

Abstract
New resveratrol–thiophene and resveratrol–maltol hybrids were synthesized as cholinesterase inhibitors and antioxidants. As with photostability experiments, biological tests also found remarkable differences in the properties and behavior of thiophene and maltol hybrids. While resveratrol–thiophene hybrids have excellent inhibitory and antioxidant properties (similar to the activity of reference drug galantamine), maltols have been proven to be weaker inhibitors and antioxidants. The molecular docking of selected active ligands gave insight into the structures of docked enzymes. It enabled the identification of interactions between the ligand and the active site of both cholinesterases. The maltols that proved to be active cholinesterase inhibitors were able to coordinate Fe3+ ion, forming complexes of 1:1 composition. Their formation constants, determined by spectrophotometry, are very similar, lgK = 11.6–12.6, suggesting that Fe3+ binds to the common hydroxy-pyranone moiety and is hardly affected by the other aromatic part of the ligand. Accordingly, the characteristic bands in their individual absorption spectra are uniformly red-shifted relative to those of the free ligands. The crystal structures of two new resveratrol–maltol hybrids were recorded, giving additional information on the molecules’ intermolecular hydrogen bonds and packing. In this way, several functionalities of these new resveratrol hybrids were examined as a necessary approach to finding more effective drugs for complicated neurodegenerative diseases.
Keywords: cholinesterase inhibitory activity, molecular docking, pyran-4-ones, thienostilbenes
1. Introduction
Resveratrol (3,4′,5-trihydroxystilbene (I), Figure 1 is a bioactive non-flavonoid polyphenol belonging to the group of trans-stilbene derivatives [1,2,3]. Resveratrol exists in the geometric isomeric forms as cis- and trans-isomers. The trans-isomer is more stable and is associated with biological properties and health benefits. It plays an important role in neurodegenerative diseases such as Alzheimer’s (AD) and Parkinson’s disease [4]. It is proven that AD is a multifactorial disease involving the aggregation of β-amyloid (Aβ) [5,6], oxidative stress [7], and metal ion dyshomeostasis [8,9].
Figure 1.

Resveratrol (I) and thienostilbenes (II and III) with improved antioxidant activity.
There is limited evidence in the literature of UV light interactions with trans-resveratrol [10,11]. Unfortunately, the trans-resveratrol isomerizes to the cis-isomer upon light or UV irradiation [12]. cis-Resveratrol is less stable and cannot be utilized commercially [13]. For this reason, there is a constant search for new heterocyclic resveratrol analogues that possess the same or even better bioactive properties as trans-resveratrol but are less sensitive to light. Oxidative stress participates in the pathology of many neurodegenerative diseases [14,15,16] and also represents an important aspect of research. In patients with AD, the brain tissues are exposed to oxidative stress, which leads to protein damage, DNA damage, and lipid oxidation, thus resulting in the growth of AD [17].
Acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are two related enzymes which are in humans, the products of different genes but share about 54% of their amino acid sequence [18]. AChE has an essential physiological role in the body as it controls the transmission of nerve impulses in the cholinergic synapses of the central and peripheral nervous system through the hydrolysis of the neurotransmitter acetylcholine. It also has a role in neuritogenesis, cell adhesion, dopamine neuronal activation, and the formation of amyloid fibers characteristic for AD [19]. The role of BChE is not physiologically essential but it could be assigned to the detoxification of xenobiotics and the bioactivation of drugs [20]. Additionally, BChE serves as a co-regulator of cholinergic neurotransmission and is capable of catalyzing the hydrolysis of acetylcholine [21]. It was found that high BChE levels are associated with neuritic plaques and neurofibrillary tangles, the neuropathologic hallmarks of AD [22]. Therefore, both cholinesterases are pharmacologically relevant targets in neurodegenerative disorders, and today’s treatment includes cholinesterase inhibitors such as donepezil, galantamine, physostigmine, rivastigmine, etc. [23].
Hence, developing cholinesterase inhibitors with antioxidant capabilities may benefit the neurotransmitter action in AD patients [24,25]. Besides the inhibitory activity, the antioxidative activity has an additional positive pleiotropic effect in increasing both the level and duration of neurotransmitter activity. For the above reason, it is worth highlighting the results of some of our previous research [26]. Heteroaromatic resveratrol analogs with a hydroxyl group(s) (compounds II and III, Figure 1) exhibited effective radical scavenging activity. The half-maximal DPPH radical scavenging concentration (IC50) for derivative II, with OH group at the ortho position and trans-geometry of the stilbene moiety, is 158.8 μM, implying that II is a potent antioxidant. For comparison, the IC50 value for resveratrol was found to be 77.9 μM [27] and 74.0 μM [28].
The configurational change to cis-isomer resulted in a decrease in antioxidant activity, but still, the IC50 value is reached in the micromolar range (IC50 782.1 μM). The same configurational effect was also observed for cis- and trans-resveratrol [29,30], indicating that the structure–activity relationship must be considered in the determination of biological activity. The introduction of an additional OH group at position 4 on the phenyl moiety increased antioxidant activity. Derivative III showed the best scavenging activity with an excellent IC50 value of 26.8 μM, three times better than resveratrol. In derivative III, both OH groups can be involved in resonance stabilization, resulting in better radical scavenging activity. Considering these results, compounds II and III may be good candidates as cholinesterase inhibitors with antioxidant capabilities.
To enhance the activity of resveratrol in the case of neurodegenerative diseases, the maltol moiety (3-hydroxypyran-4-one) was incorporated into the resveratrol-like structure to achieve resveratrol–maltol hybrids as novel multi-target-directed ligands (MTDL, structures A and B, Figure 2) and then compared with the same functionalities of thienostilbenes II and III as resveratrol derivatives (Figure 1). Among all the factors related to AD, the accumulation and aggregation of β-amyloid peptide (Aβ), the associated biometal ion dyshomeostasis, and oxidative stress have been demonstrated to be involved with the pathogenesis of AD [31,32]. There are two principal forms of Aβ in the brain tissue of AD patients, one of those responsible for the initial self-aggregation of Aβ, and the resulting β-amyloid oligomers and fibrils are toxic to neurons. High concentrations of iron ions also have been found to co-localize with amyloid deposits in AD patients compared to age-matched controls. In addition, the redox active iron ions can generate reactive oxygen species (ROS), which will cause oxidative damage and may trigger the death of neurons. The combination of metal chelation and anti-oxidant activity into one molecule may provide a promising therapeutic strategy for AD therapy. The in vitro biological evaluation of this type of compound (resveratrol–maltol hybrids) has shown their triple function: the inhibition of self-induced Aβ aggregation, antioxidation, and metal chelating activity [32]. Encouraged by this scientific knowledge, and having in mind our experience in the synthesis of (hetero)stilbene derivatives as cholinesterase inhibitors, we decided to prepare new resveratrol–maltol hybrid types combining the maltol moiety with (hetero)aryl one (structures C–E, Figure 2).
Figure 2.

Representative resveratrol–maltol target structures.
As photostability is one of the target characteristics of such resveratrol derivatives, the aim of this study was to investigate changes in UV spectra of compounds when exposed to UV light, their antioxidant and inhibitory activity against cholinesterases, as well as the capability to complex with biometals. In this way, several functionalities of these new resveratrol–maltol hybrids, as well as resveratrol–thiophene hybrids with previously improved antioxidative activity, were examined as a necessary approach to finding more effective drugs for complicated neurodegenerative diseases. As shown in our previous studies, sulfur analogs were found to be active in cholinesterase inhibition [33,34] and antioxidant activity [26]; therefore, some diheterostilbenes (maltol-heteroaromatic resveratrol analogs) are prepared in this work too (structures D and E, Figure 2).
2. Results and Discussion
2.1. Synthesis and Characterization of Resveratrol–Thiophene II and III and Resveratrol–Maltol Hybrids 1–8
The synthesis of the resveratrol–thiophene hybrids II and III (Figure 1) is presented in our previous research [26], together with their spectroscopic characterization and photostability. New 5-hydroxy-2-styryl-4H-pyran-4-ones 1–8 (Figure 2 and Scheme 1) were also prepared by the Wittig reaction from phosphonium chloride and the selected aryl- and heteroaryl-carbaldehydes. Wittig reaction provided the styryl-pyranones 1–8 as mixtures of cis- and trans-isomers (except 1 and 5, where only one isomer was formed; Scheme 1). Mixtures of the geometrical isomers of 1–8 were separated to obtain pure compounds by repeated column and thin-layer chromatography. According to 1H NMR spectroscopy, the substituent on the aryl ring strongly directs the ratio of geometric isomers in the Wittig reaction (Scheme 1). In the styryl-pyranone 1 with an ortho-OH substituent on the aromatic ring, trans-isomer predominates; however, in compound 5, corresponding cis-configuration is the only product in the reaction mixture (See also Experimental Section).
Scheme 1.

Synthesis of new 5-hydroxy-2-styryl-4H-pyran-4-ones 1–8 as chromatographically separated cis− and trans−isomers.
All isolated isomers of styryl-pyranones 1–8 are fully spectroscopically characterized (See Experimental Section and ESI). In the 1H NMR spectra of isolated geometrical isomers of 1–8 (Experimental Section and ESI), visible are the resolved patterns for ethylenic protons between 4.73 and 7.14 ppm (cis-isomers) and between 6.50 and 7.78 ppm (trans-isomers) with the characteristic coupling constants, signals for the protons on various substituents and the characteristic signals and coupling constants of protons on the thienyl and quinoxaline rings.
Styryl-pyranones 2 and 6 were additionally exposed to temperature changes from 278 to 328 K. At each temperature, the corresponding 1H NMR spectrum was recorded (Figure 3 and ESI) to examine possible changes in conformations due to the breaking of hydrogen bonds in the molecules. Only one proton of the double bond in cis-2, located at a lower chemical shift (at 6.30 ppm), moves towards a more shielded area with the temperature increase (Figure 3). There is also a noticeable shift of both protons (at 6.25 and 7.55 ppm) of the 4-pyranone ring towards the more shielded area and the hydroxyl proton due to breaking hydrogen bonds and conformational changes.
Figure 3.

