

Abstract
Introduction
The endometrial microbiota has been linked to several gynecological disorders, including infertility. It has been shown that the microbial profile of endometrium could have a role in fertilization and pregnancy outcomes. In this study we aim to assess the microbial community of endometrial tissue (ET) and endometrial fluid (EF) samples in women receiving in vitro fertilization (IVF) treatment. We also search for possible associations between chronic endometritis (CE) and endometrial microbiota.
Material and methods
This was a cohort study involving 25 women aged between 28 and 42 years with both primary and secondary infertility and with at least one IVF failure. The ET and EF sample collection was carried out between September 2016 and November 2018. Each of the participants provided two types of samples—tissue and fluid samples (50 samples in total). A 16S rRNA sequencing was performed on both of the sample types for microbial profile evaluation. CE was diagnosed based on a CD138 immunohistochemistry where CE diagnosis was confirmed in the presence of one or more plasma cells. Microbial profiles of women with and without CE were compared in both sample types separately.
Results
We report no differences in the microbial composition and alpha diversity (p Observed = 0.07, p Shannon = 0.65, p Inverse Simpson = 0.59) between the EF and ET samples of IVF patients. We show that the abundance of the genus Lactobacillus influences the variation in microbial beta diversity between and fluid samples (r 2 = 0.34; false discovery rate [FDR] <9.9 × 10−5). We report that 32% (8/25) of the participants had differences in Lactobacillus dominance in the paired samples and these samples also present a different microbial diversity (p Shannon = 0.06, FDRweighted UniFrac = 0.01). These results suggest that the microbial differences between ET and fluid samples are driven by the abundance of genus Lactobacillus. The microbiome of CE and without CE (ie non‐CE) women in our sample set of IVF patients was similar.
Conclusions
Our findings show that genus Lactobacillus dominance is an important factor influencing the microbial composition of ET and fluid samples.
Keywords: endometrial microbiome, endometrial microbiota, implantation failure, in vitro fertilization, infertility, uterine microbiota
Abbreviations
- ASV
amplicon sequence variant
- CE
chronic endometritis
- EF
endometrial fluid
- ET
endometrial tissue
- FDR
false discovery rate
- IVF
in vitro fertilization
- LH
luteinizing hormone
- PCoA
principal coordinate analysis
- SD
standard deviation
Key message.
By comparing the microbial composition of paired endometrial tissue and endometrial fluid samples we report that the differences between sample types are highly influenced by genus Lactobacillus.
1. INTRODUCTION
For a long time, it was believed that the healthy uterine cavity is sterile. With culture‐based quantification and next‐generation sequencing, it became evident that the concept of a sterile womb is mistaken. The recent progress in the microbiome field with the development of 16S rRNA and metagenome sequencing has made it possible to characterize the microbial composition of the endometrium in detail. 1 , 2 The normal vaginal microbiome is populated predominantly by the Lactobacillus genus and, compared with the gastrointestinal tract, the microbial diversity in the female reproductive tract is relatively low. 3 , 4 , 5 Similarly to the vaginal microbiome, the endometrial microbiome is also normally dominated by the genus Lactobacillus. 3 , 5 , 6 , 7 On the other hand, studies of microbiota on the material obtained after hysterectomy showed that the dominance of the genus Lactobacillus in the endometrium is not widespread and often the dominant genera are Acinetobacter, Pseudomonas and Corynebacterium. 8 , 9
The endometrial microbiota has been linked to numerous gynecological disorders, such as chronic endometritis (CE), 10 , 11 endometriosis 5 , 12 and infertility. 13 , 14 A study analyzing the impact of the endometrial microbial community on pregnancy outcome showed that the non‐Lactobacillus dominated endometrial microbiota was associated with significant decreases in implantation, pregnancy and live birth in infertile women undergoing in vitro fertilization (IVF) treatment, whereas a Lactobacillus‐dominant microbiota was advantageous for embryo implantation, 14 backing the idea that endometrial microbiota has a role in an embryo implantation process. Yet, a study comparing women with eubiotic (defined as ≥80% Lactobacillus + Bifidobacterium spp.) and dysbiotic (defined as <80% Lactobacillus + Bifidobacterium spp. with ≥20% of other bacteria) endometrium concluded that there were no differences in pregnancy rates between the two study groups. 15
Chronic endometritis is a condition of constantly recurring inflammation in the endometrial mucosa caused by bacterial pathogens in the uterine tissue. CE incidence varies greatly between studies from 2.8% up to 46% in women with infertility problems and it has been stated that CE is especially prevalent in cases of repeated IVF failure. 16 , 17 Other bacteria associated with CE include Gardnerella vaginalis, Streptococcus spp., Enterococcus faecalis, Enterobacteriaceae and Staphylococcus spp. 11
While the endometrial microbiota is an important resource of investigation when studying fertility, retrieving the endometrial tissue (ET) samples is an invasive procedure and to evaluate patients the field strives for the least intrusive method possible. In this research we aim to characterize the ET and endometrial fluid (EF) microbiome of 25 women undergoing IVF treatment in order to study whether the microbiome of these sources differ. The outcome of this study could provide important insights for future studies on the endometrial microbiome and expands our knowledge of the role of uterine microbiomes in implantation failure.
