


Abstract
Ribosomes are remarkable in their malleability to accept diverse aminoacyl-tRNA substrates from both the same organism and other organisms or domains of life. This is a critical feature of the ribosome that allows the use of orthogonal translation systems for genetic code expansion. Optimization of these orthogonal translation systems generally involves focusing on the compatibility of the tRNA, aminoacyl-tRNA synthetase, and a non-canonical amino acid with each other. As we expand the diversity of tRNAs used to include non-canonical structures, the question arises as to the tRNA suitability on the ribosome. Specifically, we investigated the ribosomal translation of allo-tRNAUTu1, a uniquely shaped (9/3) tRNA exploited for site-specific selenocysteine insertion, using single-molecule fluorescence. With this technique we identified ribosomal disassembly occurring from translocation of allo-tRNAUTu1 from the A to the P site. Using cryo-EM to capture the tRNA on the ribosome, we pinpointed a distinct tertiary interaction preventing fluid translocation. Through a single nucleotide mutation, we disrupted this tertiary interaction and relieved the translation roadblock. With the continued diversification of genetic code expansion, our work highlights a targeted approach to optimize translation by distinct tRNAs as they move through the ribosome.
Graphical Abstract
Graphical Abstract.
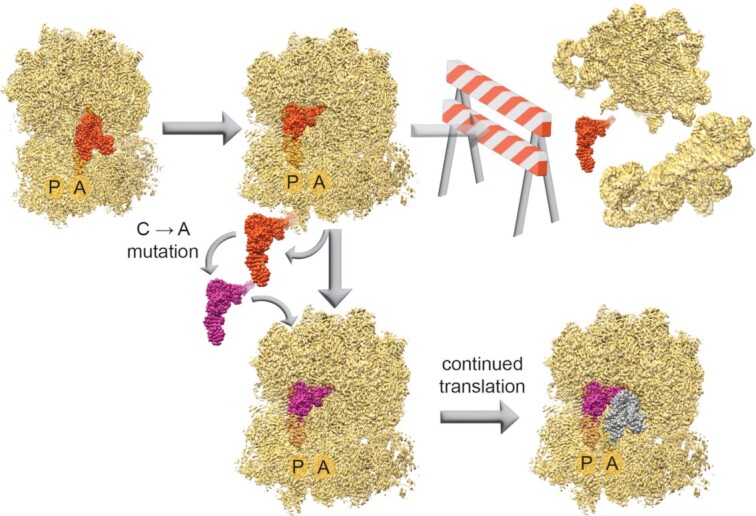
A single nucleotide mutation in tRNAUTu1 removes the translation roadblock causing ribosome disassembly and permits continued translation.
INTRODUCTION
Protein synthesis is a highly conserved process performed by the ribosome. Aminoacyl-tRNA synthetases (aaRSs) acylate the 3′-terminal CCA sequence with the cognate amino acid to produce aminoacyl-tRNAs (aa-tRNAs) that are carried to the ribosome by EF-Tu. With 46 aa-tRNA substrates in Escherichia coli (1,2), the ribosome has evolved to accommodate and discriminate between all tRNAs for efficient translation (3). Given that extensive contacts form between the ribosome and aa-tRNA, this is a remarkable feature that is only possible due to the malleability of the ribosome and conserved tRNA structure (4). The ribosome has specifically evolved for translation of aa-tRNAs from the same organism, but also has the capability to perform heterologous translation, accepting aa-tRNAs from other organisms or domains of life. The orthogonality of some tRNA:aaRS pairs between domains of life allows for translation systems from other organisms to be used for genetic recoding and insertion of non-canonical amino acids (ncAAs) (5,6). Genetic recoding typically is performed at traditional stop codons (e.g. UAG, UAA or UGA), to introduce a ncAA. The efficiency and specificity of genetic recoding relies first on the speed and accuracy of aa-tRNA synthesis, and second on the decoding and translocation function of the tRNA as it moves through the ribosome (Figure 1).
Figure 1.
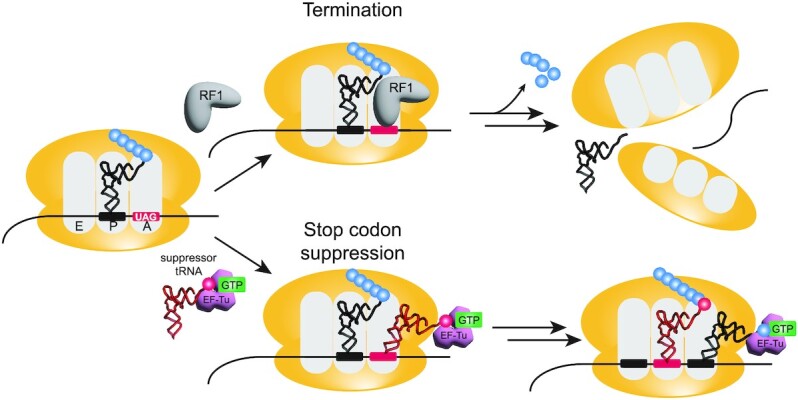
Genetic recoding at stop codons competes with release factors. In the presence of a UAG stop codon, one of two situations can occur: termination or suppression. Termination of translation occurs when release factor 1 (RF1) binds, releasing the polypeptide chain and invoking separation of the ribosomal subunits from the mRNA. Alternatively, in the presence of a suppressor tRNA, the UAG codon can be suppressed, inserting an amino acid and allowing continuation of translation.
