


Abstract
Multifunctional protein APE1/APEX1/HAP1/Ref-1 (designated as APE1) plays important roles in nuclease-mediated DNA repair and redox regulation in transcription. However, it is unclear how APE1 regulates the DNA damage response (DDR) pathways. Here we show that siRNA-mediated APE1-knockdown or APE1 inhibitor treatment attenuates the ATR–Chk1 DDR under stress conditions in multiple immortalized cell lines. Congruently, APE1 overexpression (APE1-OE) activates the ATR DDR under unperturbed conditions, which is independent of APE1 nuclease and redox functions. Structural and functional analysis reveals a direct requirement of the extreme N-terminal motif within APE1 in the assembly of distinct biomolecular condensates in vitro and DNA/RNA-independent activation of the ATR DDR. Overexpressed APE1 co-localizes with nucleolar NPM1 and assembles biomolecular condensates in nucleoli in cancer but not non-malignant cells, which recruits ATR and activator molecules TopBP1 and ETAA1. APE1 protein can directly activate ATR to phosphorylate its substrate Chk1 in in vitro kinase assays. W119R mutant of APE1 is deficient in nucleolar condensation, and is incapable of activating nucleolar ATR DDR in cells and ATR kinase in vitro. APE1-OE-induced nucleolar ATR DDR activation leads to compromised ribosomal RNA transcription and reduced cell viability. Taken together, we propose distinct mechanisms by which APE1 regulates ATR DDR pathways.
INTRODUCTION
It is estimated that >10 000 Apurinic/Apyrimidinic (AP) sites are produced per day in each mammalian cell under unperturbed conditions (1–3). AP sites are typically repaired by base excision repair (BER) and nucleotide excision repair (NER) mechanisms in normal cells (4–6). In the BER pathway, AP sites are catalyzed into single strand breaks (SSBs) with heterogenous 3' and/or 5' termini via several mechanisms including hydrolysis catalyzed by AP endonuclease 1 (APE1) and β- or β,δ-elimination by bifunctional DNA glycosylases (3,7,8). SSBs are primarily repaired via a global PARP1/XRCC1-mediated SSB repair pathway into intact DNA, while other homology-based DNA repair mechanisms may serve as auxiliary (9–11). Delayed or improper repair of AP sites and SSBs compromises DNA replication and transcription programs, which can ultimately result in cancer and neurodegenerative disorders (9,12–14). To maintain genome integrity, the ATM-Chk2 DNA damage response (DDR) pathway is activated by DNA double-strand breaks (DSBs) through a dimer dissociation mechanism of ATM and the recruitment of the Mre11-Rad50-Nbs1 (MRN) complex (15–17). As a sensor of reactive oxygen species (ROS), ATM DDR is activated by oxidative stress via a cysteine-mediated ATM dimer formation (18). In addition, stalled DNA replication forks and assorted DNA damage events can activate the ATR–Chk1 DDR pathway (2,19,20), in which TopBP1 and ETAA1 play direct roles in the activation of ATR kinase via distinct but evolutionarily conserved ATR activation domain (AAD) (21–25). RPA-coated ssDNA (RPA-ssDNA) at damage site is widely accepted as a scaffolding for ATR-ATRIP recruitment and activation (20,26). We have demonstrated recently that the ATR DDR pathway can also be activated by oxidative DNA damage and plasmid-based defined SSB structures in Xenopus egg extracts (27–29). It remains unknown whether and how AP sites and SSBs can trigger ATR-mediated DDR to maintain genome stability in mammalian cell systems.
Accumulating evidence has revealed that the ATM/ATR-mediated DDR pathways can be activated in the nucleolus, a non-membrane bound subnuclear compartment known for ribosomal RNA (rRNA) synthesis and organized by nucleolar organizing regions (NORs) located on acrosomal chromosomes (chromosome 13, 14, 15, 21 and 22 in humans) (30). Nucleoli are composed of three sub-nucleolar compartments: the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC) (31). It is generally accepted that pre-rRNA is transcribed from rDNA in the FC and/or DFC. FCs are enriched in components of the RNA Pol I machinery such as UBF, whereas the DFC harbors pre-rRNA processing factors such as FIB1. Both the FC and the DFC are enclosed by the GC, which is enriched in NPM1 for pre-ribosome subunit assembly (32). Ribosomal DNA (rDNA) in the nucleolus is transcribed into rRNA by RNA polymerase I, and such rDNA regions are often challenged by various DNA damage likely due to high rate of transcription (33). Previous work has revealed that in response to nucleolar DSBs, ATM is activated as a nucleolar DDR (nDDR) to inhibit RNA polymerase I-mediated rRNA transcription and to recruit homologous recombination (HR)-directed DNA repair machinery such as BRCA1, 53BP1, RPA32, and Rad51/Rad52 for faithful repair independent of cell cycle stages (34,35). Mechanistic studies demonstrated the implication of Nbs1, Mdc1, and Treacle (also known as TCOF1) in the ATM-mediated inhibition of rRNA transcription in nDDR (34,36–39). Interestingly, site-specific DSB generation in nucleoli by CRISPR/Cas9 or I-PpoI endonuclease also triggers the activation of an ATR-dependent nDDR, which functions downstream of the ATM/Treacle/MRN-mediated nDDR; however, DSB-induced ATR nDDR does not include Chk1/Chk2-mediated cell cycle arrest (35,39,40). In contrast, hypoosmotic stress activates TopBP1/Treacle-dependent ATR nDDR in nucleoli, due to increased R-loop stabilization and RPA-ssDNA formation on actively transcribed rDNA, which functions upstream of ATM nDDR (41). Moreover, DNA replication stress induced by hydroxyurea or aphidicolin also triggers a Treacle/TopBP1-dependent ATR-mediated nDDR activation in nucleoli (42). TopBP1 overexpression can also induce ATR nDDR and suppression of rRNA transcription in the absence of rDNA damage breaks (43). Nevertheless, our understanding on the molecular mechanisms of ATR-mediated nDDR pathways remains incomplete.
The multifunctional protein APE1/APEX1/HAP1/Ref-1 (designated as APE1) exhibits AP endonuclease, 3'-5' exonuclease, 3'-phosphodiesterase as well as 3' RNA phosphatase and 3' exoribonuclease activities and has been implicated in the BER pathway and redox regulation of transcription (44–48). Due to its cysteine residues (e.g. Cys65 and Cys93), APE1 maintains the reducing form of transcription factors (e.g. AP-1, NF-κB, HIF-1α and p53) to upregulate their DNA binding capacity (48–51). APE1 is an essential gene for early embryonic development in mice and APE1-null cells do not generally survive (52,53). APE1 protein exhibits subcellular localization in the nuclei and mitochondria which is mediated by its extreme N-terminal 33 amino acids (NT33) and its extreme C-terminal 30 amino acids (CT30), respectively (54–56). APE1 is also shown to associate with nucleolar protein Nucleiophosmin (NPM1) and participants in RNA quality control and RNA metabolism (57–59). Whereas the role of APE1 in the processing of AP sites via its endonuclease activity is widely accepted, APE1 exonuclease activity in genome integrity has not been recognized until recently (60–62). In particular, biochemical, biophysical, and structural model strongly supports the notion that APE1 preferentially senses and binds to SSB structures (60–62). We have demonstrated that APE1 plays an upstream role of APE2 in the SSB-induced ATR DDR pathway activation in Xenopus egg extracts (29,60,63). While APE2 is demonstrated as a general regulator of the global ATR DDR pathway in pancreatic cancer cells (64), it remains unclear whether APE1 plays direct roles in the activation of ATR-dependent nuclear and/or nucleolar DDR pathway in mammalian cells.
Liquid–liquid phase separation (LLPS) by biomolecular condensates has been recently reported in several cellular processes (65). At the molecular level, nucleoli in Xenopus laevis oocytes can form multiple LLPS that underlies nucleolar sub-compartments by NPM1 and fibrillarin (FIB1) (32,66,67). Inducible ectopic expression of TopBP1’s AAD domain is sufficient to activate a global ATR DDR but not ATR nDDR driving cells into p53-mediated senescence (68). Intriguingly, ectopic overexpression of full-length TopBP1 induces ATR nDDR under unperturbed conditions (43). These studies suggest that TopBP1 regions other than the AAD domain are required for the ATR nDDR. Interestingly, TopBP1 undergoes LLPS in vitro and assembles nuclear condensates to switch on ATR DDR signaling (69,70). Although ∼49 APE1-interacting proteins (e.g. NPM1, FUS, APP, LGALS3 and HNRNPA1) are involved in biomolecular condensates (71,72); however, it remains unknown whether and how APE1 forms LLPS.
Here, we provide evidence using cultured mammalian cells and reconstitution systems that APE1 plays critical functions in the ATR–Chk1 DDR signaling pathway via several distinct regulatory mechanisms. siRNA-mediated APE1-knockdown (APE1-KD) or APE1 nuclease specific inhibitors compromised the ATR–Chk1 DDR pathway under different stress conditions in several cell lines, suggesting that APE1 and its nuclease activity are important for the ATR–Chk1 DDR pathway. Overexpression of wild type (WT) or mutant APE1, that lacks nuclease or redox function, activated ATR–Chk1 DDR pathway in cultured cells under unperturbed conditions, suggesting that APE1 plays a previously unidentified role in ATR DDR activation via a catalytically independent fashion. Excess addition of recombinant APE1 protein in nuclear extracts directly activated the ATR–Chk1 DDR pathway, which required APE1 NT33 motif but not nuclease/redox functions. APE1 assembled distinct biomolecular condensates in a DNA/RNA-independent manner to associate and recruit ATR, TopBP1 and ETAA1 in nuclear extracts in vitro. APE1-OE co-localized with NPM1 and recruited ATR, TopBP1 and ETAA1 to nucleoli in cultured cells to activate ATR nDDR pathway. Elevated DNA damage load, cell cycle arrest, decreased rRNA transcription, and reduced cell viability underlie the physiological significance of the distinct ATR nDDR pathway via APE1-assembled biomolecular condensates in nucleolus. Taken together, our results shed new light on the distinct regulation of global and nucleolar ATR DDR pathway by APE1 in genome integrity maintenance.
MATERIALS AND METHODS
Cell culture and preparation of total cell lysates and nuclear extracts
MDA-MB-231, PANC1, HEK293 and U2OS cells were purchased from ATCC, and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37°C in CO2 incubator (5%). HPDE cells were gift of Dr Pinku Mukherjee and originally purchased from ATCC (73), and cultured in DMEM supplemented with 10% FBS, 1× glutamine mixture and penicillin (100 U/ml) and streptomycin (100μg/ml). For stress condition experiments, cells were treated with H2O2 (1.25 mM, Sigma Cat#HX0635), Camptothecin (CPT, 10 μM, Calbiochem Cat#208925), or methyl methanesulfonate (MMS, 0.3 mg/ml, Sigma Cat#129925) and incubated for 2 h before cell collection and further analysis. Cells were treated with VE-822 (Selleckchem Cat#S7102), KU55933 (EMD Millipore Cat#118500), or NU7441 (Selleckchem Cat#S2638), APE1iIII (Axon Medchem Cat#2137), AR03 (Axon Medchem Cat#2136) or E3330 (Novusbio Cat#MBP1-49581) to final concentrations and incubated for the periods as indicated. Generally, CPT, MMS, VE-822, KU55933, NU7441, APE1iIII, AR03 and E3330 were dissolved in DMSO as stock solutions and saved at –20°C for use.
After different treatments, cells were washed with phosphate-buffered saline (PBS) and resuspended in Lysis Buffer A (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.5% Nonidet P-40, 0.5 mM Na3V04, 5 mM NaF, 5 μg/ml of Aprotinin and 10 μg/ml of Leupeptin). The total cell lysates were isolated by centrifugation at 13 000 rpm for 30 min at 4°C, as recently described (64). The nuclear extracts were prepared as previously described (60) and briefly as follows. After washing with PBS and resuspension in Solution A (20 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2), cells were incubated on ice for 15 min, supplemented with Nonidet P-40 (a final concentration of 0.5%), and vortexed for 10 s. The samples were centrifuged at 3000 rpm for 10 min to separate permeabilized nuclei from cytoplasmic fraction. The recovered nuclei were lysed with Lysis Buffer A and centrifuged at 13 000 rpm at 4°C for 30 min to prepare nuclear extracts.
APE1 overexpression and siRNA-mediated knockdown experiments in cells
For overexpression assays, various expression plasmids (e.g. YFP, WT/mutant YFP-APE1, mCherry, mCherry-APE1, tGFP, tGFP-APE1 or △N33 tGFP-APE1, etc.) was added to cells at about 30% confluence via Lipofectamine 2000 transfection method and cultured for different times as indicated. For tGFP and tGFP-tagged protein expression, different doses of doxycycline was added 1 day after plasmid transfection and continued culture for protein expression induction for another 2 days.
For siRNA-mediated knockdown experiments, various siRNA as indicated was added to cells at 30% confluence using Lipofectamine RNAiMAX reagent transfection method. A non-target siRNA was used as CTL siRNA (5′-UGGUUUACAUGUCGACUAA-3′). The nucleotide sequences of siRNA On-Targetplus SMARTpool for APE1 include 5′-CAAAGUUUCUUACGGCAUA-3′, 5′-GAGACCAAAUGUUCAGAGA-3′, 5'-CUUCGAGCCUGGAUUAAGA-3′, and 5′-UAACAGCAUAUGUACCCUAA-3′. The nucleotide sequences of siRNA On-Targetplus SMARTpool for NPM1 are 5′-GUAGAAGACAUUAAAGCAA-3′, 5′-AAUGCAAGCAAGUAUAGAA-3′, 5′-ACAAGAAUCCUUCAAGAAA-3′, and 5′-UAAAGGCCGACAAAGAUUA-3′. The nucleotide sequences of siRNA On-Targetplus SMARTpool for TopBP1 are 5′-ACAAAUACAUGGCUGGUUA-3′, 5′-ACACUAAUCGGGAGUAUAA-3′, 5′-GAGCCGAACAUCCAGUUUA-3′, and 5′-CCACAGUAGUUGAGGCUAA-3′. The nucleotide sequences of siRNA On-Targetplus SMARTpool for ETAA1 are 5′-CAUAAUAUAGUUCCCGAAA-3′, 5′-UAGCAAUUAUGUACGGAUA-3′, 5′-GAGAAUGGCUAAAGCACGA-3′, and 5′-CGAAGACUGCUGAUAACUA-3′. The nucleotide sequences of siRNA On-Targetplus SMARTpool for ATRIP are 5′-GCUCCAGACCAGUGAACGA-3′, 5′-UGGUGAAAUUAGCCGAAAA-3′, 5′-GAAUCUGGUUGCCCGGAAU-3′, and 5′-UCACUACAUCAGACGGAAU-3′.
