

Abstract
The bacterial co-infections in SARS-CoV-2 patients remained the least explored subject of clinical manifestations that may also determine the disease severity. Nasopharyngeal microbial community structure within SARS-CoV-2 infected patients could reveal interesting microbiome dynamics that may influence the disease outcomes. Here, in this research study, we analyzed distinct nasopharyngeal microbiome profile in the deceased (n = 48) and recovered (n = 29) COVID-19 patients and compared it with control SARS-CoV-2 negative individuals (control) (n = 33). The nasal microbiome composition of the three groups varies significantly (PERMANOVA, p-value <0.001), where deceased patients showed higher species richness compared to the recovered and control groups. Pathogenic genera, including Corynebacterium (LDA score 5.51), Staphylococcus, Serratia, Klebsiella and their corresponding species were determined as biomarkers (p-value <0.05, LDA cutoff 4.0) in the deceased COVID-19 patients. Ochrobactrum (LDA score 5.79), and Burkholderia (LDA 5.29), were found in the recovered group which harbors ordinal bacteria (p-value <0.05, LDA-4.0) as biomarkers. Similarly, Pseudomonas (LDA score 6.19), and several healthy nasal cavity commensals including Veillonella, and Porphyromonas, were biomarkers for the control individuals. Healthy commensal bacteria may trigger the immune response and alter the viral infection susceptibility and thus, may play important role and possible recovery that needs to be further explored. This research finding provide vital information and have significant implications for understanding the microbial diversity of COVID-19 patients. However, additional studies are needed to address the microbiome-based therapeutics and diagnostics interventions.
Keywords: Nasopharyngeal microbiome, Opportunistic pathogens, And SARS-CoV-2
Graphical abstract

1. Introduction
The COVID-19 pandemic caused huge loss of life across the globe especially with the emergence of the new variant of concerns (VoCs) as defined by the set of mutations with higher transmissibility, pathogenesis, and virulence as compared to the reference strain. New emerging variants remains a consistent threat, such as the current outbreak of Omicron sub-variants has shaken the entire world [[1], [2], [3]]. The second wave in India was caused by the sudden rise in cases due to increase in the Delta variant (B.1.617.2 lineage) and later on it was spread across all other countries [4]. The additional risk of microbial co-infections in the COVID-19 patients remained an understudied subject until date. The concerns were raised due to the bacterial co-infections, opportunistic pathogens and fungal infections in the COVID-19 patients [[5], [6], [7]]. For example, the rise in cases of Mucormycosis, commonly known as “black fungus”, were reported from several hospitalized patients on the path to recovery and it's been reported that, imbalance in the nasal microbiota is linked with mucormycosis in COVID-19 study [8]. Recent studies demonstrated alterations in the richness and diversity of gut microbiome of SARS-CoV-2 infected patients in follow-up studies [9,10]. Similarly, few studies explored the dynamics of nasal microbiome analysis of the COVID-19 patients [6,[11], [12], [13], [14]]. The temporal association between healthy, infected and deceased patient's microbiomes is of greater significance in terms of the possible therapeutic interventions that might require additional medical treatments for complete recovery [15]. However, quantifiable data is not available to estimate the precise burden of the bacterial co-infections among the COVID-19 patients [16].
COVID-19 pandemic is one of the most complex health challenges in recent times due to several factors. Nasopharyngeal cavity is an important interface and the well-known transmission route of the SARS-CoV-2 infection. This damage in the cells is an opportunity for the pathogenic bacteria to invade and cause further disease complications in the predisposed and immunocompromised individuals. Nasopharyngeal cavity is also a rich habitat for the diversity of microbial communities [17,18]. Microbial diversity is a determinant of the persistent bacterial population within a host and has characteristic signatures of the health of an individual. The influence of the human microbiome on health and disease outcome has been recognized with the recent evidence based studies and discussed in continuum of the possible role in patient survival [19]. However, it is less studied in supplement with the viral infections and probably under hospitalized conditions.
Even the loss of smell and taste in the COVID-19 patients are reported but have not been systematically explored to identify the cause. It has been reported that, olfactory modulations such as sense of smell can be due to the altered microbiome pathophysiology of the infected patients [20]. Some research studies explored the dynamics of host genetic and environmental factors for the nasal microbiomes and colonization of Staphylococcus aureus in the nasal cavity. Further authors examined the significance and role of nasal microbiota in S. aureus nasal colonization process [21]. Similarly, another study explored the dynamics of the nasal microbiome in asthma patients and compared with the control and healthy groups. These studies found the composition differences among the different groups and argued that microbiome composition can alter the pathobiology of the diseased and control groups [22,23]. Further, these implications need to be examined in relevance to the SARS-CoV-2 infections. These studies have valuable implications for even the high-risk groups with the comorbidity and hygiene conditions, which could further help in better treatment and control measures. The health issues of the predisposed comorbid patients further deteriorate the patient survival outcomes and recovery. The modulation of host immune response by the microbiomes in early infections may certainly be examined and may have relevant implications for the susceptibility of the COVID-19. Therefore, in this research study, we explored the nasopharyngeal microbiome of the recovered and deceased COVID-19 patients and compared it with the SARS-CoV-2 negative group.
2. Materials and methods
2.1. Sample collection
For this study, a total 97 nasopharyngeal swabs samples of the COVID-19 patient's and 44 swabs from the control individuals were collected. The samples were collected during the period of April–June 2021, i.e. peak time of the second wave of COVID-19 in India. We used the diagnostics samples as well as samples used for SARS-CoV-2 genome sequencing at Gujarat Biotechnology Research Centre (GBRC). GBRC is an authorized COVID-19 testing center by RT-PCR as approved by Indian Council of Medical (ICMR) and also a member of Indian SARS-CoV-2 Genome Sequencing Consortium (INSACOG), Government of India. All the samples were collected in the viral transport medium (VTM) by the trained medical professionals by following the ICMR guideline published by the Ministry of Health and Family Welfare, Government of India. Moreover, all the samples were collected before any medical treatments were given to the patients. The confirmed COVID-19 patients were further followed till the final outcome of the disease progression i.e. either the patient recovered or deceased. The control samples were also confirmed negative for SARS-CoV-2 infection throughout RT-PCR twice i.e. the initially in the hospital where they went and further confirmed in the GBRC as described below In this way, we had total control (n = 44), recovered (SARS-CoV-2 infected but recovered, n = 43), deceased (SARS-CoV-2 infected but deceased, n = 54) samples.
2.2. Real-time PCR (RT-PCR) for SARS-CoV-2 detection in control samples
For the RNA extraction, we used QIAamp Viral RNA Mini Kit (QIAGEN, Germany). A 300 μL of the nasopharyngeal swab samples were used for the RNA extraction by following the manufacturer's instruction and RNA was eluted into a 30 μL of elution buffer. For SARS-CoV-2 RNA detection, we used QUANTIPLUS KIT from Huwel Lifesciences, (India) and samples were run on Applied Biosystems 7500 fast Dx Real-Time PCR instrument (ThermoFisher Scientific, MA USA).
2.3. DNA extraction and 16S amplicon sequencing
For total genomic DNA extraction, 250–300 μL from each nasopharyngeal swab sample were processed using QIAamp® DNA Mini Kit (QIAGEN, Germany) by following the manufacturer's instructions. DNA was eluted into a 30 μL of elution buffer supplied along with the kit. We used Qubit™ dsDNA broad range assay kit and Qubit fluorometer v4.0 (ThermoFisher Scientific, MA USA) for DNA quantification. From each sample, 20–25 ng of DNA was used for amplification of 16S rDNA V1–V3 region using 101F (5′ACTGGCGGACGGGTGAGTAA3′) and 518R (5′CGTATTACCGCGGCTGCTGG3′) universal primer which were already tagged with Ion library adaptor, key sequence and a unique barcode sequence. A 20 μL PCR reaction mixture containing 20–25 ng DNA, 10 μL, 2X EmeraldAmp® RT PCR master mix (Takara Bio, Japan), 1 μL each forward and reverse primer (5pM), 1 μL bovine serum albumin (2 mg/mL) and nuclease free water. The thermal cycling conditions were as follows; 95 °C for 5 min, followed by 25 cycles of denaturation at 95 °C for 1 min, annealing at 60 °C for 30 s, and extension at 72 °C 1 min, with final extension at 72 °C for 5 min. Each PCR reaction mixture was loaded into 1.5% agarose gel to see the amplification and positive samples were purified using Invitrogen E-Gel EX precast 2% agarose gel (Thermo Fisher Scientific, MA USA). The purified 16S amplicon libraries were quantified using Qubit™ 1X dsDNA high sensitivity assay kit and Qubit fluorometer v4.0 (Thermo Fisher Scientific, MA USA). Each library was diluted to 100 pM and pooled into equal molar concentration. The pool was further diluted to 12 pM and emulsion PCR was carried out using Ion 520™ & Ion 530™ Kit-OT2 and sequencing was performed on the Ion GeneStudio S5 Plus system with 400 bp chemistry and Ion 530 chip.
