

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), emerged in December 2019 in Wuhan, China, and rapidly spread throughout the world, threatening global public health. An animal model is a valuable and a crucial tool that allows understanding of nature in the pathogenesis of SARS-CoV-2 and its associated COVID-19 disease. Here we introduce detailed protocols of SARS-CoV-2 infection and COVID-19 disease using C57BL/6 (B6) transgenic mice expressing the human angiotensin-converting enzyme 2 (hACE2) from the human cytokeratin 18 promoter (K18 hACE2). To mimic natural SARS-CoV-2 infection, K18 hACE2 transgenic mice are infected intranasally under anesthesia. Upon infection, viral pathogenesis is determined by monitoring changes in body weight (morbidity) and monitoring survival (mortality), cytokine/chemokine responses, gross-lung pathology, histopathology, and viral replication in tissues. The presence of the virus and viral replication is evaluated by immunohistochemistry (IHC) and viral titrations, respectively, from the upper (nasal turbinate) and the lower (lungs) respiratory tracts, and nervous system (brain). Also, the immune response to SARS-CoV-2 infection is measured by cytokine/chemokine enzyme-linked immunosorbent assay (ELISA) from lung, spleen and brain homogenates to characterize the cytokine storm that hallmarks as one of the major causes of death caused by SARS-CoV-2 infection. This small rodent animal model based on the use of K18 hACE2 transgenic mice represents an excellent option to understand the pathogenicity of natural SARS-CoV-2 strains and its recently described Variants of Concern (VoC), and will be applicable to the identification and characterization of prophylactic (vaccine) and therapeutic (antiviral and/or neutralizing monoclonal antibodies) strategies for the prevention or treatment of SARS-CoV-2 infection or its associated COVID-19 disease.
Keywords: COVID-19, SARS-CoV-2, hACE2, Mouse model, K18 hACE2 transgenic mice
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the causative agent of coronavirus disease 2019 (COVID-19) which is a severe respiratory disease that first emerged in Wuhan, China, late in 2019 leading to a major threat to public health and socioeconomic activities worldwide of an unprecedented magnitude since the “Spanish flu” pandemic in 1918/1919. According to the World Health Organization (WHO), by the end of 2021, more than 272 million infections and over 5.3 million deaths were identified, with a fatality rate of approximately 1.9 % (http://covid19.who.int). Infected people show clinical manifestations such as fever, cough, shortness of breath and fatigue. Of those, people that succumbed to the infection develop acute respiratory distress syndrome (ARDS), also known as cytokine release syndrome (CRS), or cytokine storm, a major feature of the pathogenesis of COVID-19 [1, 2]. The severe morbidity and mortality of SARS-CoV-2 made it inevitable to develop effective and reliable animal models that enable understanding of the pathogenesis of SARS-CoV-2 and to evaluate prophylactic and therapeutic approaches such as vaccines, antivirals and neutralizing monoclonal antibodies for the efficient treatment of SARS-CoV-2 infection and/or associated COVID-19 disease.
During the previous SARS coronavirus (SARS-CoV) outbreak in 2002–2003, several animal models were developed using wild-type (WT) C57BL/6 (B6) mice [3], young-aged [4] and/or old-aged [5, 6] BALB/C mice, golden Syrian hamsters [7], transgenic mice expressing the human angiotensin converting enzyme 2 (hACE2) from the human cytokeratin 18 promoter (K18 hACE2 transgenic mice) [8, 9], cats [10], and ferrets [10, 11]. However, hamster and cat models did not show mortality [7], and studies using old-aged WT mice or ferrets had reproducibility problems [10, 11]. A mouse-adapted SARS-CoV (MA-15) was also developed but it took a long time for the in vivo passaging in mice, and potential concerns in virus virulence were risen [12]. Efforts have been also directed to establish a SARS-CoV-2 mouse-adapted (MA) strains [12, 13], but concerns also exist that it may not reproduce SARS-CoV-2 infection in humans [12]. Upon the establishment that both SARS-CoV and SARS-CoV-2 use hACE2 as an entry receptor, attention was given to B6.Cg-Tg(K18-ACE2) 2Prlmn/J (K18 hACE2) transgenic mice, which previously showed 100% fatality for SARS-CoV [8]. It is now established by our group and others that the K18 hACE2 transgenic mouse model can be infected with SARS-CoV-2, mimicking clinical symptoms to the ones observed in infected people developing severe COVID-19 disease (e.g., weight loss, rapid breathing, hunched posture, and inactivity) [14–17]. Additionally, SARS-CoV-2 infection in this model results in pathology lesions and inflammation in the lungs, as well as with the chemokine/cytokine storm observed in humans infected with SARS-CoV-2, and viral replication in the upper and lower respiratory tract, brain, gut and heart [14]. Thus, the K18 hACE2 transgenic mouse model represents an excellent small animal model to interrogate vaccines and/or antivirals for the prophylactic and/or therapeutic treatment, respectively, of SARS-CoV-2 infection. Moreover, K18 hACE2 transgenic mice represent an excellent option for the high throughput screening of patient convalescent plasma, monoclonal antibodies, and other biologicals for the prevention and/or treatment of SARS-CoV-2 infection, including recently identified Variants of Concern (VoC).
Here we present a detailed protocol defining how to use the K18 hACE2 transgenic mouse model for SARS-CoV-2 infection. Defining parameters such as morbidity (bodyweight loss) and mortality (survival), viral titers, cytokine/chemokine storms, histopathology, and virus tissue tropisms by immunohistochemistry (IHC). In brief, bodyweight of K18 hACE2 transgenic mice is measured and mice are then infected intranasally under isoflurane anesthesia (Fig. 1a). The body weight of infected mice, as well as control mock-infected animals, is recorded daily to determine the morbidity and survival rates (Fig. 1b). At 2 and 4 days postinfection (dpi), mice are euthanized and necropsied to collect nasal turbinate, lungs, spleen and brain (Fig. 1c). Collected lungs are analyzed for gross-pathology, homogenized for viral titration and determining immune responses, and fixed for histopathology and IHC (Fig. 1c). SARS-CoV-2 infected K18 hACE2 transgenic mice decrease their body weight progressively showing 100% fatality between 5 to 6 dpi (Fig. 2a, b, respectively). Gross-lung pathologic changes become severe over time (Fig. 3a, b), where viral titers peak by 2 dpi in the upper and lower respiratory tract (nasal turbinate and lungs, respectively), while the brain shows higher viral titers at 4 and 6 dpi (Fig. 3c). Histopathology analyses demonstrate inflammation associated with SARS-CoV-2 infection. SARS-CoV-2 tropism for specific tissues and locations is confirmed by IHC (Fig. 4). In terms of an immune response, SARS-CoV-2 infection in K18 ACE2 transgenic mice triggers a relatively localized cytokine storm at 2 dpi in the lungs, which is not apparent in the spleen (as an indicator of systemic inflammation). TH1, TH2, and TH17 cytokine responses are increased in the brain at 4 dpi, and these appear to be sex-dependent toward TH2 in females, which may contribute to the delayed viral spread in their nervous system. Similarly, a local and systemic chemokine storm is also observed, mainly driven by an increase in RANTES/CCL5 and IP10/CXCL10 in lungs (at 2 and 4 dpi) and spleen (IP10/CXCL10 at 2 dpi), while the same chemokines are significantly increased in the brain at 4 dpi when compared to mock-infected control mice (Fig. 5).
Fig. 1.
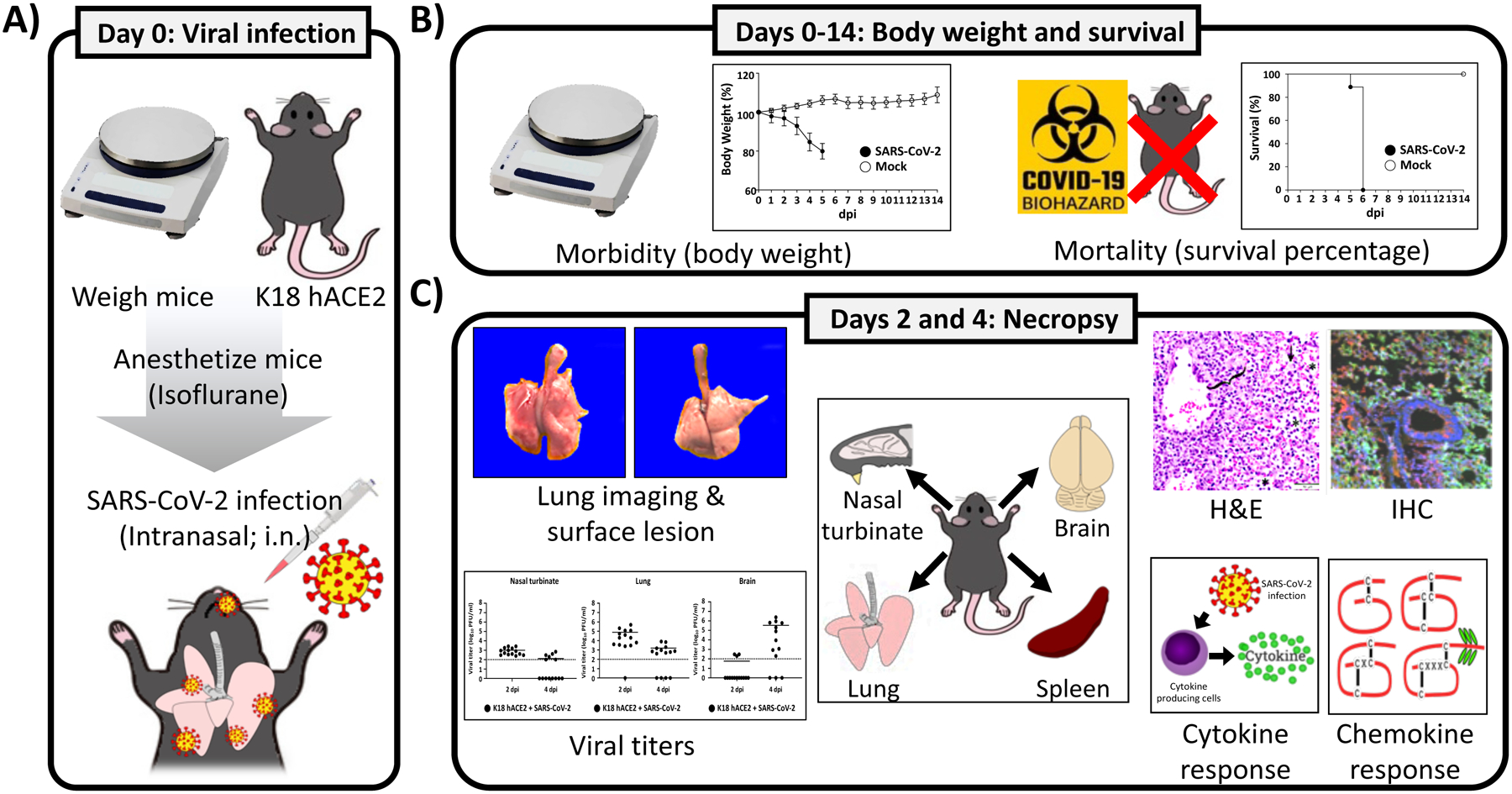
Schematic experimental procedures for SARS-CoV-2 infection in K18 hACE2 transgenic mice and parameters to measure. (a) Viral infection: K18 hACE2 transgenic mice are weighed and anesthetized using isoflurane before intranasal (i.n.) infection with 105 PFU/ mouse of SARS-CoV-2, WA-1/US strain. (b) Morbidity and mortality: From 0 to 14 dpi, mice are weighed daily to evaluate changes in body weight loss (morbidity) and mortality (survival percentage). (c) Organ lesions, viral titration, histology and cytokine changes: At 2 and 4 dpi, SARS-CoV-2 infected K18 hACE2 transgenic mice are sacrificed and internal organs such as nasal turbinate, lung, spleen, and brain are collected for gross pathology changes, viral titers, histopathology and IHC, and cytokine/chemokine analysis. All experiments to characterize SARS-CoV-2 in the K18 hACE2 transgenic mouse model are performed in ABSL3 laboratories. All experiments with SARS-CoV-2 in K18 hACE2 transgenic mice discussed are approved by the IBC and IACUC committees at Texas Biomedical Research Institute. dpi: days postinfection
Fig. 2.
