

Abstract
Objectives
As a rare and heterogeneous disease, mixed connective tissue disease (MCTD) represents a challenge. Herein, we aimed to unravel potential pitfalls including correct referral diagnosis, distinction from other connective tissue diseases (CTD) and treatment modalities.
Methods
We characterised the MCTD cohort at our tertiary referral centre. All patients were evaluated for fulfilment of classification criteria of various CTDs. SLEDAI-2 K and EUSTAR-AI were used in accordance with previous research to evaluate disease activity and treatment response.
Results
Out of 85 patients initially referred as MCTD, only one-third (33/85, 39%) fulfilled the diagnostic MCTD criteria and the other patients had undifferentiated CTD (16/85, 19%), non-MCTD overlap syndromes (11/85, 13%) and other rheumatic diseases. In our final cohort of 33 MCTD patients, 16 (48%) also met the diagnostic criteria of systemic sclerosis, 13 (39%) these of systemic lupus erythematosus, 6 (18%) these of rheumatoid arthritis and 3 (9%) these of primary myositis. Management of MCTD required immunomodulating combination therapy in most cases (15/28, 54%), whereas monotherapy was less frequent (10/28, 36%), and only a few (3/28, 11%) remained without immune modulators until the end of the follow-up period. Treatment led to a significant decline in disease activity.
Conclusions
Our study showed a high risk for misdiagnosis for patients with MCTD. As a multi-organ disease, MCTD required prolonged immunomodulating therapy to achieve remission. The establishment of an international registry with longitudinal data from observational multi-centre cohorts might represent a first step to address the many unmet needs of MCTD.
|
Key Points • This cohort study aimed to identify challenges in the highly complex management of MCTD. • Clinical presentation of MCTD significantly overlaps with that of other CTDs, leading to a high risk of misdiagnosis. • Manifestations of MCTD are highly variable and potentially life-threatening, requiring continued immunomodulating treatment in most cases. • A composite score based on SLEDAI-2 K and EUSTAR-AI measures could represent an easy applicable tool to monitor disease activity and treatment response. |
Keywords: Diagnosis, Disease activity, Mixed connective tissue disease, Treatment
Introduction
Mixed connective tissue disease (MCTD) is a systemic, immune-mediated disorder exhibiting a broad spectrum of disease manifestations including symptoms of systemic sclerosis (SSc), systemic lupus erythematosus (SLE), primary myositis (PM) or rheumatoid arthritis (RA), which underline the importance of high titres of anti-U1-snRNP autoantibodies for diagnosis. MCTD was first described in 1972 by G. C. Sharp and colleagues [1]. Since then, there has been an ongoing debate about whether it represents a distinct disease entity [2, 3] or rather an early stage of another connective tissue disease (CTD) or an unspecific overlap syndrome [4]. Evidence for MCTD as a distinct disorder has mainly come from research into genetics and immunology: HLA-typing studies identified characteristic risk alleles for MCTD compared to other CTDs [5] or healthy controls [6]. Immunologic studies have elucidated the role of anti-U1-snRNP autoantibodies in the pathogenesis of MCTD [7, 8].
Challenges in dealing with MCTD arise for many reasons. Firstly, MCTD is the most rare of all CTDs with a prevalence of 3.8/100,000 adults in a Norwegian study [9]. Secondly, in the absence of specific guidelines for MCTD, treatment is based on personal expertise from treating similar symptoms in other CTDs [2]. Thirdly, four different diagnostic and classification criteria with varying defining features and sensitivities have been proposed [10–13]. This does not only cause diagnostic uncertainty in clinical routine, but has also led to heterogeneous study cohorts in previous MCTD research endeavours [14]. Finally, some patients even fulfil classification criteria of various other CTDs due to the significant overlap between the symptoms of MCTD and SSc, SLE, PM or RA [2].
In this study, in our tertiary referral centre, we aimed to unravel potential pitfalls of the overall management of MCTD, including correct referral diagnosis, fulfilment of diagnostic and/or classification criteria, distinction from other CTDs and disease course and activity as well as treatment modalities.
Materials and methods
Study cohort
This is a retrospective study performed at the Department of Rheumatology, University Hospital Zurich, a tertiary referral centre. We searched all electronic medical reports of our department from January 2015 to December 2018 for patients diagnosed with M35.1, the code assigned to MCTD in the 10th Revision of the International Classification of Diseases (ICD-10). The search yielded 85 patients. In addition, our local cohort of SSc patients, whose data is recorded in the European Scleroderma Trials and Research (EUSTAR) database [15], was evaluated for patients fulfilling MCTD criteria. At the time of the data export from the EUSTAR database (26 July 2019), the data set of our centre consisted of 495 patients, of which 15 patients were positive for anti-U1-snRNP. The inclusion criteria used for patient selection were age ≥ 18 years, fulfilment of diagnostic MCTD criteria (either Sharp’s [10], Kasukawa’s [11], Alarcón-Segovia’s [12] or Kahn’s criteria [13]) and anti-U1-snRNP antibody titre > 100 U/ml. All participants provided informed consent and the Zurich Ethics Committee approved the study (approval number: 2020–00387).