2.4. Data processing and bioinformatics analysis
The obtained raw sequences were filtered using the PRINSEQ [24] where, the minimum quality threshold was set at Q20. The polished reads were uploaded to Metagenomic Rapid Annotations using Subsystems Technology (MG-RAST) server v4.0.3 [25] for the taxonomic analysis and annotation. For taxonomic classification of the amplicon sequencing reads, we used the SILVA SSU database [26]. The annotation results were exported with the criteria of “representative hit”, minimum e-value 10–5, and minimum identity 60%. The taxonomy profile of each read was downloaded and subjected to statistical analysis using various bioinformatics tools.
2.5. Statistical analysis
We used Statistical Analysis of Metagenomic Profiles (STAMP) tool [27], for calculating relative abundance of the taxa in each group. For this, the data was analyzed at genus level and, we used Kruskal-Wallis-H test to see statistically significant differences between three groups and Benjamini-Hochberg FDR test was applied for multiple test correction. For two group comparisons, we applied Welch's t-test (Two-side) with 0.95 CI and Benjamini-Hochberg FDR for multiple test correction. MicrobiomeAnalyst server [28] was used for the rest of the analysis where low count data (prevalence in samples = 20%) and low variance features were filtered based on inter-quantile range to improve downstream statistical analysis. Further, the data was also rarified based on the sample having lowest sequencing depth. To compare the alpha diversity between two groups and among three groups, we used observed features, Shannon, Simpson, Cho1 and Abundance-based Coverage Estimator (ACE) indices, where, Kruskal-Wallis statistics and t-test was applied for three and two group comparison, respectively. Similarly, beta diversity was estimated using Principal Coordinates Analysis (PCoA) and Non-Metric Multidimensional Scaling (NMDS) with Bray-Curtis dissimilarity and Permutation multivariate analysis of variance (PERMANOVA) statistics. Linear Discriminant Analysis (LDA) Effect Size LEfSe (p-value <0.05, LDA-4.0) was used to identify most significant taxa within each group. For dendrogram analysis we used Bray-Curtis distance measure and applied two different clustering algorithms i.e. Ward and Jaccard. Random forest classification was used to predict an error rate in each group and to evaluate the classification performance of genera. For random forest classification, 1000 trees were grown with consideration of the group as an experimental factor.
3. Results
The median age for the control group (29 ± 12.84 years), recovered group (43 ± 16.44 years) and deceased group (58 ± 13.12 years) with minimum 12 years and maximum 88 years, in which, 107 were males and 34 were females (Table 1 ). Cough, fever, and breathlessness, were the most common symptoms in the deceased COVID-19 patients while, fever, body ache, and loss of smell were most common among the recovered patients. Wherever possible, we have also collected information of the co-morbidities of the COVID-19 patients. Many of the deceased patients have comorbidities such as heart disease and hypertension. Except for two patients, none of in the recovered group have any comorbidities. Additionally, we also sequenced SARS-CoV-2 genome from all the recovered and deceased patients (n = 97), where, except eight deceased patients, all the patients were found to be infected with B.1.617.2 (Delta) variants (Supplementary Table S1). For microbiome analysis, we extracted total DNA from 141 nasopharyngeal swab samples, which included 54, 43, and 44 deceased, recovered and control samples, respectively. Out of 141 samples, 15 samples (8 deceased and 7 recovered samples) failed in PCR amplification. From the remaining 126 samples, 16 samples were discarded because having insufficient cleaned reads. Finally, a total of 110 samples which include 48 deceased, 29 recovered and 33 control samples were analyzed and reported in this study. In total, we generated 42.7 million reads with an average size of 336 bp. Details of sequencing reads generated for each sample is provided in Supplementary Table S2.