Temperature dependence of the chemical shifts of characteristic protons in the NMR experiments for cis-2.
In the case of trans-2 (Figure 4), a similar trend is visible for the same group of protons. Besides that, the transition of the signals from two aromatic doublets (at 7.72 and 7.61 ppm) to one doublet at 7.61 ppm, due to the increase in temperature, is interesting. It can be explained as a consequence of the increase in kinetic energy and rotation of the single bonds, so two aromatic protons become chemically equivalent.
Figure 4.

Temperature dependence of the chemical shifts of characteristic protons in the NMR experiments for trans-2.
This kind of examination of the change in chemical shifts under the temperature influence clearly shows that there are intermolecular hydrogen bonds between the molecules of styrylpyranones in the solutions, which break and the molecules become more flexible, taking some other conformations, which is reflected in the chemical shifts changes of individual protons that are closest to the particular changes.
2.2. Photochemical Reactivity and Photophysical Properties of New Styryl-Pyranones 1–8
To explore the photochemical reactivity of the new resveratrol–maltol hybrids 1–8, we conducted irradiations at 313 nm of representative compounds 2 and 6 in ACN, and the course of the reaction was followed by UV-Vis spectroscopy (Figure 5 and Figure 6). Upon irradiation of the cis-isomer of compound 2, the bathochromic shift was observed, without a clear isosbestic point in the spectrum (Figure 5), in the beginning, in agreement with the formation of the trans-isomer. Contrary to resveratrol–thiophene hybrids II and III [26], in the continuation of irradiation, the further bathochromic shift is noticed toward values of absorbance wavelengths higher than those of the trans-isomer, which can be attributed to secondary processes such as ring closure or phototransposition [35]. The mechanism behind these changes could be the subject of some future mechanistic computational study. It is evident that the photostability of resveratrol–maltol hybrids significantly differs from those of resveratrol–thiophene hybrids [26]. Further, the trans-2 isomer upon irradiation shows the unusual isosbestic point at 235 nm and bathochromic shift besides the hypochromic one, with the same explanation of further secondary processes, such as ring closure or phototransposition (Figure 5 and Figure 6). So, for cis-2, Z–E photoisomerization is the primary photochemical reaction, but not for trans-2.
Figure 5.

Spectral changes during the irradiation of cis-2 (A) in ACN after 0 (red), 2, 4, 8, 16, 32, 64, 128, 256, 512, 1024, 2048 and 4096 s; (λir = 313 nm, l = 1 cm) and trans-2 (B) in ACN after 0 (red), 2, 4, 8, 16, 32, 64, 128, 256, 512, 1024 and 2048 s; (λir = 313 nm, l = 1 cm).
Figure 6.

Spectral changes during the irradiation of cis-6 (A) in ACN after 0 (red), 2, 4, 8, 16, 32, 64, 128, 256, 512, 1024, 2048 and 4096 s; (λir = 313 nm, l = 1 cm) and trans-6 (B) in ACN after 0 (red), 2, 4, 8, 16, 32, 64, 128, 256, 512, 1024 and 2048 s; (λir = 313 nm, l = 1 cm).
In the case of compound 6 (Figure 6), irradiating the cis-isomer at 313 nm again resulted in the bathochromic shift to higher wavelengths than those observed for the trans-isomer, but followed by the hyperchromic one as the result of the primary Z–E photoisomerization, and secondary ring closure or phototransposition process. Upon the irradiation of trans-6, the trends are very similar to those of cis-6, only without the primary E–Z photoisomerization process and clear isosbestic point compared with trans-2.
UV-Vis spectra of cis- and trans-isomers of 1–8 in ACN (Figure 7 and Experimental section) are typical for diarylethene [36,37] with the strong absorption with a maximum at about 300 nm, respectively, for the trans-isomers, and less intense and hypsochromically shifted bands for the cis-isomers (Figure 7 and Experimental section). These absorption bands correspond to the fully symmetry allowed HOMO → LUMO transition.
Figure 7.

UV spectra (ACN) of representative trans− (A) and cis−isomers (B) of new resveratrol–maltol hybrids.
2.3. Cholinesterases Inhibitory and Antioxidative Activity of Resveratrol–Maltol and Resveratrol–Thiophene Hybrids
In continuation of the photostability testing and evidence regarding the difference between the resveratrol–maltol and resveratrol–thiophene hybrids in this sense, we tested and compared all of them (II, cis-II, III, and 1–8) as reversible inhibitors of acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) by Ellman et al. [38] modified method in a wide range of concentration to evaluate the inhibitor concentration that inhibits 50% of enzyme activity (IC50). In addition, antioxidant activity tests of the compounds have been performed using DPPH radical scavenging assay [39] to see whether they can possess both inhibitory and antioxidant activity. The half-maximal DPPH radical scavenging concentration (IC50) was calculated where applicable, and the results for all active target compounds are summarized in Table 1. Of the 12 tested resveratrol–maltol hybrids, only four showed inhibitory potency, while the rest did not show any inhibitory or antioxidation activity.
Table 1.
ChE inhibitory effects and antioxidant activities for resveratrol–thiophene and the active resveratrol–maltol hybrids.
| Resveratrol-Thiophene/ Resveratrol–Maltol Hybrids |
AChE IC50 (μM) |
BChE IC50 (μM) |
DPPH IC50 (μM) |
|---|---|---|---|
| II | 15.7 ± 1.0 | 4.6 ± 0.1 | 158.8 * |
| cis-II | ~500 | 18.9 ± 3.4 | 782.1 * |
| III | 46.6 ± 8.3 | 5.3 ± 1.8 | 26.8 * |
| trans-1 | - | 600.7 ± 34.5 | 251.3 ± 45.6 |
| cis-2 | - | 458.1 ± 30.2 | - |
| cis-4 | 1003.0 ± 10.1 | 638.1 ± 25.8 | - |
| trans-6 | - | 426.3 ± 41.8 | - |
| Galantamine | 0.15 | 7.9 |
* [26].
As with photochemical experiments, biological tests also found remarkable differences in the properties and behavior of thiophene and maltol hybrids. While resveratrol–thiophene hybrids have excellent inhibitory and antioxidant properties, maltols have been proven to be weak inhibitors and even weaker antioxidants.
The results of biological tests for resveratrol–thiophene derivatives showed the great potential of these compounds. The antioxidant effect was previously tested and compared with resveratrol [26] which showed IC50 values of 77.9 μM [27] and 74.0 μM [28]. These derivatives showed antioxidant activity, with III being the most potent antioxidant (IC50 = 26.8 μM) due to the presence of two OH groups. It has been revealed now that in addition to their antioxidant activity, they also have excellent inhibitory activity on both enzymes. Among the thiophene hybrids, II showed the highest inhibitory activity against BChE (IC50 = 4.6 μM), which is similar to the activity of reference drug galantamine used as a therapeutic drug for AD (IC50 = 7.9 μM). Derivative III also showed great inhibitory activity toward BChE (IC50 = 5.3 μM), which is still slightly better than galantamine. These derivatives performed as better potential inhibitors of BChE than AChE, where they achieved somewhat lower IC50 values but still in a good range of concentrations (IC50 = 15.7 and 46.6 μM, respectively). The combination of thiophene and phenol in trans geometry has proven to be convenient for biological properties. The earlier observation that the trans geometry is more favorable for bioactivity [29] was confirmed by examining the cis-isomer of compound II. A change in geometry to cis-II drastically decreased inhibitory activities toward AChE (IC50 > 500 μM), while the result for BChE was still in a very good range (IC50 = 18.9 μM). The configurational change was also followed by a reduction in antioxidant potency.
Of the 12 tested resveratrol–maltol hybrids, only 4 shoμwed inhibitory potency, while the rest did not show any inhibitory or antioxidation activity. These four derivatives are active only toward the BChE enzyme, with IC50 values at relatively high concentrations compared to the standard (Table 1). Antioxidant activity was recorded only for the trans-1 derivative possessing a phenolic ring, while none of the other derivatives captured the radical (IC50 >> 1000 μM). Therefore, the insertion of maltol into the resveratrol structure drastically reduces inhibition potency and antioxidant activity.
Furthermore, we can compare the structure and bioactivity of the three hybrids that possess a thiophene ring with stilbene conjunction at position 2 (II, III, and 7). While II and III showed great results, hybrid 7 completely missed any tested bioactivity. Therefore, replacing phenol with maltol completely extinguishes the biological activity of interest in this research. It is important to note that the 3-thienyl-maltol combination in the trans geometry showed some inhibitory activity toward BChE, so the position of the thiophene substitution was also crucial. Concerning these experimental results, for the resveratrol–maltol hybrids 1–8, another type of biological activity should be proposed, which will be more specific to them. Although it has been shown in previous research that resveratrol–maltol hybrids show inhibition of self-induced Aβ aggregation (which is connected with AD), antioxidation, and metal chelating activity [32], the cholinesterase inhibitory activity is not as specific for them as for the resveratrol–thiophene hybrids II and III.
2.4. Computational Study of Resveratrol–Thiophene and Resveratrol–Maltol Hybrids as Cholinesterase Inhibitors
According to Table 1, both trans-isomers of resveratrol–thiophene derivatives, II and III, showed excellent inhibitory activity toward cholinesterases. Molecular docking was performed to visualize the structure and gain insight into stabilizing interactions in the complex between the tested molecule and the enzyme’s active site. The molecular structures of the most stable complexes of II and III with the active site of AChE, respectively, are shown in Figure 8.
Figure 8.

