

Abstract
The genetic basis of many epilepsies is increasingly understood, giving rise to the possibility of precision treatments tailored to specific genetic etiologies. Despite this, current medical therapy for most epilepsies remains imprecise, aimed primarily at empirical seizure reduction rather than targeting specific disease processes. Intellectual and technological leaps in diagnosis over the past 10 years have not yet translated to routine changes in clinical practice. However, the epilepsy community is poised to make impressive gains in precision therapy, with continued innovation in gene discovery, diagnostic ability, and bioinformatics; increased access to genetic testing and counseling; fuller understanding of natural histories; agility and rigor in preclinical research, including strategic use of emerging model systems; and engagement of an evolving group of stakeholders (including patient advocates, governmental resources, and clinicians and scientists in academia and industry). In each of these areas, we highlight notable examples of recent progress, new or persistent challenges, and future directions. The future of precision medicine for genetic epilepsy looks bright if key opportunities on the horizon can be pursued with strategic and coordinated effort.
Keywords: epilepsy, exome sequencing, genomic medicine, personalized medicine, precision medicine
1 ∣. WHAT IS “PRECISION MEDICINE,” IN THE CONTEXT OF GENETIC EPILEPSY?
The National Research Council has defined precision medicine (PM) as “the ability to classify individuals into subpopulations that differ in their [disease] susceptibility, … biology and/or prognosis, or in their response to a specific treatment. …Interventions can then be concentrated on those who will benefit, sparing expense and side effects for those who will not.”1 In the epilepsies, genetic diagnoses can define treatment-relevant subgroups of patients. This is particularly true for single pathogenic variants in which a specific gain or loss of function can be targeted. Copy number variants (deletions and duplications) and polygenic risk in epilepsy with complex inheritance may also help to prognosticate in the future.2,3 Important variations in the definition of PM in the context of genetic epilepsy have suggested the need to further define therapies in terms of specific biological mechanisms4 and to consider personalized factors, such as environmental factors and chronicity of symptoms.5 In our view, there is a spectrum of increasing precision and personalization, representing advancement upon current treatment for most forms of epilepsy. The ideal precision treatment would correct a well-defined genetic mechanism in the context of individualized factors, to impart freedom from seizures and comorbidities. Recent developments with antisense oligonucleotides (ASOs) and other precision approaches have brought us closer to this ideal. A less personalized, existing antiseizure drug or newly repurposed drug with superior efficacy to improve outcomes in a genetically defined group of patients with epilepsy also represents an advance in precision from the current practice, even without full understanding of the underlying mechanisms. Here, we will consider examples from across this spectrum of precision approaches, and we will primarily focus on examples of epilepsy arising from single pathogenic variants. Pharmacogenomics is another aspect of precision medicine, involving consideration of genetic variants that do not necessarily directly contribute to the disease, but influence medication response and susceptibility to adverse reactions; this has been reviewed thoroughly.6
2 ∣. WHY DO WE NEED EPILEPSY PM, AND HOW DO WE GET THERE?
Up to one third of the 65 million people worldwide affected by epilepsy do not respond satisfactorily to available therapeutics.7 In most cases, antiseizure medications are chosen based on whether seizures are focal or generalized, and/or related to particular electroclinical syndromes. However, there is relatively little understanding of how these medications help some individuals but not others. This empirical approach can be lengthy, frustrating, and costly.
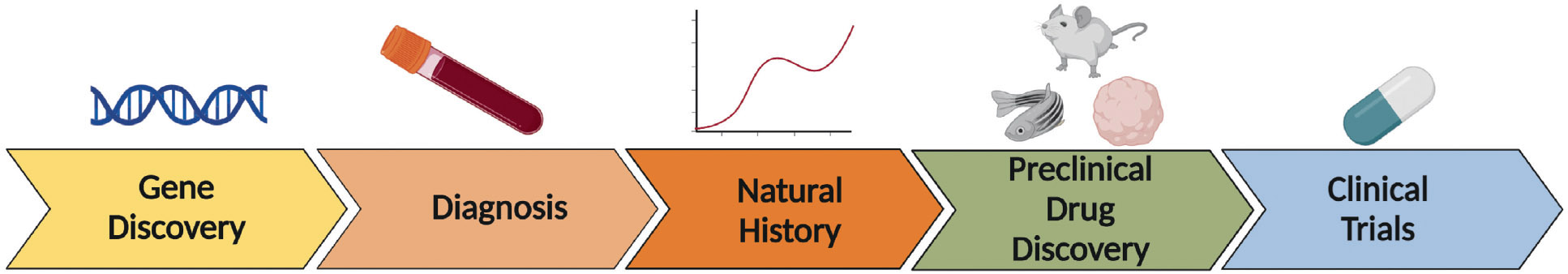
The era of gene discovery that followed completion of the Human Genome Project has explained a significant fraction of epilepsies by causes such as pathogenic variants in single genes8 or copy number variants.2 This renewed the emphasis on disease etiology for treatment approaches and led to hope for a pipeline of precision novel targets and clinical trials.9 But the ideal of PM for genetic epilepsy has been more elusive than some predicted, due to the complexity of underlying biological mechanisms and challenges in targeting them.4 Despite these challenges, considerable progress has been made in understanding mechanisms of monogenic epilepsies, some with evidence-based precision approaches emerging.10 Here, we give a state of the art survey of progress, challenges, and future directions in the major segments of the pipeline linking genetic epilepsies to precision therapies, starting with gene discovery and diagnostics, and proceeding to understanding of natural history; therapeutic discovery; preclinical testing; and clinical trials (summarized in Table 1). Concomitantly, dynamic stakeholder groups including patient advocates have increasingly facilitated progress toward epilepsy PM.
TABLE 1.
Precision medicine for genetic epilepsy: overview of progress, challenges, and future directions
|
|||||
| Key Accomplishments | International efforts (e.g. Epi4K, EuroEPINOMICS, Epi25K): new genes, new mechanisms | Increased accessibility & speed of commercial genetic testing | Natural histories provided nuanced understanding of rare diseases (e.g. cdkl5, STXBP1) |
|
|
| Key Challenges | Complexity of genotype-phenotype correlation | Unequal access to genetic testing and genetic counseling | Genetic discovery is outpacing clinical knowledge of pathogenesis and disease course |
|
|
| Strategies for continued progress |
|
|
|
|
|
Figure created with Biorender.
Abbreviations: AAV, adeno-associated virus; ASO, antisense oligonucleotide.
3 ∣. GENE DISCOVERY AND DIAGNOSIS
3.1 ∣. Progress
During the past decade, international teams such as Epi4K, the Epilepsy Phenome/Genome Project, and EuroEPINOMICS enabled increasingly powered gene identification in epilepsy.8,11 These efforts, combined with bioinformatics advances such as aggregation of large databases and matching tools (e.g., Azzariti and Hamosh12), led to a rapidly expanding number of genes implicated in epilepsy (Figure 1). The Epi25 Collaborative, seeking to sequence the exomes or genomes of 25 000 individuals with epilepsy, will be the largest epilepsy gene discovery effort to date, creating unprecedented opportunities for worldwide clinical trials (http://epi-25.org/).13
FIGURE 1.

Overview of genetic testing results from large-scale diagnostic studies in >25 000 individuals.21-23 The 50 most common genetic etiologies across all three studies are shown. lpath, likely pathogenic; path, pathogenic.
