

Abstract
Background
GNAO1 variants have been found to be associated with epileptic encephalopathies, developmental delays (DDs), and movement disorders (MDs). Therapies for patients with GNAO1 variants vary. However, treatments for GNAO1-related diseases are still in their infancy. Previous reports suggest that few pharmacological treatments are effective for patients with GNAO1 variant-related MDs. Deep brain stimulation (DBS) treatment appears to be effective, however surgical procedures and equipment failures pose risks to the patients. Effectiveness for oxcarbazepine (OXC) in GNAO1 variant-related MDs is first reported in our study, and it expand the effective drugs for MD treatment.
Case Description
We report the case of a 5-year-old male patient with a MD, who suffered from hypotonia and refractory choreoathetosis. The patient was found to have a DD and an intellectual disability. A de-novo variant of the GNAO1 gene (NM_138736: exom6: c.709G>A [p. Glu237Lys]) was identified by whole exome sequencing (WES) when he was 8 months old. The patient visited our hospital at the age of 4 years and 3 months because of fever and recurrent convulsions. Electroencephalogram (EEG) results show abnormal spikes, and magnetic resonance imaging (MRI) showed the enlargement of the lateral ventricles. The administration of tiapride hydrochloride, phenobarbital, midazolam, and hormones had no effect. OXC treatment was then initiated. No MD behaviors, such as rigidity and twisting of the limbs and trunk, or chorea, were observed after 10 days OXC treatment. Eventually, incremental doses of OXC were effective, and our patient achieved good control of his MD.
Conclusions
We are the first to demonstrate the role of OXC in alleviating MDs associated with GNAO1 mutations. This report provides a novel possibility for the clinical treatment of this rare disease. To manage MDs associated with GNAO1 mutations, we recommend that OXC treatment be attempted before invasive surgical therapy.
Keywords: GNAO1, movement disorders (MDs), development delay, oxcarbazepine (OXC), case report
Introduction
Guanine nucleotide-binding protein (Gαo) encoded by the GNAO1 gene is the α subunit of the Go (a member of the Gi/o family) heterotrimeric G-protein signal-transducing complex. It is highly expressed in the mammalian brain, especially in the cerebral cortex, hippocampus, and striatum (1). Gαo plays a key role in transducing G-protein-coupled receptor (GPCR) signals, and couples with multiple GPCRs, including dopamine, serotonin, and opioid receptors (2-4).
GNAO1 variations were first reported in 2013 with early infantile epileptiform encephalopathy and involuntary movements (5). To date, 50 GNAO1 variants have been found in patients with epileptiform encephalopathy and movement disorders (MDs). The pathogenicity of GNAO1 may be due to the inhibition of cyclic adenosine monophosphate (cAMP), which disrupts the finely tuned neurodevelopmental system, regulates neurotransmitter release, or alters neuronal maturation (6). These mechanisms have been validated in different animal models (7,8). For example, mouse and Caenorhabditis elegans models have been shown to carry orthologous mutations to clinical variants in Gαo present movement dysregulation.
The treatment of GNAO1-related diseases is in its infancy, especially in terms of MD interventions. Heretofore, pharmacotherapy, including the administration of antiepileptics and neuroleptics is a mainstay of GNAO1 mutation-related MDs treatment, which has been reported to have varying levels of success (9-11). Those who had poor response to maximal doses of medications could undergo surgical treatment. Deep brain stimulation (DBS) has also been reported to reduce the severity of MDs, although the uncertainty of long-term efficacy, complications may occur as well (12).
Oxcarbazepine (OXC) is commonly known as a sodium channels blocker and focus on the treatment for epilepsy. Gαo couples various GPCRs including adenosine receptors and regulates the neurotransmitter release, which was proposed as a theoretical basis of GNAO1-associated MDs (6). Booker et al. have demonstrated that OXC acts as an antagonist to adenosine receptors (13). Therefore, OXC might be effective for patients with GNAO1 variant-related MDs as a Gαo-coupled-receptor antagonist.
We present the case of a 5-year-old boy with a de-novo GNAO1 mutation, who had a developmental delay (DD) and MD. Whole exome sequencing (WES) identified a de-novo variant in the GNAO1 gene. OXC was administered and effectively alleviated the symptoms of dystonia in our patient. Our study confirms the role of OXC in controlling MDs, and provides a reliable basis for the clinical treatment of GNAO1-related MDs. We present the following article in accordance with the CARE reporting checklist (available at https://tp.amegroups.com/article/view/10.21037/tp-22-297/rc).
