

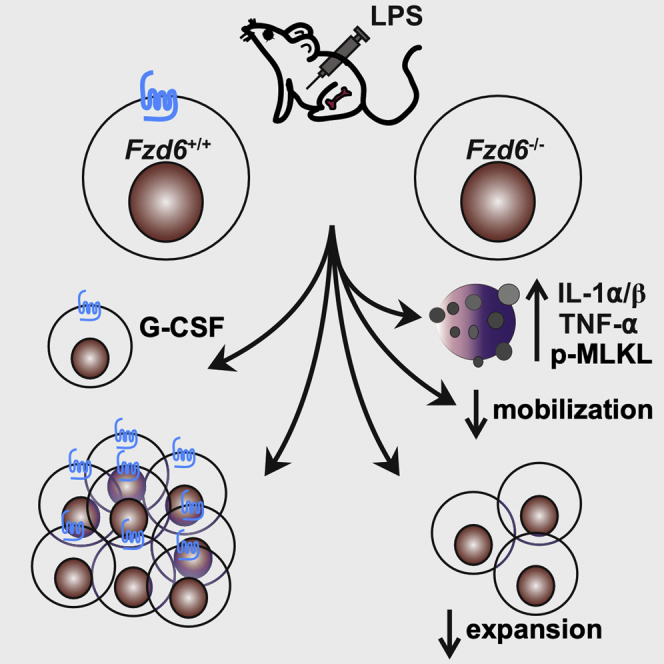
Summary
Emergency hematopoiesis involves the activation of bone marrow hematopoietic stem/progenitor cells (HSPCs) in response to systemic inflammation by a combination of cell-autonomous and stroma-dependent signals and leads to their release from bone marrow and migration to periphery. We have previously shown that FZD6 plays a pivotal role in regulating HSPC expansion and long-term maintenance. Now we sought to better understand the underlying mechanisms. Using lipopolysaccharide (LPS)-induced emergency granulopoiesis as a model, we show that failed expansion was intrinsic to FZD6-deficient HSPCs but also required a FZD6-deficient environment. FZD6-deficient HSPCs became more strongly activated, but their mobilization to peripheral blood was impaired and they were more susceptible to inflammatory cell death, leading to enhanced release of pro-inflammatory cytokines in the marrow. These studies indicate that FZD6 has a protective effect in the bone marrow to prevent an overactive inflammatory response and further suggest that mobilization improves HSPC survival during bone marrow inflammation.
Keywords: hematopoietic stem cells, emergency hematopoiesis, WNT signaling, frizzled-6, lipopolysaccharide
Graphical abstract
Highlights
-
•
Hematopoietic FZD6 is required for emergency granulopoiesis
-
•
FZD6 promotes HSPC mobilization from bone marrow to periphery
-
•
FZD6 protects bone marrow HSPCs from death by necroptosis
-
•
Fzd6−/− HSPCs are more susceptible to functional exhaustion upon acute inflammation
WNT signaling plays important roles in HSPC maintenance and expansion. In this article, Heinonen and colleagues show that the WNT/polarity pathway receptor FZD6 enhances HSPC mobilization, suppresses inflammation, and protects HSPCs from inflammation-induced cell death during emergency granulopoiesis. FZD6 could thus be targeted to promote HSPC survival and expansion under myelotoxic stress or after hematopoietic cell transplantation.
Introduction
Emergency hematopoiesis is characterized by the expansion of hematopoietic stem/progenitor cells (HSPCs) and their progeny in response to a combination of signals from their immediate environment, such as inflammatory cytokines and pathogen-associated molecular patterns (Baldridge et al., 2010; Boettcher et al., 2014; Burberry et al., 2014; Haas et al., 2015; Kwak et al., 2015; Manz and Boettcher, 2014). In the prototypical model, Toll-like receptor (TLR)-4 ligation on HSPCs by lipopolysaccharide (LPS) leads to HSPC proliferation, promotes myeloid differentiation, and ultimately results in functional HSPC exhaustion in the presence of a persistent stimulus (Liu et al., 2015; Takizawa et al., 2017). In contrast, HSPC migration to peripheral sites, such as spleen, is not dependent on direct pathogen sensing by the HSPCs but rather results from the secretion of granulocyte colony-stimulating factor (G-CSF) by the endothelial cells in the bone marrow (BM) hematopoietic niche (Boettcher et al., 2014; Burberry et al., 2014; Haas et al., 2015). The concomitant increase in granulocyte differentiation in response to G-CSF and their release to circulation serves to replace these short-lived cells that are being recruited to the site of infection or inflammation. Moreover, BM myeloid cells will also signal back to HSPCs by producing reactive oxygen species that will further stimulate HSPC proliferation and differentiation (Kwak et al., 2015; Zhu et al., 2017).
HSPC activation generally corresponds to changes in WNT signaling activity, with the various intracellular signaling pathways promoting either activation or quiescence (Abidin et al., 2015; Lento et al., 2014; Staal et al., 2016; Sugimura et al., 2012). Different WNT signaling pathways have also been linked to the regulation of inflammation: while the canonical WNT/CTNNB1 (β-catenin) signaling appears to promote the secretion of pro-inflammatory cytokines in response to LPS and to induce lung injury in sepsis (Gatica-Andrades et al., 2017; Sharma et al., 2017), the prototypical non-canonical ligand WNT5A has been shown to stimulate phagocytosis and cytokine secretion by macrophages (Maiti et al., 2012), but also to promote the differentiation of tolerogenic dendritic cells (Oderup et al., 2013; Valencia et al., 2011). Upregulation of G-CSF receptor (Csfr3) expression by HSPCs in response to infection and the activation of emergency granulopoiesis would also appear dependent on CTNNB1-TCF/LEF signaling (Danek et al., 2020). However, the precise role of non-canonical WNT signaling in emergency myelopoiesis is not well established. We have previously shown that the WNT/polarity receptor FZD6 is necessary for efficient HSPC expansion after competitive transplant and in response to LPS (Abidin et al., 2015), but the underlying cellular and molecular mechanisms remained unclear.
We now show here that FZD6 simultaneously protects HSPCs and promotes their mobilization to the periphery by a combination of cell-autonomous and stroma-dependent mechanisms. Fzd6−/− HSPCs display an efficient immediate-early response that cannot be sustained as cells become rapidly exhausted and prone to necroptotic cell death. Furthermore, they mobilize less efficiently to the spleen and generate fewer progeny in situ in the BM. Fzd6−/− BM environment contributes to the inefficient response by producing large quantities of inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin (IL)-β. These results show that hematopoietic FZD6 is required for normal emergency granulopoiesis and lead us to propose that HSPC mobilization may actually promote their survival by removing them from an inflammatory BM microenvironment.