Substantial engineering efforts have focused on the former; Methanosarcina PylRS (7) and Methanocaldococcus jannaschii TyrRS with their cognate tRNAs have become well-established orthogonal translation systems. These tRNA synthetases and their variants facilitated the successful incorporation of several hundred different ncAAs programmed by UAG or UAA (8). An alternate natural translation system with potential for genetic recoding is the insertion of selenocysteine (Sec) programmed by UGA. The high reactivity and unique chemical properties of Sec have attracted the attention of synthetic biologists for various applications. However, due to the requirement of Sec-specific translation components (mRNA hairpin recognition element and elongation factor SelB), genetic code expansion applications for Sec have been limited. Consequently, research focused on engineering a simpler translation pathway that could be applied for site-specific insertion of Sec in any protein. This strategy involves engineering tRNASec to be recognized by the natural elongation factor (EF-Tu), following the translation pathway of the other 20 canonical amino acids. Initial attempts to create a hybrid tRNA between tRNASec and tRNASer for recognition by EF-Tu resulted in low protein yields and minimal Sec insertion (9–12). Therefore, an alternate approach searched for other tRNAs that resembled tRNASec and are capable of EF-Tu recognition. A search of metagenomic sequences revealed uniquely structured tRNAs (allo-tRNAs) containing recognition elements that resembled tRNASec but with a 12 bp acceptor domain (acceptor and T-stem combined) for proper recognition by EF-Tu (13). Specifically, tRNAUTu1 was found to be a serine isoacceptor with a 9/3 (acceptor/T-stem bp) structure that allowed EF-Tu driven Sec incorporation (Figure 2).
Figure 2.

EF-Tu driven selenocysteine incorporation. (A) Cloverleaf structures of E. coli tRNASer, tRNAsupD (the amber suppressor of tRNASer) and tRNAUTu1. Residues are colored as follows: acceptor arm (green), D-arm (purple), anticodon arm (red), variable arm (blue), and T-arm (gold). (B) Translation schematic of EF-Tu driven selenocysteine incorporation with tRNAUTu1. tRNAUTu1 is first serylated by seryl-tRNA synthetase (SerRS) to generate Ser-tRNAUTu1. Selenocysteine synthase (SelA) converts the serine to selenocysteine to generate Sec-tRNAUTu1. Aminoacylated tRNAUTu1 (including Ser-tRNAUTu1 which is misincorporated) are substrates for EF-Tu, inserting at the UAG codon for continued translation.
These allo-tRNAs are non-native substrates for the E. coli ribosome, therefore their translation efficiency is unknown. For seamless translation, tRNAs must be quickly accepted into the ribosomal A site and efficiently translocated to the P site (14,15). Disturbing these processes can lead to stalling and disassembly of the translating ribosome, reducing protein synthesis. This has been observed with quadruplet tRNAs, which are poor substrates for the ribosome due to the four-base anticodon-codon interaction (16–18). This roadblock has been tackled separately by mutating either the tRNA in the anticodon loop to promote the four-base anticodon-codon interaction (19,20) or engineering an orthogonal ribosome (Ribo-Q) efficient at decoding quadruplet codons (21).
Here, we investigated the suitability of an allo-tRNA, tRNAUTu1, for translation by E. coli ribosomes, and demonstrate the importance of global tRNA structure for efficient translation. tRNAUTu1 is used for site-specific insertion of Sec by harnessing EF-Tu to avoid constraints on natural Sec translation. Using single-molecule FRET (Förster resonance energy transfer) we visualized movement of tRNAUTu1 through the ribosome and compared it to natural tRNAs (tRNASer and tRNAsupD). From this, we were able to establish the roadblocks associated with tRNAUTu1 translation through the ribosome. Implementing cryo-EM, we visualized tRNAUTu1 in complex with the E. coli ribosome to pinpoint the structural feature in tRNAUTu1 hindering translation. With this information, we engineered a single-base mutation in tRNAUTu1 that removed the rigidity of the variable arm, enhancing translational processivity on the ribosome. This was verified through in vivo and in vitro assays and led to an increase in recombinant protein production. Our results demonstrate the importance of ensuring that synthetic tRNAs used for genetic code expansion are suitable substrates for ribosomal translation and show the power of combined dynamics and structural analyses for improved tRNA engineering.
MATERIALS AND METHODS
Single-molecule translation assay
Details on preparation of reagents for the single-molecule assay are described in the Supplemental Information. Using a previously described protocol, the 30S pre-initiation complex (30S PIC) was prepared from Cy3B-labeled 30S subunit, ribosomal protein S1, initiation factor IF2, fMet-tRNAfMet, biotinylated mRNA and 4 mM GTP. Prior to immobilizing the 30S PIC, we prepared the SMRT Cell v3 from Pacific Biosciences (Menlo Park), a ZMW chip, with Neutravidin. The 30S PIC was then diluted to 15 nM and loaded into the SMRT cell (22).
Ternary complexes (TCs) of Phe(Cy5)-tRNAPhe and Ser-tRNAs were formed with EF-Tu (GTP) using a previously described protocol (23). After formation of TCs, a 2X delivery mix was created with 100–400 nM Cy5-TCPhe and 400 nM-2 μM TCSer along with 200 nM BHQ-2-50S, 100 nM EF-G, 4 mM GTP, 2.5 mM Trolox and the oxygen scavenging system (PCA and PCD). Before starting an experiment, the delivery mix was diluted into polymix mixture in the SMRT Cell and loaded into a modified PacBio RSII sequencer (24). At the start of the experiment, the instrument illuminated the SMRT cell with a green laser and then transferred 20 μl of a delivery mixture onto the cell surface at t = 10 s. All experiments were performed at 20°C, and data was collected for 6 min.
Cryo-EM
Details on preparation of grids and data processing are described in the Supplemental Information. Data was collected on a Titan Krios transmission electron microscope (Thermo Fisher Scientific) equipped with a K3 Summit direct electron detection camera (Gatan) in counting mode. Micrographs were accrued at a calibrated pixel size of 1.037 Å and with a nominal defocus range of –1 to –2 μm. Each micrograph consisted of 40 frames collected over a two second exposure at a dose rate of roughly 20 electrons per pixel per second for a total dose of roughly 40 electrons/Å2. The micrographs were acquired as dose-fractionated image stacks.
sfGFP readthrough assay
sfGFP readthrough was performed as previously described (9) using plasmids pB_sfGFP (UKGE or UGTT), pSecUAG (with tRNAsupD, tRNAUTu1 or tRNAUTu1A) and B-95.ΔAΔfabR E. coli cells (25). Each experiment was performed with a minimum of four biological replicates and background subtracted (no IPTG).