Recombinant DNA and proteins
Plasmid pET28HIS-hAPE1 (for WT His-APE1 protein) was a gift from Primo Schaer (Addgene plasmid #70757; http://n2t.net/addgene:70757; RRID:Addgene_70757) (74). Plasmid pcDNA3-YFP (for YFP protein) was a gift from Doug Golenbock (Addgene plasmid #13033; http://n2t.net/addgene:13033; RRID:Addgene_13033). Plasmid pcDNA3.1-mCherry was a gift from David Bartel (Addgene plasmid #128744; http://n2t.net/addgene:128744; RRID:Addgene_128744) (75). Plasmid Flag-ATR was a gift from Stephen Elledge (Addgene plasmid # 41909; http://n2t.net/addgene:41909; RRID:Addgene_41909) (76). Plasmid Flag-Chk1 was a gift from Yihong Ye (Addgene plasmid # 86875; http://n2t.net/addgene:86875; RRID:Addgene_86875) (77). Recombinant plasmid pcDNA3.1-mCherry-APE1 was prepared by PCR full-length APE1 from pET28HIS-hAPE1 into pcDNA3.1-mCherry at BamHI and EcoRI sites. Recombinant plasmid pET28A-Chk1 (for His-Chk1 protein) was prepared by PCR Chk1 from plasmid Flag-Chk1 and subcloned into pET28A vector at EcoRI and XhoI sites. Plasmid pcDNA3-YFP-APE1 (for WT YFP-APE1 protein) was prepared by PCR full-length APE1 from pET28HIS-hAPE1 into pcDNA3-YFP at BamHI and EcoRI sites. Recombinant pET28HIS-APE1-YFP (for His-APE1-YFP protein) was prepared by PCR WT YFP-APE1 from plasmid pcDNA3-YFP-APE1 and subcloned into pET28HIS vector at BamHI and HindIII sites. Plasmid pCMV6-AC-GFP-rtTA-APE1 (for WT tGFP-APE1 protein) was prepared by PCR full-length APE1 from pET28HIS-hAPE1 and subcloned into pCMV6-AC-GFP-rtTA vector (Origene #PS100125) at AscI and RsrII sites. Various △N33 APE1 deletion plasmids and pET28HIS-NT33-APE1-YFP were also constructed using similar approach to WT plasmids. Various point mutant plasmids were prepared using QuikChange II XL site-directed mutagenesis kit (Agilent). QIAprep spin miniprep kit was utilized to make recombinant plasmids following vendor's protocol. Various His-tagged recombinant proteins were expressed and purified in E. coli DE3/BL21. Purified recombinant proteins were examined and verified on SDS-PAGE gels with coomassie staining.
FAM-labeled DNA and RNA structures
The 39-bp FAM-labeled dsDNA-AP structure and 70-bp FAM-labeled dsDNA structure have been described recently (60). The 39-bp FAM-labeled dsDNA-AP structure was prepared by annealing of two complementary oligos (Forward #1: [FAM]-5′-TGCTCGTCAAGAGTTCGTAA[THF]ATGCCTACACTGGAGATC-3′; Reverse #1: 5′-GATCTCCAGTGTAGGCATCTTACGAACTCTTGACGAGCA-3′) as previously described (60). The 70-bp FAM-labeled dsDNA structure was prepared by annealing two complementary oligos (Forward #2: [FAM]-5′-TCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTGGCGTAATCATGGTCATAGCTGT-3′ Reverse #2: 5′-ACAGCTATGACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGA-3′). The dsDNA structure was treated with Nt.BstNBI and CIP to make the 70-bp FAM-labeled dsDNA-SSB structure (60). The 25-bp FAM-labeled ssRNA was synthesized by IDT ([FAM]-5′-GCAGCUGGCACGACAGGUAUGAAUC-3′).
Immunoblotting analysis and antibodies
Immunoblotting analysis was performed as described previously (27–29,60,64). Primary antibodies were purchased from respective vendors: APE1 (Santa Cruz Biotechnology Cat#sc-17774), ATR (Santa Cruz Biotechnology Cat#515173), ATM (GeneTex Cat#GTX70103), ATM phosphorylation at Ser1981 (Abcam Cat#ab81292), ATR phosphorylation at Thr1989 (Cell Signaling Technology Cat#D5K8W), ATRIP (Santa Cruz Biotechnology Cat#365383), Chk1 (Santa Cruz Biotechnology Cat#sc-8408), Chk1 phosphorylation at Ser345 (Cell Signaling Technology Cat#133D3), Chk1 phosphorylation at Ser317 (Cell Signaling Technology Cat#D12H3), Chk2 (Santa Cruz Biotechnology Cat#sc-9064), Chk2 phosphorylation at Thr68 (Santa Cruz Biotechnology Cat#sc-16297), eGFP (Life Technologies Corporation Cat#TA150041), ETAA1 (Abcam Cat#ab122245), H2AX (Cell Signaling Technology Cat#2D17A3), H2AX phosphorylation at Ser139 (Cell Signaling Technology Cat#2577s), NPM1 (Santa Cruz Biotechnology Cat#sc-47725), p53 phosphorylation at Ser15 (Cell Signaling Technology Cat#9284), p53 (Santa Cruz Biotechnology Cat#sc-126), PCNA (Santa Cruz Biotechnology Cat#sc-56), RPA32 (Thermo Fisher Scientific Cat#MA1-26418), RPA32 phosphorylation at Ser33 (Bethyl Laboratories Cat#A300-246A), TopBP1 (Santa Cruz Biotechnology Cat#271043), Tubulin (Santa Cruz Biotechnology Cat#sc-8035), YFP (BioVision Cat#3991–100).
In vitro pull-down assays
For the pull-down experiments, 20 μg of various His-tagged recombinant protein was added to 100 μl of cell nuclear extracts. After a 4h-incubation, an aliquot of the mixture was collected as Input and the remaining mixture was supplemented with 100 μl of Interaction Buffer (25 mM imidazole in PBS, pH 7.0) that contains 10 μl of Ni-NTA beads. After incubation with rotation for 1 h at room temperature or overnight at 4°C, bead-bound fractions were washed twice with Interaction Buffer. The Input and Pulldown (i.e. bead-bound fractions) samples were examined via immunoblotting analysis as indicated.
Immunofluorescence microscopy analysis
After 24-hour seeding on coverslide, cells were transfected with different expression plasmids for 72 h or as indicated. After washing with PBS, Cells were then fixed in 3% paraformaldehyde within PBS for 15 min and incubated with PBST (PBS supplemented with 0.2% Triton X-100) for 5 min. After 1-h incubation in blocking buffer (5% BSA in PBS), cells on coverslide were incubated with different antibodies conjugated with AF647 fluorescence in blocking buffer at 4°C overnight. After PBS wash three times, coverslips were stained with DAPI for 5 min and were mounted in ProLong Gold Antifade Mountant. Fluorescence images were acquired on a DeltaVision Elite Deconvolution microscope system equipped with a DV Elite CMOS camera and 60x objective and were further edited with Fiji-ImageJ software. Most antibodies-AF647 were purchased from Santa Cruz Biotechnology (NPM1: Cat#sc-47725 AF647; ATR: Cat#sc-515173 AF647; TopBP1: Cat#sc-271043). Anti-ETAA1 AF647 was prepared by conjugating AF647 to anti-ETAA1 antibodies (Abcam Cat#ab122245) using the Alexa Fluor 647 Conjugation Kit (fast)-Lighting-Link (Abcam Cat#ab269823) following vendor's protocol. Anti-γH2AX antibodies-AF488 was purchased from EMD Millipore (Cat#05-636-AF488). For ATM, Chk2 and Chk1-P-S345, cells on coverslide were incubated with different primary antibodies in blocking buffer at 4°C overnight, followed by incubation of second antibodies conjugated with AF488 (for ATM) or AF594 (for Chk2 and Chk1-P-S345). The second antibody Goat anti-Mouse lgG H&L-AF488 (Cat#150113) and Goat anti-Mouse lgG H&L-AF594 (Cat#150080) were purchased from Abcam.
APE1 endo/exonuclease assays in vitro
For in vitro endonuclease assays, the dsDNA-AP structure was treated with different doses of purified recombinant WT/mutant His-APE1 with/without APE1 inhibitors in APE1 reaction buffer (60 mM NaCl, 2 mM MgCl2, 2 mM DTT, 50 mM HEPES pH 7.4) at 37°C. Endonuclease assay reaction was quenched with equal volume of TBE–urea sample buffer and denatured for 5 min at 95°C. For in vitro exonuclease assays, the dsDNA-SSB structure was added with different concentrations of purified recombinant WT/mutant His-APE1 protein with/without APE1 specific inhibitors in APE1 reaction buffer at 37°C. Exonuclease assay reactions were quenched with equal volume of TBE–urea sample buffer and denatured at 95°C for 5 min. After a quick spin, samples from endo/exonuclease assays were examined on 15% TBE–urea PAGE gel and imaged with a Bio-Rad ChemiDoc MP Imaging System.
APE1 liquid–liquid phase separation (LLPS) assays in vitro
APE1 phase separation in nuclear extracts was performed in reaction mixtures containing PANC1 nuclear extract (2 μg/μl) and different doses of WT/mutant His-APE1-YFP protein in an ATR activation buffer (10 mM HEPES–KOH pH 7.6, 50 mM KCl, 0.1 mM MgCl2, 1 mM PMSF, 0.5 mM DTT, 1 mM ATP), which was modified from a previously reported TopBP1 phase separation condition (69). APE1 phase separation in a buffer was performed in reaction mixture of different doses of WT/mutant His-APE1-YFP protein in a LLPS buffer (50 mM HEPES, pH 7.5, 20 mM KCl, 10 mM MgCl2 and 2 mM DTT). Reaction mixtures were incubated at 37°C for 15 min or otherwise as indicated, followed by immunofluorescence microscopy on PEG silanized slides. Images were captured on an inverted microscope using a 10× objective.
Quantitative reverse-transcription PCR (qRT-PCR) assays
Total RNA was extracted from cells with Trizol reagent (Invitrogen Cat#15596026) according to vendor's instructions. Purified RNA was treated with Dnase I to remove any possible DNA contamination. qRT-PCR assays were performed with SuperScript III Platinum SYBR Green One-Step qTR-PCR Kit (Invitrogen Cat#11736051) according to vendor's standard procedure. qRT-PCT reactions were run in ABI 7500 FAST RT PCR system with parameters: 50°C for 5 min, 95°C for 5 min, followed by 40 cycles of PCR 95°C for 15 s and 60°C for 30 s. Primers targeting pre-r-RNA 5’ external transcribed region (5'-GGAAGGAGGTGGGTGGAC-3' and 5'-GCGGTACGAGGAAACACCT-3') and primers targeting Actin (5'-CTCTTCCAGCCTTCCTTCCT-3' and 5'-AGCACTGTGTTGGCGTACAG-3') were described recently (43). Pre-rRNA signals were normalized to Actin signals.
FACS analysis, Comet assays and MTT assays
For cell cycle profiling via FACS analysis, pelleted cells were washed by PBS and resuspended in 300 μl ice cold PBS. Then cells were mixed with 700 μl ice cold 100% ethanol and incubated at 4°C for at least 2 h. Cells were washed by PBS again and treated with RNase within PBS for 15 min at 37°C, followed by resuspension in Staining Buffer (100 mM Tris–HCl, pH7.4, 150 mM NaCl, 1 mM CaCl2, 0.5 mM MgCl2, 0.1% NP-40, 5 μg/ml DAPI) for FACS analysis. Comet assays were performed using the OxiSelect Comet Assay Kit (Cell Biolabs Cat#STA-351-5) with alkaline (pH > 7.0) or neutral condition (pH 7.0) following vendor's standard protocol. Cell nuclei were imaged using a fluorescence microscope with a FITC filter and DP Controller software. Images were analyzed using Comet Assay IV Lite software. For cell viability analysis using MTT (Thiazolyl blue tetrazolium bromide) assays, cultured cells in 96-well plate were examined using a procedure as recently described (64).
In vitro ATR kinase assays
HEK293 cells were transfected with Flag-ATR expression plasmid for 72 h. After washing with PBS, cells were suspended in lysis buffer (25 mM Tris, pH 8.0, 250 mM NaCl, 20 mM MgCl2, 0.5mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), 20% glycerol), and then Flag-ATR protein was purified by anti-Flag M2 Magnetic Beads (Sigma Cat# M8823). ATR kinase assays were performed in two steps: (i) beads coupled with Flag-ATR were firstly incubated in a stimulating buffer (50 mM HEPES, 100 mM NaCl, 1 mM MnCl2 and 2 mM dithiothreitol (DTT)) at 30°C for 60 min; (ii) after incubation, 20μl beads coupled with Flag-ATR was added to a kinase buffer (50 mM HEPES, pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM DTT, 1 mM ATP, and 10% glycerol), which was supplemented with 3 μg Chk1 as substrate, and 5 μg YFP or WT/W119R YFP-APE1 protein for another incubation at 30°C for 30 min. HEK293T cells without transfected Flag-ATR expression plasmid was a negative control. Finally, the samples were examined via immunoblotting analysis as indicated.
