

Abstract
Extracellular vesicles (EVs) are nano‐sized membranous structures involved in intercellular communication and various physiological and pathological processes. Here, we present a novel method for rapid (within 15 min), large‐scale production of high‐purity EVs using eMTDΔ4, a peptide derived from Noxa. The treatment of mesenchymal stem cells derived from human Wharton's jelly after trypsinization and subsequent eMTDΔ4 stimulation in a chemically defined sucrose buffer with orbital shaking led to a substantial increase (approximately 30‐fold) in EV production with markedly high purity (approximately 45‐fold). These EVs (TS‐eEVs) showed higher regenerative and immunomodulatory potential than natural EVs obtained from the culture media after 48 h. The calcium chelator BAPTA‐AM and calpain inhibitor ALLM, but not the natural EV biogenesis inhibitor GW4869, blocked the TS‐eEV production induced by eMTDΔ4, indicating that the eMTDΔ4‐mediated regulation of intracellular calcium levels and calpain activity are closely associated with the rapid, mass production of TS‐eEVs. The present study may lead to considerable advances in EV‐based drug development and production of stem cell‐derived EVs for cell therapy.
Keywords: anti‐inflammatory potential, drug delivery, eMTDΔ4, EV production, extracellular vesicles, mitochondrial targeting domain
1. INTRODUCTION
Extracellular vesicles (EVs) are a promising vector for the delivery of various therapeutic compounds, including RNA, proteins, and chemicals (Agrahari et al., 2019; Kalluri & LeBleu, 2020; Song et al., 2020; Van Niel et al., 2018). EVs derived from patients and stem cells are biocompatible and immunologically inert, whereas those obtained from immune cells can modulate the immune system (Van Niel et al., 2018). EVs can transmigrate through physiological barriers, such as the blood–brain barrier and placenta (Song et al., 2020). These novel characteristics have encouraged their application as drug delivery vehicles for the treatment of cancer, infections, and hypoxic damage (Lener et al., 2015; Mizrak et al., 2013; Ophelders et al., 2016; Pan et al., 2014). However, drug delivery and therapeutic strategies using EVs are less explored owing to their limited production.
Depending on their biogenesis, EVs are generally classified into two categories, namely, exosomes and microvesicles (MVs), although their highly heterogeneous nature restricts strict refinement (Van Niel et al., 2018). Exosomes are derived from intraluminal vesicles and secreted by the fusion of multivesicular endosomes with the plasma membrane. Exosome biogenesis is complex and closely associated with the endosomal system (Warren & Vales, 1972; Wolf, 1967). By contrast, MVs are derived from the outward budding of the plasma membrane. Apoptotic bodies are also generated from the plasma membrane by direct outward budding during apoptosis, showing heterogeneous sizes ranging from 50 nm to 1000 nm (Hristov et al., 2004). The genesis of MVs may be much simpler than that of exosomes, and MVs have also been described as inactive byproducts of cellular stress. An increase in intracellular calcium concentration induces membrane phospholipid arrangement by amino‐phospholipid translocases and scramblases, and the disruption of the cytoskeleton by calpain via the hydrolysis of actin‐binding proteins results in the protrusion of the plasma membrane and the formation of MVs (Al‐Nedawi et al., 2008).
Extracellular vesicles have various physiological and pathological functions (Herrera et al., 2010; Liang et al., 2020; Merino‐Gonzã¡Lez et al., 2016; Ratajczak et al., 2006; Sabin & Kikyo, 2014). In addition to their role in blood coagulation (Sims et al., 1988), EVs act as mediators of intercellular communication between various cell types, including cancer and stem cells (Al‐Nedawi et al., 2008) (Phinney & Pittenger, 2017). EVs released from stem cells can function as paracrine mediators in tissue repair, as shown in animal models of various diseases, such as myocardial infarction (Arslan et al., 2013), limb ischemia (Hu et al., 2015), and CCl4‐induced liver fibrosis (Tan et al., 2014). EVs released from cancer cells, also called oncosomes, carry oncogenic proteins, such as EGFRvII (Al‐Nedawi et al., 2008; Di Vizio et al., 2009). EVs release by tumours can promote metastasis of cancer cells by forming a pre‐metastatic niche at metastatic sites (Jaiswal & Sedger, 2019).
Noxa is a pro‐apoptotic Bcl‐2 homology 3‐only protein belonging to the Bcl‐2 family. The mitochondrial targeting domain (MTD) region of Noxa was originally identified as the delivery domain of Noxa to the mitochondria (Seo et al., 2003). In addition, the MTD region is responsible for mitochondrial fragmentation induced by Noxa (Woo et al., 2009). The MTD peptide was subsequently recognized as a strong necrosis inducer when conjugated with a cell‐penetrating peptide, such as the eight‐arginine (R8) peptide. The R8:MTD peptide causes rapid calcium spikes in the cytoplasm within a few minutes, resulting in necrotic cell death; however, the MTD peptide itself without cell‐penetrating peptide conjugation does not cause cell death (Seo et al., 2009). Furthermore, we reported that the extended MTD (eMTD) itself without cell‐penetrating peptide conjugation induced necrosis via mitochondrial catastrophe (Han et al., 2019). The eMTD and eMTDΔ4 peptide, a form of eMTD with four amino acids deleted, penetrates the cell membrane and binds to voltage‐dependent anion channels (VDACs) to open the mitochondrial permeability transition pore. eMTDΔ4 causes cell membrane blebbing, cytosolic calcium influx, and mitochondrial swelling and fragmentation. These events caused by the eMTD or eMTDΔ4 peptide can be inhibited by the VDAC inhibitor isothiocyanate groups of 4,4′‐diisothiocyanatostilbene‐2,2′‐disulfonate (Han et al., 2019; Park et al., 2019).
In this study, we serendipitously observed that high amounts of EVs could be obtained when trypsinized stem cells were treated with eMTDΔ4 in a chemically defined sucrose buffer with orbital shaking. Moreover, high‐purity EVs of similar sizes were generated very rapidly (within 15 min) with a high production efficiency (approximately 30‐fold increase) compared to the natural EVs that were obtained from the culture media of non‐treated cells after 48 h (natural EVs). Several EV markers were observed in EVs produced using eMTDΔ4 treatment (TS‐eEVs). Moreover, TS‐eEVs showed identical or improved regenerative and immunomodulatory activities compared to those of natural EVs. Time‐lapse recording using a confocal microscope showed that TS‐eEVs originated from outward budding of the plasma membrane. Furthermore, the calcium‐chelating agent BAPTA‐AM and calpain inhibitor ALLM reduced the synthesis of TS‐eEVs, suggesting that eMTDΔ4‐mediated regulation of intracellular calcium levels and calpain activity is closely associated with the rapid production of TS‐eEVs.
2. MATERIALS AND METHODS
2.1. Cell culture
HeLa and HEK293 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 10% foetal bovine serum (FBS; Gibco‐Thermo Fisher, 16000044, Waltham, MA, USA) and 1X penicillin–streptomycin (100 units/ml and 100μg/ml, respectively; Gibco‐Thermo Fisher, 15140122, Waltham, MA, USA) in a humidified incubator at 37°C with 5% CO2. The experimental protocol for human Wharton's jelly‐derived mesenchymal stem cells (hWJ‐MSCs) was approved by the Institutional Review Board of Konkuk University (7001355‐202010‐BR‐407). The hWJ‐MSCs were cultured in α‐minimum essential medium (α‐MEM) supplemented with 10% FBS (Corning, 35‐010‐CV, NY, USA) and 1x penicillin–streptomycin, in a humidified incubator at 37°C with 5% CO2. HaCaT cells, normal human dermal fibroblasts (NHDF), and RAW264.7 cells were cultured in DMEM‐high glucose (Sigma, D6046, St Louis, MO, USA) containing 10% FBS (Corning) and 1X penicillin–streptomycin.
2.2. Peptide synthesis
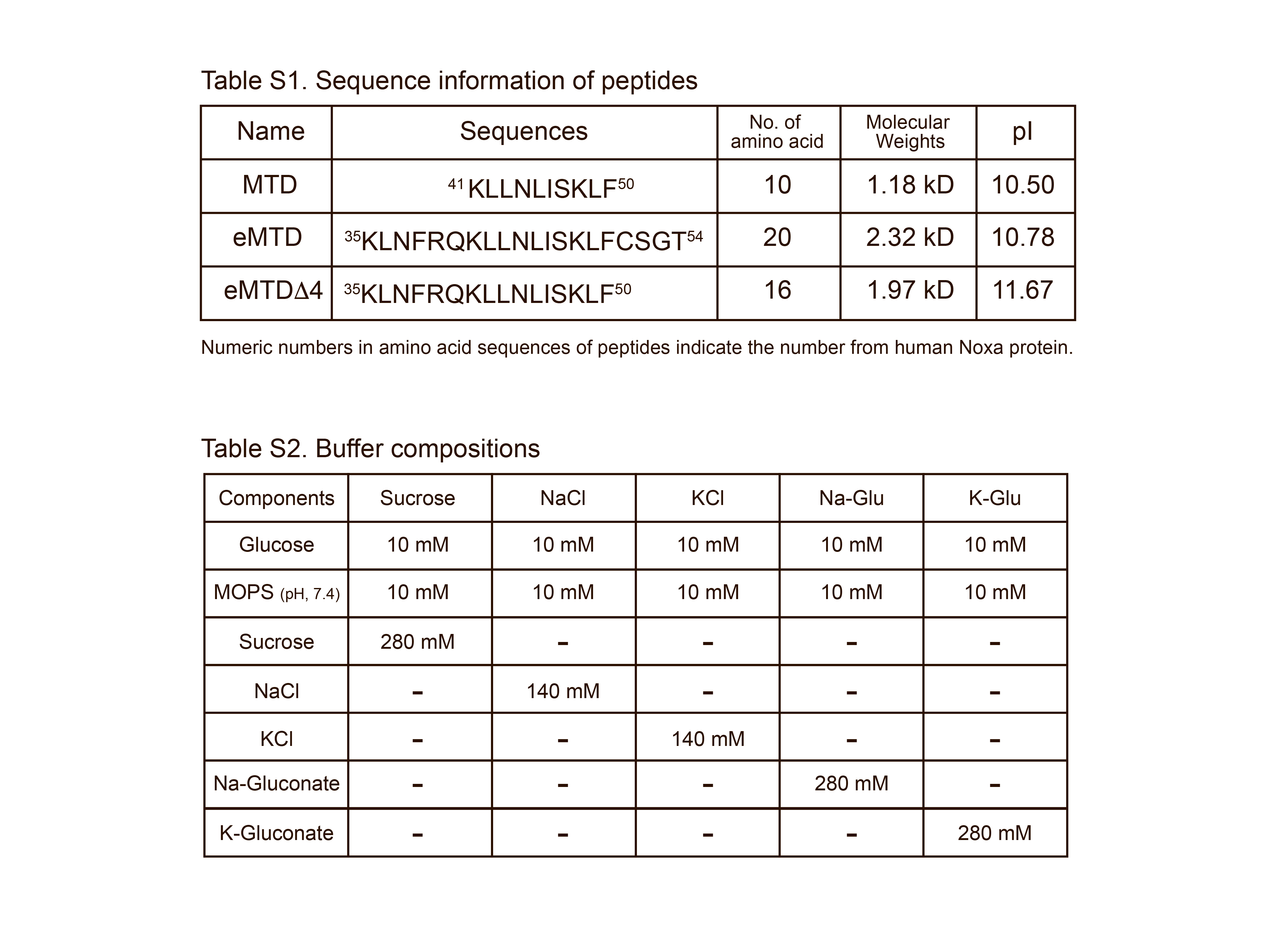
eMTDΔ4 peptide was synthesized and purified using high‐performance liquid chromatography (HPLC) at AnyGen (Gwangju, South Korea). Briefly, the Fmoc‐Phe‐wang resin was swelled with DMF for 30 min, washed twice with DMF, and then Fmoc was removed with 20% piperidine/DMF twice, followed by washing six times with DMF. For the next Fmoc‐A.A‐OH coupling, a solution of Fmoc‐A. A‐OH was added to HBTU and NMm in DMF and reacted for 1 h. The resin was washed three times with DMF, and then Fmoc was removed twice with 20% piperidine/DMF, followed by washing six times with DMF. These coupling and washing steps were repeated to synthesize eMTDΔ4. The crude synthesized eMTDΔ4 peptide was further purified using an HPLC system (Shimadzu HPLC Lab Solution, Tokyo, Japan) and SHIMADZU C18 analytical column (5 mm, 20 × 250 mm), and the final synthesized eMTDΔ4 peptide (> purity 95%) was lyophilized. The peptides were dissolved in distilled water and stored at −20°C. The peptide sequences are listed in Table S1.
