

Abstract
CD55 or decay accelerating factor (DAF), a ubiquitously expressed glycosylphosphatidylinositol (GPI)-anchored protein, confers a protective threshold against complement dysregulation which is linked to the pathogenesis of idiopathic pulmonary fibrosis (IPF). Since lung fibrosis is associated with downregulation of DAF, we hypothesize that overexpression of DAF in fibrosed lungs will limit fibrotic injury by restraining complement dysregulation. Normal primary human alveolar type II epithelial cells (AECs) exposed to exogenous complement 3a or 5a, and primary AECs purified from IPF lungs demonstrated decreased membrane-bound DAF expression with concurrent increase in the endoplasmic reticulum (ER) stress protein, ATF6. Increased loss of extracellular cleaved DAF fragments was detected in normal human AECs exposed to complement 3a or 5a, and in lungs of IPF patients. C3a-induced ATF6 expression and DAF loss was inhibited using pertussis toxin (an enzymatic inactivator of G-protein coupled receptors), in murine AECs. Treatment with soluble DAF abrogated tunicamycin-induced C3a secretion and ER stress (ATF6 and BiP expression) and restored epithelial cadherin. Bleomycin-injured fibrotic mice subjected to lentiviral overexpression of DAF demonstrated diminished levels of local collagen deposition and complement activation. Further analyses showed diminished release of DAF fragments, as well as reduction in apoptosis (TUNEL and caspase 3/7 activity), and ER stress-related transcripts. Loss-of-function studies using Daf1 siRNA demonstrated worsened lung fibrosis detected by higher mRNA levels of Col1a1 and epithelial injury-related Muc1 and Snai1, with exacerbated local deposition of C5b-9. Our studies provide a rationale for rescuing fibrotic lungs via DAF induction that will restrain complement dysregulation and lung injury.
Keywords: DAF fragments, lentiviral therapy, C5b-9, IPF, ER stress
Idiopathic pulmonary fibrosis (IPF) is a devastating, fibroproliferative chronic lung disease of unknown etiology, characterized by repeated alveolar injury and inflammatory insults, followed by progressive mesenchymal activation and matrix deposition (1). Alveolar epithelial injury is a critical initiating step in IPF (2), and loss in cellular integrity triggers molecular signatures such as endoplasmic reticulum (ER) stress (3–5) with resulting apoptosis (6), potentially due to accumulation of misfolded proteins (7). Injured pulmonary epithelial cells respond by orchestrating the local innate immunity (8, 9), including the complement cascade which is implicated in IPF pathogenesis (10–12) and normally controlled by a number of epithelial membrane-bound regulators to protect the host from inadvertent injury (13).
One of the best-characterized complement regulators in humans is CD55 or DAF (decay accelerating factor), a 70–75 kD a glycosylphosphatidylinositol (GPI)-anchored membrane protein that restricts the synthesis of C3 and C5 convertases, minimizes C3 deposition and the generation of C3a, C5a and C5b-9 (14). It has a serine/threonine-enriched spacer domain and four complement control protein (CCP) repeats that are tethered to the cell membrane by the GPI anchor (14). In an unstressed cell, the quality control of GPI-DAF is performed in the ER—which involves calnexin-mediated maturation of GPI anchors (15), and protein folding facilitated by the BiP (Binding Immunoglobin Protein) (7), before they are docked at the cell membranes. When membrane-anchored DAF binds to C3b and C4b, the CCP2/3 domains prevent C3 a formation and accelerate the decay of the C3 convertases—C3bBb (alternative pathway) and C4b2a (classical and mannose-binding lectin pathways) (16). DAF also functions as a signaling molecule by crosslinking to antibodies and triggering lymphocyte activation (17). Loss of DAF is implicated in multiple organ pathologies including retinal degeneration (18), acute renal nephritis, and renal fibrosis (19).
Complement activation has been implicated in pathogenesis of pulmonary fibrosis with pharmacologic and genetic blockade of C3a/C5a receptors mitigating complement activation and lung fibrosis due to bleomycin injury (20), and adenoviral induction of TGF-β (21). We have previously demonstrated that pro-fibrotic mediators such as TGF-β and IL-17A downregulate DAF expression in small airway epithelial cells (12, 22). Furthermore, the complement products, C3a and C5a, were also noted to lead to loss of DAF and perpetuate epithelial injury with activation of apoptosis (11). However, whether the loss of a key inhibitory molecule DAF plays a central role in driving fibrosis and if fibrosis can be rescued by DAF induction is unknown. Here, we employ gain-of-function and loss-of-function strategies targeting Daf1 expression using lentiviral overexpression and RNAi-mediated gene silencing to elucidate the role of DAF in the pathogenesis of experimental lung fibrosis and explore the link between epithelial cell injury, ER stress, DAF regulation, and complement activation.
Methods
LV Production
A third generation of replication-defective self-inactivating HIV-based lentiviral vector pCSCGW expressing EGFP under the control of an internal promoter—the immediate-early human cytomegalovirus—was used to construct the LV that can drive the overexpression of murine Daf1. The DAF sequences based on Accession number NM_010016, was inserted via LR reaction—a recombination event that occurs between the sites of attR1and attR2 of the plasmid containing a self-inactivating lentiviral vector. pCSCGW-Dest expressing eGFP from a CMV promoter was used for transgene expression. Schematic map of the lentiviral vector used in this study is shown in Figure 2B.
Figure 2.
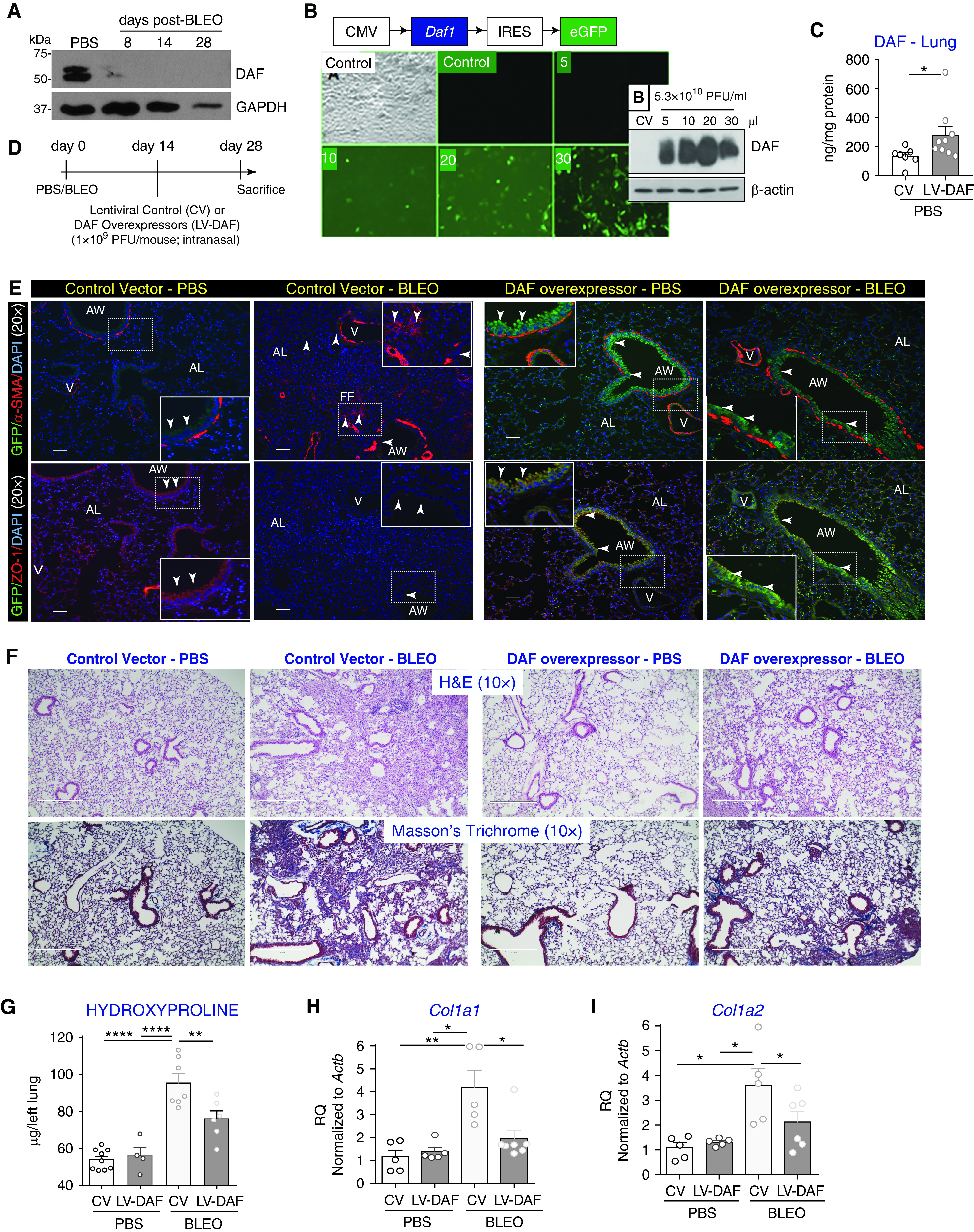