Quantification, statistical analysis and reproducibility
The data presented are representative of three biological replicates unless otherwise specified. All statistics were performed using GraphPad Prism version 8 for Windows. Statistical significance was ascertained between individual samples using a parametric unpaired t-test. Significance is denoted by asterisks in each figure: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, no significance. Error bars represent the standard deviation (SD) for three independent experiments, unless otherwise indicated.
RESULTS
APE1 is important for the ATR–Chk1 DDR pathway activation under stress conditions in mammalian cells
Our recent study has shown that APE1 plays an essential role in the ATR–Chk1 DDR pathway activation in Xenopus HSS system (60). To explore the function of APE1 in DDR pathway in mammalian cells, we first found that H2O2-induced oxidative stress triggered Chk1 phosphorylation at Ser345 (Chk1-P-S345) and Ser317 (Chk1-P-S317), ATR phosphorylation at Thr1989 (ATR-P-T1989), Chk2 phosphorylation at Thr68 (Chk2-P-T68), ATM phosphorylation at Ser1981 (ATM-P-S1981) and H2AX phosphorylation at Ser139 (γH2AX) in human breast cancer cell line MDA-MB-231, suggesting the activation of the ATR and ATM DDR pathways by oxidative stress (Figure 1A). Notably, the H2O2-induced Chk1 phosphorylation at Ser345 and Ser317 was significantly reduced in siRNA-mediated APE1-KD in MDA-MB-231 cells (Figure 1A). We observed a similar reduction of H2O2-induced Chk1 phosphorylation in human pancreatic cancer cell line PANC1 cells after APE1-KD (Figure 1D).
Figure 1.

APE1 and its nuclease activity is important for ATR–Chk1 DDR pathway in mammalian cells. (A) siRNA-mediated APE1-knockdown impaired H2O2-induced ATR–Chk1 DDR pathway in MDA-MB-231 cells. Total cell lysates were extracted and analyzed via immunoblotting analysis as indicated. (B, C) The H2O2-induced Chk1 phosphorylation was compromised by AR03 or APE1i III at different doses (5, 10, or 15 μM for 2 h) in MDA-MB-231 cells. (D) The H2O2-induced Chk1 phosphorylation was reduced in APE1-KD PANC1 cells. (E, F) The H2O2-induced Chk1 phosphorylation was impaired by AR03 or APE1i III in a dose-dependent manner (5, 10 or 15 μM for 2 h) in PANC1 cells. (A–F) ‘Chk1-P-S345/Chk1’ was quantified by the intensity of Chk1-P-S345 versus total Chk1. Means ± SD, n = 3. (G) AR03 or APE1i III but not E3330 inhibited APE1’s endonuclease activity. Different doses of AR03, APE1i III, or E3330 (1, 2, or 5 mM) was added to endonuclease assays containing 0.185 μM APE1 protein and a 5’-FAM-labeled dsDNA-AP substrate. After incubation for 30 min, samples were analyzed via 20% TBE gel. (H) AR03 or Inhibitor III could inhibit APE1’s exo-nuclease activity. Different doses of AR03, APE1i III, or E3330 (1, 2 or 5 mM) was added to exonuclease assays containing 0.185 μM APE1 protein and 5’-FAM-labeled dsDNA-SSB substrate. After 30-min incubation, samples were examined via 20% TBE gel.
Previous studies have identified and characterized AR03 and APE1 inhibitor III (APE1i III) as specific inhibitors for APE1 endonuclease activity and E3330 as an inhibitor of APE1 redox function (51,78,79). In particular, E3330 specifically binds to recombinant APE1, and selectively inhibits APE1’s ability to convert transcription factors to more reduced status but has no effect on APE1’s catalytic function (51,80,81). As expected, recombinant human full-length WT His-tagged APE1 (His-APE1) displayed AP endonuclease activity and 3′–5′ exonuclease activity in a dose-dependent manner in in vitro reconstitution systems (Supplementary Figure S1A–C). Importantly, H2O2-induced Chk1 phosphorylation at Ser345 was significantly reduced with the addition of AR03 or APE1i III in a dose-dependent manner in MDA-MB-231 cells (Figure 1B-C) and PANC1 cells (Figure 1E, F). Of note, Chk1/ATR phosphorylation but neither ATM phosphorylation nor γH2AX was downregulated by the addition of AR03 or APE1i III in MDA-MB-231 cells (Supplementary Figure S1D). However, the addition of E3330 had almost no noticeable effect on the H2O2-induced Chk1 phosphorylation in PANC1 cells (Supplementary Figure S1E). We verified that AR03 and APE1i III inhibited both AP endonuclease and 3′–5′ exonuclease activity of recombinant His-APE1 in vitro, whereas E3330 had minimal effects on APE1 nuclease activities (Figure 1G-H). These observations suggest that APE1 and its nuclease activity but not redox regulation is important for APE1’s function in oxidative stress-induced ATR–Chk1 DDR pathway in mammalian cells.
Furthermore, methyl methanesulfonate (MMS)-induced DNA alkylation damage and camptothecin (CPT)-induced DSBs and SSBs triggered Chk1 phosphorylation in MDA-MB-231 cells (Supplementary Figure S1F–I). MMS/CPT-induced Chk1 phosphorylation was also compromised by the addition of AR03 or APE1i III in a dose-dependent manner in MDA-MB-231 cells (Supplementary Figure S1F-I). AR03 and APE1i III similarly impaired MMS/CPT-induced Chk1 phosphorylation in PANC1 cells (Supplementary Figure S1J–M). These evidences indicate that APE1 plays a key role in the regulation of the ATR–Chk1 DDR pathway under stress conditions in mammalian cells.
APE1 overexpression triggers ATR–Chk1 DDR pathway activation under unperturbed conditions in mammalian cells
Meta-analysis has revealed APE1 upregulation in cancer tissues compared with normal tissues and the association of APE1 overexpression with poor survival in patients with solid tumors (82,83). To elucidate the potential role of APE1 overexpression in DDR pathway, we overexpressed YFP-tagged APE1 (YFP-APE1) to a similar level of endogenous APE1 in MDA-MB-231 cells, and found that YFP-APE1 but not YFP transfection nor control (CTL) transfection increased Chk1-P-Ser345, Chk1-P-Ser317, and ATR-P-T1989 but neither Chk2-P-T68 nor ATM-P-S1981 after 4 days of transfection (Figure 2A). Of note, the YFP-APE1-induced Chk1 phosphorylation was decreased at 7 days after transfection when YFP-APE1 expression was reduced (Figure 2A). Pre-treatment of ATR inhibitor VE-822 but not ATM inhibitor KU55933 nor DNA-PKcs inhibitor NU7441 compromised the YFP-APE1 overexpression-induced Chk1-P-S345 in MDA-MB-231 cells (Figure 2B). These observations suggest that APE1 overexpression leads to ATR-dependent Chk1 phosphorylation and DDR pathway activation in cells under unperturbed conditions. From now, we use Chk1-P-S345 as an indicator of the ATR–Chk1 DDR activation in this study.
Figure 2.

APE1 overexpression leads to the activation of ATR–Chk1 DDR pathway independent of its nuclease and redox function in mammalian cells. (A) ATR–Chk1 DDR pathway was activated by the overexpression of YFP-APE1 but not YFP nor CTL transfection (no vector transfection) in MDA-MB-231 cells. After different times of incubation (i.e. 2, 3, 4 or 7 days), total cell lysates were extracted and examined via immunoblotting analysis as indicated. (B) Chk1 phosphorylation induced by YFP-APE1 was dependent on ATR, but not ATM nor DNA-PKcs. After 4-d overexpression of YFP-APE1 or YFP, different DDR kinase inhibitors (5 μM of VE-822, KU55933 or NU7441) were added to MDA-MB-231 cells for 2 h, followed by total cell lysate extraction and immunoblotting analysis as indicated. (C) APE1 inhibitors almost had no noticeable effect on Chk1 phosphorylation by YFP-APE1 overexpression. After 2-day overexpression of YFP-APE1 or YFP, different APE1 inhibitors (1 μM of AR03, APE1i III or E3330) were added to MDA-MB-231 cells for 2 days, followed by total cell lysate extraction and immunoblotting analysis. (D) The endonuclease activity of WT or mutant (i.e. D308A, C64A, E96Q, ED) His-APE1 was examined on 20% TBE gel. (E) The exonuclease activity of WT or mutant (i.e. D308A, C64A, E96Q, ED) His-APE1 was examined on 20% TBE gel. (F) Chk1 phosphorylation was triggered by WT/mutant YFP-APE1 (i.e. D308A, C64A, E96Q, ED) overexpression in MDA-MB-231 cells. After 4-day overexpression of WT/mutant YFP-APE1 or YFP in MDA-MB-231, total cell lysates were extracted and analyzed via immunoblotting analysis. The Chk1-P-S345/Chk1 data are presented as means ± SD, n = 3.
Next, we attempted to determine whether the nuclease activities and/or redox function of APE1 play important roles for the APE1 overexpression-induced ATR DDR pathway activation. First, we utilized various APE1 inhibitors and found, surprisingly, that none of the AR03, APE1i and E3330 had noticeable effect on the APE1 overexpression-induced ATR DDR pathway (Figure 2C). Next, we constructed various mutants of His-APE1 (i.e. D308A, E96Q, E96Q/D210N (ED) and C65A) (84–87) and verified these mutants for APE1 nuclease activity in vitro. C65A His-APE1 had no noticeable deficiency in 3'-5' exonuclease activity and AP endonuclease activity compared with WT His-APE1 (Figure 2D-E and Supplementary Figure S1A). While D308A His-APE1 was deficient for 3'-5' exonuclease activity and proficient in AP endonuclease activity, E96Q and ED His-APE1 were defective for both 3′–5′ exonuclease activity and AP endonuclease activity (Figure 2D, E and Supplementary Figure S1A). We then overexpressed various mutant YFP-APE1 in MDA-MB-231 cells for 4 days and found that all mutant YFP-APE1 (D308A, E96Q, ED and C65A) still triggered Chk1 phosphorylation in a similar fashion as the WT YFP-APE1 (Figure 2F). Although we added the same amount of plasmid of WT/mutant YFP-APE1 for transfection, WT and mutant YFP-APE1 overexpression in MDA-MB-231 was slightly different between each other. Nonetheless, immunoblotting analysis and fluorescence microscopy analysis showed that all WT/mutant YFP-APE1 were indeed overexpressed as compared to endogenous APE1 levels (Figure 2F and Supplementary Figure S2A). These observations suggest that the APE1 overexpression-induced activation of the ATR–Chk1 DDR pathway under unperturbed conditions in MDA-MB-231 cells is unlikely dependent on APE1 nuclease activities and redox function.
To diversify our results, we also overexpressed WT/mutant YFP-APE1 in PANC1 cells (Supplementary Figure S2B and D) and human osteosarcoma U2OS cells (Supplementary Figure S2C and S2E). We found that Chk1 phosphorylation was upregulated by overexpression of WT YFP-APE1 to slightly different levels in different cell lines (i.e. ∼5-fold in MDA-MB-231, ∼3.5-fold in PANC1 and ∼2.5-fold in U2OS) (Figure 2F and Supplementary Figure S2D, E). Similar to WT YFP-APE1, mutant YFP-APE1 (D308A, E96Q, ED and C65A) also triggered Chk1 phosphorylation under unperturbed conditions in PANC1 cells and U2OS cells (Supplementary Figure S2B–E). These results suggest that APE1 overexpression-induced ATR–Chk1 DDR pathway under unperturbed conditions is a conserved response in cultured mammalian cell lines.
The NT33 motif of APE1 is required for its interactions with ATR, ATRIP and RPA to regulate the ATR–Chk1 DDR pathway in cells and in nuclear extracts
To test whether the different level of APE1-OE correlates with the level of ATR DDR activation, we constructed a doxycycline-inducible plasmid that induced overexpression of turbo-GFP (tGFP)-tagged APE1 (tGFP-APE1) at different levels. Overexpression of tGFP-APE1 but not tGFP by doxycycline for three days triggered Chk1 phosphorylation in PANC1 cells under unperturbed conditions (Figure 3A). It is also noted that the higher tGFP-APE1 expression was induced by doxycycline (50 μg/ml versus 10 μg/ml), the stronger Chk1-P-S345 was triggered (Figure 3A). This result suggests that the level of APE1 overexpression is important for Chk1 phosphorylation. Previous studies have shown that the extreme NT33 motif of human APE1 interacts with RNA and NPM1 (57,59,88). Interestingly, we found that overexpression of △NT33 tGFP-APE1 lacking the NT33 motif was deficient for Chk1 phosphorylation, although △NT33 tGFP-APE1 was expressed to a comparable level to WT tGFP-APE1 (Figure 3A). This observation suggests that the NT33 motif within APE1 is critical for the APE1-OE-induced ATR DDR activation. Prior research has identified a nuclear localization signal (NLS) within the NT33 motif of APE1 (55,59). Our fluorescence microscopy analysis showed that similar to tGFP, △NT33 tGFP-APE1 was not localized to specific organelles, while WT tGFP-APE1 mostly localized to nuclei (Figure 3B). This defective translocation of △NT33 tGFP-APE1 into nuclei may be one mechanism for its deficiency in ATR DDR pathway under unperturbed conditions.
Figure 3.