2. MATERIAL AND METHODS
2.1. Study participants and sample collection
Twenty‐five women with previous IVF procedure failures attending the Center of Human Reproduction Genesis clinic in St. Petersburg, Russia, were recruited in this study. The inclusion criteria included women having one or more unsuccessful IVF procedures (embryo transfer failures ranged from 1 to 10).
The sample collection was done in the middle of the secretory phase of the menstrual cycle (self‐reported menstrual cycle day 20.2 ± 2.81). Two types of samples were collected from all patients (total 50 samples): ET biopsy and paired EF. After treating the cervix with saline, a double‐lumen embryo transfer catheter (Wallace Classic, Embryo Transfer Catheter 1816; CooperSurgical) was inserted into the uterine cavity. With the help of a syringe, 50 μl of fluid was obtained. Immediately after this, an endometrial biopsy was performed using a Pipelle curette (Pipelle Mark II; CCD). The resulting material from the two samples was stored at −80°C. The sample collection was performed between September 2016 and November 2018. We also performed analysis on CE, as described previously. 18 There is no consensus on the diagnostic criteria to define what accounts for CE and there are seven or more different criteria previously reported. 19 In our study CE was diagnosed based on the presence of one or more plasma cells with positive CD138 immunohistochemistry, as these cells are typically absent in the endometrium. In our sample set there were 12 women with CE (eight women with up to four plasma cells present and four women with five or more plasma cells present), 11 with no CE and 2 with unreliable data who were excluded from the CE analyses (Table S1).
Primary infertility is defined as not having conceived a child in the past and not being able to become pregnant after 1 year or more of trying. Secondary infertility is defined as having one or more pregnancies in the past but being unable to conceive again after 1 year or more of trying.
2.2. DNA extraction and sequencing
DNA extraction from ET and EF samples was done using the DNeasy PowerSoil PRO kit (Qiagen) provided by Qiagen following the manufacturer’s instructions. The amplicon sequencing was conducted in the Institute of Genomics Core Facility, University of Tartu. Extracted DNA samples were then quantified with Qubit® 2.0 Fluorometer (Invitrogen). The genomic DNA was amplified using primers 16S_F (5′‐TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG‐3′) and 16S_R (5′‐GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC‐3′) for PCR amplification of an approximately 460‐bp region within the hypervariable (V3–V4) region of prokaryotic 16S ribosomal RNA gene. Amplicon libraries for Illumina (Illumina) next‐generation sequencing were generated by two‐step PCR. First, the region specific for 16S rRNA was amplified with 24 cycles and then the Illumina adapter and index sequences were added by 7 cycles of PCR. The quality control of amplicon libraries was performed by Agilent 2200 TapeStation analysis (Agilent Technologies) and with the Kapa Library Quantification Kit (Kapa Biosystems). Amplicon libraries were pooled in equimolar concentrations. Sequencing was carried out on an Illumina MiSeq System using MiSeq Reagent Kit v2 in paired end 2 × 250 bp mode.
After sequencing quality control steps and removing sequences belonging to the human host, a total of 2 410 402 reads were generated (on average 44 637 reads per sample). The total number of reads for ET samples was 1 029 032 with an average of 38 112 reads per sample (range: 1053–107 508), and for EF samples 1 381 332 with an average of 53 128 per sample (range: 4902–128 709).
2.3. Data analysis
Raw sequencing data were imported into an open‐source software QIIME2 2019.7 using the PairedEndSequencesWithQuality import type. The denoising step and merging the forward and reverse samples was performed with DADA2 software implemented in QIIME2. DADA2 uses a quality‐aware model of Illumina amplicon errors to achieve an abundance distribution of sequence variants with a difference of one nucleotide. Truncating and trimming the reads was done with q2‐dada2‐denoise‐paired script based on quality scores. Forward reads were truncated at position 249 and trimmed at position 14; reverse reads were truncated at position 250 and trimmed at position 13. For chimera removal the consensus filter was used; this method detects chimeras individually in each sample and removes sequences that are found to be chimeric in a fraction of samples. Sequences were BLASTed against the NCBI‐BLAST database used internally in QIIME2. Sequencing corresponding to top hit as Homo sapiens were removed (E‐value < 1 × 10−50). Amplicon sequence variant (ASV) alignment was performed using MAFFT and subsequently phylogeny was constructed using FASTTREE2. Taxonomy assignment was done using a pre‐trained naïve Bayes taxonomy classifier against the SILVA 16S V3–V4 database (v132_99) with a similarity threshold of 99%. A negative control sample (milli‐Q water, Millipore Q‐POD) was amplified and sequenced following the same protocol. The negative control yielded 38 processed reads and 9 ASVs which were removed in silico. Taxa associated with so‐called kitome contaminants or not colonizing humans were removed from the analysis. 20 , 21 , 22 , 23 , 24 , 25 In total, a unique number of 24 312 ASVs, 1594 genera, 828 families, 435 orders, 137 classes and 44 phyla were identified. After data filtering, keeping only ASVs with an abundance of at least 0.1% in at least 10% of the samples, there were 116 vs 82 ASVs, 9 vs 10 phyla, 20 vs 16 classes, 32 vs 24 orders, 42 vs 30 families, and 58 vs 43 genera for ET and EF samples, respectively (Table S2).