Lentiviral vector-mediated DAF induction mitigates bleomycin-induced lung fibrosis. (A) C57BL6/J mice were subjected to bleomycin injury and whole lung homogenates were collected at the indicated time points followed by immunoblotting against DAF and GAPDH (loading control). (B) Lentiviral vector containing cytomegalovirus (CMV) promoter that expresses murine Daf1 isoform (LV-DAF) with an internal ribosome entry site (IRES) was tagged to an enhanced green fluorescent protein (eGFP) reporter. Human small airway epithelial cells were transduced with either LV-DAF or lentiviral control vector (CV) for five days in complete media. Inset shows protein lysates immunoblotted against DAF and β-actin (loading control). (C) DAF induction was confirmed in vivo by intranasal instillation of LV-DAF or CV for two weeks in C57BL6/J. Whole lung homogenates were analyzed for murine DAF (ELISA) and normalized against protein concentrations. Values: mean ± SEM (*P < 0.05, unpaired t test; and n = 6–9/group). (D) Experimental strategy. (E) Tissue sections were immunostained against green fluorescence protein antibody (GFP—green) and mesenchymal marker—α-smooth muscle actin (α-SMA—red; top panels) or epithelial tight junctions protein—zona occludens-1 (ZO-1—red; bottom panels) and nucleus were counterstained with DAPI. White arrowheads indicate DAF expression as shown by GFP labeling with no α-SMA overlap, but a strong overlap with ZO-1. Original magnification: 20×. Scale bars, 100 μm. AW = airway; AL = alveoli. (F) Histopathological analyses using H&E and trichrome staining showed that DAF induction attenuated bleomycin-induced fibrosis and collagen deposition. Original magnification: 10×. Scale bars, 100 μm. (G) Hydroxyproline concentrations in lungs. (H and I) Real-time PCR analyses of the transcript levels of Col1a1 (H) and Col1a2 (I) in the lung. (G–I) Values: mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA, Bonferroni (G and H), and Newman-Keuls (I). n = 4–6/group.
Clinical Studies
All protocols were approved by the institutional review boards of Indiana University School of Medicine and the University of Michigan.
Animal Studies
The Animal Care and Use Committee at the University of Michigan and Indiana University School of Medicine approved the animal protocols used in this study. C57BL6/J mice (8 weeks of age) were obtained from Jackson Laboratories, Bar Harbor, ME, and oropharyngeally instilled with bleomycin (0.025 U/kg body weight) as previously described (22).
Lentiviral Transfer in vivo
Animals were anesthetized by intraperitoneal injections of a ketamine xylazine cocktail (10 mg/kg ketamine and 150 mg/kg xylazine). After anesthetization, ∼40 μl of suspension containing 1 × 109 PFU of lentiviral vectors were delivered intranasally over 20 seconds. The mice were placed under observation until they recovered consciousness. Mice were monitored daily for adverse reactions and changes in behavior.
siRNA Instillation in vivo
Anesthetized mice were intratracheally instilled with 50 μg of murine Daf1-specific siRNA or non-targeting control sequences in a final volume of 40 μl as previously described (20).
Statistical Analysis
Statistical analysis was performed using Student’s unpaired t test and one-way ANOVA with Bonferroni post hoc test using GraphPad Prism version 4.03 for Windows, GraphPad Software (www.graphpad.com); unless otherwise stated. Statistical significance was defined at P < 0.05.
Results
Diminished Cellular DAF Expression in Fibrotic Lungs Is Accompanied by DAF Fragmentation, Complement Dysregulation, and ER Stress
To investigate the role of DAF expression in the pathogenesis of fibrotic lung diseases, we first studied alveolar type II epithelial cells (AECs) purified from normal and IPF lungs. Compared with normal AECs, IPF-AECs expressed constitutively lower levels of DAF, with simultaneously higher levels of cleaved ATF6 (activating transcription factor 6) indicating significant ER stress (Figure 1A). Densitometry analyses confirmed this expression pattern (Figure 1A). DAF is cleaved by phospholipases (23, 24) and the cleaved fragments are ∼50-fold less active at restraining complement dysregulation (25); hence, we investigated the extent of DAF fragmentation in the lavage fluid of patients with IPF by ELISA. IPF patients had an average of approximately fourfold higher level of DAF fragments in the airway lumen, compared with the healthy volunteers (P < 0.002; 2.167 ± 0.828 versus 8.236 ± 1.612; Figure 1B); suggesting loss of biologically functional GPI-anchored DAF.
Figure 1.
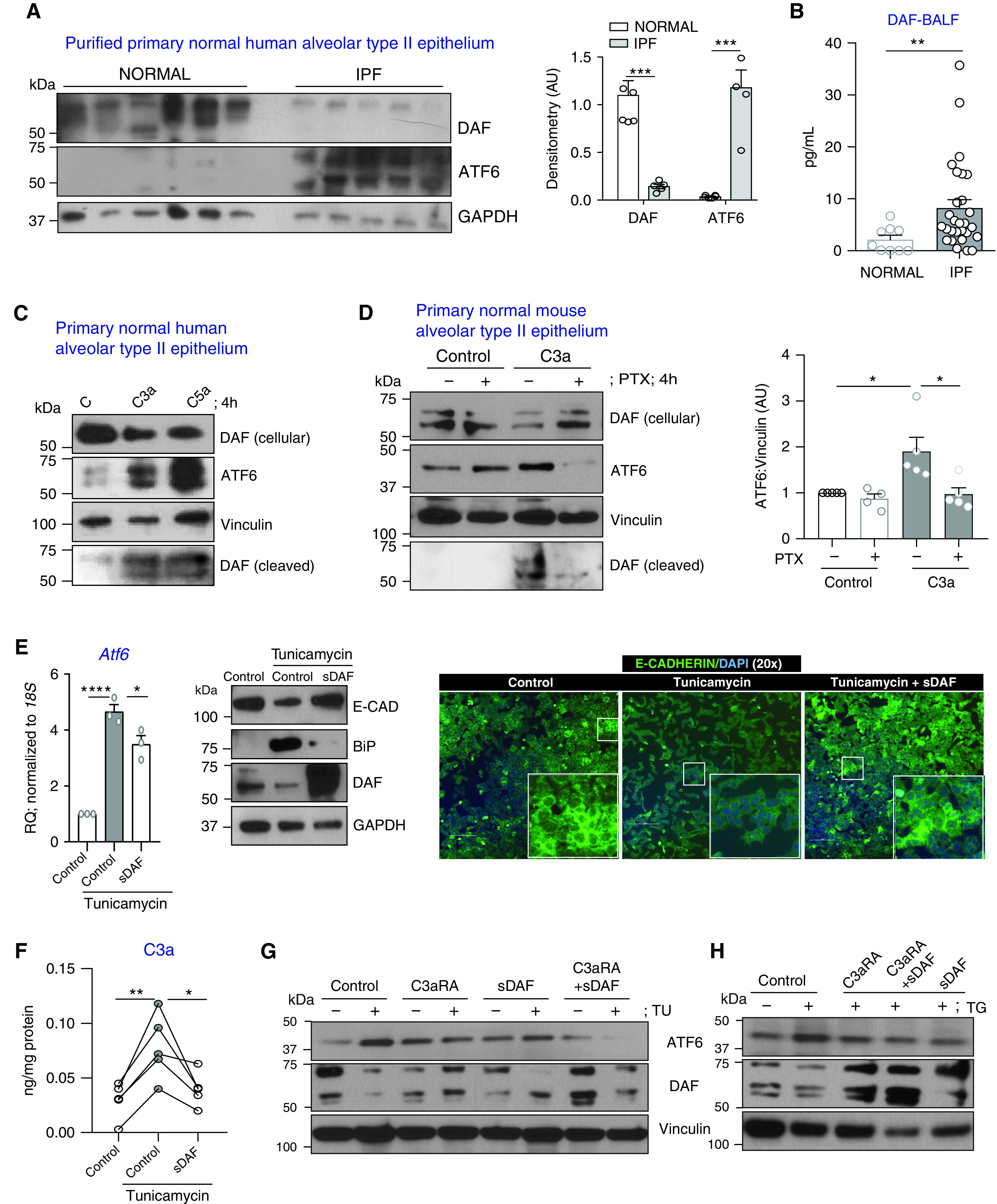
Loss of DAF (decay accelerating factor) expression and increased release of cleaved DAF fragments due to endoplasmic reticulum (ER) stress in fibrotic lungs. (A) Primary normal human alveolar type II epithelial cells (hAECs) purified from 6 normal and 5 idiopathic pulmonary fibrosis (IPF) lungs were immunoblotted with antibodies recognizing DAF, ATF6 (activating transcription factor 6), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (loading control). Immunoblots were analyzed by densitometry. Values: mean ± SEM (***P < 0.001, unpaired t test). (B) DAF levels were analyzed in BAL fluid derived from 9 normal volunteers and 28 IPF patients. Values: mean ± SEM. (**P < 0.01, unpaired t test). (C) hAECs were treated with human recombinant C3a or C5a (100 nM × 4 h). Protein lysates were immunoblotted with antibodies recognizing DAF, ATF6, and vinculin (loading control). Equal volumes of conditioned media were immunoblotted against DAF. (D) Primary normal murine AECs (mAECs) were pretreated with pertussis toxin (PTX; 100 ng/ml × 1 h) and then exposed to C3a. Protein lysates were immunblotted against DAF, ATF6, and vinculin (loading control). Equal volumes of conditioned media were immunoblotted against DAF. Immunoblots were analyzed by densitometry. Values: mean ± SEM. (*P < 0.05, one-way ANOVA, and Tukey’s). (E) mAECs were stimulated with tunicamycin (TU; 1.25 μg/ml × 4 h) and then rinsed with PBS followed by treatment with recombinant human soluble DAF (sDAF; 1.25 μg/ml × 4 h). RNA lysates were subjected to real-time PCR analyses for Atf6. Values: mean ± SEM. (*P < 0.05, ****P < 0.0001, one-way ANOVA, and Bonferroni). Protein lysates were immunoblotted against E-Cadherin (E-CAD), BiP, DAF, and GAPDH (loading control). Cells were fixed and immunostained for E-CAD. Nucleus was counterstained with DAPI. Scale bar, 100 μm. (F) Equal volumes of conditioned media were analyzed for C3a by ELISA and normalized against protein concentrations. (*P < 0.05, **P < 0.01, one-way ANOVA, and Tukey’s). (G and H) mAECs were stimulated with tunicamycin (G; TU: 1.25 μg/ml) or thapsigargin (H; TG: 20 nM) followed by treatment with recombinant human soluble DAF or C3a receptor antagonist for 4 h. Protein lysates were immunoblotted against DAF, ATF6, and vinculin (loading control).
