 393P-KL cells (I) and mouse body weight (J) after subcutaneous inoculation into syngeneic 129S2/SvPasCrl mice followed by BAY-1217389 on days 8 and 9 (as shown in red arrows) DAC from day 1 to day 7 (as shown in blue bar), and/or anti-PD1 antibody on day 1, 4, 7, and 10 (as shown in blue arrows) (n = 8).
Quantitative data are represented as mean ± SD (D, G, and J) or ±SEM (B and C). p values were calculated by two-way analysis of variance followed by Sidak’s post hoc test (B and C), an unpaired two-tailed Student’s t test (D), or the χ2 test (F and I). *p < 0.05, **p < 0.01. See also Figure S7.
To profile the immune response that develops following sequential DAC + MPS1i combination therapy in greater depth, we performed comprehensive immune profiling by flow cytometry 48 h after treatment (Figure 7A). At this early timepoint, we did not observe a significant change in absolute T, NK, or myeloid cell numbers (Figure 7F). An assessment of CD8+ T cell activation and exhaustion markers such as LAG-3, TIM-3, and PD-1 were not significantly impacted; however, we observed an alteration in the CD4+ T cell subsets with significant depletion of CD25+ Foxp3+ regulatory T cells (Tregs) (Figure 7D). These results are consistent with prior observations that type I interferon signaling can impair Tregs (Gangaplara et al., 2018), revealing that the restoration of this tumor cell STING-IFN-CXCL10 axis by this sequential combination therapy promotes both Treg depletion and CD8+ T cell infiltration in vivo.
Next, we assessed two different longer term efficacy therapeutic strategies to explore the impact on durable response in these models. We examined the impact of adding either a second MPS1i pulse after 2 weeks or a combination of the single pulse with PD-1 blockade. Following just a second MPS1i pulse, we observed durable long-term response in six of seven mice after 12 weeks with the combination therapy, in contrast with two of eight mice treated with DAC alone and 0 mice treated with BAY-1217389 or vehicle alone (p < 0.01, χ2 test) (Figures 7E and 7F). Of note, treatment of immunocompetent mice with this pulse combination therapy schedule was also very well tolerated, without any evidence of distress or body weight loss (Figure 7G). Finally, we also observed that pulse combination therapy with DAC and BAY-1217389 together with anti-PD1 treatment was also tolerable and resulted in increased durable long-term responses and significant tumor growth suppression as compared with either arm alone (p < 0.05, χ2 test) (Figures 7H–7J). In sum, these data reveal that sequential combination therapy with DAC and pulse BAY-1217389 treatment can restore durable response in Sting-silenced KL tumors and also improve sensitivity to PD-1 blockade.
DISCUSSION
LKB1 mutation is associated with an intrinsic resistance to ICB in KRAS-mutant NSCLC (Koyama et al., 2016; Rizvi et al., 2018; Skoulidis et al., 2018); therefore, novel therapeutic approaches are needed to enhance immunogenicity. We previously reported that the adaptor protein STING, which links cytoplasmic dsDNA sensing by cGAS to activation of downstream innate immune signaling, is epigenetically silenced in KL cells (Kitajima et al., 2019). These results implied that the therapeutic restoration and activation of the STING pathway could represent a targeted approach to enhance immunogenicity in KL cells. Here, using an unbiased screen of cytotoxic chemotherapies or targeted DNA-damaging agents in clinical trials, we have identified MPS1, a master regulator of the SAC, as a highly robust target to activate cGAS-STING signaling in KL cells. Mechanistically, STING agonism induced by MPS1 inhibition is related to the ability to proceed through an abnormal mitosis and generate micronuclei, which are known potent activators of cGAS (Harding et al., 2017; Mackenzie et al., 2017; Mohr et al., 2021). Pulse treatment with MPS1 inhibitors after epigenetic inhibitors such as DAC and/or GSK126 dramatically enhances STING pathway activation, leading to an increased secretion of effector cytokines and chemokines, such as CXCL10 and IFN-β, expression of MHC class I molecules, and direct STAT1-dependent cell death. Consistent with these results in vitro, combination therapy in a murine syngeneic KL model with the clinically relevant MPS1 inhibitor BAY-1217389 and DAC strongly activated cancer cell-intrinsic STING and enhanced T cell recruitment in vivo, achieving prolonged preclinical activity over 12 weeks despite using a limited pulse treatment schedule.
Recently, a wide variety of synthetic STING agonists have been developed including cyclic dinucleotide (CDN) analogues based on the 2′3′-cGAMP structure as well as non-CDN molecules (Chin et al., 2020; Kwon and Bakhoum, 2020; Pan et al., 2020). In general, synthetic STING agonists target the STING pathway most potently in surrounding non-malignant cells, especially in myeloid cells to boost antitumor immunity via an enhanced cross-presentation of neoantigen and recruitment of cytotoxic T cells (Amouzegar et al., 2021). In contrast, it is becoming increasingly clear that activity of the cancer cell-intrinsic STING pathway defines their immunogenicity and influences the efficacy of ICB. Indeed, beyond the KL cell state, it has been reported that loss of cGAS and/or STING is frequently observed in several types of cancer cells, including melanoma and colorectal cancer, leading to the escape from cancer immunosurveillance (Konno et al., 2018; Luo et al., 2020; Xia et al., 2016a, 2016b). These observations suggest that the re-activation of STING signaling in STING-silenced cancers could be a direct approach to recover their immunogenicity and sensitize to ICB therapy. However, since synthetic STING agonists in general penetrate the cancer cell membrane poorly, we took an alternate approach to increase the production of KL cancer cell intrinsic 2′3′-cGAMP by stimulation of cGAS.
Micronuclei are discrete DNA aggregates separated from the primary nucleus and recognized by cGAS as abnormal cytoplasmic DNA (Zierhut and Funabiki, 2020). Since micronuclei are formed after continuous mitotic progression along with DNA damage (Harding et al., 2017), drugs accelerating the formation of micronuclei have the potential to stimulate the STING pathway specifically in rapidly proliferating cancer cells. MPS1 is an essential SAC kinase that maintains the fidelity of chromosome segregation; it is critical for the recruitment of SAC proteins to unattached kinetochores and regulation of spindle fidelity upstream of the RZZ complex (Maciejowski et al., 2017). Since most cancer cells show rapid proliferation and chromosomal instability, they depend on the SAC to properly segregate their abnormal genome during mitosis. Thus, the abrogation of the SAC by continuous MPS1 inhibition results in intolerable levels of genomic instability and cell death. Here, we have demonstrated that transient treatment with an MPS1 inhibitor robustly generates micronuclei via chromosome missegregation in KL cells. In contrast with PARP inhibition, which is effective at activating STING in the context of synthetic lethal BRCA1/2 mutation (Ding et al., 2018; Pantelidou et al., 2019; Reislander et al., 2019), transient MPS1 inhibition may potentially engage the STING pathway in a variety of rapidly proliferating cancer cells that generate micronuclei and are capable of surviving long enough to promote an immunogenic TME. Importantly, continuous MPS1i, which favors mitotic arrest at the SAC, impaired cell proliferation and partially engaged cGAS-STING, but was not nearly as potent at activating this pathway as loading cells with micronuclei after pulse therapy. Indeed, transient MPS1 inhibition in an immunocompetent mouse model after DAC induction therapy promoted remarkable tumor shrinkage and durable response compared with prior studies using tumor cell xenografts in immunodeficient mice (Maia et al., 2018; Wengner et al., 2016). This was associated with reversal of the T cell excluded phenotype, depletion of Tregs, and dependence on CD8+ T cells, defining restoration of antitumor immunity as a key mediator of therapeutic activity.