2.4. Statistical analyses
All statistical analyses were performed in RSTUDIO v1.3.1093 (R v4.0.2) using the packages phyloseq (v1.32.0), vegan (v2.5‐6), microbiome (v1.10.0) and ALDEx2 (v1.20.0). A p‐value < 0.05 was considered to be the statistically significant and the Benjamini and Hochberg False Discovery Rate (FDR) method was used for multiple testing correction. For data visualization, ggplot2 (v3.3.2) and cowplot (v1.1.0) were used. All of the analyses were performed on data aggregated to genus‐level. Alpha and beta diversity were calculated on unfiltered data. The rest of the analyses were performed on filtered data where ASVs were present in at least 10% of the samples with a relative abundance of >0.1%. We also grouped samples into Lactobacillus dominance groups, where samples with a relative abundance of Lactobacillus ≥50% in the filtered data were considered to be Lactobacillus dominant. We chose the ≥50% cut‐off since it notes that the majority of the genera present in a sample belong to Lactobacillus, meaning that this is the most abundant genus in the current sample. Alpha (observed, Shannon’s index and inverse Simpson’s index) and beta diversity (principal coordinate analysis based on the UniFrac distance metrics) indices were calculated using the phyloseq v1.32.0 package. A paired t‐test was used for comparing alpha diversity estimators. For identifying associations with beta diversity, ADONIS‐2 function from the vegan package was used using 10 000 permutations for p‐value calculations. Differential abundance analysis on filtered centered log ratio‐transformed data was carried out using analysis of variance (ANOVA)‐Like Differential Expression tools (ALDEx2, v1.20.0) Welch’s paired t‐test.
2.5. Ethical approval
Ethics approval for this study was obtained from the Local Ethics Committee of the Federal State Budgetary Educational Institution of Higher Education of the North‐Western State Medical University named after I.I. Mechnikov Ref. No. 5‐4/И/2‐1218, on 3 March 2021. All study participants signed an informed consent on the day of sample collection.
3. RESULTS
3.1. Participant data
This study included 25 women with one or more unsuccessful IVF attempts visiting the Center of Human Reproduction Genesis clinic in St. Petersburg, Russia. All of the women had infertility issues lasting for up to 10 years. The causes of infertility were described as follows: 44% of patients with tubal factor, 36% with male factor (abnormal morphology, low sperm concentration, or poor sperm motility), 12% with endocrine factor, and 8% with endometriosis (Table S1). Ten women had primary infertility and 15 had secondary infertility. All relevant information of study participants is detailed in Table 1 and Table S1.
TABLE 1.
Characteristics of study participants
| Characteristics | Mean OR | ± SD or % |
|---|---|---|
| Age (years) | 36.28 | ± 4.51 |
| Length of infertility in years | 6.38 | ± 3.97 |
| Primary infertility (n) | 10 | 40% |
| Secondary infertility (n) | 15 | 60% |
| Chronic endometritis (n) | 12 | 48% |
| Parity | 0.24 | ±0.52 |
| Number of IVF procedures | 3.4 | ±2.2 |
| Confirmed pregnancy within 6 months after sampling (n) | 9 | 36% |
| Cycle day at sampling | 20.2 | ±2.81 |
Data are presented as mean ± standard deviation for continuous traits and as absolute proportions and prevalence (%).
Abbreviations: IVF, in vitro fertilization; n, number of individuals; SD, standard deviation.
3.2. Microbial community composition of the ET and EF samples
To investigate the bacterial composition of ET and EF samples we used 16S rRNA sequencing of the V3–V4 amplicon. We first characterized the composition and variability of endometrial microbiota in both sample types at different taxonomic levels. We focused on abundant microbes, defined as taxa with at least >0.1% relative abundance across all samples and present in at least 10% of the samples. There were 9 different phyla in the ET samples and 10 different phyla in EF samples, 9 of the phyla were represented in both sample types. Phylum Fusobacteria was present only in EF samples with mean relative abundance of 0.3% (present in eight EF samples) (Figure S1). Three most prevalent phyla in both sample types were Firmicutes (mean relative abundance of 60% in ET and 78.8% in EF), Proteobacteria (mean relative abundance of 22.9% in ET and 9% in EF) and Actinobacteria (mean relative abundance of 8.6% in ET and 5.4% in EF) (Figure S1) which is in line with previous work. 1 Consistent with gut microbiome composition, we detected considerable variability in microbiota composition across both sample types. For instance, at the phylum level the relative abundance of the most abundant phylum Firmicutes ranged between 0.4% and 99.9% in ET and 4.1% and 99.9% in EF samples (Figure S1).
Large variations in abundances were also detected at genus level. As expected, the most prevalent genus in both sample types was Lactobacillus with relative abundance of 50.2% (range 0%–99.8%) in ET and 71.8% (range 0%–99.7%) in EF samples. In total we detected 160 ASV‐s corresponding to the genus Lactobacillus. In addition to Lactobacillus there were five other genera (Enterococcus, Gardnerella, Streptococcus, Escherichia‐Shigella, and Cutibacterium) with relative abundances of at least 1% in both sample types and belonging to three phyla (Firmicutes, Actinobacteria and Proteobacteria) (Figure 1A,B). We grouped all genera with the relative abundance of <1% into “other” category. As seen in Figure 1, the fraction of “other” was 24.7% (46 genera) in ET samples and 10.7% (34 genera) in EF samples. We also grouped samples by their (non)dominance of Lactobacillus, since it has been shown that Lactobacillus is the most prevalent genus in endometrial samples. 5 For further analysis, we considered a sample to be Lactobacillus (LB) dominant if it had a relative abundance of Lactobacillus ≥50%. We report that 13 (52%) ET samples were Lactobacillus dominant and 12 (48%) ET samples were non‐Lactobacillus dominant. Among the EF samples, 20 (80%) samples were Lactobacillus dominant and 5 (20%) were non‐Lactobacillus dominant samples. Thirteen individuals had both ET and EF samples Lactobacillus dominant. Seven (28%) individuals were those in which Lactobacillus dominant was detected in both ET and EF samples.