2.3. Isolation of natural EVs and TS‐eEVs
hWJ‐MSCs were seeded onto a 150‐mm cell culture dish (SPL Life Sciences Co. Ltd., 11150, Seoul, Korea). The medium was replaced at 80%–90% confluency of MSCs, with α‐MEM supplemented with 10% exosome‐depleted FBS (Corning) according to the exosome‐depletion protocol (Shelke et al., 2014). Cells were cultured for 48 h and the medium was collected for EV isolation using the ultracentrifugation (UC) method (Thã©Ry et al., 2006). The number of cells was counted to measure the number of natural EVs produced per cell. The cells were then washed with sucrose buffer, suspended in 35 ml of sucrose buffer in a 50‐ml tube (SPL Life Sciences Co. Ltd., 50050, Seoul, Korea), treated with 1μM eMTDΔ4, and incubated in an orbital shaker (Celltron, Infors HT, Basel, Switzerland) rotating at 60 rpm for 15 min. EVs were isolated from conditioned media by differential centrifugation at 200 × g for 10 min, 2000 × g for 10 min, 10,000 × g for 30 min, and 120,000 × g for 2 h. The pellet was resuspended in 200μl of phosphate‐buffered saline (PBS).
2.4. Density gradient ultracentrifugation (DG) for EV purification
Highly purified EVs were obtained using the OptiPrep (iodixanol) DG protocol (Greening et al., 2015). An iodixanol solution (0.25 M sucrose/10 mM Tris, pH 7.5) was layered in tubes every 3 ml at 50, 30, and 10% (w/v). EVs purified using UC were loaded onto the top and centrifuged at 120,000 × g using a SW 41 Ti rotor at 4°C for 2 h. A 1‐ml aliquot of the 10%–30% layer was resuspended in 10 ml PBS and centrifuged at 120,000 × g at 4°C for 2 h to remove iodixanol. The resulting pellet was resuspended in 200μl of PBS.
2.5. Characterization of EVs
The number of particles in the purified EVs was measured using a nanoparticle tracking analyser (Nanosight NS300, Amesbery, UK), and protein quantification was performed using a BCA protein analysis kit (Thermo Fisher Scientific, 23227, MA, USA) according to the manufacturer's protocol. The number of EVs was calculated using the nanoparticle tracking analyser (Nanosight NS300) with the following settings: number of captures: 4, capture duration: 30 s, screen gain: 12, camera level: 11, threshold: 2, and temperature: 25°C. The purity of the EVs was expressed as the number of particles per microgram of protein. EVs were negatively stained with 1% uranyl acetate and visualized using a transmission electron microscope (TEM) operating at 80 kV (JEM‐1011, JEOL, Tokyo, Japan).
Extracellular vesicles were captured using exosome‐human CD9 flow detection reagent (Invitrogen, 10620D, Carlsbad, CA, USA), and surface membrane proteins were measured using a flow cytometer (Beckman Coulter/CytoFLEX) with antibodies against CD9‐BV421 (BD Bioscience, 743047, Franklin lakes, NJ, USA), CD63‐PE (BD Bioscience, 556020), and CD81‐APC (Miltenyi Biotec, 130‐119‐787, Bergisch Gladbach, Germany). EVs and whole cell lysates were separated using 4%–12% Bis‐Tris Flus Gels (Invitrogen, NW04125BOX and NW04122BOX) and transferred to a nitrocellulose membrane (Invitrogen, IB23001). The membranes were then incubated overnight at 4°C with antibodies against CD9 (Abcam, ab263023, Cambridge, UK), CD63 (Invitrogen, 10628D), CD81 (Santa Cruz, sc‐7637, Dallas, TX, USA), GM130 (Cell Signalling Technology, 12480, Danvers, MA, USA), calnexin (Santa Cruz, sc‐46669), Hsp70 (Cell Signalling Technology, #4872), Hsp90 (Cell Signalling Technology, #3350), flotilin (Cell Signalling Technology, #18634), ApoA1 (Cell Signalling Technology, #4919), cytochrome c (Cell Signalling Technology, #4280s), albumin (Cell Signalling Technology, #4872), and anti‐ApoB (Santa Cruz, sc‐393636). The membrane was washed and incubated with horseradish peroxidase‐conjugated anti‐rabbit IgG (Cell Signalling Technology, 7074) or anti‐mouse IgG (Cell Signalling Technology, 7076) at room temperature for 2 h. Protein bands were detected using enhanced chemiluminescence, captured with iBright™ (Invitrogen, CL‐1000), and analysed using the iBright analysis software. Some bands were detected with KwikQuant Imager (Kindle Biosciences, Greenwich, CT, USA), and were inverted by KwikQuant Image Analyzer 5.2.
2.6. Cellular uptake of EVs
EVs were stained with 2.5μg/ml DiR (Invitrogen, D12731) at room temperature for 1 h. Unbound dye was removed by centrifugation at 120,000 × g for 2 h. HaCaT cells were treated with 3 × 109 labelled‐EVs for 6 h; 4′, 6‐diamidino‐2‐phenylindole (DAPI; VECTASHIELD® Antifade Mounting Medium with DAPI, H‐1200) and CellMask™ (Green Plasma Membrane Stain, Invitrogen, C37608) were used for cell staining according to the manufacturer's protocol. Stained cells were observed under a confocal laser microscope (Carl Zeiss LSM 800).
2.7. Cell viability assay
HaCaT cells and NHDFs were seeded in a 96‐well plate (SPL, 30096) at a density of 1 × 104 cells/well. After 24 h, the medium was replaced with DMEM‐high glucose containing 10% exosome‐depleted FBS and 1X penicillin–streptomycin. The cells were then treated with natural EVs or TS‐eEVs (1 × 106, 1 × 107, 1 × 108, or 1 × 109 particles). After 24 h, the cells were incubated with Ez‐cytox (DOGEN, EZ‐3000, Seoul, Korea) for 1 h and the absorbance at 450 nm was measured using a Bio‐RAD x‐Mark spectrophotometer (Bio‐Rad Laboratories, Hercules, CA, USA).
2.8. In vitro scratch/wound healing assay
HaCaT cells were seeded onto a 6‐well plate (SPL, 32006) at a density of 6 × 105 cells/well and cultured to 100% confluence, followed by treatment with 10μg/ml mitomycin C (Sigma, M4287) at 37°C for 2 h to inhibit cell proliferation. The cells were scratched with a 1000‐μl micropipette tip to create a cell‐free area. Subsequently, the cells were cultured in DMEM‐high glucose supplemented with 10% exosome‐depleted FBS (control) and a medium containing natural EVs and TS‐eEVs (1 × 109 particles/ml). In vitro scratch closure was examined, and the wound closure area was assessed using TSratch software (Gebäck et al., 2009).
2.9. Transwell migration
NHDFs were seeded in the upper chamber of an 8‐μm pore Transwell (Costar, 3422, NY, USA) with DMEM‐high glucose containing 10% exosome‐depleted FBS. After 12 h, serum‐free medium containing EVs was added to the lower chamber. After 24 h, cells were washed with PBS, fixed with 4% paraformaldehyde (Biosesang, P2031, Seongnam‐si, Korea) for 15 min, and stained with 1% crystal violet (Sigma, V5265). Relative cell migration was analysed using the ImageJ software.
2.10. Western blot of EVs‐treated NHDFs
NHDFs treated with PBS or EVs were lysed using 1x RIPA buffer (LPS solution, CBR002, Daejeon, Korea) containing a protease inhibitor (Invitrogen, 87786) to assess the protein expression level. Subsequently, 10μg of protein was quantified using a BCA protein assay kit, and the primary antibodies used for western blotting were p‐STAT3 (CST, 9131S), STAT3 (CST, 9139S), p‐AKT (Santa Cruz, sc‐293125), AKT (CST, 9272S), p‐ERK (Santa Cruz, sc‐7383), ERK (CST, 9106S), p‐GSK‐3β (Santa Cruz, sc‐373800), GSK‐3β (Santa Cruz, sc‐377213), β‐catenin (Santa Cruz, sc‐7963), and β‐actin (Santa Cruz, sc‐47778). Protein signals were detected with iBright™ (Invitrogen, CL‐1000) and analyzed using the iBright analysis software.
2.11. Measurement of nitric oxide (NO) production
RAW264.7 Cells were seeded onto a 24 well plate at a density of 1.5 × 105 cells/well. After 12 h, cells in 500μl of cell culture medium containing 10% exosome‐depleted FBS were treated with 10 ng/ml LPS (Sigma, L4391) and 1 × 108 or 1 × 109 EVs. After 18 h, the medium was collected and mixed with Griess reagent [0.1% N‐(1‐naphthyl) ethylenediamine dihydrochloride and 1% sulfanilamide in 5% phosphoric acid)], and the absorbance was measured at 540 nm.