Results
Initial HSPC response to LPS is intact in Fzd6−/− BM
Our previous data showed a decreased expansion of Fzd6−/− CD150+ Lineage− SCA-1+ c-KIThi (LSK) HSPCs in the BM on day 3 after the first injection of LPS (Abidin et al., 2015). This could be explained by defective LPS sensing or lack of HSPC self-renewal, translating into exhaustion, cell differentiation, or cell death. To better discriminate between these possibilities, we analyzed HSPC activation at earlier time points, at 24 and 48 h after the first injection (Figure 1A). There was no difference in the expansion of LSKs, CD150+ CD41+ stem cell-like megakaryocyte progenitors (SL-MkP), or CD150+ CD41− hematopoietic stem cell (HSC)-like LSKs on day 1 (Figure 1B), indicating that the initial LPS sensing was likely not impaired by the absence of FZD6. However, the decline in HSPC numbers following the second injection was much more pronounced in the Fzd6−/− BM, resulting in the overall deficit in CD150+ CD41− LSK number compared with Fzd6+/+ controls (Figure 1B). Interestingly, SL-MkP numbers were not significantly reduced on day 3 (Figure 1B), suggesting functional emergency megakaryopoiesis (Haas et al., 2015). Similar results were also obtained when using CD150 and CD48 to identify CD150+CD48− HSCs and CD48+ multipotent progenitors (MPPs); there was no decrease in the number or proportion of HSCs or MPPs in the Fzd6−/− BM on day 1, but the number of CD150+CD48− HSCs as well as CD150− CD48+ MPP3s were in sharp decline by day 3 in Fzd6−/− mice (Figures 1C and S1). Once more, there was no decrease in the number of megakaryocyte-biased CD150+ CD48+ MPP2s. Together, these results indicate that the Fzd6−/− HSPCs respond to the initial LPS challenge at least as strongly as Fzd6+/+ controls. However, while the second injection stabilizes HSPC numbers in Fzd6+/+ controls, Fzd6−/− cells abruptly decline.
Figure 1.

FZD6-deficiency impairs LPS-induced HSC expansion
(A) Experimental design for LPS-induced emergency granulopoiesis.
(B) Representative flow cytometry data and gating strategy of LPS-treated BM HSPCs of Fzd6+/+ (WT) and Fzd6−/− (KO) mice at various time points. BM cells were first gated on Lineage− (B220− CD3ε− CD11b− GR1− TER119−) and identified according to SCA-1 and c-KIT (CD117) expression. Stem cell-like megakaryocyte progenitors (SL-MkP) were identified as CD41+ CD150+ within the Lineage− c-KIThi SCA-1+ (LSK) population. HSC-like cells were defined as CD150+ CD16/CD32− CD41− LSKs. Numbers on the flow cytometry plots refer to the percentage of cells in total BM (for LSKs) or within the LSK population (for SL-MkP and HSC-like cells). Graphs on the right show the numbers of different subpopulations in the BM (two tibiae + two femora).
(C) Numbers of cells in BM LSK subpopulations as defined by CD150 and CD48 expression. See also Figure S1.
(D) Frequency of cells in different phases of cell cycle as defined by KI-67/Hoechst co-staining at different time points after LPS injection. See also Figure S1.
(E) Representative cell cycle flow cytometry data for CD48− CD150+ HSCs at indicated time points. Numbers in the upper right corners refer to the percentage of cells in each quadrant. All graphs represent mean + SEM, pooled from at least three independent experiments with a total of at least five mice per group. ∗p < 0.05, ∗∗p < 0.005, WT versus KO.
Although most adult HSCs are usually in a quiescent state, the proportion of cycling cells increases in response to myeloablation or the production of inflammatory cytokines, such as seen in the emergency response to LPS (Baldridge et al., 2010; Liu et al., 2015; Pietras et al., 2016; Takizawa et al., 2017; Wilson et al., 2008). The proportion of HSCs in G0 decreased quite significantly in Fzd6+/+ controls as well as in Fzd6−/− mice in response to LPS (Figures 1D and 1E); however, the relative frequency of quiescent HSCs was decreased in the Fzd6−/− BM at 24 and 72 h. There were no significant differences in cell cycle status between Fzd6−/− and Fzd6+/+ CD48+ MPPs (Figures 1D and S1). Together with the decrease in cell numbers, these results point toward an overactivation and potential exhaustion of HSCs rather than inefficient expansion due to impaired LPS sensing.
Fzd6−/− BM HSPCs become functionally exhausted after LPS stimulation
Analysis of HSPC subpopulations during inflammation is hampered by altered expression of surface markers, such as SCA-1, that may result in contamination of HSC and MPP subsets by more committed myeloid progenitor cells (Kanayama et al., 2020). Although our analyses exclude FcγRII/III+ cells from the HSC-like cell population (Figure 1B), we wanted to determine whether Fzd6−/− HSPCs were still able to contribute to hematopoiesis after LPS stimulation using a three-way competitive transplant setting to compare Fzd6−/− and Fzd6+/+ cells in the same recipients (Figure 2A). Fzd6+/+ BM outcompeted unstimulated control cells in the periphery, most likely due to the increased frequency of HSPCs in the graft (Figure 2B). In contrast, even though Fzd6−/− BM cells were provided in excess to compensate for the decline of HSPCs on day 3, Fzd6−/− HSPCs were unable to contribute to peripheral blood reconstitution in short or long term (Figure 2B). It must be noted that Fzd6−/− HSPCs are already at a competitive disadvantage, as seen in PBS-injected controls (Figure 2C) and as we had previously reported (Abidin et al., 2015). However, LPS stimulation accentuated the difference even further, resulting in Fzd6−/− cells being found at 10- to 20-fold lower numbers in the recipient BM compared with Fzd6+/+ donor cells at 16 weeks post transplant (Figure 2C). These results provide further functional support for the idea that Fzd6−/− HSPCs fail to recover from LPS stimulation and become rapidly non-functional.
Figure 2.

LPS-stimulated Fzd6−/− BM HSCs are functionally exhausted
(A) Experimental design.
(B) Peripheral blood analysis of SSChi GR1hi granulocytes. Representative flow cytometry data are shown on the left. Graph on the right shows Fzd6+/+ (WT) and Fzd6−/− (KO) donor contribution for individual mice at different time points.
(C) Donor contribution (top) and number of donor-derived cells (bottom) in the BM for the different subpopulations. Bar graphs represent mean + SEM with five mice per group. ∗∗p < 0.005, WT versus KO.
HSC exhaustion is dependent on Fzd6−/− hematopoietic cells as well as Fzd6−/− stroma
Although repeated exposure to LPS can lead to HSC exhaustion directly via TLR4 expression on HSCs themselves (Liu et al., 2015; Takizawa et al., 2017), emergency granulopoiesis and LPS-induced extramedullary hematopoiesis in the spleen are dependent on non-hematopoietic cells, such as endothelial cells (Boettcher et al., 2012, 2014). Given that Fzd6 expression has been reported in the BM stromal compartment (Lento et al., 2013), we wished to evaluate whether the loss of HSCs in Fzd6−/− BM was due to functional deficiencies in non-hematopoietic cells. We therefore reconstituted irradiated Fzd6+/+ and Fzd6−/− recipient mice with congenic (CD45.1+ CD45.2+) BM cells (Figure 3A) and evaluated their emergency response 16 weeks after transplant. We first confirmed that there was no difference in the proportion of BM progenitor cells (Figure 3B) or peripheral blood cells (Figure 3C) before LPS injection. There was also no difference in the proportion or number of BM HSPCs between Fzd6+/+ and Fzd6−/− hosts on day 3 after the induction of emergency myelopoiesis (Figures 3D and 3E), suggesting that Fzd6−/− stroma was able to sustain both normal and emergency hematopoiesis from WT HSPCs. Although there was a slight decrease in the number of granulocyte-macrophage colony-forming units (CFU-GM) obtained from the BM of Fzd6−/− recipients compared with their Fzd6+/+ counterparts, this difference was not statistically significant (Figure 3F). We also observed a corresponding tendency toward an increase in the number of colonies obtained from the spleen of Fzd6−/− hosts (Figure 3F). The decrease in BM colony formation could thus be due to an increased release of HSPCs to the periphery. Alternatively, Fzd6−/− stroma may suppress differentiation of myeloid-biased progenitors.