Recently, the epigenetic regulation of innate immune signaling has become a major focus to promote cancer cell immunogenicity, as it represents a potentially reversible mechanism used by tumors to evade from immunosurveillance and ICB-mediated T cell killing (Topper et al., 2020). As observed with STING suppression in KL cells, genes regulating tumor cell recognition and type I interferon responses including TAP1 and MHC class I, endogenous retroviruses, and interferon-responsive genes more generally can be silenced by epigenetic mechanisms via DNA and histone lysine methylation (Canadas et al., 2018; Loo Yau et al., 2021; Mahadevan et al., 2021; Morel et al., 2021). Since cell division is required for the removal of functional epigenetic marks, DNA- or histone-demethylating agents might efficiently convert cell state from immunosuppressive to active in rapidly proliferating cancer cells compared with surrounding non-malignant cells, similar to the requirement of mitotic progression for micronuclei generation by MPS1 inhibition. Therefore, sequential epigenetic priming and pulse MPS1 inhibition not only selectively targets anticancer immunity, but also minimizes the toxicity of inhibiting each target since drugs are not given simultaneously. Furthermore, systemic administration of this regimen impacts all tumor sites, in contrast with the limitations of injectable STING agonists or the potentially narrow therapeutic window of systemic STING agonists.
Important limitations of our study include the lack of identification of specific immunogenic antigens responsible for T cell immunity in the mouse model, and the fact that combination therapy in patient KL tumors would still rely on effective priming and presentation of endogenous human tumor antigens. Additionally, since both EZH2 and DNMT1 can control STING silencing, identifying specific biomarkers to direct the need to prime with either a single agent or combination of inhibitors will be necessary. Since DNMT1 upregulation is associated with near complete silencing, developing immunohistochemistry assays to identify DNMThi/STINGabsent KL tumors would help to direct use of DAC or other DNA demethylating agents, whereas DNMTlow/STINGlow tumors may be vulnerable to EZH2 inhibition alone. Furthermore, while KL tumors are clearly enriched for this biology, they are unlikely to represent the only NSCLC genotype associated with STING silencing, and further work is necessary to examine roles for additional pathways such as KEAP-NRF2, for example (Olagnier et al., 2018). The potential role of autophagy in these phenotypes warrants further exploration, and recent work also suggests that mutant p53 may actively interfere with STING-TBK1 signaling, likely also contributing to KP cell resistance (Ghosh et al., 2021). While our preliminary findings that KRAS WT LKB1 mutated tumors are less reliant on this biology is also in agreement with recent findings from the clinic (Ricciuti et al., 2022), additional work and biomarkers are needed to refine this observation, especially since RAS signaling can be activated indirectly in many tumors.
Finally, although mouse tumor models often fail to predict durable response in humans, the ability of KL cancers to mask tumor antigens and likely avoid immune editing during tumor development may create a unique immune vulnerability. Tumor cell STING activation can also prime responses to T and NK cell therapy (Ji et al., 2021; Knelson et al., 2022; Xu et al., 2021), and export of 2′–3′ cGAMP can also prime vascular activation for immune cell extravasation (Campisi et al., 2020). Thus, immunogenic priming of KL tumors by epigenetic therapy and MPS1 inhibition could also serve to facilitate tumor infiltration by engineered T cell and/or NK cell therapies. Translating this regimen into the clinic, restoring exposure of KL tumor antigens and promoting effector cell recruitment, may have substantial potential to regenerate effective antitumor immunity for patients with treatment refractory KL tumors.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
For further information and requests for reagents generated in this study should be directed to and will be fulfilled by the lead contact, David A. Barbie (David_Barbie@DFCI.HARVARD.EDU).
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
A549, H2009, HEK293T, LLC, and CMT-167 cells were cultured in DMEM (Thermo Fisher Scientific, Cat.# 11965–118) supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-products, Cat.# 100–106), 1x penicillin-streptomycin (Gemini Bio-products, Cat# 400–109), and 2.5 μg/mL plasmocin prophylactic (Invivogen, Cat.# ant-mpp). H1944, H23, H1355, H647, H2122, A427, H1792, H441, H358, HCC44, THP-1, Jurkat, 393P, and Lacun3 cells were cultured in RPMI 1640 (Thermo Fisher Scientific, Cat.# 11875–119) supplemented with 10% FBS, 1x penicillin-streptomycin, and 2.5 μg/mL plasmocin prophylactic (Invivogen, Cat.# ant-mpp). NK-92 cells were cultured in αMEM supplemented with 0.2 mM inositol, 0.1 mM 2-mercaptoethanol, 0.02 mM folic acid, 200 U/mL recombinant IL-2, 12.5% FBS, 12.5% horse serum, and 1x penicillin-streptomycin. HUVEC cells were cultured in vascular medium (VascuLife®VEGF Endothelial Medium Complete Kit, #LL-0003). DFCI-24, DFCI-298, DFCI-316, and DFCI-332 were established as described before (DF/HCC IRB protocol 02–180) (Köhler et al., 2021). DFCI-316 cells were grown in RPMI 1640 supplemented with 10% FBS, and 1x penicillin-streptomycin. DFCI-24, DFCI-298, and DFCI-332 were grown in ACL4 media supplemented with 10% FBS, and 1x penicillin-streptomycin.CD3+ T cells were isolated from PBMCs (STEMCELL, Cat.# 70025) using EasySep™ Human T Cell Isolation Kit (STEMCELL, Cat.# 70025) according to the manufacturer’s instructions and cultured in RPMI 1640 supplemented with 10% human serum (Sigma-Aldrich, Cat.#H5667), 1x penicillin-streptomycin, 2 mM of L-Glutamine, and 100 IU/mL of IL-2, 25 ng/mL of IL-7, and 25 ng/mL of IL-15. The T cells were activated with 1% T cell Trans-Act (Miltenyi Biotec, Cat.# 130–128-758), immediately after the isolation. A549, A427, H1944, H23, H1355, H2122, H1792 and H2009 cells were originally obtained from the Broad Institute and authenticated by short tandem repeat (STR) genotyping. HEK293T, H657, H441, H358, HCC44, THP-1, Jurkat, NK-92, and LLC were purchased from ATCC. CMT-167 cells were purchased from ECACC. HUVEC cells were purchased from Lonza (Lonza, C2519A). 393P cells were established from KrasLA1/+;p53R172HΔG mice and kindly gifted from Dr. J.M. Kurie (The University of Texas, MD Anderson Cancer Center, Houston, TX). Lacun3 cells were established from a chemically induced lung adenocarcinoma and kindly gifted from Dr. L.M. Montuenga (The University of Navarra, CIMA, Spain). All experiments were performed before reaching 10 passages from the original frozen stocks. Mycoplasma infection was regularly checked by MycoAlert™ Mycoplasma Detection Kit (Lonza, Cat.# LT07–218) according to the manufacturer’s instructions.
