

Summary
Cancer immunotherapy often depends on recognition of peptide epitopes by cytotoxic T lymphocytes (CTLs). The tumor microenvironment (TME) is enriched for peroxynitrite (PNT), a potent oxidant produced by infiltrating myeloid cells and by some tumor cells. We demonstrate that PNT alters the profile of MHC class I bound peptides presented on tumor cells. Only CTLs specific for PNT-resistant peptides have a strong antitumor effect in vivo, whereas CTLs specific for PNT-sensitive peptides are not effective. Therapeutic targeting of PNT in mice reduces resistance of tumor cells to CTLs. Melanoma patients with a low PNT activity in their tumors demonstrate a better clinical response to immunotherapy than patients with a high PNT activity. Our data suggest that intra-tumoral PNT activity should be considered for the design of neoantigen-based therapy and also may be an important immunotherapeutic target.
Graphical Abstract
Introduction
Cancer immunotherapy became an attractive therapeutic option for several types of cancer. Checkpoint inhibitors demonstrated clinical efficacy in melanoma, non-small cell lung cancer, renal cancer, and others. T cell therapy (tumor infiltrating lymphocytes, T cell receptor [TCR] transduced T cells) showed encouraging clinical data (Abramson, 2020; Lee et al., 2015; Nguyen et al., 2019; Radvanyi et al., 2012). Recently, cancer vaccines against neoantigens moved from preclinical studies to the clinic (Keskin et al., 2019; Ott et al., 2017). However, despite substantial advances, the response rate of current immune therapeutics remains relatively modest. The success of cancer immunotherapy, especially vaccination, depends on the selection of appropriate T cell epitopes to target. The information about tumor associated epitopes is obtained via sequencing of tumor cell genome with subsequent analysis of selected mutant peptides for their ability to bind specific MHC class I (MHCI) alleles, or by elution and identification of peptides from MHCI molecules on tumor cell lines. It is assumed that identified peptides are expressed on tumor cells in cancer patients.
In this study, we challenge this assumption by providing evidence that the profile of peptides expressed on tumor cells can be changed by the tumor microenvironment (TME). The TME is characterized by infiltration of various myeloid cells able to produce large amounts of reactive oxygen species (ROS) and reactive nitrogen species (RNS)(Aoe et al., 1995; De Santo et al., 2005; Gabrilovich et al., 2012; Otsuji et al., 1996; Schmielau and Finn, 2001). These cells include tumor associated macrophages (TAM), polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) and neutrophils, monocytic MDSC (M-MDSC) and monocytes(Binnewies et al., 2018; Veglia et al., 2018). NADPH oxidase reduces oxygen to superoxide anion (O2.-), which readily reacts with nitric oxide (NO) resulting in the generation of peroxynitrite (PNT: ONOO-) and nitration of TCR-CD8 (Nagaraj et al., 2007). PNT is more stable than other ROS and can induce the nitration of tyrosine, cysteine, methionine and tryptophan residues (Rebrin et al., 2008). Nitrotyrosine (NT) is a marker of PNT activity(Haqqani et al., 2002). PNT has been detected in many cancers, and high intratumoral PNT levels are associated with poor prognosis (Ekmekcioglu et al., 2000; Kinnula et al., 2004; Nakamura et al., 2006). PNT is produced by infiltrating myeloid cells and by some tumor cells (Kinnula et al., 2004; Lu et al., 2011; Nakamura et al., 2006). We have previously shown that PNT could limit CTL recognition of tumor cells(Lu et al., 2011). However, the mechanism and biological consequences of this phenomenon were not clear. In this study, we tested the hypothesis that high PNT levels in the TME may render tumor cells resistant to tumor-specific cytotoxic T cells (CTLs) by changing the presentation of tumor epitopes on tumor cells.
Results
PNT alters the peptide profile in tumor cells
To assess the effect of PNT on the expression of peptides presented by MHCI, we used mouse EG.7 cells, a derivate of EL4 thymoma expressing ovalbumin (OVA). These cells provided an opportunity to use the OVA-derived H-2Kb bound epitope SIINFEKL as a reference. Treatment of EG.7 cells with PNT caused dose dependent increases in the expression of NT on cell surface (Fig. S1A). We selected a dose of PNT (2.5 mM) that did not affect cell viability, expression of MHCI H-2Kb molecules, but caused a substantial up-regulation of NT (Fig. S1B). Expression of SIINFEKL-H-2Kb complexes (OVA-Kb) on the surface was also not affected (Fig. S1C). We also evaluated the NT level (as the reflection of PNT activity) in different populations of cells isolated from Lewis Lung Carcinoma (LLC) tumors. NT was readily detectable in CD11b+ myeloid cells but not in B, T, or NK cells. Most PNT activity was found in PMN-MDSC and M-MDSC, with less but detectable level in TAM (Fig. S1D). In vitro concentration of 2.5 mM PNT resulted in NT expression levels on tumor cells similar to that were detected on TME cells from LLC mouse tumors (Fig. S1E).
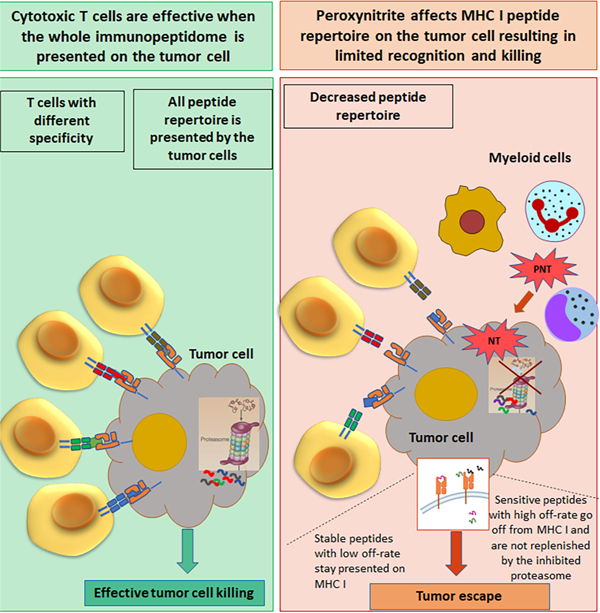
To assess the effect of PNT on the expression of MHCI peptide complexes (pMHC), tumor cells were either labelled with “heavy” media containing 13C6-lysine and 13C6,15N4-arginine or were cultured in the same medium containing non-labeled lysine and arginine. Labeled cells were then treated with 2.5 mM PNT on ice for 5 min, washed and then cultured at 37°C. At different time points after PNT treatment, untreated (unlabeled) and PNT-treated (labeled) EG.7 cells were mixed together at a 1:1 ratio, H-2Kb molecules were purified (Fig. S2A), and H-2Kb associated peptides were eluted and analyzed by LC-MS/MS mass-spectrometry (Fig. S2B). For each peptide, the ratio of labeled and non-labeled counterparts was calculated. There were no substantial changes in peptide profile right after the treatment (0 min). In contrast, 30 minutes post-treatment, about 20% of peptides in PNT-treated cells showed a greater than 2-fold decrease in yield relative to untreated EG.7 cells. Peptide profile in PNT treated cells gradually recovered after 5 h and returned to the pre-treatment level 16 h after the treatment (Fig. 1A, Table S1). For further analysis, H-2Kb bound peptides were separated into 2 groups: peptides that were minimally affected by PNT (PNT-resistant) and peptides that were reduced by at least 2-fold in PNT treated cells (PNT-sensitive). The SIINFEKL peptide falls into the PNT-resistant category. Typical examples are shown in Fig. S2C. Analysis of amino-acid sequences revealed that PNT-sensitive peptides had markedly higher representation of tyrosine in the H-2Kb anchor position 5, whereas resistant peptides had mostly phenylalanine (Fig. 1B). The search of motifs that could distinguish PNT-sensitive (PNT-S) and PNT resistant (PNT-R) peptides (Bailey and Elkan, 1994) demonstrated that Y/V3 R/E/K4F5L8 motif was enriched in PNT-R peptides and was able to distinguish 51 out of 89 resistant octamers (p<0.05). A similar consensus motif was observed in nonameric PNT-R peptides (Fig. 1C). The presence of the charged amino acids (R/K/E) adjacent to tyrosine residue can have a protective effect against nitration by PNT(Bartesaghi and Radi, 2018; Bayden et al., 2011; Souza et al., 1999). We also assessed the effect of PNT on the profile of HLA-A, -B, -C bound peptides in the human melanoma UACC903 cell line with HLA-type: A30, A74, B07, B57, C15, C18 (Fig. S3A). PNT substantially changed the peptide profile (Fig. S3B, Table S2) with substitution of anchor residues (Fig. S3C). The presence of proline motifs distinguished 24 out of 32 PNT-S 9-mer peptides, 28 out of 34 PNT-S 10-mer peptides and 46 out of 52 PNT-S 11-mer peptides (p<0.05). Thus, PNT is able to alter the MHCI peptide profile presented on tumor cells.
Figure. 1. PNT treatment changes MHC I peptide profile on tumor cells.
A. MHCI peptide profile in murine EG.7 cells after PNT treatment. The results are shown as frequency histograms of log2 (PNT/control) ratios for the abundance of the detected peptide species. Red dashed line designates 1:1 PNT/Control ratio, blue dotted line shows the decrease of PNT-treated peptide counterpart more than 2-fold. Scaled density plots for the same datasets are overlaid on the histogram to demonstrate the distribution. Results of one out of three similar experiments are shown. B. MHCI peptides from 30 min time-point are grouped according to their lengths and PNT/control ratio (less than 0.5 are designated as “PNT-sensitive” and more than 0.8 as “PNT-resistant”). The height of each bar is proportional to the degree of amino acid conservation, and the height of each letter composing the column is proportional to its frequency. Amino acids: hydrophobic (blue), polar uncharged side chains (green), with electrically charged side chains (red), proline and tyrosine - yellow and orange. For each position the amino acid content was compared between sensitive and resistant peptides and the significance of difference was tested with Fisher’s exact test with FDR adjustment, * p<0.05. C. The amino acid motif for octamers and nanomers enriched in resistant versus sensitive peptides with the corresponding E-value < 0.05 (calculated by MEME algorithm). D. Peptide binding to RMA-S cells (on the left) and the stability of pMHC complexes formed by those peptides (on the right). Three experiments with the same results were performed. Data shown as mean ± SD. P values were calculated in two-way ANOVA test. E. Effect of PNT on peptide-MHCI binding. RMA-S cells were pre-treated with PNT and then the same assays as in D were performed. Data shown as mean ± SD . P values were calculated in two-way ANOVA test. See also Figures S1-S3 and Tables S1-S3.
Mechanism of PNT mediated effect on peptide expression by MHC class I
To understand the mechanism and biological significance of the observed phenomenon we synthesized PNT-R and PNT-S mouse peptides (derived from different proteins) with high predicted binding to H-2Kb (Table S3). Using RMA-S cells we assessed the ability of these peptides to bind to MHCI. No differences were found between PNT-S and PNT-R peptides (Fig 1D). To assess the stability of peptide - MHCI (pMHCI) complexes, RMA-S cells were loaded with peptides for 2 h, washed from excess peptide and cultured for different times. PNT-S peptides demonstrated much higher dissociation rates than PNT-R peptides (Fig. 1D). Pre-treatment of RMA-S cells with 2.5 mM PNT, caused only a small decrease in the binding of the peptides and no difference was detected between resistant and stable peptides. Similarly, treatment of RMA-S cells with PNT before loading with peptides had no detectable effect on dissociation rates (Fig. 1E). These results suggested that the differences between PNT-S and PNT-R peptides were not in their MHCI binding affinity, but in the stability of pMHCI complexes. MHCI peptides with short (less than 40 min) half-life were described previously(Boulanger et al., 2018). Although most of the pMHCI complexes on tumor cells have rate of turnover of more than 6 h, a substantial proportion of peptides have a much shorter turnover (Milner et al., 2006). Thus, it appears that PNT treatment enriched the tumor immunopeptidome with pMHCI peptides with a long turnover. However, we couldn’t detect the nitrated peptides by mass-spectrometry. That could be due to the low sensitivity of the detection or decreased stability of pMHC.
We next asked if nitration of peptides by PNT may affect their binding to MHC class I molecules. We designed model peptides with high tyrosine contents and high H-2Kb binding capacity predicted in silico. These peptides should be highly susceptible to nitration by PNT. We also used several H-2Kb peptides from our dataset containing tyrosine in the sequence. Peptides were treated with PNT and then used in RMA-S binding assay. No reduction in MHC I binding capacity of PNT-pretreated peptides compared to their control counterparts was found (Fig. 2A). We also tested the possibility that PNT might affect expression of the genes encoding proteins with resistant and sensitive peptides. We found no differences in mRNA expression between control and PNT treated cells at any time point (Fig. S4).
