

Abstract
Introduction.
Sarcoma clinical outcomes have been stagnant for decades due to heterogeneity of primaries, lack of comprehensive preclinical models, and rarity of disease. We hypothesized that engineering hydrogel-based sarcoma organoids directly from the patient without xenogeneic extracellular matrices (ECMs) or growth factors is routinely feasible and allows rare tumors to remain viable as avatars for personalized research.
Methods.
Surgically resected sarcomas (angiosarcomas, leiomyosarcoma, gastrointestinal stromal tumor, liposarcoma, myxofibrosarcoma, dermatofibrosarcoma protuberans [DFSP], and pleiomorphic abdominal sarcoma) were dissociated and incorporated into a hyaluronic acid and collagen-based ECM hydrogel and screened for chemotherapy efficacy. A subset of organoids was enriched with a patient-matched immune system for screening of immunotherapy efficacy (iPTOs). Response to treatment was assessed using LIVE/DEAD staining and metabolic assays.
Results.
Sixteen sarcomas were biofabricated into three-dimensional (3D) patient-specific sarcoma organoids with a 100% success rate. Average time from organoid development to initiation of drug testing was 7 days. Enrichment of organoids with immune system components derived from either peripheral blood mononuclear cells or lymph node cells was performed in 10/16 (62.5%) patients; 4/12 (33%) organoids did not respond to chemotherapy, while response to immunotherapy was observed in 2/10 (20%) iPTOs.
Conclusions.
A large subset of sarcoma organoids does not exhibit response to chemotherapy or immunotherapy, as currently seen in clinical practice. Routine development of sarcoma hydrogel-based organoids directly from the operating room is a feasible platform, allowing for such rare tumors to remain viable for personalized translational research.
Sarcomas are rare cancers with an expected incidence of 0.7% of all adult malignancies.1 Their pathology classification incorporates heterogeneous cohorts of at least 50 subtypes with major differences in biological behavior and prognosis, hindering the development of effective drug-based therapies.2 Surgery remains the main treatment modality for the majority of sarcomas, complimented in selected cases with radiation and or systemic chemotherapy.3–5 Unfortunately, the rarity of these diseases is an obstacle for clinical trial accrual in evaluating chemotherapies and immunotherapies, while lack of reliable preclinical models has been an equal limitation for research.6
We have previously generated patient tumor organoids (PTOs) from surgically resected specimens of several tumor types, including lung, peritoneal mesothelioma, melanoma, colorectal, appendiceal, and glioma, utilizing a unique three-dimensional (3D) extracellular matrix (ECM)-derived hydrogel that is less potent than basement membrane extract (BME)-derived gels.7–11 Additionally, we have enriched PTOs with patient-matched immune cells (iPTOs) in an attempt to generate a reproducible personalized platform to assess immunotherapy efficacy.10–14 These PTOs provide results within 2 weeks from surgery, with support by a hydrogel scaffold that bypasses any interference from uncharacterized cytokines, mRNAs, and exosomes present in animal tumor-derived ECM materials (BME biomaterials).9–11,13,15–18 We employ a simple, yet supportive hydrogel ECM microenvironment based on chemically modified hyaluronic acid (HA) and denatured collagen (gelatin) utilizing a safe high-throughput manufacturing method.
In this study, we demonstrate the benefits of redirecting use of surgical specimens away from traditional tumor banks to support personalized sarcoma PTO biofabrication for a wide variety of sarcoma subtypes, eliminating the time-consuming need of cell expansion. While chemotherapies remain the primary drug-based treatment for most sarcomas, we explored the use of iPTOs to evaluate the personalized preclinical efficacy of immunotherapy where sufficiently powered clinical trials are likely not feasible. Furthermore, the benefits of testing non-expanded organoids developed from fresh specimens were exhibited by comparing them with organoids created from expanded cells and organoids from a new sarcoma cell line developed from the same patient.
MATERIALS AND METHODS
Sarcoma Biospecimen Acquisition and Processing
All biospecimens were obtained from surgically treated patients in adherence to Wake Forest Baptist Medical Center Institutional Review Board protocols. Upon resection, tumor specimens were transferred by a dedicated procurement agent to the central pathology suite within the surgical operating suites area. Specimens were inked for margins and reviewed prior to providing a piece from the center of the tumor mass. In this way, margins were not compromised. In cases where margins were not important (e.g. stage IV patients undergoing metastasectomy), our protocols allow for direct acquisition of tumor directly from the operating suite to the laboratory. The specimens were placed in RPMI (Hyclone, Logan, UT, USA) and transferred within a 2-h window post-resection.
The tumors were washed in phosphate-buffered saline (PBS; Hyclone), with 2% penicillin–streptomycin (Thermo Fisher Scientific, Waltham, MA, USA). Tumor tissues were minced and transferred to a 15 mL conical tube with Dulbecco’s modified eagle medium (DMEM) low glucose (Hyclone), supplemented with 80,000 collagenase degrading activity (CDA) collagenase HA and 22,000 neutral protease assessment (NPA) BP protease (Vitacyte, Indianapolis, IN, USA), with 3cc solution per 1cc minced tumor volume. The digestion solution was agitated at 37°C until tissue was dissolved. The digested tumor solution was neutralized using cold high glucose DMEM (Hyclone), supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific) then filtered through a 100-micron steriflip cell filter (Millipore Sigma, Burlington, MA, USA) and centrifuged. The resulting cell pellet underwent red cell lysis (RBC; BD Pharm Lyse, San Diego, CA, USA) according to the manufacturer’s protocol. Cell count was performed, and dead cells were removed using a dead cell removal kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Lymphocytes were isolated from lymph node tissue as described above, or from patient whole blood through Ficoll-Paque Plus (Cytiva, Marlborough, MA, USA) density gradient isolation, followed by RBC lysis. The cell suspension was then prepared for organoid fabrication.
Extracellular Matrix Hydrogel Preparation and Tumor Organoid Biofabrication
The HA/gelatin system (HyStem HP, ESI-BIO, Alameda, CA, USA) was utilized as previously described.7,8,10,11,17,19,20 All components were dissolved in sterile water containing 0.05% w/v Irgacure 3959 (Sigma, St. Louis, MO, USA) to make 1% w/v solution of thiol-modified heparin (Heprasil®), thiol-modified denatured collagen (Gelin-S), and thiol reactive crosslinker, PEGDA (Extralink). All components were mixed with the cell suspension at a cell density of 10 million cells/mL hydrogel precursor solution in a 2:2:1 ratio, respectively. iPTOs were created at a ratio of 1:3 tumor cells to immune cells. Organoids were formed by pipetting 5 μL of hydrogel precursor cell suspension into 96-well plates previously coated with polydimethylsiloxane (PDMS) to form round droplets, which were subsequently photo-crosslinked by ultraviolet light exposure (365 nm, 18 W cm2) for 1 s. The organoids were maintained in DMEM with 10% FBS with 1% penicillin–streptomycin, and 1% L-glutamine.
Therapy Screening Studies
Therapy screening was initiated on day 7 following organoid biofabrication. Treatments were tailored for each sarcoma subtype based on preoperative biopsies or final surgical pathology. The number of therapies and doses involved varied based on available cells. The therapies used for sarcoma organoids were doxorubicin (0.1, 1, 10 μM) [S1208, Selleckchem, Houston, TX, USA], ifosfamide (2, 20, 200 μM) [I4909, Sigma-Aldrich], temozolomide (10, 100, 1000 μM) [T2577, Sigma-Aldrich], imatinib mesylate (1, 10, 100 μM) [STI571, Selleckchem], regorafenib (0.1, 1, 10 μM) [S1178, Selleckchem], gemcitabine (1, 10, 100 μM) [G6423, Sigma-Aldrich], olaparib (0.1, 1, 10 μM) [S1060, Selleckchem], 100 nM pembrolizumab (A2005, Selleckchem,), 100 nM nivolumab (A2002, Selleckchem), and 100 nM ipilimumab (A2001, Selleckchem). Drug containing-media was added to organoid wells for 72 h, followed by viability assays.