The structure of the active site of AChE docked with II (a) and III (b). Hydrogens omitted for the sake of clarity.
Molecules II and III possess two aromatic cores, phenyl and thienyl, that can participate in stabilizing π−π stacking with aromatic residues of the AChE active site. According to the clustering histogram, ligand poses obtained by docking of II are grouped into two conformational clusters: one contains 8, and the second is populated with 17 poses (See ESI, Table S1). The structure presented in Figure 8a is the most favorably oriented binding conformation from the more populated cluster. The orientation of II enables T-shaped π−π stacking between thienyl and two residues of the anionic site (AS), Trp84 and Phe330, at distances of 4.7 and 4.9 Å from thienyl (measured from the centers of the rings). The phenyl unit of II, situated close to the acyl pocket of the active site, is engaged in π−π stacking with Phe288 at a distance of 4.1 Å.
The docking of III also results in two clusters of ligand poses; however, the more stable one is predominantly populated (24 poses), while the second cluster contains only one ligand pose. In contrast to the orientation of II, molecule III is docked so that its phenyl core faces the AS, and thienyl is engaged in stacking with residues belonging to the acyl pocket, Phe288 and Phe290, at distances of 3.8 and 5.9 Å, respectively (Figure 8b). Additional π−π stacking between thienyl of II and His440 (5.1 Å) and phenyl of III and His440 (4.5 Å) is observed. Resveratrol–thiophene derivatives have hydroxyl group(s) at phenyl, able to form hydrogen bonds (HB) within the active site. Compound II possesses one -OH group at phenyl; it forms HB with a carbonyl group of Glu327 that belongs to the esteratic subsite (Ser200, His440, Glu327). In the active site of AChE docked with III, one hydroxyl group of III makes the hydrogen bond with Glu199 (part of the anionic site), while the second -OH is approaching the Ser200, at the distance of 1.9 Å from serine oxygen.
The molecular docking of II and III into the active site of BChE again revealed the presence of π−π interactions between ligand and aromatic residues of the enzyme. It is visible from Figure 9 that thienyl in both compounds interacts with Phe329 of the anionic site, although the orientation of the two ligands is not the same. In the structure docked with III, thienyl is additionally stabilized by the π−π stacking with Tyr332 (belonging to the peripheric anionic site, PAS). In contrast, II entered the active site deeper than III; thus, the interaction with PAS is not established in the case of II.
Figure 9.

The structure of the active site of BChE docked with II (a) and III (b). Hydrogens omitted for clarity.
The phenyl core of both ligands is engaged in the π−π stacking with His438 (esteratic subsite) and Trp82 of AS. One hydrogen bond is formed between the -OH group and Glu197 in the active site of BChE docked with II. Although molecule III has two hydroxyl groups, the most favorable conformation obtained by docking results in only one H-bond; again, this is with Glu327.
Among resveratrol maltol hybrids, the only compound that shows inhibitory activity toward both cholinesterases is molecule cis-4 (Table 1). The most stable complexes obtained by docking cis-4 into the active site of AChE and BChE, respectively, are presented in Figure 10. In the complex of cis-4 docked into AChE, the π−π stacking of dichloro-substituted phenyl with residues in the anionic site, Phe330 and Trp84, occurs. In the BChE, dichloro-phenyl of the ligand interacts only with the Phe329 of the AS but also with the residue in the PAS, Tyr332. The maltol fragment is involved in π−π stacking with the residues of the acyl pocket of the AChE (Phe288, Phe190), whereas in the BChE, there is an interaction between maltol and Trp82. In AChE, histidine in the esteratic subsite (His440) interacts with the dichloro-phenyl fragment of the cis-4. In contrast, His438 from the esteratic site of BChE is situated at 4.9 Å from the maltol fragment of the ligand.
Figure 10.

The complex obtained by docking cis-4 into the active site of AChE (A), and by docking cis-4 into the active site of BChE (B). Hydrogens omitted for clarity.
The hydrogen bond in AChE is formed between the -OH group of maltol and Glu327, while in BChE, this hydroxyl forms a hydrogen bond with Glu197. The stabilizing dispersive attraction is observed between chlorine atoms and aromatic residues: in the AChE, para-Cl is placed at the distance of 3.6 Å from the center of the benzene ring of Trp84 and 3.5 Å from the closest C atom of the same residue, with the dihedral angle between the chlorine atom and the plane of the benzene ring being ~80. When the difference between these two values is equal to or less than 0.3 Å, the Cl-π interaction is defined as “face-on” [40]. Similarly, the second chlorine atom at the ortho position of the dichloro-phenyl of cis-4 is also involved in face-on Cl-π interaction with another aromatic residue in the active site, Phe330. In contrast, ortho-chlorine in the complex of cis-4 with BChE satisfies the criteria to “edge on” Cl-π interaction with the Tyr332, where chlorine is placed at distances of 4.2 and 3.5 Å to the ring center and the closest carbon, respectively. The position of para-chlorine of cis-4 docked into BChE enables edge-on Cl-π interaction with the residue Phe329: the center of the phenylalanine ring is placed at 4.2 Å from this chlorine atom, while the distance between the Cl and the nearest C atom of the phenylalanine ring is 3.6 Å.
Except for cis-4, tested resveratrol maltol hybrids did not show inhibitory activity toward AChE. However, the measurement of their activity toward BChE showed moderate inhibitory potential. The best result was achieved with compound trans-6, so we performed molecular docking to identify interactions between the ligand and the active site. Figure 11 shows the most favorable structure obtained by the molecular docking of trans-6 to the active site of BChE. Interestingly, all ligand poses belong to one conformational cluster.
Figure 11.

The complex obtained by docking trans-6 into the active site of BChE. Hydrogens omitted for clarity.
The thienyl fragment participates in T-shaped π-stacking with Trp82; on the other side of the ring plane, it interacts with His438. Maltol is placed in the acyl pocket (Leu286, Val288). The conformation and placement of trans-6 seem to block the esteratic subsite; the double CC bond (connecting maltol and thienyl) is situated at ~3 Å from the oxygen of Ser198, thus preventing the substrate from approaching it.
The docking study has been performed without taking molecules of water into account. However, given the electronic structure of ligands and their ability to form H-bonds, it could be of interest to include relevant water molecules in the proximity of the active site of both enzymes [41,42]. Therefore, we also performed the molecular docking of tested ligands into AChE and BChE, whose active sites contain water molecules that could influence the accommodation of ligands. The results, presented in detail in Supplementary Materials, show that in AChE that includes water, compounds II and III take almost the same orientation as when waters were not present. However, the phenyl core of II is rotated in a way that enables interaction of its OH group with HOH611 and HOH616 (Figure S63c). In contrast, hydroxyl groups of III prefer the interaction with Glu199 and Ser200 (Figure S63d). Docking of II and III into the BChE in the presence of five structural water molecules result in poses that are very similar to those obtained without water; however, HOH765 interacts with the OH group of maltol fragment of III, being placed at the distance of 2.2 Å (Figure S64d). The docking of di-chlorinated molecule cis-4 in the active site of AChE and BChE in the presence of the relevant water molecules results in ligand conformations that are similar to those obtained without water; the only interaction with water can be identified between HOH715 in BChE and carbonyl oxygen at maltol (Figure S65D). Finally, compound trans-6 docked in the active site of BChE containing structural waters took the opposite orientation compared to BChE without water, which resulted in the formation of H-bond between carbonyl O of maltol and HOH715 (Figure S66b).
Although the docking study provides the approximate conformations of the ligands, these results offer insight into possibilities for main interactions between the new ligands and the enzymes, helping to rationalize inhibitor activities observed by the experiment. Both enzymes accommodate tested ligands mainly through π–π stacking interactions.
2.5. Biometal Chelating Capability of Cholinesterase Inhibitory Active Pyranones
In the case of the resveratrol–maltol hybrids proven to demonstrate cholinesterase inhibitory activity, it was reasonable to investigate the complex formation with a potential biometal ion such as Fe3+. Hence, the following compounds as ligands were studied in this respect: trans-1, cis-2, cis-4, and trans-6. Since Fe3+ ions strongly hydrolyze, acidic solutions were applied for the spectrophotometric titrations to determine the complex formation constants. Both types of titrations were carried out: increasing the iron(III) concentration at constant ligand concentration and increasing ligand concentration at constant CFe(III). The previous type of titration proved to be more informative, causing a change in the spectral characteristics, while in the latter case, the increasing absorbance of the ligand was dominant in the whole spectrum.
Figure 12a well demonstrates a characteristic spectral change for the titration of trans-6 with iron(III). The spectral series clearly indicates that the spectra cannot be the linear combinations of only two absorbing species (i.e., the organic ligand (HL) and the Fe3+ ion). The third one in this equilibrium system must be a complex between the previous two species. Detailed fitting studies by application of the PSEQUAD program confirmed the existence of a complex with a 1:1 (FeL2+) composition. Furthermore, the apparent formation constant of these species (for the reaction Fe3+ + HL ⇌ FeL2+ + H+) and the individual spectrum of the species could be estimated by this procedure. Figure 12b shows the spectrum of the complex compared to those of the ligand (HL) and Fe3+. It is clearly visible that the absorbance of Fe3+ cannot disturb the determination of the spectrum of the complex species, which significantly differs from that of the ligand. The apparent formation constant determined from this spectrum series and that regarding the titration with the ligand was lgK’FeL = 4.75 ± 0.10. Using this apparent formation constant, the molar fractions of the free Fe3+ and the 1:1 complex could be calculated as functions of the (protonated) ligand concentration (pHL), as shown by the inset of Figure 12a. Since the spectrophotometric titration was carried out in an acidic solution, in order to estimate the formation constant for the Fe3+ + L− ⇌ FeL2+ reaction, the protonation constant of the deprotonated ligand (L−) was also determined (by potentiometric titration, starting from the fully deprotonated form at pH = 10, Figure 13), lgK1 = 7.81 ± 0.15. The sum of the apparent formation constant and the protonation constant gives the equilibrium constant regarding the reaction above (lgKFeL = 12.56 ± 0.15).
Figure 12.

The measured absorption spectra (a) at constant total ligand (trans-6) concentration (3.5·10−5 M) and various Fe3+ total concentration (from 0 M (blue) to 4.7·10−4 M (green)). The inset shows the molar fraction of free Fe3+ (orange) and 1:1 complex (FeL2+) (green) as functions of the protonated ligand concentration (pHL). Subfigure (b) shows the molar absorbance of Fe3+ (orange), ligand (blue), and FeL2+ adduct (green).
Figure 13.