GPx1 protein production
Expression and purification of human GPx1 was performed as previously described (9,11). Briefly, pET-GPx1(49UAG) and pSecUAG (with tRNAsupD, tRNAUTu1 or tRNAUTu1A) were transformed into B-95.ΔAΔfabR E. coli cells (25). Cultures were grown at 37°C and immediately induced with 0.1% arabinose for tRNA expression with 10 μM sodium selenite for Sec formation. Cells were grown until OD600 1.2, at which point the temperature was lowered to 20°C, and protein expression was induced for 20 hrs. Purified protein samples were sent to Bioinformatics Solutions, Inc (Canada) for intact mass spectrometry to quantify Sec incorporation. Spectra were analysed using Thermo BioPharma Finder™ 4.1 (ThermoFisher Scientific).
RESULTS
tRNAUTu1 induces low translational processivity
tRNAUTu1 was identified in a metagenomic search for use in site-specific, EF-Tu driven, Sec insertion (13,26). Since tRNAUTu1 is not a canonical substrate for E. coli translation we decided to initially explore the dynamics of tRNAUTu1 stop-codon suppression within the E. coli ribosome. This was accomplished using a single-molecule fluorescence assay and zero-mode waveguide (ZMW)-based instrumentation (24). We observed the progression of translation in real time at single-codon resolution by tracking the ribosome conformational changes underlying elongation. The ribosomal intersubunit rotational movements during elongation were monitored using a small (30S) subunit site-specifically labelled with Cy3B and a large (50S) subunit labelled with BHQ-2 (a non-fluorescent quencher), allowing for FRET between the two dyes (27). In addition to ribosomes labeled with FRET pairs, Cy5-labeled tRNAPhe was used to track the tRNA occupancy on the ribosome during translation (Figure 3A). We applied this approach to monitor decoding of the UAG stop codon by serylated tRNAUTu1 (Ser-tRNAUTu1) and compared it to a canonical serylated tRNA capable of UAG suppression (Ser-tRNAsupD).
Figure 3.

Single-molecule characterization of low translation processivity induced by tRNAUTu1. (A) In all single-molecule experiments, 30S preinitiation complexes (PIC) containing Cy3B-30S, fMet-tRNAfMet, and IF2 is immobilized on the surface of the ZMW wells through biotinylated mRNAs. The reaction is started by delivery of BHQ-2-50S, Cy5-TCPhe, EF-G, and one of the serine-charged suppressor tRNA TCs. (B) Expected sequence of fluorescence signals starting with quenching of Cy3B (green) that signals 50S subunit joining (initiation) and six cycles of changes in Cy3B intensity that signal intersubunit rotations during elongation. First and last four elongation cycles are correlated with Cy5 intensity changes that signal Cy5-tRNAPhe binding. (C) Representative single-molecule trace shows successful ribosome translation of all six codons after UAG stop codon suppression by tRNAsupD. (D) Codon survival plot of the fraction of translating ribosomes as a function of number of codons translated. In the presence of 1μM tRNAUTu1 (red, n = 223) ribosome survival (percent of codon 2 ribosomes that survive codon 3) is drastically decreased compared to the same amount of tRNAsupD (blue, n = 186) or tRNASer (black, n = 265).
This approach allowed us to monitor translation elongation and stop-codon suppression with different tRNAs. The initial low Cy3B signal in the single-molecule trace is a result of the BHQ-2-50S subunit quenching Cy3B on the 30S subunit to form the 70S non-rotated state during translation initiation (Figure 3B). Subsequent decoding of the first Phe codon prompts the arrival of Cy5-TCPhe, signaled by a burst in Cy5 fluorescence. Successful peptide bond formation is visualized by medium Cy3B intensity from a FRET transition due to the ribosome intersubunit rotation into the rotated state. This successful decoding event is then followed by EF-G-driven translocation of Cy5-tRNAPhe from the A site to the P site, placing the UAG stop codon in the A site and driving intersubunit rotation back to the non-rotated state (low Cy3B intensity). Each Cy5-tRNAPhe occupancy lasts for two cycles of elongation as it translocates from A to P and then P to E sites. Therefore, elongation of consecutive Phe codons downstream of the stop codon results in cycles of double- and single-occupancy states, making the signal for translation downstream of stop codon unequivocal (Figure 3B). Following the changes in fluorescence intensity, we can track the processivity of translation (Figure 3C).
Translation with Ser-tRNAUTu1 yielded a decoding efficiency at the UAG stop codon (codon 2) of 83%. Similar suppression efficiency was measured when translating in the presence of Ser-tRNAsupD (72%) (Figure 3D). tRNAsupD (identical in sequence to E. coli tRNASer but with an anticodon to decode UAG) provides us with information on the suppression efficiency of a natural substrate for the ribosome. Since decoding of UAG is known to be slower than a sense codon, we directly compared the two suppressor tRNAs (tRNAUTu1 to tRNAsupD) but also measured tRNASer as a control using single-molecule fluorescence. The decoding time is calculated from the dwell time of the non-rotated state prior to ribosome rotation at which point the tRNA is successfully incorporated and peptidyl transfer occurs (Supplementary Figure S1A). Decoding of tRNAUTu1 showed a similar distribution of decoding times at UAG as tRNAsupD with means of ∼7 s at 1 μM TC. This is approximately twice as long as the decoding time of tRNASer that decodes the sense codon UCG (Supplementary Figure S1B). Due to limitations in the in vitro single-molecule setup (detection times for signals and tRNA/factor concentrations), decoding times are not equivalent to those observed during cellular translation.