Organoid Viability Assays
At therapy treatment cessation, organoid viability was assessed using Promega CellTiter-Glo® (Promega, Madison, WI, USA) according to company protocol, and read on a Veritas Microplate Luminometer (Turner BioSystems). Viability was also assessed using LIVE/DEAD staining (Thermo Fisher Scientific) according to manufacturer’s instructions. Fluorescent imaging was performed using a Leica TCS LSI macro confocal microscope (Leica Microsystem Inc, Buffalo, NY, USA), with red and green channels overlaid.
Immortalization
Hygromycin B Antibody Selection
DFSP1 passage 1 tumor cells were cultured at 2×105 per well in a 24-well plate. At 80% confluence, hygromycin B (S2908, Selleckchem) was added at 0, 50, 100, 250, and 500 μg/mL, replacing the treated media every 3 days for 1 week. The culture was examined for toxicity and the lowest dose for which all cells were dead for positive selection of transfected gene was 100 μg/mL.
Virus Production
For virus particle expression, HEK-293 (ATCC® CRL-1573™) was plated overnight in a 10 cm tissue culture plate with DMEM-10. The plasmid DNA, packaging, and envelope plasmids were obtained as gifts from Dr. Baisong Lu’s laboratory (Lentiviral vector pLVHtert-IRES-hydro, plasmid #85140, pMD2.G plasmid #12259, and psPAX2 plasmid #12260; Addgene, Watertown, MA, USA). The lentiviral transfer vector DNA, together with psPAX2 packaging and pMD2.G envelope plasmid DNA were combined at 3:2:1, respectively, and the 18 μg DNA mix was added to 250 μL of Opti-MEM media. Similarly, 196 μL of Opti-MEM media is added to 54 μL of FuGENE® HD Transfection Reagent (Promega). The DNA mix and FuGENE® mix was combined and added to the plate with 7 mL of Opti-MEM. Twelve hours later, media was replaced with 8 mL of OptiMEM media. Twenty-four hours later, the virus-containing supernatant is collected and filtered. The supernatant was either used to transfect cells or frozen at −80°C for future use.
Transfection of Cells from DFSP with Sarcomatoid Changes
Patient-derived DFSP passage 1 were plated at a density of 5 × 104 cells/well in 6-well plates and cultured overnight. Media was replaced with a combination of 400μL viral supernatant and 2 mL of OptiMEM in four wells overnight, with two wells maintained as controls. The medium was changed with 1:1 DMEM-10 and Opti-MEM. The cells were maintained for three days, then 100 μg/ mL of hygromycin in DMEM-10 was used for selection and propagation of immortalized cell line.
RNAseq of Dermatofibrosarcoma Protuberans (DFSP) Cells
Immortalized and non-immortalized DFSP1 cells were collected at passages 4, 8, and 12 and snap frozen. RNA isolation was performed using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD, USA) following the manufacturer’s protocol. RNA concentration was assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
RNA (200 ng) was treated with DNase prior to constructing sequencing libraries using an NEBNext Ultra II Directional RNA Library preparation kit in conjunction with polyA mRNA selection beads. Paired-end 150-bp reads were generated on an Illumina NovaSeq 6000 to obtain >15 million reads per sample. Low-quality reads (q < 10) and adaptor sequences were eliminated using bbduk version 37.64. Reads were aligned to the GRCh38.p9 assembly of the human reference genome from the National Center for Biotechnology Information (NCBI) using version 2.6.0c of the aligner STAR.21 Features were counted using featureCounts software22 against a GFF file from Gencode (v28) and used for DESeq223 to generate a normalized count file, with all samples compared with the originating tumor biospecimen as the ‘control.’ Normalized counts were used as input for all RNA-seq analyses.
Definition of Treatment Response
Efficacy in PTOs treated by chemotherapy was defined by two criteria: (1) post-treatment adenosine triphosphate (ATP) viability of <50%; and (2) a statistically significant reduction in viability for treated organoids compared with controls. Immunotherapy efficacy required three criteria to be met by iPTOs: (1) demonstrate a statistically significant reduction in cell viability compared with iPTO control organoids (Ex: iPTO control vs. iPTO treated); (2) demonstrate a statistically significant reduction in cell viability when comparing treated organoids from immune-enhanced counter conditions and non-immune-enhanced counter conditions (Ex: nivolumab-treated iPTO vs. nivolumab-treated PTO); and (3) exhibit a post-immunotherapy ATP viability <50%. This approach was used to reduce the probability of a type 1 error occurring, which would be 0.125%, or 12.5 in 10,000. By using a similar standard, chemotherapy treatment probability of a type 1 error would be 2.5%, or 25 in 1000. In this study, we arbitrarily selected 50% killing of the tumor as the threshold suggestive of treatment response. This number is customizable according to investigators’ demands.
Statistical Analysis
All data are expressed as mean ± standard deviation for each experimental group. Each treatment and condition combination consisted of three or more organoids. Upon review of the CellTiter-Glo® results, some organoid replicates were excluded in the final analysis as their ATP levels were considered as outliers (more than two standard deviations from the mean). ATP assay values of treated organoids were standardized to condition-matched (iPTO or PTO) controls prior to statistical analysis. Two sample t-tests were used to assess differences in treatment conditions. Drug screen studies were determined to be successful if patient control PTOs demonstrated adequate viability at day 10 of culture. Adequate viability is described as blank value <1% of the control condition. Statistical analysis was performed using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) and a p value <0.05 was used as the threshold for statistical significance.
RESULTS
Sarcoma Biospecimen Procurement to Organoid Biofabrication
Sixteen tissue specimens were procured from April 2018–July 2021 (Fig. 1). Matched blood was provided for 15/16 (93.8%) specimens and lymph tissue was provided for 2/16 (12.5%) specimens. Organoid fabrication was performed for all specimens. However, due to low yield of either tumor or immune cells, chemotherapy screening was performed for 12/16 (75%) PTO sets, while immunotherapy screening was performed in 10/15 (66.7%) of PTO and iPTO matched sets (Fig. 1). Average time from organoid development to initiation of drug testing was 7 days.
FIG. 1.