Diagnostic and classification criteria
The diagnostic MCTD criteria were applied in accordance with the original publications. However, we had to adjust for the definition of the autoantibody thresholds. Since anti-U1-snRNP is measured in units per millilitre (U/ml) at our department, we regarded a titre of > 100 U/ml as sufficient instead of the anti-RNP hemagglutination titres of > 1:1600 in Alarcón-Segovia’s criteria [12], > 1:2000 in Kahn’s criteria [13] and instead of the anti-ENA titre of > 1:10,000 in Sharp’s criteria [10]. The minimum anti-U1-snRNP threshold of > 100 U/ml was chosen since this reflects exceedingly high titres based on our internal reference values (upper limit of normal = 25 U/ml). The diagnostic criteria for definite MCTD as used in this study are displayed in Fig. 1. For classification, we used the SLICC criteria for SLE [16], the ACR/EULAR criteria for SSc [17], the ACR/EULAR criteria for PM [18] and the ACR/EULAR criteria for RA [19]. Fulfilment of all mentioned classification criteria was evaluated for each patient of our local MCTD cohort.
Fig. 1.

Diagnostic criteria for definite MCTD. The criteria have been slightly modified for use in this study as described in the text. Anti-Sm, anti-Smith; DLCO, diffusion capacity for carbon monoxide; EMG, electromyography; PH, pulmonary hypertension; RP, Raynaud’s phenomenon; VC, vital capacity
Clinical parameters
Patients were assessed at their first visit to our department after referral. Clinical data were retrospectively collected by manual review of the electronic patients’ records. The collected data comprise a detailed patient’s history, detailed evaluation of clinical features and additional diagnostic exams. Results of the following diagnostic procedures were recorded: routine blood and urine testing, auto-antibody screening, nailfold capillaroscopy, pulmonary function tests, high resolution computed tomography of the chest (HRCT), conventional chest radiography, electrocardiography, echocardiography, right heart catheter examination, electromyography (EMG), whole body magnetic resonance tomography (MRI) and muscle biopsy. Nailfold capillaroscopy was evaluated for the presence of microangiopathy according to the guidelines of Smith et al. [20]. Myositis was defined as muscle symptoms associated with elevated muscle enzymes or pathologic findings on MRI or EMG.
Immunomodulatory treatment and disease activity
We also collected longitudinal data to assess for disease course, immunomodulating treatment, treatment response and drug-related adverse events (AE). All documented visits to our department from the first visit after referral to either time of data collection or loss of follow-up were manually reviewed. Patients with an observation time < 1 year were excluded from this part of the analysis.
We relied on validated disease activity measures for the assessment of disease course and treatment response. As proposed by Reiseter et al. [14], we used SLEDAI-2 K as defined by Gladman et al. [21] and EUSTAR Activity Index as defined by Valentini et al. [20], which are validated for SLE and SSc, respectively, to measure disease activity in our MCTD cohort. Immunomodulating drugs encompassed corticosteroids, antimalarials, methotrexate, sulfasalazine, azathioprine, mycophenolate mofetil, cyclophosphamide, cyclosporine A, TNFα-antagonists, rituximab and tocilizumab.
Analysis of data
Absolute and relative frequencies were calculated for nominal variables; median and interquartile range was calculated for continuous variables. Disease activity was evaluated for the first and last visit at our department based on EUSTAR-AI [21] and SLEDAI-2 K [22]. Use of immunomodulating drugs and occurrence of AEs were continually recorded through the follow-up period. Data analysis was performed on Microsoft Excel and R 4.0 [23].
Results
Identification of MCTD patients
The selection process of the study cohort (n = 33) is shown in Fig. 2. The search for patients with the ICD-10 Code M35.1, which depicts MCTD, yielded 85 patients. On detailed review by two independent reviewers (AW, BM), 29/85 (34%) patients fulfilled at least one of the four MCTD criteria sets (Sharp’s [10], Kasukawa’s [11], Alarcón-Segovia’s [12] or Kahn’s [13] criteria), whereas 56/85 (66%) patients did not fulfil any MCTD criteria set. From the Zurich SSc cohort (n = 495) at the time of the data export (26 July 2019), 15 patients were positive for anti-U1-snRNP. On detailed review (AW, BM), 10/15 patients not only were anti-U1-snRNP positive, but also exhibited findings inconsistent with SSc diagnosis and instead rather fulfilled MCTD criteria. Thus, our final MCTD dataset yielded 33 MCTD patients and comprised patients identified by search for diagnostic code alone (n = 23), by search for autoantibody alone (n = 4) or by both methods (n = 6).