International teams also brought insights into genetic mechanisms. De novo mutations play a key role in developmental and epileptic encephalopathies.8 Copy number variants confer risk for developmental and epileptic encephalopathies, generalized genetic epilepsy, and lesional focal epilepsy.2 Noncoding regions of the genome can promote disease; multiple variants outside of the annotated coding regions of SCN1A were found to promote inclusion of a poison exon, or nonsense-mediated decay, causing Dravet syndrome through reduced SCN1A expression,14 and intronic expansions in SAMD12 and other genes were identified as the cause of adult familial myoclonic epilepsy.15
Epilepsy genetics has also moved beyond Mendelian paradigms. Whereas high-risk pathogenic variants tend to be rare or ultrarare, approximately 30% of the genetic liability for generalized epilepsy is explained by common genetic variants.16 In the future, all epilepsy may need to be considered within the context of polygenic risk.3,17 It is further hypothesized that some forms of epilepsy are inherited in an oligogenic fashion, in which “modifier” genes can epistatically increase disease risk together but not individually.18
Diagnostic ability also increased, thanks to next generation sequencing. Gene panels, exome sequencing, and whole genome sequencing provide the greatest diagnostic yield, each with important advantages and caveats that determine their utility and cost-effectiveness for individual patients.19,20 In the United States, most genetic testing is done commercially. Reports from four diagnostic laboratories account for >25 000 individuals who have undergone gene panel sequencing,21-23 including >5000 individuals with 20 of the most common monogenic etiologies (Figure 1). Additional measures to clarify variants of unknown significance (such as parental testing of candidate variants) and reanalysis of exome or genome sequencing increases the likelihood of identifying an etiology. A recent meta-analysis of the diagnostic yield of genetic testing in patients with epilepsy found that exome sequencing led to a diagnosis in 24% of cases tested.20 The diagnostic yield of exome sequencing was highest in patients with developmental and epileptic encephalopathies (27%) and in patients with epilepsy and neurodevelopmental disorders (27%).20 Based on these findings, the future standard of care for suspected genetic epilepsy may be exome sequencing, or a tiered approach such as a gene panel with reflex genome sequencing.
Genetic testing is also becoming more efficient, which will be critical for timely intervention. Whereas a turnaround time of several months was standard several years ago, diagnostic laboratories are now providing test results within a few weeks or in some cases, hours.24,25
3.2 ∣. Challenges and future directions
As the science of gene discovery progresses, important challenges relate to unequal access to care. Genetic counseling and testing remain out of reach for significant numbers of individuals in the United States, including older individuals,26 those with public health insurance plans,27 and underserved populations.28 Genetic testing is not accessible for most people outside of North America, Europe, and parts of Asia.29 These disparities could impede efforts toward natural history studies and clinical trials in diverse populations. Genetic testing is particularly important for infants and children in whom timely diagnosis could determine outcomes. The meta-analysis of Sheidley et al.20 indicated that pivotal treatment decisions resulted from 12%–80% of genetic diagnoses, such as use of stiripentol in Dravet syndrome, initiation of the ketogenic diet in SLC2A1-related epilepsy, and the identification of treatable inborn errors of metabolism. Genetic diagnoses in patients with epilepsy also influenced prognosis and led to decreased hospitalizations.20 Therefore, advocacy and funding are needed to implement genetic testing as a standard of care. Greater access to expert providers will also be necessary. The recent evolution toward telemedicine, including for rare genetic diseases,30 could connect patients who are unable to travel with specialists. Further measures to increase access include increased genetics training for neurologists; training greater numbers of neurogenetics specialists, including clinicians and genetic counselors; and improved education of individuals with epilepsy about the meaning and implications of genetic testing and diagnosis.
4 ∣. UNDERSTANDING NATURAL HISTORIES
4.1 ∣. Progress
Following genetic diagnosis, management will depend on the dynamic manifestations of a genetic condition, including when signs and symptoms become evident, and variability between affected individuals. A prototype example is STXBP1, a gene initially associated with Ohtahara syndrome,31 a severe developmental and epileptic encephalopathy. With further monitoring of affected individuals, pathogenic STXBP1 variants are now associated with a range of epilepsies and neurodevelopmental disorders not involving seizures.32 Well-designed natural history studies are being performed for Rett syndrome.33 Increasingly, advocacy groups are catalysts for natural history studies, aided by larger collaboratives such as the Rare Epilepsy Network (www.rareepilepsynetwork.org) or Rare-X (www.rare-x.org/) or in collaboration with industry (e.g., Invitae, https://www.ciitizen.com/).
4.2 ∣. Challenges and future directions
Natural history studies are needed to quantify the phenotypic spectrum of genetic epilepsies, and to inform the design of relevant disease scales and outcome measures for clinical trials, including endpoints beyond seizure burden (e.g., as done for CDKL5-deficiency disorder34 and Batten disease35). Rapid increases in genetic diagnoses, natural history, and clinical information will need to be integrated efficiently. Novel approaches, such as the Human Phenotype Ontology, can exploit data extracted from electronic medical records in conjunction with large-scale data harmonization.36 The ENIGMA-Epilepsy collaborative applies innovative approaches to integrate imaging, genetic, and other clinical data.37 “Portals” integrating genetic, preclinical, and clinical data for specific neurogenetic disorders are being assembled (e.g., http://grin-portal.broadinstitute.org/). A culture of transparency and data-sharing, together with virtual “structures” streamlining integration of rapidly emerging data, will enable progress.
5 ∣. THERAPEUTICS
5.1 ∣. Repurposed drugs
5.1.1 ∣. Progress
Particularly for channelopathies, careful functional characterization has enabled strategic use of existing antiseizure medications or repurposed compounds. Published accounts of targeted treatments for individual variants are numerous; we describe a few prominent examples (a more comprehensive list can be found in Guerrini et al.10).
