

Abstract
Promoters are consensus DNA sequences located near the transcription start sites and they play an important role in transcription initiation. Due to their importance in biological processes, the identification of promoters is significantly important for characterizing the expression of the genes. Numerous computational methods have been proposed to predict promoters. However, it is difficult for these methods to achieve satisfactory performance in multiple species. In this study, we propose a novel weighted average ensemble learning model, termed iPro-WAEL, for identifying promoters in multiple species, including Human, Mouse, E.coli, Arabidopsis, B.amyloliquefaciens, B.subtilis and R.capsulatus. Extensive benchmarking experiments illustrate that iPro-WAEL has optimal performance and is superior to the current methods in promoter prediction. The experimental results also demonstrate a satisfactory prediction ability of iPro-WAEL on cross-cell lines, promoters annotated by other methods and distinguishing between promoters and enhancers. Moreover, we identify the most important transcription factor binding site (TFBS) motif in promoter regions to facilitate the study of identifying important motifs in the promoter regions. The source code of iPro-WAEL is freely available at https://github.com/HaoWuLab-Bioinformatics/iPro-WAEL.
INTRODUCTION
The transcription process mainly involves initiation, elongation and termination, in which initiation is the most complicated (1). Promoters located proximal to the transcription start sites (TSS) are non-coding DNA regions, which are essential for initiating the transcription of a particular gene by cooperating with RNA polymerase (RNAP) (2,3) and are critical for gene expression in different species. In prokaryotes, promoters are involved in many biological processes, such as the transcription of most genes (4), heat-shock response, nitrogen fixation (5), the expression of flagella (6) and so forth. In eukaryotes, the initiator region or the downstream promoter element of promoters controls the exact position where transcription starts (7). Additionally, promoters often cooperate with their distal regulatory elements via chromatin loops and tend to be involved in developmental diseases, tumorigenesis and spatiotemporal gene expression (8–10). Therefore, the identification of promoters is crucial to investigating the regulation of gene expression.
Accordingly, an experimental technique called genome-wide mapping of histone modifications has been used to identify promoters (11). However, the experimental methods are costly and time-consuming. To address this issue, several computational methods have been proposed in the last few years to predict promoters. Some of these methods, such as vw Z-curve (12), iPro54-PseKNC (13), iPro70-PseZNC (14), iPromoter-2L (15), iPSW(2L)-PseKNC (16) and Promotech (17), rely on traditional machine learning-based models; other methods utilize deep learning models to identify promoters, such as CNNProm (18) and DeePromoter (19).
Despite advances in computational methods for predicting promoters, several shortcomings in these methods remain to be addressed. Firstly, the feature encoding schemes in their studies and the architecture of their models is relatively simple, resulting in unsatisfactory performance in predicting promoters. Secondly, most of their models have validated the performance of predicting promoters on only one or few species. Therefore, it is not clear whether these methods can be implemented for the identification of promoters in multiple species. Although CNNProm (18) and DeePromoter (19) reported their performance on a few datasets, it is time-consuming to retune the parameters of the models for each species. Finally, the model interpretation is critical for exploring transcription factor binding site (TFBS) motifs in the sequences involved in promoters, but unfortunately, previous studies have not touched upon this aspect.
Therefore, we propose a comprehensive and robust framework, named iPro-WAEL (a Weighted Average Ensemble Learning-based model for identifying promoters), to identify promoters in multiple species by integrating an RF model and a CNN model. Based on 13 datasets of seven species, we compare it with the performance of eight promoter prediction models on without retuning parameters. Besides, we explore the TFBS motifs that have a significant impact on human promoter regions. Our main contributions are as follows: (i) We generate four new human datasets using stricter criteria. (ii) We propose a novel and robust weighted average ensemble learning-based model (iPro-WAEL) to identify the promoters in multiple species. (iii) We demonstrate the optimal performance of iPro-WAEL on multiple species and it is superior to state-of-the-art predictors. (iv) We identify the most important TFBS motifs in promoter regions which are consistent with the previous studies and identify many potentially important but previous underexplored motifs. The overall framework of this study is shown in Figure 1.
Figure 1.

The overall flowchart of iPro-WAEL.
MATERIALS AND METHODS
Data collection and processing
To develop an effective and robust model, it is necessary to establish the dataset using a strict criterion. Whalen et al. (20) identified human promoters using ENCODE Segway (21) and ChromHMM (22) annotations for GM12878, K562, HeLa-S3 and HUVEC cell lines. We obtain the sequences using BEDTools (23). Among these promoters, the length of most promoters is <3000-nt. Therefore, we remove promoters >3000-nt in length and remove the redundant sequences with a similarity of more than 80% in each cell line using the CD-HIT program (24). Finally, we treat them as positive samples of the benchmark dataset.
Different from previous studies in which negative samples are obtained from random genomic coordinates of non-promoter regions (15–18), this study uses the sequences with the highest similarity to the positive samples from the non-promoter regions as negative samples. Specifically, for a positive sample, assuming its length is L, the following steps are adopted to generate negative samples: (i) Utilize a window of length L to slide 1000 times upstream and downstream of the positive sequence with a stride of 1-nt, and each time a sequence whose length equals to the length of the positive sample is obtained. (ii) If the sliding range covers the genomic coordinates of other positive samples, this sample will be removed from the positive dataset to avoid the generated negative samples overlapping with other positive samples, otherwise, it is difficult to define the true category of the generated samples. (iii) If the sliding range does not cover the genome coordinates of other positive samples, the similarity between these 2000 sequence segments and the positive sequence is calculated, and the sequence segment with the highest similarity is treated as the negative sample. This ensures that the generated negative samples and positive samples have a certain similarity so that the trained model is more robust. (iv) Remove sequences containing ‘N’ in positive samples and negative samples (’N’ means that the base at this position is uncertain. Removing positive samples containing ‘N’ also removes the corresponding negative samples, and removing negative samples containing ‘N’ also removes the corresponding positive samples). Finally, to objectively evaluate the performance of the model, we use 20% of the sequences in each cell line as the independent test set. To ensure the reliability of the data, a positive sample and its corresponding negative sample are divided into a training set or test set simultaneously. The details of the human datasets are shown in Figure 2.