Fig. 2.

Search strategy for identifying MCTD patients in our department
Baseline characteristics
The patients’ demographic and clinical characteristics at first visit to our department are shown in Table 1. Most patients of our cohort were female with a median age of 53 years. Main clinical characteristics included Raynaud’s phenomenon (100%), puffy fingers (64%), scleroderma pattern in nailfold capillaroscopy (57%), oesophageal symptoms (55%), synovitis (55%), positive rheumatoid factor (45%), hypocomplementemia (42%), lung fibrosis on HRCT (39%), dyspnea (37%), sclerodactyly (36%), myositis (33%), muscle weakness (30%), CK elevation (30%) and joint contractures (29%). Median FVC was 92%; median DLCO was 67%.
Table 1.
Description of the study cohort at baseline (total n = 33)
| Item | Absolute freq. or median | Relative freq. or interquartile range |
|---|---|---|
| Age (years) | 53 | (38,59) |
| Female | 29/33 | (88) |
| Male | 4/33 | (12) |
| Raynaud’s phenomenon | 33/33 | (100) |
| Dyspnea | 11/30 | (37) |
| NYHA I | 19/30 | (63) |
| NYHA II | 7/30 | (23) |
| NYHA III | 4/30 | (13) |
| NYHA IV | 0/30 | (0) |
| Oesophageal symptoms | 18/33 | (55) |
| Stomach symptoms | 5/33 | (15) |
| Intestinal symptoms | 6/33 | (18) |
| Puffy fingers | 21/33 | (64) |
| Sclerodactyly | 12/33 | (36) |
| Pitting scars | 5/33 | (15) |
| Digital ulcers | 0/33 | (0) |
| History of digital ulcers | 2/33 | (6) |
| Gangrene | 0/33 | (0) |
| Mechanic’s hands | 0/33 | (0) |
| Modified Rodnan skin score | 0 | (0,4.25) |
| Synovitis1 | 18/33 | (55) |
| Joint contractures | 8/28 | (29) |
| Tendon friction rubs | 5/25 | (20) |
| Muscle weakness | 10/33 | (30) |
| Myositis | 11/33 | (33) |
| FVC (%) | 92 | (78,100) |
| TLC (%) | 89 | (78,100) |
| DLCO/SB (%) | 67 | (56,74) |
| Lung fibrosis on HRCT | 13/33 | (39) |
| Conduction blocks | 3/26 | (12) |
| LVEF (%) | 64 | (62,66) |
| Diastolic dysfunction | 1/32 | (3) |
| Pulmonary hypertension by Echo | 4/33 | (12) |
| Scleroderma pattern in NVC2 | 17/30 | (57) |
| ANA | 33/33 | (100) |
| ACA | 1/31 | (3) |
| Anti-Scl70 | 0/31 | (0) |
| Anti-U1-snRNP | 33/33 | (100) |
| Anti-RNA-polymerase III | 1/26 | (4) |
| Anti-PM-Scl | 1/22 | (5) |
| Anti-Jo-1 | 0/23 | (0) |
| Rheumatoid factor | 15/33 | (45) |
| ESR (mm/1 h) | 25 | (18,34) |
| CRP elevation | 4/32 | (13) |
| CK elevation | 10/33 | (30) |
| Proteinuria | 3/30 | (10) |
| Serum creatinine (mg/dL) | 0.73 | (0.64,0.80) |
| Hypocomplementemia | 14/33 | (42) |
The absolute and relative frequencies are shown for nominal variables: n/available (p%). For continuous variables, median and 1st and 3rd quartile are given: median (Q1, Q3). 1Synovitis was defined as swelling of a joint as judged by the treating physician. 2Scleroderma pattern as defined by Smith et al. (21). ACA, anti-centromere antibodies; ANA, antinuclear antibodies; CK, creatinine kinase; CRP, C reactive protein; DLCO/SB, single breath diffusion capacity of the lung for carbon monoxide; ESR, erythrocyte sedimentation rate; freq., frequency; FVC, forced vital capacity; HRCT, high-resolution computed tomography; LVEF, left ventricular ejection fraction; NVC, nailfold capillaroscopy; NYHA, New York Heart Association; TLC, total lung capacity
Diagnostic overlap between MCTD and other CTDs, performance of diagnostic criteria
Challenges arise from the fact that MCTD patients often also meet the classification criteria of other rheumatic diseases including SSc [17], SLE [16], PM [18] and RA [19]. Indeed, most patients (25/33, 76%) fulfilled the classification criteria of at least one of the mentioned diseases in addition to MCTD. The SSc (16/33 patients, 48%) and SLE criteria (13/33 patients, 39%) were the most frequently fulfilled in our MCTD cohort.