2.12. Real‐time polymerase chain reaction (RT‐PCR) and enzyme‐linked immunosorbent assay (ELISA) analysis of inflammatory cytokine expression
Total RNA was isolated from RAW264.7 cells and NHDFs were treated with PBS or EVs using Labozol reagent (Cosmo Gentech, Seoul, Korea) according to the manufacturer's protocol. The purified RNA was quantified using a NanoPhotometer (Implen, Munich, Germany). cDNA synthesis was performed with 2μg of total extracted RNA using an M‐MuLV Reverse Transcription Kit (Cosmo Gentech, Seoul, Korea). Quantitative real‐time PCR was performed using EzAMP PCR 2x Master Mix (ELPIS Biotech, Daejeon, Korea) according to the manufacturer's recommendations. The culture medium of treated RAW264.7 cells was analysed for tumour necrosis factor (TNF)‐α (Peprotech, BGK06804, Cranbury, NJ, USA) and Interleukin (IL)‐6 (Peprotech, BGK08505) production using ELISA. A Bio‐RAD x‐Mark spectrophotometer was used to measure the absorbance at 450 nm.
2.13. In vivo wound healing assay
Six‐week‐old BALB/c nude female mice were purchased from Orient Bio Animal Center (Seongnam City, Korea). The in vivo experiments were performed with approval from the Konkuk University Animal Care Committee (IACUC; approval number: KU20132‐1). The mice were anesthetized using an intraperitoneal injection of 60 mg/kg alfaxalone (Alfaxan, Careside, Gyeonggi‐do, South Korea), and two full‐thickness skin wounds were induced using a sterile biopsy punch (8‐mm diameter, Kai Industries, Tokyo, Japan) on their backs. Each mouse was injected subcutaneously at three points around each wound with PBS (control) and 1 × 109 particles suspended in 30μl of PBS. The size of the wound was measured initially (day 0) and further monitored on days 0, 3, 5, 7, and 9.
2.14. TS‐eEV biogenesis study
To study the biogenesis of TS‐eEVs, hWJ‐MSCs were pre‐treated with 50μM BAPTA‐AM (MedChem Express, HY‐100545, Monmouth Junction, NJ, USA) for 1 h, 10μM ALLM (Santa Cruz Biotechnology, sc‐201268) for 18 h, or 10μM GW4869 (Sigma, D1692) for 24 h. EVs were isolated following the method described for TS‐eEV isolation, and the number of particles was measured using nanoparticle tracking analysis (NTA).
2.15. Isolation of plasma membrane fractions
The mCherry ORF was fused with the TM region (645‐RRRHIVRKRTLRRLLQ‐660) of EGFR (Sengupta et al., 2009) in the expression vector pLenti‐x‐puro‐DEST (Addgene). HeLa cells were transfected with mCherry alone or mCherry‐TM expression plasmids using Effectene (Qiagen). Transfected HeLa cells were fractionated using the Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit (Invent Biotechnologies, Inc., Plymouth, MN, USA). Cell fractionation was performed according to the manufacturer's instructions. The transfected HeLa cells (2 × 107 cells) were washed with cold PBS, resuspended in 500μl buffer A, incubated on ice for 5 min, and vortexed for 10 s. The cells were immediately transferred to a filter cartridge and centrifuged at 16,000 × g for 30 s. Native cell lysates with intact organelles were centrifuged at 700 × g for 1 min. The supernatants, which contained ruptured membranes and cytosolic components, were transferred to fresh tubes and centrifuged at 4°C for 30 min at 16,000 × g. Thereafter, supernatants containing cytosolic fractions were collected. The pellets in which the total membrane fractions were resuspended with 200μl buffer B were centrifuged at 7800 × g for 20 min at 4°C. Upon centrifugation, the supernatants (crude plasma membrane fraction) were transferred to fresh tubes and the pellets were collected as cellular organelle fractions. The crude plasma membrane fractions were centrifuged at 16,000 × g for 1 h. The pellets were then collected as plasma membrane fractions.
2.16. Statistical analyses
Statistical analyses were performed using the GraphPad Prism (version 7). Experiments were performed independently at least three times. Data are presented as the mean ± SD. The experimental groups were compared using one‐way analysis of variance (ANOVA) or repeated‐measures analysis of variance (RMANOVA) followed by post hoc analysis to determine statistical significance (p value). In all figures, the asterisk indicates statistical significance: not significant (n.s.) when p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001.
3. RESULTS
3.1. eMTDΔ4 induces rapid, mass production of EVs
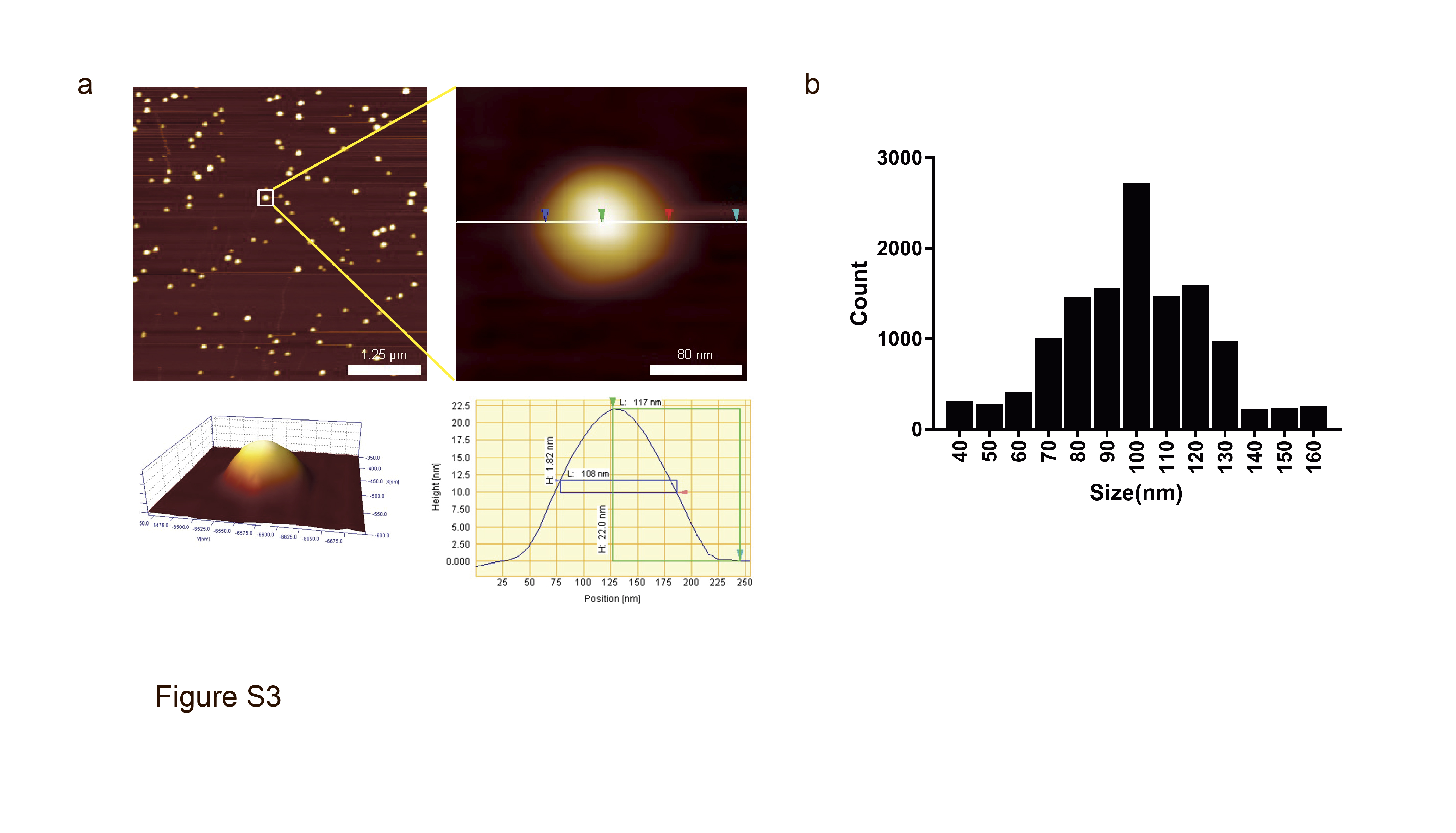
We treated HeLa cells with eMTDΔ4 in different buffers containing sucrose, NaCl, KCl, Na‐Glu, or K‐Glu and observed that treatment with eMTDΔ4 in sucrose buffer led to rapid (within 15 min) and increased production of tiny particles from the cells (Figure 1a and S1). The harvested particles were separated using SDS‐PAGE and protein sequences were determined using a MicroQ‐TOF III mass spectrometer (Figure S2c). We identified the bands as IQ motif‐containing GTPase activating protein 1, alpha‐actinin 1, valosin‐containing protein, Hsp90‐beta, and tubulin‐alpha (Figure S2a, c). The presence of Hsp90‐beta, an important EV marker, prompted us to speculate that these particles could possibly be EVs. Therefore, we tested them for CD63 expression, which is another EV marker. Western blot analysis revealed the presence of CD63 in these particles (Figure S2b). NTA and atomic force microscopy were used to determine the size range of the particles (80–120 nm; Figure 1b, c, S3a, b). These results strongly suggest that the particles produced using eMTDΔ4 treatment in sucrose buffer could be EVs [eMTDΔ4‐derived EVs, hereafter referred to as eEVs].
FIGURE 1.