Figure 2.
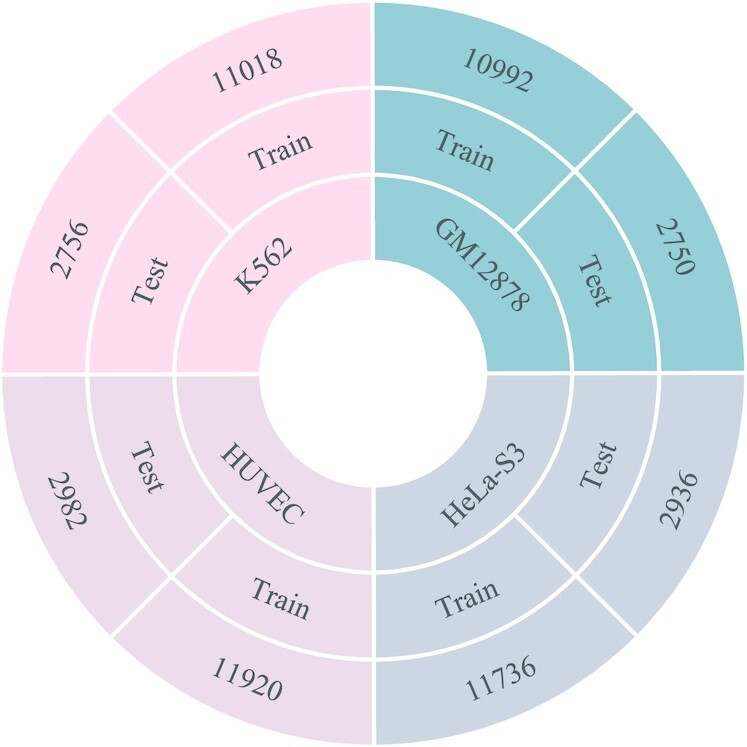
The statistical summary of the benchmark dataset. The ratio of positive and negative samples in all datasets is 1:1.
Feature encoding schemes
In this study, we employ six sequence encoding schemes to encode sequences, including reverse complement k-mer (RCKmer), mismatch k-mer (Mismatch), the composition of k-spaced nucleic acid pairs (CKSNAP), trinucleotide physicochemical properties (TPCP), pseudo trinucleotide composition (PseTNC) and Word2vec. Multiple feature encoding schemes comprehensively extract sequence information, and fusing features can effectively improve the representation ability of features. These encoding schemes are elaborated in the following sections.
Reverse complement k-mer
The RCKmer is a variant of k-mer, in which the k-mer is not strand-specific, so reverse complements are collapsed into a single feature (25,26). For instance, ‘AGGT’ is the reverse complement with ‘ACCT’; ‘TAG’ is the reverse complement with ‘CTA’, and ‘TCAGA’ is the reverse complement with ‘TCTGA’. In this study, we set k = 5 and thus the dimension of the RCKmer-based feature is 512.
Mismatch k-mer
The Mismatch is also a variant of k-mer, in which an error is allowed (26). For instance, when we calculate the occurrence number of ‘GCA’, we place ‘GCA’, ‘ACA’, ‘CCA’, ‘TCA’, ‘GAA’, ‘GGA’, ‘GTA’, ‘GCC’, ‘GCG’ and ‘GCT’. The sum of the occurrence number of these 3-tuples is treated as the occurrence number of ‘GCA’. In this study, we set k = 5, and thus the dimension of the Mismatch-based feature is 1024.
Composition of k-spaced nucleic acid pairs
The CKSNAP is used to calculate the frequency of all possible nucleotide pairs separated by any k space or less (26,27). For instance, in the sequence of ‘ANNNTNNC’, ‘AT’ is a 3-space nucleotide pair and ‘TC’ is a 2-space nucleotide pair. The CKSNAP-based feature is defined as follows:
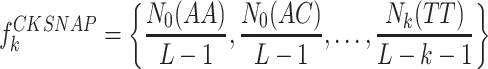 |
(1) |
where Nk(TT) represents the occurrence number of k-space nucleotide pair ‘TT’ and L is the length of the sequence. In this study, we set k = 5 and thus the dimension of the CKSNAP-based feature is 96.
Trinucleotide physicochemical properties
The TPCP encoding has been successfully applied to DNA N4-Methylcytosine site prediction (28), which is defined as follows:
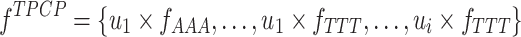 |
(2) |
where ui is the ith physicochemical property of the trinucleotide and 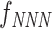 denotes the normalized frequency of trinucleotide NNN. In this study, we utilize 12 physicochemical properties (Supplementary Table S1), and thus the dimension of the TPCP-based feature is 768.
denotes the normalized frequency of trinucleotide NNN. In this study, we utilize 12 physicochemical properties (Supplementary Table S1), and thus the dimension of the TPCP-based feature is 768.
Pseudo trinucleotide composition
The PseTNC considers both the local sequence-order information and long-range sequence-order effects (26,29), which are defined as follows:
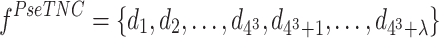 |
(3) |
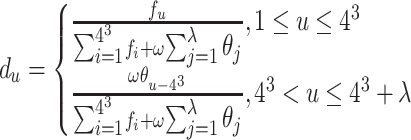 |
(4) |
where λ represents the number of the total counted ranks of the correlations along the sequence, fu represents the normalized frequency of trinucleotide, ω is the weighted factor and θj is calculated as follows:
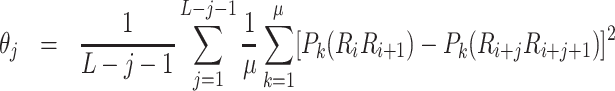 |
(5) |
where L represents the length of the sequence, Pk(RiRi+1) is the value of kth physicochemical indices of RiRi+1 at position i, and μ is the number of physicochemical indices. In this study, we set λ = 2 and utilize six physicochemical indices to predict promoters, including angular parameters (twist, tilt and roll) and translational parameters (shift, slide and rise). Therefore, the dimension of the PseTNC-based feature is 66.