Table 1.
Median age group and Ct-values with percent male/female in each study group.
| Control (n = 44) | Recovered (n = 43) | Deceased (n = 54) | Total (n = 141) | |
|---|---|---|---|---|
| Male | n = 35 (79.55%) | n = 32 (74.42%) | n = 40 (74.07%) | n = 107 (75.89%) |
| Female | n = 9 (20.45%) | n = 11 (25.58%) | n = 14 (25.93%) | n = 34 (24.11%) |
| Median age years (Std. Dev.) | 29 (12.84) | 35 (16.44) | 58 (13.12) | NA |
| Median Ct of ORF-1ab gene | NA | 21 | 18.46 | NA |
| Median Ct of N-gene | 38.42 | 20.5 | 26 | NA |
| Median Ct of E-gene | 37.6 | 21 | 24 | NA |
| Median Ct of RDRP-gene | NA | NA | 20 | NA |
3.1. Microbial diversity analysis
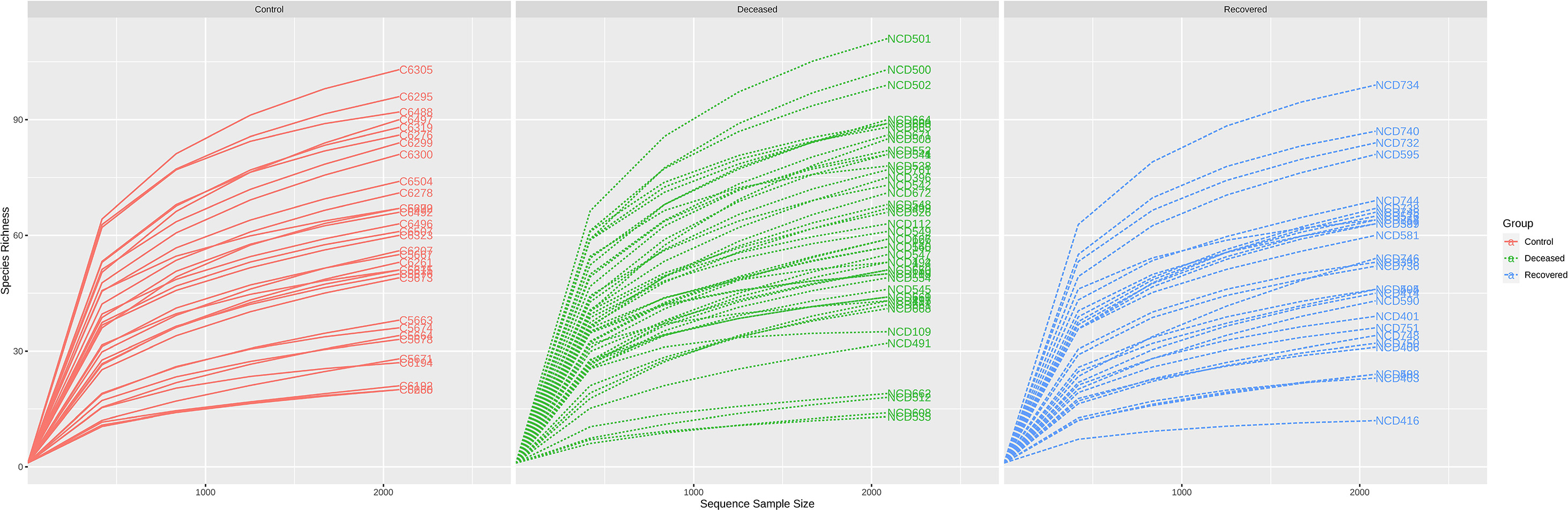
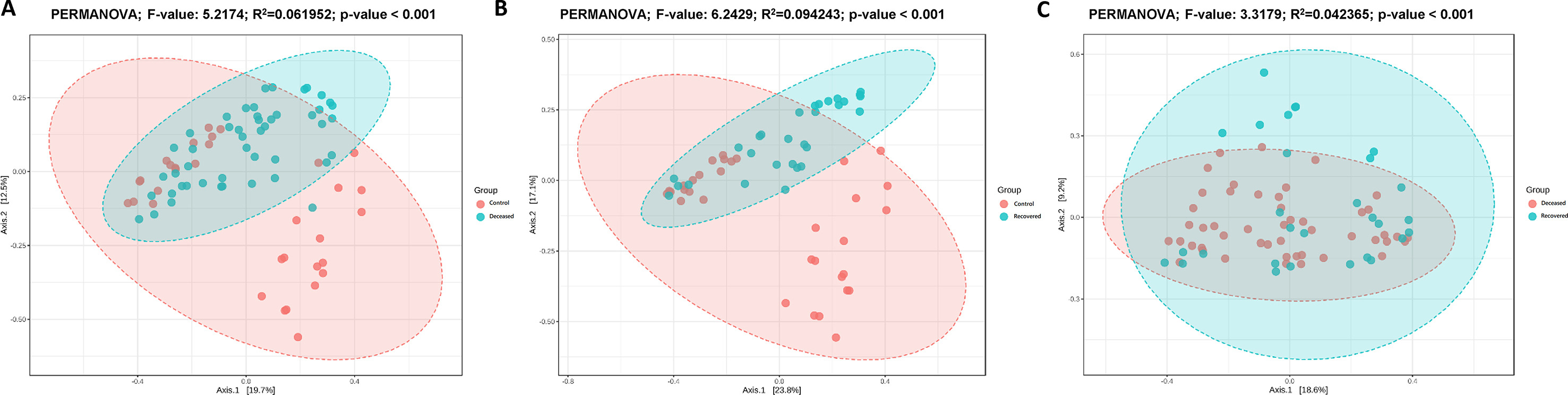
Rarefaction curve (Supplementary Fig. S1), clearly depicting that, in all the samples the curve almost reached a plateau, which means the generated reads are sufficient to capture the bacterial diversity present in the samples. Beta-diversity at genus level was determined based on PCoA (Fig. 1 (a) (PERMANOVA; F-value: 6.5886; R2: 0.10965; p-value <0.001) NMDS (Fig. 1 (b) (PERMANOVA; F-value: 6.5886; R2: 0.10965; p-value <0.001 [NMDS] Stress = 0.20839) based on Bray-Curtis distance matrix revealed significant difference in the microbiota composition among the three groups. PCoA explained overall 22.5% and 17.1% variation on Axis1 and Axis 2, respectively. PCoA plots for between two groups comparison are shown in Supplementary Fig. S2, where also we observed significant difference (p-value <0.001) between control-deceased, control-recovered and deceased-recovered groups. Except few samples, Dendrogram (Supplementary Fig. S3) also showed distinct clustering of the samples into three groups. Microbial species richness assessed based on Cho1 (Fig. 1 (c), and ACE (Fig. 1 (d), and observed features (Fig. 1 (e) index showed higher bacterial diversity in the deceased patient group, however, the difference among the three groups was not significant [Cho1: p-value: 0.41324; (Kruskal-Wallis) statistic: 1.7674; ACE: p-value: 0.39582; (Kruskal-Wallis) statistic: 1.8536 and Observed: p-value: 0.067215; (Kruskal-Wallis) statistic: 5.3997]. Also, there was no significant difference in the Shannon and Simpson indices among the three groups. However, in two group comparison, we observed a significant difference in specie richness i.e. Observed; p-value: 0.017453; [T-test] statistic: 2.4342, Chao 1: p-value: 0.030277; [T-test] statistic: 2.2084, and ACE: p-value: 0.021; [T-test] statistic: 2.3579 while comparing deceased and recovered patients group. Nonetheless, there was no significant differences between control-deceased and control-recovered COVID-19 patient group (Supplementary Fig. S4).
Fig. 1.

Alpha and Beta diversity plots (species level) for deceased, recovered and control patient group. (A) PCoA plot based on Bray-Curtis dissimilarity and with PERMANOVA statistics depicting significant differences (p-value <0.001). (B) NMDS plot based on Bray-Curtis dissimilarity and PERMANOVA statistics showing significant difference (p-value <0.001) among the three groups. (C), and (D), species richness estimated using Cho1, ACE index at the species level with Mann-Whitney/Kruskal-Walis statistical method. (E) Species richness and evenness estimated using Shannon index at species level with Mann-Whitney/Kruskal-Walis statistical method. No significant difference in the alpha diversity among the three groups.
3.2. Microbiome structure and composition
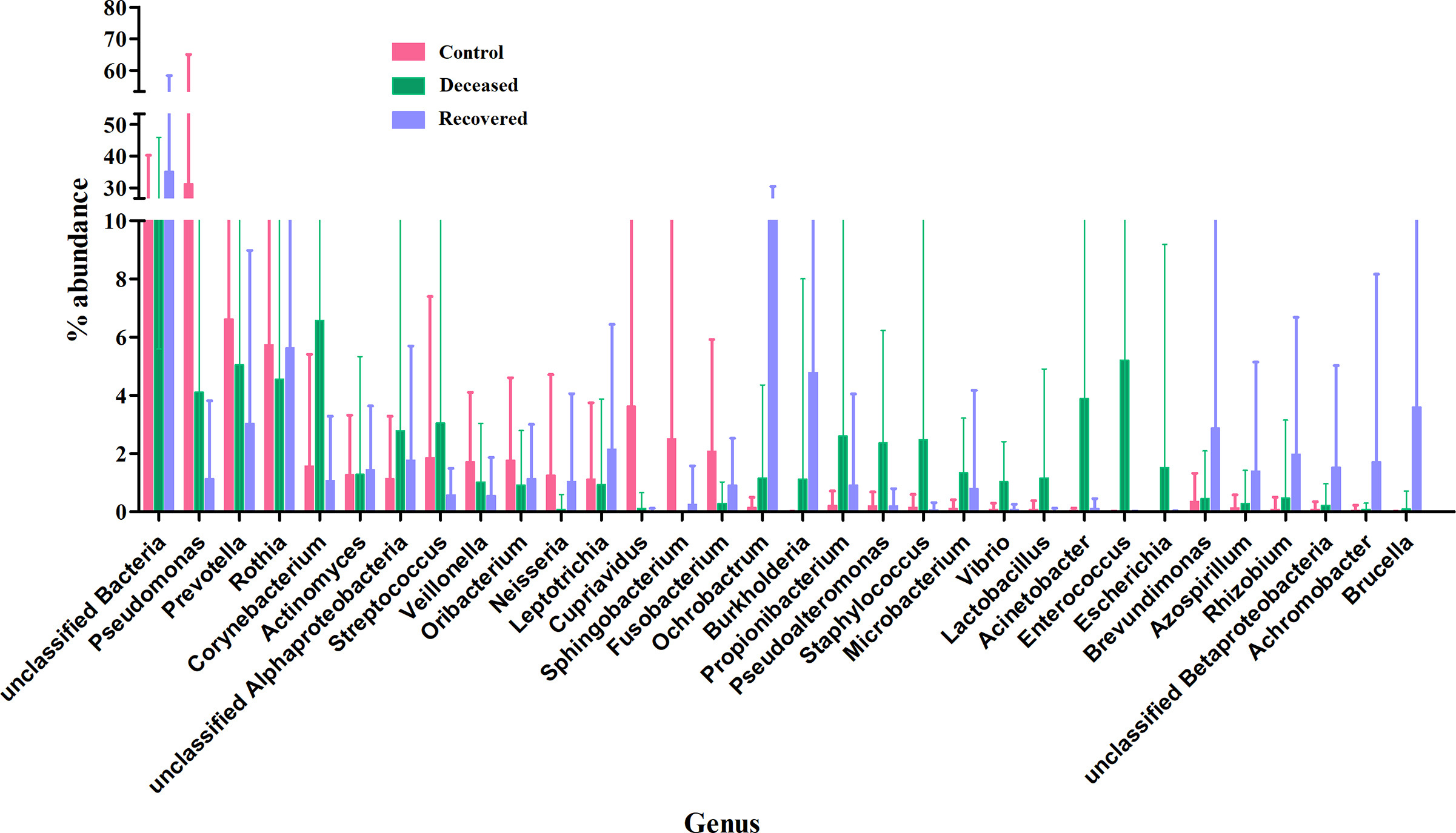
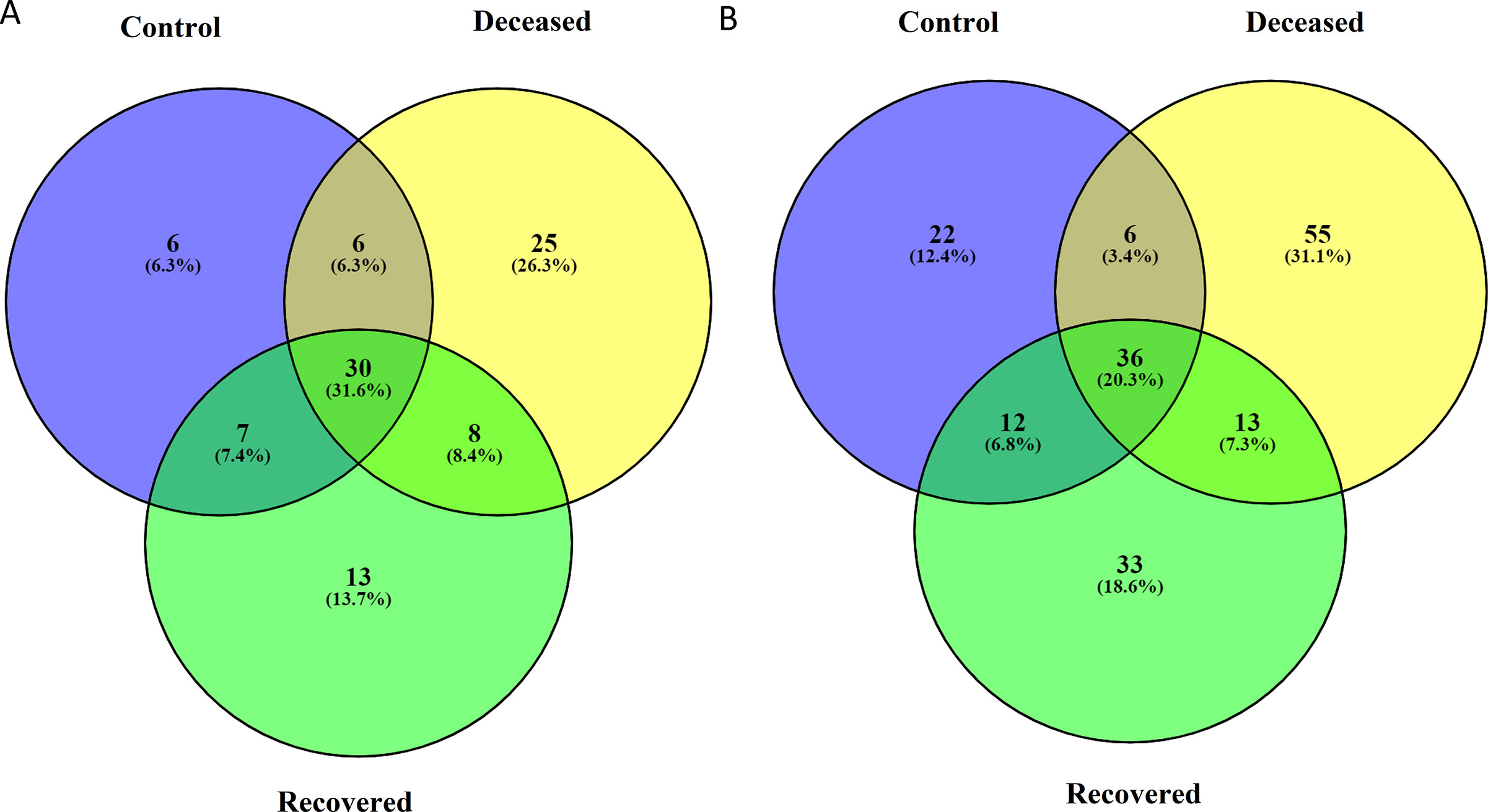