Case presentation
The boy, aged 5 years and 2 months (at the time of writing this report), is the 2nd child of non-consanguineous healthy parents and has 1 healthy brother. He was delivered by cesarean section at term and had a birth weight of 3,500 g. The developmental milestones he reached were significantly delayed. At 8 months of age, he was unable to lift his head permanently, turn his body over, or sit or crawl independently. His language development was also delayed, and at his last visit, he spoke only single words and no phrases. There was no family history of similar symptoms. On May 24, 2021, he was admitted to our hospital for recurrent fever and convulsions at the age of 4 years and 3 months. During his hospitalization, he suffered from restlessness, involuntary movements of the limbs, body writhing, profuse sweating, and unresponsiveness, lasting for 30–60 minutes each time (see Video 1). A diagnostic workup, including routine blood tests and autoimmune encephalitis antibody tests, revealed no abnormalities.
Video 1.

Before the OXC treatment, the patient exhibited body writhing while reclining in bed with a particular painful expression and an impairment of the involuntary swing movements in all limbs. OXC, oxcarbazepine.
An inter-ictal electroencephalogram (EEG) revealed spike waves, slow waves, and multi-spike and slow waves in all leads throughout the waking and sleeping phases; no epileptiform abnormalities were observed in the ictal phase of the seizures (see Figure 1A,1B). A follow-up EEG video of our patient after 4 months of treatment was taken (see Figure 1C). Magnetic resonance imaging (MRI) showed the enlargement of the lateral ventricles (see Figures 1D-1I). We initially treated the patient’s MD with tiapride hydrochloride, phenobarbital, hormones, and benzodiazepines, such as midazolam and diazepam (see Table 1), but all with no effect.
Figure 1.

Clinical examinations of the patient by MRI and EEG. (A,B) Video-EEG results for the patient during hospitalization on June 18, 2021. A shows the waking state and B shows the sleeping state. An all-conducted high-amplitude 2–3 Hz spike, slow waves and multi-spike and slow waves were observed in all stages of waking and sleeping. (C) Video-EEG results during the interictal sleep state after 4 months of regular medication on October 27, 2021. (D-F) T2-weighted scans (T2), FLAIR, and DWI sequences on May 25, 2021. (G-I) T2, FLAIR, and DWI sequences on June 7, 2021. Notably, the gray and white matter of the 2 cerebral hemispheres is demarcated, and the volume of the ventricle is slightly full. C shows that the signal in the anterior part of the right caudate nucleus is slightly higher than that of the opposite side on the +DWI sequence. No abnormal lesions related to the clinical symptoms of the child were found in the rest of the brain parenchyma. MRI, magnetic resonance imaging; EEG, Electroencephalogram; FLAIR, fluid-attenuated inversion recovery; DWI, diffusion-weighted MR imaging.
Table 1. Clinical medications administered to our patient.
| Drug | Dosage and administration | Drug use of time (2021) |
|---|---|---|
| Acyclovir | 10 mg/kg/times, q8h, ivgtt | May 25 to June 10 |
| Tiapride hydrochloride | 3.3 mg/kg/times, bid, po | May 26 to 27 |
| 5 mg/kg/times, bid, po | May 28 to June 23 | |
| 3.3 mg/kg/times, bid, po | June 24 to September | |
| 1.67 mg/kg/times, bid, po | September to October | |
| 3.3 mg/kg/times, bid, po | October to April 7 (2022) | |
| 5 mg/kg/times, bid, po | April 8 to present | |
| Changma xifeng | 0.53 g/times, tid, po | June 1 to 9 |
| Methylprednisolone sodium succinate | 1.25 mg/kg, qd, ivgtt | May 26 to 31 |
| 5 mg/kg, qd, ivgtt | June 1 to 3 | |
| 2.5 mg/kg, qd, ivgtt | June 4 to 6 | |
| 1.25 mg/kg, qd, ivgtt | June 7 to 9 | |
| 0.625 mg/kg, ivgtt | June 10 to 12 | |
| Gamma globulin | total 2 g/kg, ivgtt | May 26 to 29 |
| Midazolam | 1 μg/kg/min | May 26 |
| 2 μg/kg/min | May 27 | |
| 3 μg/kg/min | May 28 | |
| 4 μg/kg/min | May 28 to 31 | |
| 3 μg/kg/min | June 1 to 7 | |
| 2 μg/kg/min | June 8 to 9 | |
| 1 μg/kg/min | June 10 to 11 | |
| 0.5 μg/kg/min | June 12 | |
| 0.25 μg/kg/min | June 13 to 14 | |
| Phenobarbital | 5 mg/kg/times, q6h*2 times, iv | May 27 |
| 2.5 mg/kg/times, q12h, iv | May 28 to June 10 | |
| Oxcarbazepine | 5 mg/kg/times, bid, po | June 7 to 13 |
| 10 mg/kg/times, bid, po | June 14 to September | |
| 15 mg/kg/times, bid, po | September to April 7 (2022) | |
| 20 mg/kg/times, bid, po | April 7 to present |
Q8h, quaque 8 hora/every 8 hours; q12h, quaque 12 hora/every 12 hours; ivgtt, intravenously guttae; bid, bis in die/twice a day; po, peros/oral; tid, ter in die/three times a day; qd, quaque die/every day; iv, intravenous injection.