With suppression efficiency and decoding time of tRNAUTu1 matching the natural suppressor tRNAsupD, we next investigated what occurs after translocation of the suppressor tRNA from the A to the P site. After UAG suppression by tRNAUTu1, we found a significant drop in the ribosome processivity at the third codon. At low concentrations (50 nM) of Cy5-TCPhe, only 21% of ribosomes continued translating following tRNAUTu1 incorporation, compared to 84% of ribosomes that continued translating after tRNAsupD insertion (Figure 3D). The low ribosome survival after tRNAUTu1 incorporation was predominantly due to ribosome disassembly pathways. This was manifested as either a 50S subunit dissociation from the immobilized 30S complex or a 70S ribosome dissociation from the immobilized mRNA signaled by the dequenching of Cy3B signal (Supplementary Figure S2A) or disappearance of Cy3B signal (Supplementary Figure S2B), respectively. Of the ribosomes that incorporated tRNAUTu1, 9% underwent one of these two possible ribosome disassembly events before the subsequent translocation step and 64% underwent disassembly after translocation (Supplementary Figure S2C). In contrast, the frequency of this post-translocation disassembly was only 1% for ribosomes that incorporated tRNAsupD (Supplementary Figure S2D). We tested to see whether this pathway competes with the subsequent arrival of the Cy5-TCPhe to codon 3 by increasing the concentration of the Cy5-TCPhe that was delivered from 50 to 200 nM. This resulted in a 2.5-fold reduction of the frequency of post-translocation ribosome disassembly (Supplementary Figure S3A) and increased the overall processivity on codon 3 to 72% (Supplementary Figure S3B). These results suggest an intrinsic ribosome instability introduced upon successful incorporation of tRNAUTu1. This instability is predominantly present after the next translocation that places tRNAUTu1 in the P site. With a fast measured ribosome disassembly time of 9 ± 1 s at room temperature, this intrinsic instability poses a problem to the overall translational yield after stop codon suppression.
The variable arm of tRNAUTu1 forms a unique tertiary interaction
tRNAUTu1 processivity when bound in the ribosomal P site is significantly decreased (Figure 3D) as shown by the high frequency of post-translocation ribosome disassembly (Supplementary Figure S2C); we supposed that the novel structure of tRNAUTu1 may be responsible for this lack of processivity and translation complex destabilization. To visualize structures and orientations of tRNAUTu1 in the ribosome, we used single-particle cryo-EM on samples of tRNAUTu1 in complex with the E. coli 70S ribosome. Due to the high frequency of ribosome disassembly, cryo-EM was advantageous to sort out the lowly-populated ribosome–tRNAUTu1 complex from vacant ribosomes. The complex was prepared by programming tRNAUTu1 to bind in all ribosomal sites using an mRNA containing three tandem UAG codons (28). After cryo-EM image processing, we observed a class of particles with a non-rotated 70S ribosome state bound to triple tRNAUTu1 in the A/A, P/P and E/E states (PDB:7UR5, Figure 4A and Supplementary Figure S4). The global resolution of the triple tRNAUTu1 and ribosome complex is 2.6 Å, allowing atomic-level modeling (Supplementary Figure S4).
Figure 4.

Structural studies of tRNAUTu1 on the ribosome. (A) Structure of the ribosome, highlighting tRNAs in the A, P and E sites. (B) Overlay of A/A (purple, PDB:7UR5) and P/P (orange, PDB:7UR5) state tRNAUTu1 highlights the shift in the variable arm position. (C) Zoom in on the variable arm shows P/P tRNAUTu1 (orange) to rotate 17° upwards from A/A tRNAUTu1 (purple). (D) Overlay of A/A (purple) and P/P (orange) state tRNAUTu1 with A/A state tRNASec (blue, PDB:5LZE) in the context of the ribosome P site. A clash is observed with the A/A state tRNAUTu1 and the A-site finger of the ribosome. A zoom in on the variable arm with respect to the A-site finger shows the shift from the A/A to the P/P state for tRNAUTu1 and compares it with the A/A state of tRNASec (blue). (E) Cloverleaf structure of tRNAUTu1 highlights the position of the residues of interest, C8 and A45. A zoom in on the structure of those residues shows the formation of a hydrogen bond. (F) Cloverleaf structure of tRNASec highlights the corresponding residues of interest from tRNAUTu1. A zoom in on the structure of those residues (PDB:5LZE) shows that there is no base pair present. Instead, the corresponding residues point outwards, away from the tRNA core.
tRNAs form a conserved cloverleaf secondary structure with four helices: the acceptor stem, the D-arm, the anticodon arm, and the T-arm. The clover leaf folds into a three-dimensional L-shaped structure through coaxial stacking of the acceptor stem/T-stem and D-stem/anticodon stem mediated by tertiary interactions from the variable loop to the D-stem and D-loop to T-loop pairing. The length of the variable loop between the anticodon stem and the T-arm, divides the tRNAs into two classes. Class I tRNAs have a variable loop of 4–5 nucleotides, such as tRNAPhe, and class II tRNAs have a much longer variable loop of 10–24 nucleotides, such as tRNASec (29). tRNAUTu1 belongs to class II, in which the variable loop is long enough to form a fifth helix. The global structural features of the tRNAUTu1 acceptor stem and the anticodon stem of tRNAUTu1 are similar to previously published tRNA structures in the A site, including tRNAPhe (PDB: 4V6F) and tRNASec (PDB: 5LZE) (Supplementary Figure S5A–I). The distance and angles between the acceptor stem and anticodon stem are conserved, allowing tRNAUTu1 to reach into both the peptidyl transferase center and the decoding center, essential for tRNA function. The longer acceptor stem of 9 bp leads to a shift of 9.6 Å between the central loop linker G8 of tRNASec and C8 of tRNAUTu1. The compensation of a shorter T-arm of 3 bp, leads to a shift of 6.5 Å between G49 of tRNASec and U49 of tRNAUTu1 (Supplementary Figure S5J–K).
Despite the presence of a tetraloop in the D-loop, the tertiary structure of tRNAUTu1 is stabilized by canonical T-loop to D-loop pairing of G18:U55 and G19:C56. In addition, tRNAUTu1 forms a base pair between U16:U59 which is similar to C16:C59 of tRNASec but not observed in tRNAPhe between U16 and U59 (Supplementary Figure S5D–F). Furthermore, the D-arm of tRNAUTu1 consists of only Watson-Crick base pairs, whereas in both tRNASec and tRNAPhe, nucleotides from the central loop interact in the major groove of the D-arm helix to form base triples (Supplementary Figure S5G–I). Overall, the structural features of tRNAUTu1 in the ribosomal A site led to an 8.7 Å shift of the variable loop as compared to tRNASec (Supplementary Figure S5J–K).