In all the samples, unclassified bacteria were present with an abundance >23.0% with highest (35.2%) in the group of the patients those who recovered from SARS-CoV-2 infection. Genus Pseudomonas was most predominant in the control group i.e. 31.34%, followed by, 4.11% in deceased, and only 1.14% in recovered group. Similarly, Prevotella was also found to be dominant in the control group as compared to the other two groups. In COVID-19 deceased patients, Corynebacterium (6.58%), Enterococcus (5.21%), Acinetobacter (3.89%), Streptococcus (3.05%), Pseudoalteromonas (2.36%), Propionibacterium (2.6%), Staphylococcus (2.47%) and others were abundant as compared to the rest two groups. Likewise, unclassified bacteria (35.23%), Ochrobactrum (12.15%), Burkholderia (4.79%), Brevundimonas (2.88%), Leptotrichia (2.15%) and some others were abundant in the patients those who recovered from COVID-19 (Supplementary Fig. S5 and Supplementary Table S3). An extended error plots (genus level) between two group comparisons are provided in Supplementary Fig. S6. The differential abundance of various genera across two group comparison are shown as a heat-map trees as well as dot plot from LEfSe analysis in Fig. 2 and listed in Supplementary Table S4. Total 25 and 13 genera and 55 and 33 species with >0.1% abundance, were exclusively present in deceased and recovered patients, respectively (Supplementary Fig. S7, Supplementary Tables S5 and S6).
Fig. 2.

Comparison of microbial community between two groups. (A), (B), and (C) are the heat trees, depicting the significantly different genera between control vs deceased, control vs recovered and deceased vs recovered groups, respectively. Analysis was performed with non-parametric Wilcoxon Rank Sum test. The size and colour of nodes and edges are with the abundance (median) of organisms in each group. (D), (E), and (F), depicts the dot plots for Linear Discriminant Analysis (LDA) Effect Size (LEfSe) at the genus level for control vs deceased, control vs recovered and deceased vs recovered groups, respectively. LEfSe was performed with p-value cutoff 0.05 and Log LDA score cutoff 4.0.
3.3. Microbiome signature of the COVID-19 patients
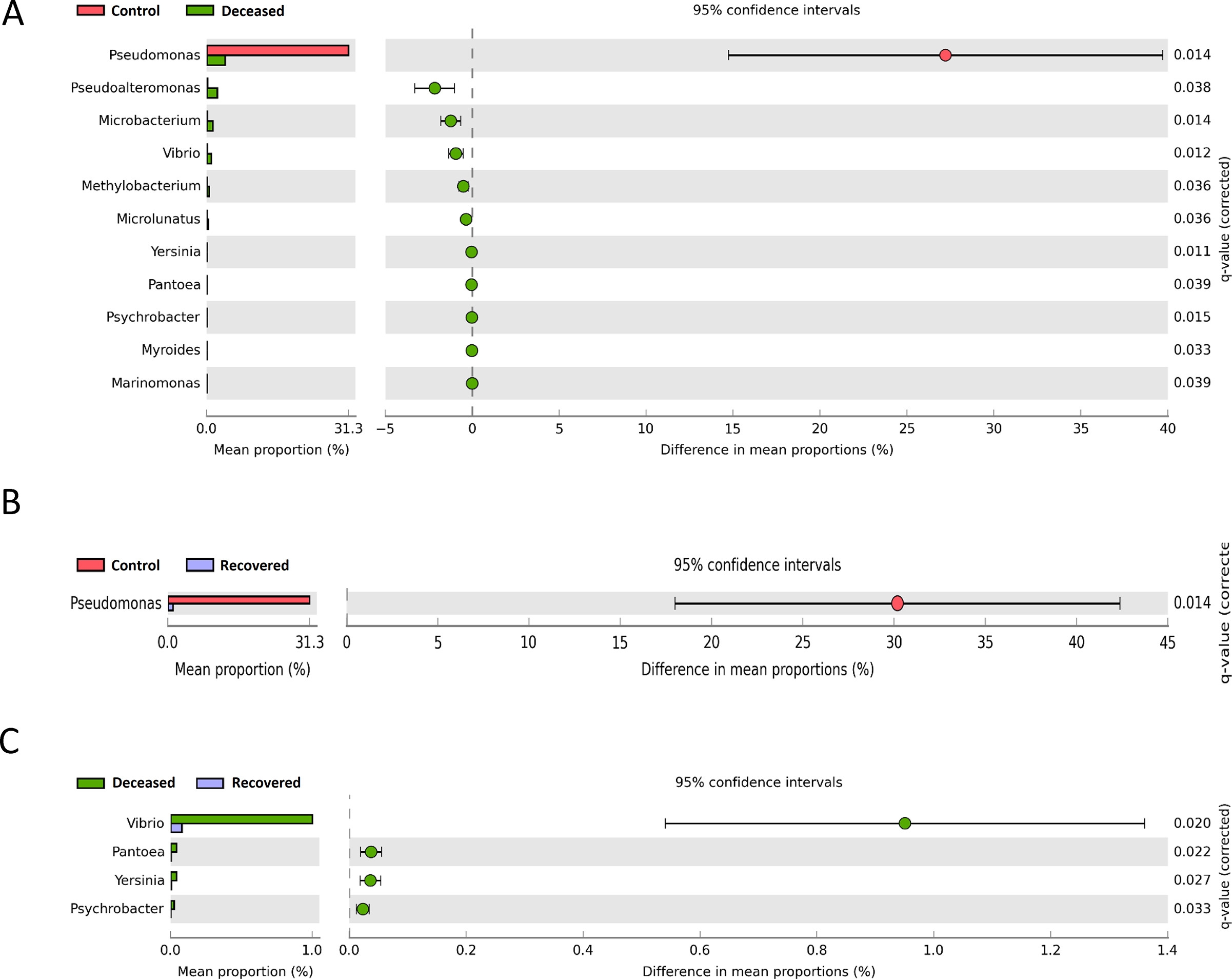
The LEfSe analysis is widely used to identify the biomarkers across the different samples. Therefore, in this study, LEfSe analysis was used to identify the biomarker genera as a signature of the nasopharyngeal microbiome in SARS-CoV-2 infected but recovered and deceased COVID-19 patients. Total 32 genera (15, 10, and 7 in deceased, recovered and control patients, respectively) and 46 species (22, 13, and 11 in deceased, recovered and control patients, respectively) were identified as biomarkers with LDA score >4.0 and p-value <0.05 (Fig. 3 and Supplementary Tables S7 and S8). Genera, Corynebacterium, with the LDA score 5.51 (highest in the deceased group) and Staphylococcus, Serratia, Micrococcus, and Klebsiella along with their, pathogenic or opportunistic pathogenic species such as C. xerosis, S. epidermidis, S. liquefaciens, K. pneumoniae were picked out as a biomarker for deceased COVID-19 patients. In a similar manner, Ochrobactrum (LDA core 5.79, highest in the recovered group), Burkholderia (LDA core 5.29), unclassified Betaproteobacteria, and their pathogenic or opportunistic pathogenic species such as O. anthropi, and O. tritici, and B. cepacia were spotted as biomarkers for recovered COVID-19 patients. Pseudomoans with highest LDA core 6.19 among the three group was picked up as a biomarker for control group. Apart from this, several healthly oral and nasal cavity commensal including Fusobacterium (F. periodonticum), Veillonella (V. parvula), Porphyromonas (P. catoniae), and Bulleidia (Bulleidia extructa) along with some pathogenic bacteria like, Neisseria (N. flavescens) were biomarkers. Fig. 4 depicts Log transformed abundance of the important biomarker genera. Random forest classification showed an excellent judgement of guessing the samples in their respective group, with 44/48, 19/29 and 29/33 correct prediction of deceased, recovered and control patients, respectively in their corresponding group with an overall 16.4% out of bag (OOB) error rate (Fig. 5 ).