Most cases of Dravet syndrome are caused by loss of function (LOF) variants in the sodium channel Nav1.1, encoded by SCN1A, leading to impaired function of inhibitory interneurons.38 Thus, antiseizure medications that block sodium channels may exacerbate seizures.39 Expert consensus and clinical trials enabled development of firstline therapies for Dravet syndrome, including valproic acid, clobazam, fenfluramine, stiripentol, topiramate, and cannabidiol.39,40 Pathogenic variants in SCN2A, which encode Nav1.2, cause a range of epilepsy syndromes. Gain of function (GOF) variants are broadly associated with neonatal presentations (benign familial infantile seizures, early infantile epileptic encephalopathy), and some LOF variants are associated with developmental delay, autism spectrum disorders, and/or epileptic encephalopathies presenting later in childhood. Accordingly, sodium channel-blocking medications such as oxcarbazepine are optimal for treatment of GOF variants, but avoided for SCN2A LOF variants.41,42 Systematic study of patients with neonatal epilepsy related to KCNQ2 LOF has indicated that sodium channel-blocking agents are more effective than other agents.43,44 For individuals with LOF variants in SLC2A1 leading to GLUT1 glucose transporter impairment, the ketogenic diet provides an alternative fuel source that dramatically decreases or eliminates seizures, and improves cognition and other disease manifestations such as movement disorders.45
In other cases, genetic diagnoses have prompted repurposing of drugs not traditionally used for antiseizure purposes. KCNA2 variants resulting in GOF in the voltage-gated potassium channel Kv1.2 were correlated with severe phenotypes, including medically refractory epilepsy, developmental delay, intellectual disability, ataxia, and other manifestations.46 Subsequently, treatment of patients with GOF KCNA2 mutations with a potassium channel blocker, 4-AP, dramatically decreased seizure burden and improved cognitive and motor function.47 As noted above, a previously US Food and Drug Administration (FDA)-approved drug promoting serotonergic signaling, fenfluramine, was approved for treatment of Dravet syndrome by the FDA and the European Commission in 2020, following clinical trials demonstrating reduced convulsive seizures.48 Memantine targets GOF variants in GRIN2A.49 Quinidine treatment of KCNT1-related epilepsy dramatically reduced seizure burden in case reports.50 Subsequent trial experience was less encouraging; challenges in the use of quinidine include heterogeneity in blood–brain barrier penetration and susceptibility to quinidine cardiotoxicity, as well as different responsiveness in patients, potentially due to different variants/electroclinical syndromes, ages, or treatment regimens.51
5.1.2 ∣. Challenges and future directions
Challenges in drug repurposing efforts, as with quinidine, illustrate useful lessons for the future. A systematic approach to drug identification may accelerate development of precision therapies that can be implemented on a larger scale. Using systems biology approaches, individually rare genetic diagnoses may be functionally classified by linking them to smaller numbers of canonical biochemical pathways, which could then serve as broadly useful therapeutic targets.52 High-throughput screening of drug libraries enables unbiased identification of compounds with the greatest efficacy and desirable pharmacological/toxicity profiles.53 The recent development of preclinical models that enable studies at larger scale may facilitate such systematic approaches (see section 6, Preclinical Models). Human clinical trials involving repurposed drugs should maximize sample size and adhere to standardized protocols, allowing for joint data analysis. Functional characterization of variants to elucidate GOF or LOF, and severity of the impairment, is obligatory. Leveraging alternative clinical trial designs for small sample sizes can increase rigor and generalizability (see section 7, Clinical Trials for Genetic Epilepsy).
Newly repurposed or novel drugs may have unknown mechanisms and/or may ultimately prove to be broadly effective, as opposed to targeted, antiseizure medications. For example, the mechanism of fenfluramine in Dravet syndrome is incompletely understood, but is thought to involve augmented serotonergic signaling and additional mechanisms, such as modulation of σ1 receptors.54 Fenfluramine may ultimately prove beneficial in multiple forms of refractory epilepsy, including Lennox–Gastaut syndrome.55
We have focused on monogenic epilepsies, which are enriched in developmental and epileptic encephalopathies but account for a small proportion of epilepsies overall.16 Ideally, the epilepsy community would also find ways to leverage information about pathogenic copy number variants and polygenic risk to increase precision in treatment.
5.2 ∣. Gene-based approaches: ASOs, adeno-associated virus vectors, and gene editing
5.2.1 ∣. Progress
Precision treatment with ASOs has become a reality in clinical neurology, including for spinal muscular atrophy (SMA),56 Duchenne muscular dystrophy,57 and familial amyloid polyneuropathy.58 In genetic epilepsies, preclinical and clinical studies of ASOs are underway. ASOs are oligonucleotides 18–30 base pairs in length that are chemically engineered to optimize pharmacokinetic and pharmacodynamic properties. ASOs correct or compensate for GOF or LOF genetic variants by targeting their mRNA transcripts and enabling modified mRNA splicing or mRNA degradation.59 ASOs decreasing gene expression reduced premature death and seizures in knockin mouse models carrying human SCN2A or SCN8A GOF variants.60,61 ASOs can also enhance gene expression by modulating nonproductive splicing events. One of these approaches, targeted augmentation of gene output, reduced seizures and mortality in a mouse model of Dravet syndrome,62 and is being tested in multiple clinical trials in the United States and United Kingdom (https://clinicaltrials.gov/ct2/show/NCT04442295). ASO approaches to reinstate UBE3A expression in Angelman syndrome have also entered clinical trials.63
On the horizon are promising approaches to directly repair mutant genes, such as gene editing with clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9. Considerable obstacles remain before these technologies can be safely deployed in humans, such as targeting these therapies to specific brain regions of interest, potential adaptive immunity to forms of Cas9, and mitigation of off-target effects. Originally, CRISPR/Cas9 technology was used to introduce insertion and deletion mutations to inactivate genes; however, more recent base editing and prime editing approaches obviate the need to cleave host cell DNA.64,65 A dCas9 (dead Cas9)-mediated promoter-enhancing strategy augmenting SCN1A expression was effective in vitro and in a mouse model of Dravet syndrome.66
Adeno-associated virus (AAV)-based approaches include gene replacement therapy and use of AAVs as vectors for ASOs and CRISPR/Cas9 systems. AAV-based approaches have not yet been tested in humans for genetic epilepsy, but have been used in preclinical models to reduce neuronal excitability in the epileptic focus, including overexpression of a potassium channel67 and replenishment of the endogenous antiseizure neuropeptide preprodynorphin.68
5.2.2 ∣. Challenges and future directions
A long-term goal will be to develop gene-based approaches that can safely be administered systemically. Currently, most gene-based approaches require intrathecal administration, due to poor central nervous system penetration and stability, in the absence of a vector that can be administered via systemic approaches, such as AAV. Strategies to overcome limitations on vector cargo size, or to decrease vector cargo size as in the example of specific Cas9 systems (e.g., dCas910,66), would enable expanded use of AAV vectors. It is important to consider that even “definitive” gene therapy may not reverse all deleterious phenotypes of a pathogenic variant, particularly those arising during early neurodevelopment.10 Furthermore, optimal titration of ASO effect is needed. For example, in studies cited above, ASOs targeting SCN2A or SCN8A GOF were not allele specific.60,61 Excessive suppression of either gene could be detrimental, as LOF in SCN2A or SCN8A is also associated with deleterious phenotypes, including epilepsy.41,69
Broader challenges relate to large-scale development of safe, ethical, and equitable gene-based approaches for the emerging multitude of rare genetic epilepsies. This will require regulation and new infrastructure. The Child Neurology Society outlined considerations for gene-targeted therapies in children, including rules and common standards regarding the choice of vectors; preclinical and clinical testing; consideration of how certain gene-based interventions might impact subsequent brain development in infants and children; long-term follow-up of individuals receiving novel therapies, given the likely emergence of new natural histories and possible long-term treatment side effects; adequate training for practitioners providing gene-based therapies; continuous ethical oversight; and sustainable and equitable economic support, which will likely vary in countries with different health care systems.70
An additional precision therapeutic modality, reviewed in Guerrini et al.10 is surgical management of focal genetic epilepsies. Emerging evidence suggests that certain medically refractory focal genetic epilepsies (such as tuberous sclerosis complex [TSC]) are likely to benefit from surgical intervention. In other cases, the presence or absence of a focal structural abnormality, in conjunction with germline versus somatic mutations, should be considered together with the usual presurgical investigations.10
6 ∣. PRECLINICAL MODELS TO UNDERSTAND PATHOGENESIS AND TEST PRECISION THERAPIES
6.1 ∣. Progress
In recent years, impressive work by numerous groups developed high- or medium-throughput preclinical models that hold promise to support rapid translation of basic biological mechanisms to precision therapies.