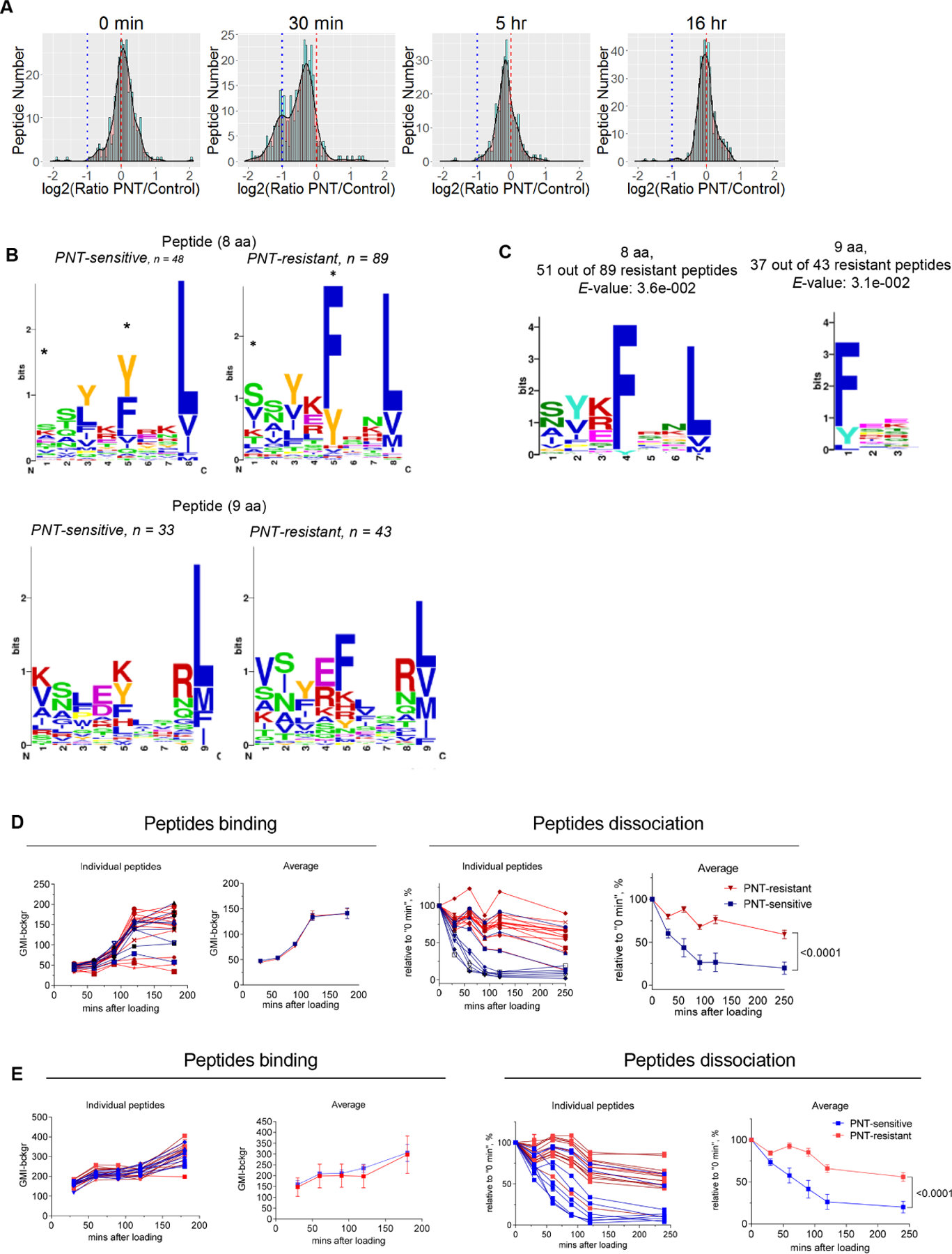
Figure 2. PNT disrupts proteasomal function and changes MHC I peptidome of tumor cells.
A. The effect of peptides pretreatment with PNT on the MHC I binding ability. 40 μg of specified peptides were either left untreated (Untr) or pretreated with PNT (2.5 mM). Reaction was stopped by diluting PNT to 0.125 mM (PNT). The binding assay was conducted with 10 μg/ml final concentration of peptide. Fluorescence of background (cells without peptides) was subtracted. Mean ± SD are shown. (n=2 for control and n=3 for PNT groups). B. Indicated tumor cells were treated with 2.5 mM PNT for 5 min and proteasomal activity was measured in the lysates (n=3). Mean ± SD are shown. ** - p<0.01; *** - p<0.001 in two-sided Student’s t-test. C. EL4 cells were co-cultured with Gr-1+ splenic cells from tumor-bearing mice for 18 h and analyzed for NT presence on cell membrane. D. Gr-1+ cells isolated from spleens and tumors of TB or control mice were cultured with EL4 tumor cells for 18 h. Gr-1+ cells were depleted by magnetic beads and proteasomal activity was measured in the tumor cells. Mean ± SD are shown. n=5, *** - p<0.001, **** - p<0.0001 in one-way ANOVA test with correction for multiple comparisons. E. EG.7 cells were treated at indicated time with vehicle (control) or 2.5 mM PNT. Chymotrypsin, trypsin and caspase-like activities were measured using the corresponding fluorogenic substrate. Mean ± SD are shown. N=3. P values were calculated in unpaired Student’s t-test. **-p<0.01; ***-p<0.001; **** - p<0.0001. F. EL4 and LLC tumor cells were treated with 2.5 mM PNT for 5 min on ice. Proteasomal activity was measured with the use of indicated fluorogenic substrates. n= 3. P values were calculated in two-sided Student’s t-test. Mean and SD are shown. See also Figure S4.
Proteasomes are critical for generating pMHCI (Milner et al., 2013; Sijts and Kloetzel, 2011). Proteasome inhibitors, epoxomicin and bortezomib, reduced the rates of synthesis pMHCI, but not all peptides were affected to a similar extent(Milner et al., 2013). The PNT inducing agent, SIN-1, has been shown to affect proteasomal activity(Osna et al., 2004). Alteration of proteasomes by PNT might change the antigen processing rate in tumor cells. In tumor cells, high PNT levels then could affect antigen processing by inhibiting proteasomal activity and as a result, peptides with low dissociation rate would remain on MHCI (PNT-R), whereas those with high dissociation rates would be lost (PNT-S). To test this hypothesis, we evaluated proteasomal activity in tumor cells treated with PNT. PNT caused a profound decrease in proteasomal activity (Fig. 2B). To determine whether a similar effect is observed in the presence of myeloid cells, tumor cells were incubated with Gr-1+ myeloid cells isolated from spleens of EL4 tumor-bearing (TB) mice. Following an 18 h co-culture, a substantial level of NT was detected in tumor cells (Fig. 2C), which was associated with a decrease in proteasomal activity (Fig. 2D). Proteasomal activity was disrupted immediately after addition of PNT, began to recover by 5 hours, and was almost completely restored by 22 hours (Fig. 2E).
Some substrates are suggested to be different between constitutive proteasomes and immunoproteasomes (Ferrington and Gregerson, 2012; Huber et al., 2012; Winter et al., 2017). We assessed the effect of PNT on different proteasomes using proteasome-specific fluorogenic substrates. In EL4 and LLC cell lines, PNT affected mostly chymotrypsin- and trypsin-like activities associated with constitutive proteasomes and to a lesser extent affected chymotrypsin-like activity associated with immunoproteasomes (Fig. 2F).
We asked if IFN-γ, which is commonly produced by activated T cells during immunotherapy, would affect PNT inhibition of proteasome. Tumor cells were pre-treated with IFN-γ followed by PNT treatment and proteasomal activity was assessed. The effect of PNT on proteasomal activity was the same in IFN-γ pre-treated and non-pre-treated cells (Fig. 3A).
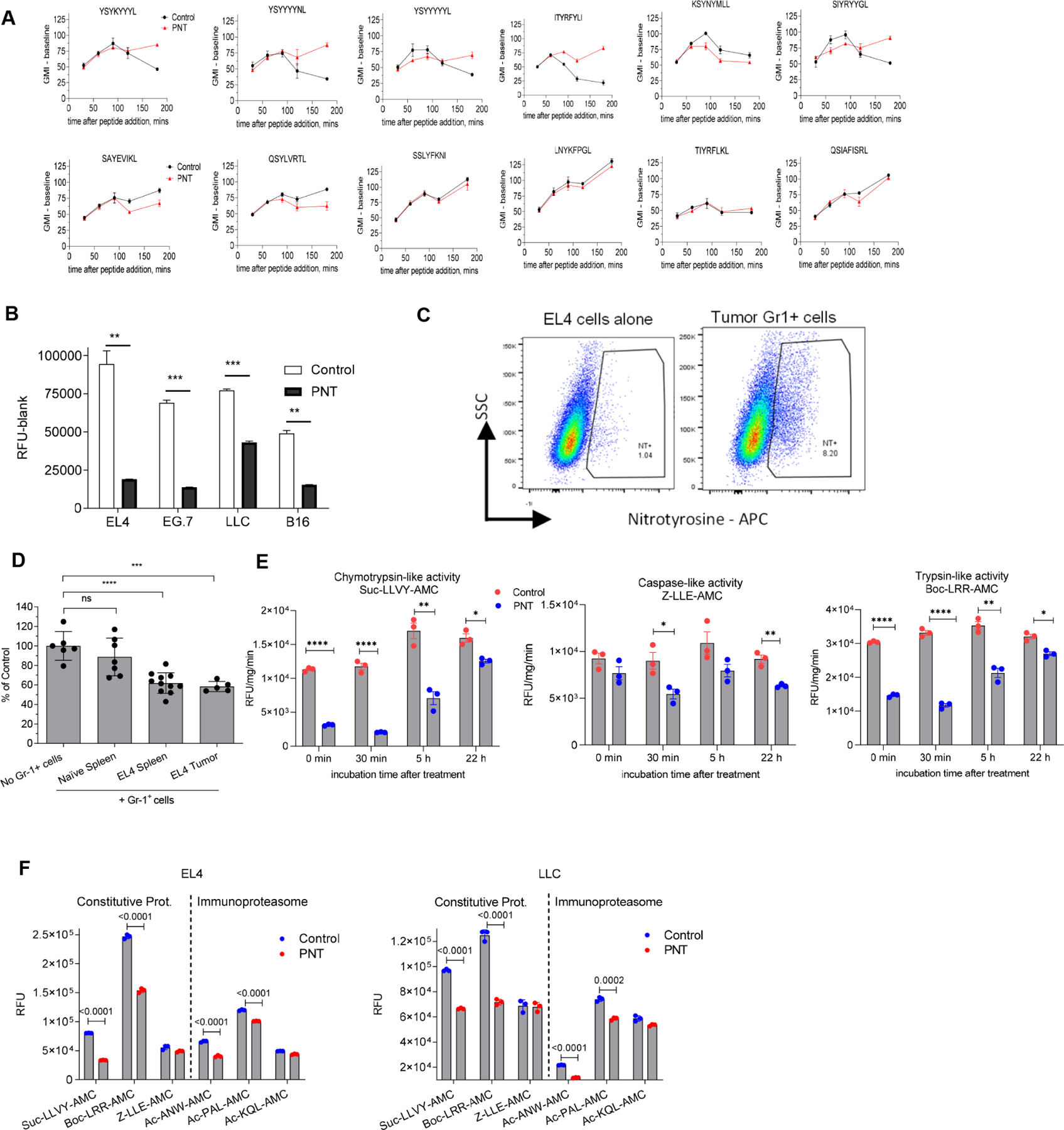
Figure 3. Effect of bortezomib on peptide repertoire.
A. PNT effect on proteasomal activity in EG7 and LLC tumor cells after the pre-treatment with 100 ng/ml IFN-γ for 24 hours . Substrates are indicated on the graph. Mean ± SD are shown. N=4, p values were calculated in two-sided unpaired Student’s t-test. *-<0.05, **-p<0.01; ***-p<0.001; ****- p<0.0001. B. EG.7 cells were incubated with 15 nM bortezomib or control media for the specified time. Proteasomal activity was measured using indicated fluorogenic substrates. Mean ± SD are shown. N=5. P values were calculated with one-way ANOVA test with corrections for multiple comparisons. **** - p<0.0001; C. MHC I peptides were isolated from EG7 cells, treated and non-treated cells with 15 nM bortezomib during the specified time periods and analyzed using SILAC technology and LC-MS/MS. Red dashed line designates 1:1 PNT/Control ratio, blue dotted line shows the decrease of PNT-treated peptide counterpart more than 2-fold. Scaled density plots for the same datasets are overlaid on the histogram to demonstrate the distribution. D. Peptides eluted from EG.7 cells from 2 experimental settings (PNT and bortezomib treatment) were compared. Sensitive peptides - ratio PNT/Control or Bortezomib/Control <0.667; resistant peptides - respective ratios > 0.75. The overlaps between different peptide groups are shown as Venn diagrams. The analysis was performed with RStudio software and VennDiagram package. E. EL4 cells were treated with PNT or vehicle, loaded with 10 μg/ml SIINFEKL peptide for a specified time at 27°C, washed and stained with antibody specific for SIINFEKL-H2Kb pMHC complexes (top panel, n=5) or total MHC class I, H2Kb (bottom panel, n=4) and evaluated by flow cytometry analysis. Mean ± SD are shown. P values were calculated in two-sided Student’s t-test. See also Figures S5, S6, and Table S4.