Word2vec
Word embedding techniques have achieved a great success in natural language processing (NLP) applications. Recently, word embedding techniques have been widely used in the bioinformatics community to tackle the limitation that the kmer-based features of different sequences may be very similar despite their orders being reversed (30–32). In this study, we divide the sequence into ‘word’ to keep the order information of sequence. Specifically, a DNA sequence described as follows:
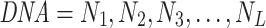 |
(6) |
where N1 denotes the nucleotide at the first position and L is the length of the sequence. We take k consecutive nucleotides as a ‘word’. Thus, the ith ‘word’ in the sequence is described as:
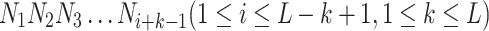 |
(7) |
After the above process, the DNA sequence is divided into sentences containing multiple words. Then we utilize the DNA sequences in the promoter dataset to form the corpus (each DNA sequence is the sentence in the corpus) and the 4k types of k-mer segments to form vocabulary (each k-mer segment is a word in the vocabulary). To guarantee complete independence of the independent test set, we only utilize sequences in the training set to form the corpus and vocabulary. We train the language model by using the continuous skip-gram (Skip-gram) model in word2vec and obtain the feature vector for each ‘word’. Then we concatenate the feature vectors of all ‘word’ in the sequence and treat them as the features of the sequence. Suppose each ‘word’ is embedded as a feature vector of dimension D, the feature dimension of each sequence is D × (L – k + 1). Therefore, if the feature dimensions obtained by sequences of different lengths in our dataset are not equal, the features cannot be trained in a deep learning model. To solve this problem, we transform the features of each sequence into equal dimensions using an adaptive pooling layer. In this study, each word has a length of 5 and is embedded as an 8-dimension feature vector. However, the sequence lengths in human datasets are unequal, which makes it impossible to train in deep learning models. Thus we additionally use adaptive pooling operations to further perform feature extraction. Given that 1000 is a centered number relative to the number of words in human promoter sequences, we set the output dimension of the adaptive pooling layer to 1000 × 8 (‘1000’ indicates 1000 words and ‘8’ indicates that the feature dimension of each word is 8). Therefore, the dimension of the Word2vec-based feature is 8000.
iPro-WAEL architecture
iPro-WAEL is a weighted average ensemble learning model that integrates a random forests (RF) model and a CNN model, in which the RF model and CNN model use different features. The framework of iPro-WAEL is shown in Figure 3, and the RF model, the CNN model and the weighted average algorithm are depicted in the following section.
Figure 3.

The architectures of iPro-WAEL. The RF model contains 300 trees and the CNN model contains three convolutional layers, three max pooling layers and two dense layers. The details of the parameters are introduced later.
Random forest
Random forest (RF) is a combination of tree predictors, which is widely used in the bioinformatics community and shows excellent performance using traditional sequence-based features for prediction (15,17,33). Therefore, in iPro-WAEL, the RF model is trained using sequence-based features, including RCKmer, Mismatch, CKSNAP, TPCP and PseTNC.
Convolutional neural network-based model
CNN has been widely used in sequence-based prediction by combining word embedding techniques (34–37). The convolutional layer performs convolution calculation using the convolution kernel to extract different features of the input, and more complex features can be extracted by stacking multiple layers. Besides, the convolutional layer is usually followed by a pooling layer to reduce the number of parameters in the network. In iPro-WAEL, the CNN model is trained using the Word2vec-based features and starts with three combinations of a convolutional layer and a max-pooling layer. We utilize ‘relu’ as the activation function in each CNN layer to enhance the nonlinear characteristics of the neural network. Then we add a dropout layer to avoid over-fitting, followed by two dense layers, which contain 64 nodes and 1 node, respectively, and utilize ‘relu’ and ‘sigmoid’ as activation functions, respectively. Finally, if the predicted value exceeds 0.5, the predicted result will be a positive example; otherwise, it will be a negative example. Besides, to obtain the model with better generalization, we divide 1/7 of the training set into the validation set and introduce an ‘early stopping’ mechanism, which is used to stop training early to avoid overfitting when the loss value of the model in the validation set does not decrease for five consecutive epochs.
Weighted averaging algorithm
The weighted averaging method obtains the combined output by averaging the outputs of individual models with different weights implying different importance (38). Compared to the simple averaging method, it can give more weight to the model with good performance. Specifically, the final output of iPro-WAEL is calculated as follows:
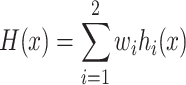 |
(8) |
where wi is the weight of the ith model, hi(x) is the output of the ith model and the weights are assumed to be constrained by
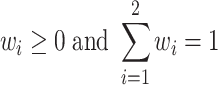 |
(9) |
To obtain the most suitable weights and complete independence of the independent test sets, we further divide the original training set into the training set and the dataset used for obtaining weights (weighted set) according to the ratio of 7:1. Therefore, the ratio of the training set, weighted set and test set is 7:1:2 in the RF model, and the ratio of the training set, weighted set, validation set and test set is 6:1:1:2 in the CNN model. Then we obtain the optimal weights by minimizing the loss values in the weighted set. Specifically, the loss value in the weighted set is calculated by the cross-entropy loss of the predicted values and the true labels, which is defined as follows:
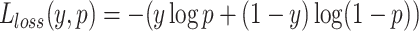 |
(10) |
where y represents the true labels, and p represents the predicted values. To minimize the loss value, we utilize Sequential Least Squares Programming (SLSQP) algorithm from the ‘scipy’ library, which uses the quasi–Newton method with a BFGS update of the B–matrix and an L1–test function in the step–length algorithm. Finally, the weights obtained with the minimum loss value are the optimal weights.