When the patient was 8 months old, genetic testing was performed to identify his etiology. No pathogenic copy number variations or mitochondrial deoxyribonucleic acid pathogenic variants were found in our patient. WES was then performed. Blood samples were collected from our patient and his parents. Sequencing was performed using an Illumina HiSeq sequencer (the reference genome was hg19). We found the heterozygous de-novo variant of NM_138736: exon6: c.709G>A (p. Glu237Lys) in the GNAO1 gene. This variant was confirmed by Sanger sequencing in the trio family (see Figure 2A,2B). Eventually, the patient’s diagnosis was determined by a genetic evaluation as ‘GNAO1-related neurodevelopmental disorder with involuntary movements (Online Mendelian Inheritance: 617493). And we summarized the previously reported GNAO1 gene variants in Figure 2C.
Figure 2.

Family pedigree—Sanger sequencing. (A) The pedigree of our patient. The affected proband is highlighted by the square and arrow. (B) The variant in the GNAO1 gene was confirmed by Sanger sequencing in the trio family. The mutated site is shown in the red box. (C) This is the schematic of the variants reported thus far. A total of 50 amino acid variations and the domain G-protein alpha subunit are shown. The variant in our patient is highlighted with red text.
Based on the above-mentioned treatment process and genetic testing results, we adjusted the patient’s treatment. Oral OXC (10 mg/kg/day) administration was initiated on day 14 of hospitalization, with the gradual withdrawal of midazolam, phenobarbital, hormones, and reductions in the dose of tiapride hydrochloride. After 2 days of OXC administration, the patient’s stiffened, twisted and chorea in limbs and trunk, and other MDs were significantly reduced compared to the period before OXC was used, and his facial expression was notably relaxed (see Video 2). On day 21 of hospitalization, the dose of OXC was increased according to the course of treatment, and the patient’s symptoms continued to improve, and he regained a clear state of consciousness (see Video 3).
Video 2.
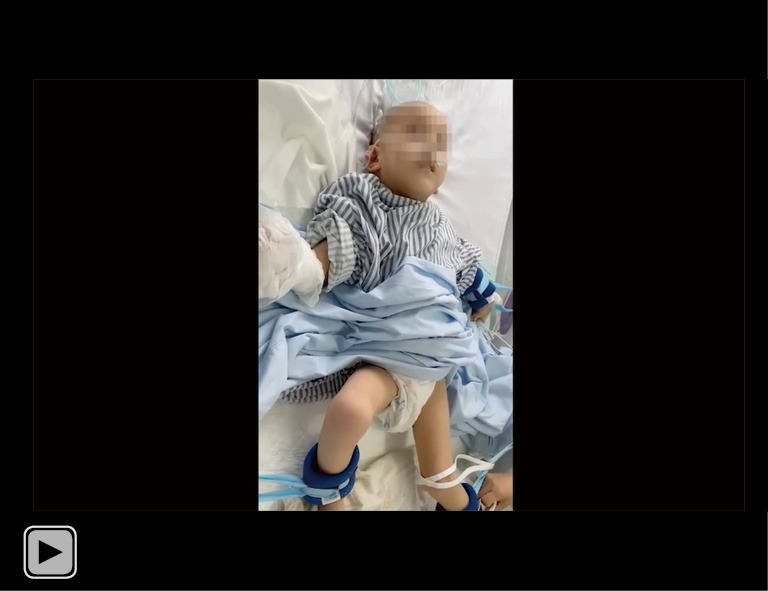
The MDs in our patient were in remission; the patient’s limbs and head shook slightly after 2 days of oral OXC administration (0.075 g bid). MD, movement disorder; OXC, oxcarbazepine. This video is published with consent from the patient’s guardian.
Video 3.