DAF prevents the assembly of new C3 convertases and decays existing convertases, thus limiting complement activation (26). The anaphylatoxins C3a and C5a are also known to regulate DAF expression, and we have reported loss of cellular DAF due to C3a and C5a in small airway epithelial cells (12). Herein, we employed IPF-relevant normal human alveolar type II epithelial cells (hAECs) to investigate C3a- and C5a-mediated regulation of bound and cleaved DAF expression along with ATF6 (Figure 1C). hAECs were treated with exogenous human recombinant C3a or C5a (100 nM × 4 h) and subjected to immunoblotting. Both C3a and C5a decreased DAF expression with concurrent upregulation of ATF6. We analyzed the conditioned media to investigate whether the decrease in DAF expression was indeed due to the release of cellular DAF in the form of cleaved fragments onto the extracellular milieu. Interestingly, higher levels of DAF expression were observed in the conditioned media in response to C3a and C5a compared with untreated controls, suggesting that C3a and C5a contribute toward loss of cellular DAF via its cleavage. To understand whether complement-mediated ER stress and DAF regulation occurs via G-protein coupled receptor binding, we pre-treated murine AECs with pertussis toxin (PTX; enzymatic inactivator of Gi protein; 100 ng/ml × 1 h) followed by exposure to C3a for 4 hours. Western blotting analyses demonstrated C3a-mediated loss of DAF and ATF6 induction which were reversed in the presence of PTX (Figure 1D). C3a induced higher levels of DAF fragments in the conditioned media, and this was abrogated by PTX (Figure 1D).
To investigate the correlation between ER stress and DAF regulation, we used tunicamycin—a known ER stress inducer (3)—and effected subsequent treatment with intact soluble recombinant human DAF in murine AECs. This serves as a proof-of-concept to test our lentiviral overexpression strategy in vivo. Our preliminary studies indicated that tunicamycin upregulated ATF6 expression comparable to the effects of C3a/C5a shown in Figure 1C. We therefore initiated ER stress responses in mAECs using tunicamycin (1.25 μg/ml x 4 h), following which the cells were rinsed with PBS and replaced with media containing sDAF (1.25 μg/ml x 4 h). Real-time PCR analyses revealed that tunicamycin induced Atf6 transcripts by fivefold, and treatment with sDAF repressed this effect by 40% (P < 0.05, Figure 1E). Western blotting analyses of murine AECs exposed to tunicamycin demonstrated diminished expression of E-CAD and DAF with concomitant upregulation of ER stress-related BiP. These effects were reversed by sDAF treatment. Further, immunostaining of these cells revealed tunicamycin-induced loss of E-CAD expression and intercellular adhesion which is less impaired by treatment with sDAF (Figure 1E). We next examined whether tunicamycin-mediated cell stress causes local secretion of C3a. To this end, we employed ELISA to measure active C3a levels in the conditioned media of murine AECs stimulated with tunicamycin, in the presence or absence of sDAF (Figure 1F). An approximately twofold increase in C3a levels was observed and was abrogated by sDAF treatment. We next investigated relative effects of treatment with sDAF and pharmacologic blockade of C3a receptor in protecting against ER stress. C3a receptor blockade reversed ATF6 induction and loss of cellular DAF, and similar protection was noted with sDAF treatment (Figure 1G). Combination of both C3a receptor blockade and sDAF showed even further reduction in ATF6 expression, suggesting an additive effect. We used another form of ER stress inducer—thapsigargin (Figure 1H)—and observed similar effects as seen with tunicamycin, wherein induction of ATF6 and loss of DAF was reversed by treatment with C3a receptor antagonist or sDAF, or by both.
DAF Overexpression Mitigates Bleomycin-induced Progression of Lung Fibrosis
We have reported diminished expression of DAF in bleomycin-injured fibrotic lungs (22). Investigation of DAF expression at various time points after bleomycin instillation showed early and persistent loss of DAF expression (Figure 2A). To test our hypothesis that re-introduction of DAF would limit complement dysregulation and alleviate injury, we generated lentiviral vectors that can overexpress murine DAF specifically, Daf1 using the schematic model of the vector shown in Figure 2B. We next confirmed the transduction efficiency of the lentiviral vector in normal primary human small airway epithelial cells that constitutively express human DAF protein as per our prior report (12), but not the protein encoded by murine Daf1 (Figure 2B). Cells were infected with either an empty lentiviral vector or lentiviral particles containing sequences that overexpress murine Daf1 for five days in culture in complete media, followed by immunoblotting analyses (Figure 2B-inset). As expected, human small airway epithelial cells infected with the control vector did not constitutively express murine DAF; however, robust expression of murine DAF was detected in the human cells transduced with the vector overexpressing Daf1. To investigate the in vivo efficiency of Daf1 lentiviral vector, normal mice were exposed to control vectors or sequences to overexpress murine Daf1 and harvested after two weeks. Lung homogenates from mice exposed to lentiviral vectors overexpressing Daf1 demonstrated approximately twofold higher levels of Daf1 protein by ELISA (P < 0.05, Figure 2C).