RESULTS
Performance evaluation of different features used for RF model
We consider more than 50 sequence features provided by IlearnPlus (26), BioSeq-Analysis2.0 (39) and BioSeq-BLM (40), but many of these feature extraction methods are not applicable for the unequal-length sequences. Therefore, we extract the applicable 29 sequence-based features of the training set in the GM12878 cell line and perform ten-fold cross-validation to evaluate the performance of these features using Area Under the Curve (AUC), Accuracy and Matthews Correlation Coefficient (MCC) (Supplementary Method). The results are shown in Supplementary Table S2. It is worth mentioning that the original one-hot encoding, DBE, TF-IDF and LDA-TFIDF cannot be used for encoding unequal-length sequences, but the fixed-length option provided by BioSeq-BLM makes it possible. Unexpectedly, however, although the good performance was achieved with fixed-length in the previous study (34), the performance of features with fixed-length in this study is poor. The first reason is that the lengths of the sequences in this study vary greatly (from tens of bp to 3000 bp), resulting in incalculable information loss caused by truncation and padding. Another reason is that the negative samples in this study have a certain similarity with the positive samples, and if the length is fixed, the similarity between the positive samples and the negative samples will be further enlarged, resulting in poor performance.
Among other 25 methods that can directly extract features of unequal-length sequences, many features have competitive performance in predicting promoters, especially Mismatch, RCKmer, CKSNAP, TPCP and PseTNC, showing that they are informative in predicting promoters. To further demonstrate the feature representation ability to fuse these five features, we visualize the distribution of the samples encoded by fused features in four cell lines using t-distributed Stochastic Neighbor Embedding (t-SNE) (41). We find that the positive samples and negative samples in the four cell lines are distributed in two clusters (Figure 4), proving that the fused features have excellent performance in distinguishing promoters and non-promoters. Therefore, we fuse and normalize these five features and use them as the input of the RF model.
Figure 4.

The feature representation map using t-SNE. (A–D) represent the results of feature visualization on GM12878, HeLa-S3, HUVEC and K562, respectively. Navy dots represent non-promoters and red dots represent promoters.
Parameter optimization
To obtain the best-performing model, we evaluate the performance of the RF model and CNN model with different parameters, including the number of trees, the learning rate, the number of kernels and the size of kernels. We determine optimal parameters using the grid search algorithm on the dataset of the GM12878 cell line. To ensure that the performance of the model is only affected by changes in parameters, we set a fixed random seed for the RF model and the CNN model. The performance of the RF model and CNN model with different parameters are shown in Supplementary Tables S3 and S4, respectively. It can be seen that the performance of models is affected by the parameters. The RF model with a tree number of 300 performs best, and the CNN model with a learning rate of 0.001, a kernel number of 32 and a kernel size of 11 performs best. Therefore, we utilize these parameters to construct the RF model and CNN model.
Ten-fold cross-validation on the training datasets
Previous studies have shown that random division of the test set may lead to a large difference in performance between the training and test sets (42,43). To comprehensively evaluate the performance of iPro-WAEL, we perform ten-fold cross-validation on the training set of four cell lines. The results are shown in Supplementary Table S5. It can be seen that iPro-WAEL achieves extremely high performance in predicting human promoters. It is worth mentioning that although the parameters are determined by parameter optimization on the GM12878 cell line, the model with the same parameters also has excellent performance on the other three cell lines. These results indicate that iPro-WAEL has splendid performance and robustness in predicting promoters.
Performance evaluation on the independent test set and comparison with the state-of-the-art predictors
To comprehensively evaluate the performance of the models, we evaluate and compare the performance of iPro-WAEL with other eight computational methods, including iPro70-PseZNC, vw Z-curve, iPro54-PseKNC, iPromoter-2L, CNNProm, Promotech, DeePromoter and iPSW(2L)-PseKNC. We apply them to seven species (13 datasets), including humans (four cell lines), four prokaryotes (E. coli, B. amyloliquefaciens, B. subtilis and R. capsulatus). According to the evaluation method in the previous study (3), E. coli contains E. coli-general and E. coli-sigma70 and two eukaryotes (Mouse and Arabidopsis, each eukaryote contains a TATA dataset and a non-TATA dataset). Given that some of these datasets are imbalanced, we randomly subsample from the class with more data to construct a balanced dataset. The details of other species datasets are shown in Supplementary Table S6. All datasets are divided into the training set and test set according to the ratio of the human promoter dataset, and all methods utilize the same training set to train these models and evaluate their performance on the same test set. Due to the limitations of some methods, these methods cannot be applied to some datasets (Supplementary Table S7), and the reasons are shown in Supplementary Information. Also of note, to demonstrate and compare the generalization of the models, all methods including iPro-WAEL keep the parameters unchanged on the datasets of different species.
We evaluate the performance of nine models on 13 datasets and calculate the average performance of the models on the species with multiple types of datasets. The results are shown in Figure 5 and the detailed results are shown in Supplementary Tables S8–S14. It can be seen that the excellent performance of some models in one species is not transferable. For instance, as far as ACC is concerned, Z-curve slightly underperforms (about 1%) our model on the human dataset, but its performance on the mouse dataset is extremely different from our model, about 10% lower than that of our model. These results indicate that some methods are limited in their ability to identify promoters in multiple species. Fortunately, the iPro-WAEL proposed in this study has satisfactory performance on multiple species. The line representing iPro-WAEL in Figure 5 is always in the highest position, showing the effectiveness of iPro-WAEL on multiple species. Specifically, iPro-WAEL achieves optimal performance on six of seven species and is <0.1% lower than the best model on the remaining one (Arabidopsis). Additionally, although the second best performing model varies to a large extent on different datasets, ipro-WAEL outperforms these methods by a comparable or significant advantage.
Figure 5.

The performance evaluation of iPro-WAEL and other predictors on the seven species. (A–C) represent the AUC, ACC and MCC of the models, respectively. Discontinuous lines represent that the method cannot be applied to these datasets. Note that the performance of Human, Arabidopsis and Mouse are the average results of multiple datasets.