6.1.1 ∣. Functional characterization with heterologous cells and cultured neuronal networks
Genetic material from humans can be expressed “heterologously” in cell lines that otherwise would not express the gene, such as Chinese hamster ovary cells or human embryonic kidney cells. This enables simplified but detailed study of protein function. Heterologous cell lines are particularly useful for characterization of ion channel function, and in numerous instances such studies have informed precision approaches (e.g., Wolff et al.,41 Masnada et al.,46 Hedrich et al.,47 Johannesen et al.69). In KCNB1-related disorders, classification of variants into distinct functional categories was performed with automated patch clamp recording.71
Neurons in culture form spontaneous networks that can be monitored on multielectrode arrays (MEAs), allowing characterization of epilepsy-related attributes such as intrinsic network excitability and synchrony.72 Thus, MEAs may complement electrophysiological studies of single neurons by revealing specific network dynamics. MEAs can be used to monitor cultured networks non-invasively for extended periods of time, have been used to study epilepsy-related genes such as CHRNB2,73 and can be used to screen compounds (which can be directly added to MEA wells).
6.1.2 ∣. Induced pluripotent stem cell-derived neurons and cerebral organoids
There are important differences between mouse and human brain development, including the presence of certain neuronal cell types that are not represented in mouse brain. Patient-derived induced pluripotent stem cells (iPSCs) are an emerging model system to understand mechanisms of neuronal excitability in humans.74 iPSCs can theoretically be differentiated in culture to any cell type in the body, including all subtypes of neurons, thus allowing the study of epilepsy variants in the context of an individual's unique genetic background. In Dravet syndrome, patient-derived neurons have been generated by several groups (e.g., Liu et al.75). iPSC-derived excitatory cortical neurons from patients with SCN8A-related disorders showed variant-specific increases in persistent or resurgent sodium current that were responsive to riluzole,76 and subsequent administration of riluzole to individuals with the specific SCN8A variants led to substantial seizure reduction. Numerous genetic epilepsies have been modeled with iPSC-derived neurons, including Rett syndrome, TSC, Angelman syndrome, developmental and epileptic encephalopathies, progressive myoclonic epilepsies, and others (reviewed in Hirose et al.77). iPSC-derived neurons have been functionally characterized using MEAs.78
Cerebral organoids are important intermediate models between traditional two-dimensional (2D) cell cultures and animals. Human embryonic stem cells or fibroblasts reprogrammed to become iPSCs can self-organize into 3D spheroids in culture.79 Cerebral organoids have been used to study multiple forms of genetic epilepsy, including Rett syndrome,80 lissencephaly,81 Angelman syndrome,82 and others. Compared to 2D iPSC neuronal cultures, organoids maximize cell–cell interactions during neural development, can be maintained for longer timelines to better recapitulate structural features and cellular heterogeneity found in brain,79 and enable assessment of neuronal cell types not found in mouse brain (e.g., radial glial cells).74,79 An organoid model of TSC allowed for recapitulation of tubers, which are not observed in mouse models.83 Current limitations, which may be surmountable with further development, include difficulty in the generation of consistent phenotypes between experiments and the current inability to generate mature cortical structures with the full repertoire of neuronal and glial cell subtypes in organoids.
6.1.3 ∣. Zebrafish
Zebrafish exhibit behavioral and electrographic changes suggestive of seizures, for example, in the setting of pentylenetetrazol or kainic acid treatment, or with genetic manipulation.84 The rapid breeding cycle, low space requirement (embryos can be grown in 96-well plates), ability to easily administer treatments directly to the water environment, ability to automate video monitoring of movements such as seizures, and other features make zebrafish particularly amenable to high-throughput drug screens.84 For example, compounds targeting serotonergic signaling were identified in zebrafish models of Dravet syndrome.85
6.2 ∣. Challenges and future directions
The models described above may aid in studying genetic conditions at greater scale. A future challenge is their strategic application to identify precision therapies. There is a need to identify robust, reproducible phenotypes that are relevant to human disease and can serve as reliable endpoints when testing potential therapeutics. These endpoints ideally would recapitulate not only across laboratories, but across modeling paradigms (e.g., consistent findings related to ion channel function in heterologous cells or neurons, network bursting activity in MEAs, seizures and behavioral abnormalities in an in vivo model).
Given the strengths and limitations of different experimental approaches, there may not be a one size fits all approach. Preclinical study design may vary, depending on whether a disease results from dysfunction of an ion channel, for example, versus a structural protein. Coordination between clinical and translational researchers to pair clinical questions with optimal experimental approaches would expedite preclinical PM efforts.
It may not be feasible to extensively characterize all pathogenic variants. Instead, one might test hypotheses about how genetic variants can be classified into functional groups, for example, using systems approaches.52
7 ∣. CLINICAL TRIALS FOR GENETIC EPILEPSY
7.1 ∣. Progress
Given the rarity of many genetic diagnoses, there has been increasing movement toward developing personalized treatments in small groups of patients. There are numerous characterizations of individual ion channel variants, along with testing of targeted treatments that correct electrophysiological abnormalities and sometimes clinical symptoms (e.g., Wolff et al.,41 Hedrich et al.,47 Tidball et al.76; see section 5 on Therapeutics). An individualized approach to ASO treatment for epilepsy has also been demonstrated. Milasen, an ASO modeled on the SMA treatment nusinersen, was rapidly created for a child with a lethal disease, neuronal ceroid lipofuscinosis (CLN7).86 Milasen was engineered to modify transcript splicing related to a unique mutation in the gene MFSD8, and was tested in the patient's own fibroblasts. Following preclinical and toxicity testing, Milasen appeared to stabilize neurological and neuropsychiatric function, and decreased seizure burden in the patient.86 Although the patient ultimately died, the remarkable creation of an individualized ASO within 1 year of patient evaluation challenged conventional assumptions about the time needed for ASO development and regulatory approval. The example of Milasen also raises issues pertaining to ethics and equity that are inherent to “n of 1” approaches, discussed below.
7.2 ∣. Challenges and future directions
Many genetic epilepsies are rare diseases. In the United States, a rare disease is defined as one that affects <200 000 people, or in which cost of therapy development and testing is not expected to be recovered following approval.87 Furthermore, pleiotropy (one variant manifesting with differing phenotypes between individuals) may obscure a beneficial effect in a traditional randomized control trial (RCT).87 Thus, adequately powered RCTs will likely be challenging for many genetic epilepsies.
Alternative trial designs can overcome issues of low sample size and high interindividual variability. Examples include small crossover trials and prospective, rigorously designed “n of 1” trials in which individuals undergo sequential treatment phases, serving as both a test and control; and adaptive designs (reviewed in Abrahamyan et al.87). A recent systematic review of “n of 1” trials for rare genetic neurodevelopmental disorders proposed methodological criteria to enhance their interpretation and generalizability, including blinding and randomization; ample description of subject baseline characteristics; statistical methods that can account for small sample size and phenotypic heterogeneity; and appropriately timed, sequential testing of conditions (intervention vs. placebo) alternating with washout periods.88 Broader challenges related to “n of 1” approaches relate to ethics, regulation, and equity. Development of a therapy for one individual, rather than a population, blurs the line between research and medical treatment, a key distinction in the ethical framework for clinical research.89 In an “n of 1” scenario, the subject and the subject's surrogates may act more as research collaborators than clinical trial participants, raising the possibility of conflicts of interest and inadequately informed consent. There is an imperative to objectively define potential risks, benefits, and criteria for stopping the trial.89,90 Minimum preclinical safety and efficacy data needed to test “n of 1” interventions, such as some ASOs, in human subject(s) remain to be defined.90 For personalized ASOs, thorough functional characterization of genetic variants and rational design of therapies, rigorous preclinical testing, and standardized toxicity testing as well as regulatory approval should be required.86
Patient advocacy groups recently highlighted the importance of nonseizure outcomes for individuals with epilepsy, including cognitive function and quality of life.91 Some genetic epilepsies give rise to complex phenotypes, including impaired neurodevelopment, ataxia, movement disorders, and progressive loss of mobility (e.g., Schreiber et al.92) that may be as important to patients as seizures. It will therefore be essential to define and measure patient-centered nonseizure outcomes.