The APE1 N33 motif is required for ATR–Chk1 DDR pathway in vivo and in vitro. (A) Overexpression of WT but not △N33 tGFP-APE1 nor tGFP activated Chk1 phosphorylation in PANC1 cells. APE1 or N33-depletion over-expression plasmid was transfected to PANC1 cells. After 1-day incubation of various overexpression plasmid transfection, different doses of doxycycline (0, 10 or 50 μg/ml) were added to PANC1 cells for another 2 days. Total cell lysates were then extracted and analyzed via immunoblotting analysis. (B) △N33 tGFP-APE1 was deficient in nuclear localization in PANC1 cells. tGFP, WT or △N33 tGFP-APE1 overexpression plasmid was transfected to PANC1 cells. After 1-day incubation of overexpression plasmid transfection, 50 μg/ml doxycycline was added to PANC1 cells for another 2 days, followed with fluorescence microscopy analysis. Scale bars = 10 μm. (C) Chk1 phosphorylation was triggered by the excess addition of recombinant His-APE1 protein in a dose-dependent manner in PANC1 nuclear extracts. Different doses of His-APE1 protein were added to PANC1 nuclear extracts for incubation for 30 min at room temperature. The samples were analyzed via immunoblotting analysis as indicated. (D) Chk1 phosphorylation induced by excess addition of recombinant His-APE1 protein in PANC1 nuclear extracts was independent of its nuclease activity and redox function. WT or mutant (i.e. D308A, C65A, E96Q, ED) His-APE1 was added to PANC1 nuclear extracts for 30-min incubation at room temperature, followed by immunoblotting analysis. (E) Pulldown assays suggest that WT/mutant His-APE1 associated with ATR, ATRIP and RPA32 in PANC1 nuclear extracts. ‘Input’ or ‘Pulldown’ samples were examined via immunoblotting analysis as indicated. (F) Chk1 phosphorylation induced by WT His-APE1-YFP protein in PANC1 nuclear extracts was inhibited by VE-822 but not KU55933 nor NU7441 (1 mM). (G) Chk1 phosphorylation was activated by excess addition of WT His-APE1-YFP but not △N33 His-APE1-YFP protein in PANC1 nuclear extracts. (H) Pulldown assays suggest that WT but not △N33 His-APE1-APE1 associated with ATR, ATRIP and RPA32 in PANC1 nuclear extracts pre-treated with DNase I (2 mg/ml). ‘Input’ or ‘Pulldown’ samples were examined via immunoblotting analysis as indicated. (A, C, D, G) Chk1-P-S345/Chk1 was quantified and examined as means ± SD (n = 3).
To directly test the role of NT33 motif of APE1 in the ATR DDR activation but not via its role in nuclear localization, we established an in vitro ATR DDR activation system using nuclear extracts isolated from PANC1 cells and found that the addition of purified WT His-APE1 protein upregulated Chk1 phosphorylation significantly in a dose-dependent manner (Figure 3C). Similar to WT His-APE1, the addition of mutant His-APE1 (D308A, C65A, E96Q and ED) also upregulated Chk1 phosphorylation in nuclear extracts (Figure 3D), consistent with the upregulation of Chk1 phosphorylation by the overexpression of WT/mutant YFP-APE1 in mammalian cells (Figure 2 and Supplementary Figure S2). We observed similar Chk1 phosphorylation through the addition of WT/mutant His-APE1 protein in nuclear extracts from U2OS cells (Supplementary Figure S3A-B). GST pulldown experiments showed that WT and mutant His-APE1 protein could pulldown ATR, ATRIP and RPA32 in nuclear extracts from PANC1 cells (Figure 3E) and from U2OS cells (Supplementary Figure S3C). Furthermore, we expressed and purified recombinant WT and △NT33 APE1 with both His-tag and YFP-tag (His-APE1-YFP) for subsequent biochemical and imaging analyses. Of note, Chk1 phosphorylation induced by excess WT His-APE1-YFP in nuclear extracts was compromised by VE-822 but not KU55933 nor NU7441 (Figure 3F). Moreover, excess addition of WT His-APE1-YFP but not △NT33 His-APE1-YFP increased Chk1 phosphorylation significantly in nuclear extracts, although both recombinant proteins were added to comparable levels (Figure 3G). Because there is no intact nuclear membrane in nuclear extracts, our observation provides the first evidence for the direct requirement of the APE1 NT33 motif in ATR DDR activation. Furthermore, GST pulldown experiments revealed that WT His-APE1-YFP but not △NT33 His-APE1-YFP associated with ATR, ATRIP and RPA32 in PANC1 nuclease extracts (Figure 3H), suggesting that the NT33 motif of APE1 is important for APE1 to interact with ATR and its associated proteins for ATR activation in nuclear extracts. The addition of NT33 motif only to nuclear extracts did not trigger Chk1 phosphorylation (Supplementary Figure S3D), suggesting that NT33 motif within APE1 is required but not sufficient for Chk1 phosphorylation in nuclear extracts at least under our experimental conditions. These data suggest that nuclear localization and interaction with ATR and association proteins are two critical functions of the APE1 NT33 motif in the ATR DDR activation.
APE1 assembles biomolecular condensates to recruit ATR, TopBP1 and ETAA for ATR DDR activation in nuclear extracts and formed LLPS in vitro
We next tested whether APE1 could form biomolecular condensates in vitro. Fluorescent microscopy analysis showed that recombinant WT His-APE1-YFP formed micrometer-sized biomolecular condensates in nuclear extracts in a dose and time-dependent manner (Figure 4A and Supplementary Figure S4A, B). In contrast, recombinant △NT33 His-APE1-YFP was deficient in the assembly of biomolecular condensates in nuclear extracts (Figure 4A and Supplementary Figure S4B). To test whether such APE1-induced condensates were dependent on DNA and/or RNA in nuclear extracts, we added DNase I and RNase A in nuclear extracts and found no noticeable change on the assembly of condensates by WT His-APE1-YFP (Figure 4B). None of APE1 specific inhibitors (AR03, APE1i III and E3330) had noticeable effects on APE1-induced biomolecular condensates (Figure 4C) and Chk1 phosphorylation status in nuclear extracts (Figure 4D). Notably, pulldown assays showed that WT but not △NT33 His-APE1-YFP formed protein complexes with ATR and its activators TopBP1 and ETAA1 in nuclear extracts, and that treatment of AR03 or E3330 had no effect on the complex formation of WT His-APE1-YFP and ATR/TopBP1/ETAA1 (Figure 4E). These results suggest that WT but not △NT33 APE1 forms biomolecular condensates which in turn recruit ATR and its activators for ATR activation in nuclear extracts.
Figure 4.

APE1 assembles biomolecular condensates in vitro to promote ATR DDR pathway. (A) WT but not △N33 His-APE1-YFP formed phase separation in PANC1 nuclear extracts. Recombinant YFP, WT or △N33 His-APE1-YFP protein (0.55 mM) was added to PANC1 nuclear extract and incubated for 15 min at room temperature, followed by fluorescence microscopy analysis. (B) DNase I and RNase A had almost no effect on the APE1-assembled phase separation in nuclear extracts. DNase I and RNase A (2 mg/ml each) was added to PANC1 nuclear extracts and incubated for 30 min at 37°C. Then WT His-APE1-YFP protein (0.55 mM) was added and incubated for 10 min at room temperature, followed by fluorescence microscopy analysis. (C) APE1 inhibitors had no noticeable effect on phase separation formed by APE1 in nuclear extracts. WT His-APE1-YFP protein (0.55 mM) was added to PANC1 supplemented with DMSO or APE1 inhibitors (AR03, APE1i III, and E3330). After 15-min incubation at room temperature, reaction mixtures were examined via fluorescence microscopy analysis. (D) APE1 inhibitors had no effect on the Chk1 phosphorylation induced by the excess addition of APE1 protein in PANC1 nuclear extracts. APE1 inhibitor (AR03, APE1i III or E3330) was added to PANC1 nuclear extracts and incubated for 10 min at room temperature, which was supplemented with WT His-APE1-YFP (0.55 mM) and incubated for another 30 min at room temperature. The samples were then analyzed via immunoblotting analysis as indicated. (E) WT but not △N33 His-APE1-YFP associated with ATR, TopBP1 and ETAA1 in PANC1 nuclear extracts. ‘Input’ and ‘Pulldown’ samples from pulldown experiments were examined via immunoblotting analysis as indicated. AR03 or E3330 was added to 1 mM. (F) WT but not △N33 His-APE1-YFP formed phase separation in a LLPS buffer. WT but not △N33 His-APE1-YFP (0.55 mM) was added to a LLPS buffer and incubated for 15 min at room temperature, followed by fluorescence microscopy analysis. (G) Phase separation formed by recombinant WT His-APE1-YFP protein was fused into large biomolecular condensates in a LLPS buffer. WT His-APE1-YFP (0.55 mM) was added to a buffer and incubated for 2 h before fluorescence microscopy analysis. BF, bright field. (H) Phase separation assembled by APE1 in a LLPS buffer was not affected by the APE1 inhibitors. WT His-APE1-YFP protein (0.55 mM) was added to LLPS buffer supplemented with DMSO or APE1 inhibitors (AR03, APE1i III and E3330). After 15-min incubation at room temperature, reaction mixtures were examined via fluorescence microscopy analysis. (I–M) The requirement of NT33 motif within APE1 for phase separation in a LLPS buffer was bypassed with the presence of different DNA or RNA. WT but not △N33 His-APE1-YFP (0.55 mM) was added to a buffer supplemented with 0.5 μM of different DNA/RNA structures (dsDNA (I), dsDNA with AP site (J), ssDNA (K), ssDNA with AP site (L) and ssRNA (M)). Reaction mixtures were incubated for 15 min at room temperature before fluorescence microscopy analysis. All scale bars = 50 μm.
To further explore whether APE1 can form LLPS in vitro, we found that recombinant WT His-APE1-YFP but not △NT33 His-APE1-YFP assembled biomolecular condensates in a dose and time-dependent manner in a LLPS buffer (Figure 4F and Supplementary Figure S4C-D). It should be noted that smaller condensates fused into larger circular condensates (Figure 4G and Supplementary Figure S4D), supporting the LLPS feature of APE1 protein in vitro. APE1-induced biomolecular condensates in the LLPS buffer are not sensitive to APE1 inhibitors (Figure 4H). Interestingly, although DNA/RNA is dispensable for the APE1-induced assembly of biomolecular condensates, the addition of different DNA/RNA structures (dsDNA, dsDNA with AP site, ssDNA, ssDNA with AP site, and ssRNA) bypassed the requirement of APE1 NT33 motif for condensate formation in vitro (Figure 4I-L).
Overexpressed APE1 colocalized with NPM1 and assembles LLPS in vivo to recruit ATR and its activator proteins for nDDR in nucleoli in cancer cells but not non-malignant cells
As WT tGFP-APE1, but neither △NT33 tGFP-APE1 nor tGFP, was found concentrated to some regions within nuclei (Figure 3B), we next asked whether and how APE1 forms biomolecular condensates within cell's nuclei in vivo. First, our fluorescent microscopy analysis showed co-localization of nucleolar NPM1 with concentrated YFP-APE1 but not YFP in PANC1 cells, suggesting that overexpressed YFP-APE1 protein is translocated to nucleoli within nuclei (Figure 5A). Furthermore, ATR and its activators TopBP1 and ETAA1 were all co-localized with YFP-APE1 but not YFP in nucleoli in PANC1 cells (Figure 5B-D). Furthermore, Chk1-P-S345 was found co-localized with YFP-APE1 but not YFP in the nucleoli of PANC1 (Figure 5E). However, γH2AX was found in the nucleoplasm but not nucleolar regions within the nucleus and didn’t colocalize with overexpressed mCherry-APE1 (Figure 5F). Neither Chk2 nor ATM was found colocalized in the nucleolar regions where YFP-APE1 or mCherry-APE1 was localized in PANC1 cells (Supplementary Figure S5A, B). We also found similar colocalization of NPM1 with YFP-APE1 but not YFP in other two cancer cells MDA-MB-231 and U2OS cells (Supplementary Figure S5C, D). These data strongly suggest that APE1-OE leads to the activation of ATR–Chk1 but not ATM-Chk2 DDR pathway in the nucleoli of cancer cells. Intriguingly, overexpressed YFP-APE1 did not form condensates in nucleoli nor activated Chk1 phosphorylation in non-malignant human pancreatic duct epithelial (HPDE) cells (Figure 5G and Supplementary Figure S5E). These observations suggest that overexpressed YFP-APE1 assembles biomolecular condensates within nucleoli and recruits ATR, TopBP1 and ETAA1 for ATR nDDR activation in cancer cells but not non-malignant cells.
Figure 5.

APE1 forms biomolecular condensates in nucleoli to recruit ATR, TopBP1, and ETAA1 for ATR activation in a NPM1-independent fashion in cancer cells but not unmalignant cells. (A) Overexpressed YFP-APE1 but not YFP colocalized with NPM1 in nucleoli in PANC1 cells. After overexpression of YFP or YFP-APE1 for 3 days, PANC1 cells were fixed and incubated with anti-NPM1-AF647 fluorescence antibody for overnight at 4°C. (B–D) ATR and its activators TopBP1 and ETAA1 were colocalized with YFP-APE1 in nucleoli in PANC1 cells. YFP or YFP-APE1 overexpression plasmid was added to PANC1 cells. After overexpression of YFP or YFP-APE1 for 3 days, PANC1 cells were fixed and incubated with anti-ATR-AF647 (B), anti-TopBP1-AF647 (C), or anti-ETAA1-AF647 (D) for overnight at 4°C. (E) After overexpression of YFP or YFP-APE1 for 3 days, PANC1 cells were fixed and incubated with anti-Chk1-P-S345 antibody for overnight at 4°C, followed by incubation with anti-Rabbit secondary antibody conjugated with AF594 at 4°C for 4 h. (F) After overexpression of mCherry or mCherry-APE1 for 3 days, PANC1 cells were fixed and incubated with anti-γH2AX-AF488 at 4°C overnight. (G) Overexpressed YFP-APE1 was not colocalized with NPM1 in nucleoli in HPDE cells. Similar experiment to Panel (A) was performed in HPDE cells. (H) Nucleolar localization of NPM1 was not affected when endogenous APE1 was knocked down via siRNA. Control (CTL) siRNA or APE1 siRNA was added and transfected to PANC1 cells for 3 days. The cells were then fixed and incubated with anti-APE1-AF488 and anti-NPM1-AF647 for overnight at 4°C. (I) YFP-APE1 still assembled condensates when endogenous NPM1 was knocked down. CTL siRNA or NPM1 siRNA was transfected to PANC1 cells and incubated for 1 day. Then YFP or YFP-APE1 overexpression plasmid was transfected and incubated for 3 days. The cells were fixed and incubated with anti-NPM1-AF647 at 4°C overnight. (A–I) All cells were examined by fluorescence microscopy analysis. All scale bars = 10 μm.