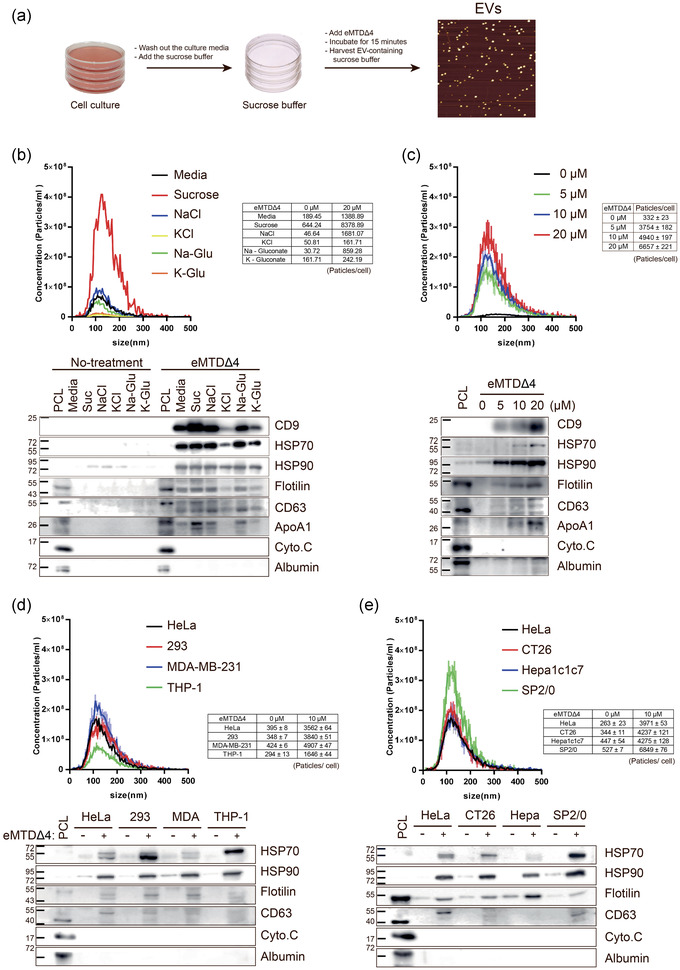
The 4 amino acid‐deleted form of extended mitochondrial targeting domain (eMTDΔ4) induces the formation of extracellular vesicles (EVs) in sucrose buffer (a) Schematic diagram of eEV production. The cells were cultured in respective media containing 10% FBS. The culture media were removed; sucrose buffer containing eMTDΔ4 was added to the cells, followed by incubation for 15 min. The sucrose buffer containing eEVs was harvested for purification. (b) HeLa cells were treated with eMTDΔ4 (20μM) in culture media or buffers containing sucrose, NaCl, KCl, Na‐gluconate, or K‐gluconate for 15 min, and eEVs were purified using Amicon Ultra centrifugal Filters (100 kDa cut‐off; Ireland Ltd., Germany). eEVs were counted using nanoparticle tracking analysis (NTA; top panel). The lysates of eEVs were subjected to SDS‐PAGE and western blot analysis. The lysates of HeLa cells cultured in DMEM (PCL) were used as positive controls. The levels of positive (CD9, Hsp70, Hsp90, Flotilin, CD63, and ApoA1) and negative markers (cytochrome C and Albumin) were assessed (bottom panel). (c) The eEVs were produced as described above using different amounts of eMTDΔ4 and subjected to NTA and western blot analysis. (d, e) eEVs produced by human (HeLa, HEK293, MDA‐MB‐231, and THP‐1) (d) or mouse cells (CT26, Hepa1c1c7, and SP2/0) (e) were characterized as described above
To determine the optimal conditions for eEV production, we determined the number of EVs produced upon eMTDΔ4 treatment in different buffers (Table S2) and analysed the expression of EV, mitochondrial, or serum markers (Figure 1b, c). The EVs produced upon eMTDΔ4 treatment in sucrose buffer had high levels of EV protein markers such as CD9, Hsp70, Hsp90, flotilin, and CD63. However, cytochrome c, a mitochondrial protein marker, was not detected in the eEVs. Because eEVs were produced in a chemically defined sucrose buffer without serum, no albumin was detected in the eEVs. We then optimized the eMTDΔ4 concentration in sucrose buffer and found that eEV production in HeLa cells increased with an increase in eMTDΔ4 concentration and reached the highest level at 20μM eMTDΔ4. The levels of EV markers such as CD9, Hsp70, Hsp90, flotilin, CD63, and ApoA1 significantly increased with an increase in eMTDΔ4 concentration (Figure 1c). Furthermore, we tested the effect of eMTDΔ4 treatment on EV production in various human cell lines, including HeLa, HEK293, MDA‐MB‐231, and THP‐1 cells (Figure 1d), and mouse cell lines, including CT26, Hepa1c1c7, and SP2/0 (Figure 1e). The results indicated that the EVs produced by eMTDΔ4‐treated human or mouse cells had a similar size (∼120 nm), although the number of EVs produced was slightly different. However, we noticed that the eEVs produced from different cells contained EV markers, such as Hsp70, Hsp90, flotilin, and CD63, but not mitochondrial (cytochrome c) or albumin (Figure 1d, e). Collectively, we demonstrated that eMTDΔ4 treatment in sucrose buffer can lead to rapid and high production of eEVs in different cells.
3.2. Isolation, characterization, and purity assessment of hWJ‐MSCs‐derived EVs
Next, we sought to determine the most efficient protocol for the production of high amounts of high‐purity EVs from hWJ‐MSCs. Cultured hWJ‐MSCs were washed with PBS and resuspended in sucrose buffer containing eMTDΔ4. In another set for comparison, cultured MSCs were trypsinized after washing and resuspended in sucrose buffer containing eMTDΔ4. MSCs were treated with chemically defined eMTDΔ4‐sucrose buffer for 15 min with or without orbital shaking (60 rpm) at 37°C and 5% CO2 (Figure 2a). We compared the production of natural EVs (EVs harvested from culture media), eEVs, and TS‐eEVs following two EV purification steps: UC and DG (Figure 2a). After UC purification, the TS‐eEV method (15 min) showed a remarkably high amount of purified EV per cell (2.39 × 104 ± 3.42 × 102 particles/cell) compared to natural EV yield (8.84 × 102 ± 2.66 × 102 particles/cell; Figure 2b). Moreover, total TS‐eEV yield (1.87 × 1012 ± 4.0 × 1010 particles/ml) was substantially higher (approximately 25‐fold) than natural EV production (7.25 × 1010 ± 2.38 × 109 particles/ml; Figure 2c). The TS‐eEV yield after successive DG (1.1 × 1010 ± 1.45 × 109 particles/ml) was still higher than that of natural EV (1.9 ×109 ± 2.16 × 108 particles/ml; Figure 2d), although the number of particles substantially decreased (160‐fold) during DG.
FIGURE 2.
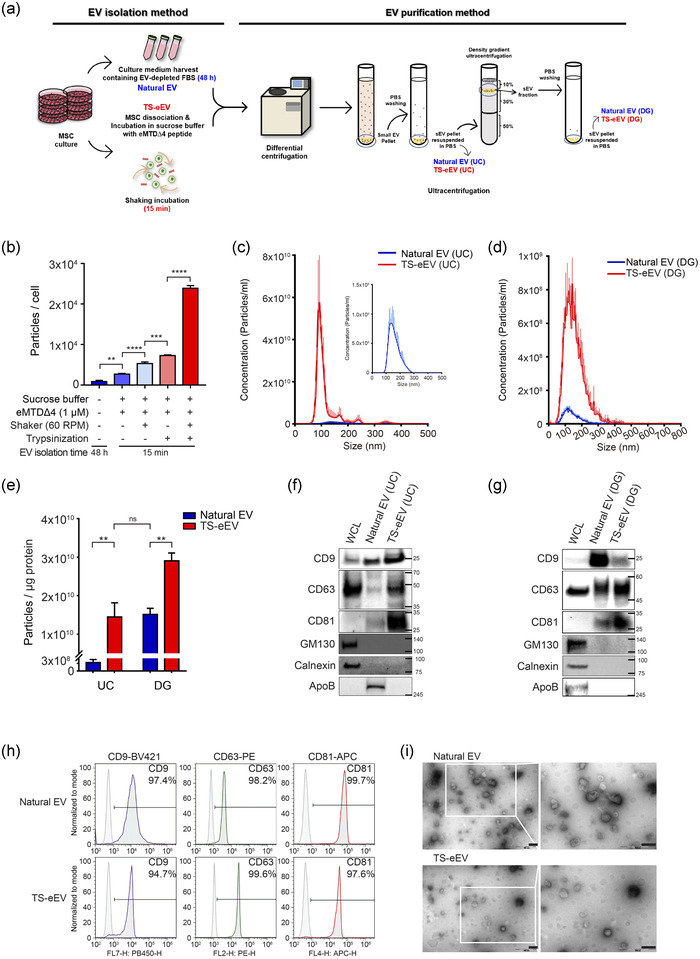
Characterization of natural EVs and TS‐eEVs derived from hWJ‐MSCs (a) Schematic diagram of the EV isolation and purification method. The Natural EV method isolated EVs from a culture medium containing exosome‐free FBS, and the TS‐eEV method isolated EVs using a sucrose buffer with defined chemical compositions. The differences in composition of EV isolation medium markedly affected EV purity. TS‐eEVs were further purified using ultracentrifugation (UC) and density gradient (DG) ultracentrifugation. (b) Bar graph showing EV yield represented by the number of EV particles per cell, produced from hWJ‐MSCs using the following methods: (1) the conventional method for natural EV production; (2) sucrose buffer and eMTDΔ4; (3) sucrose buffer, eMTDΔ4, and orbital shaking; (4) sucrose buffer, eMTDΔ4, and trypsinization; (5) sucrose buffer, eMTDΔ4, orbital shaking, and trypsinization (TS‐eEV). Data are presented as the mean ± SD of three independent experiments. Statistical significance was analysed using one‐way ANOVA. (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). (c, d) Nanoparticle tracking analysis (NTA) was used to measure the concentration of purified natural EVs (blue) and TS‐eEVs (red) using UC (c) and DG (d). (e) Purity is represented as the number of EV particles/μg protein. Statistical significance was determined using one‐way ANOVA. (n.s.: p > 0.05, **p ≤ 0.01) (f, g) Western blot analysis was used to analyse the expression levels of positive (CD9, CD63, and CD81) and negative markers (GM130, calnexin, and ApoB) in the same protein amount. The hWJ‐MSC lysate was used as the control. (h) Flow cytometry analysis of positive surface markers, including CD9, CD63, and CD81, in natural EVs and TS‐eEVs using anti‐CD9‐coated Dynabeads. (i) Morphology of natural EVs and TS‐eEVs as examined using transmission electron microscopy (TEM) with 1% phosphotungstic acid as a negative stain. Scale bar = 200 nm
Furthermore, after purification by UC, the purity of TS‐eEVs (1.44 × 1010 particles/μg protein) was markedly higher (approximately 45‐fold) than that of natural EVs (3.32 × 108 particles/μg protein). The purity of the TS‐eEVs purified using UC was comparable to that of natural EVs purified using UC and DG (1.51 × 1010 particles/μg protein; Figure 2e). Concurrently, western blot analysis of UC‐purified EVs showed that TS‐eEVs had higher amounts of EV‐positive markers, such as CD9, CD63, and CD81; however, no EV‐negative markers, including GM130 and calnexin, were detected (Figure 2f). Interestingly, the serum protein ApoB was detected in natural EVs after UC purification and disappeared after successive DG purification. However, ApoB was not detected in TS‐eEVs even after UC purification, confirming the high purity of the TS‐eEVs (Figure 2g). TS‐eEVs and natural EVs obtained from hWJ‐MSCs showed comparable levels of CD9, CD63, and CD81 expression (Figure 2h), and similar morphologies were observed in the TEM images (Figure 2i). Taken together, the TS‐eEV method for MSC‐derived EV production may markedly improve yield and EV purity compared to the conventional natural EV production method.