The use of four different diagnostic MCTD criteria may further contribute to some diagnostic uncertainty in clinical routine. Performance of MCTD criteria showed the highest sensitivities for Kasukawa’s (31/33, 94%) and Alarcón-Segovia’s criteria (30/33, 91%). Notably, most patients (32/33, 97%) fulfilled more than one set of MCTD criteria. Fulfilment of MCTD, SSc, SLE, PM and RA diagnostic and classification criteria is summarised in Table 2.
Table 2.
Patients fulfilling classification criteria of MCTD and other CTDs (total n = 33)
| Diagnostic/classification criteria | Frequency |
|---|---|
| Sharp MCTD criteria [10] | 25 (76%) |
| Kasukawa et al. MCTD criteria [11] | 31 (94%) |
| Alarcón-Segovia and Villareal MCTD criteria [12] | 30 (91%) |
| Kahn and Appelboom MCTD criteria [13] | 28 (85%) |
| ACR/EULAR systemic sclerosis criteria [17] | 16 (48%) |
| SLICC systemic lupus erythematosus criteria [16] | 13 (39%) |
| ACR/EULAR rheumatoid arthritis criteria [19] | 6 (18%) |
| ACR/EULAR primary myositis criteria [18] | 3 (9%) |
| Patients fulfilling > 1 MCTD criteria set | 32 (97%) |
| Patients fulfilling all 4 MCTD criteria sets | 20 (61%) |
| Patients fulfilling MCTD and ≥ 1 other CTD criteria | 25 (76%) |
| Patients fulfilling MCTD and ≥ 2 other CTD criteria | 11 (33%) |
The absolute and relative frequencies are shown: n (%)
High risk of misdiagnosis in clinical practice
The challenge of MCTD diagnosis was further reflected in the high percentage of patients (56/85, 66%) referred as MCTD patients, but not fulfilling any MCTD criteria set. The corrected diagnoses of these patients are shown in Table 3. Undifferentiated CTD (16/56, 29%; [24]), non-MCTD overlap syndromes (11/56, 20%; [26]) and SLE (6/56, 11%; [16]) were the most frequent diagnoses mistaken for MCTD. However, numerous other rheumatic diseases including Sjögren’s syndrome, SSc and RA had also been mistaken for MCTD.
Table 3.
Diagnoses of patients with ICD10 code M35.1 not fulfilling MCTD criteria (n = 56)
| Corrected diagnosis | Frequency |
|---|---|
| Undifferentiated connective tissue disease [24] | 16 (29%) |
| Non-MCTD overlap syndromes1 | 11 (20%) |
| Other2 | 10 (18%) |
| Systemic lupus erythematosus [16] | 6 (11%) |
| Primary Sjögren’s syndrome [25] | 5 (9%) |
| Unclassifiable connective tissue disease | 4 (7%) |
| Systemic sclerosis [17] | 2 (4%) |
| Rheumatoid arthritis [19] | 2 (4%) |
Absolute and relative frequencies are shown: n (%). 1Overlap syndromes include conditions fulfilling both serological and clinical criteria of two classic CTDs as described by Pepmueller (26). 2Other diagnoses include generalised morphea with concomitant polyarthritis, unclassified systemic inflammatory syndrome, systemic inflammation of unknown aetiology, autoimmune dacryoadenitis, chronic arthralgias with sicca syndrome, seronegative primary Sjögren’s syndrome, mechanically induced arthralgia of finger joints, somatoform pain disorder, human immunodeficiency virus infection and spondyloarthritis
Prescription of immunomodulators, disease course and adverse events
Observation time from the first visit after referral to the time of data collection or loss of follow-up was ≥ 1 year in 28/33 (85%) patients. Longitudinal data of this subcohort mirrors the management of MCTD at our department including use of immunomodulating drugs, treatment response and adverse events (AE). The median follow-up time was 7.6 (2.9, 10.3) years.
In the absence of treatment guidelines for MCTD, treatment decisions are mainly driven by the clinically dominant features and are thus often modelled to the existing recommendations for SSc [27, 28], SLE [29], PM [30] or RA [31]. We therefore examined the prescription patterns in our MCTD patients followed over ≥ 1 year (n = 28/33): Hydroxychloroquine, prednisone, methotrexate and rituximab were prescribed to 23/28 (82%), 22/28 (79%), 20/28 (71%) and 10/28 (36%) patients over the follow-up period. Mycophenolate mofetil, tocilizumab, azathioprine, leflunomide, cyclophosphamide, TNF-α-antagonists, sulfasalazine and cyclosporine A were less frequent treatment choices. The most frequent indications for treatment initiation were arthritis/arthralgia and interstitial lung disease.