When tRNAUTu1 is bound in the P site, both the acceptor stem and the anticodon stem conformations are conserved, allowing them to reach into both the peptidyl transferase center and the decoding center. The variable loop of tRNAUTu1 is shifted by 10 Å compared with its conformation in the A site. This movement creates a 17° rotation of the variable loop upwards towards the ribosome (Figure 4B, C) to avoid a clash with the A-site finger (ASF), a functional attenuator for translocation (30). The central loop and the T-loop to D-loop interaction networks in P site tRNAUTu1 are similar to A site tRNAUTu1 (Supplementary Figure S6). There are to date no published structures of tRNA with a long variable loop at the P site for comparison. Superimposing the A/A tRNASec into the P site does not result in a clash of the variable loop with the ASF (31) (Figure 4D). These results imply that while A/A tRNASec can theoretically fit into the P site as is, translocation of tRNAUTu1 requires modulation of the variable loop orientation. The additional constraint on tRNAUTu1 variable loop orientation imposed by the ribosomal scaffold could raise the energetic barrier for tRNAUTu1 translocation, and steric clash could lead to ribosome disassembly, as observed by single-molecule FRET (Supplementary Figure S2C). If this hypothesis is true, mutating tRNAUTu1 to allow for higher flexibility or alternative orientation of the variable arm would lower the energetic barrier to P site accommodation and reduce ribosome splitting.
Modulation of the flexibility or orientation of the variable arm could be achieved by redesigning the base of the variable arm, at the central loop that forms the four-way junction in the tRNA. The central loop of tRNAUTu1 does not interact with any of the stems, but A45 base paring with C8 was identified as a key stabilizing element of the central loop (Figure 4E). The A45:C8 base pair is sandwiched between A46:U48 and U9 through stacking interactions (Supplementary Figure S5G). Although uncommon, A:C mismatch base-pair exists in both RNA (e.g. initiator tRNA) (32,33) and DNA structures (34). In contrast, tRNAPhe (PDB: 4V6F) and tRNASec (PDB:5LZE) central loop nucleotides interact with the D-arm forming base triples and do not form strong interactions with each other. Specifically, the corresponding residues in tRNASec (U45 and U9) point in opposite directions with G49 of the variable arm protruding into the D-arm to stabilize the tertiary structure (31) (Figure 4F). We hypothesized that disrupting the A45:C8 interaction might destabilize the central loop, which could lead to higher flexibility of the variable loop, resulting in lower rates of ribosome splitting and improved processivity of ribosomes after suppression.
A single base mutation in tRNAUTu1 increases processivity and protein yield
To test the above hypothesis, we chose to mutate C8 since the connecting regions between the acceptor arm and D-arm are not known to interact with any of the components involved in Sec translation. We made a C8A mutation to reduce the likelihood that a base pair would form with A45 (tRNAUTu1A, Figure 5A). To test the effect of this mutation in vivo, we designed a fluorescence readthrough assay that mimics the low processivity state of tRNAUTu1 (Supplementary Figure S7A). With the observation that codon processivity was increased upon the addition of 200 nM Cy5-TCPhe compared to 50nM (Figure 3D, Supplementary Figure S3), we devised a system in which the availability of the subsequent tRNA was low, such as in a ribosomal stalling event. Following a study that investigated ribosomal stalling motifs (35), we chose to mutate the three amino acids following the amber codon (position 2 of sfGFP) to one of the top 20 stalling motifs (protein sequence GTT) (Supplementary Figure S7B). To confirm that this strategy generated an in vivo assay that reflected our in vitro data, we tested tRNAUTu1 and tRNAsupD for UAG readthrough of sfGFP with the N-terminal protein sequence UKGE (non-ribosomal stalling motif) and UGTT (ribosomal stalling motif). No significant difference was observed between the tRNAs with UKGE, however, a significant decrease in fluorescence was found for tRNAUTu1 compared to tRNAsupD with UGTT (Supplementary Figure S5C, D). These results demonstrate that we have tuned the traditional sfGFP readthrough assay (sfGFP_UGTT) to probe tRNA variants for increased P site processivity.
Figure 5.

Single nucleotide mutation enhances translation processivity. (A) Cloverleaf structure of tRNAUTu1A showing the single nucleotide C to A change from tRNAUTu1 by the red arrow. (B) sfGFP fluorescence readthrough assay (green bars) and GPx1 protein yields (purple dots) demonstrate the increased processivity of tRNAUTu1A compared to tRNAUTu1. Fluorescence reads are shown as an average of four biological replicates (± standard deviation) and protein yields are an average of two biological replicates (± standard deviation). (C) Codon survival curve plotting the fraction of translating ribosomes as a function of number of codons translated highlights the post-suppression survival (percent of codon 2 ribosomes that survive codon 3) advantage in the presence of 1 μM tRNAUTu1A (green, n = 300) over 1 μM tRNAUTu1 (red, n = 223). (D) Pathway map of ribosomes that successfully suppressed a UAG stop codon in the presence of 1 μM tRNAUTu1A.
Testing tRNAUTu1A with our sfGFP_UGTT assay, we observed a 3-fold increase in fluorescence compared to tRNAUTu1 (Figure 5B). This result was further verified when we used tRNAUTu1 and tRNAUTu1A for expression of human GPx1 (a natural selenoprotein with a GTT stalling motif after the amber codon). We found that tRNAUTu1A expressed roughly three times more protein (0.036 ± 0.004 mg/g) compared to tRNAUTu1 (0.010 ± 0.001 mg/g) (Figure 5B) with no significant change in the amount of selenocysteine (∼10%) (Supplementary Figure S8). Moreover, the protein yield achieved by tRNAUTu1A was comparable to the natural suppressor, tRNAsupD.
Real-time single-molecule FRET experiments confirmed that tRNAUTu1A processivity on the ribosome P site increased significantly (75%) relative to tRNAUTu1 (21%) (Figure 5C and D), equivalent to what was observed for tRNAsupD (Figure 3D). Results from intact mass spectrometry of purified GPx1 suggest that the effect of low processivity found for tRNAUTu1 is transferred throughout the length of the protein. ESI-MS found that tRNAUTu1 had a significant amount of truncated protein (51%) which was drastically decreased for tRNAUTu1A (15%) (Supplementary Figure S8A, B).