To compare the differential expression of miRNAs between natural EVs and TS‐eEVs, we performed the miRSeq analysis using natural EVs and TS‐eEVs. The miRNAs distribution scatter plot (upper panel of Figure S4a) and volcano plot (lower panel of Figure S4a) of natural EVs and TS‐eEVs indicated that miRNAs in natural EVs were very similar to those in TS‐eEVs. Moreover, 43 miRNAs (75.4%) of top 50 miRNAs found in natural EVs also discovered in TS‐eEVs, indicating that TS‐eEVs contain almost identical miRNAs contained in natural EVs (Figure S4b and S4c). Heatmap analysis showed similar miRNA expression patterns between natural EVs and TS‐eEVs, further supporting that the amounts of miRNAs contained in natural EVs are similar to those in TS‐eEVs. Interestingly, the miRNA expression patterns of natural EVs showed heterogenous miRNA amounts between batches (natural EV1, 2, and 3); however, TS‐eEVs showed no or little batch effects of miRNA expression patterns (Figure S4d), indicating the homogenous preparation of TS‐eEVs between baches. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed that natural EVs and TS‐eEVs contain miRNAs related to protein processing in ER, Hippo signalling, cell cycle and TGF‐beta signalling (Figure S4e), showing similar KEGG pathway found in signalling pathways for regenerative and anti‐inflammatory effect (Shirazi et al., 2021). Taken together, these results showed TS‐eEVs contain similar miRNAs contained in natural EVs.
3.3. MSC‐derived TS‐eEVs lead to significant enhancement of cell proliferation, wound healing, and anti‐inflammatory response
We extended the characterization of TS‐eEVs to test TS‐eEV internalization in vitro and biological activity of TS‐eEVs in vivo (Figure 3a). First, we observed the extent of EV internalization into HaCaT cells to investigate whether TS‐eEVs have biological functions similar to those of natural EVs. TS‐eEVs and natural EVs were labelled with the fluorescent probe DiR (red colour), and the cell membrane/cytoplasm and nucleus were labelled with CellMask (green colour) and DAPI (blue colour), respectively. EVs were detected inside the cells (Figure 3b), indicating that the cells may have similar internalization capacities for TS‐eEVs and natural EVs.
FIGURE 3.
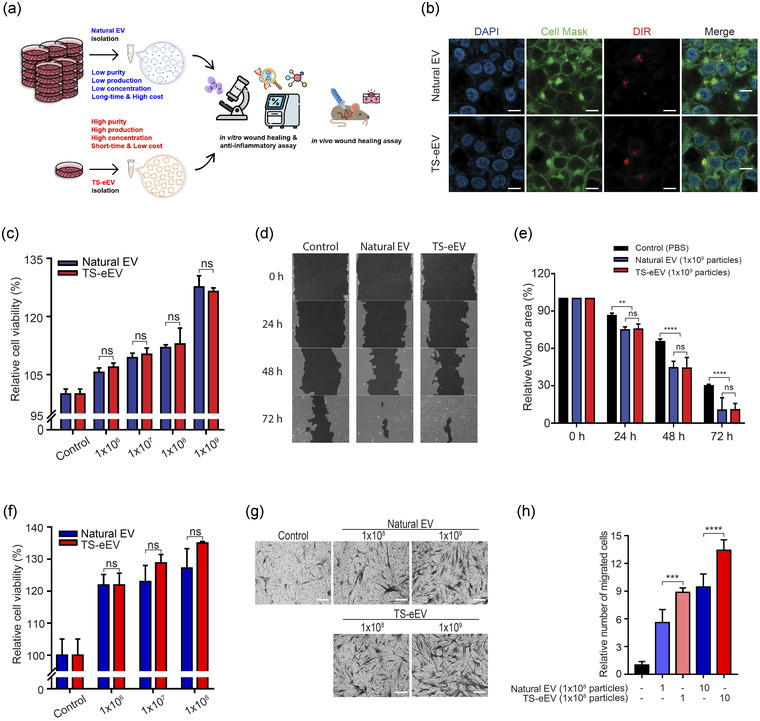
Effects of natural EVs and TS‐eEVs on the in vitro wound healing model. (a) Schematic summary of the EV isolation methods, in vitro and in vivo analysis. (b) Confocal microscopic images showing EV internalization by HaCaT cells. The internalization assay was performed at 37°C with a 6‐h incubation period. Scale bar = 40μm. (c) Cell viability assay following EV treatment of HaCaT cells. (n.s.: p > 0.05). (d) Effects of natural EVs and TS‐eEVs on the closure of in vitro wounds observed using HaCaT cells. (e) Graphical representation of the relative wound area in Figure 3d measured using the TScratch software. Statistical significance was analysed using RMANOVA with post hoc analysis. (n.s.: p > 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). (f) NHDF viability analysis of natural EVs and TS‐eEVs after treatment in a dose‐dependent manner (1 × 106, 1 × 107, and 1 × 108 particles) for 24 h. (g) Representative images of migratory capacity of EVs on NHDFs using crystal violet staining and Transwell assay. Scale bar = 250μm. (h) The relative migrated cells area was measured using the ImageJ software to determine the degree of staining. (***p ≤ 0.001, ****p ≤ 0.0001)
Furthermore, similar to natural ES, TS‐eEV treatment led to a dose‐dependent increase in HaCaT cell proliferation (Figure 3c). The exposure of HaCaT cells to 1 × 109 TS‐eEVs for 24 h resulted in an approximately 130% increase in cell proliferation, similar to that observed with natural EVs (Figure 3d). Moreover, similar to natural EVs, TS‐eEV treatment led to significant improvement in wound healing (Figure 3e). In addition, we verified the wound healing effect of the EVs produced by natural EV and TS‐eEV methods on normal human dermal fibroblasts (NHDFs). As a result of measuring the NHDF viability, it was confirmed that the cell proliferation rate was increased in a concentration‐dependent manner as in the HaCaT cells (Figure 3f). To investigate the migratory effect of EVs, NHDF cells were seeded in the upper chamber of the transwell and incubated for 24 h with the same number of EVs in the lower chamber. As a result, both the natural EVs and TS‐eEVs groups increased cell mobility, but the number of stained cells was slightly higher in the TS‐eEVs group than in the natural EVs group (Figure 3g, h).
We performed qRT‐PCR to examine changes in the mRNA expression of wound healing‐related genes in NHDF cells treated with EVs for 24 h (Figure 4a). To determine whether EVs could activate the regenerative signalling pathway in NHDF cells, we performed western blotting to evaluate the protein levels of p‐STAT3, p‐AKT, p‐ERK, p‐GSK‐3β, and β‐catenin (Figure 4b). The results indicate that both natural EVs and TS‐eEVs have wound healing effects through similar pathways.
FIGURE 4.
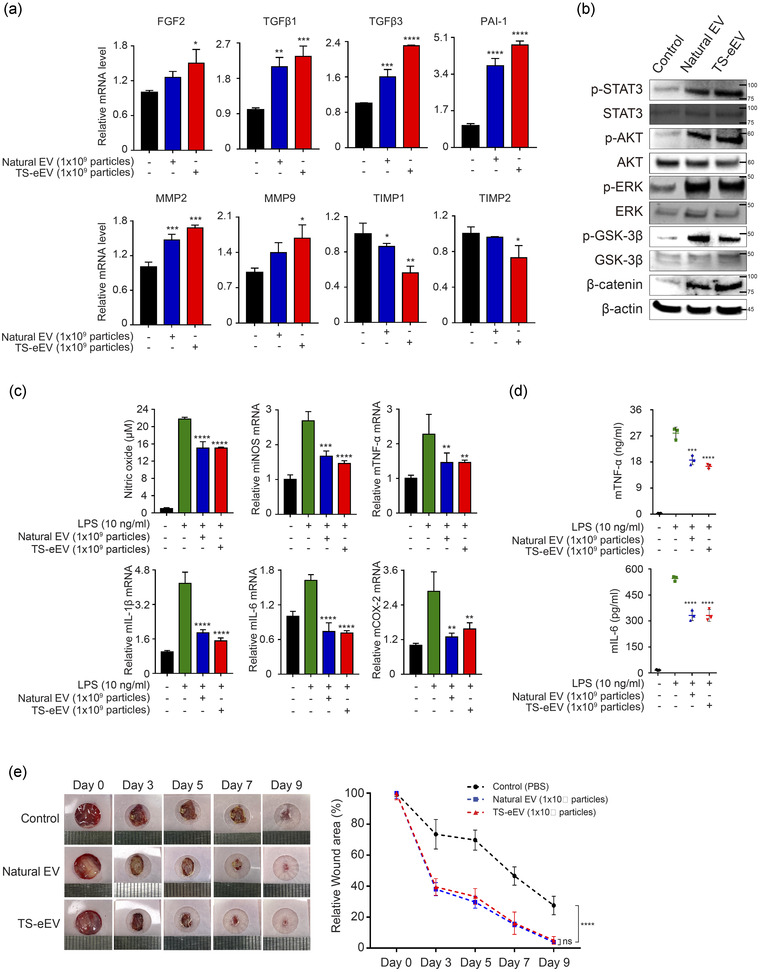
The therapeutic effects of natural EVs and TS‐eEVs on wound healing and inflammation. (a) The expression levels of genes related to wound healing were measured using qRT‐PCR. Expression levels of growth factors (FGF2, TGFβ1, TGFβ3) and genes related to cell migration ability (PAI‐1, MMP2, MMP9) were significantly increased in the EV‐treated group compared to those in the PBS control group. The expression of genes inhibiting migration ability (TIMP1 and TIMP2) decreased in the EV‐treated group. Experiments were performed in triplicate, and the results were normalized using GAPDH expression. FGF2, fibroblast growth factor 2; TGFβ1, transforming growth factor beta 1; TGFβ3, transforming growth factor beta 3; PAI‐1, plasminogen activator inhibitor‐1; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; TIMP1, TIMP metalloproteinase inhibitor 1; TIMP2, TIMP metalloproteinase inhibitor 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase. Statistical significance was analysed using one‐way ANOVA. (*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). (b) Western blot was used to determine the expression levels of STAT3, AKT, ERK, GSK‐3β, and β‐catenin proteins that play important roles in cell growth during wound healing in NHDF‐treated groups with PBS or EVs. It was established that the phosphorylated forms of STAT3, AKT, ERK, GSK‐3β were increased in the EV‐treated group compared to that in the control group. (c) An assessment of anti‐inflammatory potential of natural EVs and TS‐eEVs in RAW264.7 cells treated with PBS and LPS (10 ng/ml) for 18 h. NO content in the supernatant was measured. The relative mRNA levels of pro‐inflammatory genes including iNOS, TNF‐α, IL‐1β, IL‐6, and COX‐2 were determined using RT‐PCR; data were normalized with respect to GAPDH and were expressed as a fold change compared to untreated RAW264.7 cells (control). (n.s.: p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001). (d) ELISA data of pro‐inflammatory cytokine (TNF‐α and IL‐6) secreted by LPS‐induced RAW264.7 cells treated with PBS or EVs. (***p ≤ 0.001, ****p ≤ 0.0001). (e) Representative images of in vivo wound closure in BALB/c nude mice treated with PBS, Natural EVs, or TS‐eEVs, on days 0, 3, 5, 7, and 9. Graph representing the quantitative analysis of relative wound area after treatment with natural EVs and TS‐eEVs. Statistical significance was determined using RMANOVA with post hoc analysis. (n.s.: p > 0.05, ****p ≤ 0.0001)
We then tested the in vitro anti‐inflammatory activities of TS‐eEVs and natural EVs using RAW264.7 macrophages activated with 10 ng/ml LPS. TS‐eEV treatment markedly suppressed LPS‐induced nitric oxide (NO) production and significantly downregulated the expression of pro‐inflammatory genes, such as inducible nitric oxide synthase (iNOS), TNF‐α, IL‐1β, IL‐6, and cyclooxygenase 2 (COX‐2; Figure 4c). ELISA data showed low levels of pro‐inflammatory cytokines (TNF‐α and IL‐6) in culture supernatants from TS‐eEV‐ and natural EV‐treated macrophages (Figure 4d), confirming the high anti‐inflammatory activity of TS‐eEVs.