FIGURE 1.
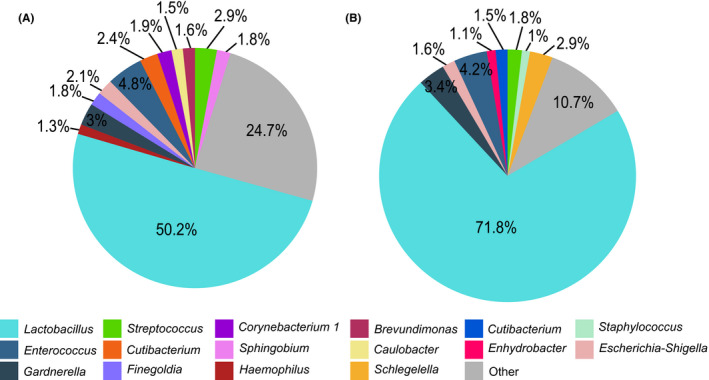
The relative abundance of genera in the (A) endometrial tissue biopsy and (B) endometrial fluid samples. Genera present in at least 10% of the samples and with relative abundance of at least 1% are plotted, genera with relative abundance of <1% are concatenated together as “Other”
Additionally, we looked at microbiome composition based on infertility type. We focused on four different infertility groups (tubal factor, male factor, endocrine and endometriosis). We found that the microbiome among infertility groups did not differ, indicating that the microbiome is not affected by the fact that individuals have different infertility reasons in our study (Table S3).
3.3. Comparison of microbiome between ET and EF samples
To detect the bacterial community differences between ET and EF samples, we conducted a differential abundance analysis (based on Welch’s paired t‐test). In this analysis we used filtered data of ASVs with a relative abundance of >0.1% and present in at least 10% of the samples and aggregated ASVs at the genus level. In total, 40 genera were used in this comparison (Table S4). The differences in Lactobacillus abundance reached a statistical significance level (p = 0.01) with higher abundances in the EF samples. However, none of the taxa tested were statistically significantly different after correcting for multiple testing (Table 2). This is likely due to the high interindividual variability and small sample size.
TABLE 2.
Top ten genera detected in the differential abundance analysis between the endometrium tissue and endometrial fluid samples
| Genus | Phyla | p‐value | FDR |
|---|---|---|---|
| Lactobacillus | Firmicutes | 0.01 | 0.33 |
| Staphylococcus | Firmicutes | 0.06 | 0.45 |
| Diaphorobacter | Proteobacteria | 0.16 | 0.54 |
| Caulobacter | Proteobacteria | 0.22 | 0.62 |
| Lawsonella | Actinobacteria | 0.23 | 0.67 |
| Acidibacter | Proteobacteria | 0.23 | 0.64 |
| Enhydrobacter | Proteobacteria | 0.25 | 0.7 |
| Chitinophagaceae uncultured | Bacteroidetes | 0.26 | 0.65 |
| Cutibacterium | Actinobacteria | 0.27 | 0.70 |
| Escherichia‐Shigella | Proteobacteria | 0.32 | 0.75 |
Abbreviation: FDR, false discovery rate.
3.4. Diversity and richness of bacterial taxa in ET and EF samples
For calculating the alpha and beta diversity indices, the unfiltered data were used. For alpha richness we looked at three different metrics: richness (ie the number of taxa observed in the sample), Shannon index (the relative abundances of different taxa) and Inverse Simpson index (taxa distribution within different sample types). In the richness analysis the ET samples had a lower observed count compared with EF (p Observed = 0.07); however, the difference did not reach statistical significance (Figure 2A). Shannon diversity showed no differences between sample types (p Shannon = 0.65) (Figure 2B). Similarly, no significant differences were observed in the Inverse Simpson index (p InverseSimpson = 0.59) (Figure 2C).
FIGURE 2.