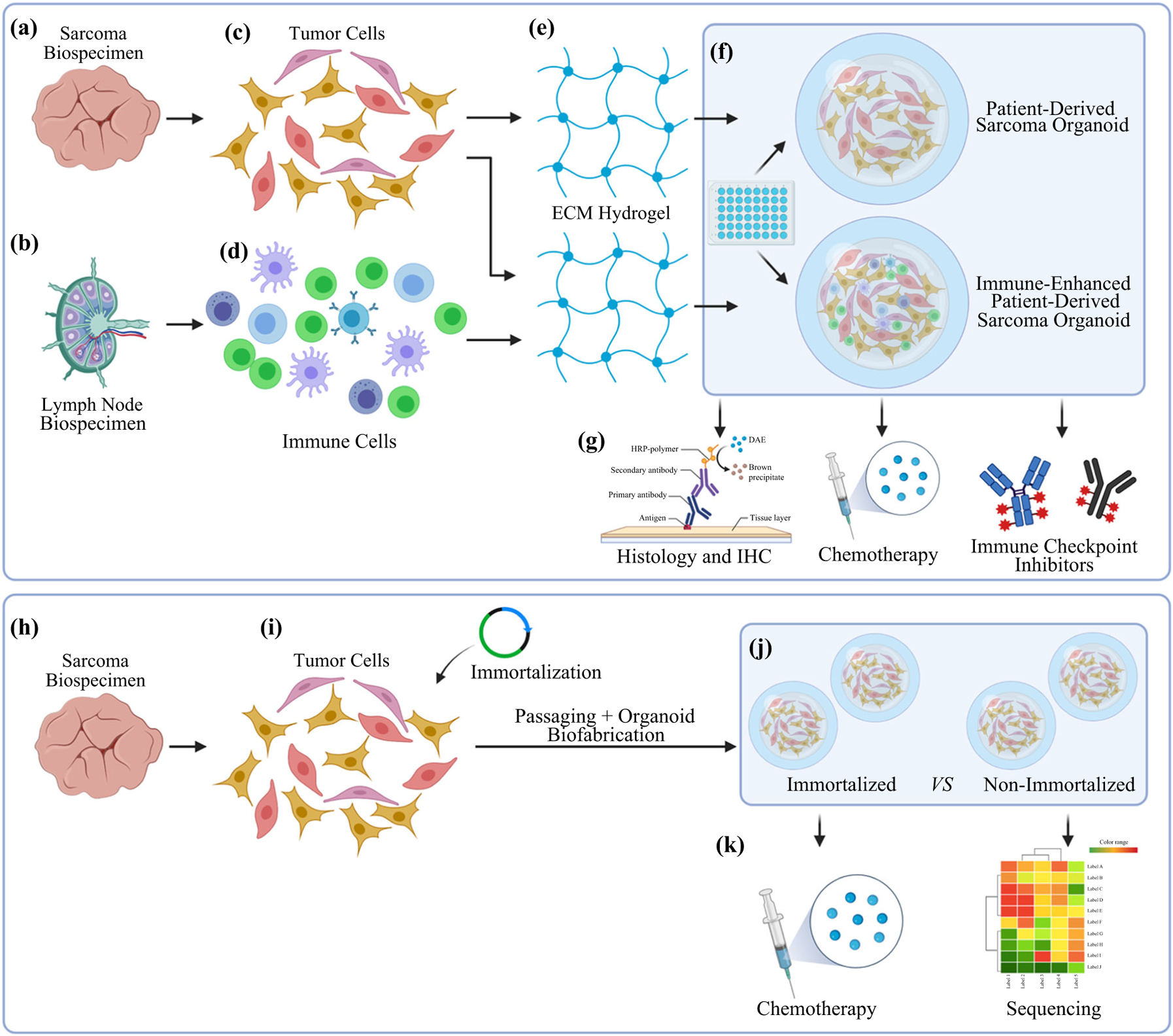
Study design and flowchart of specimen processing and the biofabrication approach. a–g PTO and iPTO construction and subsequent analyses. h–k Generation of an immortalized DFSP cell line and analyses of organoids
Viability, Histology, and Immunohistochemical Characterization of Sarcoma Organoids
To confirm viability in PTO culture, multiple viability assays were performed. LIVE/DEAD staining demonstrated robust cell viability in PTO cultures at day 7, with high numbers of viable cells (green) and few dead cells (red) [Fig. 2a]. Immunohistochemistry was performed on day 10 PTOs to analyze cell markers. By hematoxylin and eosin (H&E) stain, both the organoids and matched tumor tissues showed atypical spindle cell proliferation in LMS, PLS and DFSP patients. While organoids lost the tumor architecture, their cytomorphologies were identical to matched tumor tissues. Similarly, organoids stained positive for Ki67, indicating maintained cell proliferation in PTO culture after isolation from tissues. Tissue and organoids were stained with markers indicative of sarcoma subtypes. When compared, these patient-matched tissues and organoids displayed similar expression of markers, including CD34, PARP1, vimentin, and matrix metalloproteinases (MMP)-9 (Fig. 2b–d). Positive expression of vimentin demonstrates mesenchymal tissue origin, while highly positive expression of CD34 is an identifying marker of DFSP (Fig. 2d).24,25 Similarly, MMP9 expression has been linked to angiogenesis and metastatic behavior, while PARP1 expression suggests therapeutic sensitivity towards PARP inhibitors.26,27
FIG. 2.
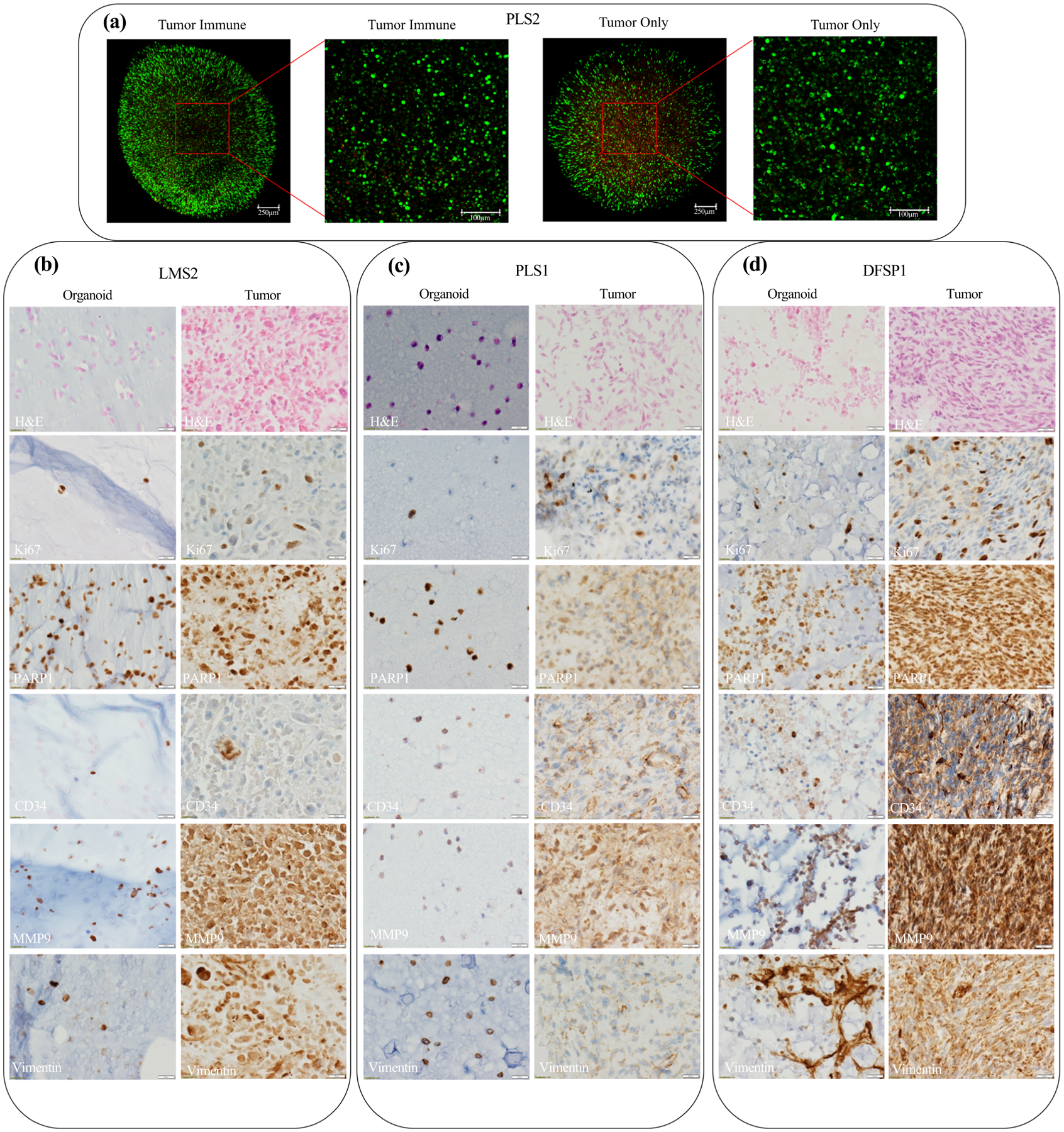
Characterization of sarcoma PTOs. a Cells in organoids demonstrated robust viability in both tumor and tumor-immune organoids when utilizing LIVE/DEAD viability staining at day 7. H&E, ki67, PARP1, CD34, MMP9, and vimentin immunohistochemistry staining of patient-matched tissues and day 10 organoids for b leiomyosarcoma patient 2, c pleiomorphic sarcoma patient 1, and d dermatofibrosarcoma protuberans patient 1 demonstrate similar cytomorphologies and expression of markers. All images taken at ×40 magnification.
Sarcoma Organoid Drug Response in Chemotherapy Screens
Sarcoma PTOs were treated with a variety of chemotherapeutics based on clinically approved treatments and number of cells. PTOs demonstrated different responses based on dose and treatments administered, with doxorubicin being the most effective tested (Table 1 and Fig. 3a, c). LIVE/DEAD assays demonstrated a decrease in viable cells, with an increase in dead cells for PTO set LMS3 (Fig. 3b) and MFS2 (Fig. 3d).
TABLE 1.