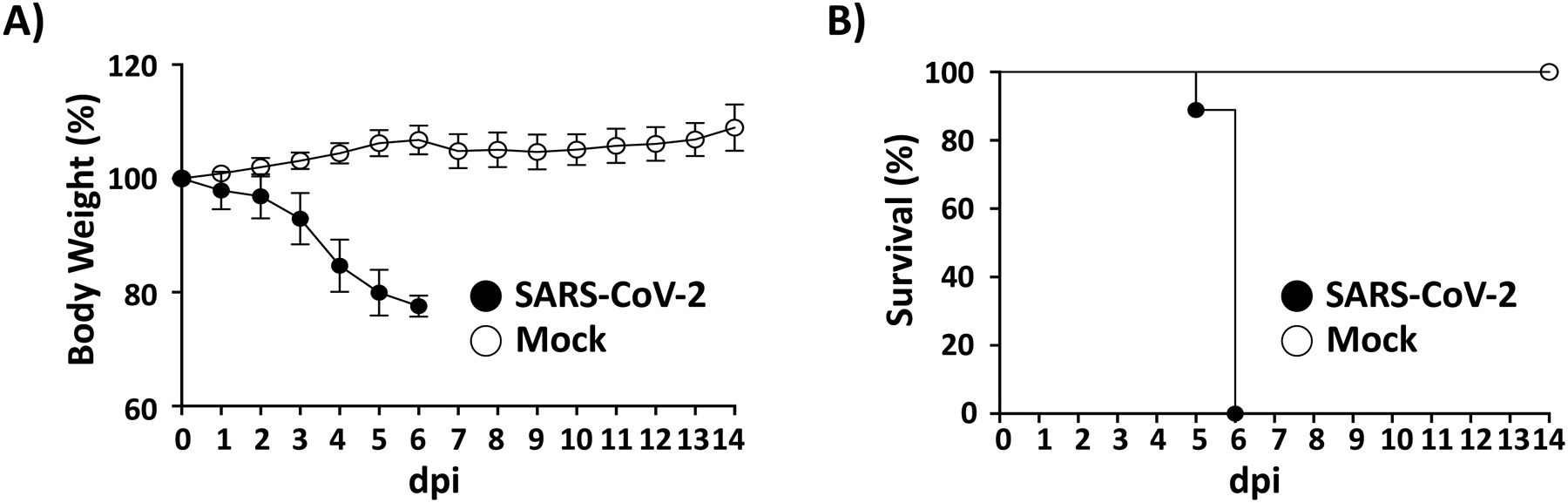
Morbidity and mortality of SARS-CoV-2 infected K18 hACE2 transgenic mice: K18 hACE2 transgenic mice are mock-infected or infected (105 PFU/mouse) with SARS-CoV-2 intranasally (n = 14). Changes in body weight (a) and survival (b) are evaluated daily for 14 days. Mice that loss more than 20% of their initial body weight are humanely euthanized. Error bars represent standard deviations (SD) of the mean for each group of mice. dpi: days postinfection
Fig. 3.
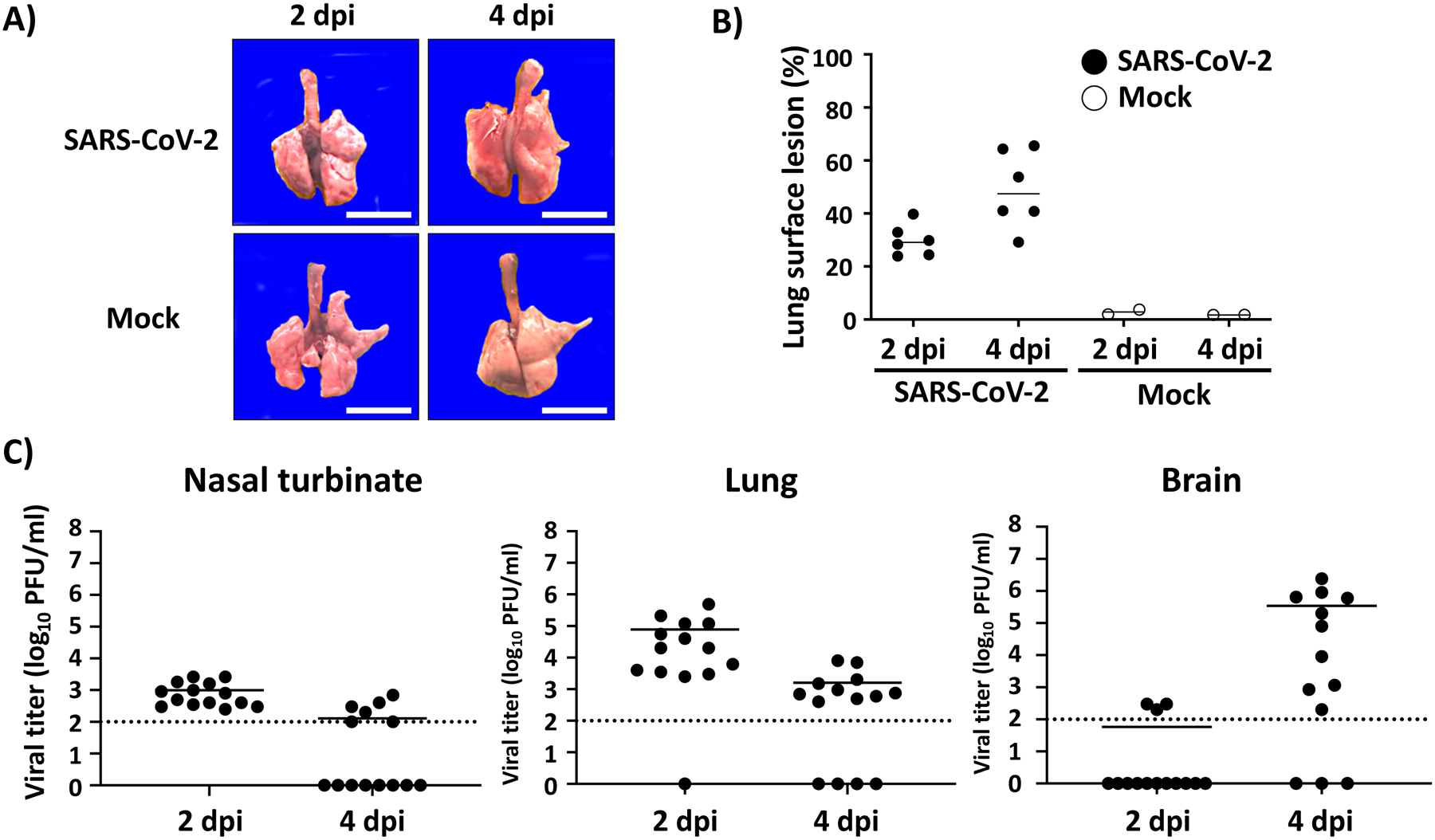
Gross lung pathology and viral titers. (a, b) Macroscopic lung pathology lesions: K18 hACE2 transgenic mice are mock-infected or infected with 105 PFU/mouse of SARS-CoV-2 and sacrificed at 2 and 4 dpi. Lungs are collected from all sacrificed mice to observe gross pathological changes (a). Scale bars = 1 cm. Macroscopic pathology scoring is determined by measuring the distributions of pathological lesions, including consolidation, congestion, and pneumonic lesions using ImageJ software (NIH) (b). (c) Viral titers: Presence of SARS-CoV-2 in the nasal turbinate (left panel), lung (middle panel) and brain (right panel) is determined by plaque assay (PFU/ml) from homogenized tissues at the indicated dpi. Dotted line indicates limit of detection (100 PFU/ml). Each symbol represents an individual animal and bars the geometric means of viral titers. dpi: days postinfection
Fig. 4.