The measured absorption spectra (a) during potentiometric titration of ligand (trans-6). The pH changes from 10.0 (blue) to 4.0 (red). Subfigure (b) shows the measured (circles) and the calculated absorbance (lines) at three characteristic wavelengths (277 nm (blue), 324 nm (red), and 380 nm (green)).
The protonation and complex formation constants were determined similarly for the other three compounds. The corresponding figures can be found in the Supplementary Materials (Figures S67 and S68 for cis-2, Figures S69 and S70 for cis-4, and Figures S71 and S72 for trans-1). The protonation and complex formation constants obtained for the four compounds are summarized in Table 2. In the case of trans-1, two protonation steps were observed due to the two OH groups. For calculating the formation constant, lgK2 was taken into account, corresponding to the proton in the OH group on the pyranone ring. This can be exchanged by the Fe3+ ion easier than the other proton in the phenolic moiety.
Table 2.
Protonation constants for the deprotonated compounds (L− or L2−) and the constants for the formation of FeL2+ or FeHL2+ complexes.
| Compounds | Protonation Constants | Formation Constants (lgKFeL or lgKFeHLa, b) | ||
|---|---|---|---|---|
| (HL or H2L) | lgK1 | lgK2 | Fe3+ + HL ⇌ FeL2+ + H+ | Fe3+ + L− ⇌ FeL2+ |
| trans-1 | 8.9 ± 0.20 | 7.25 ± 0.20 | 4.38 ± 0.20 a | 11.6 ± 0.2 b |
| cis-2 | 7.95 ± 0.10 | 4.38 ± 0.20 | 11.8 ± 0.2 | |
| cis-4 | 7.99 ± 0.15 | 4.16 ± 0.10 | 12.15 ± 0.15 | |
| trans-6 | 7.81 ± 0.15 | 4.75 ± 0.10 | 12.56 ± 0.15 | |
a lgKFeHL for Fe3+ + H2L ⇌ FeHL2+ + H+, b for Fe3+ + HL− ⇌ FeHL2+.
The similar values of the formation constants (lgK = 11.6–12.6) suggest that Fe3+ binds to the common hydroxy-pyranone moiety, and the other different aromatic part of the ligands hardly influences the coordination of the metal center. In accordance with this conclusion, the characteristic bands in the individual absorption spectra of the complexes are uniformly red-shifted compared to those in the corresponding ligand spectra.
2.6. Crystal Structure and Packing of Resveratrol–Maltol Hybrids
The molecular structures of two resveratrol–maltol hybrids, cis-2 and trans-6, are shown in Figure 14. Although the crystallographic analyses of cis-2 and trans-6 are solid, they represent useful additional information about proven active compounds that can be informative in the context of intermolecular interaction study.
Figure 14.

Molecular structure of compounds cis-2 (a) and trans-6 (b). Displacement ellipsoids are drawn for the probability of 25%, and hydrogen atoms are shown as spheres of arbitrary radii.
The crystal packing of compounds depends on a subtle interplay of weak interaction and steric effects. The molecules are held together by weak intermolecular interactions: hydrogen bonds, C–H⋯π interactions, and dispersion interactions.
In crystal packing of cis-2 intermolecular hydrogen bonds (C2–H2⋯F3 and O2–H2A⋯O1) link two molecules into zig-zag chains parallel to [1,2,3,4,5,6,7,8,9,10] (Figure 15 and Table S5). The chains are linked by hydrogen bond C7–H7⋯F1 and C–H⋯π interactions (C5–H5⋯O3→C13) extending in the direction [010], which produces a 3D structure (Figure 15 and Table S6).
Figure 15.

Crystal packing of cis-2. The chains are generated by C–H⋯F and O–H⋯O hydrogen bonds parallel to [1,2,3,4,5,6,7,8,9,10]. Hydrogen bond C7–H7⋯F1 and C–H⋯π interactions extend the chains in the direction [010].
The crystal packing of trans-6 is characterized by intermolecular hydrogen bonds (O–H⋯O) and C–H⋯π interactions (C5–H5⋯O3→C13), which generate zig-zag chains running in the direction [–1–10] (Figure 16 and Tables S5 and S6) 3D packing is achieved through stacking interactions (Table S7).
Figure 16.