Having evaluated the preferentially prescribed immunomodulators, we next examined whether these agents were used as monotherapy or in combination. Combination therapy was favoured over monotherapy in most patients with further increase over the follow-up period, probably reflecting the severity of the multi-organ disease. At the time of data collection or loss of follow-up, 15/28 (53%) patients were on combination therapy, 10/28 (36%) on monotherapy and just 3/28 (11%) off therapy as is shown in Fig. 3.
Fig. 3.

Prescription patterns and disease activity at first and last (time of data collection or loss of follow-up) visit to our department. Median, 1st and 3rd quartile of the disease activity measures SLEDAI-2 K [22] and EUSTAR-AI [21] are displayed as median (Q1, Q3). Act, activity; NI, no immunomodulators
In agreement with previous research [14], we used SLEDAI-2 K as defined by Gladman et al. [21] and EUSTAR Activity Index as defined by Valentini et al. [20], which are validated for SLE and SSc, respectively, to measure disease activity in our MCTD cohort in order to assess the disease course and treatment response in our MCTD patients. Median (Q1, Q3) SLEDAI-2 K significantly decreased from 6 (2, 8.25) at the first visit to our department to 0 (0, 4) at the time of data collection or loss of follow-up, whereas median (Q1, Q3) EUSTAR-AI hardly changed, being 1 (0, 1.8) at the first visit to our department and 1 (0, 1.0) at the time of data collection or loss of follow-up. The development of immunomodulator prescription patterns and disease activity is summarised in Fig. 3.
Finally, we assessed the frequency and nature of adverse events (AEs) in our cohort. Throughout the follow-up period, 17/28 (61%) patients experienced at least one AE, and 9/28 (32%) experienced more than one AE. There were AEs associated with methotrexate in 12/28 (43%) patients, with hydroxychloroquine in 7/28 (25%) patients, and with the concurrent use of several immune-modulating drugs in 4/28 (14%) patients. Cytopenia (8/28 patients, 29%), exanthema (6/28 patients, 21%), infections (4/28 patients, 14%) and elevated liver enzymes (4/28 patients, 14%) were the most frequently recorded AEs in our cohort.
Discussion
Our MCTD dataset comprised mainly females in their fifth or sixth decade with multi-organ involvement, particularly peripheral microangiopathy, puffy fingers, sclerodactyly, dysphagia, arthritis, interstitial lung disease and myositis (Fig. 4). A nationwide Norwegian cohort study [14] described similar frequencies of the mentioned disease manifestations, except in arthritis which occurred somewhat less frequently in our study (55% in our cohort, 76% in the Norwegian cohort).
Fig. 4.
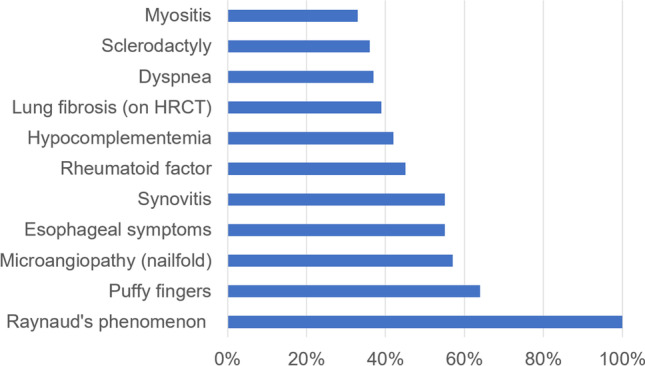
Most frequent clinical findings in our MCTD cohort (n = 33)
Since these features are not pathognomonic for MCTD, most of our patients (25/33, 76%) fulfilled not only the MCTD criteria, but also the classification criteria for SSc, SLE, RA or/and PM. This is consistent with the previously proposed view of MCTD as an overlap syndrome that is defined by its association with anti-U1-snRNP [26]. Previous studies have also described simultaneous fulfilment of MCTD and other CTD criteria [2, 32, 33].
The respective choice of the diagnostic MCTD criteria set might also influence the establishment of the diagnosis. In our cohort, 97% of the patients fulfilled several criteria sets. Sensitivity was highest for Kasukawa’s (94%; [11]), followed by 91% for Alarcón-Segovia’s [12], 85% for Kahn’s [13] and 76% for Sharp’s [10] criteria. Previous studies also reported significant differences in the performance of MCTD criteria [9, 34].