Fig. 3.

LEfSe analysis comparing three groups (deceased, recovered, and control). LEfSe analysis was performed at genus and species level with p-value cutoff 0.05 and Log LDA score cutoff 4.0 with and all the significant features i.e. 32 genera and 46 species are plotted. (A), and (C) depict the dot plots at genus and species level, respectively, and (B) and (D) depict bar plots at genus and species level, respectively.
Fig. 4.

Box plots depicting log2 transformed abundance of important genera. (A)Corynebacterium, (B)Staphylococcus, (C)Serratia, (D)Micrococcus, (E)Klebsiella, (F)Ochrobactrum, (G) Unclassified betaproteobacteria, (H)Bulkhorderia, and (I)Pseudomonas. In the Box plot, each black dot represents a single sample, the bars on the Box represent upper and lower whiskers and the median value is shown as a horizontal line in the box.
Fig. 5.

Depicting the results of Random Forest Classification where 1000 trees were grown. (A) shows the prediction of each sample in the respective group, class error and Out-of-bag (OOB), (B) classification performance, and (C) random forest plot for the importance of genera with their mean decrease accuracy, top 25 genera were plotted. The high value of mean decrease accuracy shows the importance of that genus in predicting the group.
4. Discussion
SARS-CoV-2 is an evolving pathogenic virus, as we have observed in the dominance of delta variant (B.1.617.2 lineage) during the second wave in India and now Omicron and its sub-lineages. Delta variant was more contagious, highly transmissible, and virulent than the original strain of the Wuhan virus [4]. The dysbiosis in healthy microflora of the nasopharyngeal microbiome due to the SARS-CoV-2 infections might be the key driver of the severe infection [29,30]. For example, microbiota composition of the upper respiratory track is associated with the viral load in COVID-19 [18]. Further, the drivers between the mild to the severe disease conditions at the initial stage of disease progression may also provide new avenue in terms of the therapeutic considerations before the patient might become severely ill or advance stage of the health infections and related complications. In the present study, we showed signature microbiome profile of the, deceased and recovered COVID-19 patients and compared it with SARS-CoV-2 negative controls.
The results of the present study highlighted differences in the microbial community structure, abundance and their probable functional role in COVID-19 severity and bacterial co-infections by opportunistic pathogens and thus prolonged health complications due to SARS-CoV-2 infections. The PCoA and NMDS plots clearly indicated that there is substantial shift in the nasal microbiome composition in the SARS-CoV-2 infected individuals, which is in parallel with the previous studies [[31], [32], [33]]. However, we could not observe any significant difference in the alpha diversity measured based on Shannon, Simpson, Cho1 and ACE indices among the three groups as well as between two groups. This was again in concordance with the previous study [32]. Although there was no significant difference in the different alpha diversity indices, we observed that the species richness was highest in the deceased group followed by recovered and control group. In contrast to this, some of the previous studies have reported decrease in the microbial diversity in the COVID-19 patients [31,34]. Apart from dysbiosis in the nasopharyngeal microbiota due to SARS-CoV-2 infection [33], also reported alteration in the oral and gut microbiota with respect to SARS-CoV-2 viral load in the COVID-19 patients. Collectively, all these results suggest that SARS-CoV-2 infection literally creates a dysbiosis in the healthy human nasopharyngeal microbiota which may lead to more complex disease severity and outcome especially when pathogenic microbes overwhelm the normal/beneficial bacteria.
In the deceased patients, we observed several pathogenic microbes such as Corynebacterium, Staphylococcus, Serratia, Klebsiella, Acinetobacter, and Ralstonia along with their pathogenic or opportunistic pathogenic species notably as C. xerosis, S. epidermidis, S. liquefaciens, and K. pneumoniae. Similarly, we detected total 55 species (abundance >1.0%), which were exclusively present in the deceased patients and several of them are either pathogens or opportunistic pathogens in humans [34]. also reported abundance of opportunistic pathogens in the SARS-CoV-2 infected individuals. In this study, the biomarkers species, S. epidermidis, S. liquefaciens, and K. pneumonia, which are found in deceased COVID-19 patients are well known bacterial pathogens. Similarly, C. xerosis resemble to C. amycolatum [35] which is a very close relative to C. diphtheriae may cause infection in immunocompromised patients [36]. Moreover, C. xerosis itself may cause several types of infections including endocarditis [37]. Similarly, species of Ralstonia, R. picketti and R. mannitolilytica, are also considered as an emerging pathogen and cause infection in immunocompromised patients [38]. Although Acinetobacter baumannii is not identified as a biomarker for deceased group in this study, its proportion is very high in the COVID-19 deceased patients. A. baumannii is again an opportunistic pathogen causing infection in lungs and heart [39]. The biomarkers identification is based on the LDA score, where the even if the overall proportion of the bacterial abundance is low but difference is statistically significant, it would be marked as a biomarker. Similarly, if the difference in the mean of groups is huge but within group standard deviation among the samples is high, it will not qualify for biomarker. This could be the reason although the abundance of A. baumannii high in the deceased patients, it was not called as a biomarker. We also found presence of A. calcoaceticus in the deceased COVID-19 patients which is reported to be associated with the pneumonia [40]. The presence of A. calcoaceticus is not detected in the recovered patient group however found very high in the deceased patient group and low in control group Supplementary Table S7. Research findings suggested that, human nasal and oral cavity is the residence of several opportunistic pathogens received from the air or other sources. Such pathogens may trigger respiratory diseases including asthma and dysbiosis in the nasal microbiome [17,41]. Such dysbiosis may interrupt the immune response [42], and could increase the SARS-CoV-2 susceptibility [31,43]. We further speculate, linking this with the present study, the data collectively suggests that, SARS-CoV-2 infected patients have a higher possibility of getting secondary respiratory infection from the opportunistic pathogens. Furthermore, dysbiosis in the normal nasopharyngeal microflora may lead to poor clinical outcome and simultaneously cause critical disease severity and COVID-19 outcome [34].