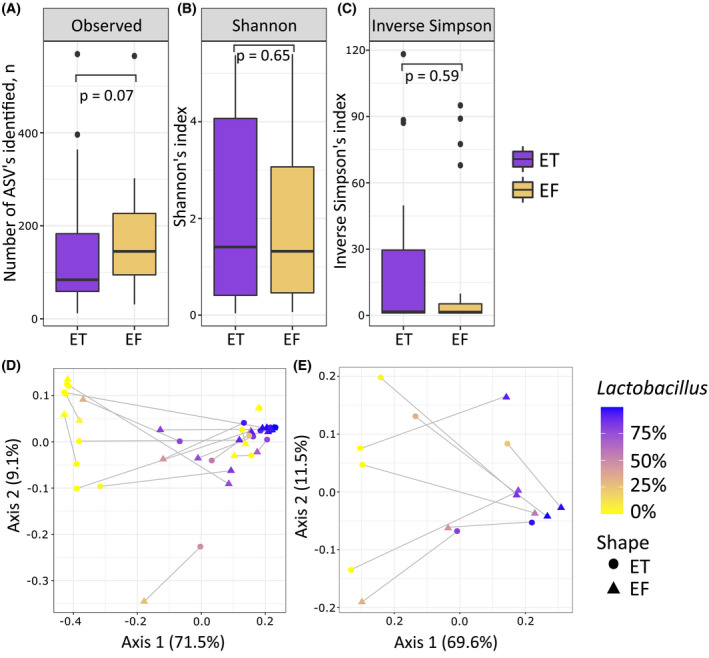
Visualizations of alpha and beta diversity analysis. Boxplots representing alpha diversity of the endometrial tissue (ET) and endometrial fluid (EF) samples: richness (A), Shannon index (B) and inverse Simpson index (C). Weighted UniFrac principal coordinate analysis (PCoA) plot (D) representing beta diversity. In (A–C) color indicates the sample type. Weighted UniFrac PCoA plot (E) representing beta diversity in individuals with sample pairs where one of the samples was Lactobacillus dominated and the other samples was non‐Lactobacillus dominated. In (D, E) color indicates the relative abundance of Lactobacillus, shape indicates sample type (triangle for EF and circle for ET) and gray lines connect the samples belonging to the same individual
In beta diversity analyses, which characterize the community changes between the two sample types, we found no differences between the ET and EF samples (r 2 = 0.02, FDR <0.27) (Figure 2D). When adding the relative abundance of Lactobacillus into the model, we observed that it had a significant effect on the variability on weighted UniFrac distances (r 2 = 0.34, FDR <9.9 × 10−5). Infertility type (primary or secondary infertility) as a covariate had no significant effect on the variability (r 2 = 1.6, FDR = 0.25). Figure 2D illustrates that the samples with similar Lactobacillus abundance cluster, whereas sample type demonstrates no clustering. Finally, we attempted to investigate further those individuals who had differences in Lactobacillus dominance in pairs of the EF–ET sample. In our dataset, there are eight (32%) individuals for whom one sample is Lactobacillus dominant and the other one is not (mean relative abundance of Lactobacillus for Lactobacillus dominated and non‐Lactobacillus dominated groups are 83.7% and 13.1%, respectively; p = 8.6 × 10−4). In six individuals, the fluid is Lactobacillus dominant and biopsy is Lactobacillus non‐dominant, and in two individuals the biopsy sample is Lactobacillus dominant and fluid Lactobacillus non‐dominant. For those individuals we saw higher alpha diversity in ET samples than in EF samples (p Shannon = 0.06, Figure S2). In addition, we identified statistically significant differences in beta diversity between sample types (r 2 = 0.25, FDR = 0.01), where samples with low and higher Lactobacillus abundance cluster differently (Figure 2E). None of the taxa tested showed statistically significant differences between different sample types (Table S5).
3.5. Endometrial microbiome and CE
Finally, we investigated the endometrial microbiome between individuals with confirmed CE diagnosis and no CE based on the presence of plasma cells with positive CD138 marker values. Twelve women had CE, 11 did not have CE and two women had CD138 marker values that were unreliable and were therefore removed (Table S1). We were unable to report any differences in diversity and taxonomical composition analyses between women with and without CE in EF or in ET samples. To investigate further whether we could identify differences in pathogens previously reported to be associated with CE, 11 , 26 we compared the relative abundance of two genera (Enterococcus and Streptococcus) present in our samples; however, we detected no differences between the CE and non‐CE groups (Figure S3, Table S6). The lack of differences observed could potentially be explained by a small sample set in these analyses.
4. DISCUSSION
A number of previous studies have suggested a possible link between the endometrial microbiome and infertility. 7 , 14 , 27 Here we performed a study comparing the microbiome from paired ET biopsy and uterine cavity fluid samples to determine whether the ET and EF samples differ from their microbial composition.
In our study, we show that both the ET and EF samples from women undergoing IVF treatment are dominated by the genus Lactobacillus, as reported previously. 6 No notable differences in alpha diversity or taxa composition were distinguished between the different sample types. However, when we consider the abundance of Lactobacillus in the beta diversity analysis, we clearly see that this genus had a significant effect on the microbiome diversity between the ET and EF samples. The abundance of Lactobacillus was higher in EF samples than in tissue samples, which probably reflects the vaginal/uterine cavity microbial environment. In our cohort, in 32% of the individuals the abundance of Lactobacillus genus between the ET and EF samples are different. Among these mismatching sample pairs, we detected a difference between sample types in their diversity profile, with ET samples having higher diversity compared with EF samples. This is consistent with previous work by Liu et al. showing higher microbial diversity in ET samples; however, our data indicate that differences between ET and EF samples are driven by Lactobacillus abundance. 6 Recent studies indicate that women with a high abundance of Lactobacillus in their vaginal and endometrial sample are most likely to have a successful embryo implantation. 28 , 29 Hence, the role of Lactobacillus in the environment of a healthy uterine microbiome appears to be an important factor and should be further investigated.