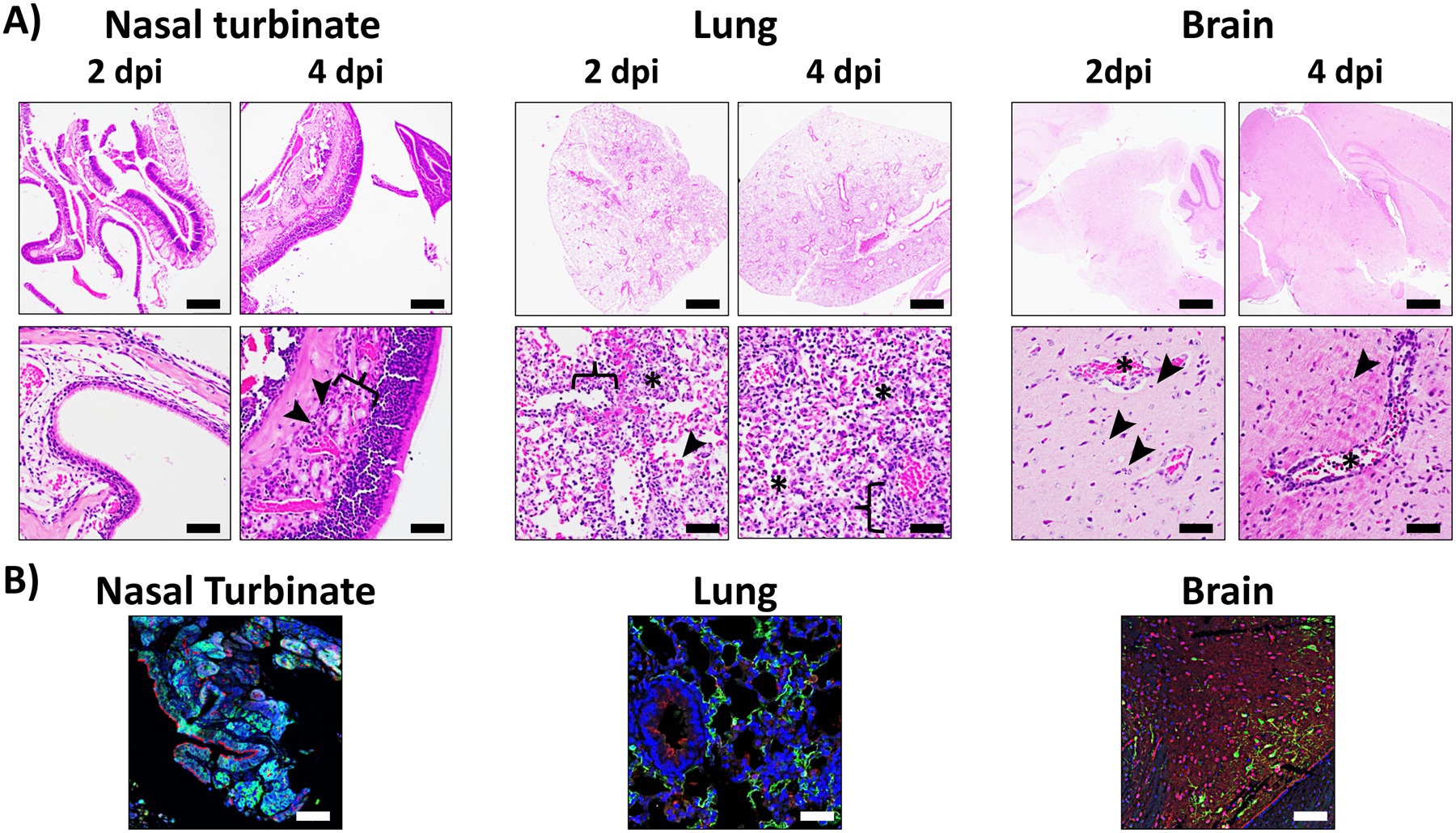
Histopathologic and IHC examination from SARS-CoV-2 infected K18 hACE2 transgenic mice: Nasal turbinate (left), lung (middle), and brain (right) tissues from K18 hACE2 transgenic mice infected with SARS-CoV-2 are fixed in 10% NBF, embedded in paraffin blocks, and sectioned for hematoxylin and eosin (H&E) (a) and confocal microscopy (b). For H&E staining, nasal turbinate, lung, and brain. Nasal turbinate at 2 dpi are microscopically unremarkable. At 4 dpi there is minimal neutrophilic inflammation (arrowheads) within the submucosa (bracket). Scale bars = 200 μm (upper row) and 50 μm (lower row). Lung at 2 dpi has mild to moderate multifocal interstitial pneumonia associated with inflammatory cell infiltrations (asterisks), mild type II pneumocyte hyperplasia (arrowhead), endothelial cells hyperplasia and vasculitis (bracket). At 4 dpi, it progressed to severe diffuse interstitial pneumonia with inflammatory cell infiltrations in alveolar spaces (asterisks) and interstitium (bracket). Brain at 2 dpi presented multifocal necrotic cellular debris within the brain parenchyma (arrowheads) and vasculitis (inflammation of blood vessels, asterisks). This phenotype extends on the brain over time (4 dpi) causing encephalitis. Scale bars = 1 mm (upper row) and 50 μm (lower row). For IHC staining, nasal turbinate, lung and brain tissues from SARS-CoV-2 infected K18 hACE2 transgenic mice at 2 and 4 dpi are immune-stained with hACE2 (red), and SARS-CoV N (green) antibodies. Cell nucleus is stained by DAPI (blue). Note of the tropism of SARS-CoV-2 for pyramidal neurons in the brain. Scale bars = 50 μm. Dpi: days postinfection
Fig. 5.
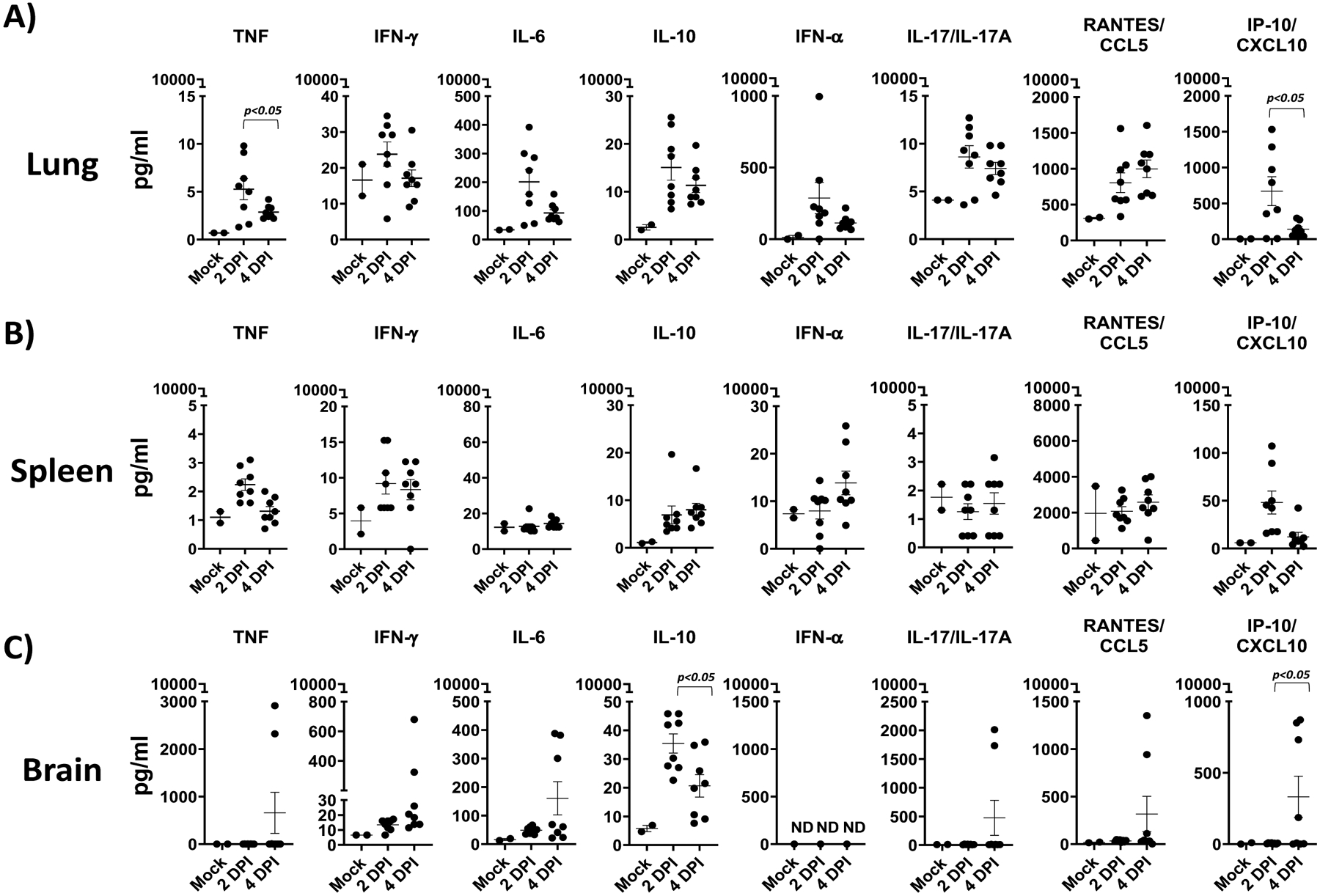
Cytokine and chemokine responses in SARS-CoV-2 infected K18 hACE2 transgenic mice: Cytokine (TNF, IFN-γ, IL-6, IL-10, IL-17/IL-17A, and IFN-α) (a) and chemokine (CCL-5 and CXCL-10) responses in SARS-CoV-2 infected K18 hACE2 transgenic mice at 2 and 4 dpi are measured in lung (a), spleen (b), and brain (c). A 2-WAY ANOVA K18 hACE2 transgenic mice over time. n = 8 (per time-point studied, except mock n =3). dpi: days postinfection
Nonhuman primates and hamsters are shown to develop mild to moderate COVID-19 clinical symptoms [18–20]. This was also observed in the mouse model expressing hACE2 by other means (e.g., using a mouse promoter, adenoviruses overexpressing mouse ACE2, or expressing hACE2 with other promoters) [8, 21–25]. The ideal SARS-CoV-2 animal model needs to exhibit clear clinical symptoms and characteristics of the severe COVID-19 disease seeing in humans, including mortality rates, viral replication in the lungs and other tissues, histopathological evidence of the disease and dysregulated immunological responses (e.g., cytokine storm). The K18 hACE2 transgenic mouse model shows all of these aspects of the virus-associated disease. Here we introduce a specific SARS-CoV-2 infection method using the K18 hACE2 transgenic mouse model, and subsequent analyses to determine the infectivity and pathogenicity of SARS-CoV-2 as well as some factors associated with COVID-19 disease in vivo.
2. Materials
2.1. Intranasal Infection
B6.Cg-Tg(K18-ACE2)2Prlmn/J (K18 hACE2) transgenic mice, The Jackson laboratory.
SARS-CoV-2 isolate USA-WA1/2020. Biodefense and Emerging Infections Research Resources Repository, BEI Resources, NR-52281: Total 50 μl of inoculum adjusted to 1 × 105 Plaque Forming Units (PFU) of SARS-CoV-2 diluted in phosphate buffer saline (PBS) per mouse.
Sterile PBS pH 7.4: 0.155 M NaCl, 0.0010 M KH2PO4, 0.0029 M Na2HPO4-7H2O without calcium and magnesium.
Anesthesia gas machine (Veterinary Anesthesia System), including small oxygen tank.
Isoflurane (Covetrus).
Heating pads.
Biohazard labels.
2% Wex-Cide (disinfectant, Wexford Labs): Dilute the Wex-Cide in tap water to 2%.
70% ethanol: Mix 7 parts of 100% ethanol and 3 parts of distilled water (dH2O).
2.2. Measuring Body Weight and Survival
Weight scale with three decimal range (0.000).
Bowl.
2% Wex-Cide (disinfectant, Wexford Labs): Dilute the Wex-Cide in tap water to 2%.
70% ethanol: Mix 7 parts of 100% ethanol and 3 parts of dH2O.
2.3. Necropsy
Necropsy tools: Necropsy board wrapped with aluminum foil, 18G needles to restrain the mice on the necropsy board, autoclaved surgical tools (including scissors, forceps and a sterile plastic 250 ml beaker to submerge and disinfect the surgical tools in 70% ethanol between animals).
Fatal-Plus (dosage >100 mg/kg): Commercially available (Vortech Phasmaceuticals Ltd.). Components: Pentobarbital Sodium 390 mg/ml, Propylene Glycol 0.01 ml/ml, Ethyl Alcohol 0.29 ml/ml, and Benzyl Alcohol (preservative) 0.02 ml/ml.