Figure 3.

Hematopoietic Fzd6 expression is sufficient to maintain emergency hematopoiesis and HSPC repopulation potential in Fzd6-deficient environment
(A) Experimental design for primary and secondary transplants.
(B) Representative flow cytometry analysis of BM aspirates 16 weeks after transplant, showing the proportion of donor-derived BM HSPCs (mean percentage within donor-derived cells).
(C) Peripheral blood analysis of recipient mice 16 weeks after transplant, showing the proportion of different blood cells within donor-derived cells (overall peripheral blood chimerism was >95% for all recipients).
(D) Numbers of donor-derived HSPCs in the BM on day 3.
(E) Representative flow cytometry analysis of the BM of LPS-injected chimeras on day 3.
(F) Numbers of myeloid colonies derived from BM (top) and spleen (bottom) of LPS-injected chimeras on day 3.
(G) Peripheral blood analysis of recipient mice 16 weeks after secondary transplant, showing the proportion of different blood cells within donor-derived cells (overall peripheral blood chimerism was >90% for all recipients).
(H) Peripheral blood donor chimerism in secondary recipients within GR1hi SSChi granulocytes at different times after transplant.
(I and J) (I) Proportion of various HSPC populations within donor-derived cells and (J) absolute numbers of donor-derived HSPCs in the BM of secondary recipients at 16 weeks post transplant. Data are pooled from two independent transplants, with similar results obtained in a third independent experiment. Each individual dot represents one mouse, and horizontal lines represent sample mean. Bar graphs represent mean +SEM (n = 6 for Fzd6+/+ and n = 8 for Fzd6−/− primary hosts; n = 9 for both groups in secondary transplant). None of the differences were statistically significant.
Given that increased mobilization could lead to the loss of BM HSCs with the ability to reconstitute secondary hosts, we further transplanted BM cells isolated from LPS-injected chimeras into congenic CD45.1+ recipients (Figure 3A). The secondary recipients showed no evidence of differentiation bias induced by Fzd6−/− environment (Figure 3G). Moreover, there was no decrease in donor chimerism within peripheral blood granulocytes, suggesting that BM cells recovered from Fzd6−/− hosts had no major repopulation defects (Figure 3H). Last, we also determined the frequency and number of donor-derived HSPCs in the BM and observed no major difference between cells derived from Fzd6+/+ and Fzd6−/− primary hosts (Figures 3I and 3J). These results demonstrate that the Fzd6−/− environment alone is not sufficient to induce defective HSC expansion and function during LPS-induced emergency hematopoiesis. They rather confirm our previous findings (Abidin et al., 2015), suggesting that the defect is dependent on hematopoietic cells.
To confirm that the Fzd6−/− environment was not required, we generated mixed chimeras (Figure 4A) using an excess of Fzd6−/− hematopoietic cells to overcome their decreased reconstitution efficiency (Abidin et al., 2015). However, we did not see any major differences in BM HSPC frequency or number between Fzd6+/+ and Fzd6−/− donors after LPS injection (Figures 4B and 4C), suggesting that either Fzd6−/− environment also played a major role in the loss of Fzd6−/− HSPCs or modifications in endothelial niches after irradiation and reconstitution altered HSPC behavior (Chen et al., 2019).
Figure 4.

Fzd6-deficient BM environment is required for the loss of Fzd6-deficient HSPCs
(A) Experimental design for mixed BM chimeras.
(B) Representative flow cytometry data of donor-derived HSPCs in the BM of LPS-treated chimeric mice.
(C) Donor contribution (top) and number of donor-derived cells (bottom) in the BM of LPS-treated chimeric mice. Data are pooled from two independent transplants, and shown as mean + SEM (n = 8 for both groups). None of the differences were statistically significant.
FZD6 promotes LPS-induced BM emergency myelopoiesis
One potential explanation for decreased HSC expansion in the Fzd6−/− BM could be their accelerated differentiation. LPS injection induces a rapid release of neutrophil granulocytes and inflammatory LY-6Chi monocytes from the BM, followed by an upregulation of myeloid differentiation programs and a shutdown of B lymphopoiesis (Boettcher et al., 2014; Liu et al., 2015). To evaluate the potential for accelerated myeloid differentiation in Fzd6−/− mice, we determined the number of granulocyte-monocyte progenitor cells (GMPs; CD16/CD32+ c-KIT+ CD41− CD150− Lin−) at different times after LPS injection. There was no difference between Fzd6+/+ and Fzd6−/− BM at steady state (Figures 5A and 5B), as we have previously published (Abidin et al., 2017). Conventional SCA-1− GMPs peaked at 48 h and returned back to baseline levels by 72 h (Figures 5A and 5B); however, the peak numbers tended to remain lower in the absence of FZD6. A similar trend could also be seen for SCA-1+ emergency GMPs, which were still detectable in Fzd6+/+ BM at 72 h, but whose numbers were approximately 3-fold lower in Fzd6−/− individuals (Figures 5A and 5B). The initial release of myeloid cells from the BM was highly similar in both groups, once more suggesting that the initial LPS sensing was intact in the absence of FZD6. However, there was a decrease in granulocyte and monocyte accumulation in the Fzd6−/− spleen (Figures 5C and 5D), and the surge of myeloid differentiation seen at 72 h in Fzd6+/+ BM was absent in Fzd6−/− mice (Figure 5D). This was not due to decreased proliferation of lineage-committed cells (Figure S2) or myeloid-biased MPP3s that include emergency GMPs (Figure 1C). These results demonstrated that the impaired emergency response in Fzd6−/− HSPCs was reflected in their downstream myeloid progeny.
Figure 5.

Decreased emergency myelopoiesis in Fzd6−/− mice
(A) Representative flow cytometry data and gating strategy for BM myeloid progenitor cells. Conventional GMPs were first gated on Lineage− SCA-1− c-KIThi and then identified as CD16/CD32+ CD150− and CD41−. Emergency GMPs (eGMPs) were defined as CD16/CD32+ CD150− CD41− LSKs. Mean percentage for each cell subset is indicated within flow cytometry plots.
(B) Numbers of GMPs and eGMPs in Fzd6+/+ (WT) and Fzd6−/− (KO) mice at various time points. See also Figure S1.
(C) Representative flow cytometry data and gating strategy for myeloid cell subsets in the spleen. The same strategy was used for BM as well. Neutrophil granulocytes were defined as SSChi GR1hi. LY-6Chi CD11b+ monocytes were selected within the “NOT granulocyte” population. Mean percentage for each cell subset is indicated within flow cytometry plots.