Due to the previously characterized interaction between APE1 and NPM1 within nucleoli in HeLa cells (57), we sought to determine the dependency of APE1 and NPM1 to nucleoli in PANC1 cells. We found that after endogenous APE1 was knocked down via siRNA, NPM1 expression was not affected and NPM1 was still localized within nucleoli (Figure 5H and Supplementary Figure S5F). Similarly, YFP-APE1 still assembled condensates, when endogenous NPM1 was knocked down (Figure 5I and Supplementary Figure S5G). NPM1-knowckdown had almost no noticeable effect on the APE1-OE-induced Chk1 phosphorylation (Supplementary Figure S5G). These observations suggest that NPM1 is dispensable for the nucleolar condensation of YFP-APE1 and associated ATR nDDR.
ATRIP has been widely accepted as a critical ATR-interacting protein for its activation at damage sites (26,76). To determine the role of ATRIP in the APE1-OE-induced ATR nDDR, we found that Chk1 phosphorylation was impaired after ATRIP was knocked down by siRNA in PANC1 cells (Supplementary Figure S5H). Furthermore, endogenous APE1 was not condensated with NPM1 in nucleoli regardless of hydrogen peroxide treatment in PANC1 cells (Supplementary Figure S6A). Oxidative DNA damage triggered partial colocalization of endogenous APE1 with ATR, TopBP1, and ETAA1 (Supplementary Figure S6B-D). Considering the pattern of endogenous APE1 complex with ATR, TopBP1, and ETAA1 in oxidative stress, our results suggest that YFP-APE1-OE-induced nDDR is distinct from oxidative stress-induced global ATR–Chk1 DDR pathway.
APE1 assembles biomolecular condensates in a TopBP1-independent manner in nucleoli to directly activate ATR–Chk1 nDDR in cells under unperturbed conditions
Previous studies have characterized biomolecular condensates through TopBP1 overexpression within nucleoli and LLPS formation by TopBP1 for ATR DDR pathway (43,69,70). Here we tested whether TopBP1 is important for nucleolar localization of YFP-APE1 and subsequent ATR–Chk1 nDDR. Although TopBP1 was recruited to nucleoli when YFP-APE1 but not YFP was expressed under unperturbed conditions, siRNA-mediated TopBP1-KD had no noticeable change for the nucleolar localization of YFP-APE1 (Figure 6A-B). Moreover, the YFP-APE1-induced Chk1 phosphorylation was reduced partially when TopBP1 was knocked down (Figure 6B). These data suggest that TopBP1 is dispensable for APE1 condensate in nucleoli and that TopBP1 may function downstream of APE1 for partial contribution to ATR-mediated Chk1 phosphorylation. In addition, ETAA1-KD had minimum to almost none effect on the YFP-APE1-induced Chk1 phosphorylation (Figure 6B). Intriguingly, Chk1 was still phosphorylated to some extent even when both TopBP1 and ETAA1 were knocked down (Figure 6B), suggesting that another activator protein may still attribute to ATR nDDR activation in the absence of TopBP1 and ETAA1.
Figure 6.

While TopBP1 is dispensable for YFP-APE1 nucleolar localization, the W119 residue within APE1’s putative ATR activation domain is required for its biomolecular condensate formation in nucleoli and nucleolar ATR activation. (A) YFP-APE1 still assembled condensates and activated ATR-chk1 pathway when endogenous TopBP1 was knocked down via siRNA. After 1-d incubation of CTL or TopBP1 siRNA in PANC1 cells, YFP or YFP-APE1 overexpression plasmid was added and incubation for another 3 d. The cells were fixed and incubated with anti-TopBP1-AF647 for fluorescence microscopy analysis. (B). After 1-d incubation of CTL, TopBP1 and/or ETAA1 siRNA in PANC1 cells, YFP or YFP-APE1 overexpression plasmid was added and incubated for another 3 d. The cells were extracted and examined by immunoblotting analysis as indicated. (C) Schematic diagrams of functional domains of hAPE1, hTopBP1 and hETAA1 as well as sequence alignment of the N-terminal domain and putative ATR activation domain (AAD) within APE1. hAPE1 (human APE1, NCBI#: P27695.1), mAPE1 (mouse APE1, NCBI#: NP_033817.1), xAPE1 (Xenopus laevis APE1, NCBI#: AAH72056.1), hTopBP1 (human TopBP1, NCBI#: Q92547.3), and hETAA1 (human ETAA1, NCBI#: NP_061875.2). * identical;: highly conserved;. low conservation. In the AAD alignment, highlighted green indicates aa with hydrophobic side chains, highlighted yellow indicates aa with polar uncharged side chains, highlighted pink indicates aa with positive charged sides chains, highlighted turquoise indicates aa with negative charged sides chains, and highlighted gray indicates the conserved WxxP/N peptide. (D) The K6, K7 and W119 residues within APE1 are important for Chk1 phosphorylation induced by YFP-APE1 in PANC1 cells. After YFP, WT or mutant (i.e. K6R/K7R, K24R/K25R, K31R/K32R, W119R) YFP-APE1 overexpression plasmid was added to PANC1 cells and incubation for 3 d, total cell lysates were extracted and examined via immunoblotting analysis as indicated. (E) APE1 forms biomolecular condensates in nucleoli dependent on its W119 residue. After WT or mutant (i.e. K6R/K7R, K24R/K25R, K31R/K32R, W119R) YFP-APE1 overexpression plasmid was added to PANC1 cells and incubation for 3 d, the cells were fixed and incubated with anti-NPM1-AF647 for overnight at 4°C, followed by fluorescence microscopy analysis. (F) WT but not W119R YFP-APE1 directly activates ATR to phosphorylate Chk1 phosphorylaton at S345 in vitro. In in vitro kinase assays, equal amount of His-YFP, WT or W119R His-APE1-YFP was added to the kinase assays (purified Flag-ATR as kinase and purified His-Chk1 as substrate). Beads bound with lysates from HEK293 cells without Flag-ATR transfection were used as a negative control (‘Control’). ‘No addition’ indicate the kinase assays without the addition of His-YFP or WT/W119R His-APE1-YFP. The samples were examined via immunoblotting analysis as indicated. (A, E) All scale bars = 10 μm.
Next, we sought to elucidate the molecular determinants within APE1 for nucleolar translocation and LLPS. We first focused on the NT33 motif within APE1 which contains eight conserved Lysine residues (Figure 6C and Supplementary Figure S7A). It is previously established that the K6, K7, K24, K25, K27, K31 and K32 within APE1 can be modified by post-translational modifications (PTMs) such as acetylation and/or ubiquitination (89,90). The conserved K24, K25, K27, K31 and K32 residues are important for interaction with RNA and NPM1 (88,91). We constructed three double KR mutations (i.e. K6R/K7R, K24R/K25R and K31R/K32R), in which such mutations remain positively charged but lose accompanying PTMs. WT, K24R/K25R, and K31R/K32R YFP-APE1, but not YFP nor K6R/K7R YFP-APE1 induced Chk1 phosphorylation in PANC1 cells (Figure 6D). Furthermore, K6R/K7R YFP-APE1 was defective in nuclear translocation as well as ATR and TopBP1 colocalization (Figure 6E and Supplementary Figure S7B, C). In contrast, K24R/K25R YFP-APE1 and K31R/K32R YFP-APE1 assembled condensates together with NPM1 within nucleoli and recruited ATR and TopBP1 in a similar fashion to WT YFP-APE1 (Figure 6E and Supplementary Figure S7B, C).
Previous studies on the AAD motifs of TopBP1 and ETAA have shown that the W1145 residue in human TopBP1 (homologue to Xenopus laevis TopBP1 W1138) and the W107 residue in human ETAA1 are critical for ATR activation23, 24, 81. Our protein sequence alignment shows that the W119 residue and its nearby ∼30-amino-acid (AA) peptide in human APE1 is conserved in humans, mouse, and Xenopus laevis, and is also conserved to the AAD of hTopBP1 and hETAA1 to some extent, considering the WxxP/N peptide (highlighted in gray, x represents any AA) and the side chain charges of flanking residues (Figure 6C). Therefore, we hypothesize the motif of AA104-134 within hAPE1 is a putative AAD domain. Notably, W119R YFP-APE1 failed to trigger Chk1 phosphorylation in PANC1 cells (Figure 6D). Although W119R YFP-APE1 was translocated into the nucleus, it was not co-localized with NPM1 and could not form biomolecular condensates within the nucleolus (Figure 6E). Consistent with its defective nucleolar localization, neither ATR nor TopBP1 was recruited to the nucleolus by W119R YFP-APE1 (Supplementary Figure S7B, C). Chk1 phosphorylation was not induced in the nucleolus by W119R YFP-APE1 overexpression (Supplementary Figure S7D). These results suggest that the W119 residue within APE1 is important for APE1 LLPS formation into nucleoli in vivo, subsequent recruitment of ATR and TopBP1, and ATR–Chk1 nDDR activation. To further test whether the W119 residue is important for APE1’s LLPS in vitro, we added WT or W119R His-APE1-YFP to nuclear extracts and found that WT but not W119R His-APE1-YFP formed biomolecular condensates in nuclear extracts (Supplementary Figure S7E). Our in vitro pulldown assays from nuclear extracts demonstrated no noticeable change in the association of W119R His-APE1-YFP with ATR, TopBP1 and ETAA1 in comparison to WT His-APE1-YFP (Supplementary Figure S7F).
To test whether a recombinant APE1 protein can directly activate ATR kinase, we established in vitro kinase assays in which Flag-ATR protein purified from HEK293 cells as kinase and His-Chk1 purified from bacteria as substrate were combined together in buffers. Notably, the addition of purified WT His-APE1-YFP but not W119R His-APE1-YFP nor His-YFP stimulated Chk1-P-S345 (Figure 6F). Although we could not exclude the possibility that ATR activator protein such as TopBP1 and ETAA1 might be co-purified onto Flag-beads coupled with Flag-ATR, such minimum amount of ATR activator proteins did not activate Flag-ATR to phosphorylate His-Chk1 (lanes 2 and 3, Figure 6F). Our control experiment also showed similar amount of His-YFP and WT/W119R His-APE1-YFP added to the in vitro kinase assays (Figure 6F). These observations suggest that APE1 protein can directly stimulate ATR kinase activity in vitro.
APE1-induced nDDR leads to rRNA transcription suppression and impaired cell viability
Based on previous study showing rRNA transcription suppression by TopBP1 overexpression-induced nDDR (43), we sought to determine the potential effect of APE1-induced nDDR in rRNA transcription and cell viability. qRT-PCR assays showed that YPF-APE1 transfection led to reduced pre-rRNA transcription compared with YFP transfection, and such rRNA suppression was sensitive to ATR inhibitor VE-822 (Figure 7A). Moreover, overexpression of K6R/K7R or W119R YFP-APE1 was defective for the rRNA transcription suppression, suggesting that APE1 nuclear and nucleolar localization and LLPS formation are important for rRNA transcription suppression (Figure 7B). These results underscore the significance of APE1-OE-induced nDDR in rRNA transcription suppression.
Figure 7.