To assess the effect of proteasome inhibition on the profile of MHC class I bound peptide, we used the proteasome inhibitor bortezomib. We selected a relatively low concentration of bortezomib (15 nM) that did not affect the viability of tumor cells or expression of MHC class I (H2Kb) (Fig. S5). This treatment resulted in modest inhibition of proteasomal activity (Fig. 3B). To verify that inhibition of proteasomes could recapitulate the effect of PNT, we performed mass spectrometry of MHCI peptides isolated from EG.7 cells treated with bortezomib. We observed a substantial decrease in representation of the corresponding peptides in cells treated with bortezomib as compared to non-treated cells (Fig. 3C, Table S4). We also compared the sequences of peptides changed in PNT and bortezomib treated EG.7 tumor cells. We found substantial overlap in the peptides reduced in both bortezomib and PNT treated cells (Fig. 3D). The treatment of cells with PNT or bortezomib did not affect the amount of 6 catalytic subunits of proteasomes in EG7 tumor cells. Five subunits were detectable and showed no changes after the treatment with PNT (Fig. S6A) or bortezomib (Fig. S6B). One subunit – PSMB10 was not detectable.
Since PNT did not affect expression of MHC class I (Fig. S1), but caused a decrease in the amount of peptides, this might result in the generation of “empty” MHC class I molecules. These molecules have low affinity peptides and are unstable and rapidly degrade at 37°C (De Silva et al., 1999). Empty MHC-I can be stabilized at 26°C and could be differentiated from peptide-MHC-I complexes (pMHC) by loading cells with high affinity peptide (SIINFEKL) and staining for pMHC with anti-H2Kb-OVA antibody. As expected, we observed time dependent increase in pMHC expression in EL4 tumor cells as a result of peptide binding (Fig. 3E). EL4 cells treated with PNT showed significantly higher expression of pMHC than the cells treated with vehicle (meaning more “empty” MHC I molecules on PNT-treated cells). In contrast, no differences in total H-2Kb expression were observed between PNT-treated and control cells (Fig. 3E). These data indicated that PNT treatment increased MHC class I molecules available for peptide binding. Thus, taken together, these results support the conclusion that inhibition of proteasomes by PNT can cause changes in the profile of peptides on the tumor cell surface.
PNT-S and PNT-R peptides are differentially recognized by CTLs
Since different tumor models share many H-2Kb bound peptides (Fig. 4A), we tested whether PNT-S and PNT-R peptides observed in EG.7 tumor cells were also present in other tumor cells. Many PNT-S and PNT-R peptides were shared among EG.7, LLC, B16F10 melanoma, and MC38 colon carcinoma cells (Fig. 4B).
Figure 4. Spontaneous anti-tumor CD8+ T cell responses develop primarily against PNT-resistant peptides.
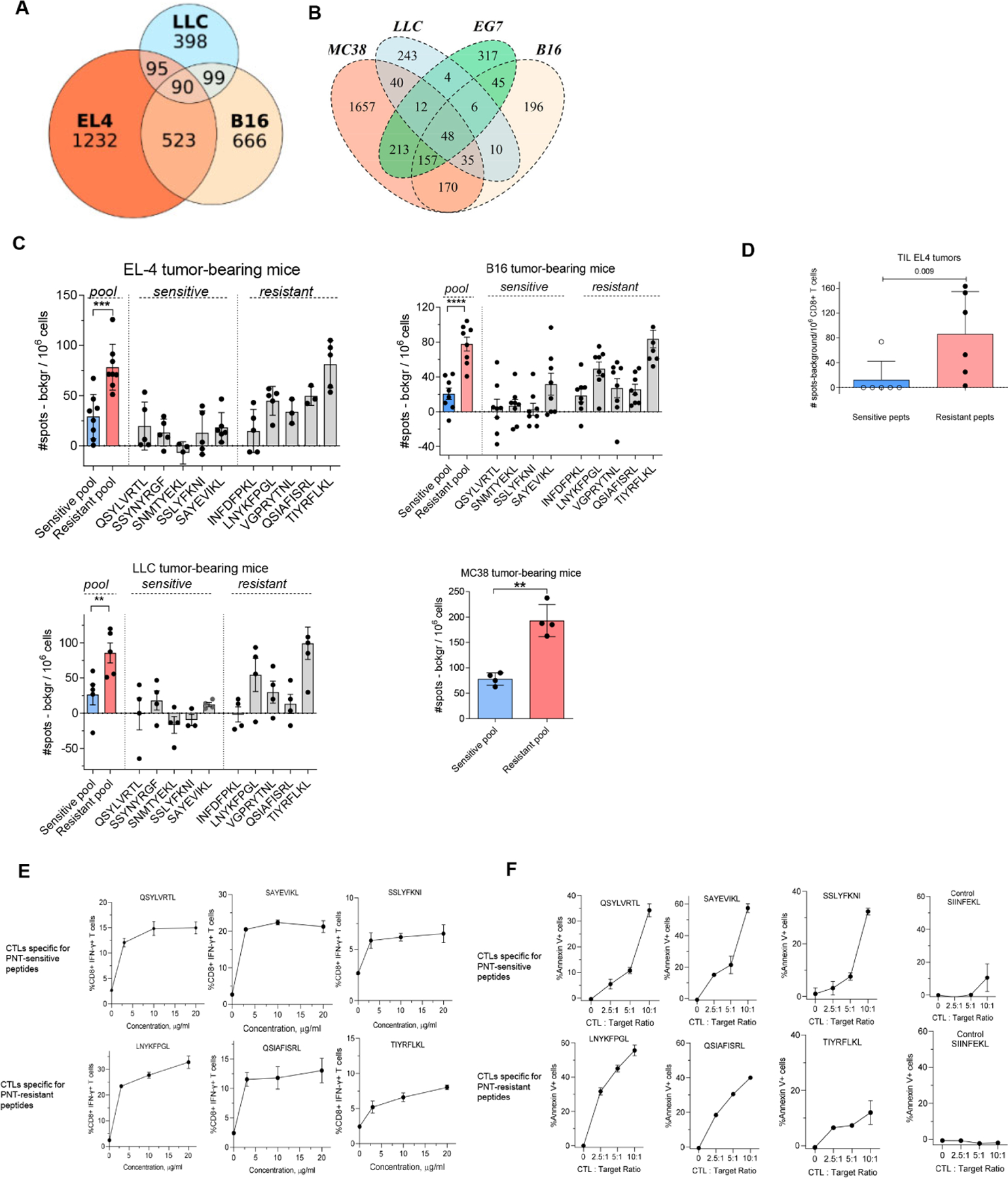
A. Datasets of MHCI peptides expressed by EL4, B16F10 and LLC were extracted from (Schuster et al., 2018) and analyzed for shared antigens and presented as a Venn diagram. B. Overlap of peptides detected in EG.7 cells with those reported (Schuster et al., 2018)(Yadav et al., 2014). C. Splenic CD8+ T cells were isolated from TB mice and re-stimulated with 2 μg/ml individual PNT-sensitive and PNT-resistant peptides or the pool of all 5 peptides for each group, 1 μg/ml each peptide. The amount of IFN-γ-producing peptide-specific CD8+ T cells was measured by ELISPOT, n= 4–8. ** - p<0.01, *** - p<0.001 in two-sided Student’s t-test. Data shown as Mean ± SD. D. Tumor CD8+ T cells were isolated from EL4 TB mice and re-stimulated with the pool of PNT-sensitive or resistant peptides. The amount of IFN-γ-producing peptide-specific CD8+ T cells was measured by ELISPOT (n=6). Mean and SD and p value in Mann-Whitney test are shown. E, F. Specificity of CTLs generated against PNT-sensitive and resistant peptides. T cells were generated by vaccinations and expansion in vitro. E. Purified CD8+ T cells were restimulated for 12 h in the presence of feeder cells (irradiated naïve splenocytes) with individual peptides used for immunization. The percentage of specific CD8+ T cells was evaluated by intracellular staining for IFN-γ. Mean and SD are shown. Each experiment was performed in duplicates. F. CTLs were tested against RMA-S cells loaded with the specific (indicated on the graph) or control (SIINFEKL) peptides. CTLs were added to the RMA-S cells in the presence of the peptides (to avoid impact of the differences in off-rate). Mean and SD are shown. Experiments were performed in duplicates. See also Figure S7.
To determine the biological significance of these observations, we assessed spontaneous development of CD8+ T cells specific for PNT-sensitive and PNT-resistant peptides in different tumor models. When tumors reached 1.5 cm in diameter, CD8+ splenic T cells were isolated and stimulated with pooled or individual PNT-R or PNT-S peptides. Responses were evaluated by IFN-γ ELISPOT assays. In all four tested models, markedly higher responses to PNT-R than to PNT-S peptides were detected (Fig. 4C). CD8+ T cells were isolated from EL4 tumors and stimulated with the pool of PNT-R or PNT-S peptides. Markedly higher numbers of CD8+ T cells specific for PNT-resistant than PNT-sensitive peptides were observed (Fig. 4D).
We assessed the expression of source proteins of PNT-sensitive and PNT resistant peptides in tumor, lung, liver, and spleen tissues. No differences between proteins producing PNT resistant and PNT-sensitive peptides were found (Fig. S7A).
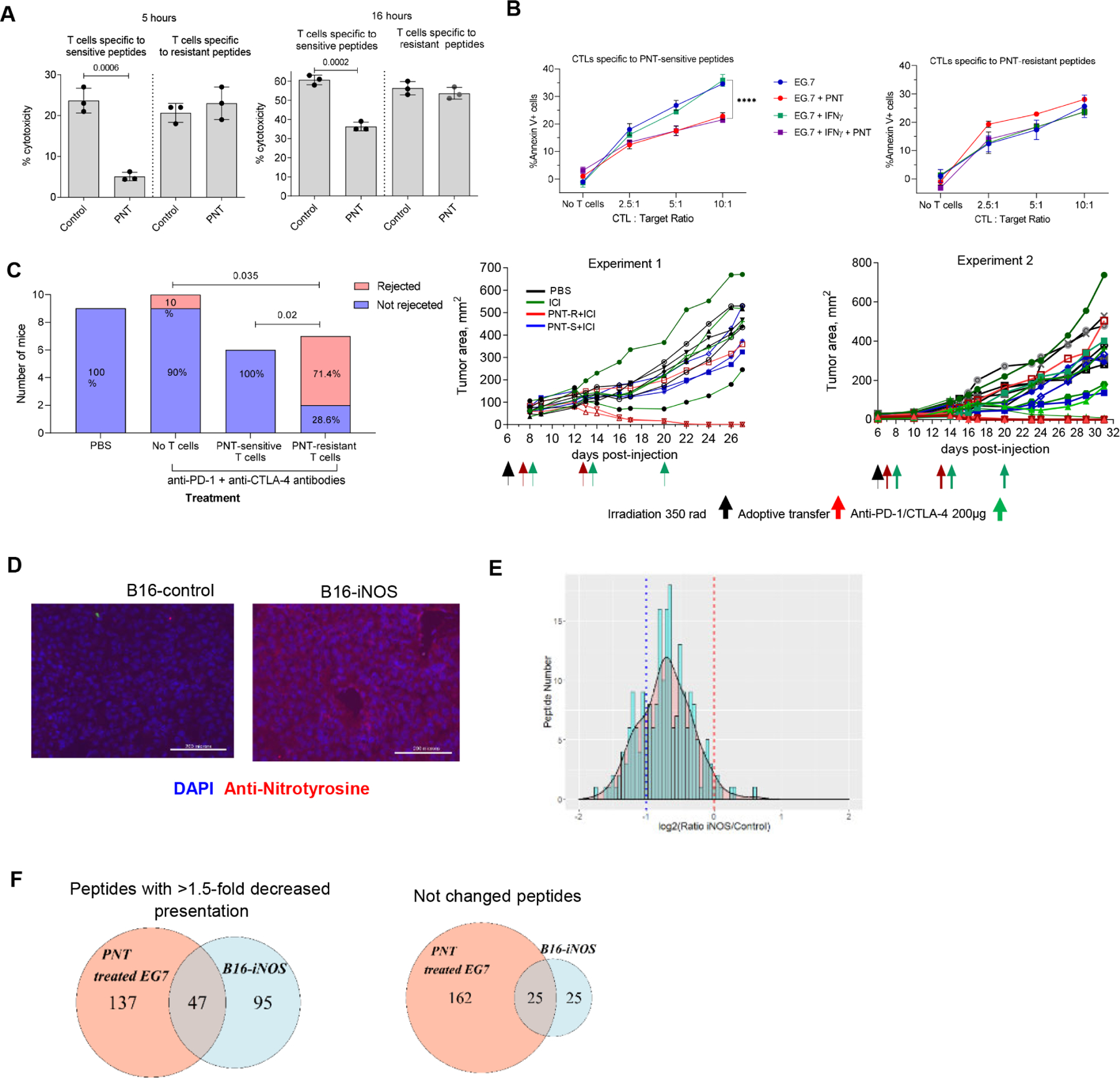
We generated CTLs specific to a pool of 3 PNT-R or 3 PNT-S peptides by sequential prime/boost vaccinations of naïve mice. T cells from spleens were then expanded in vitro by re-stimulation with specific peptides and IL-2. CD8+ T cells were isolated and used in CTL assays against EG.7 tumors. For PNT-R and PNT-S peptides, the specificity of the generated T cells was similar (Fig. S7B). To confirm these observations, we evaluated PNT-S and PNT-R peptides separately. These peptide demonstrated similar ability to induce IFN-γ producing CD8+ T cells (Fig. 4E). These CTLs also had similar capacity to kill RMA-S target cells loaded with specific peptides (Fig. 4F). Pre-treatment of tumor cells with PNT did not affect killing of tumor cells by CTLs generated against PNT-resistant peptides, whereas cytotoxicity was markedly reduced for CTLs generated against PNT-sensitive peptides (Fig. 5A). Pre-treatment of target tumor cells with IFN-γ did not change the effect of PNT on the killing of those tumor cells by CTLs (Fig. 5B). T cells specific for PNT-S and PNT-R peptides had similar proportion of naïve, effectors, and central memory T cells, and similar expression of PD-1, CTLA-4, and CD28 (Table S5).