As a disease with a broad spectrum of clinical manifestations, significant overlap with other rheumatic diseases, heterogeneous use of diagnostic criteria and low disease prevalence, MCTD represents a significant diagnostic challenge even for rheumatologists. In our study, the majority (66%) of referred “MCTD” patients did not fulfil MCTD criteria but rather had other conditions including undifferentiated CTD as defined by Mosca et al. [24] and non-MCTD overlap syndromes (meeting clinical and serological criteria of more than one classic CTD [26]).
Clinical decision-making in the absence of official recommendations and randomised controlled trials might be difficult [2, 35]. Treatment guidelines for similar features in SLE [29], SSc [27, 28] or RA [31] as well as local drug availability and personal expertise might impact treatment choices in MCTD [2, 35]. In our cohort, the most frequently prescribed agents were hydroxychloroquine, prednisone, methotrexate and rituximab. The fact that continued combination therapy (15/28 patients, 54%) was more frequent than monotherapy (10/28 patients, 36%) and that only few patients (3/28 patients, 11%) remained untreated reflects the severity of this multi-organ disease and ties in with the persistent disease activity that was observed despite immunomodulatory treatment. This observation reflects a large variance in individual outcomes [2, 36–38], including potentially fatal complications such as pulmonary hypertension [37, 38], interstitial lung disease [39] or manifest anti-phospholipid syndrome [38], and contrasts with the initial concept of MCTD as a rather mild disease [1, 14]. Measurement of disease activity using SLEDAI-2 K and EUSTAR-AI further demonstrated a significantly better response to treatment of SLE-like than of SSc-like disease manifestations, which is consistent with previous data [14, 40]. Drug-related adverse events were common and included cytopenia, exanthema, infections and elevated liver enzymes.
The limitations of our study mainly arise from the rather low cohort size. On the other hand, the relatively small number of patients even at a tertiary referral centre also reflects the low disease prevalence and our strict inclusion criteria. The patients of our cohort, however, were well-characterised and prospectively followed with a representative median observation time and minimum observation time of 1 year.
Author contribution
AW made substantial contributions to design, acquisition of data, analysis and interpretation of data and participated in drafting the article. BM made substantial contributions to conception and design, acquisition of data, analysis and interpretation of data and participated in revising the article. SJ, OD and IG were involved in the analysis and interpretation of data and participated in revising the article.
Funding
Open access funding provided by University of Bern
Data availability
Data are available upon reasonable request by contacting the corresponding author.
Declarations
Ethics approval
Ethics approval was obtained from the Zurich Ethics Committee (BASEC-Nr. 2020–00387).
Consent to participate
Informed consent was obtained from all participants.
Conflict of interests
AW, IG and SJ have no competing interests to declare. OD had consultancy relationship and/or has received research funding from Abbvie, Actelion, Acceleron Pharma, Amgen, AnaMar, Baecon Discovery, Blade Therapeutics, Bayer, Boehringer Ingelheim, Catenion, Competitive Drug Development International Ltd., CSL Behring, ChemomAb, Curzion Pharmaceuticals, Ergonex, Galapagos NV, Glenmark Pharmaceuticals, GSK, Inventiva, Italfarmaco, iQone, iQvia, Lilly, medac, Medscape, Mitsubishi Tanabe Pharma, MSD, Novartis, Pfizer, Roche, Sanofi, Target Bio Science and UCB in the area of potential treatments of scleroderma and its complications. In addition, OD has a patent mir-29 for the treatment of systemic sclerosis issued (US8247389, EP2331143). BM had grant/research support from AbbVie, Protagen and Novartis Biomedical Research and a consultancy relationship with Boehringer-Ingelheim as well as congress support from Pfizer, Roche, Actelion, mepha and MSD. In addition, BM has a patent mir-29 for the treatment of systemic sclerosis issued (US8247389, EP2331143).