To investigate if DAF overexpression in fibrotic lungs can rescue from bleomycin injury, the experimental strategy shown in Figure 2D was utilized. Since we had used an eGFP reporter tag, we immunolabeled green fluorescent protein (GFP) with mesenchymal marker, α-SMA (α-smooth muscle actin) and the epithelial tight junction marker, Zona Occludens (ZO-1). Compared with PBS- and bleomycin-instilled mice that received control vector, we observed enhanced GFP expression along the airways and in the alveoli (Figure 2E). To assess whether DAF induction was effective in protecting against bleomycin-induced ongoing lung fibrosis, histopathology analyses of the lung architecture (H&E; Figure 2F; top panels) and the collagen deposition by Masson’s blue trichrome staining (Figure 2F; bottom panels), were examined. DAF induction demonstrated a striking reduction in the extent of scarring due to bleomycin injury as revealed by H&E and Masson’s trichrome staining. Quantitative assessment of lung collagen deposition using hydroxyproline assay demonstrated ∼25% decrease in collagen content (P < 0.01, Figure 2G). mRNA analyses revealed that mice subjected to DAF overexpression demonstrated ∼50% suppression of bleomycin-induced Col1a1 and Col1a2 transcript levels (P < 0.05) as shown in Figures 2H and 2I, respectively.
DAF Induction Diminishes Complement Activation, DAF Fragmentation, and ER Stress in Fibrotic Lungs
Our previous report demonstrated complement activation—specifically, higher local levels of C3a, C5a and C5b-9 at Days 14 and 28 in response to bleomycin injury (20). We hypothesized that DAF induction would restrain complement activation and limit epithelial injury. Quantitative analyses of C3a, C5a, and C5b-9 levels in the BAL fluid (BALF) was performed by ELISA. Compared with levels induced in response to bleomycin injury, DAF induction suppressed C3a by approximately fivefold (P < 0.05, Figure 3A), C5a by approximately fourfold (P < 0.001, Figure 3A), and soluble C5b-9 by ∼50% (P < 0.05, Figure 3A). Based on our in vitro results, we hypothesized that this decreased generation of anaphylatoxins, C3a and C5a, will lead to reduced DAF fragmentation. DAF fragments were analyzed by immunoblotting (Figure 3B) and ELISA (Figure 3B) in BALF. Increased DAF expression was observed in response to bleomycin, and this was observed to be reduced by both western blotting and ELISA (P < 0.05).
Figure 3.
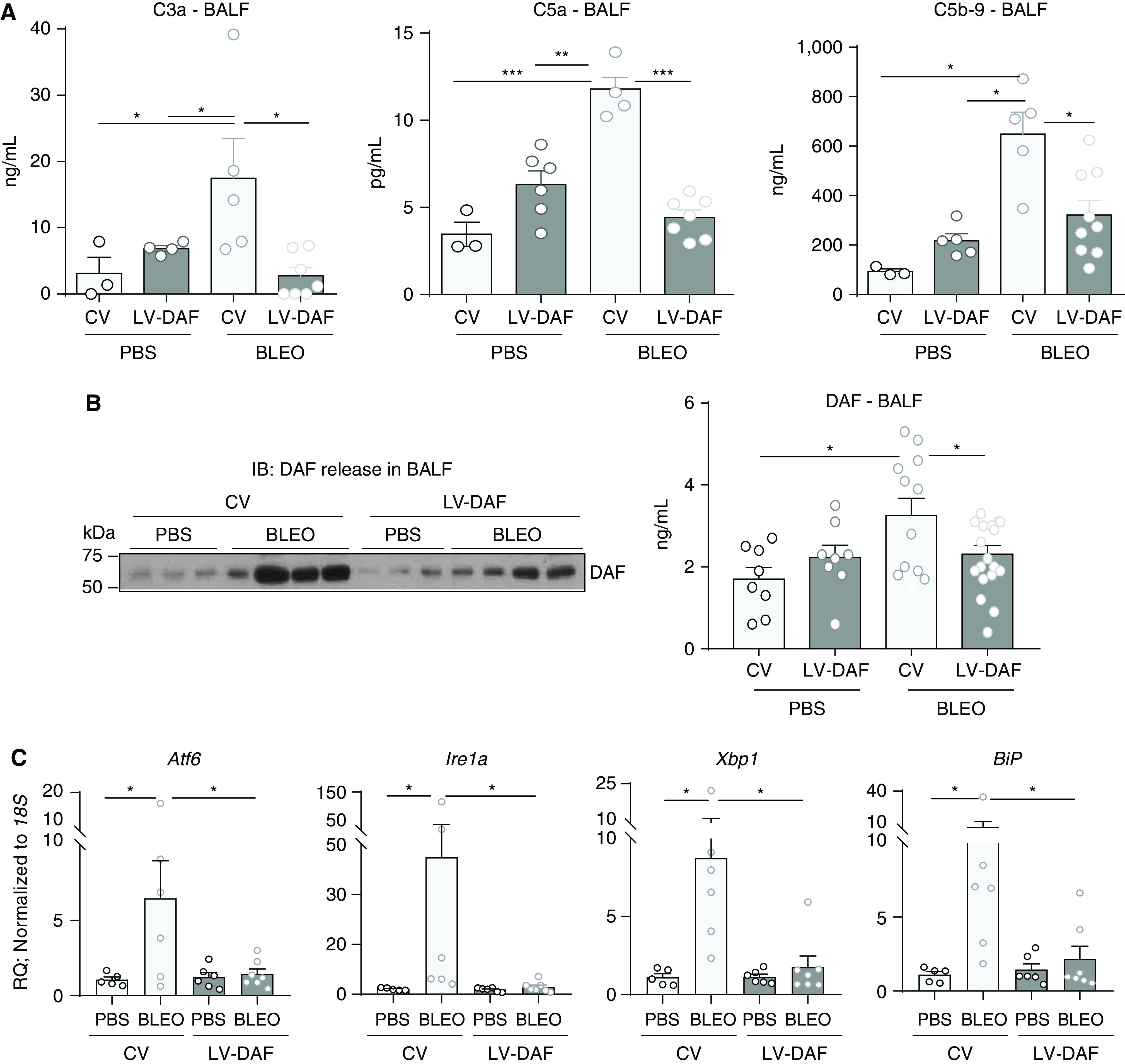
DAF induction attenuates complement activation, the release of cleaved DAF fragments, and ER stress in bleomycin-induced lung fibrosis. (A) BAL fluid (BALF) was analyzed by ELISA for C3a, C5a and C5b-9. (B) BALF was immunoblotted against DAF and β-actin, and analyzed for DAF by ELISA. (A and B) Values: mean ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. One-way ANOVA, Newman-Keuls (C3a, DAF), and Bonferroni (C5a, C5b-9). (A: n = 4–8/group), (DAF: n = 8–10/group). (C) Real-time PCR analyses of lung homogenates for ER stress-related genes. Values: mean ± SEM. (*P < 0.05, One-way ANOVA, Tukey’s (Atf6, Xbp1), Kruskal-Wallis (Ire1a, BiP), and n = 5–6/group). IB = immunoblot.
Endoplasmic reticulum stress responses have been implicated in pulmonary fibrosis (3) and when too severe are reported to cause growth arrest and cell death through apoptosis (27). ER stress-related markers– Atf6, Ire1a, Xbp1, and BiP, were examined by real-time PCR analyses in whole lung homogenates (Figure 3C). We observed decreased mRNA expression levels of these genes compared with bleomycin-injured fibrotic mice that received control vectors (P < 0.05).
DAF Induction Suppresses Cellular Apoptosis and Inflammatory Responses
Herein, we sought to examine whether bleomycin-injured mice demonstrated attenuated cellular apoptosis when subjected to DAF overexpression by TUNEL assay and quantitative caspase 3/7 activity. Figure 4A demonstrates that bleomycin injury at Day 28 has significant cellular apoptosis as indicated by TUNEL dual-labeled with α-SMA to show lung morphology. This apoptosis detected by TUNEL was suppressed by DAF induction. We next quantitatively examined cellular apoptosis in whole lung homogenates by measuring caspase 3/7 activity and found that bleomycin-induced caspase 3/7 activation was suppressed in fibrotic mice that were subjected to DAF induction (Figure 4B). DAF overexpression also led to a decrease in key inflammatory cytokines implicated in lung fibrosis, Tnfa, Ccl2 and Cxcl2 (Figure 4C).
Figure 4.