Disruption of tertiary interaction in tRNAUTu1 removes rigidity of its variable arm
To investigate how the C8A mutation boosted tRNAUTu1A processivity on the ribosome P site, we generated a cryo-EM structure using the same strategy described above (i.e. capturing A and P site tRNAUTu1A on the ribosome). Data processing and particle sorting resulted with two maps, each with either A site tRNAUTu1A or P site tRNAUTu1A alone. A site tRNAUTu1A has a similar density to tRNAUTu1 and was modelled with a similar global fold (PDB:7URI, Supplementary Figure S9). The P site tRNAUTu1A anticodon stem, acceptor arm, and elbow region density are well defined and similar to tRNAUTu1. The stabilized central loop in tRNAUTu1A is modified by C8A mutation through forming the A8 and A45 interaction while the variable loop of P site tRNAUTu1A has poor cryo-EM density and could not be traced with high confidence (PDB:7URM). Highly flexible RNA chains are often poorly defined in cryo-EM, which in turn can point to higher flexibility of the variable loop of tRNAUTu1A in accordance with our single-molecule FRET data presented above (Figure 5C). Moreover, molecular dynamic simulations of each tRNA in solution highlights the increased fluctuations of the variable arm as well as the D- and T-arms (Supplementary Figure S10). Thus, the structural and functional data support the need for flexibility in the suppressor tRNA conformation to accommodate the different ribosomal sites during translation. By engineering tRNAUTu1 to eliminate the stabilizing interactions, we were able to increase its suppression efficiency.
DISCUSSION
Here, we combined functional, dynamic and structural methods to understand translation of a synthetic tRNA (tRNAUTu1) by the E. coli ribosome. Together, our approach allowed us to identify a structural element in tRNAUTu1 that hinders processivity in the ribosome P site. A single nucleotide mutation (C8A) removed the translational roadblock, increasing protein expression to match that of a natural suppressor tRNA (tRNAsupD) (Figure 5B).
Efficient translation requires tRNAs to be optimal substrates for the ribosome with smooth transitions between the A, P and E sites. Details about this journey through the ribosome are emerging with the focus on natural tRNA substrates (36,37). Molecular dynamic simulations have found that the ASF plays a significant role in the transition of tRNAs from the A to the P site, interacting with the T-loop of the tRNA (38). Further interactions were observed with type II tRNAs containing a long variable arm (such as tRNASer, tRNALeu and tRNATyr in E. coli). These additional variable arm contacts are suggested to be the driving force which move the ASF for the tRNA to translocate between the A and P site (39). However, little is known about the details of the variable arm and ASF interactions, making it difficult to engineer a synthetic tRNASec that can efficiently translocate through the ribosome.
Movement of the ASF is only possible due to its inherent flexibility; an essential feature for tRNAs with long variable arms to be accommodated in the A/P hybrid state on the ribosome (39). As can be imagined, this flexibility must be modulated and the variable arm should move cooperatively with the ASF to reduce strain on the system. With tRNAUTu1, a synthetic tRNA species to E. coli, we observed that its long variable arm must move significantly to avoid a steric clash with the ASF for translocation from the A to the P site (Figure 4C). This uncooperative behavior likely strains the ribosome and results in disassembly and poor translational processivity (Figure 3D). We found a unique C8:A45 base pair in the core of tRNAUTu1, which may promote the stabilization of the variable arm and prevent flexibility needed for cooperative movement with the ASF.
The choice for disrupting the C8:A45 base pair was 2-fold. (i) C8 was chosen for mutagenesis over A45 to minimize any effect that a change of sequence in the variable arm would have on serylation. Although the chance of this would be small considering that SerRS specifically recognizes the structure of the long variable arm, rather than the sequence, this was avoided (40). Moreover, the similar suppression efficiencies of tRNAUTu1 compared to tRNAUTu1A (Figure 5C) suggest that serylation was not affected. (ii) The choice of base for substitution at position 8 was also carefully chosen. Uracil, being the predicted Watson–Crick pair, was an obvious exclusion while guanine is also known to commonly form a mispairs (41). It followed that mutation to an adenine would have the highest likelihood of preventing an interaction with A45. The structure of tRNAUTu1A revealed this disrupted interaction without altering the position of the variable arm in the A site. Instead, the absence of the C8:A45 mismatch base pair in the core of tRNAUTu1A created flexibility in the variable arm such that it could move cooperatively with the ASF in transition from the A to the P site (Supplementary Figure S9). This movement is likely favorable for translation and prevents ribosome disassembly.
Our discovery emphasizes one reason why it is difficult to engineer a translation system that is efficient in site-specific Sec incorporation. The initial barrier of SECIS (selenocysteine insertion sequence)-dependent translation with SelB was overcome by rewiring translation to be EF-Tu compatible. This breakthrough allowed Sec to be site-specifically incorporated into any protein (10–12). Efforts to improve the system led to the discovery of tRNAUTu1 (13), which significantly increased the protein yield compared to previous systems (9). However, as our work shows there is room for improving tRNAUTu1 as well as other synthetic tRNAs proposed for Sec insertion (26). Specifically, we highlight that the variable arm, which is necessary for serylation, can pose a problem in translation through the ribosome. Capturing the tRNA structure in the ribosome provided a map to visualize and target the tRNA regions necessary to solve this translational roadblock.