Footnotes
A previous abstract of this manuscript has been published in Supplement 1 of Annals of the Rheumatic Diseases, Volume 80, in June 2021. Wanzenried A, Garaiman A, Jordan S, Distler O, Maurer B (2021) Challenges in the management of mixed connective tissue disease: A retrospective analysis of the MCTD cohort in a tertiary referral centre. Ann Rheum Dis 80 (Suppl 1): 1233-1234.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease–an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA) Am J Med. 1972;52(2):148–159. doi: 10.1016/0002-9343(72)90064-2. [DOI] [PubMed] [Google Scholar]
- 2.Gunnarsson R, Hetlevik SO, Lilleby V, Molberg Ø. Mixed connective tissue disease. Best Pract Res Clin Rheumatol. 2016;30(1):95–111. doi: 10.1016/j.berh.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Aringer M, Steiner G, Smolen JS (2005) Does mixed connective tissue disease exist? Yes. Rheum Dis Clin North Am 31(3):411–20, v [DOI] [PubMed]
- 4.Swanton J, Isenberg D (2005) Mixed connective tissue disease: still crazy after all these years. Rheum Dis Clin North Am 31(3):421–36, v [DOI] [PubMed]
- 5.Flam ST, Gunnarsson R, Garen T, Lie BA, Molberg Ø, Group NMS The HLA profiles of mixed connective tissue disease differ distinctly from the profiles of clinically related connective tissue diseases. Rheumatology (Oxford) 2015;54(3):528–535. doi: 10.1093/rheumatology/keu310. [DOI] [PubMed] [Google Scholar]
- 6.Paradowska-Gorycka A, Stypinska B, Olesinska M, Felis-Giemza A, Manczak M, Czuszynska Z, et al. Association of HLA-DRB1 alleles with susceptibility to mixed connective tissue disease in Polish patients. HLA. 2016;87(1):13–18. doi: 10.1111/tan.12698. [DOI] [PubMed] [Google Scholar]
- 7.Greidinger EL, Hoffman RW (2005) Autoantibodies in the pathogenesis of mixed connective tissue disease. Rheum Dis Clin North Am 31(3):437–50, vi [DOI] [PubMed]
- 8.Hoffman RW, Maldonado ME. Immune pathogenesis of mixed connective tissue disease: a short analytical review. Clin Immunol. 2008;128(1):8–17. doi: 10.1016/j.clim.2008.03.461. [DOI] [PubMed] [Google Scholar]
- 9.Gunnarsson R, Molberg O, Gilboe IM, Gran JT, Group PS The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis. 2011;70(6):1047–1051. doi: 10.1136/ard.2010.143792. [DOI] [PubMed] [Google Scholar]
- 10.Sharp GC (1987) Diagnostic criteria for classification of MCTD. In: Kasukawa R, Sharp GC (eds) Mixed connective tissue disease and anti-nuclear antibodies. Elsevier Science Publishers B.V. (Biomedical Division), Amsterdam, pp 23–30
- 11.Kasukawa R, Tojo T, Miyawaki S, et al (1987) Preliminary diagnostic criteria for classification of mixed connective tissue disease. In: Kasukawa R, Sharp GC (eds) Mixed connective tissue disease and anti-nuclear antibodies. Elsevier Science Publishers B.V. (Biomedical Division), Amsterdam, pp 41–7
- 12.Alarcón-Segovia D, Villarreal M (1987) Classification and diagnostic criteria for mixed connective tissue disease. In: Kasukawa R, Sharp GC (eds) Mixed connective tissue disease and anti-nuclear antibodies. Elsevier Science Publishers B.V. (Biomedical Division), Amsterdam, pp 33–40
- 13.Kahn MF, Appelboom T, et al. Syndrome de Sharp et connectivites mixtes. In: Kahn MF, Peltier AP, Mayer O, et al., editors. Les maladies systémiques. Paris: Flammarion; 1991. pp. 545–556. [Google Scholar]
- 14.Reiseter S, Gunnarsson R, Corander J, Haydon J, Lund MB, Aaløkken TM, et al. Disease evolution in mixed connective tissue disease: results from a long-term nationwide prospective cohort study. Arthritis Res Ther. 2017;19(1):284. doi: 10.1186/s13075-017-1494-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meier FM, Frommer KW, Dinser R, Walker UA, Czirjak L, Denton CP, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis. 2012;71(8):1355–1360. doi: 10.1136/annrheumdis-2011-200742. [DOI] [PubMed] [Google Scholar]
- 16.Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. doi: 10.1002/art.34473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–1755. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 18.Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, de Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 2017;69(12):2271–2282. doi: 10.1002/art.40320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62(9):2569–2581. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 20.Smith V, Herrick AL, Ingegnoli F, Damjanov N, De Angelis R, Denton CP, et al. Standardisation of nailfold capillaroscopy for the assessment of patients with Raynaud’s phenomenon and systemic sclerosis. Autoimmun Rev. 2020;19(3):102458. doi: 10.1016/j.autrev.2020.102458. [DOI] [PubMed] [Google Scholar]