Summary of patient and tumor organoids therapy responses
| Tumor ID | Sarcoma subtype | Organoid treatments | Effective organoid treatments | Patient prior treatment(s) | Adjuvant therapy | Current clinical |
|---|---|---|---|---|---|---|
| AGS1 | Angiosarcoma (Scalp) | Dabrafenib/Trametinib, Pembrolizumab, Nivolumab | None | None | Taxol | Deceased |
| AGS2 | Angiosarcoma (Scalp) | Cisplatin, Dabrafenib/trametinib, Pembrolizumab, Ipilimumab | None | None | Taxol/Gemcitabine | Deceased |
| AGS3 | Angiosarcoma liver metastasis (Scalp primary) | Cisplatin, Dabrafenib/Trametinib, Pembrolizumab, Ipilimumab, Nivolumab | None | Paclitaxel, Doxorubicin/Olaratumab, CAD; Scalp resection,right lung metre section | None | NED |
| DFSP1 | Dermato fibrosarcoma Protuberans (Trunk) | Doxorubicin, Regorafenib, Imatinib, Pembrolizumab, Nivolumab | Doxorubicin, Regorafenib,Imatinib, Nivolumab | None | None | No follow-up |
| DFSP2 | Dermato fibrosarcoma Protuberans (UE) | Doxorubicin, Regorafenib, Imatinib | Doxorubicin, Imatinib | None | None | No follow-up |
| GIST1 | GI Stromal Tumor (RP) | None | NA | Imatinib, Sunitinib, Regorafenib; SBR | Nilotinib, Avapritinib, Repretinib | Deceased |
| LMS1 | Pleomorphic Leiomyosarcoma(LE) | Doxorubicin, Pembrolizumab, Ipilimumab,Nivolumab | Doxorubicin | Radiation | None | Deceased |
| LMS2 | Leiomyosarcoma (RP) | Doxorubicin, Pembrolizumab, Ipilimumab,Nivolumab | Doxorubicin | None | Radiation,Gemcitabine/Docetaxel | Deceased |
| LMS3 | Leiomyosarcoma (Pelvis) | Regorafenib, Gemcitabine, Olaparib,Doxorubicin, Ifosfamide, Doxorubicin/Ifosfamide, Pembrolizumab, Ipilimumab,Nivolumab | Doxorubicin, Doxorubicin/Ifosfamide, Nivolumab | None | None | NED |
| LMS4 | Leiomyosarcoma (RP) | Pembrolizumab, Ipilimumab, Nivolumab | None | None | None | NED |
| LPS1 | Liposarcoma (LE) | Pembrolizumab, Ipilimumab, Nivolumab | None | Radiation;Gemcitabine/Docetaxel | None | NED |
| MFS1 | Myxofibrosarcoma (UE) | Doxorubicin, Imatinib, Temozolomide | None | None | Radiation | NED |
| MFS2 | Myxofibrosarcoma (UE) | Doxorubicin, Ifosfamide,Doxorubicin/Ifosfamide, Vinorelbine,Gemcitabine, Vinorelbine/Gemcitabine | Doxorubicin, Doxorubicin/Ifosfamide, Gemcitabine,Vinorelbine/Gemcitabine | None | Radiation | NED |
| PLS1 | Pleomorphic Sarcoma (RP) | Doxorubicin, Irinotecan | Doxorubicin | CRS, SBR, bladder resection | None | NED |
| PLS2 | Pleomorphic Sarcoma (Trunk) | None | NA | Radiation | None | NED |
| PLS3 | Pleomorphic Sarcoma (RP) | Regorafenib, Gemcitabine, Olaparib,Imatinib, Temozolomide, Doxorubicin,Ifosfamide, Doxorubicin/Ifosfamide,Doxorubicin/Temozolomide, Pembrolizumab, Ipilimumab, Nivolumab | Gemcitabine, Imatinib,Doxorubicin/Ifosfamide,Doxorubicin/Temozolomide | None | None | Deceased |
CAD cyclophosphamide, adriamycin, dacarbazine, CRS cytoreductive surgery, SBR small bowel resection, NED no evidence of disease, UE upper extremity, LE lower extremity, RP retroperitoneum, NA not available
FIG. 3.
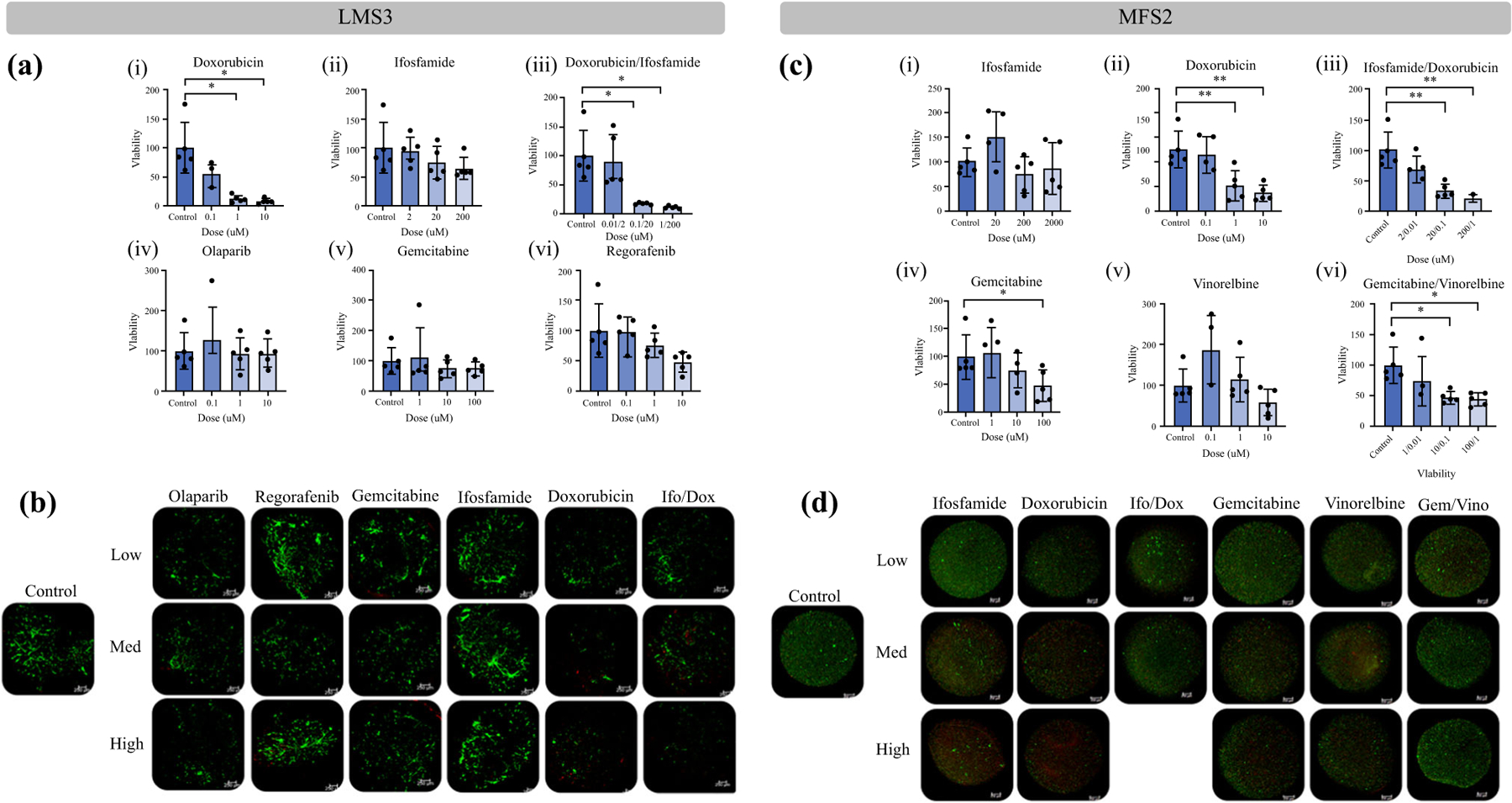
Evaluation of post-chemotherapy tumor viability in leiomyosarcoma LMS3 and myxofibrosarcoma MFS2 PTOs. a Drug screening performed on PTO set LMS3 demonstrated dose-dependent increases in drug efficacy for (i) doxorubicin (7.5% viability), (iii) doxorubicin combined with ifosfamide (10%), and (vi) regorafenib (49%), while demonstrating lack of efficacy to (ii) ifosfamide (65%), (iv) olaparib (126%), and (v) gemcitabine (74%). b LIVE/DEAD analysis further confirmed drug efficacy when comparing number of alive (green) cells with dead (red) cells. c Drug screening performed on PTO set MFS2 demonstrated dose-dependent increases in drug efficacy for (ii) doxorubicin (28%), (iii) doxorubicin combined with ifosfamide (22%), (iv) gemcitabine (47%), and (vi) gemcitabine combined with vinorelbine (44), while demonstrating lack of efficacy to (i) ifosfamide (87%) and (v) vinorelbine (57%). d LIVE/DEAD analysis further confirmed drug efficacy when comparing total number of alive (green) cells with dead (red) cells. Scale bar = 250 μM. Statistical significance: *p < 0.05. Post-treatment PTO viabilities are shown in parentheses
Immune-Enhanced Sarcoma Organoid Response to Immune Checkpoint Inhibitors
To analyze the effect of immune checkpoint blockade, iPTOs were developed using immune cells from either patient blood or resected lymph tissue. These iPTOs were tested with three agents: pembrolizumab, ipilimumab and nivolumab. Response to immunotherapy that was drug-, type of tumor-, and patient-specific was observed in a minority of specimens. More specifically, two iPTO sets (DFSP1 and leiomyosarcoma LMS3) demonstrated sensitivity towards nivolumab (2/9, 22.2%), with post-treatment iPTO viability of 7.6% and 49%, respectively (Fig. 4a, b). No iPTOs were sensitive to ipilimumab (0/9).