Sterile 1 ml syringes and 26G needles: For injection of the Fatal-Plus solution.
Precellys homogenizer tubes (Bertin Instruments): Use one Precellys tube per organ. Add 1 ml of sterile PBS to each tube.
Sterile PBS pH 7.4: 0.155 M NaCl, 0.0010 M KH2PO4, 0.0029 M Na2HPO4-7H2O without calcium and magnesium.
Sterile 1.5 ml polypropylene tubes.
Precellys tissue homogenizer (Bertin Instruments).
Conical tubes (15 ml or 50 ml) according to the dimensions of the tissue to be inactivated.
Neutral buffered formalin (NBF), 10%: Use one 15 ml or 50 ml conical tube completely full of NBF per tissue for fixation/inactivation (see Note 1).
Fine precision ruler.
Photographic camera for gross pathology.
Dissecting boards to photograph tissues after removal from each animal (Mopec).
Paper towels.
2% Wex-Cide (disinfectant, Wexford Labs): Dilute the Wex-Cide in tap water to 2%.
70% ethanol: Mix 7 parts of 100% ethanol and 3 parts of dH2O.
2.4. Viral Titration
Vero E6 cells (American Type Culture Collection, ATCC, CRL-1586).
Cell growth media: Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin, L-glutamine 100× (PSG).
Infection media: DMEM supplemented with 1% PSG.
Microcrystalline cellulose (Avicel, Sigma-Aldrich), 4%: Weigh 4 g of Avicel in 100 ml of PBS. Autoclave the bottle before use.
Postinfection media: 75 ml of DMEM supplemented with 2% FBS and 1% PSG, 25 ml of 4% microcrystalline cellulose solution.
Permeabilization solution: 0.5% of Triton X-100 in PBS.
Blocking solution: 2.5% bovine serum albumin (BSA) in PBS.
Mouse monoclonal antibody anti-SARS nucleocapsid (N) protein 1C7C7 (Leinco): Prepare 1 μg/ml of antibody in 1% BSA.
VECTASTAIN ® ABC-HRP Kit, Peroxidase, POD (Mouse IgG) (Vector Laboratory).
DAB Substrate Kit, POD (HRP), with Nickel (Vector Laboratory).
96-well cell culture plates.
Pipettes and pipette tips.
Multichannel pipette, manifold dispenser.
Vacuum aspirator-multichannel adapter (HandE-Vac Hand Controller Set, Argos Technologies).
Sterile 1.5 ml microcentrifuge tubes.
CO2 incubator at 37 °C.
CTL ImmunoSpot plate reader and counting software (Cellular Technology Limited).
2% Wex-Cide (disinfectant, Wexford Labs): Dilute the Wex-Cide in tap water to 2%.
70% ethanol: Mix 7 parts of 100% ethanol and 3 parts of dH2O.
2.5. Histopathology
Tissue processing solutions: xylene, 95% alcohol, absolute alcohol.
Microscope slides, coverslip.
Parapro XLT infiltration and embedding media (StatLab).
Hematoxylin stain (StatLab).
High Def solution (StatLab).
Reserve Bluing Reagent (StatLab).
Eosin (StatLab).
Tissue Tek VIP tissue processor.
Microm HM325 rotary microtome.
A flotation water bath set between 46 and 48 °C.
Oven at 60 °C.
Varistain Gemini AS Automated Slide Stainer (Thermo Scientific).
Bx46 microscope (Olympus).
CellSense v1.18 Life science Imaging software (Olympus).
2.6. Immunohistochemistry (IHC)
Hydration and dehydration solutions: xylene, 100%, 95%, and 80% ethanol (anhydrous denatured, histological grade).
10× Tris Buffered Saline with Triton X-100 (TBST) (Bio-Rad).
1× TBST: add 100 ml 10× TBST to 900 ml dH2O; add 0.025% Triton X-100 and mix well.
Blocking and Antibody diluent: Background punisher (BioCare Medical).
Antigen Unmasking Solution: Tris Based (Vector Laboratories).
Antigen Unmasking Solution: 0.01 M Citrate Acid Based (Vector Laboratories).
Recombinant Rabbit monoclonal antibody anti-hACE2, Thermo-Fisher, Cat#SN0754): Prepare 1:50 dilution of antibody in background punisher.
Rabbit polyclonal antibody anti-SARS N protein (homemade): Prepare 1:4000 dilution of antibody in background punisher.
Detection System: MACH 3 Rabbit or Mouse Alkaline Phosphatase Polymer Detection Kit (BioCare Medical).
Substrate: Warp Red Chromogen Kit (BioCare Medical).
Mounting Media with 4′,6-diamidino-2-phenylindole, DAPI (Thermo Scientific): Dissolve DAPI in dH2O to 1 mg/ml as stock solution. Keep the stock solution from light at −20 °C. Dilute the stock solution in PBS to 1 μg/ml for working concentration.
PBS pH 7.4: 0.155 M NaCl, 0.0010 M KH2PO4, 0.0029 M Na2HPO4-7H2O without calcium and magnesium.
PBS–Fish Skin Gelatin (PBS-FSG): 1000 parts PBS to 2 parts FSG (FSG, Sigma-Aldrich).
PBS-FSG-Tx100: 1000 parts PBS-FSG to 1-part Triton X-100 (TX-100, Sigma-Aldrich).
Normal Goat Serum (GIBCO), diluted 1:10 in PBS-FSG.
Beakers.
Coplin staining Jars.
Slide racks.
Slides incubation chamber.
Coverslips.
Microtiter pipettes and pipette tips.
A rocker platform.
2.7. Cytokines/Chemokines Assay
The Multiplex Mouse Cytokines/Chemokines assay (R&D Systems) is a commercial kit performed following manufacturer’s instructions.
Magnetic Luminex Assay, Mouse Premixed Multi-Analyte Kit (R&D Systems).
Hand-held microplate magnet or plate washer with a magnetic platform.
Microtiter pipettes and pipette tips.
Multichannel pipette, manifold dispenser, or automated dispensing unit.
Deionized H2O or dH2O.
500 ml graduated cylinder.
Horizontal orbital microplate shaker (0.12″ orbit) capable of maintaining a speed of 800 ± 50 revolutions per min (rpm).
Microcentrifuge.
Polypropylene test tubes for dilution of standards and samples.
96-well, flat bottom, polystyrene plates, Greiner Bio-One 655096, or equivalent.
1% Formaldehyde in PBS: For one 96-well plate, prepare a total volume of 18.5 ml by mixing 18 ml of PBS pH 7.4 (0.137 M NaCl, 0.0027 M KCl, 0.01 M Na2HPO4, 0.0018 M KH2PO4) and 0.5 ml of 37% formaldehyde (Sigma-Aldrich).
Plate sealer.
Aluminum foil.
1× sheath fluid (Luminex).
Luminex® MAGPIX®, Luminex® 100/200™, Luminex® FLEXMAP 3D®, or Bio-Rad Bio-Plex analyzer with X-Y platform.
2.8. Interferons Assays
2.8.1. Type I Interferon Alpha (IFN-α) Assay (See Note 2)
The Mouse IFN-α (Type I) enzyme-linked immunosorbent assay (ELISA) (PBL Assay Science) is a commercial kit developed following manufacturer’s instructions.
Mouse IFN-α ELISA Kit (PBL Assay Science).
Variable volume microtiter pipettes.
Adjustable multichannel pipette (50–300 μl).
Reagent reservoirs.
Wash bottle or plate washing system.
Distilled or deionized H2O.
Serological pipettes (1, 5, 10 or 25 ml).
Disposable pipette tips (polypropylene).
Plate shaker.
Microtiter plate reader capable of reading an optical density (OD) at a wavelength of 450 nm.
2.8.2. Type III Interferon Lambda (IFN-λ) Assay (See Note 2)
The Mouse IFN-λ (Type III) ELISA (PBL Assay Science) is a commercial kit that is performed according to the instructions provided by the manufacturer.
Mouse IFN-λ ELISA Kit (PBL Assay Science).
96-well ELISA plates.
PBS pH 7.2–7.4: 0.137 M NaCl, 0.0027 M KCl, 0.0081 M Na2HPO4, 0.0015 M KH2PO4. Filter sterilize using a 0.2 μm membrane.
Wash Buffer: 0.05% Tween 20 in PBS pH 7.2–7.4.
Reagent Diluent: 1% BSA in PBS pH 7.2–7.4. Filter sterilize using a 0.2 μm membrane (see Note 3).
Substrate Solution: 1:1 mixture of Color Reagent A (H2O2) and Color Reagent B (Tetramethylbenzidine, TMB).
Stop Solution: 2 N H2SO4.
Plate shaker.
Squirt bottle, manifold dispenser or auto-washer.
Microtiter plate reader capable of reading an OD at a wavelength of 450 nm.
3. Methods
3.1. Ethics Statement
All experimental procedures with mice were approved by the Texas Biomed Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee (IBC), and performed under Biosafety Level 3 (BSL3) and Animal BSL3 (ABSL3) laboratories.
3.2. Intranasal Infection
Transfer the specific-pathogen-free, 4–5-weeks-old, female and male K18 hACE2 transgenic mice to the ABSL3 facility. Place a maximum of 5 mice per cage and identify each animal using an ear punch code or any other approved method (e.g., tail tattoo). Label each mouse cage according to the virus and dosage to be used.
Prepare the SARS-CoV-2 inoculum under aseptic conditions. Calculate the amount of SARS-CoV-2 needed to prepare the viral dilution applying the following formula: (Titer of the virus [PFU] needed /stock viral titer) × final number of animals. Complete a total volume of 50 μl dose/mouse using sterile PBS. Keep the diluted viral inoculum on ice until use. Aliquot the total amount needed of PBS to be used for the inoculation of the mock-infected group (Fig. 1a).
Using a scale, record the bodyweight of each mouse at 0 dpi corresponding to the bodyweight/survival group (Fig. 1a).
Anaesthetize each mouse by placing the animal inside the anesthesia chamber of a Veterinary Anesthesia System and provide oxygen mixed with 3% isoflurane (Fig. 1a). Take the mouse out of the anesthesia chamber to proceed with the viral infection when the animal takes 2–3 s between breaths. Verify that the mouse is fully anaesthetized by toe pinch to see a lack of pedal withdrawal reflex. If the mouse is not fully anaesthetized, it will cough up the virus, resulting in inconsistent infection (Fig. 1a).
Place the mouse on hands in a dorsal recumbent position. Lift its head in an upright position to ensure the inoculum goes down through the airways. Infect intranasally (i.n.) the mouse using 50 μl of the viral inoculum. Make sure to eject the solution slowly but constantly and check the mouse is inhaling the inoculum by verifying the inoculum drop disappearing.