- 21.Valentini G, Iudici M, Walker UA, Jaeger VK, Baron M, Carreira P, et al. The European Scleroderma Trials and Research group (EUSTAR) task force for the development of revised activity criteria for systemic sclerosis: derivation and validation of a preliminarily revised EUSTAR activity index. Ann Rheum Dis. 2017;76(1):270–276. doi: 10.1136/annrheumdis-2016-209768. [DOI] [PubMed] [Google Scholar]
- 22.Gladman DD, Ibañez D, Urowitz MB. Systemic lupus erythematosus disease activity index 2000. J Rheumatol. 2002;29(2):288–291. [PubMed] [Google Scholar]
- 23.R Core Team (202) R: A language and environment for statistical computing. Available from: https://www.r-project.org/
- 24.Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17(5):615–620. [PubMed] [Google Scholar]
- 25.Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis. 2017;76(1):9–16. doi: 10.1136/annrheumdis-2016-210571. [DOI] [PubMed] [Google Scholar]
- 26.Pepmueller PH. Undifferentiated connective tissue disease, mixed connective tissue disease, and overlap syndromes in rheumatology. Mo Med. 2016;113(2):136–140. [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffmann-Vold A, Maher T, Philpot E, Ashrafzadeh A, Barake R, Barsotti S, et al. The identification and management of interstitial lung disease in systemic sclerosis: evidence-based European consensus statements. Lancet Rheumatol. 2020;2(2):71–83. doi: 10.1016/S2665-9913(19)30144-4. [DOI] [PubMed] [Google Scholar]
- 28.Kowal-Bielecka O, Landewé R, Avouac J, Chwiesko S, Miniati I, Czirjak L, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR) Ann Rheum Dis. 2009;68(5):620–628. doi: 10.1136/ard.2008.096677. [DOI] [PubMed] [Google Scholar]
- 29.Dörner T, Furie R. Novel paradigms in systemic lupus erythematosus. Lancet. 2019;393(10188):2344–2358. doi: 10.1016/S0140-6736(19)30546-X. [DOI] [PubMed] [Google Scholar]
- 30.Oddis CV, Aggarwal R. Treatment in myositis. Nat Rev Rheumatol. 2018;14(5):279–289. doi: 10.1038/nrrheum.2018.42. [DOI] [PubMed] [Google Scholar]
- 31.Burmester GR, Pope JE. Novel treatment strategies in rheumatoid arthritis. Lancet. 2017;389(10086):2338–2348. doi: 10.1016/S0140-6736(17)31491-5. [DOI] [PubMed] [Google Scholar]
- 32.Gendi NS, Welsh KI, Van Venrooij WJ, Vancheeswaran R, Gilroy J, Black CM. HLA type as a predictor of mixed connective tissue disease differentiation. Ten-year clinical and immunogenetic followup of 46 patients. Arthritis Rheum. 1995;38(2):259–66. doi: 10.1002/art.1780380216. [DOI] [PubMed] [Google Scholar]
- 33.van den Hoogen FH, Spronk PE, Boerbooms AM, Bootsma H, de Rooij DJ, Kallenberg CG, et al. Long-term follow-up of 46 patients with anti-(U1)snRNP antibodies. Br J Rheumatol. 1994;33(12):1117–1120. doi: 10.1093/rheumatology/33.12.1117. [DOI] [PubMed] [Google Scholar]
- 34.John KJ, Sadiq M, George T, Gunasekaran K, Francis N, Rajadurai E, et al. Clinical and immunological profile of mixed connective tissue disease and a comparison of four diagnostic criteria. Int J Rheumatol. 2020;2020:9692030. doi: 10.1155/2020/9692030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sapkota B, Al Khalili Y (2020) Mixed connective tissue disease treasure island: StatPearls Publishing; [https://www.ncbi.nlm.nih.gov/books/NBK542198/] [PubMed]
- 36.Ciang NC, Pereira N, Isenberg DA. Mixed connective tissue disease-enigma variations? Rheumatology (Oxford) 2017;56(3):326–333. doi: 10.1093/rheumatology/kew265. [DOI] [PubMed] [Google Scholar]
- 37.Hoffman RW, Greidinger EL. Mixed connective tissue disease. Curr Opin Rheumatol. 2000;12(5):386–390. doi: 10.1097/00002281-200009000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Burdt MA, Hoffman RW, Deutscher SL, Wang GS, Johnson JC, Sharp GC. Long-term outcome in mixed connective tissue disease: longitudinal clinical and serologic findings. Arthritis Rheum. 1999;42(5):899–909. doi: 10.1002/1529-0131(199905)42:5<899::AID-ANR8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 39.Gunnarsson R, Aaløkken TM, Molberg Ø, Lund MB, Mynarek GK, Lexberg AS, et al. Prevalence and severity of interstitial lung disease in mixed connective tissue disease: a nationwide, cross-sectional study. Ann Rheum Dis. 2012;71(12):1966–1972. doi: 10.1136/annrheumdis-2011-201253. [DOI] [PubMed] [Google Scholar]
- 40.Nimelstein SH, Brody S, McShane D, Holman HR. Mixed connective tissue disease: a subsequent evaluation of the original 25 patients. Med (Baltimore) 1980;59(4):239–248. doi: 10.1097/00005792-198007000-00001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon reasonable request by contacting the corresponding author.