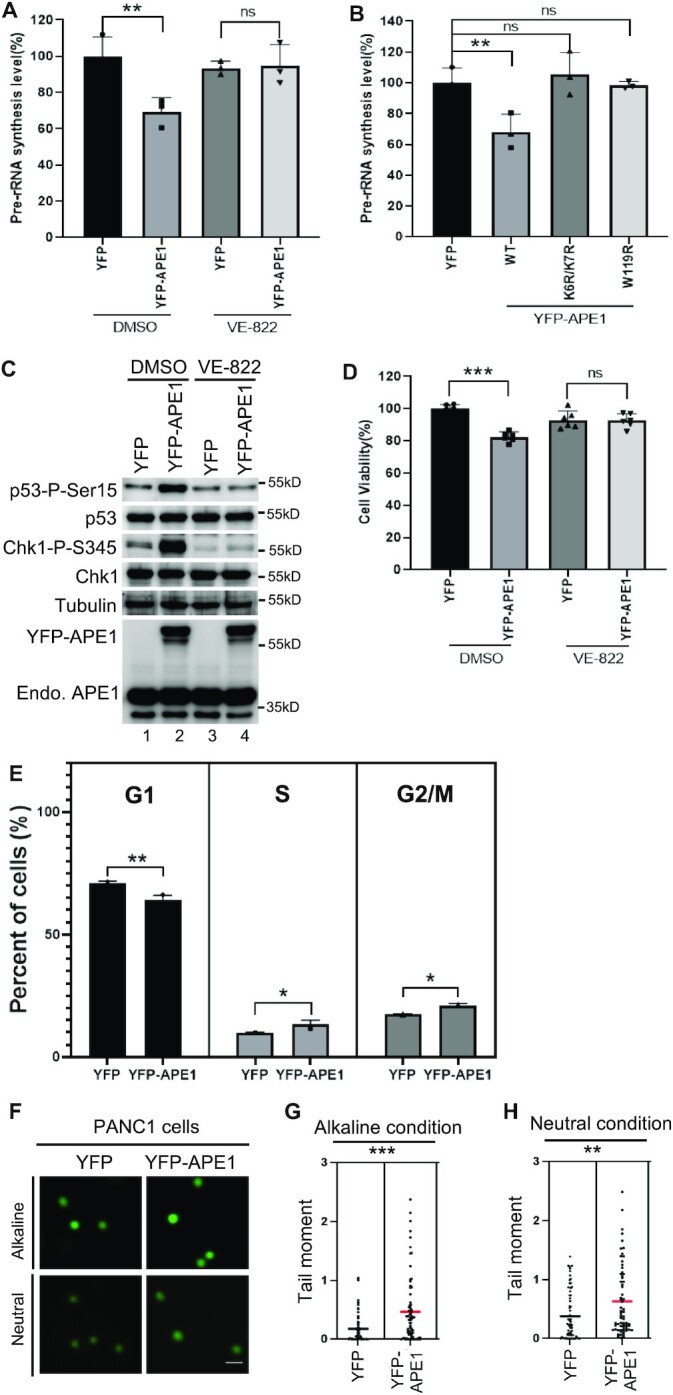
APE1-induced ATR–Chk1 nDDR leads to rRNA transcription suppression and cell viability reduction. (A) APE1 overexpression led to pre-rRNA transcription suppression in an ATR-dependent fashion. YFP or YFP-APE1 overexpression plasmid was added to PANC1 cells and incubated for 2 days. DMSO or VE-822 (1 μM) was then added and incubated for another day. The total RNA was extracted and analyzed via qRT-PCR, and pre-rRNA synthesis was normalized to beta-Actin. n = 3. (B) WT but not K6R/K7R nor W119R YFP-APE1 suppressed rRNA transcription. YFP or WT or mutant YFP-APE1 overexpression plasmid was added to PANC1 cells and incubated for 3 days, followed by total RNA extraction and qRT-PCR analysis. n = 3. (C) Overexpression of YFP-APE1 but not YFP led to p53 Ser15 phosphorylation, which was sensitive to VE-822. After YFP or YFP-APE1 overexpression plasmid was added to PANC1 cells and incubated for 3 days, DMSO or VE-822 (1 μM) was added and incubated for another 2 h, followed by total cell lysate extraction and immunoblotting analysis as indicated. (D) YFP-APE1 overexpression led to cell viability reduction. After YFP or YFP-APE1 overexpression plasmid was added to PANC1 cells and incubated for 2 days, DMSO or VE-822 (1 μM) was added and incubated for another day. The cells were analyzed via MTT assays. n = 6. (E) Overexpression of YFP-APE1 led to cell cycle arrests at S and G2/M phases in PANC1 cells. YFP or WT YFP-APE1 overexpression plasmid was added to PANC1 cells. After incubation for 4 days, the cells were collected and examined via FACS analysis. The data are presented as means ± SD, n = 3. (F–H) Overexpression APE1 led to more endogenous DNA damage in PANC1 cells. YFP or WT YFP-APE1 overexpression plasmid was added to PANC1 cells. After incubation for 4 days, the cells were collected and analyzed via comet assays under alkaline or neutral conditions. Scale bar = 100 μm. The data are presented as Scatter dot plot and the lines indicate means, n > 50.
Previous study shows that p53 with a R273H mutation is phosphorylated at Ser15 by gemcitabine-induced stalled DNA replication forks in PANC1 cells (92). To determine the p53 phosphorylation status by APE1-induced nDDR, we found that overexpression of YFP-APE1 but not YFP led to p53 phosphorylation at Ser15 (p53-P-Ser15), which was sensitive to VE-822 (Figure 7C). We speculated that cell viability might be affected by nDDR-dependent Chk1 and p53 phosphorylation. As expected, our cell viability assays showed that YFP-APE1 overexpression led to about 20% reduction in cell viability compared with YFP transfection and that such cell viability reduction by APE1-OE was reversed by the addition of VE-822 (Figure 7D). These observations suggest the significance of APE1-OE-induced nDDR in p53-mediated apoptosis and cell viability.
Next, we examined cell cycle profiling after overexpression of YFP-APE1. FACS analysis showed that compared with YFP, YFP-APE1 overexpression led to a higher percentage of PANC1 cells in S phase and G2/M phase (Figure 7E). This observation suggests that APE1 overexpression may activate S and/or G2/M checkpoint responses, consistent to the ATR nDDR pathway activation. Moreover, we analyzed endogenous DNA damage after WT YFP-APE1 overexpression in PANC1 cells. Comet assays showed moderately enhanced tail moment after overexpression of YFP-APE1 in both alkaline condition and neutral conditions, compared with YFP (Figure 7F–H). This observation suggests that endogenous DNA damage (i.e. SSBs, AP sites, or DSBs) may be upregulated mildly following APE1 overexpression, which is consistent with moderately elevated γH2AX by YFP-APE1-OE in the nucleoplasm (Figure 5F). Our APE1-OE-induced ATR nDDR, S/G2 checkpoint response, and moderately elevated DNA damage in PANC1 cells are reminiscent of a prior study demonstrating that knockdown of a ribosomal RNA processing protein named RRP15 leads to the activation of ATR–Chk1 DDR pathway, S-G2/M phase delay and cell death, as well as elevated γH2AX across the nucleus in HeLa and MCF7 cells (93). We speculate that APE1-OE-induced moderate upregulation of DNA damage is likely due to cell cycle arrest; however, the underlying mechanism warrants further investigation in the future. Overall, APE1 overexpression induces S/G2 checkpoint response and moderately elevated DNA damage in PANC1 cells.
Results from this study provide evidence showing that APE1 is important for the global regulation of the ATR–Chk1 DDR pathway under stress conditions via its nuclease activity. It is also shown that overexpressed APE1 forms biomolecular condensates within nucleoli to activate the ATR–Chk1 nDDR pathway. We propose that APE1 contributes to the genome integrity maintenance via distinct regulatory mechanisms under stressful and unperturbed conditions.
DISCUSSION
Distinct regulatory mechanisms of APE1 in the ATR–Chk1 DDR pathways
Results from this study provide direct evidence that APE1 contributes to the ATR–Chk1 DDR pathway via distinct molecular mechanisms. Under stress conditions (such as H2O2, MMS, CPT), stress-induced DNA damage such as AP sites can be recognized and processed by APE1 via its AP endonuclease activity, generating SSB structures with subsequent resection into small gap containing 1–3nt ssDNA by APE1’s 3'-5' exonuclease activity (Figure 8A). These small ssDNA gaps are further processed by other enzymes such as APE2 to enlarge the RPA-coated ssDNA for ATR/ATRIP recruitment and activation (Figure 8A) (29,60). Under unperturbed conditions, APE1 overexpression leads to the formation of biomolecular condensates in nucleolus in vivo and in nuclear extracts in vitro via APE1’s possible intermolecular and/or intramolecular associations (Figure 8B). Importantly, ATR and its activators TopBP1 and ETAA1 are recruited to APE1-mediated LLPS to promote ATR activation which leads to rRNA transcription suppression in the nucleolus, cell cycle arrest, moderately elevated DNA damage, and reduced cell viability (Figure 8B). To the best of our knowledge, this is the first evidence that APE1 forms LLPS in vivo and in vitro leading to ATR–Chk1 DDR pathway activation. Intriguingly, such APE1-dependent LLPS-mediated ATR DDR activation in nucleoli is independent of APE1 nuclease and redox function. Thereby, here we define a previously uncharacterized but significant non-catalytic function of APE1 in the ATR–Chk1 DDR pathway.
Figure 8.

A working model of distinct mechanisms of APE1 in the ATR DNA damage response. (A) Stress conditions (canonical function): APE1 promotes the ATR–Chk1 DDR pathway activation via its AP endonuclease and exonuclease activities under stress conditions, which is considered as canonical function of APE1. (B) Unperturbed conditions (non-canonical function): under unperturbed conditions, APE1 overexpression leads to biomolecular condensates (liquid–liquid phase separation) in nucleoli in vivo and in nuclear extracts in vitro via its N33 motif and/or W119 residue in AAD domain. ATR/ATRIP, TopBP1, and ETAA1 are recruited to the APE1-induced condensates for ATR activation. APE1-induced nucleolar ATR DDR suppresses rRNA transcription, arrests cell cycle, elevates DNA damage, and reduces cell viability. See text for more details.
It is recently demonstrated that ATR–Chk1 DDR pathway activation in response to defined SSB structures requires APE1, especially its exonuclease activity, in Xenopus egg extracts (60). Such requirement of APE1 and its exonuclease activity in ATR DDR pathway is also conserved in mammalian cells (Figure 1 and Supplementary Figure S1). Thus, APE1’s nuclease activity is considered a canonical function in the ATR DDR. Moreover, our data here demonstrate nucleolar ATR–Chk1 DDR pathway induction via YFP-APE1 that is independent of APE1 nuclease and redox functions. This newly defined YFP-APE1-induced nDDR is dependent on the LLPS assembled by APE1 NT33 motif and W119. This new function of APE1 LLPS in nDDR is therefore proposed as a non-canonical function.
APE1 is a direct activator of ATR DDR in vitro and in the nucleoli
It is widely accepted that TopBP1 and ETAA1 are direct activators of ATR kinase (23,24,94,95). Here we show that overexpression of APE1 in MDA-MB-231 cells and PANC1 cells can directly activate ATR, independent of APE1 nuclease activity and redox activity (Figure 2 and Supplementary Figure 2). We also provide evidence that recombinant APE1 protein interacts with ATR, ATRIP, RPA, TopBP1 and ETAA1 to directly activate ATR pathway in in vitro nuclear extracts in a NT33 motif-dependent but nuclease- and redox-independent manner (Figures 3 and 4). Moreover, we identify an APE1 AAD domain similar to TopBP1 and ETAA1’s AAD domain that is required for the translocation and condensation of YFP-APE1 to nucleoli and recruitment of ATR, TopBP1 and ETAA1 (Figure 6). Intriguingly, W1138R mutant in xTopBP1 AAD (homologue to W1145R hTopBP1) still associates with ATR but is deficient in triggering ATR kinase (94). In contrast, W107A mutant in ETAA1 AAD is defective for both ATR association and ATR activation (23). Out study shows that W119R YFP-APE1 still associates with ATR although lacks the capacity to activate ATR nDDR (Figure 6 and Supplementary Figure S7), suggesting that APE1 AAD is more conserved to TopBP1 AAD. Our in vitro kinase observations indicate that APE1 is a previously unidentified but significant direct activator of ATR kinase (Figure 6F).
How is ATR activated in the nucleoli by its three direct activator proteins TopBP1, ETAA1, and APE1? Because APE1-OE-induced ATR nDDR activation is under unperturbed conditions, this question involves two layers of regulation: recruitment and direct activation. Our data suggest that overexpressed APE1 can be relocated to the nucleoli to recruit TopBP1 and ETAA1 (Figures 5-6), and that TopBP1-KD has no effect on YFP-APE1 nucleolar localization, suggesting that overexpressed APE1 recruits TopBP1 to nucleoli but not vice versa. This point is further supported by the observations that nuclear localization mutant K6R/K7R and nucleolar localization mutant W119R YFP-APE1 failed to recruit TopBP1 into the nucleoli (Supplementary Figure S7C). For the direct activation of ATR perspective, we think that ATR is directly activated by both APE1 and TopBP1 in the nucleoli (Figure 8B). Future studies are needed to test whether APE1-mediated nucleolar ATR activation and TopBP1-mediated nucleolar ATR activation function sequentially or in parallel. Although we can’t completely rule out the possible role of ETAA1, ETAA1 may only make partial to minimal contributions to the APE1-OE-induced ATR nDDR, which needs further experimentation such as better depletion efficiency of ETAA1. Such minimal regulation of ATR nDDR by ETAA1 is reminiscent of two recent studies showing the requirement of TopBP1 but not ETAA1 in the activation of nucleolar ATR DDR pathway in response to DNA replication stress or defined DSBs in rDNA (40,42). In addition, we speculate that APE1-OE-induced minimal DNA damage in comet assays is likely the consequence of activated ATR nDDR and cell cycle arrests (Figure 7E-H). Moderately increased γH2AX in nucleoplasm but not nucleolar regions after APE1 overexpression (Figure 5F) suggests that DNA damage is mildly elevated in the nucleoplasm after APE1-OE-induced nDDR.
LLPS formation by APE1-mediated biomolecular condensates in vitro and in vivo
The N-terminal motif of APE1 can be modified by PTMs including acetylation, ubiquitination and phosphorylation and has been shown association with NPM1, XRCC1, as well as RNA (59). Our data indicate that ATR, ATRIP, TopBP1, ETAA1, and RPA32 can interact with APE1 via its N-terminal motif, although it remains unknown whether these proteins’ interaction with APE1 NT33 is direct or indirect (Figures 3 and 4). Our observation of the defective nuclear location of K6R/K7R YFP-APE1 suggests a potential role for PTMs such as acetylation at K6 and/or K7 residues in the regulation of APE1’s nuclear localization (Figure 6). Future studies are needed to better understand this in greater detail.
The data shown here is the first evidence of APE1-mediated LLPS formation in vitro (Figure 4). Our data support the recently proposed concept of APE1’s involvement in LLPS by Tell and colleagues (71,72). In vitro LLPS by APE1 was not dependent on DNA, RNA, or APE1 nuclease/redox functions (Figure 4). Importantly, NT33 motif of APE1 is indeed required for APE1 LLPS in vitro (Figure 4). LLPS is often driven by intrinsically disordered protein regions (IDRs) and/or association with nucleic acids such as RNA (96–98). Consistently, the NT33 motif is APE1’s IDR whose structure has not been characterized yet, although other, more ridged, regions of APE1 have been fully characterized (99,100). We speculate that the bypassed requirement of APE1 NT33 for LLPS in the presence of DNA/RNA is likely due to a compensatory role of another motif within APE1 (Figure 4I-M). Intriguingly, the W119R mutant APE1 leads to defective APE1 LLPS in nuclear extracts (Supplementary Figure S7E). However, future in-depth studies are needed to determine whether intramolecular interaction within APE1 between APE1 NT33 and a second motif (e.g. W119-containing motif) or intermolecular interaction of APE1 is important for APE1 LLPS (Figure 8B). Nonetheless, the newly defined requirement of APE1 NT33 in the in vitro LLPS provides new insight into how APE1’s NT33 motif folds and/or associates with other regions of APE1 or its binding partner proteins.