Crystal packing of trans-6: (a) layers generated by hydrogen bonds (O–H⋯O) and C–H⋯π interactions; (b) zig-zag chains in the direction [–1–10].
3. Materials and Methods
3.1. General Remarks
Solvents such as acetonitrile (ACN), chloroform (CHCl3), dichloromethane (DCM), diethylether (E), ethanol (EtOH), ethyl acetate (EtOAc), methanol (MeOH), and toluene were used. All enumerated solvents were purified by distillation and were commercially available. Iron(III) sulfate hexahydrate (Fe2(SO4)3·6H2O), sodium tetraborate decahydrate (Na2B4O7·10H2O), disodium hydrogen phosphate heptahydrate (Na2HPO4·7H2O), and sodium acetate trihydrate (CH3COONa·3H2O) were purchased from Reanal (Budapest, Hungary), and sodium hydroxide > 98% (NaOH) and 98% sulphuric acid from Acros Organics (Geel, Belgium). The water applied was cleaned by Millipure Elix equipment (Millipore S.A.S., Molsheim, France) and completed with a Milli-Q 50 purification system (Millipore S.A.S., Molsheim, France). The column chromatography was performed using silica gel (60 Å, technical grade). Thin-layer chromatography was performed using plates coated with silica gel (0.2 mm, Kiselgel 60 F254). Nuclear magnetic resonance (NMR) spectroscopic data for 1H and 13C nuclei were recorded at room temperature on a spectrometer at Bruker Avance 300 MHz and 600 MHz. Deuterated chloroform, CDCl3, deuterated methanol, CD3OD, and deuterated dimethyl sulfoxide, DMSO with tetramethylsilane as standard were used for recording NMR spectra. Chemical shifts were reported in parts per million. The 1H NMR spectra for four compounds were also recorded at 5, 15, 25, 35, 45, and 55 °C on a Bruker Advance 600 MHz spectrometer. UV spectra were recorded by UV/Vis spectrophotometer (Varian Cary 50 UV/Vis, Lexington, MA, USA) in ACN. Photochemical irradiations were carried out with 3.0 mL solutions in 1 mL cells at room temperature in photochemical reactor Luzchem equipped with 8 UV lamps at 313 nm. Spectral changes during the irradiation of pyranones were recorded in HPLC grade ACN. Before irradiation, the reaction mixtures were purged with nitrogen, N2, for 15 min. IR spectra were acquired using the Bruker Vertex 70 Fourier transform infrared spectrometer set on attenuated total reflectance (ATR) mode. The samples were pressed on a diamond, and the absorbance data were collected between 400 and 5000 cm−1 with a spectral resolution of 1 cm−1 and an average of 32 scans. HRMS analyses were carried out on a mass spectrometer (MALDI TOF/TOF analyzer), equipped with an Nd:YAG laser operating at 355 nm with a fitting rate of 200 Hz in the positive (H+) or negative (-H) ion reflector mode. All solvents were removed from the solutions by rotary evaporator under reduced pressure.
3.2. General Procedure for the Synthesis of 4-Pyranone Phosphonium Salt
The 4-pyranone phosphonium salt was synthesized in a three-necked flask (0.25 L) by preparing corresponding pyranone chloride. Thionyl chloride, SOCl2 (0.09 mol) was added dropwise to a cooled (0 °C) reaction flask solution of kojic acid (0.05 mol) and DCM (82 mL). The reaction mixture was stirred for 5 h, and the rest of the solvent was evaporated. Crude yellow powder was dissolved in toluene, and triphenylphosphine (0.05 mol) was added and stirred for 3 days and 4 nights at reflux temperature. The reaction mixture was filtrated under reduced pressure, and the beige salt was dried under pressure in an exicator for 12 h. Dried phosphonium salt was used in all further experiments.
3.3. General Procedure for the Synthesis of 4-Pyranone Heterostilbenes 1–8
Compounds 1–8 were obtained as mixtures of cis- and trans-isomers (except 1 and 5). The reaction apparatus was purged with N2 for 15 min before adding the reactants. The reaction is carried out in a three-necked flask (100 mL) equipped with a chlorine-calcium tube and an N2 balloon connected. Phosphonium salt (5 mmol) was added to the 40 mL of EtOH, and the mixture was stirred with a magnetic stirrer. The solutions of sodium ethoxides (5 mmol, 1.1 eq of Na dissolved in 10 mL of absolute ethanol) were added in strictly anhydrous conditions under nitrogen dropwise. The aldehyde (5 mmol) was then added to the reaction mixture, and the reaction mixture was allowed to stir for 24 h at room temperature. The reaction mixture was evaporated on a vacuum evaporator and dissolved in toluene. The mixture was then extracted with toluene (3 × 15 mL). The organic layers were dried under anhydrous magnesium sulfate, MgSO4. Products 1–8 were isolated by repeated column and thin layer chromatography on silica gel using PE/E, PE/DCM, and DCM/E solvent systems. The first isomer to eluate is cis- and then trans-isomer in the last fractions. Spectroscopic characterization of new heterostilbene derivatives of 4-pyranone is given below. The solubility of pyranone derivatives was, in some cases low, especially for corresponding trans-isomers.
5-hydroxy-2-(2-hydroxystyryl)-4H-pyran-4-one (1) (63%). Column chromatography on silica gel using EtOAc/MeOH (50%) afforded 500 mg of a mixture of trans-isomer and triphenylphosphine oxide. Repeated column chromatography on silica gel using EtOAc/MeOH (50%) afforded pure trans-isomer.
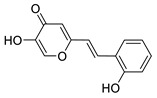
(E)-5-hydroxy-2-(2-hydroxystyryl)-4H-pyran-4-one (trans-1): 88 mg (isolated), orange powder, Rf(E) = 0.12; m.p. 185–186 °C; UV (ACN with small amount of MeOH) λmax/nm (ε/dm3mol−1cm−1) 353 (29371); IR vmax/cm−1 (NaCl): 3411, 3248, 1630, 1552, 1421, 1321, 1248, 1040, 963, 939; 1H NMR (CD3OD, 600 MHz) δ/ppm: 7.83 (s, 1H), 7.70 (d, J = 16.6 Hz, 1H), 7.51 (d, J = 7.4 Hz, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.00 (d, J = 16.3 Hz, 1H), 6.84 (t, J = 7.5 Hz, 3H); 13C NMR (DMSO, 75 MHz) δ/ppm: 144.2 (s), 136.4 (s), 131.9 (s), 130.9 (s), 128.5 (d), 124.5 (d), 124.2 (d), 119.6 (d), 116.3 (d), 116.2 (d), 105.1 (s), 107.6 (d), 99.4 (d); MS (ESI) m/z (%, fragment): 231 (100); HRMS (m/z) for C13H10O4: [M+H]+calcd = 230.0579, [M+H]+measured = 230.0583.
5-hydroxy-2-(2-(trifluoromethyl)styryl)-4H-pyran-4-one (2) (59%). Column chromatography on silica gel using PE/E (0–100%) afforded a mixture of isomers of 2 (cis-2: trans-2 = 1:1.5). Repeated thin-layer chromatography on silica gel using PE/E (30%) as eluent afforded pure cis-2 in the first fractions and trans-2 in the last fractions.
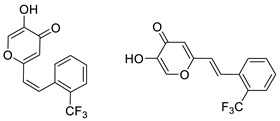
(Z)-5-hydroxy-2-(2-(trifluoromethyl)styryl)-4H-pyran-4-one (cis-2): 14.2 mg (isolated), white powder, Rf(E) = 0.45; m.p. 113–115 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 280 (11679); IR vmax/cm−1 (NaCl): 3230, 2920, 2850, 1650, 1580, 1450, 1380, 1250, 930, 860, 770, 690, 580; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.72 (d, J = 8.4 Hz, 1H), 7.50 (t, J = 7.1 Hz, 1H), 7.50 (s, 1H), 7.45 (t, J = 7.6 Hz, 2H), 7.14 (d, J = 12.3 Hz, 1H), 6.35 (d, J = 12.3 Hz, 1H), 6.21 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 173.7 (s), 161.8 (s), 145.7 (d), 137.0 (d), 135.1 (d), 134.7 (s), 131.6 (d), 130.3 (d), 128.6 (d), 126.0 (s), 125.9 (s), 123.5 (d), 112.5 (d); MS (ESI) m/z (%, fragment): 283 (100);
(E)-5-hydroxy-2-(2-(trifluoromethyl)styryl)-4H-pyran-4-one (trans-2): 21.6 mg (isolated), white powder, Rf(E) = 0.45; m.p. 117–120 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 308 (8706); IR vmax/cm−1 (NaCl): 3230, 2920, 2850, 1650, 1580, 1450, 1380, 1250, 930, 860, 770, 690, 580; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.90 (s, 1H), 7.78 (d, J = 15.9 Hz, 1H), 7.72 (d, J = 7.2 Hz, 2H), 7.61 (t, J = 7.2 Hz, 1H), 7.51 (t, J = 7.6 Hz, 1H), 6.71 (d, J = 15.9 Hz, 1H), 6.49 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 174.2 (s), 161.8 (s), 145.6 (s), 136.9 (d), 132.3 (d), 131.9 (d), 129.1 (d), 127.3 (d), 126.3 (s), 126.2 (s), 123.4 (d), 111.3 (d); MS (ESI) m/z (%, fragment): 283 (100);
HRMS (m/z) for C14H9F3O3 (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 282.0504, [M+H]+measured = 282.0505.
4-(2-(5-hydroxy-4-oxo-4H-pyran-2-yl)vinyl)benzonitrile (3) (44%). Column chromatography on silica gel using PE/E (80%) afforded a pure mixture of isomers of 3 (41 mg; cis-3:trans-3 = 1:3) without triphenylphosphine-oxide. Repeated column chromatography on silica gel using PE/E (0–70%) as eluent did not afford pure cis- and trans-isomers due to the very similar Rf values.
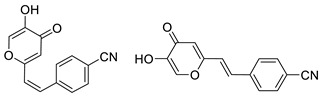
(Z)-4-(2-(5-hydroxy-4-oxo-4H-pyran-2-yl)vinyl)benzonitrile (cis-3): 1H NMR (DMSO-d6, 600 MHz) δ/ppm: 8.93 (s, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.4 (d, J = 8.1 Hz, 2H), 7.10 (d, J = 12.5 Hz, 1H), 6.84 (d, J = 12.1 Hz, 1H), 6.79 (s, 1H); MS (ESI) m/z (%, fragment): 240 (100).
(E)-4-(2-(5-hydroxy-4-oxo-4H-pyran-2-yl)vinyl)benzonitrile (trans-3): 1H NMR (DMSO-d6, 600 MHz) δ/ppm: 9.27 (s, 1H), 7.86 (d, J = 8.5 Hz, 2H), 7.76 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 16.2 Hz, 1H), 7.15 (d, J = 16.2 Hz, 1H), 6.67 (s, 1H); MS (ESI) m/z (%, fragment): 240 (100);
IR vmax/cm−1 (NaCl) (obtained for the pure mixture of geometrical isomers): 3050, 2910, 2840, 2250, 1715, 1570, 1480, 950, 840, 550; HRMS (m/z) for C14H9NO3 (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 239.0582, [M+H]+measured = 239.0588.
2-(2,4-dichlorostyryl)-5-hydroxy-4H-pyran-4-one (4) (74%). Column chromatography was performed on silica gel using PE/E (0–70%) as eluent. Repeated column chromatography on silica gel (PE/E (0–70%)) afforded pure cis-4 in the first fractions and trans-4 in the last fractions.
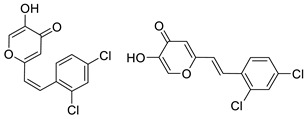
(Z)-2-(2,4-dichlorostyryl)-5-hydroxy-4H-pyran-4-one (cis-4): 250 mg (isolated), white powder, Rf(E) = 0.51; m.p. 149–151 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 291 (14966); IR vmax/cm−1 (NaCl): 3200, 3050, 1600, 1550, 1490, 1380, 1250, 1150, 950, 750, 700; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.58 (s, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.21 (d, J = 2.0 Hz, 1H), 6.90 (d, J = 12.3 Hz, 1H), 6.36–6.31 (m, 2H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 173.7 (s), 161.8 (s), 145.7 (s), 136.7 (d), 135.1 (s), 134.1 (s), 133.9 (d), 132.8 (s), 130.8 (d), 129.4 (d), 126.9 (d), 123.1 (d), 112.5 (d); MS (ESI) m/z (%, fragment): 283 (100).