FIG. 4.
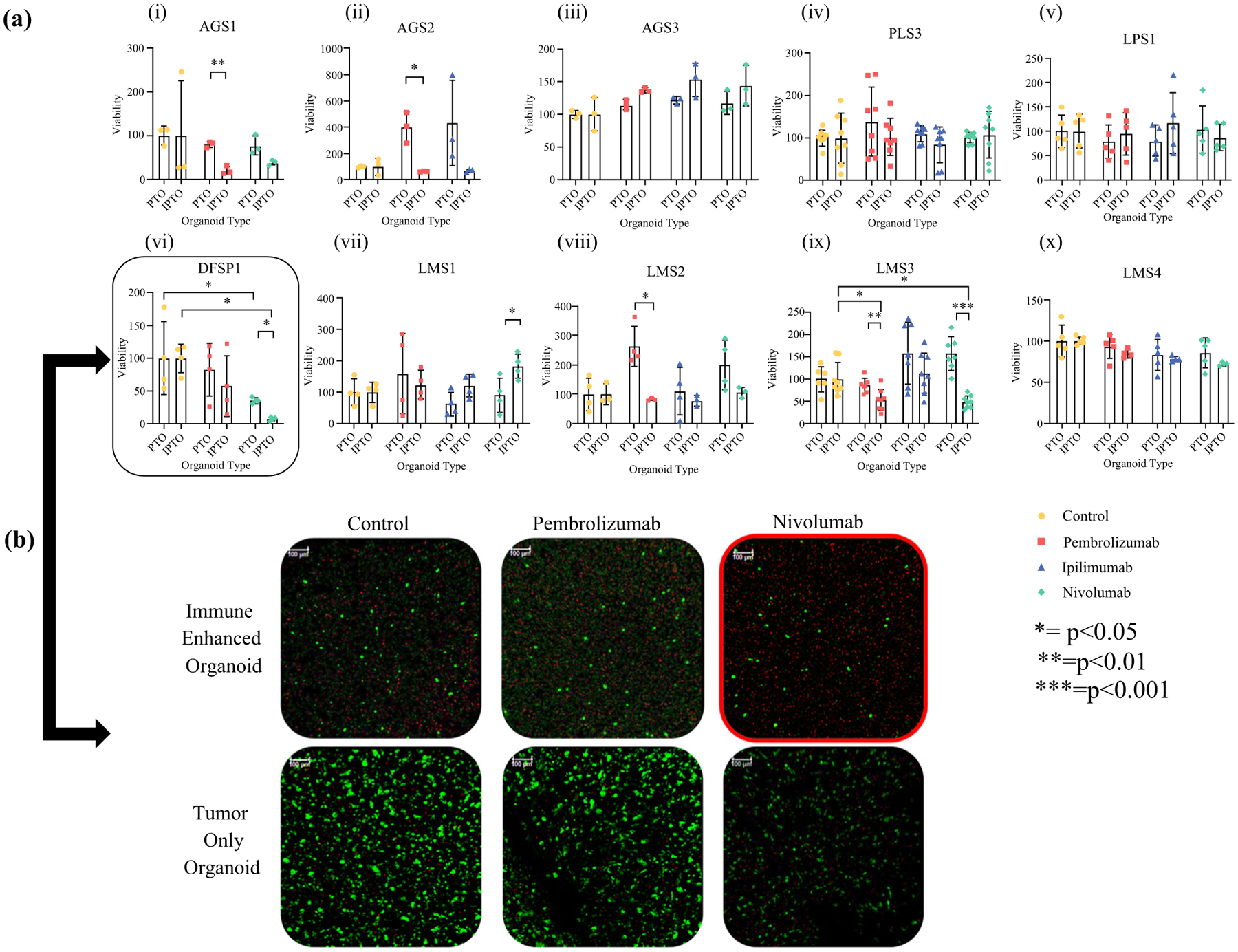
Immune system enhancement of sarcoma PTOs enables responses to immune checkpoint inhibitor treatments. a Immunotherapy screening performed on matched PTO and iPTO sets. Nivolumab (green) was effective in 2/9 (22.2%) PTO sets [(vi) DFSP1 and (ix) Leiomyosarcoma LMS3]. b LIVE/DEAD analysis further confirmed immunotherapy efficacy in DFSP1 when comparing number of living cells (green) with condition-matched controls. Scale bar = 250 μM. Statistical significance indicated within. Response to immunotherapy defined as statistical significance between condition-matched control, PTO treatment-matched condition, and >50% cell killing
In addition, four pembrolizumab-treated iPTOs (angiosarcomas AGS1/2 and leiomyosarcomas LMS2/3), demonstrated attenuated post-immunotherapy viabilities when compared with their PTO counterparts or their iPTO controls, without achieving all parameters for immunotherapy efficacy.
Genetic Drift in Sarcoma Cells Through Expansion and Immortalization
To analyze the chemotherapy response drift over time between cells obtained from the same patient, we tested cells obtained directly from a DFSP with sarcomatoid changes versus expanding cells, as well as cells from a cell line developed through hTERT immortalization. We compared the response of immortalized and post-expansion cells with the same chemotherapies used in the initial screening of the DFSP1 PTOs, immediately after tumor dissociation. The original DFSP1 tumor PTO cells responded to imatinib, doxorubicin, and regorafenib (Fig. 5a), while the immortalized tumor cells maintained imatinib and doxorubicin response through the passages but were resistant to regorafenib until passage 12 (Fig. 5b–d). Resistance to regorafenib in immortalized cells may be conferred due to increased telomerase activity through Notch-1 signaling.28 Non-immortalized cells maintained sensitivity to imatinib but became resistant to doxorubicin at passage 8 and regorafenib in passage 12. Furthermore, while both populations maintained the expression of CD34, the cellular morphology of immortalized cells retained the mesenchymal phenotype of the original tumor, whereas non-immortalized cells developed an epithelial phenotype (Fig. 6a).
FIG. 5.