Although previous studies have shown the nucleolar localization of FLAG-tagged APE1 (57,91), we propose the nucleolar condensation of YFP-APE1 as APE1-assembled LLPS in vivo which is independent of NPM1 but dependent on APE1 W119 residue (Figures 5 and 6). We also noticed the size and number of YFP-APE1-dependent LLPS in nucleoli varied from one cell to another, while NPM1 typically formed similar size condensates within the nucleoli when YFP was expressed. Whether rDNA, transcribed pre-rRNA and further processed intermediate products, or a combination of these factors are important for the APE1 LLPS in vivo remains to be determined. Our data reveal the in vivo APE1 LLPS within nucleoli subsequently recruits ATR as well as its activators TopBP1 and ETAA1 (Figure 5).
A previous study has revealed that ectopically overexpressed eGFP-TopBP1 is translocated to nucleoli and colocalized with nucleolar protein UBF and RNA Pol I and that TopBP1 overexpression leads to relocalization of NPM1 and NCL from the center region of nucleoli to the peri-nucleolar region (43). Here we show that the localization of TopBP1 induced by YFP-APE1-OE is across almost all the nucleoli (Figures 5 and 6). While YFP-APE1 is co-localized with NPM1, APE1-induced LLPS doesn’t change NPM1 localization within nucleoli, demonstrating that APE1 and NPM1 nucleolar localization are likely independent events (Figures 5 and 6). A recent study shows that TopBP1-assembled nuclear LLPS depends on ATR-mediated TopBP1 phosphorylation, serving as positive feedback to amplify the ATR–Chk1 DDR pathway (69). Our data show YFP-APE1-mediated LLPS within nucleoli is independent of TopBP1, indicating APE1 may serve as an upstream regulator of TopBP1 in the context of nucleolar LLPS (Figure 6).
Physiological significance of ATR DDR pathway activation in nucleoli
YFP-APE1 colocalization and condensation with NPM1 suggests that APE1 may be present in all three regions of nucleoli (Figures 5 and 6). YFP-APE1-induced ATR nDDR inhibits pre-rRNA transcription (Figure 7), suggesting that the activated ATR may be located in the FC or at the boundary of FC/DFC within nucleoli. However, we can’t exclude the possibility that ATR is recruited to one specific sub-nucleolar compartment by APE1 LLPS and then re-located to another sub-nucleolar compartment once activated.
In this work, we present evidence of ectopic overexpression of APE1 using different tags (YFP, tGFP, and mCherry) in nucleolar translocation, biomolecular condensate formation, and ATR nDDR. Our control experiments show no nucleolar localization and ATR nDDR by overexpression of empty tag YFP (Figure 5A), tGFP (Figure 3B), and mCherry (Figure 5F), as well as W119R YFP-APE1 (Figure 6E). These results together support the conclusion that ectopic overexpression of APE1 leads to nucleolar condensation and LLPS in vivo and ATR–Chk1 nDDR. How is endogenous APE1 regulated for nucleolar translocation and enrichment for ATR nDDR? Our experiment using PANC1 cells shows that endogenous APE1 doesn’t appear to be enriched in nucleoli and colocalized with NPM1 regardless of oxidative stress (Supplementary Figure S6A). It was noted in a previously published study that endogenous APE1 and endogenous NPM1 were colocalized within the nucleoli of HeLa cells under unperturbed conditions (57). These findings bring the question whether different protein expression levels of endogenous APE1 in different cell lines or under different conditions may determine APE1 nucleolar translocation and ATR nDDR. A prior study has demonstrated that endogenous APE1 protein in six tested pancreatic cancer cells (PANC1, BxPC3, CAPAN-1, ASPC-1, XPA1, and PK9) is highly upregulated compared with two non-neoplastic human pancreatic cell lines HPDE and HPNE (101). Although protein expression of endogenous APE1 in these six pancreatic cancer cell lines are not quantified and compared explicitly, it seems that APE1 protein displays minimal or almost no changes between these cancer cell lines. Thus, selecting different cell lines with differentially elevated APE1 protein expression or upregulating protein expression of endogenous APE1 under certain conditions are needed for future studies to examine the role and mechanism of endogenous APE1 in nucleolar translocation, biomolecular condensate formation, and ATR nDDR.
TopBP1 overexpression leads to ATR-dependent nucleolar segregation but not cell cycle arrest, as well as p53 phosphorylation but not Chk1 phosphorylation (43). Here we demonstrate that YFP-APE1-OE leads to nucleolar translocation and condensation, ATR nDDR-dependent Chk1 and p53 phosphorylation, and cell cycle arrest (Figures 5-7). This suggests that APE1-induced nDDR is distinguished from TopBP1-induced nDDR pathway although both share some similar features (e.g. rRNA transcription suppression). Of note, the nucleolar ATR DDR activation induced by YFP-APE1-OE in this study is under unperturbed conditions in cancer cells but not in normal cells (Figure 5). Considering overall APE1 up-regulation in cancer patients (82,102,103), the APE1-OE-induced ATR nDDR activation provides a new insight into a previously uncharacterized but significant mechanism of the faulty DDR activation by APE1 overexpression in nucleoli of cancer but not normal cells.
In summary, the results of this study uncover that APE1 plays canonical and non-canonical functions in the regulation of ATR–Chk1 DDR pathway (Figure 8). In particular, our study shows for the first time APE1-induced LLPS in vitro and in vivo and the requirement of APE1-assembled biomolecular condensates in the nucleolar ATR–Chk1 DDR activation. Taken together, the distinctive molecular mechanisms of APE1 in ATR–Chk1 DDR pathway may provide new avenues for future cancer therapies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Pinku Muhkerjee, Dr Primo Schaer, Dr Doug Golenbock, Dr David Bartel, Dr Stephen Elledge and Dr Yihong Ye for reagents. We are grateful for the assistance by Dr Didier Dréau, Dr Christine Richardson, Dr Paola Lopez-Duarte, Dr Richard Chi, Trey Grissom and David Gray for FACS as well as fluorescence microscopy analysis. We thank Dr Junya Tomida for the assistance in kinase assays.
Author contributions: J.L. and S.Y. designed experiments. J.L., H.Z. performed experiments. J.L., H.Z., A.M. and S.Y. analyzed the data. J.L. and S.Y. wrote the manuscript. J.L., H.Z., A.M. and S.Y. revised the manuscript. S.Y. supervised the project.
Contributor Information
Jia Li, Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
Haichao Zhao, Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
Anne McMahon, Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
Shan Yan, Department of Biological Sciences, University of North Carolina at Charlotte, Charlotte, NC 28223, USA; School of Data Science, University of North Carolina at Charlotte, Charlotte, NC 28223, USA; Center for Biomedical Engineering and Science, University of North Carolina at Charlotte, Charlotte, NC 28223, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Cancer Institute of the National Institutes of Health [R01CA225637, R01CA251141-subaward to S.Y.]; National Institutes of Environmental Health Sciences of the National Institutes of Health [R21ES032966 to S.Y.]; University of North Carolina at Charlotte [Internal Funding to S.Y.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciccia A., Elledge S.J.. The DNA damage response: making it safe to play with knives. Mol. Cell. 2010; 40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lindahl T., Barnes D.E.. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000; 65:127–133. [DOI] [PubMed] [Google Scholar]
- 4. Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993; 362:709–715. [DOI] [PubMed] [Google Scholar]
- 5. Thompson P.S., Cortez D. New insights into abasic site repair and tolerance. DNA Repair (Amst.). 2020; 90:102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torres-Ramos C.A., Johnson R.E., Prakash L., Prakash S.. Evidence for the involvement of nucleotide excision repair in the removal of abasic sites in yeast. Mol. Cell. Biol. 2000; 20:3522–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lhomme J., Constant J.F., Demeunynck M.. Abasic DNA structure, reactivity, and recognition. Biopolymers. 1999; 52:65–83. [DOI] [PubMed] [Google Scholar]
- 8. Dyrkheeva N.S., Lebedeva N.A., Lavrik O.I.. AP endonuclease 1 as a key enzyme in repair of apurinic/apyrimidinic sites. Biochemistry (Mosc). 2016; 81:951–967. [DOI] [PubMed] [Google Scholar]
- 9. Yan S., Sorrell M., Berman Z.. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci. 2014; 71:3951–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008; 9:619–631. [DOI] [PubMed] [Google Scholar]
- 11. Maizels N., Davis L.. Initiation of homologous recombination at DNA nicks. Nucleic Acids Res. 2018; 46:6962–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKinnon P.J., Caldecott K.W.. DNA strand break repair and human genetic disease. Annu. Rev. Genomics Hum. Genet. 2007; 8:37–55. [DOI] [PubMed] [Google Scholar]
- 13. Nassour J., Martien S., Martin N., Deruy E., Tomellini E., Malaquin N., Bouali F., Sabatier L., Wernert N., Pinte S.et al.. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat. Commun. 2016; 7:10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vrtis K.B., Dewar J.M., Chistol G., Wu R.A., Graham T.G.W., Walter J.C.. Single-strand DNA breaks cause replisome disassembly. Mol. Cell. 2021; 81:1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paull T.T. Mechanisms of ATM activation. Annu. Rev. Biochem. 2015; 84:711–738. [DOI] [PubMed] [Google Scholar]
- 16. Lee J.H., Paull T.T.. Direct activation of the ATM protein kinase by the mre11/rad50/nbs1 complex. Science. 2004; 304:93–96. [DOI] [PubMed] [Google Scholar]
- 17. Bakkenist C.J., Kastan M.B.. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003; 421:499–506. [DOI] [PubMed] [Google Scholar]
- 18. Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T.. ATM activation by oxidative stress. Science. 2010; 330:517–521. [DOI] [PubMed] [Google Scholar]
- 19. Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017; 66:801–817. [DOI] [PubMed] [Google Scholar]
- 20. Saldivar J.C., Cortez D., Cimprich K.A.. The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell. Biol. 2017; 18:622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kumagai A., Shevchenko A., Shevchenko A., Dunphy W.G.. Treslin collaborates with topbp1 in triggering the initiation of DNA replication. Cell. 2010; 140:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan S., Lindsay H.D., Michael W.M.. Direct requirement for xmus101 in ATR-mediated phosphorylation of claspin bound chk1 during checkpoint signaling. J. Cell Biol. 2006; 173:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bass T.E., Luzwick J.W., Kavanaugh G., Carroll C., Dungrawala H., Glick G.G., Feldkamp M.D., Putney R., Chazin W.J., Cortez D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016; 18:1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haahr P., Hoffmann S., Tollenaere M.A., Ho T., Toledo L.I., Mann M., Bekker-Jensen S., Raschle M., Mailand N.. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016; 18:1196–1207. [DOI] [PubMed] [Google Scholar]
- 25. Lee Y.C., Zhou Q., Chen J., Yuan J.. RPA-binding protein ETAA1 is an ATR activator involved in DNA replication stress response. Curr. Biol. 2016; 26:3257–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zou L., Elledge S.J.. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003; 300:1542–1548. [DOI] [PubMed] [Google Scholar]
- 27. Willis J., Patel Y., Lentz B.L., Yan S.. APE2 is required for ATR–Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:10592–10597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wallace B.D., Berman Z., Mueller G.A., Lin Y., Chang T., Andres S.N., Wojtaszek J.L., DeRose E.F., Appel C.D., London R.E.et al.. APE2 Zf-GRF facilitates 3'-5' resection of DNA damage following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:304–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lin Y., Bai L., Cupello S., Hossain M.A., Deem B., McLeod M., Raj J., Yan S.. APE2 promotes DNA damage response pathway from a single-strand break. Nucleic Acids Res. 2018; 46:2479–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weeks S.E., Metge B.J., Samant R.S.. The nucleolus: a central response hub for the stressors that drive cancer progression. Cell. Mol. Life Sci. 2019; 76:4511–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boulon S., Westman B.J., Hutten S., Boisvert F.M., Lamond A.I.. The nucleolus under stress. Mol. Cell. 2010; 40:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feric M., Vaidya N., Harmon T.S., Mitrea D.M., Zhu L., Richardson T.M., Kriwacki R.W., Pappu R.V., Brangwynne C.P.. Coexisting liquid phases underlie nucleolar subcompartments. Cell. 2016; 165:1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drygin D., Rice W.G., Grummt I.. The RNA polymerase i transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010; 50:131–156. [DOI] [PubMed] [Google Scholar]
- 34. Kruhlak M., Crouch E.E., Orlov M., Montano C., Gorski S.A., Nussenzweig A., Misteli T., Phair R.D., Casellas R.. The ATM repair pathway inhibits RNA polymerase i transcription in response to chromosome breaks. Nature. 2007; 447:730–734. [DOI] [PubMed] [Google Scholar]