Not all variants are amenable to an ASO strategy, and it may not be feasible to generate uniquely personalized ASOs for the majority of individuals. The allocation of limited resources for ASO development and testing could be based on different factors, such as the number of patients likely to benefit, disease severity, and magnitude of benefit. These questions should be addressed and ASO-related resources allocated in a transparent manner that promotes equity and avoids perpetuation of disparities.89 Standardization of vectors, ASO manufacturing, and streamlining the ASO development/testing process could increase efficiency and broaden access to gene-based approaches.70
8 ∣. EVOLVING STAKEHOLDERS IN EPILEPSY PRECISION THERAPY DEVELOPMENT
The Epilepsy Leadership Council (www.epilepsyleadershipcouncil.org), and a number of the 51 patient advocacy groups within that council, represent both common and rare epilepsies, as well as professional societies and federal agencies. These groups have emerged as an important driver of epilepsy research. Groups including the Lennox–Gastaut Syndrome Foundation (www.lgsfoundation.org), the Dravet Syndrome Foundation (www.dravetfoundation.org), and the Tuberous Sclerosis Alliance (www.tscalliance.org), as well as broader epilepsy advocacy organizations, such as the Epilepsy Foundation (www.epilepsy.org) and Citizens United for Research in Epilepsy (CURE Epilepsy; www.cureepilepsy.org), are advocating for and supporting research advances for the epilepsies. The Epilepsy Genetic Initiative supported by CURE Epilepsy, launched in 2014, reanalyzed negative diagnostic exomes and uncovered new genetic etiologies such as de novo variants in alternative exons in SCN8A.93 The Epilepsy Foundation was instrumental in establishing the Rare Epilepsy Network, capturing patient and caregiver data from >40 rare epilepsy syndromes. The American Epilepsy Society and the International League Against Epilepsy recently updated seizure and epilepsy classification to better reflect state of the art knowledge and facilitate standardized communication.94
Federally supported efforts in the United States include the Centers Without Walls' projects such as Epi4K, EpiBiosS4Rx, the Center for SUDEP Research, the Channelopathy-Associated Epilepsy Research Center, and Epilepsy Multiplatform Variant Prediction. The Undiagnosed Diseases Network has brought together national expertise and cutting edge diagnostic genetic tools, leading to increased rate of diagnosis and the definition of 31 new clinical syndromes.95 The Epilepsy Therapy Screening Program refocused its efforts in 2016 to include rare epilepsies, developing a drug screening platform using a mouse model of Dravet syndrome.96
Industry partners and startup companies are focused on specific PM approaches such as targeted ion channel modifiers. Finally, there is renewed interest in integrative academic centers in providing multidisciplinary care for rare epilepsies. Increasingly, academic collaborative networks of clinicians and researchers, such as the EuroEPINOMICS-RES Consortium, the Network for Treatment of Rare Epilepsies, and the Treat-ION network for rare neurological channelopathies (https://www.research4rare.de/en/), collaborate to improve management of rare genetic epilepsies. Emerging learning health systems are also expected to inform natural history and treatment responses in rare conditions.97
9 ∣. CONCLUSIONS
We have described tremendous progress at each stage of the epilepsy PM pipeline, including gene discovery, diagnostics, natural history studies, therapeutic strategies, preclinical models, and clinical trials, but with formidable challenges remaining (Table 1) to translate this progress into precision therapies and cures. SCN8A-related developmental and epileptic encephalopathy is a genetic epilepsy progressing in this pipeline with relative efficiency. It has been ~10 years since the discovery of SCN8A as a disease-causing gene, with significant advancement toward PM in that time (Figure 2). The examples of milasen and SCN8A-related epileptic encephalopathy are proof that teams with the necessary combination of expertise (clinicians, scientists, patient advocates, federal resources, and regulatory bodies) can efficiently shepherd particular therapeutics through the pipeline.
FIGURE 2.

Following discovery of SCN8A-related disorders, these were modeled, supported by patient advocacy, and translated into potential treatments, providing an example of relatively efficient progression through the “pipeline” from gene discovery to novel precision medicine approaches. ASO, antisense oligonucleotide.
At the same time, larger national and international efforts are likely to be required to achieve the ends outlined in Table 1, such as adoption of genetic testing as a standard of care, ethical and equitable incorporation of gene-based therapies, and increasing the number of genetic epilepsies with targeted therapies. We and others argue that the international epilepsy community is at an “inflection point” in our efforts toward epilepsy PM,91 at which coordinated and concerted efforts will be needed to translate the gains highlighted in this review into epilepsy precision therapies. A working group of the Epilepsy Leadership Council in the United States recently proposed development of a “National Plan,” modeled on efforts in pediatric oncology (e.g., ChildrensOncologyGroup.org)91 following the Curing the Epilepsies Conference in 2021.98 National and international coordination could fully integrate scientific discovery, increasing clinical knowledge, and health policy to overcome the tendency for these areas to become siloed. Important organizing forces are already in place. For example, the American Epilepsy Society/National Institute of Neurological Disorders and Stroke Epilepsy Research Benchmark Stewards Committee has effectively outlined and tracked progress in priority areas, such as understanding the causes of epilepsy, preventing epilepsy, improving treatments, and preventing adverse consequences of seizures (www.ninds.nih.gov/About-NINDS/Strategic-Plans-Evaluations/Strategic-Plans/2020-NINDS-Benchmarks-Epilepsy-Research). We argue that what is further needed is the coordinated and systematic streamlining of the epilepsy precision medicine pipeline, beginning with gene discovery and concluding with the approval of new and innovative therapies, and bringing together expert teams of clinicians, scientists, patients, and policy makers to overcome present hurdles and accomplish these ends. Although the strategies we propose are ambitious, the resulting gains could bring us to a new age in the care of epilepsy, in which treatment shifts from loosely informed empiricism to data-driven and patient-centered precision therapy. In the words of one patient advocate, “Time is brain and we've lost too much of both. It's time for Covid-level collaboration that includes a National Strategy to cure the epilepsies.”91
Key Points.