This knowledge can apply to other synthetic tRNAs that are used for translation in E. coli. tRNAPyl and M. jannaschii tRNATyr, which are both highly used for genetic code expansion applications, have small variable arms, similar in size to tRNAPhe. The movement required of the ASF is minimal for the tRNA to reach the A/P hybrid state, interacting only with the T-loop of the tRNA (38,39). However, quadruplet tRNAs are being engineered from tRNAs with and without variable arms (19,42–44) Therefore, consideration as to the suitability of their variable arms in the E. coli ribosome is imperative to ensure efficient A to P site translocation. Moving forward with designing synthetic tRNAs, care should be taken when considering the tRNA scaffold to maximize protein yield and prevent translational roadblocks.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported cryo-EM structures have been deposited with the Protein Data Bank under accession numbers 7UR5, 7URI and 7URM and the Electron Microscopy Data Bank under accession numbers EMD-26705, EMD-26713 and EMD-26714.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Takahito Mukai for help in the early stage of this work as well as Leonard Liu, Alessandra Villa, and Rebecca Howard for molecular discussions. The molecular simulation work was supported by the Swedish e-Science Research Centre, with computational resources provided by the Swedish National Infrastructure of Computing (SNIC 2021/3–39). We are grateful for the technical support from the Stanford-SLAC Cryo-EM Center (S2C2) facility staff: Elizabeth Montabana, Dong-Hua Chen, and Chensong Zhang. We thank Stanford University and the Stanford Research Computing Center for providing the Sherlock cluster resources.
Notes
Present address: Arjun Prabhakar, Pacific Biosciences, Inc., Menlo Park, CA 94025, USA.
Present address: Ana Crnković, Laboratory for Molecular Biology and Nanobiotechnology, National Institute of Chemistry, Hajdrihova 19, Ljubljana, Slovenia.
Contributor Information
Arjun Prabhakar, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA; Program in Biophysics, Stanford University, Stanford, CA 94305-5126, USA.
Natalie Krahn, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA.
Jingji Zhang, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA.
Oscar Vargas-Rodriguez, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA.
Miri Krupkin, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA.
Ziao Fu, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Francisco J Acosta-Reyes, Department of Biochemistry and Molecular Biophysics, Columbia University, New York, NY 10032, USA.
Xueliang Ge, Department of Cell and Molecular Biology, Uppsala University, Uppsala 751 24, Sweden.
Junhong Choi, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA.
Ana Crnković, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA.
Måns Ehrenberg, Department of Cell and Molecular Biology, Uppsala University, Uppsala 751 24, Sweden.
Elisabetta Viani Puglisi, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA.
Dieter Söll, Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA; Department of Chemistry, Yale University, New Haven, CT 06511, USA.
Joseph Puglisi, Department of Structural Biology, Stanford University, Stanford, CA 94305-5126, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [R35 GM122560 and R35 GM122560-05S1 to D.S., GM51266 to J.D.P., AI15046 to E.V.P., T32-GM008294 to A.P.]; Stanford Interdisciplinary Graduate Fellowship [to A.P.]; Department of Energy Office of Basic Energy Sciences [DE-FG0298ER2031 to D.S.]; Cystic Fibrosis Foundation [PUGLIS20G0 to J.D.P]; Knut and Alice Wallenberg Foundation postdoctoral scholarship [KAW 2016.0488 to J.Z.] Funding for open access charge: NIH [AI15046].
Conflict of interest statement. None declared.
REFERENCES
- 1. Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S.. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998; 26:148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmeing T.M., Ramakrishnan V.. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009; 461:1234–1242. [DOI] [PubMed] [Google Scholar]
- 3. Rozov A., Westhof E., Yusupov M., Yusupova G.. The ribosome prohibits the G*U Wobble geometry at the first position of the codon-anticodon helix. Nucleic Acids Res. 2016; 44:6434–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pavlov M.Y., Ehrenberg M.. Substrate-induced formation of ribosomal decoding center for accurate and rapid genetic code translation. Annu. Rev. Biophys. 2018; 47:525–548. [DOI] [PubMed] [Google Scholar]
- 5. Wang L., Brock A., Herberich B., Schultz P.G.. Expanding the genetic code of Escherichiacoli. Science. 2001; 292:498–500. [DOI] [PubMed] [Google Scholar]
- 6. Arranz-Gibert P., Patel J.R., Isaacs F.J.. The role of orthogonality in genetic code expansion. Life (Basel.). 2019; 9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wan W., Tharp J.M., Liu W.R.. Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta. 2014; 1844:1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krahn N., Tharp J.M., Crnković A., Söll D. Engineering aminoacyl-tRNA synthetases for use in synthetic biology. Enzymes. 2020; 48:351–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung C.Z., Krahn N., Crnković A., Söll D. Intein-based design expands diversity of selenocysteine reporters. J. Mol. Biol. 2021; 434:167199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thyer R., Robotham S.A., Brodbelt J.S., Ellington A.D.. Evolving tRNASec for efficient canonical incorporation of selenocysteine. J. Am. Chem. Soc. 2015; 137:46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aldag C., Bröcker M.J., Hohn M.J., Prat L., Hammond G., Plummer A., Söll D. Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew. Chem. Int. Ed. Engl. 2013; 52:1441–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller C., Bröcker M.J., Prat L., Ip K., Chirathivat N., Feiock A., Veszpremi M., Söll D. A synthetic tRNA for EF-Tu mediated selenocysteine incorporation in vivo and in vitro. FEBS Lett. 2015; 589:2194–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mukai T., Vargas-Rodriguez O., Englert M., Tripp H.J., Ivanova N.N., Rubin E.M., Kyrpides N.C., Söll D. Transfer RNAs with novel cloverleaf structures. Nucleic Acids Res. 2017; 45:2776–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodnina M.V., Gromadski K.B., Kothe U., Wieden H.J.. Recognition and selection of tRNA in translation. FEBS Lett. 2005; 579:938–942. [DOI] [PubMed] [Google Scholar]