(E)-2-(2,4-dichlorostyryl)-5-hydroxy-4H-pyran-4-one (trans-4): 40 mg (isolated), white powder, Rf(E) = 0.51; m.p. 103–105 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 325 (23654), 288 (20467); 1H NMR (CDCl3, 600 MHz) δ/ppm: 8.06 (s, 1H), 7.93 (s, 1H), 7.74 (d, J = 16.2 Hz, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 6.68 (d, J =16.2 Hz, 1H), 6.56 (s, 1H); 13C NMR (CDCl3, 75 MHz) δ/ppm: 174.1 (s), 161.8 (s), 145.7 (s), 137.5 (s), 135.2 (s), 131.1 (d), 130.2 (d), 130.0 (d), 128.4 (d), 127.7 (d), 127.6 (d), 122.1 (s), 111.3 (d); MS (ESI) m/z (%, fragment): 283 (100);
HRMS (m/z) for C13H8Cl2O3 (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 281.9850, [M+H]+measured = 281.9852.
N-(4-(2-(5-hydroxy-4-oxo-4H-pyran-2-yl)vinyl)phenyl)acetamide (5) (53%). Repeated column chromatography was performed on silica gel with solvent system EtOAc/MeOH (20%). This derivative gave only cis-isomer as a product of the Wittig reaction, but repeated chromatography was necessary to divide the triphenylphosphine-oxide from cis-5.
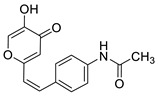
(Z)-N-(4-(2-(5-hydroxy-4-oxo-4H-pyran-2-yl)vinyl)phenyl)acetamide (cis-5): 100 mg (isolated), orange powder, Rf(DCM:MeOH = 7:3) = 0.35; m.p. 154–156 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 338 (10321); IR vmax/cm−1 (NaCl): 3372, 1639, 1582, 1534, 1321, 1210, 1178, 1040, 970, 930, 825; 1H NMR (CD3OD, 600 MHz) δ/ppm: 6.28 (s, 1H), 6.04 (d, J = 8.2 Hz, 3H), 5.76 (d, J = 8.0 Hz, 2H), 5.40 (d, J = 12.3 Hz, 1H), 4.73 (d, J = 12.3 Hz, 1H), 3.34 (s, 3H); 13C NMR (CD3OD, 75 MHz) δ/ppm: 174.4 (s), 166.9 (s), 141.7 (s), 139.8 (s), 134.9 (s), 133.2 (d), 132.0 (d), 123.6 (d), 123.2 (d), 121.7 (d), 116.6 (d), 114.6 (s), 26.5 (q); MS (ESI) m/z (%, fragment): 272 (100);
HRMS (m/z) for C15H13NO4: [M+H]+calcd = 271.0845, [M+H]+measured = 271.0850.
5-hydroxy-2-(2-(thiophen-3-yl)vinyl)-4H-pyran-4-one (6) (38%). Repeated column chromatography was performed on silica gel using PE/DCM (0–90%), DCM/E (30%), and DCM/MeOH (10%) as solvent systems in combination with preparative thin-layer chromatography in a solvent system PE/E (30%). Pure cis-6 was isolated in the first fractions and trans-6 in the last fractions.
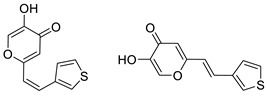
(Z)-5-hydroxy-2-(2-(thiophen-3-yl)vinyl)-4H-pyran-4-one (cis-6): 7 mg (isolated), white powder, Rf(E) = 0.55; m.p. 143–146 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 311 (11679); IR vmax/cm−1 (NaCl): 3220, 2925, 2840, 1740, 1650, 1580, 1470, 1435, 1380, 950, 825, 770, 725; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.77 (s, 1H), 7.39 (dd, J = 2.9 Hz, 1H), 7.29 (dd, J = 3.0 Hz, 1H), 7.09 (dd, J = 5.0 Hz, 1H), 6.81 (d, J = 12.6 Hz, 1H), 6.47 (s, 1H), 6.10 (d, J = 12.5 Hz, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 173.9 (s), 162.9 (s), 145.9 (s), 136.7 (d), 136.3 (s), 132.8 (d), 127.8 (d), 127.4 (d), 125.9 (d), 119.2 (d), 111.9 (d); MS (ESI) m/z (%, fragment): 221 (100).
(E)-5-hydroxy-2-(2-(thiophen-3-yl)vinyl)-4H-pyran-4-one (trans-6): 9.1 mg (isolated), yellow powder, Rf(E) = 0.55; m.p. 111–115 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 314 (20340); IR vmax/cm−1 (NaCl): 3220, 2925, 2840, 1740, 1650, 1580, 1470, 1435, 1380, 950, 825, 770, 725; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.83 (s, 1H), 7.48 (d, J = 16.3 Hz, 1H), 7.37 (dd, J = 5.2 Hz, 1H), 7.32 (dd, J = 5.2 Hz, 1H), 7.29 (dd, J = 5.4 Hz, 1H), 6.53 (d, J = 16.3 Hz, 1H), 6.48 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 174.2 (s), 162.8 (s), 145.5 (s), 138.0 (s), 136.8 (d), 130.3 (d), 127.2 (d), 126.9 (d), 124.8 (d), 119.1 (d), 110.1 (d); MS (ESI) m/z (%, fragment): 221 (100);
HRMS (m/z) for C11H8O3S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 220.0194, [M+H]+measured = 220.0199.
5-hydroxy-2-(2-(thiophen-2-yl) vinyl)-4H-pyran-4-one (7) (42%). Repeated column chromatography was performed on silica gel with solvent system PE/E (0–50%). After that, repeated thin-layer chromatography of some enriched fractions was performed with a solvent system E/CHCl3 (80%). Only the isolation of the pure trans-7 in the last fractions was successful, while the obtained pure mixture of isomers (13 mg) without triphenylphosphine-oxide confirmed the formation of cis-7 (cis-7:trans-7 = 1:1).
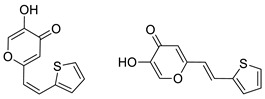
(Z)-5-hydroxy-2-(2-(thiophen-2-yl)vinyl)-4H-pyran-4-one (cis-7): 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.91 (s, 1H), 7.44 (d, J = 5.2 Hz, 1H), 7.25 (d, J = 3.5 Hz, 1H), 7.06 (dd, J = 5.2 Hz, 1H), 6.90 (d, J = 12.8 Hz, 1H), 6.47 (s, 1H), 6.0 (d, J = 12.8 Hz, 1H); MS (ESI) m/z (%, fragment): 221 (100).
(E)-5-hydroxy-2-(2-(thiophen-2-yl)vinyl)-4H-pyran-4-one (trans-7): 8 mg (isolated), white powder, Rf(E) = 0.35; m.p. 113–116 °C; UV (ACN) λmax/nm (ε/dm3mol−1cm−1) 340 (30518); IR vmax/cm−1 (NaCl): 3210, 2920, 2840, 1730, 1660, 1580, 1470, 1435, 1390, 1210, 950, 850, 760, 680; 1H NMR (CDCl3, 600 MHz) δ/ppm: 7.83 (s, 1H), 7.52 (d, J = 16.5 Hz, 1H), 7.36 (d, J = 5.7 Hz, 1H), 7.22 (d, J = 3.5 Hz, 1H), 7.05 (dd, J = 5.0 Hz, 1H), 6.50 (d, J = 16.5 Hz, 1H), 6.39 (s, 1H); 13C NMR (CDCl3, 150 MHz) δ/ppm: 174.2 (s), 162.8 (s), 145.5 (s), 140.1 (s), 136.8 (d), 129.9 (d), 129.3 (d), 128.2 (d), 127.2 (d), 118.3 (d), 110.1 (d); MS (ESI) m/z (%, fragment): 221 (100);
HRMS (m/z) for C11H8O3S (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 220.0194, [M+H]+measured = 220.0200.
5-hydroxy-2-(2-(quinoxalin-2-yl)vinyl)-4H-pyran-4-one (8) (35%). Repeated column and thin-layer chromatographies (E/EtOAc (0–70%) and EtOAc/MeOH (0–90%)) were performed on silica gel. The pure mixture of isomers was obtained (12 mg) without the presence of triphenylphosphine-oxide (cis-8:trans-8 = 1.3:1).
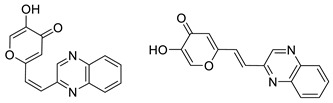
(Z)-5-hydroxy-2-(2-(quinoxalin-2-yl)vinyl)-4H-pyran-4-one (cis-8): 1H NMR (DMSO-d6, 600 MHz) δ/ppm: 8.93 (s, 1H), 8.13–8.08 (m, 2H), 7.92–7.86 (m, 3H), 6.84 (d, J = 12.7 Hz, 1H), 6.79 (d, J = 12.7 Hz, 1H), 6.79 (s, 1H); MS (ESI) m/z (%, fragment): 267 (100).
(E)-5-hydroxy-2-(2-(quinoxalin-2-yl)vinyl)-4H-pyran-4-one (trans-8): 1H NMR (DMSO-d6, 600 MHz) δ/ppm: 9.27 (s, 1H), 8.13–8.08 (m, 2H), 8.02 (d, J = 9.5 Hz, 1H), 7.92–7.86 (m, 2H), 7.75 (d, J = 16.0 Hz, 1H), 7.68 (d, J = 16.0 Hz, 1H), 6.67 (s, 1H); MS (ESI) m/z (%, fragment): 267 (100);
IR vmax/cm−1 (NaCl) (obtained for the pure mixture of geometrical isomers): 3320, 2959, 2917, 2847, 1584, 1568, 1377, 1261, 1088, 1014, 794, 749; HRMS (m/z) for C15H10N2O3 (obtained for the pure mixture of geometrical isomers): [M+H]+calcd = 266.0691, [M+H]+measured = 266.0697.
3.4. Cholinesterase Inhibitory Activity
Acetylcholinesterase (AChE, E.C. from electric eel), butyrylcholinesterase (BChE, E.C. from equine serum), Tris-HCl buffer, acetylthiocholine iodide (ATChI), S-butyrylthiocholine iodide (BTChI) and galantamine were purchased from Sigma-Aldrich (St. Louis, MO, USA), 5,50-dithiobis-(2-nitrobenzoic acid) (Ellman’s reagent, DTNB) was purchased from Zwijndrecht (Belgium). The AChE/BChE inhibitory potentials were determined using modified Ellman’s method [38]. Galantamine was used as a reference standard. Briefly, 180 µL Tris HCl buffer (50 mM, pH 8.0), 10 µL of AChE/BChE (final concentration 0.03 U/mL, prepared in 20 mM Tris HCl buffer, pH 7.5), and 10 µL tested solution (final concentrations, depending on solubility and activity, in a range 1–1000 μM) were mixed and pre-incubated for 5 min at 4 °C. The reaction was then initiated with the addition of 10 μL of DTNB (final concentration 0.3 mM prepared in Tris buffer) and 10 μL of ATChI/BTChI (final concentration of 0.5 mM prepared in Tris buffer). AChE/BChE activity was measured using a 96-well microplate reader (IRE 96, SFRI Medical Diagnostics, Bordeaux, France) at room temperature at 405 nm over 6 min. Non-enzymatic hydrolysis was measured as blank for control measurement without inhibitors. The non-enzymatic hydrolysis reaction with added inhibitor was used as a blank for the samples. An equivalent buffer amount replaced the enzyme. The experiment was run in triplicate. Percentage enzyme inhibition was calculated according to the equation:
| Inhibition (%) = [(AC − AT)/AC] × 100 | (1) |
where AC is the enzyme activity without the test sample, and AT is the enzyme activity with the test sample. Data were used to calculate the IC50 value by a nonlinear fit of compound concentration (log) values vs. response. The results are represented as mean values ± standard deviation. Samples were dissolved in ethanol. The inhibitory activity of ethanol was measured, and its contribution to inhibition was subtracted.
Antioxidant activity of the tested compounds was measured in terms of hydrogen donating or radical scavenging ability using the stable radical 2,2′-diphenyl-1-picrylhydrazyl (DPPH) [39]. The 50 μL of various concentrations of tested solutions (5 μM–1000 μM) were placed in a cuvette, 1 mL of ethanolic solution of DPPH was added, and the final DPPH concentration was kept constant (c = 8 × 10−4 M). The mixture was incubated for 30 min at 25 °C. The absorbance was measured at 517 nm (UV-1800 UV/Vis Spectrophotometer, Shimadzu, Kyoto, Japan). All determinations were performed in triplicate. The concentrations of tested compounds were expressed as final concentrations. Inhibition of DPPH expressed in percentage was calculated according to the equation:
| (2) |
where AC(0) is the absorbance of the control at t = 0 min, and AC(t) is the absorbance of the antioxidant at t = 30 min. Data were used to calculate the IC50 value by a nonlinear fit of compound concentration values vs. inhibition percentage. The results are represented as mean values ± standard deviation.
3.5. Molecular Docking