Figure 5. PNT-resistant peptides are a primary target for effective CTL responses.
A. CTL activity of CD8+ T cells specific to a pool of 3 PNT-sensitive or 3 PNT-resistant peptides. T cells were cultured with EL4 cells at 5:1 ratio. The killing was assessed by annexin V/DAPI staining after 5 and 16 hours of culture, n=3. Mean ± SD are shown. B. EG7 tumor cells were pre-treated with 100 ng/ml IFN-γ for 24 hours and then treated with 2.5 mM PNT for 5 mins on ice. Cells were washed and the cytotoxicity assay with CTLs specific to PNT-resistant and sensitive peptides was performed by calculating the proportion of Annexin V+ cells. The baseline (no CTL present) values were subtracted (n=3). P values were calculated in one-way AVOVA with correction for multiple comparisons. ****-p<0.0001. Data shown as Mean ± SD. C. CD8+ T cells specific to the pool of 3 PNT-sensitive or resistant peptides were used for adoptive transfer to EL4-bearing mice. Treatment schedule is shown on graphs on the right. The results of two independent experiments are shown. Left panel - cumulative result showing proportion of mice rejecting tumors. Right panels – results of individuals mice each indicated by different color. P values on the graph were calculated by exact Fisher’s test. D. Immunofluorescent NT staining of B16-F10 and B16F10-iNOS tumor tissues. Scale bar = 200 μm. E. The effect of iNOS overexpression on B16 melanoma cells. The ratio of peptide representation on MHCI of B16-iNOS vs B16-Control cells was measured by SILAC LC-MS/MS. B16-iNOS and B16-Control cells were cultured with 10 ng/ml IFN-γ for 12 hours before the experiment in order to sufficiently up-regulate H-2Kb expression. Results for 2 independent experiments are shown. Red dashed line designates 1:1 PNT/Control ratio, blue dotted line shows the decrease of PNT-treated peptide counterpart more than 2-fold. F. Peptides from 2 experimental settings (PNT treatment of EG.7 cells and B16F10-iNOS overexpressing cells) were compared for the presence of similar sequences of the peptides with ratio PNT/Control or iNOS/Control <0.667 and in the group of resistant peptides with respective ratio > 0.75. The overlap is shown as Venn diagrams. See also Tables S5 and S6.
To evaluate the therapeutic potential of CTLs generated against these two types of peptides, CD8+ T cells specific for a pool of PNT-S or PNT-R peptides were transferred to EL4-bearing recipient mice followed by the treatment with immune checkpoint inhibitors (ICI), anti-PD-1 and anti-CTLA-4 antibodies. For PNT-S and PNT-R peptides, the specificity of the generated T cells was similar (about 20%) (Fig. S7C). EL4 tumor growth was not sensitive to ICI alone. Adoptive transfer of CTLs specific to PNT-S peptides plus ICI did not have a significant therapeutic effect. In striking contrast, treatment with PNT-R peptide specific CTLs and ICI resulted in tumor rejection in 70% of mice (Fig. 5C).
To determine the role of PNT in CTL recognition of tumor cells, we used B16F10 mouse melanoma model. In contrast to human melanomas, which are characterized by high PNT activity(Ekmekcioglu et al., 2005; Ekmekcioglu et al., 2000), B16F10 displayed little NT staining (Fig. 5D). To overcome this limitation, we overexpressed Nos2 (iNOS) in B16F10 cells. This resulted in marked up-regulation of NT expression reflecting PNT activity (Fig. 5D). Control vector B16F10 and B16F10-iNOS cells expressed similar levels of H-2Kb. We assessed how the overexpression of iNOS affected the immunopeptidome using SILAC. We compared H-2Kb immunopeptidome of B16F10-iNOS cells with B16F10 cells expressing control vector. We observed substantial decrease in the representation of peptides in B16F10-iNOS cells as compared to corresponding peptides in B16F10 cells (24% peptides decreased at least two-fold, 63% at least 1.5-fold, with median iNOS/control ratio 0.62) (Fig. 5E, Table S6). We compared the identity of the specific peptides in PNT treated EG.7 and B16F10-iNOS cells. There was about 70% overlap between these tumor cells in all H-2Kb bound peptides. When we compared sequences of peptides with more than 1.5-fold decreased representation in PNT treated EG.7 cells versus 1.5-fold decreased in B16F10-iNOS tumor cells, we observed 30% overlap. 50% overlap was observed in peptides that were not changed more than 1.3-fold in both experimental settings (Fig. 5F).
B16F10 tumor cells express the H-2Db gp100 derived epitope EGSRNQDWL, which is recognized by TCR transgenic Pmel CD8+ T cells. We investigated the dissociation rate of this peptide. We found that dissociation pattern of the peptide was similar to that of PNT-sensitive peptides. EGSRNQDWL dissociated significantly faster (Fig. 6A) than H-2Db LCMV gp33 peptide known for its slow dissociating rate (Cho et al., 2012). Thus, gp100 derived peptide has a dissociation pattern similar to PNT-S H-2Kb peptides.
Figure 6. PNT inhibition recovers CD8+ T cell responses to PNT-sensitive peptides and improves immunotherapy outcome.
A. Dissociation rate for gp100 and control LCMV gp33 peptide was measured in RMA-S cell assay, H-2Db expression was measured by flow cytometry. n= 4. Experiment was performed twice with the same results. Data shown as means and SEM. B. B16F10 cells expressing control vector or iNOS were labeled with 0.5 μM and 5 μM CFSE, respectively, mixed at 1:1 ratio and co-cultured for 6 hours with activated Pmel-1 cells at indicated ratios and evaluated by flow cytometry. One of three representative experiments is shown. C. B16.F10 tumor cells expressing control construct (B16-C) cells were cocultured with the activated CD8+ Pmel-1 T cells at different ratios with or without B16-iNOS separated by 0.45 μm Transwell. Viability of target cells was assessed after 6 hours. Experiments were performed in triplicates. Mean and SD are shown. P values were calculated in one-way Anova test with correction for multiple comparisons. ** - p<0.01, *** - p<0.001; **** p<0.0001. D. B16F10-control and B16F10-iNOS cells were labeled with 0.5 μM and 5 μM CFSE, respectively, mixed at 1:1 ratio and were either left untreated (top panels) or pre-treated with RTA408 (Omaveloxolone, bottom panels). Cells were cultured with activated Pmel-1 cells at indicated ratios. One of three representative experiments is shown E. Immunofluorescent NT and Gr-1 staining of LLC tumor tissue from non-treated (left) or RTA408-treated animals. Scale bar – 200 μm. F. LLC tumor-bearing mice were treated with 1 mg/kg RTA408 starting from day 10 alone or in combination with 200 μg/ml anti-PD-1 antibody twice a week. N=8, p values were calculated in two-way ANOVA test. Data shown as means and SD. G. LLC tumor-bearing mice were treated as described above. For the depletion of CD8+ T cells mice received 100 μg/mouse anti-CD8 antibody twice a week starting from day 3. N=5 in treatment groups and n=4 in control group (vehicle+isotype). Statistics was calculated with two-way ANOVA test. Data shown as mean and SD.
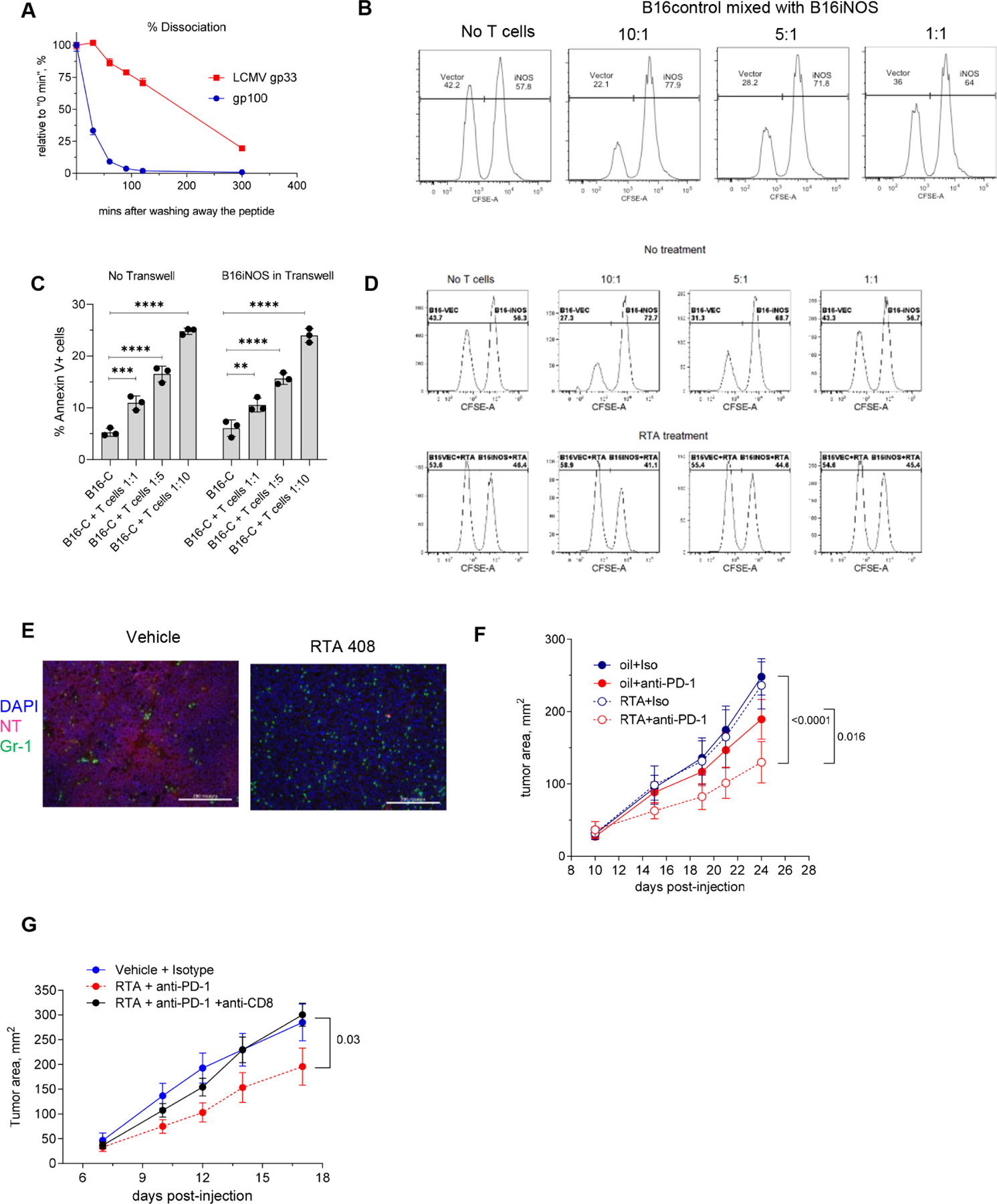
To account for possible effects of PNT or NO directly on T cells, we mixed B16F10 cells expressing a control vector with B16F10-iNOS at 1:1 ratio and used them as targets in the CTL assay. To distinguish these cells, B16F10-control cells were labeled with 0.5 μM CFSE and B16F10-iNOS cells with 5 μM CFSE. Activated transgenic Pmel-1 CD8+ CTLs were used as effector cells. Killing of B16F10-control cells was substantially higher than that of B16F10-iNOS cells (Fig. 6B). Further, we separated B16-iNOS cells with transwell membrane and found similar killing of B16 WT cells by Pmel-1 CD8+ CTLs in the presence or absence of transwell-separated B16-iNOS cells (Fig. 6C). Thereby, PNT-induced alteration of tumor recognition by CTLs was not due to targeting CTLs by diffused NO or PNT .
Effect of PNT inhibition of peptide presentation by tumor cells
Next we assessed whether inhibition of PNT would reverse this phenomenon. To inhibit PNT production, we used triterpenoid RTA408 (Omaveloxolone) (Probst et al., 2015). B16F10 cells expressing control vector or B16F10-iNOS were mixed at 1:1 ratio and incubated with activated effector cells in the absence or presence of RTA408. Treatment with RTA408 completely abrogated resistance of B16F10-iNOS cells to killing by CTLs (Fig. 6D). Using the LLC tumor model with high PNT activity in tumors derived from myeloid cells(Nagaraj et al., 2010), we tested the effect of combining RTA408 with anti-PD-1 antibody. Treatment of mice with RTA408 reduced NT staining in tumor tissues (Fig. 6E). In contrast to single treatment arms where no effect on tumor progression was observed, a combination of anti-PD-1 antibody and RTA408 caused substantial delay in tumor progression (Fig. 6F). This effect was abrogated when CD8+ T cells were depleted (Fig.6G).