(D) Numbers of GR1hi granulocytes and LY-6Chi monocytes in BM and spleen in Fzd6+/+ (WT) and Fzd6−/− (KO) mice at various time points. See also Figure S2. All graphs represent mean + SEM pooled from at least three independent experiments with a total of at least five mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, WT versus KO.
FZD6 is necessary for efficient HSC mobilization in response to G-CSF
LPS-induced emergency granulopoiesis and HSPC mobilization is largely mediated by G-CSF, secreted by endothelial cells in response to TLR4-mediated signals (Boettcher et al., 2012, 2014). Therefore, a deficient emergency response in Fzd6−/− BM could be due to a decrease in G-CSF production or an impaired reaction to G-CSF. Given that the Fzd6−/− environment was able to support emergency hematopoiesis from WT HSPCs, the former possibility seemed unlikely. We rather administered G-CSF intraperitoneally on three consecutive days, and then analyzed HSPC activation and mobilization to evaluate the potential of Fzd6−/− BM cells to respond to G-CSF.
There was no difference in the relative proportion or absolute number of HSPC subsets in the BM between Fzd6+/+ and Fzd6−/− mice (Figures 6A and 6B), suggesting that they were able to respond to G-CSF-mediated activation. However, the proportion of HSCs that were mobilized to the peripheral blood was decreased in Fzd6−/− mice compared with controls (Figures 6C and S3). This difference was even further accentuated in the spleen, where the number of Fzd6−/− HSCs was 2-fold lower after G-CSF treatment (Figure 6B). A similar pattern was also seen for downstream MPPs (Figure 6B) and for myeloid and erythroid colony-forming cells (Figure 6D), whose recruitment to the spleen was significantly reduced in the absence of FZD6. These results indicate that Fzd6−/− HSPCs become normally activated in the BM but are then less efficiently mobilized to the periphery.
Figure 6.

FZD6 promotes G-CSF-induced HSC mobilization to peripheral blood and spleen
(A) Representative flow cytometry data for LSKs and HSPC subsets as identified by CD150 and CD48 staining in the BM of PBS- and G-CSF-treated Fzd6+/+ (WT) and Fzd6−/− (KO) mice. Numbers within panels represent mean percentage in total BM.
(B) Numbers of HSPCs in the BM (top) and spleen (bottom) from PBS- and G-CSF-treated mice.
(C) Relative frequency of HSCs in peripheral blood of PBS- and G-CSF-treated mice. See also Figure S3.
(D) Myeloid colony-forming assay from BM and spleen of G-CSF-treated Fzd6+/+ and Fzd6−/− mice.
(E) Imaging flow cytometry analysis of pyknotic nuclei with condensed chromatin as shown by morphology and DAPI staining intensity. All bar graphs represent mean + SEM with three mice per group for PBS and five per group for G-CSF-treated mice pooled from three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
Inflammation promotes Fzd6−/− HSPC necroptosis and prevents HSPC expansion
Our previous results after BM transplant suggested that increased CTNNB1 activation could promote apoptosis in expanding Fzd6−/− HSPCs (Abidin et al., 2015), and we observed an increase in the frequency of cells with pyknotic nuclei within Fzd6−/− HSPCs after G-CSF treatment (Figure 6E). We thus hypothesized that the decreased accumulation of Fzd6−/− HSPCs could be due to enhanced cell death. We detected no CASP3 activation in HSPCs after LPS injection, independent of genotype (Figure S4). However, pyknotic nuclei are associated not only with apoptosis but also with necroptosis, a form of cell death induced by inflammation. We used nuclear phospho-mixed lineage kinase domain like pseudokinase (MLKL) as readout for early necroptosis (Yoon et al., 2016), and observed an increase in necroptosis in Fzd6−/− HSPCs in the BM but not in the spleen (Figure 7A). Moreover, the frequency of necroptotic HSPCs in the spleen was not increased by LPS injection (Figure 7A), suggesting that BM HSPCs were more susceptible to inflammation-induced cell death.
Figure 7.

Fzd6−/− BM environment is highly inflammatory in response to LPS and promotes HSPC necroptosis
(A) Imaging flow cytometry analysis of phospho-MLKL staining in BM and spleen HSPCs from Fzd6+/+ (WT) and Fzd6−/− (KO) mice. Representative images are shown on top. Graphs represent the frequency of HSPCs with nuclear pMLKL staining.
(B) Analysis of soluble pro-inflammatory factors from BM of LPS-injected mice by protein array. Results are presented as ratio over naive BM. Factors in bold show statistically significant differences between Fzd6+/+ and Fzd6−/−. See also Figure S5.
(C) Quantification of TNF-α and IL-1β from BM of PBS- and LPS-treated mice using ELISA.
(D) c-KIT+ cell numbers from IL-3-dependent colony assays in the presence of BM supernatants. Cells were treated with MLKL-inhibitor (GW806742X) or a pan-caspase inhibitor (Q-VD-Oph). See also Figure S4.
(E) IL-3-dependent colony formation by WT BM progenitors in the presence of supernatants obtained from LPS-stimulated macrophages. Graphs represent mean + SEM from at least three independent experiments with a total of at least five mice per group. ∗p < 0.05, ∗∗p < 0.005, WT versus KO.
The decrease in HSPC survival could be due to HSPC-intrinsic loss of quiescence, as suggested by the cell cycle analysis in Figure 1. An alternative, but not mutually exclusive, explanation for the loss of Fzd6−/− HSPCs could be the presence of myelosuppressive factors in the BM environment (Hockendorf et al., 2016; Pietras et al., 2016; Wagner et al., 2019; Yamashita and Passegue, 2019). To address this question, we analyzed the soluble factors present in freshly harvested BM suspensions from LPS-treated Fzd6+/+ and Fzd6−/− mice by protein arrays (Figures 7B and S5). We divided these factors into three broad categories: (1) chemokines upregulated in both groups, such as CCL2 and G-CSF; (2) cytokines and chemokines, including most myeloid growth factors, that were not significantly altered in either group; and (3) factors that were preferentially upregulated in Fzd6−/− mice. The last group included IL-1, TNF-α, IFN-γ, and related factors (Figures 7B and 7C). To determine the potential contribution of these factors in suppressing HSPC expansion and myeloid differentiation, we supplemented IL-3-containing culture medium with BM plasma from control (BM PBS) or LPS-injected mice (wild-type [WT] LPS, knockout [KO] LPS) and evaluated myeloid differentiation and c-KIT+ cell recovery. There was no difference in the number of Fzd6+/+ c-KIT+ cells recovered from cultures treated with BM plasma from control or LPS-injected mice (Figures 7D and S4). In contrast, when Fzd6−/− cells were cultured with soluble factors derived from LPS-injected mice, c-KIT+ cell recovery was significantly decreased compared with Fzd6−/− cultures exposed to PBS-injected controls, or compared with Fzd6+/+ cells under the same conditions (Figures 7D and S5). Inhibition of MLKL activity fully restored Fzd6−/− c-KIT+ cell numbers (Figure 7D), while a pan-caspase inhibitor provided a milder recovery, suggesting that both necroptosis and apoptosis may be involved. Moreover, while using conditioned medium from LPS-stimulated BM-derived macrophages (BMDMs), we saw that the Fzd6−/− BMDM-conditioned medium was more effective in suppressing colony formation in response to IL-3 (Figure 7E).