Animal study
All mouse experiments were conducted according to a Dana-Farber Cancer Institute approved protocol. Five million cells (393P-KL, 393P-STING KO) in PBS with 30% Matrigel (Corning, Cat.#356231, NY) were subcutaneously injected into the flank of 8-week-old female 129-Elite mice (129S2/SvPasCrl, Strain code 476, Charles River Laboratories) or NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) female mice (The Jackson Laboratory, ME). Tumor volume was determined from caliper measurements of tumor length (L) and width (W) according to the formula (L × W2)/2. Animals were randomized (Studylog software, CA) into various treatment groups once tumor volumes were in the range of 110–190 mm3 for efficacy studies and in the range of 280–410 mm3 for the PD study before treatment initiation. The durable response was defined as mice with tumor volume less than 250 mm3 for at least 50 days after treatment was completed. Both tumor size and body weight were measured twice per week. BAY-1217389 was formulated in 50% PEG 400, 10% ethanol and 40% water and dosed at 5 mg/kg twice daily by oral gavage. Decitabine (purchased from DFCI pharmacy) was dissolved in saline and dosed at 0.5 mg/kg once per day by intraperitoneal injection. For CD8 depletion study, tumor bearing mice were injected intraperitoneally with CD8+ T cell depleting antibody (Clone 53–6.7 from BioXcell, NH) diluted in PBS at a concentration of 250 μg/mouse. In satellite animals, spleen and tumor tissue was isolated 24 h after the last treatment and subjected to FACS analysis.
METHOD DETAILS
ELISA
Human IFN-β (Thermo Fisher Scientific, Cat.# 414101), human CXCL10 (R&D systems, Cat.# DIP100), mouse CXCL10 (R&D systems, Cat.# DY466), and 2′3′-cGAMP (Cayman Chemical, Cat.# 501700) ELISAs were performed according to the manufacturer’s instructions. Conditioned media from each cell lines was collected after 24 h culture. Values represent the average of four replicates from at least two independent experiments (biological replicates).
Generation of lentivirus
3 × 106 HEK293T cells were plated onto a 60-mm dish and transfected using X-tremeGENE HP DNA Transfection Reagent (Roche, Cat.# 06366236001) with 1 μg of lentivirus-based expression vectors together with 1 μg of pCMV-dR8.91 and 1 μg of pCMV-VSV-G. After 48 h incubation, the media containing lentivirus particles were collected, passed through a 0.45 μm filter, and concentrated using Lenti-X Concentrator (Clontech, Cat.# 631231). For selection of virally infected cells, 1–2 μg/mL of puromycin (pCRISPR-v2 sgRNAs, plx307-hCXCR3) or 1.5–8 μg/mL of blasticidin (plx304-NanoLuc, plx304-hLKB1, plx304-STING, plx304-cGAS) was used 24 h post-infection.
Immunoblotting
Cells were lysed in RIPA buffer containing 1x protease inhibitors (Roche, Cat# 11-836-145-001) and phosphatase inhibitors (50 mmol/L NaF and 100 mmol/L Na3VO4). Immunoblotting was performed as described (Kitajima et al., 2018) using following antibodies to: cGAS (#15102, Cell Signaling Technology), STING (#13647, Cell Signaling Technology), STING (Rodent preferred) (#50494, Cell Signaling Technology), phospho-STAT1 (#9167, Cell Signaling Technology), STAT1 (#9172, Cell Signaling Technology), LKB1 (#3047, Cell Signaling Technology), cleaved PARP (#5625, Cell Signaling Technology), IFNAR1 (A304-290A, Thermo Fisher), phospho-Histone H2A.X (#9718, Cell Signaling Technology), Histone H3 (#4499, Cell Signaling Technology), phospho-TBK1 (#5483, Cell Signaling Technology), TBK1 (#3013, Cell Signaling Technology), DNMT1 (#5032, Cell Signaling Technology), PCNA (#M0879, DAKO), Aurora B (#3094, Cell Signaling Technology), MPS1 (#ab11108, Abcam), Lamin B2 (#ab151735, Abcam), and β-Actin (#3700, Cell Signaling Technology). Secondary antibodies were from LICOR Biosciences: IRDye 680LT Goat anti-Mouse IgG (#926–68020), IRDye 800CW Goat anti-Rabbit IgG (#926–32211), or Cell Signaling Technology: anti-mouse IgG HRP-linked antibody (#7076), anti-rabbit IgG HRP-linked antibody (#7074). Imaging of blots and quantitation of bands was performed using the LICOR Odyssey system, or LAS-3000 (Fujifilm).
CRISPR-Cas9 system
Target sequences for CRISPR interference were designed using the sgRNA designer (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). A non-targeting sgRNA from the Gecko library v2 was used as a scramble sgRNA. sgRNA target sequences are listed in Table S2 sgRNAs were cloned into pCRISPRv2-puro.
dsDNA stimulation
3 × 105 cells were plated onto a 6-well plate and transfected using X-tremeGENE HP DNA Transfection Reagent (Roche, Cat.# 06366236001) with the indicated amount of poly (dA:dT) (Invivogen, Cat.# tlrl-patn).
Cell viability assay to determine an IC50
3000 cells were plated onto 96-well plates, and then cultured for 72 h in the presence of each DNA-damaging agent at the indicated concentration. Values of CellTiter-Glo Luminescent Cell Viability assay (Promega) after 96 h were normalized to vehicle treated cells. Plates were read on a Tecan Infinite M200 Pro plate reader and analysis was performed using Prism7 (GraphPad Software). All conditions were tested in triplicate.
siRNA transfection
siRNAs targeting MPS1 (s121), AURKB (s17611), and negative control (AM4611) were purchased from Thermo Fisher Scientific. Cells were transfected with 100 nM of the respective siRNAs using XtremeGENE siRNA transfection reagent (Roche, Cat.#4476093001) following the manufacturer’s instructions, and collected after 96 h culture.
Quantification of micronucleus formation
Cells were plated onto chamber slides (CellTreat, Cat# 229168), and treated with DNA-damaging agents for 48 h. And then, cells were cultured for 24 h in normal growth medium after drug withdrawal, fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Cat# 15700) for 15 min at room temperature (RT), permeabilized with 0.1% v/v Triton X-100 for 5 min at RT, and stained with 1 μg/mL DAPI for 5 min at RT. Treated cells were imaged using an Olympus spinning disk confocal Imaging System (IX3-SPIN) equipped with a 60× silicon oil-immersion objective. Each image was taken with z stack at 0.43 μm interval to cover the entire cells of interest. z stack images were subjected to maximum projection followed by quantitative analysis using CellSens. All samples were imaged and analyzed with the same setting throughout the experiments. The number of micronucleus were counted from three different fields for each sample. To examine colocalization of cGAS with micronuclei, cells were washed twice by PBS and fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Cat# 15700) for 15 min at RT. Cells were then permeabilized with 0.1% v/v Triton X-100 for 10 min at RT and washed twice by PBS. After blocking with 1% BSA (Sigma-Aldrich, cat# A4503–100G) in PBS for 1 h at RT, cells were stained using following antibody to: cGAS (#15102, Cell Signaling Technology) Secondary antibody was anti-rabbit Alexa Fluor 488 (1:1,000; Invitrogen Cat. #A11034). Cells were then subjected to 5 min of DAPI (D9542, Sigma-Aldrich) staining and washed twice by PBS before cover slides were mounted with Vectashield hardset mounting medium (H-1400–10, Vector Laboratories). Slides were imaged with Zeiss LSM 880 confocal microscopy, and colocalization was determined using ImageJ.