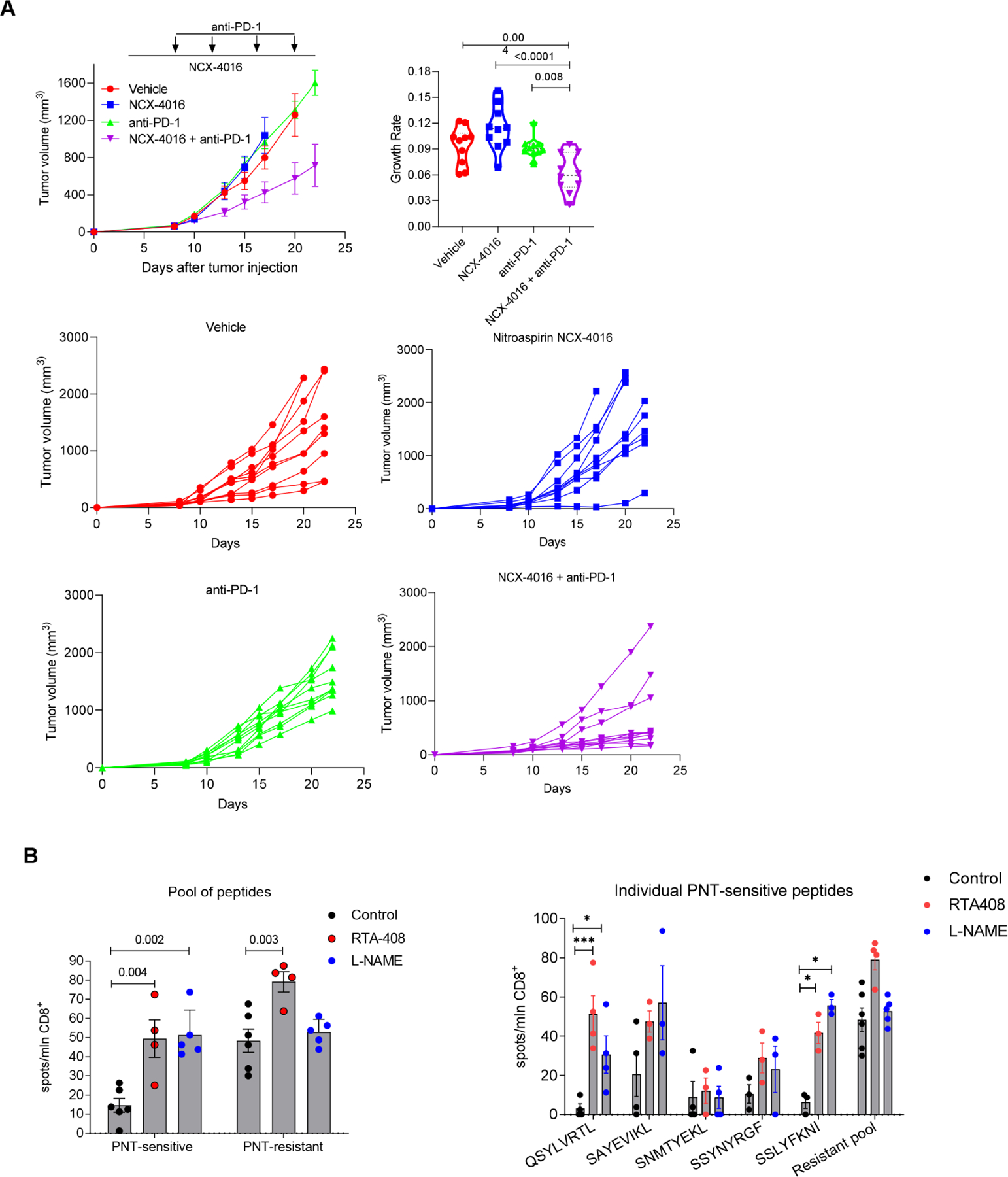
To expand these observations to a different model and different PNT inhibitor, we used the colon carinomoa CT26 tumor model on Balb/c background and nitroaspirin (NCX-4016) previously implicated in inhibition of PNT (De Santo et al., 2005). Combination of nitroaspirin with PD-1 demonstrated marked antitumor activity (Fig. 7A).
Figure 7. Antitumor effect of combination of nitroaspirin and PD-1 antibody.
A. CT26 TB mice were treated with Nitroaspirin (NCX-4016) 12.5mg/kg PO every day from day 4 and/or PD-1 antibody 200μg/mouse twice a week from day 7. N=10. Top left panel – cumulative results of tumor size in the groups, data shown as mean and SD. Top right panel – tumor growth rate calculation, violin plots show median and quartiles as well as all data points. Bottom four panels results of individual mice in indicated treatment groups. P values were calculated in one-way ANOVA with correction for multiple comparisons. B. EL4 tumor-bearing mice were treated with 1 mg/kg RTA 408 daily or with 2 g/L L-NAME in drinking water ad libitum starting from day 4 after tumor injection. On day 17 mice were sacrificed, splenic CD8+ T cells were isolated and stimulated with 2 μg/ml individual peptides or the pool of all 5 specified peptides for each group (1 μg/ml of each peptide). The IFN-γ producing CD8+ T cells were measured in ELISPOT assay and calculated per 106 CD8+ T cells. Mean and SEM are shown. n=6 in control, 4 in RTA408 treated mice, and 5 in L-NAME treated mice. P values were calculated in one-way ANOVA test with correction for multiple comtarisons and are shown for pool of peptides. For individual peptides * - p<0.05, ** -p<0.01, ***-p<0.001.
To investigate how PNT inhibition affects T cell responses to PNT-S peptides we treated EL4-bearing mice with RTA408 or with NOS inhibitor Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME). After 13 days of daily treatment we isolated CD8+ splenic T cells and re-stimulated them with PNT-S or PNT-R peptides. T cell responses were evaluated in IFN-γ ELISPOT assays. As expected, the background responses to PNT-S peptides was substantially lower those to PNT-resistant peptides. For both RTA408 and L-NAME treatments, we found almost 5-fold increase in CD8+ T cell responses to the pool of PNT-S peptides (p=0.004 and p=0.002, respectively) (Fig. 7B). The effect on the response to PNT-R peptides was much less pronounced (Fig. 7B). Thus, the recognition of tumor cells and expansion of tumor-specific CTLs highly depend on the sensitivity of a given MHCI epitope to PNT.
Association of PNT amount in tumors with clinical effect of cancer immunotherapy
To assess a possible link of high PNT levels with clinical outcome to immunotherapy, we analyzed two cohorts of patients. The first cohort included metastatic melanoma patients vaccinated with Melan-A peptide at University of Lausanne Hospital (Switzerland) (Table S7). Archived tumor tissues were collected before the start of the treatment. Samples were pre-selected by clinical investigators to form two groups of patients with favorable and unfavorable response to therapy and then were stained for NT blindly by laboratory investigators without knowledge of clinical outcome. After breaking the code, patients were split into two groups: one with positive NT staining and the other one with negative NT staining (the criteria are described in STAR Methods). After the treatment, CD8+ T cell response to Melan-A peptide was the same in both groups (Fig. 8A). The proportion of Melan-A peptide specific effector memory T cells (CD45RA-CCR7-) as well as CD28+ Melan-A specific T cells were the same in patients with NT positive and negative tumors. No differences were found in other population of peptide-specific T cells (Fig. 8B). In contrast, patients with NT negative tumors had significantly better overall survival and progression free survival than patients with NT positive tumors (Fig. 8C). We assessed dissociation of Melan A peptide from HLA-A2 molecules using T2 cells. For a control we used the peptide of CEF1 Influenza matrix protein M1 (58–66), known for its high binding capacity and formation of relatively stable complex with HLA-A2. Melan A peptide demonstrated rapid dissociation, which was similar to PNT-sensitive mouse H-2Kb peptides (Fig. 8D).
Figure 8. Nitrotyrosine in tumor tissues is associated with worse response to immunotherapy.
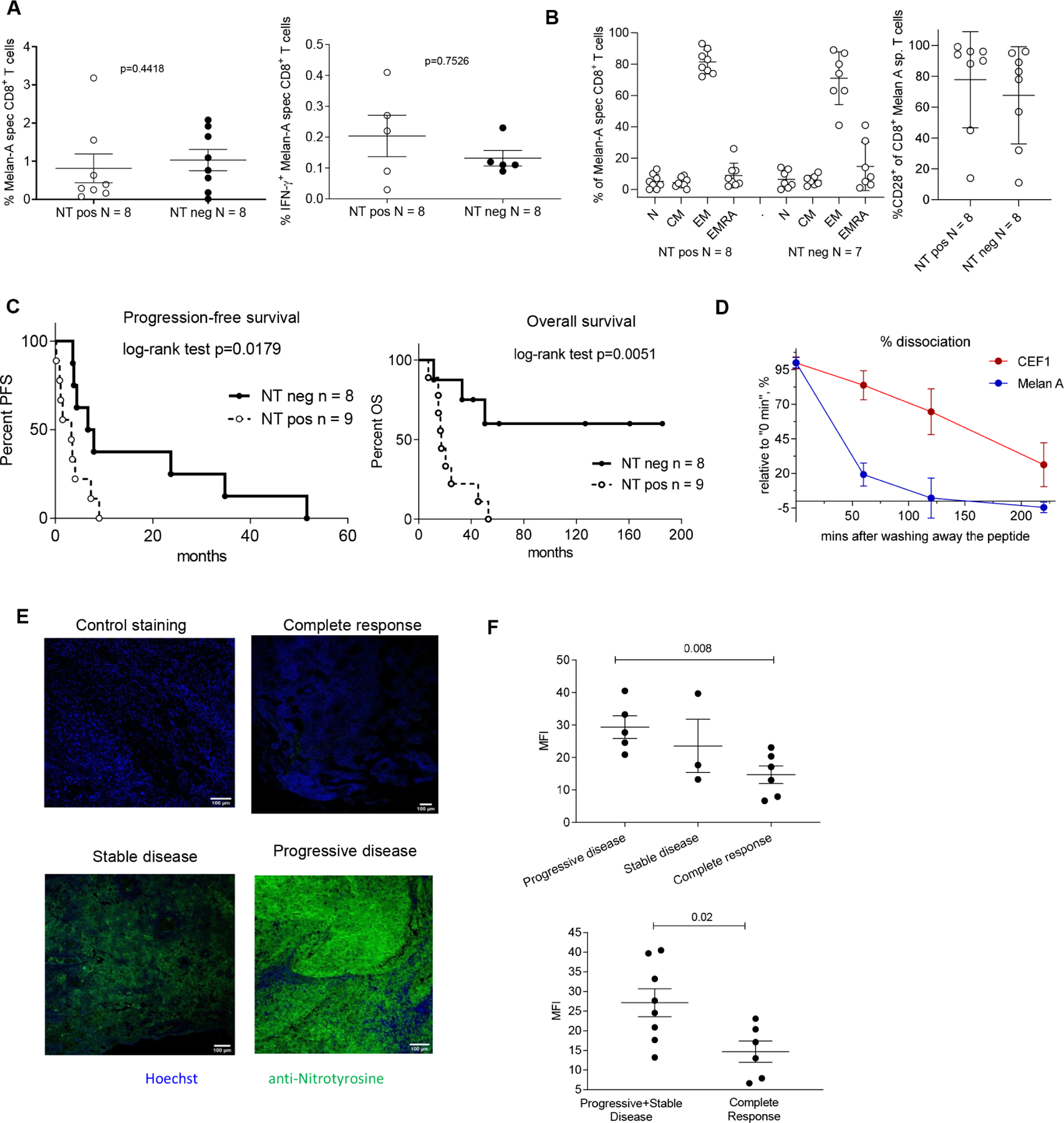
A-C. Patients with metastatic melanoma were vaccinated with Melan-A peptide and CpG 7909. NT staining was performed in tumor tissue obtained prior the start of the treatment. A. The frequency of Melan-A specific CD8+ T cells (left panel) and IFN-γ producing Melan-A specific CD8+ T cells (right panel) after the treatment. B. MelanA specific T cell after the treatment: Frequency of subpopulations of Melan-A specific T cells according to differentiation marker expression. Left panel. N (naïve) CD45RA+CCR7+, CM (central memory) CD45RA-CCR7+, EM (effector memory) CD45RA-CCR7-, EMRA (terminally differentiated effector memory) CD45RA+CCR7-. Right panel: CD28+T cells. In A and B, data shown as mean ± SD. C. Kaplan-Meier curves of progression-free survival and overall survival of the melanoma patients. Statistical analysis was performed with the Log-rank (Mantel-Cox) test. D. Dissociation rate for Melan A and control CEF1 influenza matrix protein-M158–66, GILGFVFTL was measured with T2 cells by flow cytometry, n= 4. Two experiments with the same results were performed. Data shown as mean ± SD. E, F. Metastatic melanoma patients were treated with anti-PD-1 antibody (pembrolizumab). Tumors tissues were collected prior to the start of the treatment and analyzed for NT presence by immunofluorescent microscopy. E. Representative images of the tumor tissue from the patients with different clinical outcome. Scale bar – 100 μm. F. Cumulative data of mean fluorescence intensity (MFI) for NT staining of tumor tissues from patients with progressive & stable disease, or complete response. P values were calculated in two-tailed Student’s t-test. Data shown as mean ± SD. See also Tables S7 and S8.