Collectively these results suggest that Fzd6−/− HSPCs were more susceptible to necroptosis and that this increased susceptibility to cell death was largely cell autonomous. It did not appear to be due to either overly strong or inefficient activation of CTNNB1-TCF/LEF signaling, as there was no significant difference in CTNNB1 phosphorylation on serine 552 between Fzd6+/+ and Fzd6−/− HSPCs (Figure S4). In contrast, Fzd6−/− HSPCs stained less strongly for phalloidin than their Fzd6+/+ counterparts in LPS-treated BM (Figure S4), suggesting that FZD6 was necessary for actin reorganization, which is one of the hallmarks of non-canonical WNT signaling, and could thus also play a direct role in HSPC motility.
Discussion
Emergency myelopoiesis in acute infections serves to replace short-lived myeloid cells that have been recruited to peripheral tissues to counter the infectious agent. It can be conceptually divided into three parts: (1) mobilization of granulocytes and monocytes from the BM, (2) activation and mobilization of BM HSPCs, and (3) preferential differentiation of HSPCs into myeloid cells. Each part is regulated by a combination of signals from microbial products, immune cells, and non-immune cells. We have examined here the implication of WNT/FZD6 signaling in LPS-induced emergency granulopoiesis. Our results show that Fzd6−/− HSPC expansion, survival, and mobilization to peripheral blood were impaired. The defect was dependent on hematopoietic cells as well as on Fzd6−/− stroma, and it most likely stemmed from HSPC-intrinsic overactivation and defective actin reorganization combined with excessive production of inflammatory cytokines. These results support the hypothesis that non-canonical WNT signaling protects BM HSPCs from an overactive inflammatory response.
The impact of WNT signaling on HSPC proliferation and self-renewal is dose dependent (Luis et al., 2011; Sugimura et al., 2012). Moderate levels of canonical CTNNB1-dependent signaling enhance HSC self-renewal and BM repopulation (Lento et al., 2014; Luis et al., 2011), while sustained CTNNB1 activity promotes myeloid proliferation and differentiation but leads to HSC exhaustion (Hérault et al., 2017; Luis et al., 2011; Scheller et al., 2006). Increased WNT/Ca2+ signaling also results in HSC activation, loss of quiescence, and functional aging (Florian et al., 2013; Sugimura et al., 2012). Our previous data show that FZD6 is a negative regulator of both CTNNB1 and WNT/Ca2+/CDC42/JNK pathways (Abidin et al., 2015). Lack of FZD6 could thus result in loss of quiescence due to a decreased HSC activation threshold. Our results herein indicate that Fzd6−/− HSCs and progenitor cells become readily activated and enter cell cycle after LPS stimulation, but their capacity to maintain the response is impaired, and they become prematurely exhausted. Fzd6−/− HSPCs were more likely to express nuclear phospho-MLKL, a sign of early necroptosis, and MLKL inhibition rescued Fzd6−/− c-KIT+ cell recovery in culture. Necroptosis leads to the release of cellular components known as death-associated molecular patterns. Their recognition by immune cells stimulates the release of inflammatory cytokines, such as TNF-α and IL-1β, which may then further contribute to decreased HSPC function and ultimately lead to BM failure (Hockendorf et al., 2016; Pietras et al., 2016; Wagner et al., 2019; Yamashita and Passegue, 2019). The combination of increased inflammation and increased cell death in the Fzd6−/− BM is thus likely to form a feedforward loop that leads to the loss of HSCs with full repopulation capacity. Increased basal JNK activity in Fzd6−/− cells (Abidin et al., 2015) could further promote both necroptosis and LPS-induced inflammatory response (Cao et al., 2018). Although we did not detect caspase-3 activation in HSPCs from LPS-injected mice, treatment with a pan-caspase inhibitor also partially restored Fzd6−/− c-KIT+ cell numbers in culture. Apoptosis and necroptosis may thus both contribute to the loss of Fzd6−/− HSPCs in an inflammatory environment.
Circulating HSPCs have been suggested to patrol extramedullary tissues and have the capacity to either settle down to differentiate or return to the BM (Massberg et al., 2007). Upon stimulation with LPS, the increase in BM G-CSF concentrations together with the concomitant decrease in CXCL12 will accentuate HSPC and granulocyte release into circulation (Boettcher et al., 2014), but the functional importance of HSPC mobilization in emergency hematopoiesis is still unknown. Fzd6−/− BM HSPCs were maintained after G-CSF administration, but their decreased accumulation in peripheral blood and spleen suggests that their mobilization was impaired. This mobilization defect could be HSPC intrinsic or dependent on the environment. We detected no deficit in G-CSF production, and our results with chimeric mice suggest that Fzd6−/− radioresistant cells promote mobilization at least as efficiently as Fzd6+/+ BM. The number of myeloid colony-forming cells recovered in the spleen was slightly higher in Fzd6−/− mice reconstituted with WT BM compared with control chimeras, which supports the capacity of Fzd6−/− stromal cells to sustain mobilization. The initial release of Fzd6−/− myeloid cells was also normal on day 1. Collectively, these observations indicate that the defect is restricted to Fzd6−/− HSPCs, although we cannot exclude the potential contribution of an intact Fzd6−/− stroma or BM cells of hematopoietic origin. Fzd6 is most strongly expressed in HSPCs, with relatively low levels in most mature immune cells. However, although Fzd6−/− hematopoietic cells were not sufficient to induce HSPC death in chimeric mice, the presence of increased amounts pro-inflammatory cytokines, such as TNF and IL-1β, in Fzd6−/− BM suggest that they also contribute to the feedforward mechanism.
WNT5a/CDC42-mediated F-actin reorganization regulates HSPC adhesion and migration toward CXCL12 (Schreck et al., 2017), and we have previously reported enhanced CDC42 clustering in Fzd6−/− HSPCs (Abidin et al., 2015). Fzd6−/− cells displayed decreased phalloidin staining in response to LPS in vivo, suggesting that their actin reorganization was altered. It is thus conceivable that FZD6 directly influences HSPC mobilization by regulating their association with BM niches. Although we cannot exclude the contribution of cell death to the decreased numbers of HSPCs found in the periphery, the fact that we do not see any decrease in the number of BM HSPCs between G-CSF-treated Fzd6−/− and Fzd6+/+ mice supports the thesis of a true mobilization defect. There is no accumulation of HSPCs in the BM of G-CSF-treated mice, indicating that they are mostly released to the circulation and then migrate to the spleen. If Fzd6−/− HSPCs were to simply undergo cell death without any mobilization defect, we would expect to see at least a tendency toward fewer cells in the BM. Instead, we observe comparable colony formation and cell numbers. Decreased mobilization of Fzd6−/− HSPCs during LPS-induced inflammation could thus increase their exposure to pro-inflammatory cytokines and reactive oxygen species present in the BM (Ito et al., 2006; Matatall et al., 2016; Pietras et al., 2016; Zhu et al., 2017) and contribute to their decreased expansion, function, and survival. Interestingly, HSPC survival in the spleen was much better than in the BM, irrespective of genotype, suggesting that HSPC mobilization during emergency hematopoiesis directly contributes to their survival.