- 35. van Sluis M., McStay B.. A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev. 2015; 29:1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larsen D.H., Hari F., Clapperton J.A., Gwerder M., Gutsche K., Altmeyer M., Jungmichel S., Toledo L.I., Fink D., Rask M.B.et al.. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat. Cell Biol. 2014; 16:792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Korsholm L.M., Gal Z., Nieto B., Quevedo O., Boukoura S., Lund C.C., Larsen D.H.. Recent advances in the nucleolar responses to DNA double-strand breaks. Nucleic Acids Res. 2020; 48:9449–9461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ciccia A., Huang J.W., Izhar L., Sowa M.E., Harper J.W., Elledge S.J.. Treacher collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:18631–18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Korsholm L.M., Gal Z., Lin L., Quevedo O., Ahmad D.A., Dulina E., Luo Y., Bartek J., Larsen D.H.. Double-strand breaks in ribosomal RNA genes activate a distinct signaling and chromatin response to facilitate nucleolar restructuring and repair. Nucleic Acids Res. 2019; 47:8019–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mooser C., Symeonidou I.E., Leimbacher P.A., Ribeiro A., Shorrocks A.K., Jungmichel S., Larsen S.C., Knechtle K., Jasrotia A., Zurbriggen D.et al.. Treacle controls the nucleolar response to rDNA breaks via TOPBP1 recruitment and ATR activation. Nat. Commun. 2020; 11:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Velichko A.K., Petrova N.V., Luzhin A.V., Strelkova O.S., Ovsyannikova N., Kireev II, Petrova N.V., Razin S.V., Kantidze O.L. Hypoosmotic stress induces r loop formation in nucleoli and ATR/ATM-dependent silencing of nucleolar transcription. Nucleic Acids Res. 2019; 47:6811–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Velichko A.K., Ovsyannikova N., Petrova N.V., Luzhin A.V., Vorobjeva M., Gavrikov A.S., Mishin A.S., Kireev II, Razin S.V., Kantidze O.L. Treacle and TOPBP1 control replication stress response in the nucleolus. J. Cell Biol. 2021; 220:e202008085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sokka M., Rilla K., Miinalainen I., Pospiech H., Syvaoja J.E.. High levels of topbp1 induce ATR-dependent shut-down of rRNA transcription and nucleolar segregation. Nucleic Acids Res. 2015; 43:4975–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hadi M.Z., Ginalski K., Nguyen L.H., Wilson D.M. 3rd. Determinants in nuclease specificity of ape1 and ape2, human homologues of escherichia coli exonuclease III. J. Mol. Biol. 2002; 316:853–866. [DOI] [PubMed] [Google Scholar]
- 45. Chohan M., Mackedenski S., Li W.M., Lee C.H.. Human apurinic/apyrimidinic endonuclease 1 (APE1) has 3' RNA phosphatase and 3' exoribonuclease activities. J. Mol. Biol. 2015; 427:298–311. [DOI] [PubMed] [Google Scholar]
- 46. Whitaker A.M., Freudenthal B.D.. APE1: a skilled nucleic acid surgeon. DNA Repair (Amst.). 2018; 71:93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li M., Wilson D.M. 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid. Redox Signal. 2014; 20:678–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tell G., Quadrifoglio F., Tiribelli C., Kelley M.R.. The many functions of APE1/Ref-1: not only a DNA repair enzyme. Antioxid. Redox Signal. 2009; 11:601–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Georgiadis M.M., Luo M., Gaur R.K., Delaplane S., Li X., Kelley M.R.. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008; 643:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Walker L.J., Robson C.N., Black E., Gillespie D., Hickson I.D.. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of jun DNA binding. Mol. Cell. Biol. 1993; 13:5370–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kelley M.R., Georgiadis M.M., Fishel M.L.. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr. Mol. Pharmacol. 2012; 5:36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xanthoudakis S., Smeyne R.J., Wallace J.D., Curran T.. The redox/DNA repair protein, ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:8919–8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fung H., Demple B.. A vital role for ape1/ref1 protein in repairing spontaneous DNA damage in human cells. Mol. Cell. 2005; 17:463–470. [DOI] [PubMed] [Google Scholar]
- 54. Chattopadhyay R., Wiederhold L., Szczesny B., Boldogh I., Hazra T.K., Izumi T., Mitra S.. Identification and characterization of mitochondrial abasic (AP)-endonuclease in mammalian cells. Nucleic Acids Res. 2006; 34:2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jackson E.B., Theriot C.A., Chattopadhyay R., Mitra S., Izumi T.. Analysis of nuclear transport signals in the human apurinic/apyrimidinic endonuclease (APE1/Ref1). Nucleic Acids Res. 2005; 33:3303–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li M., Zhong Z., Zhu J., Xiang D., Dai N., Cao X., Qing Y., Yang Z., Xie J., Li Z.et al.. Identification and characterization of mitochondrial targeting sequence of human apurinic/apyrimidinic endonuclease 1. J. Biol. Chem. 2010; 285:14871–14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vascotto C., Fantini D., Romanello M., Cesaratto L., Deganuto M., Leonardi A., Radicella J.P., Kelley M.R., D’Ambrosio C., Scaloni A.et al.. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell. Biol. 2009; 29:1834–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Antoniali G., Serra F., Lirussi L., Tanaka M., D’Ambrosio C., Zhang S., Radovic S., Dalla E., Ciani Y., Scaloni A.et al.. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nat. Commun. 2017; 8:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lopez D.J., Rodriguez J.A., Banuelos S.. Molecular mechanisms regulating the DNA repair protein APE1: a focus on its flexible N-terminal tail domain. Int. J. Mol. Sci. 2021; 22:6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin Y., Raj J., Li J., Ha A., Hossain M.A., Richardson C., Mukherjee P., Yan S.. APE1 senses DNA single-strand breaks for repair and signaling. Nucleic Acids Res. 2020; 48:1925–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Liu T.C., Lin C.T., Chang K.C., Guo K.W., Wang S., Chu J.W., Hsiao Y.Y.. APE1 distinguishes DNA substrates in exonucleolytic cleavage by induced space-filling. Nat. Commun. 2021; 12:601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu T.C., Guo K.W., Chu J.W., Hsiao Y.Y.. Understanding APE1 cellular functions by the structural preference of exonuclease activities. Comput. Struct. Biotechnol. J. 2021; 19:3682–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin Y., McMahon A., Driscoll G., Bullock S., Zhao J., Yan S.. Function and molecular mechanisms of APE2 in genome and epigenome integrity. Mutat. Res. Rev. Mutat. Res. 2021; 787:108347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hossain M.A., Lin Y., Driscoll G., Li J., McMahon A., Matos J., Zhao H., Tsuchimoto D., Nakabeppu Y., Zhao J.et al.. APE2 is a general regulator of the ATR–Chk1 DNA damage response pathway to maintain genome integrity in pancreatic cancer cells. Front. Cell Dev. Biol. 2021; 9:738502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lyon A.S., Peeples W.B., Rosen M.K.. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell. Biol. 2021; 22:215–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van Sluis M., McStay B.. Nucleolar DNA double-strand break responses underpinning rDNA genomic stability. Trends Genet. 2019; 35:743–753. [DOI] [PubMed] [Google Scholar]
- 67. Brangwynne C.P., Mitchison T.J., Hyman A.A.. Active liquid-like behavior of nucleoli determines their size and shape in xenopus laevis oocytes. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:4334–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Toledo L.I., Murga M., Gutierrez-Martinez P., Soria R., Fernandez-Capetillo O.. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008; 22:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Frattini C., Promonet A., Alghoul E., Vidal-Eychenie S., Lamarque M., Blanchard M.P., Urbach S., Basbous J., Constantinou A.. TopBP1 assembles nuclear condensates to switch on ATR signaling. Mol. Cell. 2021; 81:1231–1245. [DOI] [PubMed] [Google Scholar]
- 70. Dunphy W.G. TopBP1 comes into focus. Mol. Cell. 2021; 81:1126–1127. [DOI] [PubMed] [Google Scholar]
- 71. Tosolini D., Antoniali G., Dalla E., Tell G.. Role of phase partitioning in coordinating DNA damage response: focus on the apurinic apyrimidinic endonuclease 1 interactome. Biomol. Concepts. 2020; 11:209–220. [DOI] [PubMed] [Google Scholar]
- 72. Malfatti M.C., Antoniali G., Codrich M., Tell G.. Coping with RNA damage with a focus on APE1, a BER enzyme at the crossroad between DNA damage repair and RNA processing/decay. DNA Repair (Amst.). 2021; 104:103133. [DOI] [PubMed] [Google Scholar]
- 73. Yazdanifar M., Zhou R., Grover P., Williams C., Bose M., Moore L.J., Wu S.T., Maher J., Dreau D., Mukherjee A.P.. Overcoming immunological resistance enhances the efficacy of a novel anti-tMUC1-CAR t cell treatment against pancreatic ductal adenocarcinoma. Cells. 2019; 8:1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schuermann D., Scheidegger S.P., Weber A.R., Bjoras M., Leumann C.J., Schar P.. 3CAPS - a structural AP-site analogue as a tool to investigate DNA base excision repair. Nucleic Acids Res. 2016; 44:2187–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kleaveland B., Shi C.Y., Stefano J., Bartel D.P.. A network of noncoding regulatory RNAs acts in the mammalian brain. Cell. 2018; 174:350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cortez D., Guntuku S., Qin J., Elledge S.J.. ATR and ATRIP: partners in checkpoint signaling. Science. 2001; 294:1713–1716. [DOI] [PubMed] [Google Scholar]
- 77. Xu Y., Anderson D.E., Ye Y.. The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov. 2016; 2:16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rai G., Vyjayanti V.N., Dorjsuren D., Simeonov A., Jadhav A., Wilson D.M. 3rd, Maloney D.J.. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 2012; 55:3101–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bapat A., Glass L.S., Luo M., Fishel M.L., Long E.C., Georgiadis M.M., Kelley M.R.. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J. Pharmacol. Exp. Ther. 2010; 334:988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shimizu N., Sugimoto K., Tang J., Nishi T., Sato I., Hiramoto M., Aizawa S., Hatakeyama M., Ohba R., Hatori H.et al.. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000; 18:877–881. [DOI] [PubMed] [Google Scholar]
- 81. Zou G.M., Luo M.H., Reed A., Kelley M.R., Yoder M.C.. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007; 109:1917–1922. [DOI] [PubMed] [Google Scholar]
- 82. Jensen K.A., Shi X., Yan S.. Genomic alterations and abnormal expression of APE2 in multiple cancers. Sci. Rep. 2020; 10:3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yuan C.L., He F., Ye J.Z., Wu H.N., Zhang J.Y., Liu Z.H., Li Y.Q., Luo X.L., Lin Y., Liang R.. APE1 overexpression is associated with poor survival in patients with solid tumors: a meta-analysis. Oncotarget. 2017; 8:59720–59728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wang Z., Ayoub E., Mazouzi A., Grin I., Ishchenko A.A., Fan J., Yang X., Harihar T., Saparbaev M., Ramotar D. Functional variants of human APE1 rescue the DNA repair defects of the yeast AP endonuclease/3'-diesterase-deficient strain. DNA Repair (Amst.). 2014; 22:53–66. [DOI] [PubMed] [Google Scholar]
- 85. McNeill D.R., Wilson D.M. 3rd. A dominant-negative form of the major human abasic endonuclease enhances cellular sensitivity to laboratory and clinical DNA-damaging agents. Mol. Cancer Res. 2007; 5:61–70. [DOI] [PubMed] [Google Scholar]
- 86. Gelin A., Redrejo-Rodriguez M., Laval J., Fedorova O.S., Saparbaev M., Ishchenko A.A.. Genetic and biochemical characterization of human AP endonuclease 1 mutants deficient in nucleotide incision repair activity. PLoS One. 2010; 5:e12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Freudenthal B.D., Beard W.A., Cuneo M.J., Dyrkheeva N.S., Wilson S.H.. Capturing snapshots of APE1 processing DNA damage. Nat. Struct. Mol. Biol. 2015; 22:924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fantini D., Vascotto C., Marasco D., D’Ambrosio C., Romanello M., Vitagliano L., Pedone C., Poletto M., Cesaratto L., Quadrifoglio F.et al.. Critical lysine residues within the overlooked N-terminal domain of human APE1 regulate its biological functions. Nucleic Acids Res. 2010; 38:8239–8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Busso C.S., Iwakuma T., Izumi T.. Ubiquitination of mammalian AP endonuclease (APE1) regulated by the p53-MDM2 signaling pathway. Oncogene. 2009; 28:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Busso C.S., Lake M.W., Izumi T.. Posttranslational modification of mammalian AP endonuclease (APE1). Cell. Mol. Life. Sci. 2010; 67:3609–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lirussi L., Antoniali G., Vascotto C., D’Ambrosio C., Poletto M., Romanello M., Marasco D., Leone M., Quadrifoglio F., Bhakat K.K.et al.. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell. 2012; 23:4079–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fiorini C., Cordani M., Padroni C., Blandino G., Di Agostino S., Donadelli M.. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta. 2015; 1853:89–100. [DOI] [PubMed] [Google Scholar]
- 93. Dong Z., Zhu C., Zhan Q., Jiang W.. The roles of RRP15 in nucleolar formation, ribosome biogenesis and checkpoint control in human cells. Oncotarget. 2017; 8:13240–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kumagai A., Lee J., Yoo H.Y., Dunphy W.G.. TopBP1 activates the ATR-ATRIP complex. Cell. 2006; 124:943–955. [DOI] [PubMed] [Google Scholar]
- 95. Yan S., Michael W.M.. TopBP1 and DNA polymerase-alpha directly recruit the 9-1-1 complex to stalled DNA replication forks. J. Cell Biol. 2009; 184:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brocca S., Grandori R., Longhi S., Uversky V.. Liquid-Liquid phase separation by intrinsically disordered protein regions of viruses: roles in viral life cycle and control of virus-host interactions. Int. J Mol. Sci. 2020; 21:9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Clerc I., Sagar A., Barducci A., Sibille N., Bernado P., Cortes J.. The diversity of molecular interactions involving intrinsically disordered proteins: a molecular modeling perspective. Comput. Struct. Biotechnol. J. 2021; 19:3817–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wiedner H.J., Giudice J.. It's not just a phase: function and characteristics of RNA-binding proteins in phase separation. Nat. Struct. Mol. Biol. 2021; 28:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mol C.D., Izumi T., Mitra S., Tainer J.A.. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination (corrected). Nature. 2000; 403:451–456. [DOI] [PubMed] [Google Scholar]
- 100. Whitaker A.M., Flynn T.S., Freudenthal B.D.. Molecular snapshots of APE1 proofreading mismatches and removing DNA damage. Nat. Commun. 2018; 9:399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zou G.M., Maitra A.. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 2008; 7:2012–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Manoel-Caetano F.S., Rossi A.F.T., Calvet de Morais G., Severino F.E., Silva A.E.. Upregulation of the APE1 and H2AX genes and miRNAs involved in DNA damage response and repair in gastric cancer. Genes Dis. 2019; 6:176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Song H., Zeng J., Lele S., LaGrange C.A., Bhakat K.K.. APE1 and SSRP1 is overexpressed in muscle invasive bladder cancer and associated with poor survival. Heliyon. 2021; 7:e06756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.