Quantitative RT-PCR
RNA extraction was performed using RNeasy Mini Kit (Qiagen, Cat.# 74106). RNA samples (1 μg) were reverse-transcribed using SuperScript® III First-Strand Synthesis Super-Mix (Thermo Fisher Scientific, Cat.# 1683483). Quantitative real-time PCR was performed using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat.# 4367659). The sequences of the primers used for qRT-PCR are listed in Table S2. Values represent the average of four technical replicates from at least two independent experiments (biological replicates).
Analysis of cell cycle and cell viability
For cell cycle analysis, cells were stained by BD CycletestTM Plus DNA according to the manufacturer’s instructions, and then analyzed by FACSCanto ll (BD Biosciences). For cell viability analysis, cells were stained by propidium iodide (PI) according to the manufacturer’s instructions of Annexin V using Alexa Fluor 488 Annexin V dead cell apoptosis kit (Thermo Fisher Scientific, Cat.#V13245), and then analyzed by FACSCanto ll (BD Biosciences).
Immunofluorescence staining
After being grown on chamber slides (CellTreat, Cat# 229168) and subjected to various treatment conditions, cells were fixed and permeabilized according to standard protocols. In brief, cells were washed twice by PBS and fixed in 4% paraformaldehyde (PFA, Electron Microscopy Sciences, Cat# 15,700) for 15 min at RT. Cells were then permeabilized with 0.1% v/v Triton X-100 for 10 min at RT and washed twice by PBS. After blocking with 1% BSA (Sigma-Aldrich, cat# A4503-100G) in PBS for 1 h at RT, cells were stained using following antibodies to: STING (PA5-23381, Invitrogen), and ERGIC53 (PSC-PM-7213-C100, Axxora). Secondary antibodies were anti-mouse Alexa Fluor 488 (1:1,000; Invitrogen Cat. #A21202), or anti-rabbit Alexa Fluor 555 (1:1,000; Invitrogen Cat. #A21428). Cells were then subjected to 20 min of DAPI (D9542, Sigma-Aldrich) staining and washed twice by PBS before cover slides were mounted with Vectashield hardset mounting medium (H-1400-10, Vector Laboratories). Slides were imaged with the Nikon Eclipse 80i microscope, and colocalization was determined using CoLoc2 in ImageJ.
Immune profiling by flow cytometry
Fresh tumor tissue was placed in dissociation buffer consisting of RPMI (Life Technologies, Carlsbad, CA) +10% FBS (HyClone, Logan, UT), 100 U/mL collagenase type IV (Life Technologies, Carlsbad, CA), and 50 μg/mL DNase I (Roche, Indianapolis, IN) at a ratio of 5 mL of dissociation buffer per 500 mg of sample and mechanically separated using gentleMACS C Tubes and gentleMACS Octo Dissociator system according to manufacturer’s protocol (Miltenyi, San Diego, CA). Suspension was incubated at 37°C for 45 min. Red blood cells were removed from samples using red blood cell lysis buffer (BioLegend, San Diego, CA). Samples were pelleted and then resuspended in fresh RPMI +10% FBS and strained over a 70 μm filter. Cells were incubated with Live/Dead Fixable Zombie NIR™ (Biolegend, San Diego, CA) for 5 min in the dark at room temperature in PBS. Fc receptors were blocked prior to surface antibody staining using mouse TruStain FcX blocking reagent (Biolegend, San Diego, CA). Cells were stained with pre-conjugated antibodies for 15 min on ice in FBS +2% FBS and washed prior to analysis on a BD LSRFortessa with FACSDiva software (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo (Ashland, OR) software version 10.7.1. Antibodies were specific for the following mouse markers: CD3 (17A2), CD4 (GK1.5), CD8 (53–6.7), CD11b (M1/70), CD19 (6D5), CD25 (PC61), CD45 (30-F11), CD49b (DX5), Ly6G (1A8), LAG-3 (C9B7W), PD-1 (29F.1A12), TIM-3 (B8.2C12), all from Biolegend (San Diego, CA), and Foxp3 (FJK-16s) from Thermo Fisher Scientific. To analyze cell surface markers in vitro, 2 × 105 cells resuspended in 100 μL PBS containing 3% FBS were stained by FITC-conjugated anti-CXCR3 antibody (Biolegend, Cat.# 353703), FITC-conjugated anti-HLA-A.B.C antibody (Biolegend, Cat.# 311404), or PerCP/Cy5.5-conjugated anti-PD-L1 antibody (Biolegend, Cat.# 329738) for 30 min at room temperature, washed by PBS containing 3% FBS, and then analyzed by FACSCanto ll (BD Biosciences) or FACSLyric (BD Biosciences). FITC-conjugated mouse IgG2a (Biolegend, Cat.# 400208) or PerCP/Cy5.5-conjugated mouse IgG2b (Biolegend, Cat.# 400338) was used as isotype control antibody.
Immune cell migration assay
Immune cell migration assay was performed as previously described (Kitajima et al., 2019; Ritter et al., 2020). Briefly, cancer cell spheroids (h1944) were generated by seeding 5 × 105 cells in suspension in an ultra-low attachment dish (Corning, Cat.# 3471) for 24 h. Samples were pelleted and then resuspended in type I rat tail collagen (Corning) at a concentration of 2.5 mg/mL following the addition of 10× PBS with phenol red with pH adjusted using NaOH. pH 7.0–7.5 was confirmed using PANPEHA Whatman paper (Sigma-Aldrich). Cells and collagen are kept on ice. The spheroids-collagen suspension was then injected into the central gel region of the 3D DAX-1 3D microfluidic cell culture chip (AIM Biotech, Singapore, Cat.# DAX-1). Microfluidic devices were designed as previously described (Aref et al., 2018), with a central region containing the cell-collagen mixture in a 3D microenvironment, surrounded by 2 media channels located on either side. After injection, collagen hydrogels containing cells were incubated 40 min at 37°C in humidity chambers, then hydrated with culture media, with 5 × 104 CXCR3-overexpressing Jurkat cells in one of the side media channels. CXCR3-overexpressing Jurkat cells were labeled with Cell Tracker Red (Thermo Fisher Scientific, Cat.#C34552) following manufacturer’s instructions. After 72–96 h of incubation, cancer cell spheroids and infiltrated immune cells were stained for 15 min with Acridine orange (AO) diluted 1:1 in culture media, (ViaStain™ AO Staining Solution - CS1–0108-5mL, nexcelom). For NK cells migrations, NK-92 cells (ATCC) were cultured as previously described. 5 × 104 were stained with cell Blue dye (cell proliferation dye eFluor 450, Invitrogen, Cat.# 65–0842) and cultured in the device for 3 days with IL-2 deprivation, followed by culture in the device with a full complete media for a total of 144 h. For PBMC-derived CD3+ T cells migrations, 5 × 104 were stained with cell blue dye and cultured in the device with RPMI 1640 supplemented with 10% human serum, 2mM of L-Glutamine, 1x penicillin-streptomycin, and 100 IU/mL of IL-2, 25 ng/mL of IL-7, and 25 ng/mL of IL-15 for a total of 72 h.