The overall role of WNT signaling in the polarization of the immune response depends on the model system as well as the cell of origin. For example, WNT5a has been shown to enhance pro-inflammatory cytokine secretion by peritoneal macrophages in vitro (Maiti et al., 2012), but its reported in vivo effects vary from tolerogenic to bactericidal (Bergenfelz et al., 2012; Jati et al., 2018; Valencia et al., 2011). In contrast, WNT4, another putative ligand for FZD6 (Heinonen et al., 2011), has been linked to type 1 differentiation in dendritic cells (Hung et al., 2019), but its expression protects BM from age-associated or LPS-induced inflammation (Hetu-Arbour et al., 2021; Yu et al., 2014). Our results suggest that FZD6 is more likely to play a tolerogenic role, at least in the context of sterile, LPS-induced inflammation, and pharmacological activation of FZD6 could thus be an interesting option for limiting systemic inflammation. However, response to LPS does not predict susceptibility to infection (Fensterheim et al., 2017), and it would be necessary to verify the importance of FZD6 signaling in sepsis, for example, by using infectious models.
In summary, we have shown here that FZD6 not only protects HSPCs during LPS-induced emergency hematopoiesis from inflammation-induced cell death but also enhances their mobilization to the periphery. Collectively these data are in line with previous results demonstrating that non-canonical WNT signaling protects BM HSPCs from undue activation. They further prompt us to hypothesize that mobilization may serve to promote HSPC survival during BM inflammation.
Experimental procedures
Mice
C57BL/6 (B6; CD45.2+) and B6.SJL-PtprcaPep3b/BoyJ (Ly5a) (B6.SJL; CD45.1+) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Fzd6−/− mice were first backcrossed onto C57BL/6 background and maintained as Fzd6+/− intercrosses as previously detailed (Abidin et al., 2015). Mice were kept under specific pathogen-free conditions in sterile ventilated racks at the animal facility of Institut National de la Recherche Scientifique (INRS). Sex-matched Fzd6+/+ littermates were used as controls. All mice used for the experiments were between 7 and 12 weeks of age. All procedures involving animals were done according to the Canadian Council on Animal Care guidelines and approved by the Comité institutionnel de protection des animaux of INRS.
Transplantation assays
Total BM cells were injected intravenously via the lateral tail vein into lethally irradiated (two times 450 rad) congenic recipient mice. For secondary transplants, 5 × 106 total BM cells from pooled primary recipients were injected into lethally irradiated B6.SJL (CD45.1+) secondary recipients. Blood was collected from the mandibular vein every 4 weeks to determine peripheral donor chimerism by flow cytometry. BM and spleen reconstitution were evaluated 16–20 weeks after transplant. Further details of the various transplant assays can be found in the figures.
LPS and G-CSF injections
Mice were injected intraperitoneally with γ-irradiated LPS (Escherichia coli 0111:B4; Sigma-Aldrich, Oakville, ON, Canada) twice with a 48-h interval at a dose of 1 mg/kg body weight (Fzd6−/− and Fzd6+/+ mice) or 0.5 mg/kg body weight (chimeras) per injection. Mice were euthanized at indicated time points with CO2 and their BM and spleen harvested for analysis.
Recombinant murine G-CSF (Stem Cell Technologies, Vancouver, BC, Canada) was injected intraperitoneally for three consecutive days at a dose of 25 μg per mouse per day. Peripheral blood was collected 24 h after last injection by cardiac puncture under isoflurane-induced anesthesia into EDTA tubes. Mice were then euthanized and their BM and spleen harvested for analysis.
Flow cytometry
BM was harvested by flushing femora and tibiae with sterile PBS using a 25-gauge needle to obtain a single cell suspension. Spleen was ground in PBS with a sterile plunger and cell suspension passed through a 100-μm filter mesh. PBS was supplemented with 0.1% BSA and 0.5 mM EDTA for flow cytometry staining. See Table S1 for a complete list of antibodies. For intracellular staining, surface-stained cells were fixed and permeabilized using the Foxp3 staining kit (eBioscience, San Diego, CA) and then incubated with appropriate antibodies. For cell cycle analysis, cells were first incubated for 30 min at 37°C with Hoechst #33342 (Sigma-Aldrich) in DMEM supplemented with 10% Premium FBS (Wisent Bioproducts, St-Bruno, QC, Canada) and 1 mM HEPES (Life Technologies, Burlington, ON, Canada), followed by staining with surface antibodies and intracellular anti-Ki67 as described above. Samples were acquired with a four-laser LSR Fortessa flow cytometer (BD Biosciences, Mountain View, CA) and analyzed using BD FACS Diva software (BD Biosciences) or FlowJo (for histogram overlays; Tree Star).
Imaging flow cytometry
BM cells were first enriched for HSPCs using EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit (catalog, 19856, Stem Cell Technologies, Vancouver, BC), and then stained with surface antibodies, after which stained cells were fixed and permeabilized as described above before staining with anti-phospho-MLKL (Abcam, Cambridge, UK). Finally, cells were stained with DAPI (Life Technologies), and image acquisition was performed with Amnis ImageStream Mark II imaging flow cytometer (Luminex, Austin, TX). Raw data were analyzed with IDEAS v6.1 software for co-localization.
BM-derived macrophage cultures
BM suspension was prepared in sterile HBSS (Life Technologies) and 1.5 × 106 cells were plated in 10 mL of DMEM (Life Technologies) supplemented with 10% Premium FBS and 20% L929-conditioned medium in non-adherent Petri dishes as previously described (Heinonen et al., 2006). The cultures were incubated at 37°C, 5% CO2 for 7 days with a full medium change on day 4. Macrophages were harvested with PBS/EDTA on day 7, seeded at 1.2–1.5 × 106 cells/well in DMEM/10% FBS in a six-well plate, and stimulated with 100 ng/mL LPS for 24 h. Supernatants were collected by centrifugation and stored at −80°C until use.
Colony-forming unit assays
Freshly isolated BM cells were diluted in Iscove's modified Dulbecco's medium (IMDM) (Life Technologies) containing 10% Premium FBS, and seeded into 35-mm non-adherent Petri dishes in semi-solid methylcellulose medium at a concentration of 104 cell/dish. Dishes were incubated at 37°C, 5% CO2 for 7–10 days, and colonies were counted based on morphology under an inverted microscope.
Methocult GF M3434 (Stem Cell Technologies) was used for standard myeloerythroid CFU assays. For growth inhibition and to evaluate necroptosis and apoptosis, base methylcellulose medium (Methocult GF M3231; Stem Cell Technologies) was supplemented with 10 ng/mL recombinant IL-3 (Peprotech, Rocky Hill, NJ) and 20% conditioned culture medium or BM supernatant collected from Fzd6−/− or Fzd6+/+ mice as indicated. Cultures were further supplemented with 100 ng/mL LPS, 100 ng/mL TNFα (Peprotech, Rocky Hill, NJ), 100 nM GW806742X, 10 μM Q-VD-OPh, or 500 nM BV6 Calbiochem (Millipore Sigma) as indicated. Cells were recovered by diluting methylcellulose in PBS and analyzed by flow cytometry on day 10.