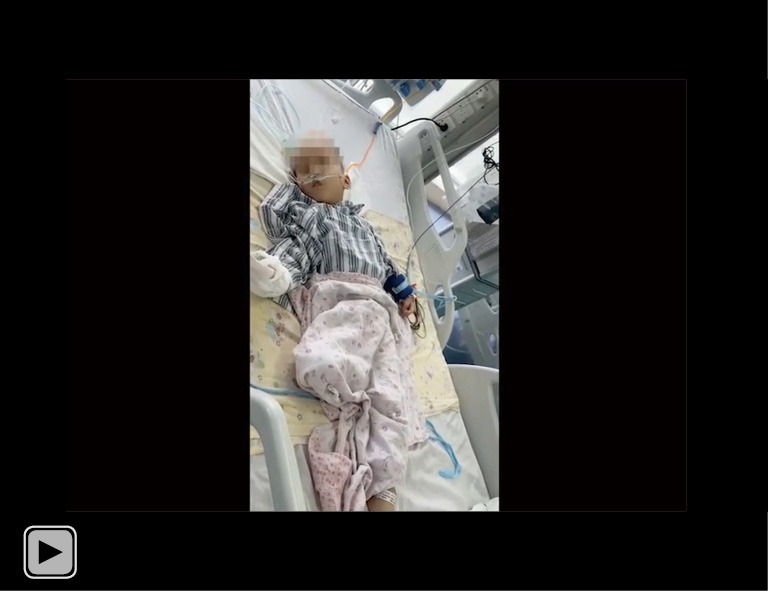
The patient regained a clear state of consciousness, and the frequency and amplitude of body shaking in our patient continued to improve clinically after 4 days of oral OXC administration (0.075 g bid). OXC, oxcarbazepine. This video is published with consent from the patient’s guardian.
After 10 days of treatment with OXC, no MD behaviors, such as rigidity and twisting of the limbs and trunk, or chorea, were observed clinically, but occasional clenching and frowning movements were observed (see Video 4). After 2 weeks of treatment with OXC, the patient’s involuntary movements completely disappeared (see Video 5). On June 23, the patient was discharged from the hospital with a prescription of oral OXC (20 mg/kg/day) and tiapride hydrochloride (6.6 mg/kg/day) for maintenance therapy. The patient was still unable to walk. He attended regular follow-ups and achieved good control of his MD. A follow-up EEG video taken 4 months after the treatment demonstrated a lower amplitude and frequency of abnormal wave activity compared to the previous video (see Figure 1C). The clinical course of treatment for MDs was shown in Figure 3.
Video 4.

The movement of the limbs was still a little bit stiff; however, the rigidity and twisting of limbs and trunk, and chorea could barely be observed clinically after 10 days of oral OXC administration (0.15 g bid). OXC, oxcarbazepine. This video is published with consent from the patient’s guardian.
Video 5.
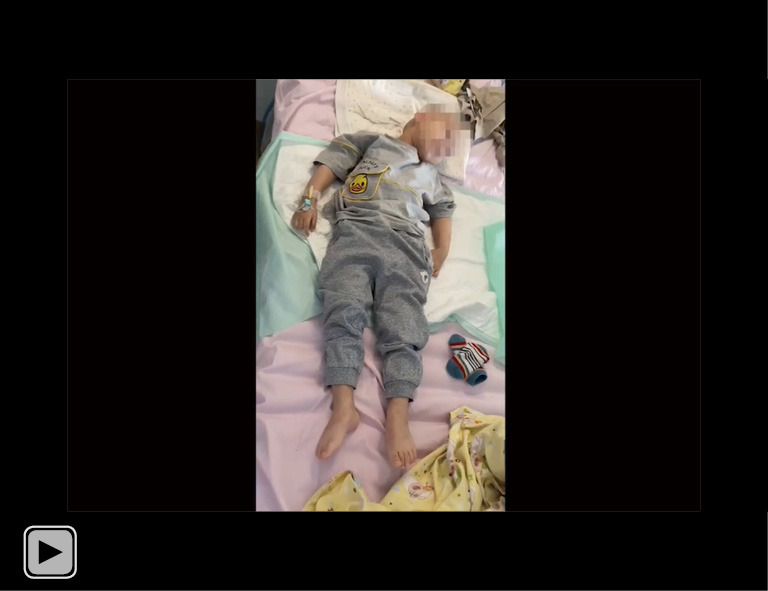
The patient’s involuntary movements in the clinic completely disappeared after 14 days of oral OXC administration (0.15 g bid). OXC, oxcarbazepine. This video is published with consent from the patient’s guardian.
Figure 3.