We also assessed the in vivo regenerative capacity of TS‐eEVs in a mouse model of wound healing. TS‐eEV treatment led to a significant improvement in wound healing compared to that in the PBS‐treated control group on day 9 after wound formation (Figure 4e). Therefore, TS‐eEV treatment significantly enhanced cell proliferation, wound healing capacity, and anti‐inflammatory activity.
Importantly, even though TS‐eEVs and natural EVs showed comparable potential in improving wound healing and anti‐inflammatory potential, the TS‐eEV method resulted in EVs with a much higher yield and purity within a shorter time than those generated with the natural EV method. As the TS‐eEV method generates a much higher number of EVs, its relative efficacy may be considered much higher than that of the natural EV method. (Figure 4f).
3.4. eMTDΔ4‐mediated regulation of intracellular calcium level and calpain activity is closely associated with rapid production of TS‐eEVs by outward budding of cell membrane
We investigated the mechanism underlying the eMTDΔ4‐mediated EV production. To visualize the cell membrane using fluorescent proteins, we generated mCherry‐TM protein, in which mCherry is fused to the transmembrane domain (TM) of EGFR (Sengupta et al., 2009). HeLa cells transfected with mCherry‐TM‐expressing vector showed that mCherry‐TM was well localized on the cell membrane; however, mCherry without TM was dispersed in the cytosol (Figure S5a). Furthermore, mEmerald‐CD9 was localized on the cell membrane as well as in other intracellular compartments (Figure S5a). The localization of mCherry‐TM in the cell membrane was further confirmed by cell membrane fractionation. The cell membrane fraction separated using a biochemical method and differential centrifugation contained mCherry‐TM protein, ERBB‐2, and CD‐9, but not mCherry alone, GM130, and HSP90 (Figure 5a), although there was some residual proportion of mCherry‐TM. ERBB‐2, CD9, and GM130 was detected in the organelle membrane fractions. These results confirmed that mCherry‐TM is localized to the cell membrane.
FIGURE 5.
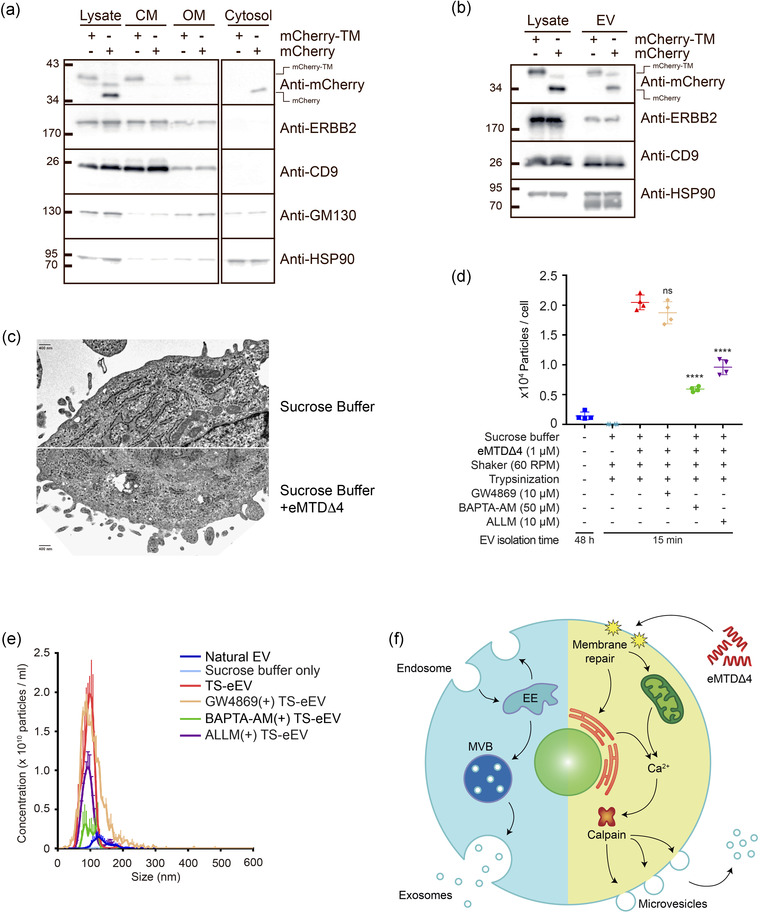
eMTDΔ4 induced formation of MVs (a) HeLa cells expressing mCherry‐TM or mCherry alone were fractionated using differential centrifugation. Western blot analysis was performed with anti‐mCherry, anti‐ERBB2, anti‐CD9, anti‐GM130, and anti‐HSP90 antibodies. (b) EVs and lysates were prepared from HeLa cells expressing mCherry‐TM or mCherry alone, and western blot analysis was performed with anti‐mCherry, anti‐ERBB2, anti‐CD9, and anti‐HSP90 antibodies. (c) Morphology of cell membranes treated with sucrose buffer alone or sucrose buffer containing eMTDΔ4 was examined using transmission electron microscopy (TEM) with 1% phosphotungstic acid as a negative stain. Scale bar = 400 nm. (d) Calcium dependence of TS‐eEV production was assessed using the calcium chelator BAPTA‐AM (50μM), the calpain inhibitor ALLM (10μM), and the nSMase inhibitor GW4869 (10μM). (n.s.: p > 0.05, ****p ≤ 0.0001). (e) NTA histogram showing EV concentrations and size distribution: natural EVs (blue), sucrose buffer only (sky blue), TS‐eEVs (red), TS‐eEVs treated with GW4869 (orange), TS‐eEVs treated with BAPTA‐AM (green), and TS‐eEVs treated with ALLM (purple). (d) Schematic diagram of MVs generated using eMTDΔ4 treatment
To address the question of whether EVs generated by eMTDΔ4 originate from the cell membrane, eEVs were prepared from HeLa cells expressing mCherry‐TM or mCherry alone. The mCherry‐TM protein, as well as other membrane proteins such as ERBB2 and CD9, were present in eEVs, and eEVs also contained cytosolic proteins such as HSP90 and mCherry alone (Figure 5b). These results indicate that eEVs are derived from the cell membrane and contain cytosolic proteins.
Thus, we assumed that eEVs were derived from the cell membrane via outward budding. To strengthen this assumption, we acquired time‐lapse images of the cell membranes of HeLa cells expressing mEmerald‐CD9 and mCherry‐TM every 2 s using confocal microscopy. In the time‐lapse images of mEmerald‐CD9‐ and mCherry‐TM‐expressing HeLa cells treated with eMTDΔ4, the eEVs (white arrows) appeared to be generated from the cell membrane (red arrows); however, the EVs were not found in sucrose buffer alone (Figure S5b). Although these results are suggestive, they are still inconclusive because the resolution of time‐lapse images was not high enough to visualize the moment of eEVs outward budding from the cell membrane. Thus, we used TEM images of hWJ‐MSCs treated with eMTDΔ4 to visualize the morphological changes in the cell membranes in response to eMTDΔ4. The cell membrane of hWJ‐MSCs in sucrose buffer alone showed no outward budding; however, hWJ‐MSCs treated with eMTDΔ4 in sucrose buffer clearly showed outward budding of cell membranes of various sizes (Figure 5c). Thus, we believe that eEVs in response to eMTDΔ4 were generated by outward budding of the plasma membrane, a mechanism similar to that of MV biogenesis.
Intracellular calcium levels and calpain activity play key roles in MV biogenesis through outward budding of the plasma membrane (Al‐Nedawi et al., 2008; Minciacchi et al., 2015). We previously reported that eMTDΔ4 induced intracellular calcium spikes in cells within minutes of treatment (Han et al., 2019). Therefore, we assumed that the mechanism of eEV production might be associated with intracellular calcium spikes and calpain activity. hWJ‐MSCs were pre‐treated with the calcium‐chelating agent BABTA‐AM, the calpain inhibitor ALLM, or the natural EV biogenesis inhibitor GW4869, and eEV production was monitored (Figure 5d, e). Our results clearly confirmed that we could not produce a significant amount of TS‐eEVs when the cells were treated with sucrose buffer only, without eMTDΔ4 treatment, or when the cells were treated with eMTDΔ4 in sucrose buffer after pre‐treatment with inhibitors such as BAPTA‐AM and ALLM. Treatment with inhibitors modulating the intracellular calcium level and calpain activity led to a significant decrease in TS‐eEVs production; by contrast, treatment with a natural EV biogenesis inhibitor, GW4869 (an nSMase inhibitor), did not have any significant effect on TS‐eEV production, indicating that TS‐eEVs production may be closely related to calcium spike in the cytosol but not to a natural EV generation process. Taken together, these results suggest that eMTDΔ4 causes cell membrane damage, calcium spike in cytosol released from possibly mitochondria and/or ER, and outward budding of cell membranes to produce TS‐eEVs (Figure 5f).
4. DISCUSSION
The application of EVs for efficient drug delivery, diagnosis, and intractable disease therapy is of considerable research interest (Zipkin, 2020). EVs derived from MSCs are biocompatible and immunologically inert, and have regenerative and immunomodulatory capacities (Van Niel et al., 2018). EVs have the ability to enter cells and target specific tissues and transverse physiological barriers, including the blood–brain barrier, which may encourage their potential application as drug delivery vehicles in therapeutics (Lener et al., 2015; Mizrak et al., 2013; Ophelders et al., 2016; Pan et al., 2014). EVs have the potential to become next‐generation drug delivery systems and have evolved into off‐the‐shelf therapeutics targeting the global market. Nevertheless, several challenges, including cell selection, heterogeneity, large‐scale production, and drug‐loading strategies limit the use of EVs.