Co-culture patient-derived tumor cells with PBMC-derived T cell
1 × 105 cancer cells/well and PBMC derived 1 × 105 CD3+ T cells/well were seeded in 96-well plates. Twenty-four hours after seeding the cells, conditioned media from each wells were collected and human Granzyme B (R&D systems, Cat.# DGZB00) ELISA was performed according to the manufacturer’s instructions.
Cytokine profiling
Multiplex assays were performed utilizing the Mouse Cytokine/Chemokine Magnetic Bead Panel (Cat.# MCYTMAG-70K-PX32) on a Luminex MAGPIX system (Merck Millipore). Fold changes relative to the corresponding control were calculated and plotted as log2FC. Lower and upper limits of quantitation (LLOQ/ULOQ) were imputed from standard curves for cytokines above or below detection.
IHC staining and analysis
Immunohistochemistry was performed on the Leica Bond III automated staining platform. The antibody for CD3ε (Cell Signaling Technology #99940, clone D4V8L) was run at 1:150 dilution using the Leica Biosystems Refine Detection Kit with EDTA antigen. The antibody for CD8α (Cell Signaling Technology #98941, clone D4W2Z) was run at 1:200 dilution using the Leica Biosystems Refine Detection Kit with EDTA antigen. CD3 IHC staining was quantified using QuPath software (0.2.0-m4) (Bankhead et al., 2017). Positive Pixel Detection analysis was used with default settings for DAB staining to detect and quantify positive pixels in each of three individual, randomly selected fields from the center of each mouse tumor.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was assessed using unpaired two-tailed Student’s t-test, one-way ANOVA followed by Tukey’s post-hoc test or by Dunnet’s post-hoc test, or two-way ANOVA followed by Sidak’s post-hoc test. p values less than 0.05 were considered significant. Asterisks used to indicate significance correspond with: *p < 0.05, **p < 0.01. Columns represent means ± SD(SD). We showed mouse tumor volume data with means ± SE(S.E.). In one-way or two-way ANOVA followed by post-hoc tests, we showed asterisks only in pairs of our interest. GraphPad Prism7 was used for all statistical analysis.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-cleaved PARP | Cell signaling technology | Cat# 5625S RRID: AB_10699459 |
| Mouse monoclonal anti-β-actin | Cell signaling technology | Cat# 3700S RRID: AB_2242334 |
| Rabbit monoclonal anti-LKB1 | Cell signaling technology | Cat# 3047S RRID: AB_2198327 |
| Rabbit monoclonal anti-cGAS | Cell signaling technology | Cat#15102S RRID: AB_2732795 |
| Rabbit monoclonal anti-STING | Cell signaling technology | Cat#13647S AB 732796 |
| Rabbit monoclonal anti-STING (Rodent preferred) | Cell signaling technology | Cat#50494S RRID: AB_2799375 |
| Rabbit monoclonal anti-phospho STAT1 (Tyr701) | Cell signaling technology | Cat#9167S RRID: AB_561284 |
| Rabbit polyclonal anti-STAT1 | Cell signaling technology | Cat#9172S RRID: AB_2198300 |
| Rabbit polyclonal anti-IFNAR1 | Thermo Fisher Scientific | Cat#A304-290A RRID: AB_2620486 |
| Rabbit monoclonal anti-phospho-Histone H2A.X (Ser139) | Cell signaling technology | Cat#9718S RRID: AB_2118009 |
| Rabbit monoclonal anti-Histone H3 | Cell signaling technology | Cat#4499S RRID: AB_10544537 |
| Rabbit monoclonal anti-phospho-TBK1/NAK (Ser172) | Cell signaling technology | Cat#5483S RRID: AB_10693472 |
| Rabbit monoclonal anti-TBK1/NAK | Cell signaling technology | Cat#3013S RRID: AB_2199749 |
| Rabbit monoclonal anti-DNMT1 | Cell signaling technology | Cat#5032S RRID: AB_10548197 |
| Mouse monoclonal anti- PCNA | Dako | Cat#M0879 RRID: AB_2160651 |
| Rabbit monoclonal anti-Aurora B/AIM1 | Cell signaling technology | Cat# 3094 RRID: AB_10695307 |
| Mouse monoclonal anti-TTK/MPS1 | abcam | Cat# ab11108 RRID: AB_297757 |
| Rabbit monoclonal anti-Lamin B2 | abcam | Cat#ab151735 RRID: AB_2827514 |
| Alexa Fluor 700 anti-mouse CD3 | Biolegend | Cat#100216 RRID: AB_493697 |
| Brilliant Violet 711 anti-mouse CD4 | Biolegend | Cat# 100550 RRID: AB_2562099 |
| PE/Dazzle 594 anti-mouse CD8a | Biolegend | Cat# 100762 RRID: AB_2564026 |
| Brilliant Violet 711 anti-mouse/human CD11b | Biolegend | Cat# 101242 RRID: AB_11218791 |
| Brilliant Violet 605 anti-mouse CD19 | Biolegend | Cat# 115540 RRID: AB_11203538 |
| Brilliant Violet 785 anti-mouse CD25 | Biolegend | Cat# 102051 RRID: AB_2564131 |
| Brilliant Violet 510™ anti-mouse CD45 | Biolegend | Cat# 103138 RRID: AB_2561392 |
| PerCP/Cyanine5.5 anti-mouse CD49b | Biolegend | Cat# 108916 RRID: AB_2129358 |
| PerCP/Cyanine5.5 anti-mouse Ly-6G | Biolegend | Cat# 127616 RRID: AB_1877271 |
| PE anti-mouse CD223 (LAG-3) | Biolegend | Cat# 125208 RRID: AB_2133343 |
| Brilliant Violet 421 anti-mouse CD279 (PD-1) | Biolegend | Cat# 135218 RRID: AB_2561447 |
| APC anti-mouse CD366 (Tim-3) Antibody | Biolegend | Cat# 134008 RRID: AB_2562998 |
| FOXP3 Monoclonal Antibody (FJK-16s), PE-Cyanine7 | Thermo Fisher Scientific | Cat# 25-5773-82 RRID: AB_891552 |
| FITC anti-human CD183 (CXCR3) | Biolegend | Cat# 353703 RRID: AB_10962910 |
| FITC anti-human HLA-A,B,C | Biolegend | Cat# 311404 RRID: AB_314873 |
| PE/Cyanine7 anti-human CD274 (B7-H1, PD-L1) | Biolegend | Cat# 329738 RRID: AB_2617010 |
| PerCP/Cyanine5.5 Mouse IgG2b, κ Isotype Ctrl | Biolegend | Cat# 400338 RRID: AB_10765682 |
| FITC Mouse IgG2a, κ Isotype Ctrl | Biolegend | Cat# 400208 RRID: AB_10764135 |