To intuitively compare the performance of iPro-WAEL and the methods proposed by previous studies, we compare iPro-WAEL and these methods individually based on the maximum intersection of the dataset to which these models could be applied. For instance, only Human, Arabidopsis and Mouse are applicable to both DeePromoter and iPro-WAEL. Therefore, we compare the average performance of the two models on these three species. For another example, all species are applicable to iPromoter-2L, CNNProm, Promotech, iPSW(2L)-PseKNC, Z-curve and iPro-WAEL, so we compare the average performance of these models on all species. Based on this, we divide all datasets into three subsets according to their applicable conditions and compare iPro-WAEL and the methods proposed in previous studies. It can be seen from Supplementary Table S15 that the performance of iPro-WAEL is significantly better than other methods. As far as ACC is concerned, iPro-WAEL obtains 1.7–18.6% higher than the second best performing model on the three subsets. However, it cannot be ignored that DeePromoter, which has the smallest difference from iPro-WAEL in performance, has very strict conditions of use. Due to its model architecture, DeePromoter has a very high limit on sequence length. Therefore, DeePromoter is difficult to be applied to prokaryotic promoter detection (the length of datasets in most prokaryotic is 81 bp or less, but DeePromoter can only predict sequences over 140 bp), and thus they also only report the performance on eukaryotic in their paper. Among other promoter predictors that can apply to both prokaryotes and eukaryotes, iPromoter-2L and iPro-WAEL have the smallest gap. But even so, iPro-WAEL still obtains 2.3% higher than iPromoter-2L. Overall, these results demonstrate that iPro-WAEL is an effective tool in predicting promoters and can be widely used to predict promoters in different species with satisfactory performance.
Cross cell lines validation
To explore the potential relationship between promoters in different cell lines, we further employ iPro-WAEL in the cross-cell lines and compare its performance with Z-curve, which is the best-performing method on Human datasets in the previous studies. We train the model on the training set of one cell line and predict the test set of the other three cell lines. The results are shown in Supplementary Tables S16 and S17. It can be seen that the performance of iPro-WAEL on cross-cell lines validation outperforms the best-performing method (Z-curve) in previous studies and is not significantly different from that on the same cell line. It is noted that the data used to obtain the weights is from the training set, which ensures the independence of the test set when conducting cross-cell lines validation. These results indicate that iPro-WAEL is effective and robust for effectively predicting promoters in different cell lines and indicate potential similarity in sequence structure between promoters in different cell lines.
Identification of promoters and enhancers
Recently, some studies have shown that sequence architectures of enhancers and promoters are remarkably similar (44–48). Therefore, it is natural to ask whether iPro-WAEL can distinguish between promoters and enhancers. Whalen et al. (20) identified enhancers using the same method as promoters, most of which are less than 1000-nt in length. Therefore, we obtain the sequence using BEDTools (23) and remove enhancers over 1000-nt in length. Then we remove the redundant sequences with >80% similarity in each cell line using the CD-hit program (24). Given that the number of enhancers is much larger than that of promoters, we randomly subsample from the enhancer sequences to obtain the same number of sequences as the promoter sequences to construct a balanced dataset. The enhancer sequences are used as negative samples, and the promoter sequences are used as positive samples. The details are shown in Supplementary Table S18. Similarly, we use 20% of the sequences in each cell line as the independent test set. We keep the parameters of iPro-WAEL unchanged and the results of identifying promoters and enhancers are shown in Supplementary Table S19. It can be seen that iPro-WAEL achieves exceptionally high performance in distinguishing between promoters and enhancers, even though sequence architectures of enhancers and promoters are remarkably similar. These results demonstrate the superior robustness of iPro-WAEL and some sequence-level differences in the sequence architectures of enhancers and promoters.
Prediction ability on the promoters annotated in another method
Identification of promoters annotated with different methods may vary. Therefore, we further verify whether iPro-WAEL can predict promoters annotated by another method. Whalen et al. (20) identified promoters using Roadmap Epigenomics ChromHMM annotations for IMR90 and NHEK cell lines. We utilize the same method (see Method) to obtain negative sequences for these two cell lines and utilize 20% of the sequences in each cell line as the independent test set. Finally, the number of training sets for IMR90 and NHEK cell lines is 8192 and 8170, respectively, and the number of test sets for IMR90 and NHEK cell lines is 2048 and 2044 respectively. Similarly, we keep the parameters of iPro-WAEL unchanged. Then we evaluate the performance of iPro-WAEL on these two cell lines. It can be seen from Supplementary Table S20 that the performance of iPro-WAEL in predicting promoters annotated by different methods slightly decreased but is still satisfactory, which demonstrates that iPro-WAEL can capture potential features of promoters annotated by different methods and is of high capacity of generalization.
Estimate of important TFBS motifs in human promoter regions
Transcription factors play an important role in gene transcription through direct binding to their motifs in the genome and are involved in a large number of human diseases (49,50). To identify the important TFBS motifs that tend to bind in promoter regions, we propose a method to calculate the importance scores of the TFBS motifs. Specifically, given that the RCKmer and Mismatch used in the RF model can effectively reflect the importance of motifs, we integrate three methods to calculate the importance scores of the subsequence segment by interpreting these two features, including permutation-based importance, Gini importance and SHAP value. To integrate the three methods, we normalize and sum the importance scores of these two features obtained by each method. Then we perform a summation of importance scores representing the same subsequence segments, which is calculated as follows:
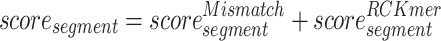 |
(11) |
where scoresegment represents the importance score of the segment, 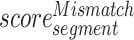 represents the importance score of the Mismatch-based feature corresponding to the ‘segment’ and
represents the importance score of the Mismatch-based feature corresponding to the ‘segment’ and 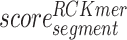 represents the importance score of the RCKmer-based feature corresponding to the ‘segment’. For instance, the importance score of segment ‘AAAAA’ is calculated by the sum of the importance scores of the segment ‘AAAAA’ in the Mismatch-based feature and RCKmer-based feature. It is noted that reverse complements are collapsed into a single feature. Therefore, the RCKmer-based feature importance of a segment corresponds to the RCKmer-based feature importance of its reverse complementary segment. For example, the RCKmer-based importance score of ‘TTTTT’ corresponds to the RCKmer-based importance score of ‘AAAAA’.
represents the importance score of the RCKmer-based feature corresponding to the ‘segment’. For instance, the importance score of segment ‘AAAAA’ is calculated by the sum of the importance scores of the segment ‘AAAAA’ in the Mismatch-based feature and RCKmer-based feature. It is noted that reverse complements are collapsed into a single feature. Therefore, the RCKmer-based feature importance of a segment corresponds to the RCKmer-based feature importance of its reverse complementary segment. For example, the RCKmer-based importance score of ‘TTTTT’ corresponds to the RCKmer-based importance score of ‘AAAAA’.