In this study, we propose a method for producing large amounts of homogeneous EVs from different cells within a very short time. We accidentally and experimentally developed the TS‐eEV method, a novel EV production method for treating cells with eMTDΔ4 in a chemically defined sucrose buffer with orbital shaking after cell trypsinization (Figure 1 and 2). Using this TS‐eEV method, we were able to obtain EVs with 45‐fold higher purity and up to 30‐fold higher quantity within 15 min than those obtained with the conventional EV production method (EVs obtained the culture media of the non‐treated cells after 48 h; Figure 2). In particular, the EV purity of TS‐eEVs immediately after the UC purification step was similar to that of native EVs after the UC and DG purification step, as TS‐eEV were prepared in a relatively small amount of chemically defined sucrose buffer in a short time (15 min). The high purity of TS‐eEVs will be very advantageous for the industrial application of the TS‐eEV method as the number of EV particles is significantly reduced in successive DG purification steps. Indeed, in this study, the number of EVs was reduced by more than 160‐fold during the successive DG purification steps (Figure 2d). EVs prepared using the TS‐eEV method exhibited similar EV size, EV marker expression and similar (or better) regenerative and immunomodulatory activities to those generated using the conventional methods (Figures 2, 3, 4). Importantly, the total efficacy of total TS‐eEVs was much higher (up to 30 times) than that of total natural EVs (Figures 3 and 4). The high yield, short time period, and easy purification procedure are the strengths of this method, and scaling up may find potential applications at the industry level. Moreover, high purity, anti‐inflammatory effects, and regenerative potential may render TS‐eEVs suitable for disease treatment.
As mentioned, eMTDΔ4 is a strong necrosis inducer via mitochondrial catastrophe, raising the question of whether the EVs produced by eMTDΔ4 in sucrose buffer are apoptotic or necrotic particles. Previously, we showed that cell death induced by R8:MTD, a peptide similar to eMTDΔ4, does not cause caspase‐8 activation or cytochrome c release from mitochondria, implying that eMTDΔ4 does not induce apoptosis (Seo et al., 2009). In addition, TEM images of R8:MTD‐treated HeLa cells showed no outward budding of the cell membrane, suggesting that eMTDΔ4 or R8:MTD does not produce typical apoptotic bodies (Seo et al., 2009).
Consistent with the findings of previous studies (Han et al., 2019; Park et al., 2019), HeLa cells treated with eMTDΔ4 in DMEM demonstrated rapid necrotic cell death (Figure 1S); however, the number of EV particles in DMEM was not increased by treatment with eMTDΔ4 (Figure 1b). HeLa cells treated with eMTDΔ4 in NaCl, KCl, Na‐Glu, or K‐Glu buffer showed exacerbated necrotic cell death (Figure 1S) but no increase in EV particles (Figure 1b). By contrast, in the case of sucrose buffer, eMTDΔ4 produced massive amounts of EVs (Figure 1), despite showing some damages of intracellular organelles such as mitochondria and ER within 15 min (Figure 1S and 5c). However, these damages of mitochondria and ER in eMTDΔ4‐treated cells in sucrose buffer (Figure 5c) were not as severe as mitochondrial damages and swollen cell membrane observed in Jurkat cell treated with R8:MTD (Seo et al., 2009), indicating that eMTDΔ4‐treated cells in sucrose buffer do not cause the typical MTD‐induced necrosis. As shown in a previous report (Seo et al., 2009), TEM images of cell membranes in R8:MTD‐treated cells in culture media showed a swollen cell membrane but no outward budding on the cell membrane and no production of EVs. Thus, we speculated that damages induced by eMTDΔ4 in sucrose buffer may not be strong enough to cause the typical MTD‐induced necrosis and that the eMTDΔ4‐treated cells in sucrose buffer but not in culture media and other buffers may maintain cell membrane integrity and produce out‐budding small vesicles. Therefore, sucrose buffer is a key player to maintain cell membrane integrity, supporting the eMTDΔ4‐induced massive EVs production. The detailed mechanism by which eMTDΔ4 in sucrose buffer triggers EVs production will be elucidated in the further study.
Despite of the large amount of EV production by eMTDΔ4 in sucrose buffer, this method raises several safety concerns. For example, residual eMTDΔ4 in/on EVs may cause cell death in target cells or animals. Since eMTDΔ4 is a potent necrosis inducer, safety concerns against residual eMTDΔ4 in/on TS‐eEV are understandable. Despite these facts, we believe that residual eMTDΔ4 in/on TS‐eEV may not induce necrosis, as the amounts of residual eMTDΔ4 may not be high enough to induce necrosis and that residual eMTDΔ4 may be susceptible to serum or intracellular proteases. Another safety concern would be the toxic molecules released from EV‐producing cells, since eMTDΔ4 causes some damages of EV‐producing cells while producing EVs. Since the EV‐producing cells are neither apoptotic nor necrotic, the toxic molecules released from EV‐producing cells or loaded in TS‐eEV would be negligible. Future proteomics analysis should be performed to address these safety issues in future.
The mechanism of EV generation using eMTDΔ4 remains unclear; however, we provide evidence that EVs generated by eMTDΔ4 treatment are derived from the plasma membrane through the outward budding process. The present method needs only 10–15 min for large‐scale EV production via outward budding of the plasma membrane (Figure 5). Interestingly, the inhibition of eMTDΔ4‐induced EV generation by the calcium chelator and calpain inhibitor indicated the role of intracellular calcium and calpain activity in EV generation by eMTDΔ4. We previously reported that eMTDΔ4 treatment induces intracellular calcium influx from the mitochondria and/or extracellular space and forms tiny holes that disrupt the integrity of the plasma membrane (Han et al., 2019). Therefore, we speculate that eMTDΔ4‐induced intracellular calcium influx leads to disruption of plasma membrane integrity, thereby creating tiny holes to facilitate the passive diffusion of extracellular materials into the cytosol. Therefore, extracellular materials in sucrose buffer that diffuse into the cytosol through the holes may be encapsulated in EVs during their formation from the plasma membrane. Ts‐eEVs can be used to load different reagents, proteins, antioxidants, and drugs into target cells.
We believe that the utilization of sucrose buffer for EV production using eMTDΔ4 could provide several advantages. First, it eliminates the possibility of contamination, such as serum‐originated EVs, proteins, and any unwanted materials from serum or conditioned media, because the culture media must be replaced with sucrose buffer prior to EV production and the sucrose buffer does not contain serum. This advantage is supported by the fact that the major serum protein albumin was not detected in EVs produced in sucrose buffer using eMTDΔ4, as shown in Figure 1.
Second, the EVs produced in sucrose buffer are highly homogeneous (Figure S4), because the EV production will use the cells from the same batch culture and will be completed within 15 min. Conventional EV production is mostly achieved by collecting multiple conditioned media harvested at different time points during cell culture. This conventional method introduces various unwanted factors into culture conditions, such as changes in cultured cell number at collection time points, fresh serum‐containing media, and culture temperature; thus, it causes heterogeneous EV production, a major hurdle for clinical application. The use of sucrose buffer removes all these unwanted factors from EVs production.
Finally, the utilization of sucrose buffers can reduce the processing volume during EVs production. Conventional methods of EV production require the collection of a large volume of conditioned medium, which makes EV production difficult. The sucrose buffer can control the initial volume of EV production, thus reducing the processing volume for EVs production. Taken together, the method presented in this study may be a suitable alternative to overcome the challenges that limit the clinical use of EVs, maximize their therapeutic potential of EVs, and optimize their inherent properties.
AUTHOR CONTRIBUTIONS
Kyung Min Lim: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Ji‐Hye Han: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Yoonjoo Lee: Data curation; Formal analysis; Investigation. Junghee Park: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Visualization. Ahmed Abdal Dayem: Data curation; Formal analysis; Methodology. Jongyub An: Data curation; Formal analysis; Investigation; Methodology. Geun‐Ho Kang: Data curation; Formal analysis; Investigation; Methodology. Sejong Kim: Data curation; Formal analysis; Investigation; Methodology. Sangwoo Kwon: Data curation; Formal analysis; Investigation; Methodology; Visualization. Ssang‐Goo Cho: Conceptualization; Funding acquisition; Investigation; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Tae‐Hyoung Kim: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing.
CONFLICT OF INTEREST
Ssang‐Goo Cho was a StemExOne stockholder. Geun‐Ho Kang and Sejong Kim are employed by StemExOne Ltd. Tae‐Hyoung Kim, Ji‐Hye Han, Junghee Park, and Seung‐Hyun Myung are stockholders of ExoCalibre Ltd. The other authors declare no conflicts of interest.
ETHICS STATEMENT
The in vivo experiments were performed with approval from the Konkuk University Animal Care Committee (IACUC; approval number: KU20132‐1). The Institutional Review Board of Konkuk University (7001355‐202010‐BR‐407) approved the experimental protocol for human Wharton's jelly‐derived mesenchymal stem cells for this study.
Supporting information
Supporting Information
Supporting Information
{kind=link}
Supporting Information
{kind=link}
Supporting Information
{kind=link}
Supporting Information
{kind=link}
Supporting Information
{kind=link}
Supporting Information
{kind=link}
ACKNOWLEDGEMENTS
This research was supported by the Ministry of Education (Grant Nos. NRF‐2018R1D1A1A02043850 and NRF‐2021R1I1A3047317 to TH Kim and Grant No. NRF‐2019M3A9H1030682 to SG Cho) and Samsung Research Funding and Incubation Center for Future Technology (Grant No. SRFCIT1802‐03 to SG Cho).
Lim, K. M. , Han, J. ‐H. , Lee, Y. , Park, J. , Dayem, A. A. , Myung, S. H. , An, J. , Song, K. , Kang, G. ‐H. , Kim, S. , Kwon, S. , Kim, K. S. , Cho, S. G. , & Kim, T. H. (2022). Rapid production method with increased yield of high‐purity extracellular vesicles obtained using extended mitochondrial targeting domain peptide. Journal of Extracellular Vesicles, 11, e12274. 10.1002/jev2.12274
Kyung Min Lim and Ji‐Hye Han contributed equally to this work.
Contributor Information
Ssang‐Goo Cho, Email: ssangoo@konkuk.ac.kr.
Tae‐Hyoung Kim, Email: thkim65@chosun.ac.kr.