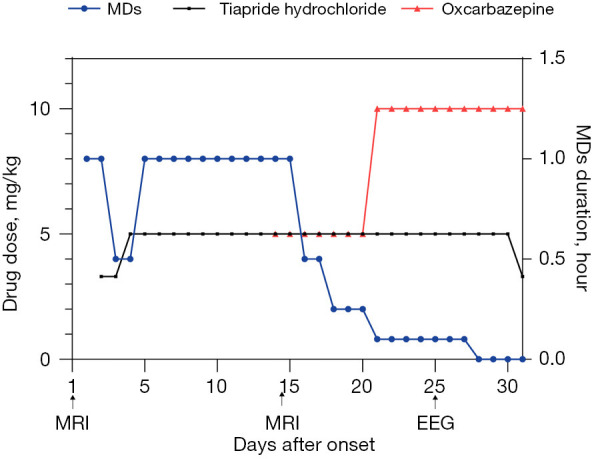
Clinical course of treatment for movement disorders. Antidyskinetic dose-response curves for Tiapride hydrochloride and Oxcarbazepine as the duration of the movement disorders during hospitalization before and after oral administration and increasing doses. MDs, movement disorders; MRI, magnetic resonance imaging; EEG, electroencephalogram.
All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient’s guardians for publication of this case report and accompanying images. A copy of the written consent is available for review by the editorial office of this journal.
Discussion
We described the case of a male patient with a GNAO1 mutation who progressively presented with MDs and DDs without seizures from 8 months of age. A de-novo missense mutation in exon 6 of GNAO1 was identified. To date, 50 variants (see Figure 2C) of the GNAO1 gene have been found in patients with epileptic encephalopathy and MDs. Variant c.709G>A (p. Glu237Lys) in GNAO1 has been reported in 8 other patients (6,12,14-16), and the clinical features are summarized in Table 2. All the 9 patients (include our patient) to have Glu237Lys presented with MDs, but none of them had epilepsy. Brain atrophy occurred in 2 of the patients, but MRI did not show any abnormalities in the remaining patients. The MDs were progressive and almost all of the patients had to be admitted to the intensive care unit. Of the 9 patients, 4 developed choreoathetosis, including our patient.
Table 2. The clinical characteristics of patients with the GNAO1 variant (c.709G>A, p.Glu237Lys).
| Ref (PMID) | Variant | Gender | Age | Inheritance | Diagnosis | MRI | Seizures | DD | ID | MD | Hypotonia | Chorea/athetosis | Treatment | Adverse effect | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 28668776 | C.709G>A, p.Glu237Lys | Male | 3 months | De novo | Dyskinetic cerebral palsy | Progressive global atrophy | – | + | Chorea, dystonia and orofaciolingual dyskinesia | + | DBS. No exacerbations requiring intensive care unit admission were observed following DBS. Decrease of orofaciolingual dyskinesia, chorea and dystonia; no more hyperkinetic exacerbations; improvement of function | Postoperative complications included stimulator site infection, and lead displacement requiring reinsertion | DBS did not result in complete remission for movements; patients with short exacerbations were managed medically | ||
| 29758257 | C.709G>A, p.Glu237Lys | Male | 3 months | De novo | – | + | Tetrabenazine | ||||||||
| C.709G>A, p.Glu237Lys | Male | 4 months | De novo | – | + | Levetiracetam | |||||||||
| 29935962 | C.709G>A, p.Glu237Lys | Female | 4 years | De novo | MD | Normal | – | + | Decreased spontaneous movement; mild dystonic features | + | |||||
| 30103967 | C.709G>A, p.Glu237Lys | Male | – | De novo | Severe hyperkinesia | Small medio-putaminal atrophy | – | + | Recurrent episodes of hyperkinesia with dystonia; choreoathetosis, ballism, severe orofacial dyskinesia and dysphagia with concomitant autonomic features | + | + | DBS. Almost complete remission of hyperkinesia and dystonia at rest; improvement of non-verbal communication, hand function, and mobility | 6.5 y reimplantation due to hardware infection | DBS almost complete remission | |
| C.709G>A, p.Glu237Lys | Male | – | Unknown | Severe hyperkinesia | Normal | – | + | + | DBS. Significant effects on the hyperkinetic, choreatic features | Patient suffered from recurrent loss of beneficial effects due to dysfunctions of the DBS system requiring several hardware replacements | At 14.8 y, the child died due to the refractory worsening of the hyperkinesia | ||||
| 34441836 | C.709G>A, p.Glu237Lys | Female | 15 years | Not shown | Dystonia | – | + | + | + | DBS, trihexyphenidyl | |||||
| C.709G>A, p.Glu237Lys | Female | 3 years | Not shown | Dystonia | – | + | + | Trihexyphenidyl, Tetrabenazine | |||||||
| This report | C.709G>A, p.Glu237Lys | Male | 5 years | De novo | MD | Normal | – | + | + | + | + | OXC | The complex MDs were reduced significantly |
y, years; MRI, magnetic resonance imaging; DD, developmental delay; ID, intellectual disability; MD, movement disorder; DBS, deep brain stimulation; OXC, oxcarbazepine; ‘+’, positive; ‘–’, negative.