Of the main genera detected in EF and ET samples, with a relative abundance of at least 1% and a presence in ≥10% of the samples, Lactobacillus, Gardnerella, Streptococcus and Finegoldia have also been reported in previous studies as being the most prevalent genera in endometrial samples. 5 , 6 , 27 , 30 , 31 , 32 , 33 The rest of the most prevalent genera in our samples (Enterococcus, Cutibacterium, Escherichia‐Shigella, Corynebacterium_1, Sphingobium, Brevundimonas, Caulobacter, Haemophilus, and Enhydrobacter) have not been previously reported among the top genera seen in endometrial samples. Also, in our samples we were unable to detect Atopobium, Prevotella or Snethia among the predominant genera, although these have previously been shown to be common genera in endometrial samples. 6 , 14 The discrepancy between our findings and the previous studies may arise from technical differences (eg usage of different sequencing regions, sample collection methods) and also from differences in the samples’ characteristics. It is known that the vaginal microbiota changes throughout the menstrual cycle; greater microbiota stability is associated with the estradiol peak at ovulation and progesterone rise in the midluteal phase, the same could be true for endometrial microbiota. 34 In our study the sample collection was done in the middle of the secretory phase, around 20th day of cycle, but the exact day after luteinizing hormone (LH) surge was not documented. In a study by Moreno et al., the samples were collected on days LH+2 and LH+7 in early‐ and mid‐secretory phase, while Liu et al. performed sample collection on day LH+7. Therefore, it is possible that differences in the results are due to different cycle days of sample collection.
The human microbiome is very unique, having many species with very low relative abundance. With our sequencing depth (average read count of 44 637 per sample) there were 46 genera in ET and 34 genera in EF samples with a relative abundance of <1% in the samples (Figure 1). These results are comparable to the previous finding by Chen et al. that reported that in the endometrial samples the fraction of taxa with relative abundance of <1% was 11.04%. 5 In this study, we reported these percentages to be 10.7% for EF and 24.7% for ET samples.
In CE analysis we did not identify any differences in endometrial microbiome between CE and non‐CE IVF women. However, quantitative determination of CE‐causing bacterial pathogens was not performed for our study participants. Still, interestingly, some of the most common and known bacteria for CE, such as Streptococcus spp. (genus Streptococcus), Gardnerella vaginalis (genus Gardnerella), as well as Enterococcus faecalis (genus Enterococcus), belong to genera that were among the most abundant ones in our study (Figure 1) of IVF patients who are known to be more prone to developing CE. 35 However, as no major differences in the microbiome of CE and non‐CE samples were found in microbiota analysis in our study, it is still possible that the microbial composition is at least to some extent reasonable for the development of CE as shown in previous studies. 11 , 26 , 36 The role of microbes in CE is also supported by the fact that antibiotic treatment ameliorates the reproductive outcomes of CE women. 26 , 37 , 38 , 39
As previously noted, one of the reasons why similar microbes and/or their abundances cannot be found when compared with previous studies is the usage of V3–V4 regions of 16S rRNA, which are commonly used in gut microbiome studies. Using V3–V4 regions might not give a good resolution of Lactobacillus species, a grounding species of the cervicovaginal microbiome, whereas V1–V2 is known to differentiate between Lactobacillus well. 40 Therefore, preferring one 16S rRNA region to another may lead to a bias when studying endometrial samples. In 2018, Kyono et al. evaluated whether some target regions can represent the endometrial microbiome better from other regions for which they sequenced 10 EF samples with three different regions (V4, V1–V2 and V3–V5). 27 They were able to show that regions V4 and V3–V5 were able to detect Gardnerella and Bifidobacterium, known genera in the cervicovaginal tract, but V1–V2 could not. Based on these results they decided to carry out their work on infertile women in the Japanese population using the V4 region. 27 We used V3–V4 regions as well, based on the fact that previous studies had used these regions successfully when working with endometrial samples. 6 , 14 , 27 , 30 The microbiome field is a rapidly changing field with many new study methods and pipelines being introduced yearly; this profits the analysis greatly but also complicates the benchmarking process. Future work in studying the endometrial microbiome could benefit from deep sequencing metagenome studies, which are likely to eliminate the bias from the 16S rRNA region selection for sequencing as well as benefit from detecting the microbial residents of the endometrium on a strain level.
Limitations of our study include lack of non‐IVF control samples, which would provide an understanding of the differences in endometrial microbiome between the IVF patients and fertile women and might help to link specific taxa to fertility problems. Also, control samples from the vagina might potentially show a distinct cervicovaginal microbial profile of these women, which in turn could give an insight into the possible contamination via the transcervical approach. Another noticeable limitation of the study is the small sample size, which may make it difficult to determine whether the outcome is a true finding. However, other studies on similar topics also have small sample sizes. 6 , 33 In future, studies with a larger sample set would undoubtedly be beneficial to the field.
Strengths of our study include matching samples of ET and EF taken from the same woman, and the usage of sampling methods that minimize the risk of contamination from the cervicovaginal tract.
5. CONCLUSION
It is suggested that the endometrial microbiome may play an important role in female infertility and modulate the success of IVF procedure. Here we report that the microbial composition of ET and EF samples of 25 women undergoing IVF treatment is significantly influenced by the abundance of Lactobacillus. To maximize the probability of finding useful microbial markers we acknowledge the possible need to continue examining the endometrial microbiome by including both ET and EF samples. As stated before, future studies might greatly benefit from metagenomics research which would eliminate the 16S region bias and make it possible to investigate the endometrial microbiome on a strain level. It would also be interesting to investigate the changes of endometrial microbiome throughout the menstrual cycle as well as to perform longitudinal studies to investigate the stability of the endometrial microbiome in humans.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
EO, AS, MP, MS and KB planned and supervised the study. KB, EK and AZ oversaw collection of samples and collected the study participants’ data. KL prepared microbiome DNA extraction and performed all data analysis. KL and EO wrote the first draft of the manuscript. AS, MP, MS and KB participated in the discussion of the findings and revised the manuscript.