REFERENCES
- Agrahari, V. , Agrahari, V. , Burnouf, P. A. , Chew, C. , & Burnouf, T. (2019). Extracellular microvesicles as new industrial therapeutic frontiers. Trends in Biotechnology, 37(7), 707–729. 10.1016/j.tibtech.2018.11.012 [DOI] [PubMed] [Google Scholar]
- Al‐Nedawi, K. , Meehan, B. , Micallef, J. , Lhotak, V. , May, L. , Guha, A. , & Rak, J. (2008). Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nature Cell Biology, 10(5), 619–624. 10.1038/ncb1725 [DOI] [PubMed] [Google Scholar]
- Arslan, F. , Lai, R. C. , Smeets, M. B. , Akeroyd, L. , Choo, A. , Aguor, E. N. E. , Timmers, L. , Van Rijen, H. V. , Doevendans, P. A. , Pasterkamp, G. , Lim, S. K. , & De Kleijn, D. P. (2013). Mesenchymal stem cell‐derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Research, 10(3), 301–312. [DOI] [PubMed] [Google Scholar]
- Di Vizio, D. , Kim, J. , Hager, M H. , Morello, M. , Yang, W. , Lafargue, C J. , True, L D. , Rubin, M A. , Adam, R M. , Beroukhim, R. , Demichelis, F. , & Freeman, M R. (2009). Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Research, 69(13), 5601–5609. 10.1158/0008-5472.CAN-08-3860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebäck, T. , Schulz, M. M. P. , Koumoutsakos, P. , & Detmar, M. (2009). TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays: Short technical reports. Biotechniques, 46(4), 265–274. [DOI] [PubMed] [Google Scholar]
- Greening, D. W. , Xu, R. , Ji, H. , Tauro, B. J. , & Simpson, R. J. (2015). A protocol for exosome isolation and characterization: Evaluation of ultracentrifugation, density‐gradient separation, and immunoaffinity capture methods. In Proteomic profiling (pp. 179–209): Springer. [DOI] [PubMed] [Google Scholar]
- Han, Ji‐H. , Park, J. , Myung, S. ‐H. , Lee, S. H. , Kim, H. ‐Y. , Kim, K. S. , Seo, Y. ‐W. , & Kim, T. ‐H. (2019). Noxa mitochondrial targeting domain induces necrosis via VDAC2 and mitochondrial catastrophe. Cell Death & Disease, 10(7), 519. 10.1038/s41419-019-1753-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera, M. B. , Fonsato, V. , Gatti, S. , Deregibus, M. C. , Sordi, A. , Cantarella, D. , Calogero, R. , Bussolati, B. , Tetta, C. , & Camussi, G. (2010). Human liver stem cell‐derived microvesicles accelerate hepatic regeneration in hepatectomized rats. Journal of Cellular and Molecular Medicine, 14(6b), 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov, M. , Erl, W. , Linder, S. , & Weber, P. C. (2004). Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood, 104(9), 2761–2766. 10.1182/blood-2003-10-3614 [DOI] [PubMed] [Google Scholar]
- Hu, G. , Li, Q. , Niu, X. , Hu, B. , Liu, J. , Zhou, S. , & Wang, Y. (2015). Exosomes secreted by human‐induced pluripotent stem cell‐derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Research & Therapy, 6(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal, R. , & Sedger, L. M. (2019). Intercellular vesicular transfer by exosomes, microparticles and oncosomes ‐ Implications for cancer biology and treatments. Frontiers in Oncology, 9, 125. 10.3389/fonc.2019.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri, R. , & Lebleu, V. S. (2020). The biology, function, and biomedical applications of exosomes. Science, 367(6478), 10.1126/science.aau6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener, T. , Gimona, M. , Aigner, L. , Börger, V. , Buzas, E. , Camussi, G. , Chaput, N. , Chatterjee, D. , Court, F. A. , Portillo, H. A. D. , O'driscoll, L. , Fais, S. , Falcon‐Perez, J. M. , Felderhoff‐Mueser, U. , Fraile, L. , Gho, Y. S. , Görgens, A. , Gupta, R. C. , Hendrix, A. , … Giebel, B. (2015). Applying extracellular vesicles based therapeutics in clinical trials ‐ An ISEV position paper. Journal of Extracellular Vesicles, 4, 30087. 10.3402/jev.v4.30087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, Z. , Luo, Y. , & Lv, Y. (2020). Mesenchymal stem cell‐derived microvesicles mediate BMP2 gene delivery and enhance bone regeneration. Journal of Materials Chemistry B, 8(30), 6378–6389. [DOI] [PubMed] [Google Scholar]
- Merino‐González, C. , Zuñiga, F. A. , Escudero, C. , Ormazabal, V. , Reyes, C. , Nova‐Lamperti, E. A. , Salomón, C. , & Aguayo, C. (2016). Mesenchymal stem cell‐derived extracellular vesicles promote angiogenesis: Potential clinical application. Frontiers in Physiology, 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minciacchi, V. R. , Freeman, M. R. , & Di Vizio, D. (2015). Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Seminars in Cell and Developmental Biology, 40, 41–51. 10.1016/j.semcdb.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrak, A. , Bolukbasi, M. F. , Ozdener, G. B. , Brenner, G. J. , Madlener, S. , Erkan, E. P. , Ströbel, T. , Breakefield, X. O. , & Saydam, O. (2013). Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Molecular Therapy, 21(1), 101–108. 10.1038/mt.2012.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophelders, D. R. M. G. , Wolfs, T. G. A. M. , Jellema, R. K. , Zwanenburg, A. , Andriessen, P. , Delhaas, T. , Ludwig, A. ‐. K. , Radtke, S. , Peters, V. , Janssen, L. , Giebel, B. , & Kramer, B. W. (2016). Mesenchymal stromal cell‐derived extracellular vesicles protect the fetal brain after hypoxia‐ischemia. Stem Cells Translational Medicine, 5(6), 754–763. 10.5966/sctm.2015-0197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, S. , Yang, X. , Jia, Y. , Li, R. , & Zhao, R. (2014). Microvesicle‐shuttled miR‐130b reduces fat deposition in recipient primary cultured porcine adipocytes by inhibiting PPAR‐g expression. Journal of Cellular Biochemistry, 229(5), 631–639. 10.1002/jcp.24486 [DOI] [PubMed] [Google Scholar]
- Park, J. , Han, Ji‐H. , Myung, S. ‐. H. , Kang, H. , Cho, Ju‐Y. , & Kim, T. ‐H. (2019). A peptide containing Noxa mitochondrial‐targeting domain induces cell death via mitochondrial and endoplasmic reticulum disruption. Biochemical and Biophysical Research Communications, 518(1), 80–86. 10.1016/j.bbrc.2019.08.011 [DOI] [PubMed] [Google Scholar]
- Phinney, D. G. , & Pittenger, M. F. (2017). Concise review: MSC‐derived exosomes for cell‐free therapy. Stem Cells, 35(4), 851–858. [DOI] [PubMed] [Google Scholar]
- Ratajczak, J. , Miekus, K. , Kucia, M. , Zhang, J. , Reca, R. , Dvorak, P. , & Ratajczak, M. Z. (2006). Embryonic stem cell‐derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia, 20(5), 847–856. [DOI] [PubMed] [Google Scholar]
- Sabin, K. , & Kikyo, N. (2014). Microvesicles as mediators of tissue regeneration. Translational Research, 163(4), 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta, P. , Bosis, E. , Nachliel, E. , Gutman, M. , Smith, S. O. , Mihalyne, G. N. , Zaitseva, I. , & Mclaughlin, S. (2009). EGFR juxtamembrane domain, membranes, and calmodulin: Kinetics of their interaction. Biophysics Journal, 96(12), 4887–4895. 10.1016/j.bpj.2009.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, Y. ‐W. , Shin, J. N. a. , Ko, K. H. , Cha, J. H. , Park, J. Y. , Lee, B. R. , Yun, C. ‐W. , Kim, Y. M. , Seol, D. W. , Kim, D. ‐W. , Yin, X. M. , & Kim, T. H. (2003). The molecular mechanism of Noxa‐induced mitochondrial dysfunction in p53‐mediated cell death. Journal of Biological Chemistry, 278(48), 48292–48299. 10.1074/jbc.M308785200 [DOI] [PubMed] [Google Scholar]
- Seo, Y. W. , Woo, H. , Piya, S. , Moon, A. , Oh, J. ‐W. , Yun, C. ‐W. , Kim, K. ‐K. , Min, J. , Jeong, S. Y. , Chung, S. , Song, P. I. , Jeong, S. ‐Y. , Choi, E. K. , Seol, D. , & Kim, T. ‐H. (2009). The cell death‐inducing activity of the peptide containing Noxa mitochondrial‐targeting domain is associated with calcium release. Cancer Research, 69(21), 8356–8365. 10.1158/0008-5472.Can-09-0349 [DOI] [PubMed] [Google Scholar]
- Shelke, G. V. , Lasser, C. , Gho, Y. S. , & Lotvall, J. (2014). Importance of exosome depletion protocols to eliminate functional and RNA‐containing extracellular vesicles from fetal bovine serum. Journal of Extracellular Vesicles, 3(1), 24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirazi, S. , Huang, C. ‐C. , Kang, M. , Lu, Y. , Ravindran, S. , & Cooper, L. F. (2021). The importance of cellular and exosomal miRNAs in mesenchymal stem cell osteoblastic differentiation. Scientific Reports, 11(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims, P. J. , Faioni, E. M. , Wiedmer, T. , & Shattil, S. J. (1988). Complement proteins C5b‐9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. Journal of Biological Chemistry, 263(34), 18205–18212. [PubMed] [Google Scholar]
- Song, Y. , Kim, Y. , Ha, S. , Sheller‐Miller, S. , Yoo, J. , Choi, C. , & Park, C. H. (2020). The emerging role of exosomes as novel therapeutics: Biology, technologies, clinical applications, and the next. American Journal of Reproductive Immunology, e13329. 10.1111/aji.13329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, C. Y. , Lai, R. C. , Wong, W. , Dan, Y. Y. , Lim, S. K. , & Ho, H. K. (2014). Mesenchymal stem cell‐derived exosomes promote hepatic regeneration in drug‐induced liver injury models. Stem Cell Research & Therapy, 5(3), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Amigorena, S. , Raposo, G. A. , & Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Current Protocols in Cell Biology, 30(1), 3.22. 21‐23.22. 29. [DOI] [PubMed] [Google Scholar]
- Van Niel, G. , D'angelo, G. , & Raposo, G. A. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology, 19(4), 213–228. 10.1038/nrm.2017.125 [DOI] [PubMed] [Google Scholar]
- Warren, B. A. , & Vales, O. (1972). The release of vesicles from platelets following adhesion to vessel walls in vitro. British Journal of Experimental Pathology, 53(2), 206–215. [PMC free article] [PubMed] [Google Scholar]
- Wolf, P. (1967). The nature and significance of platelet products in human plasma. British Journal of Haematology, 13(3), 269–288. 10.1111/j.1365-2141.1967.tb08741.x [DOI] [PubMed] [Google Scholar]
- Woo, H. , Seo, Y. W. , Moon, A. , Jeong, S. Y. , Jeong, S. Y. , Choi, E. K. , & Kim, T. H. (2009). Effects of the BH3‐only protein human Noxa on mitochondrial dynamics. FEBS Letters, 583(14), 2349–2354. 10.1016/j.febslet.2009.06.029 [DOI] [PubMed] [Google Scholar]
- Zipkin, M. (2020). Big pharma buys into exosomes for drug delivery. Nature Biotechnology, 38(11), 1226–1228. 10.1038/s41587-020-0725-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information