The mechanisms of the GNAO1 mutation related to MDs have been reported in the dysregulation of cAMP signaling, regulation of neurotransmitter release, and altered neuronal maturation (6). The discovery of these mechanisms is based on the simulation of an animal MD phenotype and molecular mechanism research (7,16,17). An early study (6) used a heterologous cell-based assay to evaluate Gαo and placed pathological mutations in the following 3 categories: loss of function, gain of function (GOF), and normal function (NF), different functional alterations can lead to phenotypic heterogeneity. Additionally, this research probed into the phenotypic heterogeneity of GNAO1 mutations and revealed that the GOF or NF mutations were associated with the MD phenotype (6).
The variant Glu237Lys found in our patient was reported to be pathogenic for MD by affecting guanine-nucleotide binding (15). A structural estimation for Glu237Lys in the Ga-containing complex was performed with the patient in active and inactive states. The Glu237 is located in the switch III region and is responsible for the binding of guanine nucleotides and the activation of downstream effectors. The Glu237Lys mutation could thus destabilize the active state complexes. The importance of the locus also demonstrates the pathogenicity of this variant in GNAO1.
Various medications have been used to treat GNAO1-related MDs (see Table 3). Patients with MDs appear to be more responsive to tetrabenazine than other drugs (see Table 3). Tetrabenazine binds to and inhibits the type 2 vesicular monoamine transporter, which is responsible for the introduction of neurotransmitters from the cytosol into the vesicles of neuronal cells. Additionally, trihexyphenidyl, which selectively blocks the striatal cholinergic pathways, has been reported to effectively treat GNAO1 related MDs (9). Subsequently, some antiepileptic drugs that act on ion channels (6,18) and central synapses (10,11,19), such as topiramate and levetiracetam (see Table 3), have also been shown to be effective in patients with MDs. As Table 2 shows, 4 patients with Glu237Lys received DBS, which reduced dyskinesia, chorea, and dystonia, and improved their movement function (12,14). DBS appears to be effective at reducing life-threatening exacerbations; however, its long-term efficacy in treating MDs is unknown. Due to the dysfunction of DBS systems, which require multiple hardware replacements, some patients have died as a result of the refractory worsening of the hyperkinesia (12).
Table 3. The positive treatment for GNAO1 related MD.
| Ref (PMID/DOI) | Positive treatment | GNAO1 variant | Sex | Inheritance | Age of onset | Seizures | MD |
|---|---|---|---|---|---|---|---|
| 28357411 | Tetrabenazine | p.Glu246Gly | Female | De novo | 6 months | + | + |
| 28357411 | Tetrabenazine | p.Ser47Gly | Male | De novo | 5 months | + | + |
| 30838255 | Tetrabenazine | p.Arg209His | Male | De novo | 10 months | + | |
| 29758257 | Tetrabenazine | p.Glu237Lys | Male | De novo | 4 months | + | |
| 28668776 | Tetrabenazine | p.Glu237Lys | Male | De novo | 3 months | + | |
| 27068059 | Tetrabenazine | p.Glu246Lys | Female | De novo | 4 years | + | |
| 27068059 | Tetrabenazine | p.Glu246Lys | Female | De novo | 6 months | + | |
| 27068059 | Tetrabenazine | p.Glu246Lys | Male | De novo | 14 years | + | |
| 28668776 | Tetrabenazine | p.Glu246Lys | Female | De novo | 3 months | + | |
| DOI: 10.1055/s-0036-1597627 | Levetiracetam | p.Gly45Arg | Male | De novo | unknown | + | + |
| 29758257 | Levetiracetam | p.Glu237Lys | Male | De novo | 4 months | + | |
| 25966631/27916449 | Topiramate | p.Arg209Cys | Female | De novo | 11 months | + | |
| 30838255 | Trihexyphenidyl | p.Arg209His | Male | De novo | 10 months | + | |
| 33358199 | Gabapentin | p.Arg209Cys | Female | De novo | 2 years | + | |
| Our study | oxcarbazepine | p.Glu237Lys | Male | De novo | 4 years | + | |
| 32044685 | DBS + levodopa | p.Ser207Phe | Female | De novo | 16 years | + | |
| 31076915 | DBS | p.Glu246Lys | Female | Unknown | 13 months | + | |
| 31076915 | DBS | p.Glu246Lys | Female | Unknown | 4 years | + | + |
| 30103967 | DBS | c.723+1G>T | Female | De novo | 3 years | + | |
| 30103967 | DBS | p.Arg209Cys | Female | De novo | 0 month | + | + |
| 30103967 | DBS | p.Glu237Lys | Male | De novo | 0 month | + | |