In the other cohort we evaluated patients with metastatic melanoma treated with anti-PD-1 antibody (pembrolizumab) at the University of Pennsylvania (Table S8). Tumor tissues collected prior to the start of therapy were pre-selected by clinical investigators to include patients with complete response, and progressive or stable disease. Samples were evaluated blindly for NT expression by laboratory investigators without knowledge of clinical outcome. We observed significantly higher NT expression in tumors from patients with progressive or stable disease than in those from patients with complete responses (Fig. 8E,F). Thus, high PNT activity in melanoma was associated with poor clinical outcomes in two different clinical trials, and specifically, a lack of responsiveness to anti-PD-1 therapy.
Discussion
Our study demonstrated that PNT can be an important factor limiting the efficacy of T cell-based cancer immunotherapy, especially in tumors, like melanoma, with substantial PNT activity. Although it is known that PNT can affect T cell function, primarily during close interaction with myeloid cells (Nagaraj et al., 2007), our results, consistent with our previous observations(Lu et al., 2011), indicate that even potent CTLs may fail to recognize tumor cells if they are raised against PNT-sensitive peptide epitopes. Consistent with these findings were the results of generation of spontaneous CD8+ T cells in TB-mice. It is established that a small number of tumor-specific T cells can be generated in TB hosts. These cells can be amplified further with cancer vaccines or ICI. Our data demonstrated that spontaneous responses are largely generated against PNT-resistant peptides rather than PNT-sensitive ones. This can be explained by the different representation of these peptides on tumor cells. Over-expression of iNOS in tumor cells resulted in decreased presentation of MHCI bound peptides on tumor cells surface. Many of those peptides were the same as the ones lost after PNT treatment, which supported the concept that high PNT levels affect the tumor cell surface peptide repertoire.
We found that PNT-R and PNT-S peptides had similar binding affinity to MHCI. However, PNT-S peptides had much higher dissociation rates. This effect was not changed when we pre-treated cells with PNT before formation of pMHCI complexes. Expression of MHCI molecules also was not affected. This suggested that the direct effect on MHCI or peptides may not be the mechanism of PNT effects. Proteasomes are critical components for generating pMHCI (Milner et al., 2013; Sijts and Kloetzel, 2011). As it was shown previously, proteasome inhibitors reduced the rates of synthesis pMHCI, but different peptides were affected differently by the treatment(Milner et al., 2013). In addition, PNT inducing agent, SIN-1 has been shown to affect the proteasomal activity(Osna et al., 2004). We found that PNT inhibited proteasomes. Moreover, co-culture of tumor cells with myeloid cells recapitulated that phenomenon. Bortezomib reduced presentation of many of the same peptides, which were reduced by treatment with PNT. Substantial number of the same peptides were down-regulated by both, PNT and bortezomib treatment of tumor cells. However, there were also numbers of peptides, which were down-regulated by each treatment alone. We believe that the reason for this could be in the degree and specificity of proteasomal inhibition. PNT and bortezomib concentrations were selected based on lack of the effect on viability and MHC class I expression. As a result, at the selected concentration bortezomib only partially blocked proteasomal activity. In addition, bortezomib is known to be specific to only some of the types of proteasomal activity (Teicher and Tomaszewski, 2015). This might result in somewhat different profile of affected peptides between PNT and bortezomib treated cells. Peptides constantly generated in tumor cells have different stability in pMHCI complexes. Inhibition of proteasomes by PNT reduced formation of new peptides. More stable peptides remained on the surface in the complex with MHC class I molecules much longer than the peptides with high dissociation rates. As a result, the PNT-stable peptides should be much better targets for CTLs.
These findings have direct clinical relevance. Inhibition of PNT production by down-regulation of ROS or NOS, markedly improved CTL response against PNT-resistant peptides. More importantly, it resulted in dramatic improvement of anti-tumor effect of T cell therapy in combination with ICI. This was further supported by correlative clinical studies in two cohorts of patients treated with immunotherapy. Our results suggest that the selection of MHCI-binding peptides for neoantigen or shared antigen vaccines based on their resistance to PNT may improve clinical responses. Targeting PNT production may be a valuable approach to limit tumor cell resistance to CTLs and enhance the therapeutic efficacy of immunotherapies such as ICI and adoptive T cell therapy.
STAR Methods
Resource Availability
Lead contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Dmitry Gabrilovich, ICC, Oncology R&D, AstraZeneca, Gaithersburg, MD, 20878, USA, email: Dmitry.gabrilovich@astrazeneca.com
Material availability:
Newly generated cell line is available upon request
Data and code availability:
The mass spectrometry proteomics data have been deposited into the MassIVE (http://massive.ucsd.edu) and ProteomeXchange (http://www.proteomexchange.org) data repositories with the accession number MSV000087990 and PXD027956, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and clinical protocols.
Clinical study at University of Lausanne.
HLA-A2-positive patients with histologically proven metastatic (stage III/IV) melanoma of the skin were included in phase I prospective trials (ClinicalTrials.gov; Identifiers: NCT00112229 and NCT00112242)(Speiser et al., 2008; Speiser et al., 2005). Study protocols (LUD00–018 and LUD01–003) were approved and conducted according to the relevant regulatory standards from (i) the ethical commission of the University of Lausanne (Lausanne, Switzerland), (ii) the Protocol Review Committee of the Ludwig Institute for Cancer Research (New York) and (iii) Swissmedic (the Swiss agency for therapeutic products, Bern, Switzerland). Patient recruitment, study procedures, and blood withdrawal were carried out upon obtaining written informed consent. Eligible patients received monthly vaccinations injected subcutaneously. Vaccine emulsions were prepared with Melan-A/MART-126–35 peptide, IFA (Montanide ISA-51; Seppic), and CpG-B 7909/PF-3512676 (Pfizer and Coley Pharmaceutical Group) as described(Speiser et al., 2005). NT staining of paraffin embedded tissue slides were performed as described previously. Allred scoring system was used to evaluate the percentage of positive stained cells and staining intensity. Average intensity of staining in positive cells was assigned an intensity score. Samples with score 0 (no staining and score 1 (weak staining) were considered NT negative, and scores 2 and 3 (intermediate and strong staining) NT positive. Evaluation of NT staining was performed blindly by investigator on coded samples without access to clinical information. Results of staining were sent to clinical investigators who broke the codes and evaluated the association with clinical outcome.
Clinical study at University of Pennsylvania.
Paraffin-embedded tumor samples were collected from stage III/IV melanoma patients prior to anti-PD-1 (pembrolizumab) therapy, under the University of Pennsylvania Abramson Cancer Center’s melanoma research program tissue collection protocol UPCC 08607 and IRB 703001 in accordance with the Institutional Review Board. Samples collection was performed after obtaining written consent prior to study initiation. Tumor tissues were processed for immunofluorescent (IF) staining with the NT antibody (Millipore-Sigma) and secondary antibody (Invitrogen). Cell nuclei were counterstained with Hoechst 33342 dye (Thermo Scientific). Digital images were captured using Leica TCS SP8 laser scanning confocal system with LasX Life Science software. Mean fluorescent intensity of randomly selected 15 – 30 fields in every sample was measured using Fiji software. Evaluation of NT staining was performed blindly by investigator on coded samples without access to clinical information. Results were assessed together with clinical investigators after breaking the codes.
Mice.
Mouse experiments conform to the relevant regulatory standards and were approved by the Institutional Animal Care and Use Committee (IACUC) of The Wistar Institute and AstraZeneca. C57BL/6 mice (female, 6–10 weeks old) and BALB/c mice (female, 6–10 weeks old) were obtained from Charles River and Envigo. Mice were housed 5 per cage and fed with standard diet Littermates were randomly assigned to experimental groups. PMEL transgenic mice (female 6–8 weeks old) (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) were obtained from Jackson Lab.
Cell lines.
EG7, B16F10, T2, IL-4, LLC, and CT26 cell lines were obtained from ATCC and authenticated there. UACC-903 human melanoma cell line was obtained from University of Arizona Cell Collection and provided by Dr. Herlyn, Wistar Institute. RMA-S cells were provided by Dr. Eisenlohr (University of Pennsylvania). The specific characteristics of this cell line were confirmed in functional studies (peptide binding). Cell lines were cultured in RPMI-1640 or DMEM media supplemented with 10% FBS and antibiotics. Cell lines were regularly tested on the absence of mycoplasma contamination.
METHOD DETAILS
Study design.
This is exploratory study assessing possible effect of PNT on immune response in cancer. In mouse studies the readouts were tumor size. Experiments were stopped if tumor size exceeded the one permitted by IACUC. No values were excluded. In mouse study simple randomization was performed. Experiments were performed multiple times. The details for each experiments are provided in figure legends. Clinical studies were retrospective analysis of samples collected from patients. Samples were pre-selected by participating physicians to form statistically meaningful groups of responders and non-responders to the therapies. Sample size was calculated based on expected parameters of nitrotyrosine expression and power of 80%. Each sample was coded and evaluated blindly in laboratory. Code was open after all information was received by participating physicians.
Analysis of tumor-specific CD8+ T cell responses in patients.
Peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque (Pharmacia), cryopreserved in 10% DMSO (Sigma-Aldrich) and stored in liquid nitrogen until further use. Flow cytometry analysis was done with unstimulated PBMCs that were positively enriched using anti-CD8-coated magnetic microbeads (Miltenyi Biotec). CD8-positive T cell fractions were stained in PBS, 0.2% BSA, and 50 mM EDTA with PE-labeled HLA-A*0201 multimers (“tetramers”) loaded with analog Melan-A 26–25 (A27L) (Peptide and Tetramer Core Facility, Ludwig Cancer Research, UNIL CHUV, Lausanne, Switzerland) at 4°C for 45 minutes, followed with CD8-specific and further antibodies at 4°C for 30 minutes. Samples were acquired on a LSRII cytometer (BD Biosciences) and analyzed using FlowJo 9.7.6 software (TreeStar, Inc). The IFN-γ Elispot was performed using total and unstimulated PBMC, using the ELISpotPRO kit for Human IFN-γ from MABTECH (3420–2APT-10), following the standard supplier instructions. CD8 T cells were thawed, and their numbers per well were adjusted to have maximum 3% of cytokine producing cells per well to avoid saturation of the membrane (values taken from the multimer analysis). The frequency of the resulting colored spots corresponding to the cytokine producing cells was counted using the Elispot Bioreader 5000.
MHCI peptide isolation.
EL4-OVA (EG.7) murine thymoma, B16-iNOS murine melanoma or UACC903 human melanoma cells were labeled with “heavy” lysine (13C6-Lys) and arginine (13C6,15N4-Arg) with the use of SILAC kit (cells were cultured for at least 8 cell doublings). Control cells were cultured in the same medium containing non-labeled (“light”) lysine and arginine. In the experiments with PNT treatment the cells were treated for 5 min with 2.5 mM PNT (labeled cells) or vehicle solution (non-labeled cells) on ice, washed three times and cultured for the designated time (0 min – no culture, 30 min, 5h or 16h). In the similar experiments with bortezomib treatment the cells were treated for the specified time with 15 nM bortezomib (labeled cells) or equivalent amount of DMSO solution (non-labeled cells). Then the treated and control cells were collected, washed with PBS twice and mixed in 1:1 ratio (108 cells total), pelleted and lysed in 0.25% sodium deoxycholate, 0.2 mM iodoacetamide, 1 mM EDTA, 1∶200 Protease Inhibitors Cocktail (Sigma), 1 mM PMSF, 1% octyl-β-D glucopyranoside (Sigma) in 50 mM Tris-HCl, pH 8.0 on ice for 1 h with the following sonication. The lysate was cleared by 30 min centrifugation at 20 000 × g. MHCI H-2-Kb molecules for mouse EG.7 and B16 cells or HLA-A, -B, -C molecules for human UACC903 melanoma cells were immunoprecipitated from the lysates with the specific antibody bound to Protein G magnetic beads (Dynabeads, Life Technology, Y3 clone antibody specific to murine H-2Kb molecules and TP25.99.8.4 clone antibody specific to human HLA-A,B,C molecules were used). The beads were washed twice with 150 mM NaCl/Tris-HCl, once in 400 mM NaCl/Tris-HCl, and once in 50 mM Tris-HCl, pH 7.5, then the peptides were eluted by 2% acetic acid, desalted by C18 tips (Pierce) and submitted for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis.
LC-MS/MS Analysis and Data Processing.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis was performed using a Q Exactive HF mass spectrometer (ThermoFisher Scientific) coupled with a Nano-ACQUITY UPLC system (Waters). Samples were injected onto a UPLC Symmetry trap column (180 μm i.d. x 2 cm packed with 5 μm C18 resin; Waters), and peptides were separated by reversed phase HPLC on a BEH C18 nanocapillary analytical column (75 μm i.d. x 25 cm, 1.7 μm particle size; Waters) using a 90 min gradient formed by solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). Eluted peptides were analyzed by the mass spectrometer set to repetitively scan m/z from 300 to 2000 in positive ion mode. The full MS scan was collected at 60,000 resolution followed by data-dependent MS/MS scans at 15,000 resolution on the 20 most abundant ions exceeding a minimum threshold of 10,000. Peptide match was set as preferred, exclude isotope option was enabled and no charge exclusion was performed.