Hold the mouse in an upright position until its breathing rhythm gradually returns to normal. Make sure it is breathing as the viral inoculum can block the respiratory tract. Return the mouse to its home cage in a dorsal recumbent position. If any sign of respiratory distress is observed, help the mouse to breathe by holding the animal vertically and induce impact-driven respiration. Keep monitoring until the mouse regains consciousness (2–5 min after being anaesthetized). Place some heating pads underneath the cage to keep the animals warmed while the anesthesia effect disappears.
3.3. Characterization of Viral Pathogenesis
3.3.1. Evaluation of Morbidity and Mortality (Fig. 1b)
After viral infection, record the weight of mice once a day during the next 14 consecutive days at approximately the same time that the viral infection was performed, minimizing weight fluctuations such as food ingestion/water uptake, and keep monitoring for any clinical signs. Humanely euthanize mice that lose over 20% of their initial body weight or show severe clinical signs such as lethargic behavior with rough fur, hunched appearance, immobile, social distancing, and/or if it does not eat or drink (Fig. 1b).
After 14 days (end-point), humanely euthanize the mice that survive the viral infection according to the standard procedures at your institution/your local IACUC (Fig. 2).
3.3.2. Necropsy (Fig. 1c)
At the established end-points (2 and 4 dpi, or at the end of your study), euthanize mice according to IACUC approved protocol, such as a lethal dose of Fatal-Plus (200 μl) via intraperitoneal route (i.p.) injection. (see Note 4).
Lay the euthanized mouse in a dorsal recumbent position on the clean necropsy board and disinfect the fur over the thorax and abdomen spraying 70% ethanol on it evenly. Using 18G needles, restrain the animal on the necropsy board by the extremities. Take the scissors and cut the center skin of the body from the base of the abdomen to the chin, detaching the skin from the membrane that supports internal organs (do not tear apart the membrane). Make 1 cm incision from the abdomen and open the rib cage by cutting the ribs laterally from the costal cartilage through the diaphragm to the top of the sternum. Keep cutting until reaching the middle line of the animal toward the mouth without damaging the trachea. Remove the ribs and sternum and examine the thoracic cavity including all the lobes of the lung (see Note 5).
Lung isolation: Remove the whole lower respiratory tract from the start of the trachea to the lungs and place it on a dissecting board. Trim the collected lower respiratory tract to remove heart, fat, bronchus-associated lymphoid tissue (BALT) and unnecessary connective tissue.
Spleen isolation: Using forceps, gently pull the spleen free of the peritoneum, tearing or cutting the connective tissue attached to it. Avoid cutting into the intestine as this organ is not sterile.
Brain isolation: Using scissors, remove the head from the mouse body by cutting between cranium and atlas. Cut to remove also the skin of the head. Hold the cranium with the dissecting forceps and cut the cranium along the sagittal suture with scissors. Collect the brain from the inside part of the two halves of the cranium and place it on a dissecting board.
Nasal turbinate: Cut the junctions between the maxilla and the mandible, discard the mandible. With the scissors, make two cuts at the ends of the zygomatic arcs and remove them. Remove the eyes with the help of the dissecting forceps. The nasal turbinates are surrounded by the anterior bones of the skull including the nasal, maxilla, palatine, zygomatic, and ethmoid bones. Using the scissors, collect the whole anterior bones and nasal turbinate and place them on a dissecting board.
3.3.3. Gross Pathology
Lung Images: Place the lungs on a clean dissecting board for photographing and obtain the lung images from dorsal and ventral view to evaluate gross lung pathology (Fig. 3a, b).
3.3.4. Distribution of Tissues for Fixation and Homogenization
Transfer a half of each organ (left lung lobe, nasal turbinate and brain) to a fixative solution to inactivate following Subheading 3.5 (see below Histopathology section).
Place the other half of the lung (right lobes), nasal turbinate and brain, and the whole spleen in the corresponding sterile Precellys homogenizer tube filled with 1 ml of PBS.
Homogenize tissues in the Precellys tissue homogenizer using the following parameters: 5000 × g, 20 s, once. This allows a well-homogenized tissue to be obtained. If a different tissue homogenizer is used, the appropriate parameters to homogenize tissues must be standardized according to the case.
Transfer the tissue homogenates to sterile 1.5 ml microcentrifuge polypropylene tubes and centrifuge at 6000 × g, 5 min, 4 °C.
Collect the supernatant and transfer it to a new 1.5 ml polypropylene tube. Store at −80 °C until use for viral titers determination or cytokines/chemokines evaluation.
3.4. Viral Titration
The day before the in vitro infection for the viral titration, seed 96-well plates with Vero E6 cells (1 × 104 cells/well) with cell growth media. Maintain the cells at 37 °C, 5% CO2. On the same day, thaw the homogenized tissue supernatant to be tested for the viral titration.
On the day of the viral titration, and before starting the in vitro viral infection, check the confluence of Vero E6 cells under the microscope to confirm a monolayer is formed.
In a different 96-well plate, prepare a ten-fold dilution of homogenized tissue supernatant in an empty, sterile 96-well plate using infection media. Briefly, add 90 μl of infection media to the 96-well plate rows A to H, and add 10 μl of the supernatant of each homogenized sample to wells of the row A. Using a multichannel micro dispenser, transfer 10 μl from row A to B, and mix ~10 times. Repeat this process from row B to row C, and subsequently until row H, changing the tips between dilutions to prevent the transfer of residual virus in the tips. Discard 10 μl from the dilution already mixed in row H so that each of the 96-well plate wells has 90 μl of diluted sample. Each sample should be tested at least in duplicate.
On the 96-well plate containing Vero E6 cells seeded, remove the tissue culture medium using a vacuum aspirator-multichannel adapter.
Proceed with the in vitro infection transferring 50 μl/well of the serially diluted tissue homogenized supernatants to the Vero E6 cells seeded in the 96-well plate starting from the lower dilutions (row H) to the higher dilutions (row A), so that it is not necessary to change the tips between different rows.
Incubate the 96-well plates with infected cells for 1 h at 37 °C in 5% CO2 to allow viral adsorption. After this period of time, remove the inoculum using a vacuum aspirator-multichannel adapter and overlay with postinfection media containing 1% Avicel. Then, incubate infected Vero E6 cells for 24 h at 37 °C, 5% CO2.
At 24 h postinfection (hpi), remove infectious media and completely submerge the plate in 10% NBF for 24 h at 4 °C for fixation/inactivation (shown to inactivate SARS-CoV-2).
Upon fixation/inactivation, transfer the plate from the BSL3 to a BSL2 laboratory following approved standard procedures, remove residual 10% NBF by gently washing with dH2O before staining.
Proceed with the staining protocol [26]. Wash cells gently 3× with 100 μl/well of PBS and permeabilize the cells with 100 μl/well of 0.5% Triton X-100 dissolved in PBS. Incubate at room temperature for 15 min in the biosafety cabinet.
Wash the cells three times with 100 μl/well of PBS and incubate fixed cells with blocking solution (100 μl/well of 2.5% BSA in PBS) at 37 °C for 1 h.
Remove the blocking solution. Add 50 μl/well of 1 μg/ml primary antibody solution (anti-NP monoclonal antibody 1C7C7) diluted in 1% BSA in PBS. Incubate the cells at 37 °C for 1 h.
Remove the primary antibody solution, wash with 100 μl/well of PBS, three times. Prepare the biotinylated anti-mouse antibody [VECTASTAIN® ABC-HRP Kit, Peroxidase (Mouse IgG); Vector Laboratory] following the manufacturer’s instructions. Briefly, add 75 μl of normal blocking serum stock and 25 μl of biotinylated secondary antibody stock to 5 ml of PBS per 96-well plate. Add 50 μl/well of biotinylated antibody solution to each well and incubate for 30 min at 37 °C.
Remove the secondary biotinylated antibody solution and wash each well three times with 100 μl/well of PBS. Prepare VECTASTAIN ABC Reagent as described in manufacturer’s instructions (VECTASTAIN ® ABC-HRP Kit, Peroxidase (Mouse IgG; Vector Laboratory). Add 50 μl each of Reagent A (Avidin, ABC) and Reagent B (Biotinylated HRP, ABC) to 5 ml of PBS for one 96-well plate. Mix the solution thoroughly and add 50 μl/well of VECTASTAIN ABC Reagent and incubate for 30 min at 37 °C.
Wash cells three times with 100 μl/well of PBS. Dry the plate by tapping on paper towels. Prepare developing solution by following manufacturer’s instructions [DAB Substrate Kit, Peroxidase (HRP), with Nickel; Vector Laboratory]. Add 50 μl of developing a solution to each well and wait for ~3–5 min to visualize viral plaques.
Remove the developing solution to stop the reaction and wash with PBS. Make sure not to develop for too long once the plaques are evident to prevent the cells from turning black.
Count the stained positive cells using a CTL ImmunoSpot plate reader and counting software (or your alternative method) to calculate the viral titer. Calculate the viral titer expressed in PFU/ml from the dilution with 30 to 50 positive stained cells using the formula: [(Mean No of plaques] × 20 × 1/dilution (Fig. 3c).
3.5. Histopathology
This protocol describes methods corresponding to the preparation of tissues after necropsies of animals and their histopathological analysis.
As mentioned above (in the distribution of tissues for fixation and homogenization section), separate half of each organ (left lung lobe, nasal turbinate and brain) intended to be inactivated. Determine the dimension of each tissue using a precision ruler to calculate the volume of 10% NBF to be used. Determine the length, width, and thickness of the tissue. The sample cannot exceed the following dimensions: 2.5 cm × 2.5 cm × 0.6 cm (thickness) due to the decontamination protocol approved using 10% NBF which allows that the 10% NBF completely penetrates the tissue.
Determine the volume of 10% NBF solution using the following formula: Length × Width × Thickness = Tissue Volume × 15 (Volume of NBF per volume of tissue) (see Note 6).
Fully submerge the infected tissue in a conical tube using the appropriate volume of 10% NBF solution according to the formula described above. If the volume needed is less than the maximum capacity of the conical tube, such tube must be completely full of NBF. Coat tube threads and gasket with 10% NBF prior to closing the tube’s cap.
Store samples at the BSL3 laboratory submerged in 10% NBF for 7 days at room temperature prior to removal to a lower BSL laboratory (see Note 7).
Follow your approved institutional biosafety standard procedures for removal of tissues in 10% NBF from the BSL3 and transfer them to the corresponding Pathology Laboratory for subsequent histopathological analysis.
Proceed with the paraffin processing and embedding of tissues. Fixed tissues are processed using a Tissue Tek VIP tissue processor by dehydration through a series of graded alcohols, clearing through one change of 50:50 absolute alcohol–xylene mixture and two changes of xylene, concluding with the infiltration of tissues with paraffin wax using Parapro XLT infiltration and embedding media.
The paraffin-embedded blocks are then cut using a Microm HM325 rotary microtome at a thickness of 4 microns and mounted onto microscope slides by use of a flotation water bath set between 46 °C and 48 °C. Unstained slides are dried in a 60 °C oven for 1 h before proceeding with Hematoxylin and Eosin (H&E) staining protocol.