Given that the segments are relatively short and thus may match multiple motifs at the same time, for each segment, we obtain the three motifs with the highest matching score with the motif position weight matrices (PWMs) from the HOCOMOCO Human v11 database (51) and assign them the same importance score. If multiple subsequences match the same motif, the importance scores of these subsequences are summed as the importance score of the motif. Then we categorize the motifs into different importance levels based on quantiles spaced at 20% of the importance score. We display the most important motifs (top 20%) in Figure 6. It can be seen from Figure 6 that the distribution of motifs’ importance score in different cell lines is remarkably consistent, which can explain the reason for the high performance of iPro-WAEL on cross-cell lines validation and applicability of iPro-WAEL in predicting the promoters annotated in another method.
Figure 6.

The most important TFBS motifs identified by iPro-WAEL.
Besides, we find that many estimated important motifs are highly consistent with the previous studies. Among the most important motifs identified by iPro-WAEL, both the CTCF motif and CTCFL motif play a significant role in mediating chromatin loops (52,53); YY1 contributes to enhancer-promoter structural interactions and forms dimers to promote promoter-involved DNA interactions (54,55); ZNF143 is directly involved in chromatin looping (20) and provides sequence specificity to cohesin-associated chromatin loops at promoters (56), where ZNF143 directly binds to promoters and facilitates chromatin interactions that link promoters to distal regulatory elements (57); CUX1 is significantly enriched at promoters and contributes to predicting promoter-enhancer interactions (20); SP1 regulates chromatin looping between enhancer and distal promoter, and restores DNA looping and transcriptional activity by inserting at the promoter (58,59); REST is a transcriptional repressor with high predictive importance, as pointed out in TargetFinder (20); TFAP2C is a pioneer factor and is identified by TargetFinder as the most predictive feature for predicting enhancer-promoter interactions (20). In addition, some next most important (top20%-top40%) motifs are consistent with some critical but underestimated motifs in promoter-involved interactions, as pointed out in TargetFinder (20), including SRF, MAX, TBP, CEBPB and RUNX. These results indicate that the formation of promoters is a complicated procedure and tends to be affected by a variety of factors.
DISCUSSION AND CONCLUSION
Identification of promoters can be achieved by some annotation methods such as genome-wide mapping of histone modifications (11), ENCODE Segway (21) and ChromHMM (22), which are time-consuming. In addition, it remains unclear what the potentially important TFBS motifs exist in human promoters. In this study, we propose a comprehensive and robust framework, named iPro-WAEL, to answer this question. We integrate two different models in iPro-WAEL, including the RF model, which utilizes traditional sequence-based features, and the CNN model, which utilizes word embedding techniques to extract the sequence features. From the analysis of a series of examinations on multiple datasets, it is concluded that iPro-WAEL has optimal performance and robustness, and it is superior to the existing methods. Besides, some previous methods have limitations in applying certain datasets. Fortunately, the iPro-WAEL proposed in this study does not suffer from this limitation. It is worth mentioning that iPromoter-2L noticed this problem, thus their webserver splits the sequence into multiple 81-nt segments when predicting sequences with length over 81-nt, but still cannot make predictions when the sequence length is less than 81-nt. Overall, iPro-WAEL is a competitive and robust model for predicting promoters in multiple species and can be widely applied to datasets with different sequence lengths.
In addition, we estimate potentially important TFBS motifs in promoter regions using the computational method we designed. We identify many critical sequence motifs for predicting promoters, including ZNF143, YY1, CTCF, CUX1, SP1, REST, TFAP2C, SRF, MAX, TBP, CEBPB and RUNX, which are consistent with the previous study. However, to the best of our knowledge, several potentially important motifs identified by iPro-WAEL have received little or no attention. Therefore, these motifs may be involved in the formation of promoters and gene regulation through underappreciated or potentially new biological interactions with certain proteins, and may be the key to analyzing the difference and association of promoters in different cell lines of humans.
There are also some areas in which our method can be improved. For instance, we identify the importance of motifs by the importance of features reflecting the frequency of 5-mer segments, but indeed, the Word2vec-based features also reflect the infor-mation of the 5-mer segment because its window size is 5. However, we cannot interpret Word2vec-based features due to the difficulty of interpreting the embedding space intuitively. Therefore, we do not integrate word2vec information when calculating the 5-mer segment importance scores. Indeed, the field of NLP also faces this dilemma, although word embedding techniques are widely used in the NLP field. Although the RF model has better performance and higher weights than the CNN model on human cells, which indicates that interpreting the features used by the RF model may have higher confidence, our experimental results also show that integrating the RF model and CNN model can further improve the performance in predicting promoters. Therefore, if the importance scores of the Word2vec-based features can be calculated reasonably, the reliability of the method for analyzing the importance of motifs may be further improved when being combined with the importance scores of the features used in this study. Additionally, we have demonstrated a satisfactory performance when using iPro-WAEL trained on one cell line to predict promoters in another cell line, indicating that promoters from different cell lines may be similar at the motif level and related in some way. However, the performance of using models trained on prokaryotes to predict the promoters of eukaryotes is unsatisfactory, indicating that promoters of different species may be associated with different motifs. Overall, it will be interesting to explore the differences and associations between promoters of different cell lines and different species by analyzing the importance of motifs, which will provide a novel way to analyze cell specificity and species specificity.
DATA AVAILABILITY
The source code of iPro-WAEL is freely available at https://github.com/HaoWuLab-Bioinformatics/iPro-WAEL.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the group for valuable discussions and comments. The scientific calculations in this paper have been done on the HPC Cloud Platform of Shandong University. The funders did not play any role in the design of the study, the collection, analysis, and interpretation of data, or the writing of the manuscript.