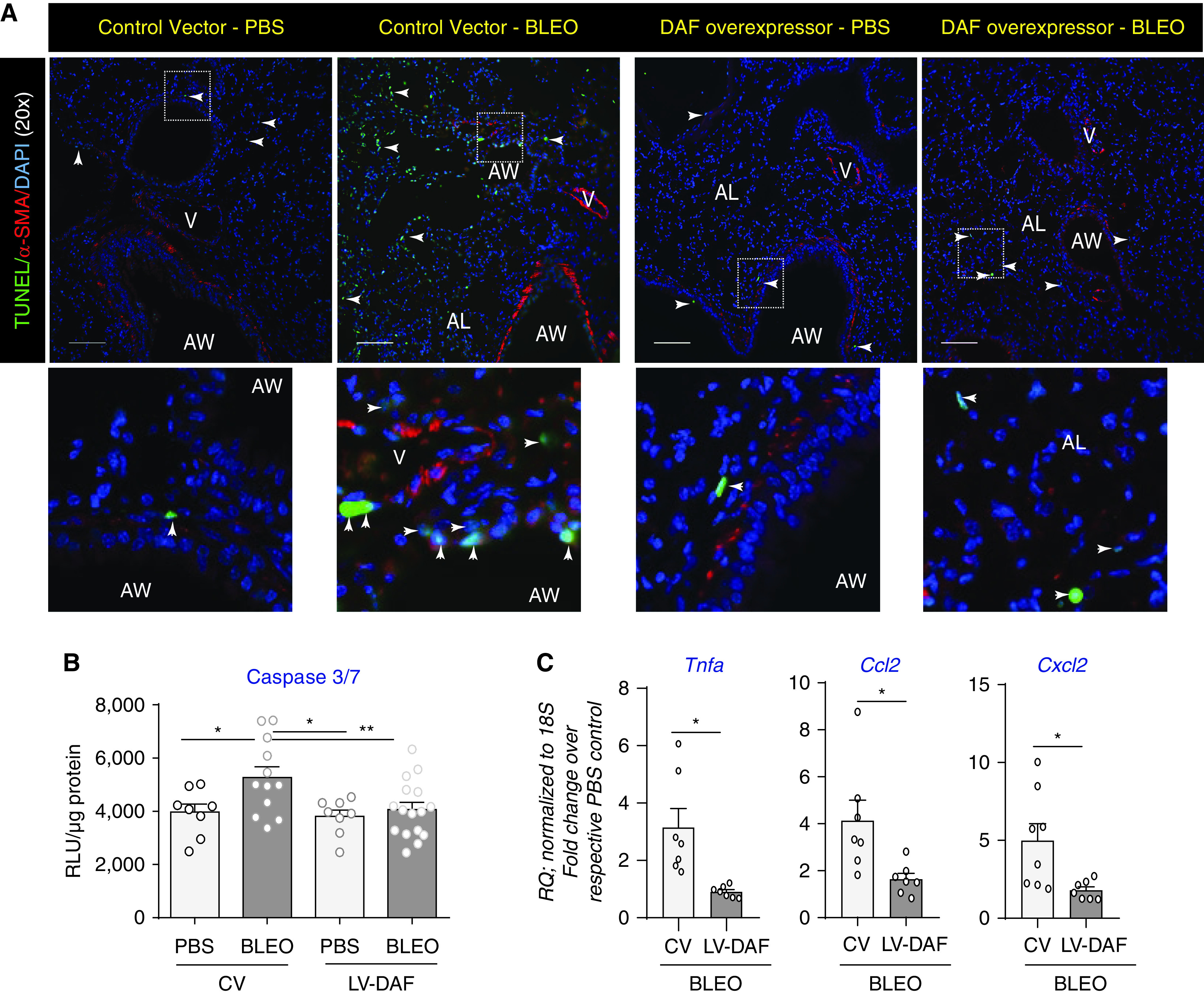
DAF induction suppresses cellular apoptosis and inflammatory responses. (A) Representative micrographs show tissue sections from mice described in Figure 2D were subjected to dual immunolabeling for TUNEL (green) and α-SMA (red) with nucleus counterstaining by DAPI. Images were captured at 20× magnification. Scale bar, 100 μm. (B) Analyses of caspase 3/7 levels by luminescence assay. Values: mean ± SEM. (*P < 0.05, **P < 0.01, One-way ANOVA, Newman-Keuls), and n = 10–12/group. (C) Real-time PCR analyses of lung homogenates for inflammation genes—Tnfa, Ccl2, Cxcl2, normalized to respective PBS controls. Values: Mean ± SEM. (*P < 0.05 and unpaired t test). n = 6–8/group.
RNAi-mediated Daf1 Silencing Exacerbates Bleomycin-induced Lung Injury
As shown in the schema in Figure 5A, Daf1-specific siRNA or non-targeting control sequences were intratracheally instilled into mice 3 days prior to bleomycin injury and killed at Day 28 post-injury. Histopathological analyses revealed that bleomycin-injured mice silenced with Daf1 siRNA have more lung injury/fibrosis (Figure 5B). We hypothesized that since DAF restrains complement activation, Daf1 silencing would aggravate complement deposition. The downstream functions due to Daf1 silencing demonstrated elevated levels of C5b-9 in bleomycin-injured lungs that was exacerbated in Daf1-silenced mice with bleomycin injury by 30% (P < 0.001, Figure 5C). Using real-time PCR analyses, the efficiency of Daf1 silencing was confirmed by analyzing Daf1 transcript levels which showed ∼65% decrease (P < 0.05, Figure 5D). While histopathology findings and C5b-9 ELISA suggest that Daf1 silencing exacerbated lung fibrosis along with unrestrained complement deposition, and loss of DAF is attributed to epithelial injury as observed in our findings in Figures 1–4, we investigated epithelial injury due to DAF silencing. To this end, real-time PCR analyses was performed to determine mRNA expression levels of alveolar epithelial injury-associated Muc1 (28), Col1a1 to measure fibrosis, and mRNA levels of Snail (Snai1) - a transcriptional repressor of E-cadherin (29). Bleomycin injury increased Muc1, Col1a1, and Snai1 transcript levels by ∼1.75-fold (P < 0.001, P < 0.01, P < 0.0001, Figure 5D). These levels were further exacerbated due to Daf1 silencing by a ∼25% increase (P < 0.05, Figure 5D). We next determined if there is a possibility of aggravated lung injury in Daf1-silenced mice subjected to bleomycin injury by investigating markers of ER stress and apoptosis using real-time PCR analyses and TUNEL assay, respectively. Findings from real-time PCR analyses of ER stress markers indicate that Atf6, Ire1a and Xbp1 were unaffected by Daf1 siRNA silencing with bleomycin injury (Figure 5E). Analyses of the lung homogenates for ER stress mediated apoptosis marker, CHOP, by western blotting revealed strong upregulation due to bleomycin injury and sustained levels in Daf1-silenced mice with bleomycin injury (Figure 5F). TUNEL assay which indicates apoptosis on these tissue sections show comparable cellular apoptosis in Daf1-silenced lung tissues when compared with bleomycin injured controls (Figure 5G). Collectively, our loss-of-function studies on the role of DAF in the progression of fibrosis indicate aggravated injury and fibrosis due to Daf1 silencing in bleomycin-injured lungs.
Figure 5.