| 29661126 | DBS | p.Arg209Leu | Male | De novo | 18 months | ||
| 28357411 | DBS | p.Glu246Gly | Female | De novo | 6 months | + | + |
| 28668776 | DBS | p.Glu237Lys | Male | De novo | 3 months | + | |
| 28668776 | DBS | p.Glu246Lys | Female | De novo | 3 months | + | |
| 26060304 | DBS | p.Arg209His | Male | De novo | 18 months | + | |
| 26060304 | DBS | p.Arg209His | Male | De novo | 2 years | + | |
| 28668776 | DBS | p.Arg209Cys | Female | De novo | 6 months | + | + |
| 29661126 | DBS | p.Arg209Leu | Male | De novo | 2 years | + | |
| 27278281 | DBS | p.Gln233Phe | Female | De novo | 13 months | + |
MD, movement disorder; DBS, deep brain stimulation. ‘+’, positive.
Adenosine A1 receptor, which is an important GPCR coupled with Gαo, plays a key role in regulating neurotransmitter release, movement, and neural development. Thus, Gαo-coupled-receptor antagonists, including adenosine receptor antagonists, have been deemed to be beneficial for patients with MDs, as they decrease the signals from hyperactive GOF GNAO1 mutants (6). An in-vitro experiment was conducted in wild-type mice to investigate the effects of carbamazepine (CBZ) and OXC on fast excitatory and inhibitory synaptic transmission in the hippocampal cornu ammonis 1 (CA1) area. According to this study, a low-therapeutical dose of the 2 drugs enhanced excitatory postsynaptic currents (EPSCs), which could be blocked by a selective agonist of the adenosine A1 receptor called 2-chloro-N6-cyclopentyladenosine (CCPA). Additionally, the increase in EPSCs induced by CBZ and OXC was also abolished when the enzyme adenosine deaminase was applied to reduce endogenous adenosine. Thus, OXC acts as an antagonist to native adenosine receptors (13), and we conjecture that this provides the mechanism of action that produces its beneficial effects on MDs associated with GNAO1.
To our knowledge, no study has examined the efficacy of OXC with GNAO1-associated epilepsy or MDs. Our study confirmed the positive effect of OXC in alleviating GNAO1-related MDs and provides a reliable basis for the clinical treatment of this rare disease. The limitation of our report is that it comprises a single case report. Longer follow-up and additional studies with more patients might provide further insights into the efficacy and mechanisms of OXC treatment.
Conclusions
We reported the case of a male patient with MD, who presented with DDs, hypotonia, and choreoathetosis. Genetic testing revealed the Glu237Lys variant in GNAO1, which clarified the diagnosis and etiology of the patient. The innovative OXC treatment proved to effectively alleviate the GNAO1-related MDs. Due to limited cases, the effectiveness of OXC for GNAO1-related MD treatment needs more clinical practice and exploration. Effective medication should be focused to develop, and could be considered before invasive surgical therapy, including DBS, in patients with MDs due to GNAO1 mutations.
Supplementary
The article’s supplementary files as
Acknowledgments
The authors would like to thank the patient and his parents for allowing us to publish this case report.
Funding: The costs of the publication and molecular analyses of this research were supported by the National Natural Science Foundation of China (No. 82171441), the Natural Science Foundation of Jiangsu Province (No. BK20201175), and the Project of Science and Technology Development Plan in Suzhou (Technology of People’s Livelihood) (No. SYSD2020122). The funding bodies played no role in the design of the study, the collection, analysis, and interpretation of the data, or the writing the manuscript.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in this study were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient’s guardians for publication of this case report and accompanying images. A copy of the written consent is available for review by the editorial office of this journal.
Footnotes
Reporting Checklist: The authors have completed the CARE reporting checklist. Available at https://tp.amegroups.com/article/view/10.21037/tp-22-297/rc
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://tp.amegroups.com/article/view/10.21037/tp-22-297/coif). FY and ZY are employed by the company Cipher Gene LLC. The other authors have no conflicts of interest to declare.