In the recovered COVID-19 patients, we observed very few pathogenic bacteria such as Ochrobactrum (O. anthropi, and O. tritici) and B. cepacia. Instead of bacterial pathogens, in this group, we found abundance of several common oral or nasal and air microflora. In contrast to the present study [44], authors have reported presence of Burkholderia cepacia complex in critically ill COVID-19 patients. Surprisingly, we observed the presence of Brucella in the recovered patients that was unusual, as infection of Brucella in the respiratory tract is very rare [45,46]. However, a case study from Saudi Arabia has reported Brucella bacteremia in a 41 year old COVD-19 patient and comparison of respiratory track microbiome in hospitalized patients in China [47,48] and brucellosis mimics the COVID-19 symptoms [49]. Normal microflora in the human nasal cavity play an important role in the evasion of pathogens via local immune response [17,50,51] and thus may help in preventing disease complexity which may result in healthy outcomes like what healthy gut microbiota do. In summary, absence of pathogens/opportunistic pathogens and presence of normal flora in the recovered COVID-19 patients highlight less disease severity and possible recovery in the SARS-CoV-2 infected patients. The present data also support the very recent hypothesis of transplantation of oral microbiota to combat COVID-19 and other inflammatory diseases [52] as, dysbiosis in the oral microbiota is also responsible for COVID-19 disease severity as it may disrupt the local immune responses [34,53,54].
The control group samples were represented by the RT-PCR SARS-CoV-2 negative patients without any prior COVID-19 infections. In the control group, Pseudomonas, Veillonella, Porphyromonas, Gemella,and Bulleidia were abundant as compared to the deceased and recovered patients [31] have reported higher abundance of Veillonella in COVD-19 patients however, in this study it was a biomarker for control patients. Veillonella species are normal human flora of the human oral cavity [55]. Likewise, in contrast to the current study, Prevotella has been reported in the COVID-19 patients [32,56,57] however, although it was abundant in this study, it was not a biomarker for the control group may be because its proportion in the control samples vary. As the author mentioned [58], oropharyngeal and nasopharyngeal swabs for diagnosis of SARS-CoV-2 were performed, and oral swab specimens touching the tongue, palatum and cheeks were additionally collected for oral microbiota and local immune response profiling. Again, Neisseria was predicted as a biomarker for the control group which is again a part of normal oral microflora [33,59,60]. Gemella sanguinis, which is a nasopharyngeal commensal bacterium [5], is also predicated as biomarkers in the control group in this study. The presence of Pseudomonas might have protective role as Pseudomonas in the respiratory tract produce mucin, which may serve as a physical barrier against viral infection [61]. In summary, the control group in this study mostly harbors normal human commensal as well as high abundance of Pseudomonas, which may prevent the SARS-CoV-2 infection. However, as described by Ref. [61], the health respiratory microbiota is still undefined, the role of Pseudomonas in respiratory diseases still required more insights, as, Pseudomonas aeruginosa is also an opportunistic pathogen for the respiratory disease especially in the sinus [62,63].
In summary, the present study demonstrated comparative analysis of nasopharyngeal microbiome that might be the key driver of the patient outcome and disease using 16S ribosomal RNA (rRNA) gene amplicon sequencing. However, further experiments needed to study the specific functional role of dominant bacterial communities in the deceased and recovered patients in order to draw larger conclusions. Furthermore, clinical profiles including the co-morbidity, age, sampling location, gender etc. need to be further examined with sufficient coverage and sequencing depth in randomized controlled experiments. Overall, this research study provides a glimpse of the composite microbial diversity and signature microbiome profile among the different groups of COVID-19 patients.
5. Conclusions
With the emergence of the new SARS-CoV-2 variants, the global impact of the pandemics has certainly taken a toll on human health, disease susceptibility and recovery. The present research study examined the critical aspects of the nasopharyngeal microbiome of the SARS-CoV-2 negative, recovered and deceased COVID-19 patients from Gujarat, India and revealed the signature microbiome within the three different groups. Dysbiosis in nasopharyngeal microbiota, especially due to infection of opportunistic pathogens could lead to more complex and severe COVID-19 disease dynamics. Whereas, healthy commensal bacteria may trigger the immune response and alter the viral infection susceptibility and thus, may help in preventing the viral infection and possible recovery which is yet to be fully explored. The median age group for the deceased patients was higher compared to the control and recovered patient group, therefore dysbiosis in microbiota profile could vary due to the factors which are beyond the scope of this research study and should be considered for further investigation for clinical significance. However, our observations through a light from a lower number of samples under investigation. The findings in the present study provide key considerations and have significant implications for the COVID-19 treatment and control measures. However, further study is required to address the therapeutic aspects of the microbiome-based interventions.
Ethical approval
GBRC is an Indian Council of Medical Research (ICMR) approved COVID-19 testing and diagnosis research laboratory. Institutional Ethical Committee of Gujarat Biotechnology Research Centre (GBRC), Gandhinagar, B. J. Medical College and Civil hospital, Ahmedabad, reference No: EC/Approval/38/2020 and GMERS medical College Gandhinagar, reference No. GMERS/MCG/IEC/06/2020.
Funding
This research was funded by Department of Science and Technology (DST), Government of Gujarat, Gandhinagar, Gujarat, India.
CRediT authorship contribution statement
Dinesh Kumar: Visualization, Methodology, Formal analysis, Data curation. Ramesh Pandit: Writing – review & editing, Visualization, Software, Methodology, Formal analysis, Data curation. Sonal Sharma: Methodology, Data curation. Janvi Raval: Methodology, Data curation. Zarna Patel: Methodology, Data curation. Madhvi Joshi: Writing – review & editing, Supervision, Resources, Investigation, Funding acquisition, Conceptualization. Chaitanya G. Joshi: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Professor Chaitanya G Joshi reports financial support was provided by Government of Gujarat Department of Science and Technology.
Acknowledgments
Authors would like to acknowledge the Department of Science and Technology (DST), Government of Gujarat for the financial assistance and infrastructure support for the research work. Authors also would like to acknowledge and highly appreciate the support of the Commissionerate of Health, Government of Gujarat, Gujarat Medical Education and Research Society (GMERS) and associated medical colleges for providing the clinical samples.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.micpath.2022.105829.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Mahase E. Covid-19: is the UK heading for another omicron wave? Bmj. 2022:o738. doi: 10.1136/bmj.o738. [DOI] [PubMed] [Google Scholar]
- 2.Brandal L.T., MacDonald E., Veneti L., Ravlo T., Lange H., Naseer U., Feruglio S., Bragstad K., Hungnes O., Ødeskaug L.E. Others, outbreak caused by the SARS-CoV-2 omicron variant in Norway, November to December 2021. Euro Surveill. 2021;26 doi: 10.2807/1560-7917.ES.2021.26.50.2101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assessment R.R. 2021. Assessment of the Further Emergence and Potential Impact of the SARS-CoV-2 Omicron Variant of Concern in the Context of Ongoing Transmission of the Delta Variant of Concern in the EU/EEA, 18th Update. [Google Scholar]