Peptide sequences were identified using MaxQuant 1.5.2.8 (Cox and Mann, 2008). Heavy 13C6-lysine and 13C615N4-arginine were also considered in the search for SILAC samples. Consensus identification lists were generated with false discovery rates set at 1% for peptide identifications. The ratio of labeled vs non-labeled peptide counterparts (PNT-treated vs non-treated ratio – PNT/ Control Ratio) for cell-derived peptide species was estimated.
Peptide analysis.
To assess the effect of PNT treatment on MHCI peptide presentation the obtained PNT versus Control ratios for all the detected peptide species were plotted as histograms of log2(PNT/Control Ratio) with overlaid scaled density plot using R Studio and ggplot2 package. Based on PNT/Control Ratio from 30 min time-point peptides were assigned to “PNT-S” (PNT/ Control Ratio less than 0.5) or “PNT-R” (PNT/ Control Ratio more than 0.8) type. “PNT-S” and “PNT-R” peptides were grouped according to their size and compared for amino acid composition in their sequence. For each position the amino acid content was compared between “sensitive” and “resistant” peptides and the significance of difference was tested with Fisher’s exact test. The P values were adjusted for multiple comparisons using FDR(Benjamini and Yekutieli, 2001). Logos for amino acid conservation were created by https://weblogo.berkeley.edu/logo.cgi website. The search of unique motifs that distinguishes between “PNT-sensitive” and “resistant” peptides was conducted by MEME (Multiple Em for Motif Elicitation) tool used in Differential Enrichment mode (http://meme-suite.org/tools/meme(Bailey and Elkan, 1994)). Motifs with E-value < 0.05 were considered significant in accordance with MEME algorithm guidelines. The comparison of our peptide datasets with each other as well as with the datasets published previously(Schuster et al., 2018; Yadav et al., 2014) was conducted using R Studio soft with the use of VennDiagram package.
RMA-S binding and dissociation assays.
Peptide MHCI binding capacity was assessed with RMA-S cell assay. RMA-S cells were loaded with 10 μg/ml of the synthesized peptide, cultured for the specified time, washed and total H2-Kb expression was measured by flow cytometer using FITC-conjugated H2-Kb antibody (clone AF6–88.5). To characterize the stability of MHC I complex with the given peptide (pMHC stability) RMA-S cells were loaded with 10 μg/ml peptide for 2 hours, washed, cultured for designated time-points and the H2-Kb expression was measured by flow cytometry. MFI relative to initial MFI (“0” time-point) was calculating as following: (observed MFI - background MFI)*100%/(MFI at 0 min - background MFI).
Analogous experiments were conducted for H-2Db measurement with the use of APC-conjugated anti-H-2Db antibody (clone KH95).
T2 binding and dissociation assays.
Peptide HLA-A2 binding capacity was assessed with T2 (174x CEM.T2) (ATCC® CRL-1992™) cells. T2 cells were loaded with 10 μg/ml of the tested peptide for 2h, washed, cultured for the specified time and HLA-A2 expression was measured by flow cytometry using PerCP/Cy5.5-conjugated anti-HLA-A2 antibody (clone BB7.2). MFI relative to initial MFI (“0” time-point) was calculated as following: (observed MFI - background MFI)*100%/(MFI at 0 min - background MFI).
Proteasomal activity.
The total activity of tumor cell proteasome was measured by proteasome activity kit (Millipore-Sigma). Briefly, for different experiments cells were either treated with PNT or co-cultured with isolated Gr-1+ myeloid cells, myeloid cells were depleted by magnetic beads (Miltenyi), tumor cells were washed with PBS and lysed in RIPA buffer, normalized based on total protein concentration and proteasomal activity was measured in the accordance to manufacturer recommendations.
To measure specific activities of constitutive proteasome and immunoproteasome the respective kits from UBPBio were used. Briefly, lysates were prepared as described above and used for the reaction with specific fluorogenic substrates followed by fluorescence measurements. Suc-LLVY-AMC, Boc-LRR-AMC and Z-LLE-AMC were used to measure chymotrypsin-like, trypsin-like and caspase-like activities of constitutive proteasomes, respectively. Ac-ANW-AMC, Ac-KQL-AMC, Ac-PAL-AMC were used to measure chymotrypsin-like, trypsin-like and branched amino acid preferring activities of immunoproteasomes, respectively, according to UBPBio manufacturer.
Nitroaspirin and anti-PD-1 treatment in vivo.
BALB/C mice were purchased by Envigo and implanted respectively with 5×105 CT26 tumor cells subcutaneously in 200ul of PBS. Nitroaspirin (NCX-4016 Sigma Aldrich cat # SML1669–25MG) resuspended in DMSO at 25 mg/ml and further resuspended in Methylcellulose for oral gavage administration. Mice were dosed with 12.5mg/kg every day starting at day 4 post tumor implantation. Ani-PD-1 (BioXcell clone RMP1–14) was administered IP diluted in PBS starting at day 7 twice a week.
Flow cytometry.
Flow cytometry was conducted with the use of BD LSR II flow cytometer (BD). Intracellular staining was performed with the use of BD Cytofix/Cytoperm kit according to manufacturer recommendations.
Western Blot.
Tissues were homogenized and lysed in RIPA buffer (Thermo Fisher) + protease inhibitors (Thermo Fisher cat A32963) using Lysing Matrix A tubes and FastPrep Instrument. Tissue and cell lysates were fractionated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane using a transfer apparatus according to the manufacturer’s protocols (Bio-Rad). After blocking with 5% nonfat milk in PBST (PBS, pH 7.5, 0.5% Tween 20) for 60 min, the membrane was washed once with PBST and incubated 2 h with antibodies against mouse or human MHC I (rabbit polyclonal antibody for mouse cells, Abcam, or W6/32 clone for human cells, BioLegend) with the following 1 h incubation with the corresponding secondary antibody and ECL development (Amersham Biosciences). For peptide source proteins the antibodies listed below were used. After overnight incubation, membranes were washed in TBST-tween 20 and then corresponding horseradish peroxidase conjugated secondary antibodies were added to membrane at a concentration of 1:2000 and incubated for 1 hour at room temperature. Immunoblots were visualized using Odyssey Imagers system (LI-COR).
| Company | Antibody | Clone | Catalog number | Dilution used in the study |
|---|---|---|---|---|
| ThermoFisher | DDX6 | Polyclonal/Rabbit | PA555012 | 1/500 |
| ThermoFisher | PRICKLE1 | Polyclonal/Rabbit | 22589-1-AP | 1/1000 |
| ThermoFisher | ELF3 | Polyclonal/Rabbit | PA589261 | 1/1000 |
| ThermoFisher | LDHB | Polyclonal/Rabbit | PA596736 | 1/1000 |
| ThermoFisher | IFT25 | Polyclonal/Rabbit | 15732-1-AP | 1/1000 |
| ThermoFisher | PICK1 | Polyclonal/Rabbit | PA1073 | 1/500 |
| ThermoFisher | STT3A | Polyclonal/Rabbit | 12034–1-AP | 1/1000 |
| ThermoFisher | RPS15A | Polyclonal/Rabbit | PA5103532 | 1/1000 |
| ThermoFisher | beta Actin (BA3R) |
BA3R | MA515739 | 1/1000 |
Quantitative real-time PCR.
Total RNA was extracted with total RNA microkit (Zymo) according to manufacturer’s protocol. Complementary DNA (cDNA) was synthesized with the use of High-Capacity cDNA Reverse Transcription Kit (AppliedBiosystems). qRT-PCR was performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems). The sequences of oligonucleotides used in the study are provided in Table S9
ELISPOT assays.
To estimate in vivo presence of T cells specific to representative peptides from our dataset general ELISPOT assay was performed. Briefly, mice bearing EL4, B16, LLC or MC38 tumors (1.5 cm in diameter) were sacrificed and their spleens were harvested. CD8+ T cells were isolated from the spleen using magnetic beads (Miltenyi), mixed with irradiated total splenocytes from the naïve mouse (as the source of antigen-presenting cells) and re-stimulated with 2 μg/ml individual synthetic peptide or 1 μg/ml pool of “PNT-sensitive” or “resistant” peptides (all the peptides used in this study were synthesized by GenScript). Cells were cultured for 48 h in the PVDF plates (Millipore-Sigma) pre-coated with IFN-γ capture antibody (Mabtech), then the plates were washed and developed according to manufacturer procedure (Mabtech). Plates were counted with the use of ImmunoSpot reader (CTL, Cleveland, OH). Values obtained for the cells cultured without peptide restimulation were used as a background and were subtracted from the corresponding values for peptide restimulation.
For isolation of tumor CD8+ T cells, tumors were dissociated with Miltenyi tumor dissociation kit with the following isolation of CD8+ T cells using magnetic beads (Miltenyi). The purity of isolated splenic and tumor CD8+ T cells was >95%.
Generation of specific T cells.
Naïve mice were immunized with the pool of 3 PNT-sensitive (QSYLVRTL, SAYEVIKL, SSLYFKNI) or resistant (LNYKFPGL, TIYRFLKL, QSIAFISRL) peptides (50 μg per injection) in combination with CpG (20 μg/injection s.c.) or poly-ICLC (50 μg/injection i.v.) adjuvant and on day 12 after immunization mice were boosted with the same peptide pool, adjuvant and 50 μg anti-CD40 antibody. On day 23–28 after immunization splenic cells were collected and re-stimulated with 2 μg/ml of specific peptide pool in the presence of 5 ng/ml murine IL-2. On day 4 medium was replaced and on day 6 CD8+ T cells were isolated using magnetic beads (Miltenyi) and used for in vitro killing assays or in vivo adoptive transfer experiments.
CTL assay and T cell adoptive transfer experiments.
CD8+ T cells specific to a pool of PNT-sensitive or PNT-resistant peptides were generated as described above. For both groups the specificity of the generated T cells was about 20% measured by intracellular staining for IFN-γ production. EL4 tumor cells were treated with PNT or vehicle and co-cultured with different ratios of CD8+ T cells. The killing of target tumor cells was assessed by annexin V/DAPI staining by flow cytometry.
In another setting, B16F10-control vector or B16F10-iNOS cells were stained with low (0.5 μg/ml) and high (5 μg/ml) concentrations of CFSE (BioLegend), mixed together in 1:1 ratio and co-cultured for 6 h with Pmel splenocytes at different ratios. The results were assessed by flow cytometry.
CD8+ T cells specific to the PNT-sensitive or PNT-resistant peptides were used for adoptive transfer into EL4-bearing recipient mice. Recipients were injected with 3×105 EL4 cells s.c. on day 0, sub-lethally irradiated with 350 rad on day 6 and on day 7 were injected with 5×106 CD8+ T cells. On day 7 mice were treated with 200 μg anti-PD1 (clone RMP1–14, BioXcell) and 200 μg anti-CTLA4 (clone 9D9, BioXcell) antibodies. T cell transfer and antibodies treatment was repeated on day 13 and day 14 respectively. Mice received one more treatment with anti-PD1/anti-CTLA4 antibodies on day 20. The kinetics of tumor growth was measured.
RTA408 and L-NAME treatment.
Mice were injected s.c. with the specified tumor cell line. In experiments with RTA408 treatment, mice were treated with 1 mg/kg RTA408 (Reata Pharmaceutical) by oral gavage daily alone or in combination with 200 μg anti-PD1 (clone RMP1–14, BioXcell), twice a week. For L-NAME treatment mice received 2g/L L-NAME solution in drinking water ad libitum, pH was adjusted to pH 7.0.
Immunofluorescence (IF) staining and analysis of tumor tissues.
Human tissues were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Mouse tumor tissues were frozen in OCT medium. 5-μm sections of tumor tissues were processed for immunofluorescent (IF) staining with the NT antibody (Sigma) and the following secondary antibody (Invitrogen). Cell nuclei were counterstained with Hoechst 33342 dye (Thermo Scientific). Digital images were captured using Leica TCS SP8 laser scanning confocal system with LasX Life Science software. The comparison of NT in different patient samples was conducted by the measurement of mean fluorescent intensity of randomly selected 15 – 30 fields in every sample by Fiji software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using unpaired two-tailed Student’s t-test with significance determined at 0.05. Estimation of variation within each group of data was performed and variance was similar between groups that were compared. In experiments with more than 2 groups assessed simultaneously one-way ANOVA test was used with Tukey’s post-test evaluation of multiple comparisons. Assessment of clinical samples was blinded. Animal experiments were not blinded. Tumor growth was evaluated using two-way ANOVA test with Bonferroni correction for multiple comparisons. The rate of tumor growth for each animal is estimated based on fitting each tumor’s growth curve to an exponential model (Hather et al., 2014): log10(tumor volume)=a+b⋅time+errorlog10(tumor volume)=a+b⋅time+error where a and b are parameters that correspond to the log initial volume and growth rate, respectively. The model assumes the error terms are normally distributed. The analysis compares each treatment group to the reference treatment.