| STING Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5-23381 RRID: AB_2540904 |
| ERGIC53 Monoclonal monoclonal antibody | AXXORA | Cat# PSC-PM-7213-C100 |
| CD3ε (D4V8L) Rabbit mAb | Cell signaling technology | Cat# 99940 RRID: AB_2755035 |
| CD8α (D4W2Z) XP Rabbit mAb (Mouse Specific) | Cell signaling technology | Cat# 98941 AB 2756376 |
| InVivoMAb anti-mouse CD8α | Bio X cell | Cat# BE0004-1 RRID: AB_1107671 |
| Biological Samples | ||
| Patient-derived cell lines | Köhler et al. 2021 | N/A |
| PBMC | STEMCELL | Cat#70025 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2’3’-cGAMP | Invivogene | Cat# tlrl-nacga23 CAS#:1441190-66-4 |
| ADU-S100 | Chemietek | Cat# CT-ADUS100 CAS#:1638750-95-4 |
| ruxolitinib | Selleck chemicals | Cat# S1378 CAS#:941678-49-5 |
| cisplatin | Sigma Aldrich | Cat# 232120 CAS#: 15663-27-1 |
| docetaxel | Selleck chemicals | Cat# S1148 CAS#:114977-28-5 |
| etoposide | Sigma Aldrich | Cat# 341205 CAS#:33419-42-0 |
| vinorelbine | Sigma Aldrich | Cat# V2264 CAS#:125317-39-7 |
| pemetrexed | Selleck chemicals | Cat# S1135 CAS#:150399-23-8 |
| methotrexate | Sigma Aldrich | Cat# A6770 CAS#:133073-73-1 |
| aminopterin | Sigma Aldrich | Cat# A1784 CAS#:54-62-6 |
| nocodazole | Sigma Aldrich | Cat# M1404 CAS#:31430-18-9 |
| hydroxyurea | Sigma Aldrich | Cat# H8627 CAS#:127-07-1 |
| olaparib | Selleck chemicals | Cat# S1060 CAS#:763113-22-0 |
| barasertib | Cayman Chemical | Cat# 11602 CAS#:722544-51-6 |
| MK5108 | Cayman Chemical | Cat# 21266 CAS#:1010085-13-8 |
| ceralasertib | Cayman Chemical | Cat# 21035 CAS#:1352226-88-0 |
| prexasertib | Cayman Chemical | Cat# 21490 CAS#:1234015-52-1 |
| CFI-402257 | Cayman Chemical | Cat# 21960 CAS#:1610759-22-2 |
| volasertib | Cayman Chemical | Cat# 18193 CAS#:755038-65-4 |
| BAY-1217389 | Selleck chemicals | Cat# S8215 CAS#:1554458-53-5 |
| CC-671 | MedChemExpress | Cat# HY-108709 CAS :1618658-88-0 |
| Human IFN-γ | R&D systems | Cat# 285-IF-100 |
| Human IL-2 | Thermo Fisher Scientific | Cat# PHC0021 |
| Human IL-7 | Thermo Fisher Scientific | Cat# PHC0075 |
| Human IL-15 | Thermo Fisher Scientific | Cat# PHC9154 |
| decitabine | Selleck chemicals | Cat# S1200 CAS#:2353-33-5 |
| GSK126 | Selleck chemicals | Cat# S7061 CAS#:1346574-57-9 |
| Poly(dA:dT) | InvivoGen | Cat# tlrl-patn CAS#:86828-69-5 |
| Critical Commercial Assays | ||
| Human IFN-β ELISA kit | Thermo Fisher Scientific | Cat# 414101 |
| HumanCXCL10 ELISA kit | R&D systems | Cat# DIP100 |
| Human 2’3’-cGAMPv ELISA kit | Cayman Chemical | Cat# 501700 |
| Human Granzyme B ELISA kit | R&D systems | Cat# DGZB00 |
| Mouse Cytokine/Chemokine Magnetic Bead Panel | Merck Millipore | Cat# MCYTMAG-70K-PX32 |
| CellTiter-Glo luminescent cell viability assay kit | Promega | Cat# G7572 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: A549 | ATCC | CCL-185 |
| Human: HCC44 | Broad Institute | N/A |
| Human: NCI-H23 | ATCC | CRL-5800 |
| Human: NCI-H1355 | ATCC | CRL-5865 |
| Human: NCI-H2122 | ATCC | CRL-5985 |
| Human: NCI-H2030 | ATCC | CRL-5914 |
| Human: NCI-H2009 | ATCC | CRL-5911 |
| Human: NCI-H1792 | ATCC | CRL-5895 |
| Human: NCI-H441 | ATCC | HTB-174 |
| Human: NCI-H358 | ATCC | CRL-5807 |
| Human: NCI-H1944 | ATCC | CRL-5907 |
| Human: NCI-H647 | ATCC | CRL-5834 |
| Human: A-427 | ATCC | HTB-53 |
| Human: Jurkat, Clone E6-1 | ATCC | TIB-152 |
| Human: HEK293T | ATCC | CRL-3216 |
| Human: NK-92 | ATCC | CRL-2407 |
| Human: THP-1 | ATCC | TIB-202 |
| Human: DFCI-24 | Köhler et. al. 2021 | N/A |
| Human: DFCI-298 | Köhler et. al. 2021 | N/A |
| Human: DFCI-316 | Köhler et. al. 2021 | N/A |
| Human: DFCI-332 | Köhler et. al. 2021 | N/A |
| Mouse: LLC | ATCC | CRL-1642 |
| Mouse: CMT167 | ECACC | 10032302 |
| Mouse: 393P | Gibbons et. al. 2009 | N/A |
| Mouse: Lacun3 | Bleau et. al. 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: 129S2/SvPasCrl | Charles River Laboratories | Strain #: 476 |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Strain #: 005557 |
| Recombinant DNA | ||
| pLX304-NanoLuc | Kitajima et al 2019 | N/A |
| pLX304-human LKB1 | Kitajima et al 2019 | N/A |
| pLX304-human STING | Kitajima et al 2019 | |
| pLX304-human cGAS | Campisi et al 2020 | |
| pLX307-human CXCR3 | Kitajima et al 2019 | |
| pCRISPRv2-puro-scramble sgRNA control, see Table S2 | Broad Institute | N/A |
| pCRISPRv2-puro-human LKB1 sgRNA, see Table S2 | Kitajima et al 2019 | |
| pCRISPRv2-puro-human cGAS sgRNA, see Table S2 | Campisi et al 2020 | |
| pCRISPRv2-puro-human STING sgRNA, see Table S2 | Campisi et al 2020 | |
| pCRISPRv2-puro-human IFNAR1 sgRNA, see Table S2 | This paper | |
| pCRISPRv2-puro-human STAT1 sgRNA, see Table S2 | Campisi et al 2020 | |
| pCRISPRv2-puro-human ATG5 sgRNA, see Table S2 | This paper | |
| pCRISPRv2-puro-mouse scramble sgRNA control, see Table S2 | This paper | |
| pCRISPRv2-puro-mouse LKB1 sgRNA, see Table S2 | This paper | |
| pCRISPRv2-blast-mouse STING sgRNA, see Table S2 | This paper | |
| pCMV-dR8.91 (lentivirus packaging) | Broad Institute | N/A |
| pCMV-VSV-G | Broad Institute | N/A |
| Sequence-Based Reagents | N/A | |
| Primers for qRT-PCR, see Table S2 | This paper | |
| Software and Algorithms | ||
| GraphPad Prism7 | GraphPad Software, Inc. | http://www.graphpad.com/ |
| sgRNA designer | Broad Institute | http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design |
| Other | ||
Highlights.