The conformations of selected ligands were examined at the M06-2X/6-31G(d) level of theory using the Gaussian16 program suite [43]. The optimized structures of the most stable conformers were then used for a molecular docking study. Molecular docking was performed using the Autodock program package [44], with crystal structures of cholinesterases taken from the Protein Data Bank: for AChE, crystal structure 1EEA.pdb [45], and for BChE, crystal structure 1P0I.pdb [46] was used. The docking results were obtained using the Lamarckian Genetic Algorithm, with 25 requested genetic algorithm dockings with 25 binding poses for each ligand.
3.6. Metal Chelating Affinity
Two titration series were used to determine the stability constants of equilibrium systems containing Fe3+ and ligand. The iron(III) content was increased at constant ligand content, the ligand concentration was increased at constant Fe(III) content, and the spectral changes were recorded with a Specord S600 spectrophotometer (Analytic Jena GmbH, Jena, Germany) in the 190–900 nm wavelength range. Since the molar absorption coefficient of the ligand is significantly higher than that of Fe(III), the former series provides more information for the determination of the spectra of the new complex formed, while the latter series is necessary for a more accurate determination of the stability constant. The two series were evaluated together using PSEQUAD software [47] in the 210–430 nm range.
A 0.01–0.01 M phosphate–borate–acetate buffer solution was used for potentiometric titration. A 50 cm3 beaker was filled with 25 cm3 of buffer solution containing the appropriate amount of ligand, and the pH was adjusted to 10.0 by adding 2 M NaOH solution. The pH was determined using a Consort C3010 multi-parameter analyzer (Consort bvba, Turnhout, Belgium) equipped with a combined glass electrode. The solution was circulated with an IKA PA-SF (IKA Labortechnik, Staufen, Germany) peristaltic pump through a Hellma (176.000-QS 10 mm) flow through the cuvette. Titration was carried out with 6 M H2SO4 solution. The spectra were recorded with a Specord S600 spectrophotometer after pH stabilization.
3.7. X-Ray Crystallography
Single crystal measurements were performed on an XtaLAB Synergy diffractometer, using micro-focus sealed X-ray tube CuKα (1.54184 Å) radiation at room temperature [293(2) K]. The CrysAlisPRO package (Rigaku OD, 2018) was used for data reduction and numerical absorption correction. The structure was solved with SHELXS97 [48] and refined with SHELXL2018 [49]. The model was refined using the full matrix least squares refinement; all non-hydrogen atoms were refined anisotropically. Hydrogen atoms were located in a difference Fourier map and refined as a mixture of free and riding entities. Molecular geometry calculations were performed by PLATON [50], and the molecular graphics were prepared using ORTEP-3 [51] and CCDC-Mercury [52]. Crystallographic and structure refinement data for the structure reported in this paper are shown in Table 3. Supplementary crystallographic data for this paper can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html accessed on 2 September 2022 (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; or deposit@ccdc.cam.ac.uk). CCDC-2204641 and CCDC-2204642 contain supplementary crystallographic data for this paper.
Table 3.
Crystallographic data and structure refinement details for cis-2 and trans-6.
| Compound | cis-2 | trans-6 |
|---|---|---|
| Empirical formula | C14H9F3O3 | C11H8O3S |
| Formula wt./g mol−1 | 282.21 | 220.23 |
| Crystal dimensions/mm | 0.4 × 0.25 × 0.2 | 0.4 × 0.3 × 0.25 |
| Space group | P21/c | P21/c |
| a/Å | 18.0019 (10) | 15.3582 (2) |
| b/Å | 4.8342 (3) | 5.14770 (10) |
| c/Å | 14.7412 (8) | 25.9362 (5) |
| α/° | 90 | 90 |
| β/° | 107.425 (6) | 102.554 (2) |
| γ/° | 90 | 90 |
| Z | 4 | 8 |
| V/Å3 | 1223.98 (13) | 2001.48 (6) |
| Dcalc/g cm−3 | 1.531 | 1.462 |
| μ/mm−1 | 1.2 | 2.748 |
| Θ range/° | 5.15–79.384 | 3.492–79.837 |
| T(K) | 293 (2) | 299 (2) |
| Radiation wavelength | 1.54184 (CuKα) | 1.54184 (CuKα) |
| Diffractometer type | XtaLAB Synergy, Dualflex, HyPix |
XtaLAB Synergy, Dualflex, HyPix |
| Range of h, k, l | −22 > h > 19; −5 > k > 5; −18 > l > 18 |
−19 > h > 16; −6 > k > 5; −31 > l > 33 |
| Reflections collected | 6269 | 13048 |
| Independent reflections | 2389 | 4146 |
| Observed reflections (I ≥ 2σ) |
1886 | 3366 |
| Rint | 0.0839 | 0.039 |
| R (F) | 0.1161 | 0.099 |
| Rw (F2) | 0.3576 | 0.3512 |
| No. of parameters, restraints | 181, 0 | 272, 0 |
| Goodness of fit | 1.495 | 1.539 |
| Δρmax, Δρmin (eÅ−3) | 0.654; −0.38 | 2.09; −1.034 |
4. Conclusions
New resveratrol–thiophene and resveratrol–maltol hybrids were synthesized as cholinesterase inhibitors, metal-chelating agents, and antioxidants. As photostability is the favorable property of resveratrol derivatives, the aim of this study was also to investigate changes in the UV spectra of compounds when exposed to UV light, as well as the capability to complex with biometals. As with photostability experiments, biological tests also found remarkable differences in the properties and behavior of thiophene and maltol hybrids. While resveratrol–thiophene hybrids have excellent inhibitory and antioxidant properties (similar to the activity of the reference drug galantamine), maltols have been proven to be weaker inhibitors and antioxidants. The results of the docking study indicate that all docked ligands are accommodated by both enzymes, AChE and BChE, mainly through π–π stacking interactions. The compounds proved to be active cholinesterase inhibitors, also prone to coordination with the Fe3+ ion as biometals. In the crystal packing, they are held together by weak intermolecular interactions: hydrogen bonds, C–H⋯π interactions, and dispersion interactions.
Abbreviations
| Aβ | aggregation of β-amyloid |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s disease |
| ACN | acetonitrile |
| AS | anionic site |
| BChE | butyrylcholinesterase |
| DCM | dichloromethane |
| DPPH | 2,2′-diphenyl-1-picrylhydrazyl |
| HB | hydrogen bond |
| E | ether |
| EtOH | ethanol |
| NMR | nuclear magnetic resonance |
| EtOAc | ethyl acetate |
| MeOH | methanol |
| PAS | peripheric anionic site |
| PE | petroleum ether, |
| UV | ultraviolet spectrophotometry |
| s | singlet |
| d | doublet |
| t | triplet |
| q | quartet |
| dd | doublet of doublets |
| m | multiplet |
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27196379/s1, Figures S1–S34: NMR spectra of pure compounds; Figures S35–S46: UV spectra of pure compounds; Figures S47–S54: IR ATR spectra of pure compounds; Figures S55–S62: HRMS analyses, Figures S63–S66: Data for molecular docking; Figures S67–S72: Biometal chelating capability of cholinesterase inhibitory active pyranones; Tables S1–S4: Data for molecular docking; Tables S5 and S6: Crystal data. Table S7: Geometric parameters of π interactions in the compound trans-6. [53,54].
Author Contributions
Conceptualization, I.O. and I.Š.; Formal analysis, M.M., L.F., M.J.L., D.B., V.M., Ž.M. and Z.L.; Investigation, M.M.; Methodology, L.F. and K.M.; Resources, I.O., O.H., D.B., K.M. and I.Š.; Writing—original draft, I.O., O.H., D.B., V.M. and I.Š.; Writing—review & editing, D.B. and I.Š. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not available.
Informed Consent Statement
Not available.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was supported by grants from the University of Zagreb for short-term scientific support for 2021 under the title Synthesis and photochemistry of various new heterostilbene derivatives. We also acknowledge the NMR Centre at RBI for recording all the NMR spectra. We thank the University of Zagreb Computing Centre (SRCE) for granting computational time on the ISABELLA cluster. This work has been implemented by the TKP2021-NKTA-21 project with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the 2021 Thematic Excellence Programme funding scheme.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Belwal T., Nabavi S.M., Nabavi S.F., Dehpour A.R., Shirooie S. Naturally Occurring Chemicals against Alzheimer’s Disease. Andre Gerhard Wolff; Cambridge, MA, USA: 2020. pp. 33–42. [Google Scholar]
- 2.Anekonda T.S. Resveratrol—A boon for treating Alzheimer’s disease? Brain Res. Rev. 2006;52:316–326. doi: 10.1016/j.brainresrev.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 3.Anekonda T.S., Wadsworth T.L., Sabin R., Frahler K., Harris C., Petriko B., Ralle M., Woltjer R., Quinn J.F. Phytic acid as a potential treatment for Alzheimer’s pathology: Evidence from animal and in vitro models. J. Alzheimer’s Dis. 2011;23:21–35. doi: 10.3233/JAD-2010-101287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J., Qiu Z., Zhao G.R. Modular engineering of E. coli coculture for efficient production of resveratrol from glucose and arabinose mixture. Synth. Syst. Biotechnol. 2022;7:718–729. doi: 10.1016/j.synbio.2022.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reitz C. Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int. J. Alzheimer’s Dis. 2012;2012:369808. doi: 10.1155/2012/369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rauk A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009;38:2698. doi: 10.1039/b807980n. [DOI] [PubMed] [Google Scholar]
- 7.Darvesh A.S., Carroll R.T., Bishayee A., Geldenhuys W.J., Schyf C.J.V.D. Oxidative stress and Alzheimer’s disease: Dietary polyphenols as potential therapeutic agents. Expert Rev. Neurother. 2010;10:729–745. doi: 10.1586/ern.10.42. [DOI] [PubMed] [Google Scholar]
- 8.Citron M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- 9.Savelieff M.G., Lee S., Liu Y., Lim M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013;8:856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Hernandez J., Paseiro-Losada P., Sanches-Silva A.T., Lage-Yusty M.A. Study of the changes of trans-resveratrol caused by ultraviolet light and determination of trans- and cis-resveratrol in Spanish white wines. Eur. Food Res. Technol. 2007;225:789–796. doi: 10.1007/s00217-006-0483-x. [DOI] [Google Scholar]