Supporting information
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
ACKNOWLEDGMENTS
Data analyses were carried out in part in the High‐Performance Computing Center of University of Tartu.
Lüll K, Saare M, Peters M, et al. Differences in microbial profile of endometrial fluid and tissue samples in women with in vitro fertilization failure are driven by Lactobacillus abundance. Acta Obstet Gynecol Scand. 2022;101:212–220. doi: 10.1111/aogs.14297
Funding information
This work was funded by Estonian Research Council grants PUT 1371 (to E.O. and supported K.L.) and PRG 1076 (to A.S., M.P., M.S.), Enterprise Estonia grant No. EU48695 (to A.S.), and EMBO Installation grant 3573 (to E.O. and supported K.L.). E.O. was supported by European Regional Development Fund Project No. 15‐0012 GENTRANSMED, Estonian Center of Genomics/Roadmap II project No. 16‐0125 and Estonian Research Council grant PRG 687. K.L. was supported by The European Regional Development Fund. A.S., M.P. and M.S. were supported by the Horizon 2020 innovation grant no. EU952516 (ERIN).
DATA AVAILABILITY STATEMENT
The 16S rRNA data underlying this article is available in the Sequence Read Archive (SRA) under the reference PRJNA771346.
REFERENCES
- 1. Baker JM, Chase DM, Herbst‐Kralovetz MM. Uterine microbiota: residents, tourists, or invaders? Front Immunol. 2018;9:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Einenkel R, Zygmunt M, Muzzio DO. Microorganisms in the healthy upper reproductive tract: from denial to beneficial assignments for reproductive biology. Reprod Biol. 2019;19:113‐118. [DOI] [PubMed] [Google Scholar]
- 3. Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive‐age women. Proc Natl Acad Sci U S A. 2011;108(Suppl_1):4680‐4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bracewell‐Milnes T, Saso S, Nikolaou D, Norman‐Taylor J, Johnson M, Thum MY. Investigating the effect of an abnormal cervico‐vaginal and endometrial microbiome on assisted reproductive technologies: a systematic review. Am J Reprod Immunol. 2018;80:1‐17. [DOI] [PubMed] [Google Scholar]
- 5. Chen C, Song X, Wei W, et al. The microbiota continuum along the female reproductive tract and its relation to uterine‐related diseases. Nat Commun. 2017;8:875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu Y, Wong K‐W, Ko E‐L, et al. Systematic comparison of bacterial colonization of endometrial tissue and fluid samples in recurrent miscarriage patients: implications for future endometrial microbiome studies. Clin Chem. 2018;64:1743‐1752. [DOI] [PubMed] [Google Scholar]
- 7. Moreno I, Simon C. Relevance of assessing the uterine microbiota in infertility. Fertil Steril. 2018;110:337‐343. [DOI] [PubMed] [Google Scholar]
- 8. Miles SM, Hardy BL, Merrell DS. Investigation of the microbiota of the reproductive tract in women undergoing a total hysterectomy and bilateral salpingo‐oopherectomy. Fertil Steril. 2017;107:813‐820.e1. [DOI] [PubMed] [Google Scholar]
- 9. Winters AD, Romero R, Gervasi MT, et al. Does the endometrial cavity have a molecular microbial signature? Sci Rep. 2019;9:9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Ko E‐L, Wong K‐W, et al. Endometrial microbiota in infertile women with and without chronic endometritis as diagnosed using a quantitative and reference range‐based method. Fertil Steril. 2019;112:707‐717.e1. [DOI] [PubMed] [Google Scholar]
- 11. Moreno I, Cicinelli E, Garcia‐Grau I, et al. The diagnosis of chronic endometritis in infertile asymptomatic women: a comparative study of histology, microbial cultures, hysteroscopy, and molecular microbiology. Am J Obstet Gynecol. 2018;218:602.e1‐602.e16. [DOI] [PubMed] [Google Scholar]
- 12. Hernandes C, Silveira P, Rodrigues Sereia AF, et al. Microbiome profile of deep endometriosis patients: comparison of vaginal fluid, endometrium and lesion. Diagnostics. 2020;10:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benner M, Ferwerda G, Joosten I, van der Molen RG. How uterine microbiota might be responsible for a receptive, fertile endometrium. Hum Reprod Update. 2018;24:393‐415. [DOI] [PubMed] [Google Scholar]
- 14. Moreno I, Codoñer FM, Vilella F, et al. Evidence that the endometrial microbiota has an effect on implantation success or failure. Am J Obstet Gynecol. 2016;215:684‐703. [DOI] [PubMed] [Google Scholar]
- 15. Hashimoto T, Kyono K. Does dysbiotic endometrium affect blastocyst implantation in IVF patients? J Assist Reprod Genet. 2019;36:2471‐2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takebayashi A, Kimura F, Kishi Y, et al. The association between endometriosis and chronic endometritis. PLoS One. 2014;9:e88354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kasius JC, Broekmans FJM, Sie‐Go DMDS, et al. The reliability of the histological diagnosis of endometritis in asymptomatic IVF cases: a multicenter observer study. Hum Reprod. 2012;27:153‐158. [DOI] [PubMed] [Google Scholar]
- 18. Radzinsky VE, Kostin IN, Petrov YA, Polina ML, Gasanova BM. Diagnostic significance of chronic endometritis macrotypes differentiation among women with reproductive losses. Gynecol Endocrinol. 2017;33(Suppl 1):36‐40. [DOI] [PubMed] [Google Scholar]