Proceed with the H&E staining using a Varistain Gemini Automated Slide Stainer. Deparaffinization of tissue sections is performed through three changes of xylene (incubation time for each xylene step is 3 min, 2 min, and 2 min, respectively), two changes of absolute alcohol (incubation time for each absolute alcohol step is 1 min), two changes of 95% alcohol (incubation time for each 95% alcohol step is 1 min) and rinse in running water for 30 s.
Perform the hematoxylin staining for 2 ½ min, followed by rinsing in running water for 1 min. Remove the excess of stain using High-Def solution for 45 s, rinsing with running water for 1 min. Continue bluing the hematoxylin stain using Reserve Bluing Reagent for 1 min. Rinse in running water for 1 min and add 95% alcohol for 15 s. Tissue sections are then stained with eosin for 2 ½ min.
Dehydration is performed through three changes of absolute alcohol (incubation time for each absolute alcohol step is 1 min), a 50:50 absolute alcohol–xylene (v/v) mixture for 30 s and cleared in three changes of xylene (incubation time for each xylene step is 1 min) before coverslipping.
Upon completion of the H&E staining, tissue sections are evaluated using light microscopy in a blinded manner by a board-certified veterinary pathologist. Images are acquired using a Bx46 microscope and the CellSense vr. 1.18 Life science Imaging software.
3.6. Immunohistochemistry
To perform antibody staining, paraffin wax must be removed from the sample and the sample must be rehydrated (see Note 8).
Place the slides in a 56–60 °C oven for 1 h (see Note 9).
Transfer slides to the xylene bath and perform three changes of xylene for 5 min each.
Drain off excess liquid and rehydrate slides in three changes of fresh absolute ethanol for 2 min each.
Drain off excess liquid and place slides in fresh 95% ethanol for 2 min.
Drain off excess liquid and place slides in fresh 80% ethanol for 2 min.
Place the slides in deionized H2O.
Microwave double antigen retrieval procedure: For the first antigen retrieval, place slides rack in a microwave-resistant 250 ml glass beaker containing TRIS-based Antigen unmasking solution, pH 9.0 (Vector Labs H-3301), that also contains 0.01% Tween-20. Make sure slides are fully covered with the solution.
Operate the microwave oven on 20% power (700 watts) for 11–12 min for one beaker and for about 20 min for 2 beakers (see Note 10). Leave slides in the beaker before proceeding to the next step.
Second Antigen Retrieval: Take two 250 ml size glass beakers, one filled with deionized H2O and another filled with 0.01 M Citrate-based Antigen unmasking solution, pH 6.0 (Vector Laboratories, H-3300), each up to 200 ml volume. Place deionized H2O and a 0.01 M citrate-based solution containing beakers in the microwave oven.
Operate the microwave oven on high power about 3–4 min (see Note 10).
Take out slides from the antigen unmasking Tris-based solution beaker, drain off excess solution, and transfer it to the microwave boiled deionized H2O beaker to wash the slides only (e.g., 30 s).
Next, transfer slides from the deionized H2O beaker to the microwave boiled Antigen unmasking Citrate-based solution beaker and let cool slowly at room temperature for at least 30 min.
After this period, transfer slides into a beaker containing Tris Buffered Saline (TBS).
For chromogenic staining, wash sections in TBS for 5 min.
Use a hydrophobic pen to draw a large circle around the sample, taking care not to touch the sample. This creates a hydrophobic boundary so that a smaller volume of antibody solution can be used and, if desired, allows multiple sections on one slide to be stained with different antibodies.
To prevent nonspecific binding of the antibody to tissues, block each section with 100–400 μl Background punisher (blocking solution) for 40 min at room temperature in a humidified chamber.
To stain hACE2, use anti-hACE2 as a primary antibody: Remove blocking solution and add 100–400 μl primary antibody diluted in recommended antibody diluent (by the manufacturer) to each section for 1 h at room temperature in a humidified chamber.
Equilibrate MACH 3 detection reagent kept at 4 °C to room temperature. (Use detection kit of the same species as the source of the primary antibody).
Remove primary antibody solution and wash sections in wash buffer three times for 10 min each.
Cover section with 1 to 3 drops of MACH 3 probe as needed. Incubate in a humidified chamber for 20 min at room temperature.
Wash sections 3× with wash buffer for 10 min each.
Cover section with 1 to 3 drops of MACH 3 alkaline phosphatase polymer as needed. Incubate in a humidified chamber for 20 min at room temperature.
Wash sections 3× with wash buffer for 10 min each.
Development: Add 1 drop of Warp Red Chromogen to 2.5 ml of Warp Red Buffer mix well before use. Apply the Warp Red mixture to the tissue section. Visualize staining of tissue under a microscope using a bright-field illumination. Incubate 5–20 min, stop reaction when you observe a red coloration in the cells indicating a positive reaction.
Wash sections in deionized water 2× for 5 min each.
Stained tissue mounted with mounting media with DAPI, fluorescent stain that binds strongly to adenine–thymine-rich regions in DNA, used to visualize nucleus and distinct cells).
Warp Red is highly fluorescent and will not fade. Use with a Texas Red filter.
Repeat antigen retrieval procedures from steps 7 to 13, for fluorescence IHC.
For fluorescence staining, dry slides as much as possible without touching the cells or tissues.
Circle tissue with a wax pen, put slides inside a slide Coplin jar, and add PBS-FSG.
Place slides in humidifying chamber (such as a black slide box, cover the bottom with a shallow layer of tap water) for the following steps.
Cover the tissues with 10% normal goat serum for 60 min (approx. 200 μl/slide).
Prepare primary antibody in optimal concentration. Use 10% normal goat serum to dilute the antibody; for example, if you have 5 slides and you need 200 μl of liquid to cover each one, you will need 1 ml total. Adjust numbers to reflect the quantity of slides you are staining and be certain about all calculations.
Centrifuge the primary antibody diluted in 10% normal goat serum at 13,000 × g for 1 min (use microfuge).
Remove normal goat serum from slides (e.g., dump it onto a paper towel). Do not rinse tissue!
Add the appropriate concentration of primary antibody, diluted in 10% normal goat serum.
Incubate at room temperature for 60 min, in black slide box.
Fill a Coplin jar with PBS-FSG-TX100.
Using a transfer pipette, rinse tissue thoroughly and place slides in the Coplin jar.
Place on the rocker for 10 min.
Repeat 3×.
Fill another Coplin jar with PBS-FSG. Using a transfer pipette, rinse the tissue thoroughly and place slides in the Coplin jar.
Place on the rocker for 10 min.
Centrifuge the secondary antibody diluted in 10% Normal Goat Serum at 13,000 × g for 1 min.
Apply enough of the secondary antibody solution (approximately 200 μl) to all slides.
Incubate at room temperature, for 60 min, in a black slide box.
Fill a Coplin jar with PBS-FSG-Tx100. Using a transfer pipette, rinse tissue thoroughly and place slides in the Coplin jar.
Place on the rocker for 10 min.
Repeat 3 ×.
Fill another Coplin jar with PBS-FSG. Using a transfer pipette, rinse the tissue thoroughly and place slides in the Coplin jar until you can check them under the microscope.
Record your results and suggestions, as you examine each slide.
If you desire more antigen colors for these slides, you may return to step 5 and repeat the protocol. Be sure that the antibodies are compatible to be distinguished by secondary antibody detection.
Once your slides have all desired antigens labelled, mount slides in antiquenching media.
3.7. Determination of Cytokines/Chemokines
The determination of cytokines and chemokines is performed using a custom multiplex panel mouse magnetic bead Luminex assay, which is summarized below according to the instructions provided by the manufacturer.
Prepare reagents and standards provided in the kit following the instructions of the manufacturer (see Note 11). Use the supernatant from homogenized tissues (nasal turbinate, lung, brain and spleen) to prepare a dilution 1:2 using the Calibrator Diluent RD6–52 provided in the kit (see Note 12).
Add 50 μl per well of standard or diluted tissue supernatant to the 96-well microplate provided in the kit (see Note 13).
Resuspend the diluted Microparticle Cocktail (provided in the kit and diluted according to the manufacturer’s instructions) by inversion or vortex (see Note 14). Add 50 μl to each well of the Microparticle Cocktail (see Note 13). Cover with a foil plate sealer and incubate for 2 h at room temperature on a horizontal orbital microplate shaker set at 800 ± 50 rpm (see Note 15).
Wash the 96-well microplate by applying a magnetic device to the bottom of such plate and allow 1 min before removing the liquid. Fill each well with 100 μl of Wash Buffer (provided in the kit and diluted according to the manufacturer’s instructions) and allow 1 min before removing the liquid again. Perform this step 3× (see Note 16).
Add 50 μl per well of diluted Biotin-Antibody Cocktail (provided in the kit and diluted according to manufacturer’s instructions) (see Note 13). Cover with a foil plate sealer and incubate for 1 h at room temperature on a shaker set at 800 ± 50 rpm (see Note 15).
Repeat once the wash step (see Note 16).
Add 50 μl to each well of diluted Streptavidin-PE (Streptavidin–phycoerythrin provided in the kit and diluted according to the manufacturer’s instructions) (see Note 14). Cover with a foil plate sealer and incubate for 30 min at room temperature on a shaker set at 800 ± 50 rpm (see Note 15).
Repeat the wash step (see Note 16).
Proceed with the decontamination process of the Luminex plate for removal of the BSL3 laboratory. Bring into the cabinet a brand-new plastic black 96-well plate and label it specifying the assay name and date (see Note 17).
Remove the Luminex plate from the magnetic device (after the final wash step) and resuspend the beads by adding 150 μl per well of 1% Formaldehyde in PBS. Mix content immediately by pipetting up and down 4 to 6 times to prevent bead agglutination.
Transfer the resuspended beads to the brand-new labelled 96-well plate keeping the same well position. Use different tips for each row/column and keep the same order as the initial plate to avoid confusions.
Cover the plate with a plate sealer or film and aluminum foil. Incubate overnight inside the biosafety cabinet in the dark at room temperature.
After overnight decontamination, wipe the external surface of the plate with 70% ethanol and place it inside a sealable plastic bag.
Follow approved institutional biosafety standard procedures for removal of the plate from the BSL3 laboratory and bring it to the BSL2 laboratory.
Proceed with washing process of the plate and data acquisition. Wipe the external surface of the 96-well plate again. Bring the Washing Buffer (provided in the Luminex kit) to room temperature.
Wash the plate adding 200 μl per well of Washing Buffer and using the magnetic device as previously described. Remove and transfer the liquid to a container with 1% bleach solution.
Repeat the wash step once.
Resuspend the microparticles by adding 125 μl per well of Luminex 1× sheath fluid or appropriate buffer and volume described in the commercial kit protocol. Mix the beads by pipetting up and down 5 to 6 times, using different tips for each column.
3.8. Type I IFN-α Assay
The determination of IFN-α (Type I) is performed using a Mouse IFN-α (Type I) ELISA assay, which is summarized below according to the manufacturer’s instructions (see Note 2).