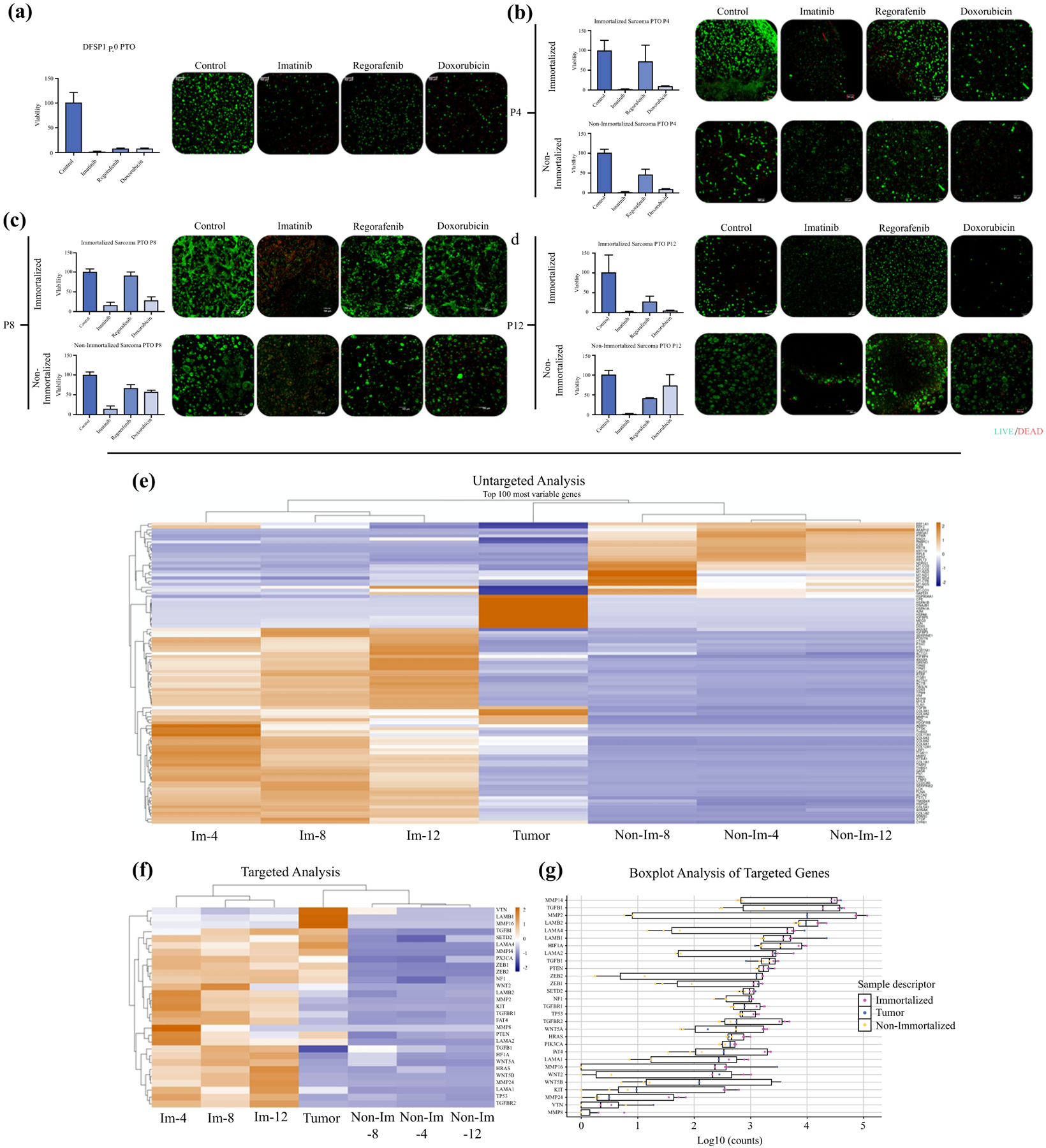
Evaluation of drug screening of immortalized versus non-immortalized cells versus originating PTOs. Drug response to imatinib, regorafenib, and doxorubicin in a original PTOs, passage b 4, c 8, and d 12 organoids prepared with immortalized or non-immortalized cells. e–g Genomic analysis of immortalized versus non-immortalized DFSP cells via RNAseq. e Untargeted cluster analysis of the top 100 variably expressed genes. f Targeted cluster analysis of a subset of genes of interest. g Boxplot analysis of the same subset of genes of interest. Statistical significance: *p < 0.05
FIG. 6.
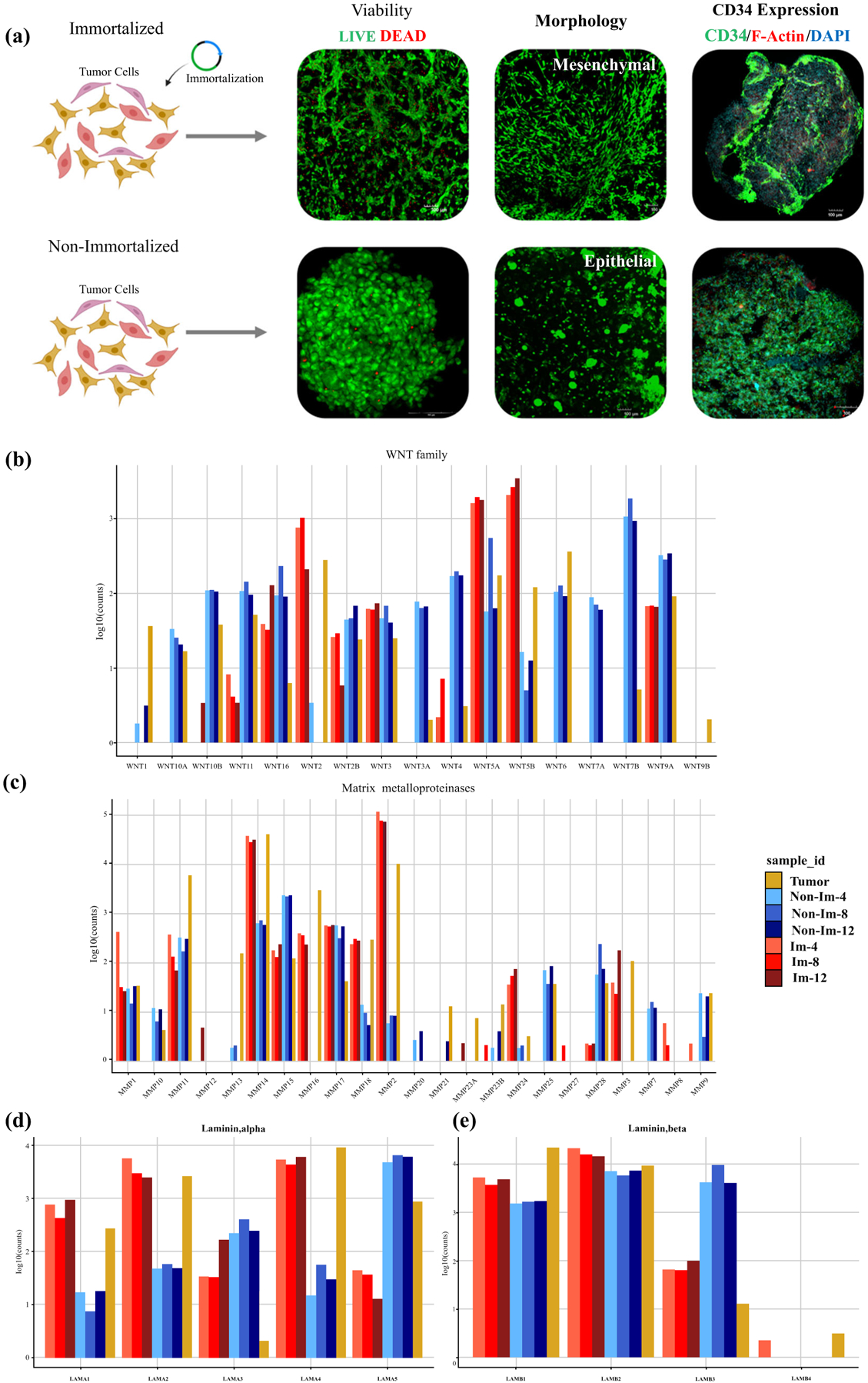
Evaluation of immortalized and non-immortalized DFSP1 organoids showing a viability (green—calcein AM-stained viable cells; red—dead ethidium homodimer-stained nuclei), morphology, and expression of CD34 (green), F-actin (red), and DAPI (blue). Relative gene expression values of b Wnt family genes, c MMPs, and laminin d alpha and e beta subunits. MMPs matrix metalloproteinases
RNAseq Analysis of Immortalized Versus Non-immortalized DFSP Cells
To evaluate the impact of immortalization on the genomic profile of DFSP cells, we performed bulk RNAseq on cells from the original tumor and after 4, 8, or 12 passages in immortalized and non-immortalized cells. First, an untargeted cluster analysis was performed on the 100 most variably expressed genes (Fig. 5e). This analysis shows a stark difference between immortalized cells and non-immortalized cells at each passage assessed. Next, we used a targeted analysis of 29 genes implicated to ECM composition (vitronectin, laminins, MMPs), Wnt pathway, TP53, and tumor progression (Fig. 5f). This analysis indicates the non-immortalized cells exhibit a decreased expression, while the immortalized cells exhibit an increase of these genes. Figure 5g further shows a boxplot analysis of the gene expression relative changes in relation to originating tumor biospecimen.