- 4.Chaudhari A.M., Joshi M., Kumar D., Patel A., Lokhande K.B., Krishnan A., Hanack K., Filipek S., Liepmann D., Renugopalakrishnan V., Paulmurugan R., Joshi C. Evaluation of immune evasion in SARS-CoV-2 Delta and Omicron variants. Comput. Struct. Biotechnol. J. 2022;20:4501–4516. doi: 10.1016/j.csbj.2022.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu J., Liu S., Zhang Z., Lee X., Wu W., Huang Z., Lei Z., Xu W., Chen D., Wu X., Guo Y., Peng L., Lin B., Chong Y., Mou X., Shi M., Lan P., Chen T., Zhao W., Gao Z. Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synth. Syst. Biotechnol. 2021;6:135–143. doi: 10.1016/j.synbio.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mostafa H.H., Fissel J.A., Fanelli B., Bergman Y., Gniazdowski V., Dadlani M., Carroll K.C., Colwell R.R., Simner P.J. Metagenomic next-generation sequencing of nasopharyngeal specimens collected from confirmed and suspect covid-19 patients. mBio. 2020;11:1–13. doi: 10.1128/mBio.01969-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X., Liao B., Cheng L., Peng X., Xu X., Li Y., Hu T., Li J., Zhou X., Ren B. The microbial coinfection in COVID-19. Appl. Microbiol. Biotechnol. 2020;104:7777–7785. doi: 10.1007/s00253-020-10814-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh R., Kumari A. Nasal microbiota imbalance as a contributory Link in the emergence of COVID-19 associated mucormycosis. ACS Infect. Dis. 2021;7:2211–2213. doi: 10.1021/acsinfecdis.1c00371. [DOI] [PubMed] [Google Scholar]
- 9.Gu S., Chen Y., Wu Z., Chen Y., Gao H., Lv L., Guo F., Zhang X., Luo R., Huang C., Lu H., Zheng B., Zhang J., Yan R., Zhang H., Jiang H., Xu Q., Guo J., Gong Y., Tang L., Li L. Alterations of the gut microbiota in patients with coronavirus disease 2019 or H1N1 influenza. Clin. Infect. Dis. 2020;71:2669–2678. doi: 10.1093/cid/ciaa709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu R., Lu R., Zhang T., Wu Q., Cai W., Han X., Wan Z., Jin X., Zhang Z., Zhang C. Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults. Commun. Biol. 2021;4:1–11. doi: 10.1038/s42003-021-01796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rueca M., Fontana A., Bartolini B., Piselli P., Mazzarelli A., Copetti M., Binda E., Perri F., Gruber C.E.M., Nicastri E., Marchioni L., Ippolito G., Capobianchi M.R., Di Caro A., Pazienza V. Investigation of nasal/oropharyngeal microbial community of covid-19 patients by 16s rdna sequencing. Int. J. Environ. Res. Publ. Health. 2021;18:1–12. doi: 10.3390/ijerph18042174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhong H., Wang Y., Shi Z., Zhang L., Ren H., He W., Zhang Z., Zhu A., Zhao J., Xiao F., Yang F., Liang T., Ye F., Zhong B., Ruan S., Gan M., Zhu J., Li F., Li F., Wang D., Li J., Ren P., Zhu S., Yang H., Wang J., Kristiansen K., Tun H.M., Chen W., Zhong N., Xu X., Li Y.-M., Li J., Zhao J. Characterization of respiratory microbial dysbiosis in hospitalized COVID-19 patients. Cell Discov. 2021;7:23. doi: 10.1038/s41421-021-00257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu X., Wang L., Zheng X.M., Wen Y., Zhang Z., Fan L., Zhou Q., Yang X., Xue B., Lin Y. Moraxella occupied the largest proportion in the nasal microbiome in healthy children, which potential protect them from COVID-19. Microb. Pathog. 2022;170 doi: 10.1016/j.micpath.2022.105685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoque M.N., Akter S., Mishu I.D., Islam M.R., Rahman M.S., Akhter M., Islam I., Hasan M.M., Rahaman M.M., Sultana M., Islam T., Hossain M.A. Microbial co-infections in COVID-19: associated microbiota and underlying mechanisms of pathogenesis. Microb. Pathog. 2021;156 doi: 10.1016/j.micpath.2021.104941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giugliano R., Sellitto A., Ferravante C., Rocco T., D'Agostino Y., Alexandrova E., Lamberti J., Palumbo D., Galdiero M., Vaccaro E., Pagliano P., Weisz A., Giurato G., Franci G., Rizzo F. NGS analysis of nasopharyngeal microbiota in SARS-CoV-2 positive patients during the first year of the pandemic in the Campania Region of Italy. Microb. Pathog. 2022;165 doi: 10.1016/j.micpath.2022.105506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu H., Shi D., Guo J., Yi P., Liu J., Tao J., Zhang H. 2021. Month Follow- up of Gut Microbiota Richness in Patients with COVID-19; pp. 1–3. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esposito S., Principi N. Impact of nasopharyngeal microbiota on the development of respiratory tract diseases. Eur. J. Clin. Microbiol. Infect. Dis. 2018;37:1–7. doi: 10.1007/s10096-017-3076-7. [DOI] [PubMed] [Google Scholar]
- 18.Rosas-Salazar C., Kimura K.S., Shilts M.H., Strickland B.A., Freeman M.H., Wessinger B.C., Gupta V., Brown H.M., Rajagopala S.V., Turner J.H., Das S.R. SARS-CoV-2 infection and viral load are associated with the upper respiratory tract microbiome. J. Allergy Clin. Immunol. 2021;147:1226–1233. doi: 10.1016/j.jaci.2021.02.001. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finlay B.B., Amato K.R., Azad M., Blaser M.J., Bosch T.C.G., Chu H., Dominguez-Bello M.G., Ehrlich S.D., Elinav E., Geva-Zatorsky N., Gros P., Guillemin K., Keck F., Korem T., McFall-Ngai M.J., Melby M.K., Nichter M., Pettersson S., Poinar H., Rees T., Tropini C., Zhao L., Giles-Vernick T. vol. 118. 2021. (Erratum: the Hygiene Hypothesis, the COVID Pandemic, and Consequences for the Human Microbiome). Proceedings of the National Academy of Sciences of the United States of America. Proc. Natl. Acad. Sci. U. S. A. 118 (2021). https://doi.org/10.1073/pnas.2102333118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koskinen K., Reichert J.L., Hoier S., Schachenreiter J., Duller S., Moissl-Eichinger C., Schöpf V. The nasal microbiome mirrors and potentially shapes olfactory function. Sci. Rep. 2018;8:1–11. doi: 10.1038/s41598-018-19438-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C.M., Price L.B., Hungate B.A., Abraham A.G., Larsen L.A., Christensen K., Stegger M., Skov R., Andersen P.S. Staphylococcus aureus and the ecology of the nasal microbiome. Sci. Adv. 2015;1:1–8. doi: 10.1126/sciadv.1400216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soltani S., Zakeri A., Zandi M., Kesheh M.M., Tabibzadeh A., Dastranj M., Faramarzi S., Didehdar M., Hafezi H., Hosseini P., Farahani A. The role of bacterial and fungal human respiratory microbiota in COVID-19 patients. BioMed Res. Int. 2021 doi: 10.1155/2021/6670798. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fazlollahi M., Lee T.D., Andrade J., Oguntuyo K., Chun Y., Grishina G., Grishin A., Bunyavanich S. The nasal microbiome in asthma. J. Allergy Clin. Immunol. 2018;142:834–843. doi: 10.1016/j.jaci.2018.02.020. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer F., Paarmann D., D'Souza M., Olson R., Glass E.M., Kubal M., Paczian T., Rodriguez A., Stevens R., Wilke A., Wilkening J., Edwards R.A. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf. 2008;9:1–8. doi: 10.1186/1471-2105-9-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quast C., Pruesse E., Yilmaz P., Gerken J., Schweer T., Yarza P., Peplies J., Glöckner F.O. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parks D.H., Tyson G.W., Hugenholtz P., Beiko R.G. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhariwal, 2017, MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data, Academic.Oup.Com. (n.d.). https://academic.oup.com/nar/article-abstract/45/W1/W180/3760191 (accessed March 29, 2021). [DOI] [PMC free article] [PubMed]
- 29.Kolhe R., Sahajpal N.S., Vyavahare S., Dhanani A.S., Adusumilli S., Ananth S., Mondal A.K., Patterson G.T., Kumar S., Rojiani A.M., Isales C.M., Fulzele S. Alteration in nasopharyngeal microbiota profile in aged patients with covid-19. Diagnostics. 2021:11. doi: 10.3390/diagnostics11091622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jochems S.P., Ferreira D.M., Smits H.H. Microbiota and compartment matter in the COVID-19 response. Nat. Immunol. 2021;22:1350–1352. doi: 10.1038/s41590-021-01041-w. [DOI] [PubMed] [Google Scholar]