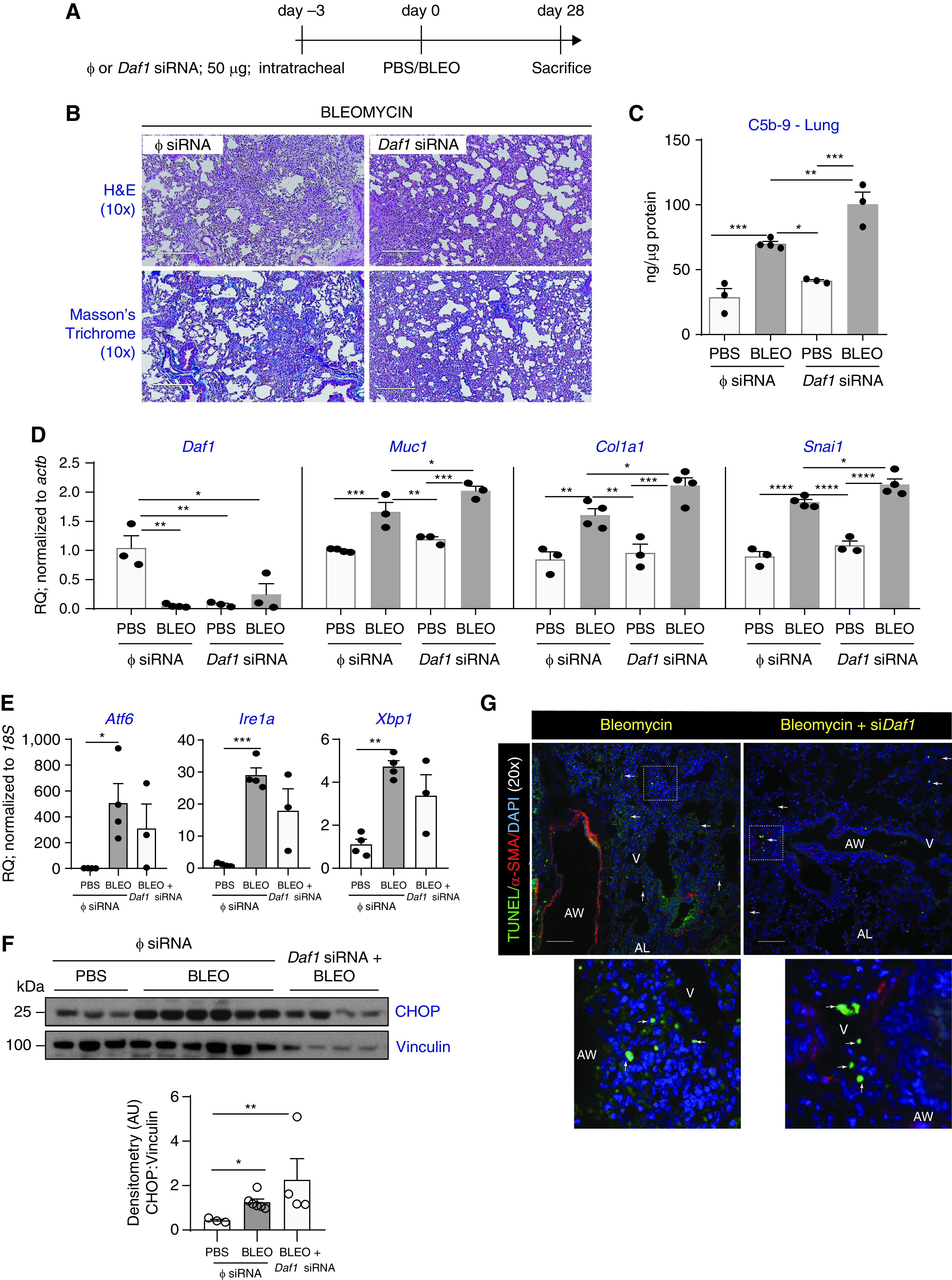
RNAi-mediated DAF silencing exacerbates lung fibrosis, complement deposition, and lung injury. (A) C57BL6/J mice were subjected to the experimental strategy as shown in the schema. (B) Histopathological analyses using HandE and Masson’s trichrome staining shows exacerbation of fibrosis and collagen deposition. (C) Lung homogenates were analyzed for C5b-9 deposition by ELISA. Values: mean ± SEM. (**P < 0.01, ***P < 0.001, one-way ANOVA, Bonferroni, and n = 3–4/group). (D) RNA isolated from lung homogenates were analyzed for the indicated genes. Values: mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA, Bonferroni (Daf1), Newman-Keuls (Muc1, Col1a1, Snai1), and n = 3–4/group. (E) ER stress-related markers were analyzed by real-time PCR analyses. Values: Means ± SEM. (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA, Tukey’s test, and n = 3–4/group). (F) Whole lung tissue homogenates were immunoblotted against an ER stress-induced apoptosis marker CHOP, and vinculin (loading control). Densitometry of blots show significantly higher upregulation in injured mice with Daf1 silencing versus PBS, than the injured controls versus PBS. Values: mean ± SEM. (*P < 0.05, **P < 0.01, One-way ANOVA, Tukey’s test), and n = 3–6/group. (G) Tissue sections from mice described in Figure 5A were subjected to dual immunolabeling for TUNEL (green) and α-SMA (red) and nucleus counterstaining by DAPI. Images were captured at 20× magnification. Scale bar, 100 μm.
Discussion
While complement dysregulation is implicated in the pathogenesis of a plethora of diseases, the concept of restraining complement activation as a potential therapeutic option in lung fibrosis is yet to be ascertained. For clinical relevance, this is likely due to the uncertainty regarding the optimal timing of intervention and the optimal target among the several potential candidates in the complement cascade. Contribution of complement to IPF pathogenesis was demonstrated in a 1978 report showing circulating complement-activating immune complexes in patients (10), and a recent report suggested that MUC5B promoter variant, the strongest risk factor of IPF, is associated with higher C3 expression (11). Others demonstrated efficacy of complement intervention by demonstrating that blocking C3 convertase (30) and C5 (releases C5a via convertase) (31) prevented sepsis- and LPS-induced lung fibrosis, respectively. Our prior reports demonstrate evidence of complement activation in IPF (12), role of complement in lung epithelial injury (12) and fibroblast activation (20) in vitro, and the therapeutic efficacy in restraining complement activation and arresting lung fibrosis by blocking receptors for C3a/C5a (20, 21) or by neutralizing IL-17A (22) in experimental models of lung fibrosis.
To our knowledge, our study is the first to demonstrate a central role of DAF in the pathogenesis of lung injury and fibrosis. This study demonstrates that restoration of DAF expression abrogates progression of fibrosis in the bleomycin injury model. Our studies demonstrate loss of DAF expression in alveolar epithelial cells isolated from IPF patients and murine lungs exposed to bleomycin injury. Restoration of DAF utilizing lentiviral vectors is sufficient to abrogate ongoing bleomycin-induced fibrogenesis, and restrain local complement components—C3a, C5a and C5b-9. Further, we detected reduced ER stress and cellular apoptosis as shown by caspase 3/7 and TUNEL, along with reduction in noncomplement-associated inflammation molecules implicated in IPF such as CCL2/MCP-1 possibly derived from injured AECs (32), the macrophage inflammation molecule CXCL2/MIP2 (33), and the inflammation mediator TNF-α, which are suppressed with DAF induction. Complementing our studies on DAF induction, we utilized Daf1 siRNA to perform loss-of-function studies and demonstrated the exacerbation of fibrosis and complement deposition with sustained ER stress and apoptosis in the injured mice. These findings are consistent with the prior knowledge that loss of the biological function of DAF (which prevents the formation of C5b-9) leads to complement activation. Our experiments showing gain-of-function and loss-of-function studies suggest that DAF is a central player in the pathogenesis of lung fibrosis by curtailing complement activation and protecting against epithelial injury.
While DAF is studied extensively in regulating the complement cascade, Daf1 induction has only been utilized to delay the onset of murine experimental autoimmune encephalitis (34), and protect against renal ischemia reperfusion injury (35), but not against ongoing lung fibrosis. In the current report, we utilized lentiviral vectors tagged with enhanced GFP reporter to restore DAF expression in the lung epithelial cells of these mice and thus, mitigate lung fibrosis. Lentiviral-mediated gene therapy has been reported in very few studies in combating the pathogenesis of lung diseases. Hirayama and colleagues have reported the local long-term expression of IL-10 in mitigating the obliteration of intrapulmonary allograft airways in an experimental tracheal implant model (36). Ikawa and colleagues demonstrated the functional correction of Hermansky-Pudlak Syndrome type-1 by lentiviral-mediated gene transfer in patient dermal melanocytes (37). DAF is abundantly expressed in the respiratory epithelia, and to our knowledge, we are the first to demonstrate that therapeutic inhibition of the complement cascade via DAF induction mitigates lung fibrosis and blunts the local cleavage of membrane-bound DAF. Further, our studies establish the therapeutic potential of DAF induction that suppresses complement activation and deposition, and therefore collagen deposition that triggers fibrogenesis. This is consistent with our previous observations that show complement inhibition by means of pharmacologic or genetic blockade of receptors against C3a- or C5a-attenuated collagen content and deposition (20).
A novel finding of our study was the presence of increased DAF fragments in lavage fluid from patients with IPF whereas cellular DAF expression was decreased indicating the cleavage of membrane-bound DAF. This was mimicked in vitro when epithelial cells were exposed to ER stress. Moran and colleagues have demonstrated the efficacy of membrane-bound DAF and the inefficacy of released DAF fragments in reducing the severity of immune complex-mediated inflammatory reactions (38). It should be noted that anchorless forms are ∼50-fold less active than GPI-anchored DAF at inhibiting complement activation (38). A recent report demonstrated the requirement of calnexin-mediated maturation of the GPI anchors in the ER to allow stability. Calnexin directly binds the N-glycan on the proteins to extend the retention time in the ER for proper folding and maturation of the GPI anchors. With ER stress, they demonstrated increased surface expression of misfolded DAF with immature GPI anchors (15). Our findings are consistent with prior reports showing that ER stress is strongly linked to the development of IPF (39). Accumulation of misfolded proteins in the ER activates the unfolded protein response pathways (UPR) that initially protect the cells and eventually leads to apoptosis. UPR comprises signaling by three ER transmembrane proteins: inositol-requiring element 1 (IRE1), protein kinase RNA-like ER kinase (PERK), and ATF6, which remain inactive under nonstress conditions through binding with glucose-regulated protein 78/immunoglobulin heavy-chain-binding protein (GRP78/BiP) (40). This BiP binds to and translocates either newly translated proteins into the ER for subsequent folding, or aberrant proteins to the proteasome for degradation. Tunicamycin induces ER stress in cells by inhibiting the first step in the biosynthesis of N-linked glycans in proteins resulting in many misfolded proteins (41), while thapsigargin induces ER stress in cells by the potent inhibition of the ER Ca2 + ATPase (42). We have presented downregulation of DAF and ER stress induction due to both tunicamycin and thapsigargin and found that treatment with sDAF protected against both stimulants of ER stress. Our data provides evidence linking DAF expression in AECs to ER stress and therefore to epithelial injury. This link between loss of membrane-bound DAF has not been previously described. This provides a novel pathway by which ER stress leads to the loss of epithelial protective mechanisms.