Contributor Information
Pengyu Zhang, School of Software, Shandong University, Jinan, 250101, Shandong, China; College of Information Engineering, Northwest A&F University, Yangling, 712100, Shaanxi, China.
Hongming Zhang, College of Information Engineering, Northwest A&F University, Yangling, 712100, Shaanxi, China.
Hao Wu, School of Software, Shandong University, Jinan, 250101, Shandong, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [62272278 & 61972322]; National Key Research and Development Program [2021YFF0704103]; Natural Science Foundation of Shaanxi Province [2021JM110]; Fundamental Research Funds of Shandong University. Funding for open access charge: National Natural Science Foundation of China [61972322]; National Key Research and Development Program [2021YFF0704103]; Natural Science Foundation of Shaanxi Province [2021JM110]; Fundamental Research Funds of Shandong University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wang Q., Lei M., Wu J.. A structural perspective of human RNA polymerase III. RNA Biol. 2022; 19:246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramprakash J., Schwarz F.P.. Energetic contributions to the initiation of transcription in E. coli. Biophys. Chem. 2008; 138:91–98. [DOI] [PubMed] [Google Scholar]
- 3. Zhang M., Jia C., Li F., Li C., Zhu Y., Akutsu T., Webb G.I., Zou Q., Coin L.J.M., Song J.. Critical assessment of computational tools for prokaryotic and eukaryotic promoter prediction. Brief. Bioinform. 2022; 23:bbab551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Potvin E., Sanschagrin F., Levesque R.C.. Sigma factors in Pseudomonas aeruginosa. FEMS Microbiol. Rev. 2008; 32:38–55. [DOI] [PubMed] [Google Scholar]
- 5. Kustu S., Santero E., Keener J., Popham D., Weiss D.. Expression of sigma 54 (ntrA)-dependent genes is probably united by a common mechanism. Microbiol. Rev. 1989; 53:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arora S.K., Ritchings B.W., Almira E.C., Lory S., Ramphal R.. A transcriptional activator, FleQ, regulates mucin adhesion and flagellar gene expression in Pseudomonas aeruginosa in a cascade manner. J. Bacteriol. 1997; 179:5574–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sandelin A., Carninci P., Lenhard B., Ponjavic J., Hayashizaki Y., Hume D.A.. Mammalian RNA polymerase II core promoters: Insights from genome-wide studies. Nat. Rev. Genet. 2007; 8:424–436. [DOI] [PubMed] [Google Scholar]
- 8. Carleton J.B., Berrett K.C., Gertz J.. Dissection of enhancer function using multiplex CRISPR-based enhancer interference in cell lines. J. Vis. Exp. 2018; 2018:57883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dixon J.R., Xu J., Dileep V., Zhan Y., Song F., Le V.T., Yardımcı G.G., Chakraborty A., Bann D. V., Wang Y.et al.. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet. 2018; 50:1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spielmann M., Lupiáñez D.G., Mundlos S.. Structural variation in the 3D genome. Nat. Rev. Genet. 2018; 19:453–467. [DOI] [PubMed] [Google Scholar]
- 11. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M.et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song K. Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res. 2012; 40:963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin H., Deng E.Z., Ding H., Chen W., Chou K.C.. IPro54-PseKNC: A sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014; 42:12961–12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin H., Liang Z.Y., Tang H., Chen W.. Identifying Sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2019; 16:1316–1321. [DOI] [PubMed] [Google Scholar]
- 15. Liu B., Yang F., Huang D.S., Chou K.C.. IPromoter-2L: A two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2018; 34:33–40. [DOI] [PubMed] [Google Scholar]
- 16. Xiao X., Xu Z.C., Qiu W.R., Wang P., Ge H.T., Chou K.C.. iPSW(2L)-PseKNC: A two-layer predictor for identifying promoters and their strength by hybrid features via pseudo K-tuple nucleotide composition. Genomics. 2019; 111:1785–1793. [DOI] [PubMed] [Google Scholar]
- 17. Chevez-Guardado R., Peña-Castillo L.. Promotech: a general tool for bacterial promoter recognition. Genome Biol. 2021; 22:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Umarov R.K., Solovyev V.V.. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks. PLoS One. 2017; 12:e0171410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oubounyt M., Louadi Z., Tayara H., To Chong K.. Deepromoter: robust promoter predictor using deep learning. Front. Genet. 2019; 10:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Whalen S., Truty R.M., Pollard K.S.. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016; 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., Noble W.S.. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012; 9:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li W., Godzik A.. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006; 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- 25. Noble W.S., Kuehn S., Thurman R., Yu M., Stamatoyannopoulos J.. Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics. 2005; 21:i338–i343. [DOI] [PubMed] [Google Scholar]
- 26. Chen Z., Zhao P., Li C., Li F., Xiang D., Chen Y.Z., Akutsu T., Daly R.J., Webb G.I., Zhao Q.et al.. ILearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res. 2021; 49:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lv H., Dao F.-Y., Zulfiqar H., Su W., Ding H., Liu L., Lin H.. A sequence-based deep learning approach to predict CTCF-mediated chromatin loop. Brief. Bioinform. 2021; 22:bbab031. [DOI] [PubMed] [Google Scholar]
- 28. Manavalan B., Basith S., Shin T.H., Lee D.Y., Wei L., Lee G.. 4mCpred-EL: an ensemble learning framework for identification of DNA N4-methylcytosine sites in the mouse genome. Cells. 2019; 8:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu B., Liu F., Fang L., Wang X., Chou K.C.. RepDNA: A Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–1309. [DOI] [PubMed] [Google Scholar]
- 30. Le N.Q.K., Yapp E.K.Y., Ho Q.T., Nagasundaram N., Ou Y.Y., Yeh H.Y.. iEnhancer-5Step: identifying enhancers using hidden information of DNA sequences via Chou’s 5-step rule and word embedding. Anal. Biochem. 2019; 571:53–61. [DOI] [PubMed] [Google Scholar]