- 31.Ma S., Zhang F., Zhou F., Li H., Ge W., Gan R., Nie H., Li B., Wang Y., Wu M., Li D., Wang D., Wang Z., You Y., Huang Z. Metagenomic analysis reveals oropharyngeal microbiota alterations in patients with COVID-19. Signal Transduct. Targeted Ther. 2021;6 doi: 10.1038/s41392-021-00614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J., Liu S., Zhang Z., Lee X., Wu W., Huang Z., Lei Z., Xu W., Chen D., Wu X., Guo Y., Peng L., Lin B., Chong Y., Mou X., Shi M., Lan P., Chen T., Zhao W., Gao Z. Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synth. Syst. Biotechnol. 2021;6:135–143. doi: 10.1016/j.synbio.2021.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y., Cheng X., Jiang G., Tang H., Ming S., Tang L., Lu J., Guo C., Shan H., Huang X. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. Npj Biofilms Microbiomes. 2021;7 doi: 10.1038/s41522-021-00232-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta A., Karyakarte R., Joshi S., Das R., Jani K., Shouche Y., Sharma A. Nasopharyngeal microbiome reveals the prevalence of opportunistic pathogens in SARS-CoV-2 infected individuals and their association with host types. Microb. Infect. 2021 doi: 10.1016/j.micinf.2021.104880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Funke G., Lawson P.A., Bernard K.A., Collins M.D. Most Corynebacterium xerosis strains identified in the routine clinical laboratory correspond to Corynebacterium amycolatum. J. Clin. Microbiol. 1996;34:1124–1128. doi: 10.1128/jcm.34.5.1124-1128.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith K.F., Oram D.M. Corynebacteria (including diphtheria) Encycl. Microbiol. 2009:94–106. doi: 10.1016/B978-012373944-5.00216-9. [DOI] [Google Scholar]
- 37.Belmares J., Detterline S., Pak J.B., Parada J.P. Corynebacterium endocarditis species-specific risk factors and outcomes. BMC Infect. Dis. 2007;7:4. doi: 10.1186/1471-2334-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang Q., Feng Y., Feng P., Wang X., Zong Z. Nosocomial bloodstream infection and the emerging carbapenem-resistant pathogen Ralstonia insidiosa. BMC Infect. Dis. 2019;19:1–9. doi: 10.1186/s12879-019-3985-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer L.D., Green E.R., Sheldon J.R., Skaar E.P. Assessing acinetobacter baumannii virulence and persistence in a murine model of lung infection. Methods Mol. Biol. 2019;1946:289–305. doi: 10.1007/978-1-4939-9118-1_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.El Gharib K., Masri K., Daoud Z., Sfeir T. Community-acquired Acinetobacter calcoaceticus pneumonia in a patient with agammaglobulinaemia. New Microbes New Infect. 2021;41 doi: 10.1016/j.nmni.2021.100870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang Y.J. Nasopharyngeal microbiota: gatekeepers or fortune tellers of susceptibility to respiratory tract infections? Am. J. Respir. Crit. Care Med. 2017;196:1504–1505. doi: 10.1164/rccm.201707-1470ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salzano F.A., Marino L., Salzano G., Botta R.M., Cascone G., D'Agostino Fiorenza U., Selleri C., Casolaro V. Microbiota composition and the integration of exogenous and endogenous signals in reactive nasal inflammation. J. Immunol. Res. 2018 doi: 10.1155/2018/2724951. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedland R.P., Haribabu B. The role for the metagenome in the pathogenesis of COVID-19. EBioMedicine. 2020;61:61–62. doi: 10.1016/j.ebiom.2020.103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong H., Wang Y., Shi Z., Zhang L., Ren H., He W., Zhang Z., Zhu A., Zhao J., Xiao F., Yang F., Liang T., Ye F., Zhong B., Ruan S., Gan M., Zhu J., Li F., Li F., Wang D., Li J., Ren P., Zhu S., Yang H., Wang J., Kristiansen K., Tun H.M., Chen W., Zhong N., Xu X., min Li Y., Li J., Zhao J. Characterization of respiratory microbial dysbiosis in hospitalized COVID-19 patients. Cell Discov. 2021;7 doi: 10.1038/s41421-021-00257-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hatipoglu C.A., Bilgin G., Tulek N., Kosar U. Pulmonary involvement in brucellosis. J. Infect. 2005;51:116–119. doi: 10.1016/j.jinf.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Pappas G., Bosilkovski M., Akritidis N., Mastora M., Krteva L., Tsianos E. Brucellosis and the respiratory system. Clin. Infect. Dis. an Off. Publ. Infect. Dis. Soc. Am. 2003;37:e95–e99. doi: 10.1086/378125. [DOI] [PubMed] [Google Scholar]
- 47.Elzein F., Alsherbeeni N., Almatrafi K., Shosha D., Naoufel K. COVID-19 co-infection in a patient with brucella bacteremia. Respir. Med. Case Reports. 2020;31 doi: 10.1016/j.rmcr.2020.101183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen J., Liu X., Liu W., Yang C., Jia R., Ke Y., Guo J., Jia L., Wang C., Chen Y. 2022. Comparison of the Respiratory Tractmicrobiome in Hospitalized COVID-19 Patients with Different Disease Severity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kucuk G.O., Gorgun S. Brucellosis mimicking COVID-19: a point of view on differential diagnosis in patients with fever, Dry cough, arthralgia, and hepatosplenomegaly. Cureus. 2021;13 doi: 10.7759/cureus.15848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dimitri-Pinheiro S., Soares R., Barata P. The microbiome of the Nose-friend or foe? Allergy Rhinol. (Providence) 2020;11 doi: 10.1177/2152656720911605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shukla S.D., Budden K.F., Neal R., Hansbro P.M. Microbiome effects on immunity, health and disease in the lung. Clin. Transl. Immunol. 2017;6:e133. doi: 10.1038/cti.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.AbdelMassih A., Gadalla M., Hussein E., Elahmady M., Zahra N., Eid M.A., Hussein M., Hassan A.A., Abou-Zeid A.S., Hassan A., El Nahhas N., Emad N., Aboushadi N., Ibrahim N., Mokhtar S., El-Husseiny N., Kamel A., Hozaien R., Menshawey E., Ismail H.A., Mokhles M., Menshawey R., Fouda R. The forgotten oral microbial transplantation for improving the outcomes of COVID-19. New Microbes New Infect. 2021;43 doi: 10.1016/j.nmni.2021.100923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soffritti I., D'Accolti M., Fabbri C., Passaro A., Manfredini R., Zuliani G., Libanore M., Franchi M., Contini C., Caselli E. Oral microbiome dysbiosis is associated with symptoms severity and local immune/inflammatory response in COVID-19 patients: a cross-sectional study. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.687513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel J., Sampson V. The role of oral bacteria in COVID-19. The Lancet. Microbe. 2020;1:e105. doi: 10.1016/S2666-5247(20)30057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knapp S., Brodal C., Peterson J., Qi F., Kreth J., Merritt J. Natural competence is common among clinical isolates of Veillonella parvula and is useful for genetic manipulation of this key member of the oral microbiome. Front. Cell. Infect. Microbiol. 2017;7:139. doi: 10.3389/fcimb.2017.00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiong D., Muema C., Zhang X., Pan X., Xiong J., Yang H., Yu J., Wei H. Enriched opportunistic pathogens revealed by metagenomic sequencing hint potential Linkages between pharyngeal microbiota and COVID-19. Virol. Sin. 2021:1–10. doi: 10.1007/s12250-021-00391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ventero M.P., Cuadrat R.R.C., Vidal I., Andrade B.G.N., Molina-Pardines C., Haro-Moreno J.M., Coutinho F.H., Merino E., Regitano L.C.A., Silveira C.B., Afli H., López-Pérez M., Rodríguez J.C. Nasopharyngeal microbial communities of patients infected with SARS-CoV-2 that Developed COVID-19. Front. Microbiol. 2021;12 doi: 10.3389/fmicb.2021.637430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iebba V., Zanotta N., Campisciano G., Zerbato V., Di Bella S., Cason C., Luzzati R., Confalonieri M., Palamara A.T., Comar M. Profiling of oral microbiota and cytokines in COVID-19 patients. Front. Microbiol. 2021:1603. doi: 10.3389/fmicb.2021.671813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu G., Tang C.M., Exley R.M. Non-pathogenic Neisseria: members of an abundant, multi-habitat, diverse genus. Microbiology. 2015;161:1297–1312. doi: 10.1099/mic.0.000086. [DOI] [PubMed] [Google Scholar]
- 60.Weyand N.J. Neisseria models of infection and persistence in the upper respiratory tract. Pathog. Dis. 2017;75 doi: 10.1093/femspd/ftx031. [DOI] [PubMed] [Google Scholar]
- 61.Lanaspa M., Bassat Q., Medeiros M.M., Muñoz-Almagro C. Respiratory microbiota and lower respiratory tract disease. Expert Rev. Anti Infect. Ther. 2017;15:703–711. doi: 10.1080/14787210.2017.1349609. [DOI] [PubMed] [Google Scholar]
- 62.Koltai P.J., Maisel B.O., Goldstein J.C. Pseudomonas aeruginosa in chronic maxillary sinusitis. Laryngoscope. 1985;95:34–37. doi: 10.1288/00005537-198501000-00010. [DOI] [PubMed] [Google Scholar]
- 63.Fothergill J.L., Neill D.R., Loman N., Winstanley C., Kadioglu A. Pseudomonas aeruginosa adaptation in the nasopharyngeal reservoir leads to migration and persistence in the lungs. Nat. Commun. 2014;5:4780. doi: 10.1038/ncomms5780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.