Prepare the reagents and standards provided in the kit following the manufacturer’s instructions (see Notes 20–22). Use the supernatant from homogenized tissues (nasal turbinate, lung, brain and spleen) to prepare a dilution 1:2 using the Sample Buffer provided in the kit (see Notes 23 and 24).
Determine the number of microplate strips required to test the desired number of samples, besides the number of wells needed to run blanks and standards (see Note 25). Remove extra microtiter strips from the frame, seal in the foil bag provided and store at 2–8 °C. Unused strips can be used in later assays.
Add 100 μl per well of standard, diluted samples or blank to the precoated microtiter plate (provided in the kit).
Add 50 μl per well of diluted Antibody Solution (provided in the kit and diluted according to the manufacturer’s instructions) completing a total volume of 150 μl per well, changing pipette tips between each addition.
Cover with a plate sealer (provided in the kit) and incubate for 1 h at room temperature (22–25 °C) on a shaker at 450 rpm (see Note 26).
Transfer the plate to 4 °C and incubate 20–24 h without shaking.
After 20–24 h, empty the contents of the plate by inverting and shaking over a sink and blotting the plate on lint-free absorbent paper; tap the plate. Wash the wells four times by adding a minimum of 300 μl of diluted Wash Solution (provided in the kit and diluted according to the manufacturer’s instructions) for each wash step.
Add 100 μl per well of diluted horseradish peroxidase (HRP) Solution (provided in the kit and diluted according to the manufacturer’s instructions). Cover with a plate sealer (provided in the kit) and incubate for 2 h at room temperature (22–25 °C) on a shaker at 450 rpm (see Notes 26 and 27).
Repeat the wash step once.
Add 100 μl per well of the Tetramethyl-benzidine (TMB) Substrate Solution (provided in the kit). Incubate 15 min in the dark at room temperature (22–25 °C). Do not use a plate sealer during the incubation. Do not shake.
Without emptying the wells and without washing them, add 100 μl per well of Stop Solution (provided in the kit).
Read the plate absorbance at 450 nm within 5 min using a microplate reader.
The interferon titer in the samples can be determined by plotting obtained optical densities (ODs) using a 4-parameter fit for the standard curve. Blank ODs should be subtracted from standards and sample ODs to eliminate the background.
3.9. Type III IFN-λ Assay
The determination of IFN-λ (Type III) is performed using a Mouse IFN-λ (Type III) ELISA assay, which is summarized below according to the manufacturer’s instructions (see Notes 2 and 28).
Coat a 96-well microplate with 100 μl per well of the Capture Antibody (provided in the kit and diluted according to the manufacturer). Seal the plate and incubate overnight at room temperature (22–25 °C) (see Notes 20, 21, 24, 25, 29 and 30).
Aspirate each well and wash with Wash Buffer by adding 400 μl per well. Repeat this process 2× to complete three total washes. After the last wash, remove any remaining Wash Buffer by aspirating or by inverting the plate and shaking over a sink and blotting the plate on lint-free absorbent paper; tap the plate (see Note 31).
Block the plate by adding 300 μl per well of Reagent Diluent. Incubate at room temperature (22–25 °C) for a minimum of 1 h (see Note 30).
Repeat the wash step once (see Note 31).
Prepare the standards provided in the kit following the manufacturer’s instructions (see Note 22). Use the supernatant from homogenized tissues (nasal turbinate, lung, brain and spleen) to prepare a dilution 1:2 using the Reagent Diluent recommended (see Note 32).
Add 100 μl per well of sample or standards diluted in Reagent Diluent. Cover with a plate sealer and incubate 2 h at room temperature (22–25 °C).
Repeat the wash step once (see Note 31).
Add 100 μl per well of the Detection Antibody (provided in the kit and diluted according to the instructions of the manufacturer). Cover with a new plate sealer and incubate 2 h at room temperature (22–25 °C).
Repeat the wash step once (see Note 31).
Add 100 μl per well of the streptavidin–horseradish peroxidase (HRP, provided in the kit and diluted according to the manufacturer’s instructions). Cover the plate with a new plate sealer and incubate for 20 min at room temperature (22–25 °C). Avoid placing the plate in direct light.
Repeat the wash step once (see Note 31).
Add 100 μl per well of Substrate Solution to each well. Incubate for 20 min at room temperature (22–25 °C). Avoid placing the plate in direct light.
Add 50 μl of Stop Solution to each well. Gently tap the plate to ensure thorough mixing.
Determine ODs of each well immediately, using a microplate reader set to 450 nm. If wavelength correction is available, set to 540 nm or 570 nm. If wavelength correction is unavailable, subtract readings at 540 nm or 570 nm from the readings at 450 nm (see Note 33).
Average the duplicate readings for each standard, control, and sample and subtract the average zero standard OD. Create a standard curve by reducing the data using computer software capable of generating a four-parameter logistic (4-PL) curve-fit. If samples have been diluted, the concentration read from the standard curve must be multiplied by the dilution factor.
4. Notes
The volume of the 10% NBF solution must be 15–20 times the volume of tissue. For inactivation, tissues must be incubated with NBF solution for 7 days before removing from the BSL3 facility [27].
The detection of IFN-α (Type I) and IFN-λ (Type III) is performed using separated commercial kits because such analytes are not part of the Luminex custom multiplex panel.
-The use of high quality BSA for the Reagent Diluent is critical for the optimum performance of the kit. Impurities such as proteases, binding proteins, soluble receptors or other interfering substances can be found to varying degrees in virtually all BSA preparations and can inhibit or interfere with the detection of certain analytes. If the standard curve appears suppressed, consider evaluating a different preparation of BSA.
Euthanasia using carbon dioxide (CO2) overdose is not recommended because it induces artifact lung pathological changes such as pulmonary congestions and hemorrhage, and perivascular and peribronchiolar edema [28, 29].
To avoid contamination of samples, clean and disinfect the dissecting tools between tissues and between animals by submerging the tools in the 250 ml beaker with 70% ethanol. When removed from the beaker, let them air-dry each time before use.
Examples of calculation of NBF volume per sample dimensions/volume for rodent tissues: Example 1: 1 cm (length) × 1 cm (width) × 0.6 cm (thickness) = 0.6 cm3=0.6 ml (tissue volume) × 15 (volume of NBF per volume of tissue) = 9 ml of 10% NBF solution. Recommended to use a 15 ml conical tube and fill it up to 15 ml of 10% NBF to assure the tissue is fully submerged. Example 2 (maximum tissue volume allowed): 2.5 cm (length) × 2.5 cm (width) × 0.6 cm (thickness) = 3.75 cm3 = 3.75 ml (tissue volume) × 15 (volume of NBF per volume of tissue) = 57 ml of 10% NBF solution.
According to the literature, 4% formaldehyde solution penetrates at a rate of 2.4 mm/24 h. Given a sample thickness of 6 mm, complete penetration by the fixating agent will be obtained 6 mm/2.4 mm per 24 h = 2.5 days [30].
Do not allow slides to dry at any time during this procedure as this can lead to inconsistent staining.
The oven temperature must not exceed 60 °C.
Stop when the solution starts boiling and use care with hot solutions.
Bring all reagents to room temperature before use. When mixing or reconstituting protein solutions, always avoid foaming.
The kit provides a plate layout to record standards and samples assayed.
To avoid cross-contamination, change pipette tips between additions of each standard level, between sample additions and between reagent additions. Also, use separate reservoirs for each reagent.
Protect microparticles and Streptavidin-PE from light at all times to prevent photobleaching.
To ensure accurate results, proper adhesion of plate sealers during incubation steps is necessary.
Complete removal of liquid during washing steps is essential for good performance. Do NOT blot; this may cause a loss of microparticles).
Before proceeding with this step, decontaminate the whole surface of the biosafety cabinet with 2% Wex-Cide solution and 70% ethanol, or with the approved disinfectants available in your BSL3 facility.
Adjust the probe height setting on the analyzer to avoid puncturing the plate as well as other parameters such as sample volume, bead type, microparticle region, and count/region, according to the manufacturer’s instructions. Calibrate the analyzer using the proper reagents for superparamagnetic microparticles (refer to instrument manual).
If samples have been diluted, the concentration read from the standard curve must be multiplied by the dilution factor. If samples generate values higher than the highest standard, further dilute the samples with calibrator diluent and repeat the assay.
For retention of full activity, all reagents should be kept at 2–8 °C in the dark.
Deionized or distilled H2O should be used for the preparation of all reagents.
All dilutions should be made with polypropylene tubes and pipette tips. Pipette tips should be changed between each dilution tube.
It is important to run the standard curve in the same matrix as your samples unless you have demonstrated that the matrix does not affect the signal.
The kit provides a plate layout to record standards and samples assayed.
It is recommended to run standards, blanks, and samples in duplicate. At least two control wells (wells with Sample Buffer only) should be used for each assay; these control values should be subtracted from all readings prior to any calculations or plots of the data.
All incubations should be performed in a closed chamber at room temperature (22–25 °C) keeping the plate away from drafts and other temperature fluctuations.
Prepare the HRP Solution within 15 min prior to use.
This kit detects both Lambda 2 and 3 with similar reactivity.
Bring all reagents to room temperature (22–25 °C), before use. Avoid microbial contamination of reagents and buffers because it may interfere with the sensitivity of the assay. Buffers containing a large quantity of protein should be made under sterile conditions and stored at 2–8 °C or be prepared fresh daily. The type of enzyme, substrate, and the concentrations of capture/detection antibodies used can be varied to create an immunoassay with different sensitivity and dynamic range.
Use a fresh reagent reservoir and new pipette tips for each step.
Complete removal of liquid at each step is essential for good performance.
It is important that the diluents selected for reconstitution and for dilution of the standard reflect the environment of the samples being measured. The diluent suggested in this protocol should be suitable for most cell culture supernatant samples. Validate diluents for specific sample types prior to use.
The reading subtraction will correct for optical imperfections in the plate. Readings made directly at 450 nm without correction may be higher and less accurate.
Acknowledgments
We thank Texas Biomedical Research Institute Pathology Laboratory members, specially Dolores Renee Escalona and Drs. Olga Gonzalez, Anna Allue Guardia, and Shalini Gautam, for their support providing detailed information about the discussed protocols. Jun-Gyu Park and Paula A. Pino contributed equally to this study.