Despite rapid discovery of genetic causes of epilepsy, precision therapies are not yet available for the majority of genetic epilepsies
Progress has been made in diagnosis, understanding natural histories, therapeutic development, preclinical models, and clinical trials
We provide an overview of progress and key remaining challenges for precision medicine for genetic epilepsy
We argue for coordinated and systematic streamlining of the epilepsy precision medicine pipeline, from gene discovery to clinical trials
Collaborative efforts of clinicians, scientists, patient advocates, and policy makers, as in the Epilepsy Leadership Council, are needed
ACKNOWLEDGMENTS
Many ideas in this review were inspired by the Epilepsy Precision Medicine Meeting in Washington, DC, September 2019, and we are grateful to the attendees for stimulating discussion. We gratefully acknowledge organizations who supported the meeting, including: CURE Epilepsy Foundation, Dravet Syndrome Foundation, Epilepsy Foundation, GeneDx, Invitae, Jonathan Mugar, Knopp Biosciences, Lennox Gastaut Syndrome Foundation, Praxis Precision Medicines, Syngap Research Fund, TESS Research Foundation, UCB, and Xenon. The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health (NIH) or the NIH Institutes and Centers. We would like to thank Eryn Fitch and Michael Kaufman for their efforts in coordination and contribution to the manuscript.
Funding information
I.H. was supported by the Hartwell Foundation through an Individual Biomedical Research Award. This work was also supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS; K02 NS112600), including support through the Channelopathy-Associated Epilepsy Research Center Without Walls (U54 NS108874), the Eunice Kennedy Shriver National Institute of Child Health and Human Development through the Intellectual and Developmental Disabilities Research Center at Children's Hospital of Philadelphia and the University of Pennsylvania (U54 HD086984), and intramural funds of the Children's Hospital of Philadelphia through the Epilepsy NeuroGenetics Initiative. Research reported in this publication was also supported by the NIH National Center for Advancing Translational Sciences under award number UL1TR001878. This project was also supported in part by the Institute for Translational Medicine and Therapeutics' Transdisciplinary Program in Translational Medicine and Therapeutics at the Perelman School of Medicine of the University of Pennsylvania. C.S.M. is supported by HHSN271201600048C, Screening of Investigational Compounds to Treat, Modify or Prevent Epilepsy for the NIH/NINDS Epilepsy Therapy Screening Program. J.K.K. is supported by NIH/NINDS K08NS119800, NIH/NINDS K12NS098482, the Taking Flight and Research Continuity awards from the CURE Epilepsy Foundation, the Elterman Award from the Child Neurology Society/Child Neurology Foundation, and the Stanford Maternal and Child Health Research Institute. E.M.G. is supported by NIH/NINDS K08 NS097633 and the Burroughs Wellcome Fund Career Award for Medical Scientists. A.L.G. is supported by NIH/NINDS U54 NS108874. L.L.I. is supported by NIH/NINDS R37 NS076752 and by a research grant to the University of Michigan from Stoke Therapeutics. H.L. is supported by grants from the German Research Foundation (Research Unit For-2715, grant Le1030/23-1), and the Federal Ministry of Education and Research in Germany (Treat-ION, 01GM1907A). S.F.B. is supported by a National Health and Medical Research Council Program Grant (ID: 1091593). S.D. has funding from NIH/NINDS U01NS114312, International Foundation for CDKL5 Research, Project 8P Foundation, and Mila's Miracle Foundation. S.W. is supported by grants from the Fonds Wetenschappelijk Onderzoek Fund and European Joint Program on Rare Diseases (1861419 N, G041821N, TreatKCNQ). L.S.L, V.W., and D.H.L. have no funding sources to report.
Footnotes
CONFLICT OF INTEREST
L.L.I. serves as cochair of the scientific advisory board of the Dravet Syndrome Foundation and served on the Board of the American Epilepsy Society. A portion of her research is funded by a grant to the University of Michigan from Stoke Therapeutics. S.D. has consulted for Upsher-Smith, BioMarin, Neurogene, Marinus, Tysha, and Ovid Therapeutics. S.D. also serves on the advisory board for the nonprofit foundations SLC6A1 Connect, Ring14 USA, Project 8P, and FamilieSCN2A. None of the other authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
REFERENCES
- 1.National Research Council of the National Academies. Toward precision medicine: building a knowledge network for biomedical research and a new taxonomy of disease. Washington, DC: The National Academies Press; 2011. [PubMed] [Google Scholar]
- 2.Niestroj LM, Perez-Palma E, Howrigan DP, Zhou Y, Cheng F, Saarentaus E, et al. Epilepsy subtype-specific copy number burden observed in a genome-wide study of 17 458 subjects. Brain. 2020;143:2106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leu C, Stevelink R, Smith AW, Goleva SB, Kanai M, Ferguson L, et al. Polygenic burden in focal and generalized epilepsies. Brain. 2019;142:3473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sisodiya SM. Precision medicine and therapies of the future. Epilepsia. 2021;62(Suppl 2):S90–S105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kearney H, Byrne S, Cavalleri GL, Delanty N. Tackling epilepsy with high-definition precision medicine: a review. JAMA Neurol. 2019;76:1109–16. [DOI] [PubMed] [Google Scholar]
- 6.Balestrini S, Sisodiya SM. Pharmacogenomics in epilepsy. Neurosci Lett. 2018;667:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol. 2018;75:279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Epi4K Consortium, Epilepsy Phenome/Genome Project. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.EpiPM Consortium. A roadmap for precision medicine in the epilepsies. Lancet Neurol. 2015;14:1219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guerrini R, Balestrini S, Wirrell EC, Walker MC. Monogenic epilepsies: disease mechanisms, clinical phenotypes, and targeted therapies. Neurology. 2021;97:817–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epi4K Consortium, Epilepsy Phenome/Genome Project. Ultrarare genetic variation in common epilepsies: a case-control sequencing study. Lancet Neurol. 2017;16:135–43. [DOI] [PubMed] [Google Scholar]
- 12.Azzariti DR, Hamosh A. Genomic data sharing for novel mendelian disease gene discovery: the matchmaker exchange. Annu Rev Genomics Hum Genet. 2020;21:305–26. [DOI] [PubMed] [Google Scholar]
- 13.Epi25 Collaborative. Ultra-rare genetic variation in the epilepsies: a whole-exome sequencing study of 17,606 individuals. Am J Hum Genet. 2019;105:267–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvill GL, Engel KL, Ramamurthy A, Cochran JN, Roovers J, Stamberger H, et al. Aberrant inclusion of a poison exon causes dravet syndrome and related SCN1A-associated genetic epilepsies. Am J Hum Genet. 2018;103:1022–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581–90. [DOI] [PubMed] [Google Scholar]
- 16.International League Against Epilepsy Consortium on Complex Epilepsies. Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat Commun. 2018;9:5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis CA, Petrovski S, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol. 2020;19:93–100. [DOI] [PubMed] [Google Scholar]
- 18.Perucca P, Bahlo M, Berkovic SF. The genetics of epilepsy. Annu Rev Genomics Hum Genet. 2020;21:205–30. [DOI] [PubMed] [Google Scholar]