- 11.Montsko G., Nikfardjam M.S.P., Szabo Z., Boddi K., Lorand T., Ohmacht R., Mark L. Determination of products derived from trans-resveratrol UV photoisomerisation by means of HPLC–APCI-MS. J. Photochem. Photobiol. A. 2008;196:44–50. doi: 10.1016/j.jphotochem.2007.11.011. [DOI] [Google Scholar]
- 12.Silva C.G., Monteiro J., Marques R.R.N., Silva A.M.T., Martínez C., Canle M., Faria J.L. Photochemical and photocatalytic degradation of trans-resveratrol †‡. Photochem. Photobiol. Sci. 2013;12:638. doi: 10.1039/C2PP25239B. [DOI] [PubMed] [Google Scholar]
- 13.Cottart C.H., Nivet-Antoine C., Laguillier-Morizot C., Beaudeux J.L. Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food Res. 2010;54:7–16. doi: 10.1002/mnfr.200900437. [DOI] [PubMed] [Google Scholar]
- 14.Jie L., Wuliji O., Wei L., Zhi-Gang J., Hossein A. Oxidative Stress and Neurodegenerative Disorders. Int. J. Mol. Sci. 2013;14:24438–24475. doi: 10.3390/ijms141224438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaikh S., Dhavan P., Ramana M.M.V., Jadhav B.L. Design, synthesis and evaluation of new chromone-derived aminophosphonates as potential acetylcholinesterase inhibitor. Mol. Divers. 2021;25:811–825. doi: 10.1007/s11030-020-10060-y. [DOI] [PubMed] [Google Scholar]
- 16.Jalili-Baleh L., Nadri H., Forootanfar H., Küçükkılınç T.T., Ayazgök B., Sharifzadeh M., Rahimifard M., Baeeri M., Abdollahi M., Foroumadi A., et al. Chromone–lipoic acid conjugate: Neuroprotective agent having acceptable butyrylcholinesterase inhibition, antioxidant and copper-chelation activities. J. Pharm. Sci. 2021;29:23–38. doi: 10.1007/s40199-020-00378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucas M., Freitas M., Silva M.S., Fernandes E., Ribeiro D. Styrylchromones: Biological Activities and Structure-Activity Relationship. Oxid. Med. Cell. Longev. 2021;2021:2804521. doi: 10.1155/2021/2804521. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Taylor P., Radić Z. The cholinesterases: From genes to proteins. Annu. Rev. Pharmacol. Toxicol. 1994;33:281–320. doi: 10.1146/annurev.pa.34.040194.001433. [DOI] [PubMed] [Google Scholar]
- 19.Soreq H., Seidman S. Acetylcholinesterase—new roles for an old actor. Nat. Rev. Neurosci. 2001;2:294–302. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- 20.Çokugras A.N. Butyrylcholinesterase: Structure and physiological importance. Turk. J. Biochem. 2003;28:54–61. [Google Scholar]
- 21.Mesulam M.M., Guillozet A., Shaw P., Levey A., Duysen E.G., Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 2002;110:627–639. doi: 10.1016/S0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- 22.Guillozet J.A.L., Smiley F., Mash D.C. Butyrylcholinesterase in the life cycle of amyloid plaques. Ann. Neurol. 1997;42:909–918. doi: 10.1002/ana.410420613. [DOI] [PubMed] [Google Scholar]
- 23.Darvesh S., Walsh R., Kumar R., Caines A., Roberts S., Magee D., Rockwood K., Martin E. Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2003;17:117–126. doi: 10.1097/00002093-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Hu C., Jiang L., Tang L., Zhang M., Sheng R. Design, synthesis and biological evaluation of 2-styryl-5-hydroxy-4-pyrone derivatives and analogues as multiple functional agents with the potential for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2021;44:116306. doi: 10.1016/j.bmc.2021.116306. [DOI] [PubMed] [Google Scholar]
- 25.Guzior N., Wieckowska A., Panek D., Malawska B. Recent development of multifunctional agents as potential drug candidates for the treatment of Alzheimer’s disease. Curr. Med. Chem. 2014;22:373–404. doi: 10.2174/0929867321666141106122628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mlakić M., Rajič L., Ljubić A., Vušak V., Zelić B., Gojun M., Odak I., Čule I., Šagud I., Šalić A., et al. Synthesis of new heterocyclic resveratrol analogues in milli- and microreactors: Intensification of the Wittig reaction. J. Flow Chem. 2022 doi: 10.1007/s41981-022-00239-9. [DOI] [Google Scholar]
- 27.Gülçin I. Antioxidant properties of resveratrol: A structure–activity insight. Innov. Food Sci. Emerg. Technol. 2010;11:210–218. doi: 10.1016/j.ifset.2009.07.002. [DOI] [Google Scholar]
- 28.Fauconneau B., Waffo-Teguo P., Huguet F., Barrier L., Decendit A., Merillon J.M. Comparative study of radical scavenger and antioxidant properties of phenolic compounds from vitis vinifera cell cultures using in vitro tests. Life Sci. 1997;61:2103–2110. doi: 10.1016/S0024-3205(97)00883-7. [DOI] [PubMed] [Google Scholar]
- 29.Francioso A., Mastromarino P., Masci A., d’Erme M., Mosca L. Chemistry, stability and bioavailability of resveratrol. Med. Chem. 2014;10:237–245. doi: 10.2174/15734064113096660053. [DOI] [PubMed] [Google Scholar]
- 30.Kulkarni S.S., Canto C. The molecular targets of resveratrol. Biochim. Biophys. Acta. 2015;1852:1114–1123. doi: 10.1016/j.bbadis.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Cheng G., Xu P., Zhang M., Chen J., Sheng R., Ma Y. Resveratrol-maltol hybrids as multi-target-directed agents for Alzheimer’s disease. Bioorganic Med. Chem. 2018;26:5759–5765. doi: 10.1016/j.bmc.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Xu P., Zhang M., Sheng R., Ma Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Aβ1–42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017;127:174–186. doi: 10.1016/j.ejmech.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 33.Mlakić M., Faraho I., Odak I., Talić S., Vukovinski A., Raspudić A., Bosnar M., Zadravec R., Ratković A., Lasić K., et al. Synthesis, photochemistry and computational study of novel 1,2,3-triazole heterostilbenes: Expressed biological activity of their electrocyclization photoproducts. Bioorganic Chem. 2022;121:105701. doi: 10.1016/j.bioorg.2022.105701. [DOI] [PubMed] [Google Scholar]
- 34.Šagud I., Maček Hrvat N., Grgičević A., Čadež T., Hodak J., Dragojević M., Lasić K., Kovarik Z., Škorić I. Design, synthesis and cholinesterase inhibitory properties of new oxazole benzylamine derivatives. J. Enzym. Inhib. Med. Chem. 2020;35:460–467. doi: 10.1080/14756366.2019.1707197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Škorić I., Flegar I., Marinić Ž., Šindler-Kulyk M. Synthesis of the novel conjugated ω,ω′-diaryl/heteroaryl hexatriene system with the central double bond in a heteroaromatic ring: Photochemical transformation of 2,3-divinylfuran derivatives. Tetrahedron. 2006;62:7396–7407. doi: 10.1016/j.tet.2006.05.034. [DOI] [Google Scholar]
- 36.Horspool W.M., Song P.S. CRC Handbook of Organic Photochemistry and Photobiology. CRC Press; Boca Raton, FL, USA: 1995. [Google Scholar]
- 37.Griesbeck A., Oelgemöller M., Ghetti F. CRC Handbook of Organic Photochemistry and Photobiology. CRC Press; Boca Raton, FL, USA: 2012. [Google Scholar]
- 38.Ellman G.L., Courtnex K.D., Andres V., Featherstone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 39.Brand-Williams W., Cuvelier M.E., Berset C. Use of a free radical method to evaluate antioxidant activity. Food Sci. Technol. 1995;28:25–30. doi: 10.1016/S0023-6438(95)80008-5. [DOI] [Google Scholar]
- 40.Imai Y.N., Inoue Y., Nakanishi I., Kitaura K. Cl–π interactions in protein–ligand complexes. Protein Sci. 2008;17:1129–1137. doi: 10.1110/ps.033910.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koellner G., Kryger G., Millard C.B., Silman I., Sussman J.L., Steiner T. Active-site Gorge and Buried Water Molecules in Crystal Structures of Acetylcholinesterase from Torpedo californica. Mol. Biol. 2000;296:713–735. doi: 10.1006/jmbi.1999.3468. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Y., Lu X., Yang H., Chen Y., Wang F., Li J., Tang Z., Cheng X., Yang Y., Xu L., et al. Discovery of Selective Butyrylcholinesterase (BChE) Inhibitors through a Combination of Computational Studies and Biological Evaluations. Molecules. 2019;24:4217. doi: 10.3390/molecules24234217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., et al. Gaussian 16. Gaussian, Inc.; Wallingford, CT, USA: 2016. Revision C.01. [Google Scholar]
- 44.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDock-823 Tools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raves M.L., Giles K., Schrag J.D., Schmid M.F., Phillips G.N., Jr., Wah C., Howard A.J., Silman I., Sussman J.L., Doctor B.P., et al. Structure and Function of Cholinesterases and Related Proteins. Springer; Berlin/Heidelberg, Germany: 1998. Quaternary Structure of Tetrameric Acetylcholinesterase. [DOI] [Google Scholar]
- 46.Nicolet Y., Lockridge O., Masson P., Fontecilla-Camps J.C., Nachon F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 47.Zékány L., Nagypál I., Peintler G. PSEQUAD for Chemical Equilibria. Technical Distributors; Baltimore, MD, USA: 1991. [Google Scholar]
- 48.Farrugia L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999;32:837–838. doi: 10.1107/S0021889899006020. [DOI] [Google Scholar]
- 49.Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 50.Spek A.L. Structure validation in chemical crystallography. Acta Crystallogr. D. 2009;D65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farrugia L.J. It ORTEP-3 for Windows—A Version of It ORTEP-III with a Graphical User Interface (GUI) J. Appl. Cryst. 1997;30:565. doi: 10.1107/S0021889897003117. [DOI] [Google Scholar]
- 52.Macrae C.F., Edgington P.R., McCabe P., Pidcock E., Shields G., Taylor R., Towler M., van de Streek J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006;39:453–457. doi: 10.1107/S002188980600731X. [DOI] [Google Scholar]
- 53.Košak U., Brus B., Knez D., Šink R., Žakel S., Trontelj J., Pišlar A., Šlenc J., Gobec M., Živin M., et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016;6:39495. doi: 10.1038/srep39495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raves M.L., Harel M., Pang Y.P., Silman I., Kozikowski A.P., Sussman J.L. Structure of acetylcholinesterase complexed with the nootropic alkaloid, (−)-huperzine A. Nat. Struct. Biol. 1997;4:57. doi: 10.1038/nsb0197-57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to privacy.