KL mutant cells are sensitive to intracellular 2′–3′cGAMP accumulation
MPS1i potently generates 2′–3′-cGAMP via cGAS sensing of micronuclei
De-repression of STING further co-opts this vulnerability and recruits T/NK cells
Sequential DNMT-MPS1 inhibitor treatment effectively restores immunogenicity in vivo
ACKNOWLEDGMENTS
We thank Drs J.M. Kurie, R. Pio, and L.M. Montuenga for providing materials, and Drs R. Yao, T. Hirota, H. Yamanaka, and M. Fukuda for scientific discussion and support. We also thank Dana-Farber/Harvard Cancer Center in Boston, MA, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry service.
Dana-Farber/Harvard Cancer Center is supported in part by an NCI Cancer Center Support Grant # NsluIH 5 P30 CA06516. This work was supported by NIH R01CA190294 (D.A.B), the Ludwig Center at Harvard Medical School (D.A.B), the Mark Foundation (D.A.B.), Heerwagen, Candice Bagby, Ming and Polly Tsai Funds for Lung Cancer Research (D.A.B.), and DFCI/Northeastern University Joint Program in Cancer Drug Development (D.A.B). Research was also supported by JSPS Postdoctoral Fellowship for Research Abroad (S.K.), and Developmental Research Project Award in Lung Cancer Research (S.K., E.H.K.), the program of Leading Initiative for Excellent Young Researchers (S.K.), JSPS KAKENHI Scientific Research (B) (#20H03521) (S.K.), AMED P-CREATE (20cm0106705h0001, 21cm0106705h0002) (S.K.), Takeda Science Foundation (S.K.), Princess Takamatsu Cancer Research Fund (S.K.), Uehara Memorial Foundation Research Fellowship (T.T.), Lilly Oncology Fellowship Program (T.T.), AIRC fellowship for Abroad (M.C.), Gross-Loh Fellowship (E.H.K.), and Expect Miracles Foundation and the Rob- ert A. and Renee E. Belfer Family Foundation (L.P.). Graphic abstract is created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
S.K., T.T., D.A.B., C.P.P, and P.G. are inventors on a pending patent for combination DAC and MPS1 inhibitor therapy to prime cancer immunogenicity. D.A.B. is a consultant for N of One/Qiagen and Tango Therapeutics; is a founder and shareholder in Xsphera Biosciences; has received honoraria from Merck, H3 Biomedicine/Esai, EMD Serono, Gilead Sciences, Abbvie, and Madalon Consulting; and has received research grants from BMS, Takeda, Novartis, Gilead, and Lilly. T.U.B is a consultant for N of One/Qiagen. P.C.G. has sponsored research agreements with Marengo Therapeutics, Epizyme, Daiichi Sankyo, and Foghorn Therapeutics. C.P.P is a consultant for DropWorks and Xsphera Biosciences; has stock and other ownership interests in Xsphera Biosciences; received honoraria from Bio-Rad; and has sponsored research agreements with Daiichi Sankyo, Bicycle Therapeutics, Transcenta, Bicara Therapeutics, AstraZeneca, Intellia Therapeutics, Janssen Pharmaceuticals, and Array Biopharma. S.K. has a sponsored research agreement with Boehringer-Ingelheim. E.H.K. is an employee and stockholder of Merck & Co. and previously had a Sponsored Research Agreement with Takeda Pharmaceuticals. H.K. has a Sponsored Research Agreement with Takeda Pharmaceuticals. P.A.J. has received consulting fees from AstraZeneca, Boehringer-Ingelheim, Pfizer, Roche/Genentech, Takeda Oncology, ACEA Biosciences, Eli Lilly and Company, Araxes Pharma, Ignyta, Mirati Therapeutics, Novartis, LOXO Oncology, Daiichi Sankyo, Sanofi Oncology, Voronoi, SFJ Pharmaceuticals, Takeda Oncology, Transcenta, Silicon Therapeutics, Syndax, Nuvalent, Bayer, Esai, Biocartis, Allorion Therapeutics, Accutar Biotech, and Abbvie; receives post-marketing royalties from DFCI; owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, DaichiSankyo, PUMA, Boehringer Ingelheim, Eli Lilly and Company, Revolution Medicines; and Astellas Pharmaceuticals; and has stock ownership in LOXO Oncology and Gatekeeper Pharmaceuticals.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2022.08.015.
REFERENCES
- Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, and Luke JJ (2021). STING agonists as cancer therapeutics. Cancers (Basel) 13, 2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, Mathur N, Zhou C, Coakley RV, Bartels A, et al. (2018). 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip 18, 3129–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi M, Sundararaman SK, Shelton SE, Knelson EH, Mahadevan NR, Yoshida R, Tani T, Ivanova E, Canadas I, Osaki T, et al. (2020). Tumor-derived cGAMP regulates activation of the vasculature. Front. Immunol. 11, 2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi M, Kuang Y, Zhang Y, Gjini E, et al. (2018). Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat. Med. 24, 1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin EN, Yu C, Vartabedian VF, Jia Y, Kumar M, Gamo AM, Vernier W, Ali SH, Kissai M, Lazar DC, et al. (2020). Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999. [DOI] [PubMed] [Google Scholar]
- Cordova AF, Ritchie C, Bohnert V, and Li L (2021). Human SLC46A2 is the dominant cGAMP importer in extracellular cGAMP-sensing macrophages and monocytes. ACS Cent. Sci. 7, 1073–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Huang YJ, He X, Zhao M, Wang X, Liu ZS, Xue W, Cai H, Zhan XY, Huang SY, et al. (2019). Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 176, 1447–1460.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, Poirier JT, Peng D, Bulatovic M, Mukhopadhyay S, et al. (2021). ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat. Cancer 2, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH, et al. (2018). PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 25, 2972–2980.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahat R, Berglund A, Putney RM, Perez-Villarroel P, Aoyama S, Pilon-Thomas S, Barber GN, and Mule JJ (2021). Epigenetic reprogramming of tumor cell-intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA 118. e2013598118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falahat R, Perez-Villarroel P, Mailloux AW, Zhu G, Pilon-Thomas S, Barber GN, and Mule JJ (2019). STING signaling in melanoma cells shapes antigenicity and can promote antitumor T-cell activity. Cancer Immunol. Res. 7, 1837–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD, Lopez-Ocasio M, Baur R, Kanakabandi K, Porcella SF, and Shevach EM (2018). Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 14, e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, van der Velden AWM, and Martinez LA (2021). Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 39, 494–508.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabosch S, Bulatovic M, Zeng F, Ma T, Zhang L, Ross M, Brozick J, Fang Y, Tseng G, Kim E, et al. (2019). Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 38, 2380–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan J, Lu C, Jin Q, Lu H, Chen X, Tian L, Zhang Y, Ortega J, Zhang J, Siteni S, et al. (2021). MLH1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cGAS-STING pathway. Cancer Cell 39, 109–121.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch EM, Fischer AH, Deerinck TJ, and Hetzer MW (2013). Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, and Hornung V (2020). Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 21, 501–521. [DOI] [PubMed] [Google Scholar]