References
- 1.Worley PF, Baraban JM, Van Dop C, et al. Go, a guanine nucleotide-binding protein: immunohistochemical localization in rat brain resembles distribution of second messenger systems. Proc Natl Acad Sci U S A 1986;83:4561-5. 10.1073/pnas.83.12.4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hescheler J, Rosenthal W, Trautwein W, et al. The GTP-binding protein, Go, regulates neuronal calcium channels. Nature 1987;325:445-7. 10.1038/325445a0 [DOI] [PubMed] [Google Scholar]
- 3.Masuho I, Ostrovskaya O, Kramer GM, et al. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal 2015;8:ra123. 10.1126/scisignal.aab4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Oliveira PG, Ramos MLS, Amaro AJ, et al. Gi/o-Protein Coupled Receptors in the Aging Brain. Front Aging Neurosci 2019;11:89. 10.3389/fnagi.2019.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura K, Kodera H, Akita T, et al. De Novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013;93:496-505. 10.1016/j.ajhg.2013.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng H, Khalil S, Neubig RR, et al. A mechanistic review on GNAO1-associated movement disorder. Neurobiol Dis 2018;116:131-41. 10.1016/j.nbd.2018.05.005 [DOI] [PubMed] [Google Scholar]
- 7.Di Rocco M, Galosi S, Lanza E, et al. Caenorhabditis elegans provides an efficient drug screening platform for GNAO1-related disorders and highlights the potential role of caffeine in controlling dyskinesia. Hum Mol Genet 2022;31:929-41. 10.1093/hmg/ddab296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muntean BS, Masuho I, Dao M, et al. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep 2021;34:108718. 10.1016/j.celrep.2021.108718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dhamija R, Mink JW, Shah BB, et al. GNAO1-Associated Movement Disorder. Mov Disord Clin Pract 2016;3:615-7. 10.1002/mdc3.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akasaka M, Kamei A, Tanifuji S, et al. GNAO1 mutation-related severe involuntary movements treated with gabapentin. Brain Dev 2021;43:576-9. 10.1016/j.braindev.2020.12.002 [DOI] [PubMed] [Google Scholar]
- 11.Sakamoto S, Monden Y, Fukai R, et al. A case of severe movement disorder with GNAO1 mutation responsive to topiramate. Brain Dev 2017;39:439-43. 10.1016/j.braindev.2016.11.009 [DOI] [PubMed] [Google Scholar]
- 12.Koy A, Cirak S, Gonzalez V, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci 2018;391:31-9. 10.1016/j.jns.2018.05.018 [DOI] [PubMed] [Google Scholar]
- 13.Booker SA, Pires N, Cobb S, et al. Carbamazepine and oxcarbazepine, but not eslicarbazepine, enhance excitatory synaptic transmission onto hippocampal CA1 pyramidal cells through an antagonist action at adenosine A1 receptors. Neuropharmacology 2015;93:103-15. 10.1016/j.neuropharm.2015.01.019 [DOI] [PubMed] [Google Scholar]
- 14.Waak M, Mohammad SS, Coman D, et al. GNAO1-related movement disorder with life-threatening exacerbations: movement phenomenology and response to DBS. J Neurol Neurosurg Psychiatry 2018;89:221-2. 10.1136/jnnp-2017-315653 [DOI] [PubMed] [Google Scholar]
- 15.Okumura A, Maruyama K, Shibata M, et al. A patient with a GNAO1 mutation with decreased spontaneous movements, hypotonia, and dystonic features. Brain Dev 2018;40:926-30. 10.1016/j.braindev.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 16.Graziola F, Garone G, Grasso M, et al. Cognitive Assessment in GNAO1 Neurodevelopmental Disorder Using an Eye Tracking System. J Clin Med 2021;10:3541. 10.3390/jcm10163541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Dao M, Muntean BS, et al. Genetic modeling of GNAO1 disorder delineates mechanisms of Gαo dysfunction. Hum Mol Genet 2022;31:510-22. 10.1093/hmg/ddab235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda K, Serajee F, Huq AM. Exome Sequencing Identifying Dual Mutations in Calcium Signaling Genes GNAO1 and ATP2B3 in a Patient with Early Infantile Epileptic Encephalopathy. J Pediatr Neurol 2017;15:183-6. 10.1055/s-0036-1597627 [DOI] [Google Scholar]
- 19.Saitsu H, Fukai R, Ben-Zeev B, et al. Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur J Hum Genet 2016;24:129-34. 10.1038/ejhg.2015.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as