References
- 1.Rokni M, Ghasemi V, Tavakoli Z (2020) Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: comparison with SARS and MERS. Rev Med Virol 30(3): e2107. 10.1002/rmv.2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, Wu Y, Zhang L, Yu Z, Fang M, Yu T, Wang Y, Pan S, Zou X, Yuan S, Shang Y (2020) Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med 8(5):475–481. 10.1016/S2213-2600(20)30079-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glass WG, Subbarao K, Murphy B, Murphy PM (2004) Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J Immunol 173(6):4030–4039. 10.4049/jimmunol.173.6.4030 [DOI] [PubMed] [Google Scholar]
- 4.Subbarao K, McAuliffe J, Vogel L, Fahle G, Fischer S, Tatti K, Packard M, Shieh WJ, Zaki S, Murphy B (2004) Prior infection and passive transfer of neutralizing antibody prevent replication of severe acute respiratory syndrome coronavirus in the respiratory tract of mice. J Virol 78(7):3572–3577. 10.1128/jvi.78.7.3572-3577.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts A, Paddock C, Vogel L, Butler E, Zaki S, Subbarao K (2005) Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J Virol 79(9):5833–5838. 10.1128/JVI.79.9.5833-5838.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, Subbarao K (2010) Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol 84(3):1289–1301. 10.1128/JVI.01281-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts A, Vogel L, Guarner J, Hayes N, Murphy B, Zaki S, Subbarao K (2005) Severe acute respiratory syndrome coronavirus infection of golden Syrian hamsters. J Virol 79(1): 503–511. 10.1128/JVI.79.1.503-511.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCray PB Jr, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S (2007) Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol 81(2):813–821. 10.1128/JVI.02012-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tseng CT, Huang C, Newman P, Wang N, Narayanan K, Watts DM, Makino S, Packard MM, Zaki SR, Chan TS, Peters CJ (2007) Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human angiotensin-converting enzyme 2 virus receptor. J Virol 81(3):1162–1173. 10.1128/JVI.01702-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Brand JM, Haagmans BL, Leijten L, van Riel D, Martina BE, Osterhaus AD, Kuiken T (2008) Pathology of experimental SARS coronavirus infection in cats and ferrets. Vet Pathol 45(4):551–562. 10.1354/vp.45-4-551 [DOI] [PubMed] [Google Scholar]
- 11.Chu YK, Ali GD, Jia F, Li Q, Kelvin D, Couch RC, Harrod KS, Hutt JA, Cameron C, Weiss SR, Jonsson CB (2008) The SARS-CoV ferret model in an infection-challenge study. Virology 374(1):151–163. 10.1016/j.virol.2007.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K (2007) A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog 3(1):e5. 10.1371/journal.ppat.0030005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu H, Chen Q, Yang G, He L, Fan H, Deng YQ, Wang Y, Teng Y, Zhao Z, Cui Y, Li Y, Li XF, Li J, Zhang NN, Yang X, Chen S, Guo Y, Zhao G, Wang X, Luo DY, Wang H, Yang X, Li Y, Han G, He Y, Zhou X, Geng S, Sheng X, Jiang S, Sun S, Qin CF, Zhou Y (2020) Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 369(6511): 1603–1607. 10.1126/science.abc4730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oladunni FS, Park JG, Pino PA, Gonzalez O, Akhter A, Allue-Guardia A, Olmo-Fontanez A, Gautam S, Garcia-Vilanova A, Ye C, Chiem K, Headley C, Dwivedi V, Parodi LM, Alfson KJ, Staples HM, Schami A, Garcia JI, Whigham A, Platt RN 2nd, Gazi M, Martinez J, Chuba C, Earley S, Rodriguez OH, Mdaki SD, Kavelish KN, Escalona R, Hallam CRA, Christie C, Patterson JL, Anderson TJC, Carrion R Jr, Dick EJ Jr, Hall-Ursone S, Schlesinger LS, Alvarez X, Kaushal D, Giavedoni LD, Turner J, Martinez-Sobrido L, Torrelles JB (2020) Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat Commun 11(1):6122. 10.1038/s41467-020-19891-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreau GB, Burgess SL, Sturek JM, Donlan AN, Petri WA, Mann BJ (2020) Evaluation of K18-hACE2 mice as a model of SARS-CoV-2 infection. Am J Trop Med Hyg 103(3): 1215–1219. 10.4269/ajtmh.20-0762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang RD, Liu MQ, Chen Y, Shan C, Zhou YW, Shen XR, Li Q, Zhang L, Zhu Y, Si HR, Wang Q, Min J, Wang X, Zhang W, Li B, Zhang HJ, Baric RS, Zhou P, Yang XL, Shi ZL (2020) Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell 182(1):50–58.e8. 10.1016/j.cell.2020.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun SH, Chen Q, Gu HJ, Yang G, Wang YX, Huang XY, Liu SS, Zhang NN, Li XF, Xiong R, Guo Y, Deng YQ, Huang WJ, Liu Q, Liu QM, Shen YL, Zhou Y, Yang X, Zhao TY, Fan CF, Zhou YS, Qin CF, Wang YC (2020) A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe 28(1):124–133. e4. 10.1016/j.chom.2020.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandrashekar A, Liu J, Martinot AJ, McMahan K, Mercado NB, Peter L, Tostanoski LH, Yu J, Maliga Z, Nekorchuk M, Busman-Sahay K, Terry M, Wrijil LM, Ducat S, Martinez DR, Atyeo C, Fischinger S, Burke JS, Slein MD, Pessaint L, Van Ry A, Greenhouse J, Taylor T, Blade K, Cook A, Finneyfrock B, Brown R, Teow E, Velasco J, Zahn R, Wegmann F, Abbink P, Bondzie EA, Dagotto G, Gebre MS, He X, Jacob-Dolan C, Kordana N, Li Z, Lifton MA, Mahrokhian SH, Maxfield LF, Nityanandam R, Nkolola JP, Schmidt AG, Miller AD, Baric RS, Alter G, Sorger PK, Estes JD, Andersen H, Lewis MG, Barouch DH (2020) SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science 369(6505):812–817. 10.1126/science.abc4776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh DK, Singh B, Ganatra SR, Gazi M, Cole J, Thippeshappa R, Alfson KJ, Clemmons E, Gonzalez O, Escobedo R, Lee TH, Chatterjee A, Goez-Gazi Y, Sharan R, Gough M, Alvarez C, Blakley A, Ferdin J, Bartley C, Staples H, Parodi L, Callery J, Mannino A, Klaffke B, Escareno P, Platt RN 2nd, Hodara V, Scordo J, Gautam S, Vilanova AG, Olmo-Fontanez A, Schami A, Oyejide A, Ajithdoss DK, Copin R, Baum A, Kyratsous C, Alvarez X, Ahmed M, Rosa B, Goodroe A, Dutton J, Hall-Ursone S, Frost PA, Voges AK, Ross CN, Sayers K, Chen C, Hallam C, Khader SA, Mitreva M, Anderson TJC, Martinez-Sobrido L, Patterson JL, Turner J, Torrelles JB, Dick EJ Jr, Brasky K, Schlesinger LS, Giavedoni LD, Carrion R Jr, Kaushal D (2021) Author correction: responses to acute infection with SARS-CoV-2 in the lungs of rhesus macaques, baboons and marmosets. Nat Microbiol 6(3):413. 10.1038/s41564-021-00867-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu J, Tostanoski LH, Peter L, Mercado NB, McMahan K, Mahrokhian SH, Nkolola JP, Liu J, Li Z, Chandrashekar A, Martinez DR, Loos C, Atyeo C, Fischinger S, Burke JS, Slein MD, Chen Y, Zuiani A, Lelis FJN, Travers M, Habibi S, Pessaint L, Van Ry A, Blade K, Brown R, Cook A, Finneyfrock B, Dodson A, Teow E, Velasco J, Zahn R, Wegmann F, Bondzie EA, Dagotto G, Gebre MS, He X, Jacob-Dolan C, Kirilova M, Kordana N, Lin Z, Maxfield LF, Nampanya F, Nityanandam R, Ventura JD, Wan H, Cai Y, Chen B, Schmidt AG, Wesemann DR, Baric RS, Alter G, Andersen H, Lewis MG, Barouch DH (2020) DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 369(6505): 806–811. 10.1126/science.abc6284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rathnasinghe R, Strohmeier S, Amanat F, Gillespie VL, Krammer F, Garcia-Sastre A, Coughlan L, Schotsaert M, Uccellini M (2020) Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. bioRxiv. 10.1101/2020.07.06.190066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, Wei Q, Yu P, Xu Y, Qi F, Qu Y, Li F, Lv Q, Wang W, Xue J, Gong S, Liu M, Wang G, Wang S, Song Z, Zhao L, Liu P, Zhao L, Ye F, Wang H, Zhou W, Zhu N, Zhen W, Yu H, Zhang X, Guo L, Chen L, Wang C, Wang Y, Wang X, Xiao Y, Sun Q, Liu H, Zhu F, Ma C, Yan L, Yang M, Han J, Xu W, Tan W, Peng X, Jin Q, Wu G, Qin C (2020) The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 583(7818):830–833. 10.1038/s41586-020-2312-y [DOI] [PubMed] [Google Scholar]
- 23.Zheng J, Wong LR, Li K, Verma AK, Ortiz ME, Wohlford-Lenane C, Leidinger MR, Knudson CM, Meyerholz DK, McCray PB Jr, Perlman S (2021) COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589(7843): 603–607. 10.1038/s41586-020-2943-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Golden JW, Cline CR, Zeng X, Garrison AR, Carey BD, Mucker EM, White LE, Shamblin JD, Brocato RL, Liu J, Babka AM, Rauch HB, Smith JM, Hollidge BS, Fitzpatrick C, Badger CV, Hooper JW (2020) Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 5(19): e142032. 10.1172/jci.insight.142032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winkler ES, Bailey AL, Kafai NM, Nair S, McCune BT, Yu J, Fox JM, Chen RE, Earnest JT, Keeler SP, Ritter JH, Kang LI, Dort S, Robichaud A, Head R, Holtzman MJ, Diamond MS (2020) Publisher correction: SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat Immunol 21(11):1470. 10.1038/s41590-020-0794-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JG, Oladunni FS, Chiem K, Ye C, Pipenbrink M, Moran T, Walter MR, Kobie J, Martinez-Sobrido L (2020) Rapid in vitro assays for screening neutralizing antibodies and antivirals against SARS-CoV-2. J Virol Methods 287:113995. 10.1016/j.jviromet.2020.113995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alfson KJ, Griffiths A (2018) Development and testing of a method for validating chemical inactivation of Ebola virus. Viruses 10(3):126. 10.3390/v10030126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boivin GP, Bottomley MA, Schiml PA, Goss L, Grobe N (2017) Physiologic, Behavioral, and histologic responses to various euthanasia methods in C57BL/6NTac male mice. J Am Assoc Lab Anim Sci 56(1):69–78 [PMC free article] [PubMed] [Google Scholar]
- 29.Fawell JK, Thomson C, Cooke L (1972) Respiratory artefact produced by carbon dioxide and pentobarbitone sodium euthanasia in rats. Lab Anim 6(3):321–326. 10.1258/002367772781006185 [DOI] [PubMed] [Google Scholar]
- 30.Start RD, Layton CM, Cross SS, Smith JH (1992) Reassessment of the rate of fixative diffusion. J Clin Pathol 45(12):1120–1121. 10.1136/jcp.45.12.1120 [DOI] [PMC free article] [PubMed] [Google Scholar]