BM cytokine/chemokine analysis
BM supernatant was collected by harvesting cells from both hind legs in 2 mL of sterile PBS. Cells were removed by centrifugation, and the supernatant aliquoted, snap-frozen, and stored at −80°C until use. Conditioned culture medium from LPS-treated macrophages was collected and stored at −80°C until use. Supernatants were pooled from at least four mice per sample and analyzed using a membrane-based proteome profiler mouse cytokine/chemokine array kit (R&D Systems). Array images were further analyzed using the NIH ImageJ image analysis software. Samples were normalized by subtracting pixel intensities from negative controls, and the fold changes for treated mice were determined as a ratio over untreated mice of the same genotype. TNF-α and IL-1β concentration in BM supernatant was determined using respective mouse cytokine ELISA kits (R&D system) following the manufacturer’s procedures.
Statistical analysis
Two-tailed Student’s t test was used to compare Fzd6−/− and Fzd6+/+ mice unless otherwise specified. p value <0.05 was considered significant.
Author contributions
Conceptualization, B.M.A. and K.M.H.; methodology, B.M.A. and K.M.H.; validation and formal analysis, N.H.T., B.M.A., and K.M.H.; investigation, N.H.T. and B.M.A.; writing, K.M.H.; visualization, N.H.T., B.M.A., and K.M.H.; supervision and funding, K.M.H.
Acknowledgments
We are grateful to Roxann Hétu-Arbour and the staff of the LNBE animal care facility for technical assistance. This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery grants #RGPIN-419226-2012 and #RGPIN-2018-05258), Canadian Institutes of Health Research (CIHR Project grant PJT-148614), the Fonds de Recherche du Québec - Santé (grant #32598), and the Canada Foundation for Innovation (CFI Leaders Fund #31377). K.M.H. is a Chercheur-Boursier Junior of the Fonds de recherche du Québec – Santé.
Conflict of interest
The authors declare no competing interests.
Published: September 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2022.08.004.
Supplemental information
References
- Abidin B.M., Hammami A., Stäger S., Heinonen K.M. Infection-adapted emergency hematopoiesis promotes visceral leishmaniasis. PLoS Pathog. 2017;13:e1006422. doi: 10.1371/journal.ppat.1006422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abidin B.M., Owusu Kwarteng E., Heinonen K.M. Frizzled-6 regulates hematopoietic stem/progenitor cell survival and self-renewal. J. Immunol. 2015;195:2168–2176. doi: 10.4049/jimmunol.1403213. [DOI] [PubMed] [Google Scholar]
- Baldridge M.T., King K.Y., Boles N.C., Weksberg D.C., Goodell M.A. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465:793–797. doi: 10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergenfelz C., Medrek C., Ekström E., Jirström K., Janols H., Wullt M., Bredberg A., Leandersson K. Wnt5a induces a tolerogenic phenotype of macrophages in sepsis and breast cancer patients. J. Immunol. 2012;188:5448–5458. doi: 10.4049/jimmunol.1103378. [DOI] [PubMed] [Google Scholar]
- Boettcher S., Gerosa R.C., Radpour R., Bauer J., Ampenberger F., Heikenwalder M., Kopf M., Manz M.G. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. 2014;124:1393–1403. doi: 10.1182/blood-2014-04-570762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher S., Ziegler P., Schmid M.A., Takizawa H., van Rooijen N., Kopf M., Heikenwalder M., Manz M.G. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J. Immunol. 2012;188:5824–5828. doi: 10.4049/jimmunol.1103253. [DOI] [PubMed] [Google Scholar]
- Burberry A., Zeng M.Y., Ding L., Wicks I., Inohara N., Morrison S.J., Núñez G. Infection mobilizes hematopoietic stem cells through cooperative NOD-like receptor and Toll-like receptor signaling. Cell Host Microbe. 2014;15:779–791. doi: 10.1016/j.chom.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M., Chen F., Xie N., Cao M.Y., Chen P., Lou Q., Zhao Y., He C., Zhang S., Song X., et al. c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis. 2018;9:1140. doi: 10.1038/s41419-018-1189-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Liu Y., Jeong H.W., Stehling M., Dinh V.V., Zhou B., Adams R.H. Apelin(+) endothelial niche cells control hematopoiesis and mediate vascular regeneration after myeloablative injury. Cell Stem Cell. 2019;25:768–783.e6. doi: 10.1016/j.stem.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danek P., Kardosova M., Janeckova L., Karkoulia E., Vanickova K., Fabisik M., Lozano-Asencio C., Benoukraf T., Tirado-Magallanes R., Zhou Q., et al. beta-Catenin-TCF/LEF signaling promotes steady-state and emergency granulopoiesis via G-CSF receptor upregulation. Blood. 2020;136:2574–2587. doi: 10.1182/blood.2019004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterheim B.A., Guo Y., Sherwood E.R., Bohannon J.K. The cytokine response to lipopolysaccharide does not predict the host response to infection. J. Immunol. 2017;198:3264–3273. doi: 10.4049/jimmunol.1602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian M.C., Nattamai K.J., Dörr K., Marka G., Uberle B., Vas V., Eckl C., Andrä I., Schiemann M., Oostendorp R.A.J., et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503:392–396. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica-Andrades M., Vagenas D., Kling J., Nguyen T.T.K., Benham H., Thomas R., Körner H., Venkatesh B., Cohen J., Blumenthal A. WNT ligands contribute to the immune response during septic shock and amplify endotoxemia-driven inflammation in mice. Blood Adv. 2017;1:1274–1286. doi: 10.1182/bloodadvances.2017006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S., Hansson J., Klimmeck D., Loeffler D., Velten L., Uckelmann H., Wurzer S., Prendergast Á.M., Schnell A., Hexel K., et al. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. 2015;17:422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Heinonen K.M., Dubé N., Bourdeau A., Lapp W.S., Tremblay M.L. Protein tyrosine phosphatase 1B negatively regulates macrophage development through CSF-1 signaling. Proc. Natl. Acad. Sci. USA. 2006;103:2776–2781. doi: 10.1073/pnas.0508563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen K.M., Vanegas J.R., Lew D., Krosl J., Perreault C. Wnt4 enhances murine hematopoietic progenitor cell expansion through a planar cell polarity-like pathway. PLoS One. 2011;6:e19279. doi: 10.1371/journal.pone.0019279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hérault A., Binnewies M., Leong S., Calero-Nieto F.J., Zhang S.Y., Kang Y.-A., Wang X., Pietras E.M., Chu S.H., Barry-Holson K., et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature. 2017;544:53–58. doi: 10.1038/nature21693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hétu-Arbour R., Tlili M., Bandeira Ferreira F.L., Abidin B.M., Kwarteng E.O., Heinonen K.M. Cell-intrinsic Wnt4 promotes hematopoietic stem and progenitor cell self-renewal. Stem Cell. 2021;39:1207–1220. doi: 10.1002/stem.3385. [DOI] [PubMed] [Google Scholar]