- 31. Inayat N., Khan M., Iqbal N., Khan S., Raza M., Khan D.M., Khan A., Wei D.Q.. IEnhancer-DHF: identification of enhancers and their strengths using optimize deep neural network with multiple features extraction methods. IEEE Access. 2021; 9:40783–40796. [Google Scholar]
- 32. Zeng M., Wu Y., Lu C., Zhang F., Wu F.-X., Li M.. DeepLncLoc: a deep learning framework for long non-coding RNA subcellular localization prediction based on subsequence embedding. Bioinformatics. 2022; 23:bbab360. [DOI] [PubMed] [Google Scholar]
- 33. Wu H., Zhang P., Ai Z., Wei L., Zhang H., Yang F., Cui L.. StackTADB: a stacking-based ensemble learning model for predicting the boundaries of topologically associating domains (TADs) accurately in fruit flies. Brief. Bioinform. 2022; 23:bbac023. [DOI] [PubMed] [Google Scholar]
- 34. Lin Y., Pan X., Shen H. Bin. LncLocator 2.0: A cell-line-specific subcellular localization predictor for long non-coding RNAs with interpretable deep learning. Bioinformatics. 2021; 37:2308–2316. [DOI] [PubMed] [Google Scholar]
- 35. Ji Y., Zhou Z., Liu H., Davuluri R.V.. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021; 37:2112–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Le N.Q.K., Ho Q.T., Nguyen T.T.D., Ou Y.Y.. A transformer architecture based on BERT and 2D convolutional neural network to identify DNA enhancers from sequence information. Brief. Bioinform. 2021; 22:bbab005. [DOI] [PubMed] [Google Scholar]
- 37. Khanal J., Tayara H., Chong K.T.. Identifying Enhancers and Their Strength by the Integration of Word Embedding and Convolution Neural Network. IEEE Access. 2020; 8:58369–58376. [Google Scholar]
- 38. RE M., Valentini G.. Ensemble Methods[M]. Advances in Machine Learning and Data Mining for Astronomy. 2012; [Google Scholar]
- 39. Liu B., Gao X., Zhang H.. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019; 47:E127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li H.L., Pang Y.H., Liu B.. BioSeq-BLM: a platform for analyzing DNA, RNA and protein sequences based on biological language models. Nucleic Acids Res. 2021; 49:e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van der Maaten L., Hinton G.. Visualizing data using t-SNE. J. Mach Learn Res. 2008; 9:2579–2605. [Google Scholar]
- 42. Lim D.Y., Khanal J., Tayara H., Chong K.T.. iEnhancer-RF: Identifying enhancers and their strength by enhanced feature representation using random forest. Chemom. Intell. Lab. Syst. 2021; 212:104284. [Google Scholar]
- 43. Cai L., Ren X., Fu X., Peng L., Gao M., Zeng X.. IEnhancer-XG: Interpretable sequence-based enhancers and their strength predictor. Bioinformatics. 2021; 37:1060–1067. [DOI] [PubMed] [Google Scholar]
- 44. Andersson R., Sandelin A.. Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 2020; 21:71–87. [DOI] [PubMed] [Google Scholar]
- 45. Core L.J., Martins A.L., Danko C.G., Waters C.T., Siepel A., Lis J.T.. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 2014; 46:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koch F., Fenouil R., Gut M., Cauchy P., Albert T.K., Zacarias-Cabeza J., Spicuglia S., De La Chapelle A.L., Heidemann M., Hintermair C.et al.. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol. 2011; 18:956–963. [DOI] [PubMed] [Google Scholar]
- 47. Chen Y., Pai A.A., Herudek J., Lubas M., Meola N., Järvelin A.I., Andersson R., Pelechano V., Steinmetz L.M., Jensen T.H.et al.. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat. Genet. 2016; 48:984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Andersson R., Chen Y., Core L., Lis J.T., Sandelin A., Jensen T.H.. Human gene promoters are intrinsically bidirectional. Mol. Cell. 2015; 60:346–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lambert M., Jambon S., Depauw S., David-Cordonnier M.H.. Targeting transcription factors for cancer treatment. Molecules. 2018; 23:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim Y.W., Kim A.R.. Deletion of transcription factor binding motifs using the CRISPR/spCas9 system in the β-globin LCR. Biosci. Rep. 2017; 3:BSR20170976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kulakovskiy I. V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A.et al.. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bonev B., Cavalli G.. Organization and function of the 3D genome. Nat. Rev. Genet. 2016; 17:661–678. [DOI] [PubMed] [Google Scholar]
- 53. Salameh T.J., Wang X., Song F., Zhang B., Wright S.M., Khunsriraksakul C., Ruan Y., Yue F.. A supervised learning framework for chromatin loop detection in genome-wide contact maps. Nat. Commun. 2020; 11:3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Weintraub A.S., Li C.H., Zamudio A.V., Sigova A.A., Hannett N.M., Day D.S., Abraham B.J., Cohen M.A., Nabet B., Buckley D.L.et al.. YY1 is a structural regulator of enhancer-promoter loops. Cell. 2017; 171:1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dao F.Y., Lv H., Zhang D., Zhang Z.M., Liu L., Lin H.. DeepYY1: a deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. 2021; 22:bbaa356. [DOI] [PubMed] [Google Scholar]
- 56. Bailey S.D., Zhang X., Desai K., Aid M., Corradin O., Cowper-Sallari R., Akhtar-Zaidi B., Scacheri P.C., Haibe-Kains B., Lupien M.. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nat. Commun. 2018; 9:16194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang Y., Zhang R., Singh S., Ma J.. Exploiting sequence-based features for predicting enhancer-promoter interactions. Bioinformatics. 2017; 33:i252–i260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nolis I.K., McKay D.J., Mantouvalou E., Lomvardas S., Merika M., Thanos D.. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:20222–20227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Deshane J., Kim J., Bolisetty S., Hock T.D., Hill-Kapturczak N., Agarwal A.. Sp1 regulates chromatin looping between an intronic enhancer and distal promoter of the human heme oxygenase-1 gene in renal cells. J. Biol. Chem. 2010; 285:16476–16486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code of iPro-WAEL is freely available at https://github.com/HaoWuLab-Bioinformatics/iPro-WAEL.