Next, we quantified relative expression of Wnt family genes, MMP family genes, and laminin family genes (Fig. 6b).29,30 Visualization of relative expression of each Wnt gene indicated large differences between expression levels, with Wnt2, Wnt5A, and Wnt 5B being expressed at higher levels in immortalized cells, better resembling the original tumor (Fig. 6b).
As with Wnt, MMP expression is crucial for tumor progression and metastasis through degradation of ECM components. MMP14, MMP 2, and MMP9 varied significantly between immortalized versus non-immortalized cells (Fig. 6c). Laminin is comprised of alpha, beta, and gamma subunits and is important in ECM remodeling. The combinations of alpha and beta subunits account for major differences in specificity for cell adhesions.31 In Fig. 6d, we see a clear divergence between laminin alpha subunit genes, with LAMA1, LAMA2, and LAMA4 nearer to original tumor tissue, while LAMA5 is in the converse relationship. In terms of laminin beta (Fig. 6e), we see little differences in expression between all groups in LAMB1, LAMB2, LAMB4; however, we see a striking difference in LAMB3, where the immortalized cells show decreased expression compared with the elevated profile of non-immortalized cells.
DISCUSSION
The rarity and heterogeneity of sarcoma subtypes have proven to be impeding factors in clinical trial accrual, as demonstrated by the almost stagnant sarcoma clinical outcomes after multimodality treatment. The above is unfortunately true for most rare primaries. We have previously shown the ability to test both clinically available and experimental therapies utilizing PTO cultures in a variety of tumors, including rare primaries.7–11,13,14,17 In this study, we describe the generation of PTOs derived from a variety of surgically resected sarcoma subtypes to be applied in personalized translational research. In addition, a new DFSP cell line was generated and applied to demonstrate differences between PTOs and traditional research tools such as cell line or expanded cells when all three cell construct types were developed from tissue from the same patient.
In the current work, 33% of sarcoma PTOs exhibited no response to chemotherapy, while a smaller minority showed response to immunotherapy that is aligned with clinical observations. Personalized iPTO efficacy with immune checkpoint inhibitors (ICIs) was observed in angiosarcoma, leiomyosarcoma, and DFSP subtypes. Immunotherapy has become an increasingly effective therapeutic option in many cancers, and its application in sarcomas is being explored but is currently recommended only for the treatment of tumors with increased PD-1, PDL1 expression, deficiency in mismatch repair proteins, or increased tumor mutational burden (TMB).32 The Alliance A0911401 study recently demonstrated responses with combination immunotherapy in unresectable or metastatic angiosarcoma and leiomyosarcoma subtypes that is aligned with our findings.33 Extensive clinical data on immunotherapy efficacy in these specific sarcoma subtypes do not exist and are unlikely to be generated from cohort analysis. Our work herein builds on our previous work with integrating immune cells into melanoma and appendiceal cancer PTOs to enable assessment of likelihood of response to ICIs.10–12 The implications of a platform to test therapies for not only T-cell targeting but also other components of the immune-tumor microenvironment, such as natural killer (NK) cells, antigen-presenting cells, B cells, and regulatory T cells, can revolutionize treatment of tumors not previously targeted through immunotherapy due to lack of indication of actionable targets.34,35
In addition, our studies describe PTOs that do not utilize BME-derived biomaterials, such as Matrigel. The decision to not utilize Matrigel like most laboratories lies within the confidence of the hydrogel platform with which we have generated viable organoids or 3D cultures across cancer types.7–11,14,17,19,20,36
Surgical specialties have a major advantage in incorporating similar work within already existing clinical practice line frameworks, resulting in the generation of results within 10 days from resection. As demonstrated here, avoiding expansion of cells preserves the characteristics of the resected specimens without the genetic drift observed in expanding cells or cells lines even after a limited number of passages. Redirecting specimens away from traditional ‘dead tissue’ tumor banks into personalized living tissue repositories fits well in the clinical setting where decisions on appropriate treatment needs to be implemented within 2–4 weeks. The presented PTO establishment rate of 100% and therapeutic testing of 85% is above reported values, ranging from 50 to 80% for organoids from more common tumors, including breast, colon and gastric.37,38 Although the differences in establishment rate are due to many factors, the hydrogel employed for cellular encapsulation and support is vitally important. Creating collagen and HA-based, tissue-specific ECM with the accurate physical and chemical properties allows for cells to maintain ex vivo properties similar to human tissue.39,40
There is a rapidly increasing volume of data where organoids employed either in the setting of prospective randomized trials or retrospective analysis demonstrated 100% negative predictive value in a variety of different primaries.9,11,37,41–46 In other words, when the organoid does not respond to chemotherapy the patient also does not respond. Whether or not the same is true for positive predictive value is too early to say due to the impact of intra-tumor heterogeneity and sampling that may miss a non-responding malignant clone. However, it is logical that the survival of any cancer patient is ultimately determined by non-responding cells. Therefore, we ask if the time has arrived to develop and test a framework (especially in tumors such as sarcomas with a high percentage of poor response to chemotherapy), where organoids will be used to pre-emptively identify patients who will not respond to chemotherapy or immunotherapy, so they can be spared from the morbidity and costs associated with non-effective and unnecessary treatments.
To our knowledge, this is the first study utilizing sarcoma PTOs to study personalized treatment responses, yet it comes with several limitations. More specifically, clinical correlation with patient outcomes was not feasible, especially for immunotherapy, because it is not currently within sarcoma treatment algorithms due to a paucity of clinical efficacy data. Thus, it is uncertain to what degree the PTOs in this study correlate with patient results, although others have shown promising efforts in relating PTO response to patient outcomes.10,37 Finally, radiation in PTOs is an emerging field, but our study was not designed to incorporate this therapy.47
CONCLUSION
Despite the apparent benefits of incorporating organoid technology in transforming tissue biobanks into a library of living tissue, we do not claim organoids are ready to be applied in clinical practice without the backing of sufficiently powered correlative data with patient outcomes. However, this work shows the potential benefits and challenges ahead of us. There is only one way to proceed: to incorporate PTOs as a correlative companion tool into existing prospective randomized trials along with extensive genomic and proteomic characterization. Until then, we will continue to apply cohort analysis data on individual patients who often will not respond to systemic therapy.
ACKNOWLEDGMENT
The authors graciously acknowledge the work of Libby McWilliams and Kathleen Perry (Wake Forest Baptist Medical Center) for their assistance in tissue procurement and data management, and Parisa Javidi-Parsijani and Baisong Lu (Wake Forest Institute for Regenerative Medicine) for their assistance in our cell immortalization studies.
FUNDING
Steven D. Forsythe is supported by a grant from the National Institutes of Health (T32CA247819). Konstantinos I. Votanopoulos acknowledges funding from the NIH/NCI R01CA249087, R01CA258692, R01A158569, and Wake Forest Comprehensive Cancer Center. Aleksander Skardal acknowledges funding from the National Institutes of Health and National Cancer Institute grant R21CA229027, the Wake Forest Comprehensive Cancer Center, the Ohio State University Comprehensive Cancer Center, and the Pelotonia Foundation. Hemamylammal Sivakumar, Richard A. Erali, Nadeem Wajih, Wencheng Li, Perry Shen, Edward A. Levine, and Katherine E. Miller have no conflicts of interest to declare.