- Ji F, Zhang F, Zhang M, Long K, Xia M, Lu F, Li E, Chen J, Li J, Chen Z, et al. (2021). Targeting the DNA damage response enhances CD70 CAR-T cell therapy for renal carcinoma by activating the cGAS-STING pathway. J. Hematol. Oncol. 14, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan TE, Burke KP, and Van Allen EM (2019). Genomic correlates of response to immune checkpoint blockade. Nat. Med. 25, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima S, Asahina H, Chen T, Guo S, Quiceno LG, Cavanaugh JD, Merlino AA, Tange S, Terai H, Kim JW, et al. (2018). Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS. Cancer Cell 34, 439–452.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, Tange S, Mitsuishi Y, Thai TC, Masuda S, et al. (2019). Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 9, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knelson EH, Ivanova EV, Tarannum M, Campisi M, Lizotte PH, Booker MA, Ozgenc I, Noureddine M, Meisenheimer B, Chen M, et al. (2022). Activation of tumor-cell STING primes NK-cell therapy. Cancer Immunol. Res. 10, 947–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler J, Zhao Y, Li J, Gokhale PC, Tiv HL, Knott AR, Wilkens MK, Soroko KM, Lin M, Ambrogio C, et al. (2021). ERK inhibitor LY3214996-based treatment strategies for RAS-driven lung cancer. Mol. Cancer Ther. 20, 641–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Yamauchi S, Berglund A, Putney RM, Mule JJ, and Barber GN (2018). Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 37, 2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. (2016). STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 76, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, and Bakhoum SF (2020). The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, and Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, and Biggins S (2014). Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell Biol. 15, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo Yau H, Bell E, Ettayebi I, de Almeida FC, Boukhaled GM, Shen SY, Allard D, Morancho B, Marhon SA, Ishak CA, et al. (2021). DNA hypomethylating agents increase activation and cytolytic activity of CD8(+) T cells. Mol. Cell 81, 1469–1483.e8. [DOI] [PubMed] [Google Scholar]
- Lu C, Guan J, Lu S, Jin Q, Rousseau B, Lu T, Stephens D, Zhang H, Zhu J, Yang M, et al. (2021). DNA sensing in mismatch repair-deficient tumor cells is essential for anti-tumor immunity. Cancer Cell 39, 96–108.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, Moghbeli T, Tan YS, Lin X, Bellile E, et al. (2020). HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J. Clin. Invest. 130, 1635–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, McWhirter SM, Katibah GE, Corn JE, Woodward JJ, and Raulet DH (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Drechsler H, Grundner-Culemann K, Ballister ER, Rodriguez-Rodriguez JA, Rodriguez-Bravo V, Jones MJK, Foley E, Lampson MA, Daub H, et al. (2017). Mps1 regulates kinetochore-microtubule attachment stability via the Ska complex to ensure error-free chromosome segregation. Dev. Cell 41, 143–156.e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigi M, Campisi M, Hong D, Thai TC, Piel B, Han S, et al. (2021). Intrinsic immunogenicity of small cell lung carcinoma revealed by its cellular plasticity. Cancer Discov. 11, 1952–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia ARR, Linder S, Song JY, Vaarting C, Boon U, Pritchard CEJ, Velds A, Huijbers IJ, van Tellingen O, Jonkers J, and Medema RH (2018). Mps1 inhibitors synergise with low doses of taxanes in promoting tumour cell death by enhancement of errors in cell division. Br. J. Cancer 118, 1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason JM, Wei X, Fletcher GC, Kiarash R, Brokx R, Hodgson R, Beletskaya I, Bray MR, and Mak TW (2017). Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc. Natl. Acad. Sci. USA 114, 3127–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr L, Toufektchan E, von Morgen P, Chu K, Kapoor A, and Maciejowski J (2021). ER-directed TREX1 limits cGAS activation at micronuclei. Mol. Cell 81, 724–738.e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, Qiu X, Canadas I, Roehle K, Heckler M, et al. (2021). EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, Laustsen A, Peri S, Hansen AL, Bonefeld L, et al. (2018). Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 9, 3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan BS, Perera SA, Piesvaux JA, Presland JP, Schroeder GK, Cumming JN, Trotter BW, Altman MD, Buevich AV, Cash B, et al. (2020). An orally available non-nucleotide STING agonist with antitumor activity. Science 369. eaba6098. [DOI] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reislander T, Lombardi EP, Groelly FJ, Miar A, Porru M, Di Vito S, Wr.ight B, Lockstone H, Biroccio A, Harris A, et al. (2019). BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 10, 3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reislander T, Groelly FJ, and Tarsounas M (2020). DNA damage and cancer immunotherapy: a STING in the tale. Mol. Cell 80, 21–28. [DOI] [PubMed] [Google Scholar]
- Ricciuti B, Arbour KC, Lin JJ, Vajdi A, Vokes N, Hong L, Zhang J, Tolstorukov MY, Li YY, Spurr LF, et al. (2022). Diminished efficacy of programmed death-(Ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J. Thorac. Oncol. 17, 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 75, 372–381.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter JL, Zhu Z, Thai TC, Mahadevan NR, Mertins P, Knelson EH, Piel BP, Han S, Jaffe JD, Carr SA, et al. (2020). Phosphorylation of RAB7 by TBK1/IKKepsilon regulates innate immune signaling in triple-negative breast cancer. Cancer Res. 80, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N, et al. (2018). Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 36, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. (2018). STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger S, and Gajewski TF (2018). Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 18, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper MJ, Vaz M, Marrone KA, Brahmer JR, and Baylin SB (2020). The emerging role of epigenetic therapeutics in immuno-oncology. Nat. Rev. Clin. Oncol. 17, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wengner AM, Siemeister G, Koppitz M, Schulze V, Kosemund D, Klar U, Stoeckigt D, Neuhaus R, Lienau P, Bader B, et al. (2016). Novel Mps1 kinase inhibitors with potent antitumor activity. Mol. Cancer Ther. 15, 583–592. [DOI] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, and Barber GN (2016a). Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep. 14, 282–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, and Barber GN (2016b). Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 76, 6747–6759. [DOI] [PubMed] [Google Scholar]
- Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, Willis C, Dotti G, Vincent BG, Restifo NP, and Serody JS (2021). STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp. Med. 218. e20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bai XC, and Chen ZJ (2020). Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity 53, 43–53. [DOI] [PubMed] [Google Scholar]
- Zhao M, Wang F, Wu J, Cheng Y, Cao Y, Wu X, Ma M, Tang F, Liu Z, Liu H, and Ge B (2021). CGAS is a micronucleophagy receptor for the clearance of micronuclei. Autophagy 17, 3976–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, and Funabiki H (2020). Regulation and consequences of cGAS activation by self-DNA. Trends Cell Biol. 30, 594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, and Funabiki H (2019). The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 178, 302–315.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to re-analyze the data reported in this paper is available from the lead contact upon request.