- 19. Liu Y, Chen X, Huang J, et al. Comparison of the prevalence of chronic endometritis as determined by means of different diagnostic methods in women with and without reproductive failure. Fertil Steril. 2018;109:832‐839. [DOI] [PubMed] [Google Scholar]
- 20. Grahn N, Olofsson M, Ellnebo‐Svedlund K, Monstein H‐J, Jonasson J. Identification of mixed bacterial DNA contamination in broad‐range PCR amplification of 16S rDNA V1 and V3 variable regions by pyrosequencing of cloned amplicons. FEMS Microbiol Lett. 2003;219:87‐91. [DOI] [PubMed] [Google Scholar]
- 21. Salter SJ, Cox MJ, Turek EM, et al. Reagent and laboratory contamination can critically impact sequence‐based microbiome analyses. BMC Biol. 2014;12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laurence M, Hatzis C, Brash DE. Common contaminants in next‐generation sequencing that hinder discovery of low‐abundance microbes. PLoS One. 2014;9:e97876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glassing A, Dowd SE, Galandiuk S, Davis B, Chiodini RJ. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016;8:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weyrich LS, Farrer AG, Eisenhofer R, et al. Laboratory contamination over time during low‐biomass sample analysis. Mol Ecol Resour. 2019;19:982‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, Weyrich LS. Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 2019;27:105‐117. [DOI] [PubMed] [Google Scholar]
- 26. Kitaya K, Takeuchi T, Mizuta S, Matsubayashi H, Ishikawa T. Endometritis: new time, new concepts. Fertil Steril. 2018;110:344‐350. [DOI] [PubMed] [Google Scholar]
- 27. Kyono K, Hashimoto T, Nagai Y, Sakuraba Y. Analysis of endometrial microbiota by 16S ribosomal RNA gene sequencing among infertile patients: a single‐center pilot study. Reprod Med Biol. 2018;17:297‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kyono K, Hashimoto T, Kikuchi S, Nagai Y, Sakuraba Y. A pilot study and case reports on endometrial microbiota and pregnancy outcome: an analysis using 16S rRNA gene sequencing among IVF patients, and trial therapeutic intervention for dysbiotic endometrium. Reprod Med Biol. 2019;18:72‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koedooder R, Singer M, Schoenmakers S, et al. The vaginal microbiome as a predictor for outcome of in vitro fertilization with or without intracytoplasmic sperm injection: a prospective study. Hum Reprod. 2019;34:1042‐1054. [DOI] [PubMed] [Google Scholar]
- 30. Fang RL, Chen LX, Shu WS, Yao SZ, Wang SW, Chen YQ. Barcoded sequencing reveals diverse intrauterine microbiomes in patients suffering with endometrial polyps. Am J Transl Res. 2016;8:1581‐1592. [PMC free article] [PubMed] [Google Scholar]
- 31. Franasiak JM, Werner MD, Juneau CR, et al. Endometrial microbiome at the time of embryo transfer: next‐generation sequencing of the 16S ribosomal subunit. J Assist Reprod Genet. 2016;33:129‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreno‐Indias I, Sánchez‐Alcoholado L, Sánchez‐Garrido MÁ, et al. Neonatal androgen exposure causes persistent gut microbiota dysbiosis related to metabolic disease in adult female rats. Endocrinology. 2016;157:4888‐4898. [DOI] [PubMed] [Google Scholar]
- 33. Kitaya K, Nagai Y, Arai W, Sakuraba Y, Ishikawa T. Characterization of microbiota in endometrial fluid and vaginal secretions in infertile women with repeated implantation failure. Mediators Inflamm. 2019;2019:4893437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gajer P, Brotman RM, Bai G, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Molina N, Sola‐Leyva A, Saez‐Lara M, et al. New opportunities for endometrial health by modifying uterine microbial composition: present or future? Biomolecules. 2020;10:593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cicinelli E, De Ziegler D, Nicoletti R, et al. Chronic endometritis: correlation among hysteroscopic, histologic, and bacteriologic findings in a prospective trial with 2190 consecutive office hysteroscopies. Fertil Steril. 2008;89:677‐684. [DOI] [PubMed] [Google Scholar]
- 37. McQueen DB, Bernardi LA, Stephenson MD. Chronic endometritis in women with recurrent early pregnancy loss and/or fetal demise. Fertil Steril. 2014;101:1026‐1030. [DOI] [PubMed] [Google Scholar]
- 38. Cicinelli E, Matteo M, Tinelli R, et al. Chronic endometritis due to common bacteria is prevalent in women with recurrent miscarriage as confirmed by improved pregnancy outcome after antibiotic treatment. Reprod Sci. 2014;21:640‐647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cicinelli E, Matteo M, Trojano G, et al. Chronic endometritis in patients with unexplained infertility: prevalence and effects of antibiotic treatment on spontaneous conception. Am J Reprod Immunol. 2018;79:e12782. [DOI] [PubMed] [Google Scholar]
- 40. Bayar E, Bennett PR, Chan D, Sykes L, MacIntyre DA. The pregnancy microbiome and preterm birth. Semin Immunopathol. 2020;42:487‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Data Availability Statement
The 16S rRNA data underlying this article is available in the Sequence Read Archive (SRA) under the reference PRJNA771346.