- Höckendorf U., Yabal M., Herold T., Munkhbaatar E., Rott S., Jilg S., Kauschinger J., Magnani G., Reisinger F., Heuser M., et al. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell. 2016;30:75–91. doi: 10.1016/j.ccell.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Hung L.Y., Johnson J.L., Ji Y., Christian D.A., Herbine K.R., Pastore C.F., Herbert D.R. Cell-intrinsic Wnt4 influences conventional dendritic cell fate determination to suppress type 2 immunity. J. Immunol. 2019;203:511–519. doi: 10.4049/jimmunol.1900363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Hirao A., Arai F., Takubo K., Matsuoka S., Miyamoto K., Ohmura M., Naka K., Hosokawa K., Ikeda Y., Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- Jati S., Kundu S., Chakraborty A., Mahata S.K., Nizet V., Sen M. Wnt5A signaling promotes defense against bacterial pathogens by activating a host autophagy circuit. Front. Immunol. 2018;9:679. doi: 10.3389/fimmu.2018.00679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama M., Izumi Y., Yamauchi Y., Kuroda S., Shin T., Ishikawa S., Sato T., Kajita M., Ohteki T. CD86-based analysis enables observation of bona fide hematopoietic responses. Blood. 2020;136:1144–1154. doi: 10.1182/blood.2020004923. [DOI] [PubMed] [Google Scholar]
- Kwak H.J., Liu P., Bajrami B., Xu Y., Park S.Y., Nombela-Arrieta C., Mondal S., Sun Y., Zhu H., Chai L., et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity. 2015;42:159–171. doi: 10.1016/j.immuni.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lento W., Congdon K., Voermans C., Kritzik M., Reya T. Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb. Perspect. Biol. 2013;5:a008011. doi: 10.1101/cshperspect.a008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lento W., Ito T., Zhao C., Harris J.R., Huang W., Jiang C., Owzar K., Piryani S., Racioppi L., Chao N., Reya T. Loss of beta-catenin triggers oxidative stress and impairs hematopoietic regeneration. Genes Dev. 2014;28:995–1004. doi: 10.1101/gad.231944.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A., Wang Y., Ding Y., Baez I., Payne K.J., Borghesi L. Cutting Edge: hematopoietic stem cell expansion and common lymphoid progenitor depletion require hematopoietic-derived, cell-autonomous TLR4 in a model of chronic endotoxin. J. Immunol. 2015;195:2524–2528. doi: 10.4049/jimmunol.1501231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luis T.C., Naber B.A.E., Roozen P.P.C., Brugman M.H., de Haas E.F.E., Ghazvini M., Fibbe W.E., van Dongen J.J.M., Fodde R., Staal F.J.T. Canonical Wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell. 2011;9:345–356. doi: 10.1016/j.stem.2011.07.017. [DOI] [PubMed] [Google Scholar]
- Maiti G., Naskar D., Sen M. The Wingless homolog Wnt5a stimulates phagocytosis but not bacterial killing. Proc. Natl. Acad. Sci. USA. 2012;109:16600–16605. doi: 10.1073/pnas.1207789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manz M.G., Boettcher S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- Massberg S., Schaerli P., Knezevic-Maramica I., Köllnberger M., Tubo N., Moseman E.A., Huff I.V., Junt T., Wagers A.J., Mazo I.B., von Andrian U.H. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008. doi: 10.1016/j.cell.2007.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matatall K.A., Jeong M., Chen S., Sun D., Chen F., Mo Q., Kimmel M., King K.Y. Chronic infection depletes hematopoietic stem cells through stress-induced terminal differentiation. Cell Rep. 2016;17:2584–2595. doi: 10.1016/j.celrep.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderup C., LaJevic M., Butcher E.C. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J. Immunol. 2013;190:6126–6134. doi: 10.4049/jimmunol.1203002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras E.M., Mirantes-Barbeito C., Fong S., Loeffler D., Kovtonyuk L.V., Zhang S., Lakshminarasimhan R., Chin C.P., Techner J.M., Will B., et al. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat. Cell Biol. 2016;18:607–618. doi: 10.1038/ncb3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller M., Huelsken J., Rosenbauer F., Taketo M.M., Birchmeier W., Tenen D.G., Leutz A. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat. Immunol. 2006;7:1037–1047. doi: 10.1038/ni1387. [DOI] [PubMed] [Google Scholar]
- Schreck C., Istvánffy R., Ziegenhain C., Sippenauer T., Ruf F., Henkel L., Gärtner F., Vieth B., Florian M.C., Mende N., et al. Niche WNT5A regulates the actin cytoskeleton during regeneration of hematopoietic stem cells. J. Exp. Med. 2017;214:165–181. doi: 10.1084/jem.20151414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A., Yang W.L., Ochani M., Wang P. Mitigation of sepsis-induced inflammatory responses and organ injury through targeting Wnt/beta-catenin signaling. Sci. Rep. 2017;7:9235. doi: 10.1038/s41598-017-08711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal F.J., Chhatta A., Mikkers H. Caught in a Wnt storm: complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016;44:451–457. doi: 10.1016/j.exphem.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Sugimura R., He X.C., Venkatraman A., Arai F., Box A., Semerad C., Haug J.S., Peng L., Zhong X.B., Suda T., Li L. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150:351–365. doi: 10.1016/j.cell.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa H., Fritsch K., Kovtonyuk L.V., Saito Y., Yakkala C., Jacobs K., Ahuja A.K., Lopes M., Hausmann A., Hardt W.D., et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell. 2017;21:225–240.e5. doi: 10.1016/j.stem.2017.06.013. [DOI] [PubMed] [Google Scholar]
- Valencia J., Hernández-López C., Martínez V.G., Hidalgo L., Zapata A.G., Vicente Á., Varas A., Sacedón R. Wnt5a skews dendritic cell differentiation to an unconventional phenotype with tolerogenic features. J. Immunol. 2011;187:4129–4139. doi: 10.4049/jimmunol.1101243. [DOI] [PubMed] [Google Scholar]
- Wagner P.N., Shi Q., Salisbury-Ruf C.T., Zou J., Savona M.R., Fedoriw Y., Zinkel S.S. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood. 2019;133:107–120. doi: 10.1182/blood-2018-05-847335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E., et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Yamashita M., Passegué E. TNF-alpha coordinates hematopoietic stem cell survival and myeloid regeneration. Cell Stem Cell. 2019;25:357–372.e7. doi: 10.1016/j.stem.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S., Bogdanov K., Kovalenko A., Wallach D. Necroptosis is preceded by nuclear translocation of the signaling proteins that induce it. Cell Death Differ. 2016;23:253–260. doi: 10.1038/cdd.2015.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., Chang J., Liu Y., Li J., Kevork K., Al-Hezaimi K., Graves D.T., Park N.H., Wang C.Y. Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat. Med. 2014;20:1009–1017. doi: 10.1038/nm.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Kwak H.J., Liu P., Bajrami B., Xu Y., Park S.Y., Nombela-Arrieta C., Mondal S., Kambara H., Yu H., et al. Reactive oxygen species-producing myeloid cells act as a bone marrow niche for sterile inflammation-induced reactive granulopoiesis. J. Immunol. 2017;198:2854–2864. doi: 10.4049/jimmunol.1602006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.