Our studies in vitro and in vivo suggest a feed forward loop with DAF expression and complement components regulating each other. We and others have shown decreased expression of cellular DAF in response to IL-17A (22, 43), C3a/C5a or TGF-β via p38MAPK/Snail signaling axis (12), and hypoxia-mediated regulation by HIF-1α (44), over an extended period of at least 24 hours in normal primary human small airway epithelial cells. In stark contrast, our current observations in alveolar type II epithelia that form the relevant cellular compartment affected by IPF pathogenesis, demonstrate a strikingly swift and early downregulation of DAF due to C3a and C5a within 4 hours. This decrease in membrane DAF by C3a and C5a is accompanied by increased DAF in the cell supernatant suggesting cleavage. This effect was mediated by GPCR with inhibition of C3a and C5a-mediated signaling by pertussis toxin. These findings suggest a feedback loop where loss of DAF indicates leading to an increase in C3a and C5a, in turn leads to further decrease in DAF. While the mechanism leading to this effect remains unknown, a potential mechanism could be ER stress as shown by treatment with tunicamycin and thapsigargin, leading to loss of DAF. This hypothesis was strengthened by experiments in vivo where overexpression of DAF in a fibrotic lung was accompanied by decrease in the complement componentsC3a, C5a, and C5b-9; and lower DAF fragments in lavage fluid from the lungs of these mice.
Our in vitro and in vivo studies suggest a novel feedforward loop involving ER stress, membrane-bound DAF, and complement components that contributes to lung injury and fibrosis. ER stress in the epithelium was linked to the release of membrane-DAF proteins, rendering them biologically inactive to sequester the complement components needed to form C3 convertases. The consequent increase in C3 convertase activity drives increase in anaphylatoxin C3a that further induces ER stress and loss of DAF via G-protein receptor-ligand complex. This perpetuates a vicious cycle that is prevented by treatment with soluble DAF or C3a receptor blockade. Several key pro-fibrotic mediators associated with pulmonary fibrosis, including IL-17A and TGF-β, have also been shown to regulate DAF (12, 22). Studies demonstrate that TGF-β induces the three arms of ER stress-related UPR pathways during injury and fibrosis: PERK/ATF4 induction in lung fibroblasts (45); ATF6 upregulation in renal fibroblasts which stabilizes TGF-β receptor I (46); and finally, IRE-1α/XBP1 induction with XBP1 blockade inhibiting TGF-β-induced collagen synthesis in IPF lung fibroblasts (47). TGF-β also induces ER stress via endothelin-1/phosphatidylinositol-specific phospholipase C (PI-PLC) (48), and staphylococcal PI-PLC aggravates acute respiratory distress syndrome by causing shedding of DAF in endothelial cells with unleashing of complement activity (49). While the specific role of pro-fibrotic mediators in temporally regulating DAF during the pathogenesis of fibrosis with respect to ER stress remains to be investigated, it is intriguing that TGF-β and ER stress could potentially interact to cause lung injury via DAF regulation. Our study has some potential limitations. First, although we have shown lower levels of membrane-bound DAF expression in AECs derived from IPF patients, the post-transcriptional mechanisms underlying the cleaved release of GPI-anchored membrane DAF are unknown. Second, while we show cleaved DAF fragments in BALF from patients with IPF, low sample size precludes us in assessing correlations with clinical parameters. These limitations will be in part addressed in our future studies.
In conclusion, our results suggest that complement inhibition via DAF induction mitigates the progression of fibrosis by mitigating complement activation and associated cellular apoptosis accompanied with ER stress. Additionally, we present clinical evidence demonstrating local release of cleaved DAF fragments, and the possibilities of restoring DAF expression in vivo that can contribute to host defense and tissue repair. Finally, investigating the complement cascade represents an important unexploited pathway in IPF, and there are several U.S. Food and Drug Administration-approved medications currently available to target several components of the complement cascade including C1 esterase inhibitor (Berinert, Cinryze), C3 inhibitor (Pegcetacoplan), and C5 antibody (Eculizumab, Ravulizumab). Herein, we present a viable therapeutic strategy in deterring complement dysregulation and associated tissue remodeling in IPF pathogenesis.
Acknowledgments
Acknowledgment
The graphical abstract was created with BioRender.com.
Footnotes
Supported by National Institutes of Health–National Heart, Lung, and Blood Institute grants R01; HL109288 (R.V.), HL153056 and HL108904 (K.K.K.), HL119682 and HL127805 (B.B.M.), and HL118017 and HL094622 (V.N.L.), as well as by the Cystic Fibrosis Foundation Grant 16XX0 and Taubman Institute Scholarship (V.N.L.).
Author Contributions: Conception and study design (R.V. and V.N.L.). Conduct of the experiments and data acquisition (A.J.F., E.L.T., H.G., E.M.C., E.A.M., A.V., and M.A.). Data interpretation and analyses (K.K.K., M.R.V., B.B.M., V.N.L., and R.V.). Drafting the manuscript (R.V. and V.N.L.). Revising the manuscript critically for important intellectual content (R.V. and V.N.L.).