Supplementary Material
Table S1.List of peptides evaluated in EG.7 cells. Related to Figure 1.
Table S2. List of peptides evaluated in UACC903 cells. Related to Figure 1.
Table S4. List of peptides evaluated in EG.7 cells after bortezomib treatment. Related to Figure 3.
Table S6. List of peptides evaluated in B16-iNOS cells versus B16-Control cells. Related to Figure 5
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Protein G magnetic beads conjugated with antibodies | Dynabeads, Life Technology | Y13 clone antibody specific to murine H-2Kb TP25.99.8.4 clone antibody specific to human HLA-A,B,C |
| FITC-conjugated H2-Kb antibody | Biologend | clone AF6–88.5 |
| APC-conjugated anti-H-2Db antibody | Biolegend | clone KH95 |
| PerCP/Cy5.5-conjugated anti-HLA-A2 antibody | BD Biosciences | clone BB7.2 |
| immunoproteasome detection kit | UPBIO | J4160 |
| classical proteasome detection kit | UPBIO | J4120 |
| Anti-nitrotyrosine antibody | Sigma | N0409 |
| Ani-PD-1 | BioXcell | clone RMP1–14 |
| Anti-CTLA-4 | BioXcell | clone 9D9 |
| Anti-DDX6 | ThermoFisher | PA555012 |
| Anti-PRICKLE1 | ThermoFisher | 22589–1-AP |
| Anti-ELF3 | ThermoFisher | PA589261 |
| Anti-LDHB | ThermoFisher | PA596736 |
| Anti-IFT25 | ThermoFisher | 15732–1-AP |
| Anti-PICK1 | ThermoFisher | PA1073 |
| Anti-STT3A | ThermoFisher | 12034–1-AP |
| Anti-RPS15A | ThermoFisher | PA5103532 |
| Anti-beta Actin (BA3R) | ThermoFisher | MA515739 |
| 5-(and 6)-Carboxyfluorescein diacetate succinimidyl ester Kit, CFDA SE Kit | Biolegend | 423801 |
| Anti-mouse IFN-γ mAb (AN18) | Mabtech | 3321–3-250 |
| Chemicals, peptides, and recombinant proteins | ||
| Peptides used in the study are listed in Figure S1 | GenScript | N/A |
| Tumor Dissociation Kit, mouse | Miltenyi | 130–096-730 |
| CD8a (Ly-2) MicroBeads, mouse | Miltenyi | 130–117-044 |
| Omaveloxolone (RTA-408) | Reata Pharmaceutical | N/A |
| Nitroaspirin (NCX-4016) | Sigma | SML1669 |
| Hoechst 33342 dye | ThermoFisher | H3570 |
| N-Nitro-L-arginine methylester (L-NAME) | Sellechem | S2877 |
| Deposited data | ||
| The mass spectrometry proteomics data | MassIVE (http://massive.ucsd.edu) | MSV000087990 |
| The mass spectrometry proteomics data | ProteomeXchange (http://www.proteomexchange.org) | PXD027956 |
| Experimental models: Cell lines | ||
| UACC-903 | Originally from University of Arizona Cell Collection Provided by Dr. M. Herlyn, Wistar Institute | RRID:CVCL_4052 |
| EG7 | ATCC | CRL-2113 |
| B16F10 | ATCC | CRL-6475 |
| B16F10-iNOS | This manuscript | This manuscript |
| T2 cells | ATCC | CRL-1992 |
| RMA-S cells | Gift from Dr. Eisenlohr | RRID: CVCL_2180 |
| EL-4 | ATCC | TIB-39 |
| LLC | ATCC | CRL-1642 |
| CT26 | ATCC | CRL-2638 |
| Experimental models: Organisms/strains | ||
| PMEL transgenic mice B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J | Jackson Lab | RRID:IMSR_JAX:005023 |
| BALB/c mice | Charles River | |
| C57BL/6 mice | Charles River | |
| C57BL/6 mice | Envigo | |
| Oligonucleotides See Table S9 | ||
| Biological Samples | ||
| Tissue samples | University of Lausanne | Cancer Immun. 2004 May 19;4:4. PMID: 15149168 |
| Biological Samples | ||
| Tissue samples | University of Pennsylvania | Nature. 2017 May 4;545(7652):60–65. 10.1038/nature22079 |
| Software and algorithms | ||
| GraphPad Prizm | Dotmatics | 8.4.3 |
PNT alters the profile of MHC class I peptides on tumor cells;
Only CTLs specific for PNT-resistant peptides have a strong antitumor effect;
Therapeutic targeting of PNT in mice reduces resistance of tumor cells to CTLs;
High PNT in tumors is associated with worse clinical response to immunotherapy;
Tcyganov et al. demonstrate that peroxynitrite (PNT) in the tumor microenvironment alters the profile of MHC class I bound peptides on tumor cells. This makes recognition of tumor cells by cytotoxic T lymphocytes (CTLs) much less efficient and negatively impacts antitumor response. This suggests that PNT-resistant peptides are better suited for vaccination and that targeting of tumor cell resistance may include inhibition of PNT production.
Acknowledgements
This study was supported by NIH grant RO1 CA177646, by animal, proteomics, and flow cytometry core facilities of Wistar Institute (NIH grant P30 CA010815), by NIH grants CA219603 and DE028172 to S.F, and CA165065 to LJM. Clinical sample acquisition was supported by NIH grant P50 CA174523 SPORE on Skin Cancer to the University of Pennsylvania and the Wistar Institute, and the Tara Miller Foundation. We thank Reata Pharmaceuticals for providing RTA408 and support for part of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
EC is paid consultant for Oncovir, Inc. EM, QZ, AM, and DG are employee and shareholders of AstraZeneca. All other authors declare no competing interests.
References
- Abramson JS (2020). Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus Med Rev 34, 29–33. [DOI] [PubMed] [Google Scholar]
- Aoe T, Okamoto Y, and Saito T (1995). Activated macrophages induce structural abnormalities of the T cell receptor-CD3 complex. JExpMed 181, 1881–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, and Elkan C (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2, 28–36. [PubMed] [Google Scholar]
- Bartesaghi S, and Radi R (2018). Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol 14, 618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayden AS, Yakovlev VA, Graves PR, Mikkelsen RB, and Kellogg GE (2011). Factors influencing protein tyrosine nitration--structure-based predictive models. Free Radic Biol Med 50, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Yekutieli D (2001). The control of the false discovery rate in multiple testing under dependency. Ann Satist 29, 1165–1188. [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger DSM, Eccleston RC, Phillips A, Coveney PV, Elliott T, and Dalchau N (2018). A Mechanistic Model for Predicting Cell Surface Presentation of Competing Peptides by MHC Class I Molecules. Frontiers in immunology 9, 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HI, Reyes-Vargas E, Delgado JC, and Celis E (2012). A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res 72, 1986–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- De Santo C, Serafini P, Marigo I, Dolcetti L, Bolla M, Del Soldato P, Melani C, Guiducci C, Colombo MP, Iezzi M, et al. (2005). Nitroaspirin corrects immune dysfunction in tumor-bearing hosts and promotes tumor eradication by cancer vaccination. Proc Natl Acad Sci U S A 102, 4185–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva AD, Boesteanu A, Song R, Nagy N, Harhaj E, Harding CV, and Joyce S (1999). Thermolabile H-2Kb molecules expressed by transporter associated with antigen processing-deficient RMA-S cells are occupied by low-affinity peptides. J Immunol 163, 4413–4420. [PubMed] [Google Scholar]
- Ekmekcioglu S, Ellerhorst J, Prieto V, Johnson M, Broemeling L, and Grimm EA (2005). Early Detection and Diagnosis Tumor iNOS predicts poor survival for stage III melanoma patients. Int J Cancer 119, 861–866. [DOI] [PubMed] [Google Scholar]
- Ekmekcioglu S, Ellerhorst J, Smid CM, Prieto VG, Munsell M, Buzaid AC, and Grimm EA (2000). Inducible nitric oxide synthase and nitrotyrosine in human metastatic melanoma tumors correlate with poor survival. Clin Cancer Res 6, 4768–4775. [PubMed] [Google Scholar]
- Ferrington DA, and Gregerson DS (2012). Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci 109, 75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V (2012). Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haqqani AS, Kelly JF, and Birnboim HC (2002). Selective nitration of histone tyrosine residues in vivo in mutatect tumors. J Biol Chem 277, 3614–3621. [DOI] [PubMed] [Google Scholar]
- Hather G, Liu R, Bandi S, Mettetal J, Manfredi M, Shyu WC, Donelan J, and Chakravarty A (2014). Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform 13, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, and Groll M (2012). Immuno- and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148, 727–738. [DOI] [PubMed] [Google Scholar]
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. (2019). Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnula VL, Torkkeli T, Kristo P, Sormunen R, Soini Y, Paakko P, Ollikainen T, Kahlos K, Hirvonen A, and Knuutila S (2004). Ultrastructural and chromosomal studies on manganese superoxide dismutase in malignant mesothelioma. Am J Respir Cell Mol Biol 31, 147–153. [DOI] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. (2015). T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Ramakrishnan R, Altiok S, Youn JI, Cheng P, Celis E, Pisarev V, Sherman S, Sporn MB, and Gabrilovich D (2011). Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J Clin Invest 121, 4015–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner E, Barnea E, Beer I, and Admon A (2006). The turnover kinetics of major histocompatibility complex peptides of human cancer cells. Mol Cell Proteomics 5, 357–365. [DOI] [PubMed] [Google Scholar]
- Milner E, Gutter-Kapon L, Bassani-Strenberg M, Barnea E, Beer I, and Admon A (2013). The effect of proteasome inhibition on the generation of the human leukocyte antigen (HLA) peptidome. Mol Cell Proteomics 12, 1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, and Gabrilovich DI (2007). Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med 13, 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, et al. (2010). Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res 16, 1812–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Yasuoka H, Tsujimoto M, Yoshidome K, Nakahara M, Nakao K, Nakamura M, and Kakudo K (2006). Nitric oxide in breast cancer: induction of vascular endothelial growth factor-C and correlation with metastasis and poor prognosis. Clin Cancer Res 12, 1201–1207. [DOI] [PubMed] [Google Scholar]
- Nguyen LT, Saibil SD, Sotov V, Le MX, Khoja L, Ghazarian D, Bonilla L, Majeed H, Hogg D, Joshua AM, et al. (2019). Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol Immunother 68, 773–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osna NA, Haorah J, Krutik VM, and Donohue TM Jr. (2004). Peroxynitrite alters the catalytic activity of rodent liver proteasome in vitro and in vivo. Hepatology 40, 574–582. [DOI] [PubMed] [Google Scholar]
- Otsuji M, Kimura Y, Aoe T, Okamoto Y, and Saito T (1996). Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci USA 93, 13119–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, and Ferguson DA (2015). RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PloS one 10, e0122942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, et al. (2012). Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res 18, 6758–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebrin I, Bregere C, Gallaher TK, and Sohal RS (2008). Detection and characterization of peroxynitrite-induced modifications of tyrosine, tryptophan, and methionine residues by tandem mass spectrometry. Methods Enzymol 441, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmielau J, and Finn OJ (2001). Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res 61, 4756–4760. [PubMed] [Google Scholar]
- Schuster H, Shao W, Weiss T, Pedrioli PGA, Roth P, Weller M, Campbell DS, Deutsch EW, Moritz RL, Planz O, et al. (2018). A tissue-based draft map of the murine MHC class I immunopeptidome. Sci Data 5, 180157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijts EJ, and Kloetzel PM (2011). The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci 68, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza JM, Daikhin E, Yudkoff M, Raman CS, and Ischiropoulos H (1999). Factors determining the selectivity of protein tyrosine nitration. Arch Biochem Biophys 371, 169–178. [DOI] [PubMed] [Google Scholar]
- Speiser DE, Baumgaertner P, Voelter V, Devevre E, Barbey C, Rufer N, and Romero P (2008). Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc Natl Acad Sci U S A 105, 3849–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F, Krieg AM, Cerottini JC, and Romero P (2005). Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest 115, 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teicher BA, and Tomaszewski JE (2015). Proteasome inhibitors. Biochem Pharmacol 96, 1–9. [DOI] [PubMed] [Google Scholar]
- Veglia F, Perego M, and Gabrilovich D (2018). Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter MB, La Greca F, Arastu-Kapur S, Caiazza F, Cimermancic P, Buchholz TJ, Anderl JL, Ravalin M, Bohn MF, Sali A, et al. (2017). Immunoproteasome functions explained by divergence in cleavage specificity and regulation. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.List of peptides evaluated in EG.7 cells. Related to Figure 1.
Table S2. List of peptides evaluated in UACC903 cells. Related to Figure 1.
Table S4. List of peptides evaluated in EG.7 cells after bortezomib treatment. Related to Figure 3.
Table S6. List of peptides evaluated in B16-iNOS cells versus B16-Control cells. Related to Figure 5
Data Availability Statement
The mass spectrometry proteomics data have been deposited into the MassIVE (http://massive.ucsd.edu) and ProteomeXchange (http://www.proteomexchange.org) data repositories with the accession number MSV000087990 and PXD027956, respectively.