- 19.Dunn P, Albury CL, Maksemous N, Benton MC, Sutherland HG, Smith RA, et al. Next generation sequencing methods for diagnosis of epilepsy syndromes. Front Genet. 2018;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheidley BR, Malinowski J, Bergner AL, Bier L, Gloss DS, Mu W, et al. Genetic testing for the epilepsies: a systematic review. Epilepsia. 2022;63:375–87. [DOI] [PubMed] [Google Scholar]
- 21.Lindy AS, Stosser MB, Butler E, Downtain-Pickersgill C, Shanmugham A, Retterer K, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59:1062–71. [DOI] [PubMed] [Google Scholar]
- 22.Truty R, Patil N, Sankar R, Sullivan J, Millichap J, Carvill G, et al. Possible precision medicine implications from genetic testing using combined detection of sequence and intragenic copy number variants in a large cohort with childhood epilepsy. Epilepsia Open. 2019;4:397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heyne HO, Artomov M, Battke F, Bianchini C, Smith DR, Liebmann N, et al. Targeted gene sequencing in 6994 individuals with neurodevelopmental disorder with epilepsy. Genet Med. 2019;21:2496–503. [DOI] [PubMed] [Google Scholar]
- 24.Gorzynski JE, Goenka SD, Shafin K, Jensen TD, Fisk DG, Grove ME, et al. Ultrarapid nanopore genome sequencing in a critical care setting. N Engl J Med. 2022;386:700–2. [DOI] [PubMed] [Google Scholar]
- 25.Australian Genomics Health Alliance Acute Care Flagship. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian public health care system. JAMA. 2020;323:2503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardakjian TM, Helbig I, Quinn C, Elman LB, McCluskey LF, Scherer SS, et al. Genetic test utilization and diagnostic yield in adult patients with neurological disorders. Neurogenetics. 2018;19:105–10. [DOI] [PubMed] [Google Scholar]
- 27.Kutscher EJ, Joshi SM, Patel AD, Hafeez B, Grinspan ZM. Barriers to genetic testing for pediatric medicaid beneficiaries with epilepsy. Pediatr Neurol. 2017;73:28–35. [DOI] [PubMed] [Google Scholar]
- 28.Erwin DJ, LaMaire C, Espana A, Eble TN, Dhar SU. Financial barriers in a county genetics clinic: problems and solutions. J Genet Couns. 2020;29:678–88. [DOI] [PubMed] [Google Scholar]
- 29.Gatto EM, Walker RH, Gonzalez C, Cesarini M, Cossu G, Stephen CD, et al. Worldwide barriers to genetic testing for movement disorders. Eur J Neurol. 2021;28:1901–9. [DOI] [PubMed] [Google Scholar]
- 30.Shur N, Atabaki SM, Kisling MS, Tabarani A, Williams C, Fraser JL, et al. Rapid deployment of a telemedicine care model for genetics and metabolism during COVID-19. Am J Med Genet A. 2021;185:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–8. [DOI] [PubMed] [Google Scholar]
- 32.Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, et al. STXBP1 encephalopathy: a neurodevelopmental disorder including epilepsy. Neurology. 2016;86:954–62. [DOI] [PubMed] [Google Scholar]
- 33.Tarquinio DC, Hou W, Berg A, Kaufmann WE, Lane JB, Skinner SA, et al. Longitudinal course of epilepsy in Rett syndrome and related disorders. Brain. 2017;140:306–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Demarest S, Pestana-Knight EM, Olson HE, Downs J, Marsh ED, Kaufmann WE, et al. Severity assessment in CDKL5 deficiency disorder. Pediatr Neurol. 2019;97:38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masten MC, Williams JD, Vermilion J, Adams HR, Vierhile A, Collins A, et al. The CLN3 disease staging system: a new tool for clinical research in Batten disease. Neurology. 2020;94:e2436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis-Smith D, Galer PD, Balagura G, Kearney H, Ganesan S, Cosico M, et al. Modeling seizures in the human phenotype ontology according to contemporary ILAE concepts makes big phenotypic data tractable. Epilepsia. 2021;62:1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sisodiya SM, Whelan CD, Hatton SN, Huynh K, Altmann A, Ryten M, et al. The ENIGMA-Epilepsy working group: mapping disease from large data sets. Hum Brain Mapp. 2020;43(1):113–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–9. [DOI] [PubMed] [Google Scholar]
- 39.Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American Consensus Panel. Pediatr Neurol. 2017;68:18–34.e13. [DOI] [PubMed] [Google Scholar]
- 40.Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: treatment options and management of prolonged seizures. Epilepsia. 2019;60(Suppl 3):S39–48. [DOI] [PubMed] [Google Scholar]
- 41.Wolff M, Johannesen KM, Hedrich UBS, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–36. [DOI] [PubMed] [Google Scholar]
- 42.Wolff M, Brunklaus A, Zuberi SM. Phenotypic spectrum and genetics of SCN2A-related disorders, treatment options, and outcomes in epilepsy and beyond. Epilepsia. 2019;60(Suppl 3):S59–67. [DOI] [PubMed] [Google Scholar]
- 43.Pisano T, Numis AL, Heavin SB, Weckhuysen S, Angriman M, Suls A, et al. Early and effective treatment of KCNQ2 encephalopathy. Epilepsia. 2015;56:685–91. [DOI] [PubMed] [Google Scholar]
- 44.Kuersten M, Tacke M, Gerstl L, Hoelz H, Stülpnagel C, Borggraefe I. Antiepileptic therapy approaches in KCNQ2 related epilepsy: a systematic review. Eur J Med Genet. 2020;63:103628. [DOI] [PubMed] [Google Scholar]
- 45.Klepper J. Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia. 2008;49(Suppl 8):46–9. [DOI] [PubMed] [Google Scholar]
- 46.Masnada S, Hedrich UBS, Gardella E, Schubert J, Kaiwar C, Klee EW, et al. Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain. 2017;140:2337–54. [DOI] [PubMed] [Google Scholar]
- 47.Hedrich UBS, Lauxmann S, Wolff M, Synofzik M, Bast T, Binelli A, et al. 4-Aminopyridine is a promising treatment option for patients with gain-of-function KCNA2-encephalopathy. Sci Transl Med. 2021;13:eaaz4957. [DOI] [PubMed] [Google Scholar]
- 48.Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol. 2020;77:300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pierson TM, Yuan H, Marsh ED, Fuentes-Fajardo K, Adams DR, Markello T, et al. GRIN2A mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol. 2014;1:190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol. 2014;76:457–61. [DOI] [PubMed] [Google Scholar]
- 51.Chong PF, Nakamura R, Saitsu H, Matsumoto N, Kira R. Ineffective quinidine therapy in early onset epileptic encephalopathy with KCNT1 mutation. Ann Neurol. 2016;79:502–3. [DOI] [PubMed] [Google Scholar]
- 52.Delahaye-Duriez A, Srivastava P, Shkura K, Langley SR, Laaniste L, Moreno-Moral A, et al. Rare and common epilepsies converge on a shared gene regulatory network providing opportunities for novel antiepileptic drug discovery. Genome Biol. 2016;17:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Atkin TA, Maher CM, Gerlach AC, Gay BC, Antonio BM, Santos SC, et al. A comprehensive approach to identifying repurposed drugs to treat SCN8A epilepsy. Epilepsia. 2018;59:802–13. [DOI] [PubMed] [Google Scholar]
- 54.Martin P, de Witte PAM, Maurice T, Gammaitoni A, Farfel G, Galer B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020;105:106989. [DOI] [PubMed] [Google Scholar]
- 55.Lagae L, Schoonjans AS, Gammaitoni AR, Galer BS, Ceulemans B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia. 2018;59:1881–8. [DOI] [PubMed] [Google Scholar]
- 56.Mercuri E, Darras BT, Chiriboga CA, Day JW, Campbell C, Connolly AM, et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med. 2018;378:625–35. [DOI] [PubMed] [Google Scholar]
- 57.Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22–31. [DOI] [PubMed] [Google Scholar]