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2021-0463OC on July 27, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol . 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, et al. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med . 2010;181:254–263. doi: 10.1164/rccm.200810-1615OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem . 2011;286:30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest . 2015;125:521–538. doi: 10.1172/JCI74942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kropski JA, Blackwell TS. Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest . 2018;128:64–73. doi: 10.1172/JCI93560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Katzen J, Beers MF. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J Clin Invest . 2020;130:5088–5099. doi: 10.1172/JCI139519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burman A, Tanjore H, Blackwell TS. Endoplasmic reticulum stress in pulmonary fibrosis. Matrix Biol . 2018;68-69:355–365. doi: 10.1016/j.matbio.2018.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Whitsett JA, Alenghat T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat Immunol . 2015;16:27–35. doi: 10.1038/ni.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wills-Karp M. Complement activation pathways: a bridge between innate and adaptive immune responses in asthma. Proc Am Thorac Soc . 2007;4:247–251. doi: 10.1513/pats.200704-046AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dreisin RB, Schwarz MI, Theofilopoulos AN, Stanford RE. Circulating immune complexes in the idiopathic interstitial pneumonias. N Engl J Med . 1978;298:353–357. doi: 10.1056/NEJM197802162980701. [DOI] [PubMed] [Google Scholar]
- 11. Okamoto T, Mathai SK, Hennessy CE, Hancock LA, Walts AD, Stefanski AL, et al. The relationship between complement C3 expression and the MUC5B genotype in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol . 2018;315:L1–L10. doi: 10.1152/ajplung.00395.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gu H, Mickler EA, Cummings OW, Sandusky GE, Weber DJ, Gracon A, et al. Crosstalk between TGF-β1 and complement activation augments epithelial injury in pulmonary fibrosis. FASEB J . 2014;28:4223–4234. doi: 10.1096/fj.13-247650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Varsano S, Frolkis I, Ophir D. Expression and distribution of cell-membrane complement regulatory glycoproteins along the human respiratory tract. Am J Respir Crit Care Med . 1995;152:1087–1093. doi: 10.1164/ajrccm.152.3.7545058. [DOI] [PubMed] [Google Scholar]
- 14. Lublin DM, Atkinson JP. Decay-accelerating factor: biochemistry, molecular biology, and function. Annu Rev Immunol . 1989;7:35–58. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- 15. Guo XY, Liu YS, Gao XD, Kinoshita T, Fujita M. Calnexin mediates the maturation of GPI-anchors through ER retention. J Biol Chem . 2020;295:16393–16410. doi: 10.1074/jbc.RA120.015577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuttner-Kondo L, Hourcade DE, Anderson VE, Muqim N, Mitchell L, Soares DC, et al. Structure-based mapping of DAF active site residues that accelerate the decay of C3 convertases. J Biol Chem . 2007;282:18552–18562. doi: 10.1074/jbc.M611650200. [DOI] [PubMed] [Google Scholar]
- 17. Davis LS, Patel SS, Atkinson JP, Lipsky PE. Decay-accelerating factor functions as a signal transducing molecule for human T cells. J Immunol . 1988;141:2246–2252. [PubMed] [Google Scholar]
- 18. Yu M, Kang K, Bu P, Bell BA, Kaul C, Qiao JB, et al. Deficiency of CC chemokine ligand 2 and decay-accelerating factor causes retinal degeneration in mice. Exp Eye Res . 2015;138:126–133. doi: 10.1016/j.exer.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrer MF, Scharrig E, Alberdi L, Cedola M, Pretre G, Drut R, et al. Decay-accelerating factor 1 deficiency exacerbates leptospiral-induced murine chronic nephritis and renal fibrosis. PLoS One . 2014;9:e102860. doi: 10.1371/journal.pone.0102860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gu H, Fisher AJ, Mickler EA, Duerson F, III, Cummings OW, Peters-Golden M, et al. Contribution of the anaphylatoxin receptors, C3aR and C5aR, to the pathogenesis of pulmonary fibrosis. FASEB J . 2016;30:2336–2350. doi: 10.1096/fj.201500044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fisher AJ, Cipolla E, Varre A, Gu H, Mickler EA, Vittal R. Potential mechanisms underlying TGF-β-mediated complement activation in lung fibrosis. Cell Mol Med Open Access . 2017;3:14. doi: 10.21767/2573-5365.100037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cipolla E, Fisher AJ, Gu H, Mickler EA, Agarwal M, Wilke CA, et al. IL-17A deficiency mitigates bleomycin-induced complement activation during lung fibrosis. FASEB J . 2017;31:5543–5556. doi: 10.1096/fj.201700289R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Angeletti A, Cantarelli C, Petrosyan A, Andrighetto S, Budge K, D’Agati VD, et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J Exp Med . 2020;217:e20191699. doi: 10.1084/jem.20191699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mueller M, Herzog C, Larmann J, Schmitz M, Hilfiker-Kleiner D, Gessner JE, et al. The receptor for activated complement factor 5 (C5aR) conveys myocardial ischemic damage by mediating neutrophil transmigration. Immunobiology . 2013;218:1131–1138. doi: 10.1016/j.imbio.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 25. Moran P, Raab H, Kohr WJ, Caras IW. Glycophospholipid membrane anchor attachment. Molecular analysis of the cleavage/attachment site. J Biol Chem . 1991;266:1250–1257. [PubMed] [Google Scholar]
- 26. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res . 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 27. Delbrel E, Soumare A, Naguez A, Label R, Bernard O, Bruhat A, et al. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci Rep . 2018;8:17939. doi: 10.1038/s41598-018-36063-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borensztajn K, Crestani B, Kolb M. Idiopathic pulmonary fibrosis: from epithelial injury to biomarkers--insights from the bench side. Respiration . 2013;86:441–452. doi: 10.1159/000357598. [DOI] [PubMed] [Google Scholar]
- 29. Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol . 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 30. Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, et al. Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med . 2015;19:2549–2563. doi: 10.1111/jcmm.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Addis-Lieser E, Köhl J, Chiaramonte MG. Opposing regulatory roles of complement factor 5 in the development of bleomycin-induced pulmonary fibrosis. J Immunol . 2005;175:1894–1902. doi: 10.4049/jimmunol.175.3.1894. [DOI] [PubMed] [Google Scholar]
- 32. Yang J, Agarwal M, Ling S, Teitz-Tennenbaum S, Zemans RL, Osterholzer JJ, et al. Diverse injury pathways induce alveolar epithelial cell ccl2/12, which promotes lung fibrosis. Am J Respir Cell Mol Biol . 2020;62:622–632. doi: 10.1165/rcmb.2019-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hagiwara SI, Ishii Y, Kitamura S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med . 2000;162:225–231. doi: 10.1164/ajrccm.162.1.9903129. [DOI] [PubMed] [Google Scholar]
- 34. Li Q, Huang D, Nacion K, Bu H, Lin F. Augmenting DAF levels in vivo ameliorates experimental autoimmune encephalomyelitis. Mol Immunol . 2009;46:2885–2891. doi: 10.1016/j.molimm.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bongoni AK, Lu B, Salvaris EJ, Roberts V, Fang D, McRae JL, et al. Overexpression of human cd55 and cd59 or treatment with human cd55 protects against renal ischemia-reperfusion injury in mice. J Immunol . 2017;198:4837–4845. doi: 10.4049/jimmunol.1601943. [DOI] [PubMed] [Google Scholar]
- 36. Hirayama S, Sato M, Liu M, Loisel-Meyer S, Yeung JC, Wagnetz D, et al. Local long-term expression of lentivirally delivered IL-10 in the lung attenuates obliteration of intrapulmonary allograft airways. Hum Gene Ther . 2011;22:1453–1460. doi: 10.1089/hum.2010.225. [DOI] [PubMed] [Google Scholar]
- 37. Ikawa Y, Hess R, Dorward H, Cullinane AR, Huizing M, Gochuico BR, et al. In vitro functional correction of Hermansky-Pudlak Syndrome type-1 by lentiviral-mediated gene transfer. Mol Genet Metab . 2015;114:62–65. doi: 10.1016/j.ymgme.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moran P, Beasley H, Gorrell A, Martin E, Gribling P, Fuchs H, et al. Human recombinant soluble decay accelerating factor inhibits complement activation in vitro and in vivo. J Immunol . 1992;149:1736–1743. [PubMed] [Google Scholar]
- 39. Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA . 2011;108:10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell . 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 41. Dawood AA, Altobje MA. Inhibition of n-linked glycosylation by tunicamycin may contribute to the treatment of sars-cov-2. Microb Pathog . 2020;149:104586. doi: 10.1016/j.micpath.2020.104586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ito H, Yamashita Y, Tanaka T, Takaki M, Le MN, Yoshida LM, et al. Cigarette smoke induces endoplasmic reticulum stress and suppresses efferocytosis through the activation of RhoA. Sci Rep . 2020;10:12620. doi: 10.1038/s41598-020-69610-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki H, Lasbury ME, Fan L, Vittal R, Mickler EA, Benson HL, et al. Role of complement activation in obliterative bronchiolitis post-lung transplantation. J Immunol . 2013;191:4431–4439. doi: 10.4049/jimmunol.1202242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pandya PH, Fisher AJ, Mickler EA, Temm CJ, Lipking KP, Gracon A, et al. Hypoxia-inducible factor-1α regulates cd55 in airway epithelium. Am J Respir Cell Mol Biol . 2016;55:889–898. doi: 10.1165/rcmb.2015-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O’Leary EM, Tian Y, Nigdelioglu R, Witt LJ, Cetin-Atalay R, Meliton AY, et al. TGF-β promotes metabolic reprogramming in lung fibroblasts via mtorc1-dependent atf4 activation. Am J Respir Cell Mol Biol . 2020;63:601–612. doi: 10.1165/rcmb.2020-0143OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen YT, Jhao PY, Hung CT, Wu YF, Lin SJ, Chiang WC, et al. Endoplasmic reticulum protein TXNDC5 promotes renal fibrosis by enforcing TGF-β signaling in kidney fibroblasts. J Clin Invest . 2021;131:e143645. doi: 10.1172/JCI143645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ghavami S, Yeganeh B, Zeki AA, Shojaei S, Kenyon NJ, Ott S, et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol . 2018;314:L493–L504. doi: 10.1152/ajplung.00372.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jain A, Olovsson M, Burton GJ, Yung HW. Endothelin-1 induces endoplasmic reticulum stress by activating the PLC-IP(3) pathway: implications for placental pathophysiology in preeclampsia. Am J Pathol . 2012;180:2309–2320. doi: 10.1016/j.ajpath.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 49. Lin YC, Liao YJ, Lee YH, Tseng SF, Liu JY, Chen YS, et al. Staphylococcal phosphatidylinositol-specific phospholipase C potentiates lung injury via complement sensitisation. Cell Microbiol . 2019;21:e13085. doi: 10.1111/cmi.13085. [DOI] [PubMed] [Google Scholar]