- 15. Wohlgemuth I., Pohl C., Rodnina M.V.. Optimization of speed and accuracy of decoding in translation. EMBO J. 2010; 29:3701–3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang K., Schmied W.H., Chin J.W.. Reprogramming the genetic code: from triplet to quadruplet codes. Angew. Chem. Int. Ed. Engl. 2012; 51:2288–2297. [DOI] [PubMed] [Google Scholar]
- 17. Choi J., O’Loughlin S., Atkins J.F., Puglisi J.D.. The energy landscape of -1 ribosomal frameshifting. Sci. Adv. 2020; 6:eaax6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gamper H., Li H., Masuda I., Miklos Robkis D., Christian T., Conn A.B., Blaha G., Petersson E.J., Gonzalez R.L. Jr, Hou Y.M.. Insights into genome recoding from the mechanism of a classic +1-frameshifting tRNA. Nat. Commun. 2021; 12:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anderson J.C., Wu N., Santoro S.W., Lakshman V., King D.S., Schultz P.G.. An expanded genetic code with a functional quadruplet codon. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:7566–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang N., Shang X., Cerny R., Niu W., Guo J.. Systematic evolution and study of UAGN decoding tRNAs in a genomically recoded bacteria. Sci. Rep. 2016; 6:21898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neumann H., Wang K., Davis L., Garcia-Alai M., Chin J.W.. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010; 464:441–444. [DOI] [PubMed] [Google Scholar]
- 22. Prabhakar A., Capece M.C., Petrov A., Choi J., Puglisi J.D.. Post-termination ribosome intermediate acts as the gateway to ribosome recycling. Cell Rep. 2017; 20:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aitken C.E., Marshall R.A., Puglisi J.D.. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophys. J. 2008; 94:1826–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen J., Dalal R.V., Petrov A.N., Tsai A., O’Leary S.E., Chapin K., Cheng J., Ewan M., Hsiung P.L., Lundquist P.et al.. High-throughput platform for real-time monitoring of biological processes by multicolor single-molecule fluorescence. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:664–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mukai T., Hoshi H., Ohtake K., Takahashi M., Yamaguchi A., Hayashi A., Yokoyama S., Sakamoto K.. Highly reproductive escherichiacoli cells with no specific assignment to the UAG codon. Sci. Rep. 2015; 5:9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mukai T., Sevostyanova A., Suzuki T., Fu X., Söll D. A facile method for producing selenocysteine-containing proteins. Angew. Chem. Int. Ed. Engl. 2018; 57:7215–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J., Zhang P., Fang G., Yi P., Zeng F., Wu S.. Design and synthesis of FRET-mediated multicolor and photoswitchable fluorescent polymer nanoparticles with tunable emission properties. J. Phys. Chem. B. 2012; 116:4354–4362. [DOI] [PubMed] [Google Scholar]
- 28. Voorhees R.M., Weixlbaumer A., Loakes D., Kelley A.C., Ramakrishnan V.. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009; 16:528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Auffinger P., Westhof E.. An extended structural signature for the tRNA anticodon loop. RNA. 2001; 7:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Komoda T., Sato N.S., Phelps S.S., Namba N., Joseph S., Suzuki T.. The A-site finger in 23 s rRNA acts as a functional attenuator for translocation. J. Biol. Chem. 2006; 281:32303–32309. [DOI] [PubMed] [Google Scholar]
- 31. Fischer N., Neumann P., Bock L.V., Maracci C., Wang Z., Paleskava A., Konevega A.L., Schröder G.F., Grubmüller H., Ficner R.et al.. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature. 2016; 540:80–85. [DOI] [PubMed] [Google Scholar]
- 32. Zuleeg T., Vogtherr M., Schubel H., Limmer S.. The C-A mismatch base pair and the single-strand terminus in the E. coli initiator tRNAfMet acceptor stem adopt unusual conformations. FEBS Lett. 2000; 472:247–253. [DOI] [PubMed] [Google Scholar]
- 33. Puglisi J.D., Wyatt J.R., Tinoco I. Jr. Solution conformation of an RNA hairpin loop. Biochemistry. 1990; 29:4215–4226. [DOI] [PubMed] [Google Scholar]
- 34. Hunter W.N., Brown T., Anand N.N., Kennard O.. Structure of an adenine-cytosine base pair in DNA and its implications for mismatch repair. Nature. 1986; 320:552–555. [DOI] [PubMed] [Google Scholar]
- 35. Woolstenhulme C.J., Parajuli S., Healey D.W., Valverde D.P., Petersen E.N., Starosta A.L., Guydosh N.R., Johnson W.E., Wilson D.N., Buskirk A.R.. Nascent peptides that block protein synthesis in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E878–E887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Frank J., Sengupta J., Gao H., Li W., Valle M., Zavialov A., Ehrenberg M.. The role of tRNA as a molecular spring in decoding, accommodation, and peptidyl transfer. FEBS Lett. 2005; 579:959–962. [DOI] [PubMed] [Google Scholar]
- 37. Zhou J., Lancaster L., Donohue J.P., Noller H.F.. How the ribosome hands the A-site tRNA to the P site during EF-G-catalyzed translocation. Science. 2014; 345:1188–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nguyen K., Whitford P.C.. Capturing transition states for tRNA hybrid-state formation in the ribosome. J. Phys. Chem. B. 2016; 120:8768–8775. [DOI] [PubMed] [Google Scholar]
- 39. Nguyen K., Yang H., Whitford P.C.. How the ribosomal A-site finger can lead to tRNA species-dependent dynamics. J. Phys. Chem. B. 2017; 121:2767–2775. [DOI] [PubMed] [Google Scholar]
- 40. Krahn N., Fischer J.T., Söll D. 2020) Naturally occurring tRNAs with non-canonical structures. Front. Microbiol. 11:596914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ananth P., Goldsmith G., Yathindra N.. An innate twist between Crick's Wobble and Watson-Crick base pairs. RNA. 2013; 19:1038–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore B., Nelson C.C., Persson B.C., Gesteland R.F., Atkins J.F.. Decoding of tandem quadruplets by adjacent tRNAs with eight-base anticodon loops. Nucleic Acids Res. 2000; 28:3615–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DeBenedictis E.A., Carver G.D., Chung C.Z., Söll D., Badran A.H.. Multiplex suppression of four quadruplet codons via tRNA directed evolution. Nat. Commun. 2021; 12:5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DeBenedictis E.A., Söll D., Esvelt K.M.. Measuring the tolerance of the genetic code to altered codon size. Elife. 2022; 11:e76941. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported cryo-EM structures have been deposited with the Protein Data Bank under accession numbers 7UR5, 7URI and 7URM and the Electron Microscopy Data Bank under accession numbers EMD-26705, EMD-26713 and EMD-26714.