- 59.Scoles DR, Minikel EV, Pulst SM. Antisense oligonucleotides: a primer. Neurol Genet. 2019;5:e323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lenk GM, Jafar-Nejad P, Hill SF, Huffman LD, Smolen CE, Wagnon JL, et al. Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann Neurol. 2020;87:339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li M, Jancovski N, Jafar-Nejad P, Burbano LE, Rollo B, Richards K, et al. Antisense oligonucleotide therapy reduces seizures and extends life span in an SCN2A gain-of-function epilepsy model. J Clin Invest. 2021;131:e152079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12:eaaz6100. [DOI] [PubMed] [Google Scholar]
- 63.Elgersma Y, Sonzogni M. UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. 2021;63:802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Turner TJ, Zourray C, Schorge S, Lignani G. Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J Neurochem. 2021;157:229–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colasante G, Lignani G, Brusco S, di Berardino C, Carpenter J, Giannelli S, et al. dCas9-based scn1a gene activation restores inhibitory interneuron excitability and attenuates seizures in Dravet syndrome mice. Mol Ther. 2020;28:235–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dey D, Eckle VS, Vitko I, Sullivan KA, Lasiecka ZM, Winckler B, et al. A potassium leak channel silences hyper-active neurons and ameliorates status epilepticus. Epilepsia. 2014;55:203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Agostinho AS, Mietzsch M, Zangrandi L, Kmiec I, Mutti A, Kraus L, et al. Dynorphin-based "release on demand" gene therapy for drug-resistant temporal lobe epilepsy. EMBO Mol Med. 2019;11:e9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Johannesen KM, Liu Y, Koko M, Gjerulfsen CE, Sonnenberg L, Schubert J, et al. Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain. 2021;awab321. 10.1093/brain/awab321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shellhaas RA, de Veber G, Bonkowsky JL, Child Neurology Society Research, Committee. Gene-targeted therapies in pediatric neurology: challenges and opportunities in diagnosis and delivery. Pediatr Neurol. 2021;125:53–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang SK, Vanoye CG, Misra SN, Echevarria DM, Calhoun JD, O'Connor JB, et al. Spectrum of KV 2.1 dysfunction in KCNB1-associated neurodevelopmental disorders. Ann Neurol. 2019;86:899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhindsa RS, Goldstein DB. Genetic discoveries drive molecular analyses and targeted therapeutic options in the epilepsies. Curr Neurol Neurosci Rep. 2015;15:70. [DOI] [PubMed] [Google Scholar]
- 73.Gullo F, Manfredi I, Lecchi M, Casari G, Wanke E, Becchetti A. Multi-electrode array study of neuronal cultures expressing nicotinic beta2-V287L subunits, linked to autosomal dominant nocturnal frontal lobe epilepsy. An in vitro model of spontaneous epilepsy. Front Neural Circuits. 2014;8:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parent JM, Anderson SA. Reprogramming patient-derived cells to study the epilepsies. Nat Neurosci. 2015;18:360–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O'Malley HA, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol. 2013;74:128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tidball AM, Lopez-Santiago LF, Yuan Y, Glenn TW, Margolis JL, Clayton Walker J, et al. Variant-specific changes in persistent or resurgent sodium current in SCN8A-related epilepsy patient-derived neurons. Brain. 2020;143:3025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirose S, Tanaka Y, Shibata M, Kimura Y, Ishikawa M, Higurashi N, et al. Application of induced pluripotent stem cells in epilepsy. Mol Cell Neurosci. 2020;108:103535. [DOI] [PubMed] [Google Scholar]
- 78.Odawara A, Matsuda N, Ishibashi Y, Yokoi R, Suzuki I. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Sci Rep. 2018;8:10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niu W, Parent JM. Modeling genetic epilepsies in a dish. Dev Dyn. 2020;249:56–75. [DOI] [PubMed] [Google Scholar]
- 80.Mellios N, Feldman DA, Sheridan SD, Ip JPK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry. 2018;23:1051–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bershteyn M, Nowakowski TJ, Pollen AA, di Lullo E, Nene A, Wynshaw-Boris A, et al. Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell. 2017;20:435–449.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun AX, Yuan Q, Fukuda M, Yu W, Yan H, Lim GGY, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 2019;366:1486–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blair JD, Hockemeyer D, Bateup HS. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med. 2018;24:1568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fontana BD, Mezzomo NJ, Kalueff AV, Rosemberg DB. The developing utility of zebrafish models of neurological and neuropsychiatric disorders: a critical review. Exp Neurol. 2018;299:157–71. [DOI] [PubMed] [Google Scholar]
- 85.Griffin A, Hamling KR, Knupp K, Hong S, Lee LP, Baraban SC. Clemizole and modulators of serotonin signalling suppress seizures in Dravet syndrome. Brain. 2017;140:669–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim J, Hu C, Moufawad el Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381:1644–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abrahamyan L, Feldman BM, Tomlinson G, Faughnan ME, Johnson SR, Diamond IR, et al. Alternative designs for clinical trials in rare diseases. Am J Med Genet C Semin Med Genet. 2016;172:313–31. [DOI] [PubMed] [Google Scholar]
- 88.Muller AR, MMMG, van de Ven PM, KCB R, Cornel MC, van CDM K, et al. Systematic review of N-of-1 studies in rare genetic neurodevelopmental disorders: the power of 1. Neurology. 2021;96:529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bateman-House A, Kearns L. Individualized therapeutics development for rare diseases: the current ethical landscape and policy responses. Nucleic Acid Ther. 2022;32:111–7. [DOI] [PubMed] [Google Scholar]
- 90.Woodcock J, Marks P. Drug regulation in the era of individualized therapies. N Engl J Med. 2019;381:1678–80. [DOI] [PubMed] [Google Scholar]
- 91.Penn Miller I, Hecker JE, Fureman B, Meskis MA, Roberds S, Jones M, et al. Epilepsy community at an inflection point: translating research toward curing the epilepsies and improving patient outcomes. Epilepsy Curr. 2021;21:385–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schreiber JM, Tochen L, Brown M, Evans S, Ball LJ, Bumbut A, et al. A multi-disciplinary clinic for SCN8A-related epilepsy. Epilepsy Res. 2020;159:106261. [DOI] [PubMed] [Google Scholar]
- 93.Epilepsy Genetics Initiative. The Epilepsy Genetics Initiative: systematic reanalysis of diagnostic exomes increases yield. Epilepsia. 2019;60:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Splinter K, Adams DR, Bacino CA, Bellen HJ, Bernstein JA, Cheatle-Jarvela AM, et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N Engl J Med. 2018;379:2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pernici CD, Mensah JA, Dahle EJ, Johnson KJ, Handy L, Buxton L, et al. Development of an antiseizure drug screening platform for Dravet syndrome at the NINDS contract site for the Epilepsy Therapy Screening Program. Epilepsia. 2021;62:1665–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grinspan ZM, Patel AD, Shellhaas RA, Berg AT, Axeen ET, Bolton J, et al. Design and implementation of electronic health record common data elements for pediatric epilepsy: foundations for a learning health care system. Epilepsia. 2021;62:198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marsh ED, Whittemore V, Leenders M, Poduri A, American Epilepsy Society Epilepsy Research Benchmark Stewards Committee. The 2021 epilepsy research benchmarks-respecting core principles, reflecting evolving community priorities. Epilepsy Curr. 2021;21:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]