Footnotes
DISCLOSURE Aleksander Skardal is an inventor on several patents pertaining to bioengineered tumor organoids derived from clinical biospecimens. Konstantinos Votanopoulos holds organoid technology patents not currently licensed. Steven D. Forsythe, Hemamylammal Sivakumar, Richard A. Erali, Nadeem Wajih, Wencheng Li, Perry Shen, Edward A. Levine, and Katherine E. Miller have no conflicts of interest to declare
REFERENCES
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Kallen ME, Hornick JL. The 2020 WHO classification: what’s new in soft tissue tumor pathology? Am J Surg Pathol. 2021;45:e1–23. 10.1097/PAS.0000000000001552. [DOI] [PubMed] [Google Scholar]
- 3.Chouliaras K, et al. Role of radiation therapy for retroperitoneal sarcomas: an eight-institution study from the US Sarcoma Collaborative. J Surg Oncol. 2019;120:1227–34. 10.1002/jso.25694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chouliaras K, et al. Recurrence patterns after resection of retroperitoneal sarcomas: an eight-institution study from the US Sarcoma Collaborative. J Surg Oncol. 2019;120:340–7. 10.1002/jso.25606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gortzak E, et al. A randomised phase II study on neo-adjuvant chemotherapy for “high-risk” adult soft-tissue sarcoma. Eur J Cancer. 2001;37:1096–103. 10.1016/s0959-8049(01)00083-1. [DOI] [PubMed] [Google Scholar]
- 6.Ratan R, Patel SR. Chemotherapy for soft tissue sarcoma. Cancer. 2016;122:2952–60. 10.1002/cncr.30191. [DOI] [PubMed] [Google Scholar]
- 7.Maloney E, et al. Immersion bioprinting of tumor organoids in multi-well plates for increasing chemotherapy screening throughput. Micromachines (Basel). 2020. 10.3390/mi11020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazzocchi A, et al. Pleural effusion aspirate for use in 3D lung cancer modeling and chemotherapy screening. ACS Biomater Sci Eng. 2019;5:1937–43. 10.1021/acsbiomaterials.8b01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazzocchi AR, Rajan SAP, Votanopoulos KI, Hall AR, Skardal A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci Rep. 2018;8:2886. 10.1038/s41598-018-21200-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Votanopoulos KI, et al. Appendiceal cancer patient-specific tumor organoid model for predicting chemotherapy efficacy prior to initiation of treatment: a feasibility study. Ann Surg Oncol. 2019;26:139–47. 10.1245/s10434-018-7008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Votanopoulos KI, et al. Model of patient-specific immune-enhanced organoids for immunotherapy screening: feasibility study. Ann Surg Oncol. 2020;27:1956–67. 10.1245/s10434-019-08143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Votanopoulos KI, Skardal A. ASO Author Reflections: Co-cultured lymph node and tumor organoids as a platform for the creation of adaptive immunity and predict response to immunotherapy. Ann Surg Oncol. 2020. 10.1245/s10434-020-08351-7. [DOI] [PubMed] [Google Scholar]
- 13.Forsythe SD, et al. Organoid platform in preclinical investigation of personalized immunotherapy efficacy in appendiceal cancer: feasibility study. Clin Cancer Res. 2021;27:5141–50. 10.1158/1078-0432.CCR-21-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forsythe S, et al. Development of a colorectal cancer 3D micro-tumor construct platform from cell lines and patient tumor biospecimens for standard-of-care and experimental drug screening. Ann Biomed Eng. 2020;48:940–52. 10.1007/s10439-019-02269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aisenbrey EA, Murphy WL. Synthetic alternatives to Matrigel. Nat Rev Mater. 2020;5:539–51. 10.1038/s41578-020-0199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devarasetty M, Forsythe SD, Shelkey E, Soker S. In vitro modeling of the tumor microenvironment in tumor organoids. Tissue Eng Regen Med. 2020;17:759–71. 10.1007/s13770-020-00258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsythe SD, et al. Personalized identification of optimal HIPEC perfusion protocol in patient-derived tumor organoid platform. Ann Surg Oncol. 2020;27:4950–60. 10.1245/s10434-020-08790-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clark CC, Aleman J, Mutkus L, Skardal A. A mechanically robust thixotropic collagen and hyaluronic acid bioink supplemented with gelatin nanoparticles. Bioprinting. 2019;16:e00058. 10.1016/j.bprint.2019.e00058. [DOI] [Google Scholar]
- 19.Aleman J, Skardal A. A multi-site metastasis-on-a-chip micro-physiological system for assessing metastatic preference of cancer cells. Biotechnol Bioeng. 2018. 10.1002/bit.26871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forsythe SD, et al. Environmental toxin screening using human-derived 3D bioengineered liver and cardiac organoids. Front Public Health. 2018;6:103. 10.3389/fpubh.2018.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–30. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lisovsky M, et al. Apolipoprotein D in CD34-positive and CD34-negative cutaneous neoplasms: a useful marker in differentiating superficial acral fibromyxoma from dermatofibrosarcoma protuberans. Mod Pathol. 2008;21:31–8. 10.1038/modpathol.3800971. [DOI] [PubMed] [Google Scholar]
- 25.Leader M, Collins M, Patel J, Henry K. Vimentin: an evaluation of its role as a tumour marker. Histopathology. 1987;11:63–72. 10.1111/j.1365-2559.1987.tb02609.x. [DOI] [PubMed] [Google Scholar]
- 26.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 27.Pignochino Y, et al. PARP1 expression drives the synergistic antitumor activity of trabectedin and PARP1 inhibitors in sarcoma preclinical models. Mol Cancer. 2017;16:86. 10.1186/s12943-017-0652-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirone G, Perna S, Shukla A, Marfe G. Involvement of Notch-1 in resistance to Regorafenib in colon cancer cells. J Cell Physiol. 2016;231:1097–105. 10.1002/jcp.25206. [DOI] [PubMed] [Google Scholar]
- 29.Skardal A, Devarasetty M, Rodman C, Atala A, Soker S. Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Ann Biomed Eng. 2015;43:2361–73. 10.1007/s10439-015-1298-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36:1461–73. 10.1038/onc.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dogic D, Rousselle P, Aumailley M. Cell adhesion to laminin 1 or 5 induces isoform-specific clustering of integrins and other focal adhesion components. J Cell Sci. 1998;111(Pt 6):793–802. [DOI] [PubMed] [Google Scholar]
- 32.FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. 2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication.
- 33.D’Angelo SP, et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018;19:416–26. 10.1016/S1470-2045(18)30006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wculek SK, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 35.Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507–19. 10.1038/s41422-020-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aleman J, et al. Deconstructed microfluidic bone marrow on-a-chip to study normal and malignant hemopoietic cell-niche interactions. Small. 2019;15:e1902971. 10.1002/smll.201902971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlachogiannis G, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920–6. 10.1126/science.aao2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachs N, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172:373–386.e310. 10.1016/j.cell.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 39.Skardal A, Devarasetty M, Forsythe S, Atala A, Soker S. A reductionist metastasis-on-a-chip platform for in vitro tumor progression modeling and drug screening. Biotechnol Bioeng. 2016;113:2020–32. 10.1002/bit.25950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mazzocchi A, Devarasetty M, Huntwork R, Soker S, Skardal A. Optimization of collagen type I-hyaluronan hybrid bioink for 3D bioprinted liver microenvironments. Biofabrication. 2018;11:015003. 10.1088/1758-5090/aae543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jacob F, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 2020;180:188–204.e122. 10.1016/j.cell.2019.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steele NG, et al. An organoid-based preclinical model of human gastric cancer. Cell Mol Gastroenterol Hepatol. 2019;7:161–84. 10.1016/j.jcmgh.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiriac H, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018;8:1112–29. 10.1158/2159-8290.CD-18-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharick JT, et al. Metabolic heterogeneity in patient tumor-derived organoids by primary site and drug treatment. Front Oncol. 2020;10:553. 10.3389/fonc.2020.00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat Commun. 2018;9:2983. 10.1038/s41467-018-05190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Phan N, et al. A simple high-throughput approach identifies actionable drug sensitivities in patient-derived tumor organoids. Commun Biol. 2019;2:78. 10.1038/s42003-019-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nagle PW, Coppes RP. Current and future perspectives of the use of organoids in radiobiology. Cells. 2020;9:2649. 10.3390/cells9122649. [DOI] [PMC free article] [PubMed] [Google Scholar]