

Abstract
Each heartbeat is initiated by the action potential, an electrical signal that depolarizes the plasma membrane and activates a cycle of calcium influx via voltage-gated calcium channels, calcium release via ryanodine receptors, and calcium reuptake and efflux via calcium-ATPase pumps and sodium-calcium exchangers. Agonists of the sympathetic nervous system bind to adrenergic receptors in cardiomyocytes, which, via cascading signal transduction pathways and protein kinase A (PKA), increase the heart rate (chronotropy), the strength of myocardial contraction (inotropy), and the rate of myocardial relaxation (lusitropy). These effects correlate with increased intracellular concentration of calcium, which is required for the augmentation of cardiomyocyte contraction. Despite extensive investigations, the molecular mechanisms underlying sympathetic nervous system regulation of calcium influx in cardiomyocytes have remained elusive over the last 40 years. Recent studies have uncovered the mechanisms underlying this fundamental biologic process, namely that PKA phosphorylates a calcium channel inhibitor, Rad, thereby releasing inhibition and increasing calcium influx. Here, we describe an updated model for how signals from adrenergic agonists are transduced to stimulate calcium influx and contractility in the heart.
Keywords: calcium channel, sympathetic nervous system, phosphorylation, heart, excitation-contraction coupling
INTRODUCTION
Sidney Ringer, in the late nineteenth century, recognized that calcium (Ca2+) was essential for cardiac contraction. While perfusing isolated frog hearts, removal of Ca2+ from the perfusion buffer stopped the heart from contracting (1), demonstrating that external Ca2+ is required. Cardiac excitation-contraction coupling occurs when initiation of the cardiac action potential activates voltage-gated L-type Ca2+ (CaV1.2) channels, which reside in the transverse tubules (T-tubules) (2–4). The Ca2+ influx via CaV1.2 triggers opening of ryanodine receptor 2 (RyR2), leading to Ca2+ release from the sarcoplasmic reticulum (SR). Calcium then binds troponin C, enabling myofilament cross-linking and contraction. Both voltage-gated Ca2+ channels and RyR2 channels close, and relaxation of the heart ensues as Ca2+ is removed from the cytosol via the sarcoplasmic-endoplasmic reticulum Ca2+-ATPase (SERCA), modulated by phospholamban (PLB), and the Na+-Ca2+ exchanger (NCX) (Figure 1).
Figure 1.
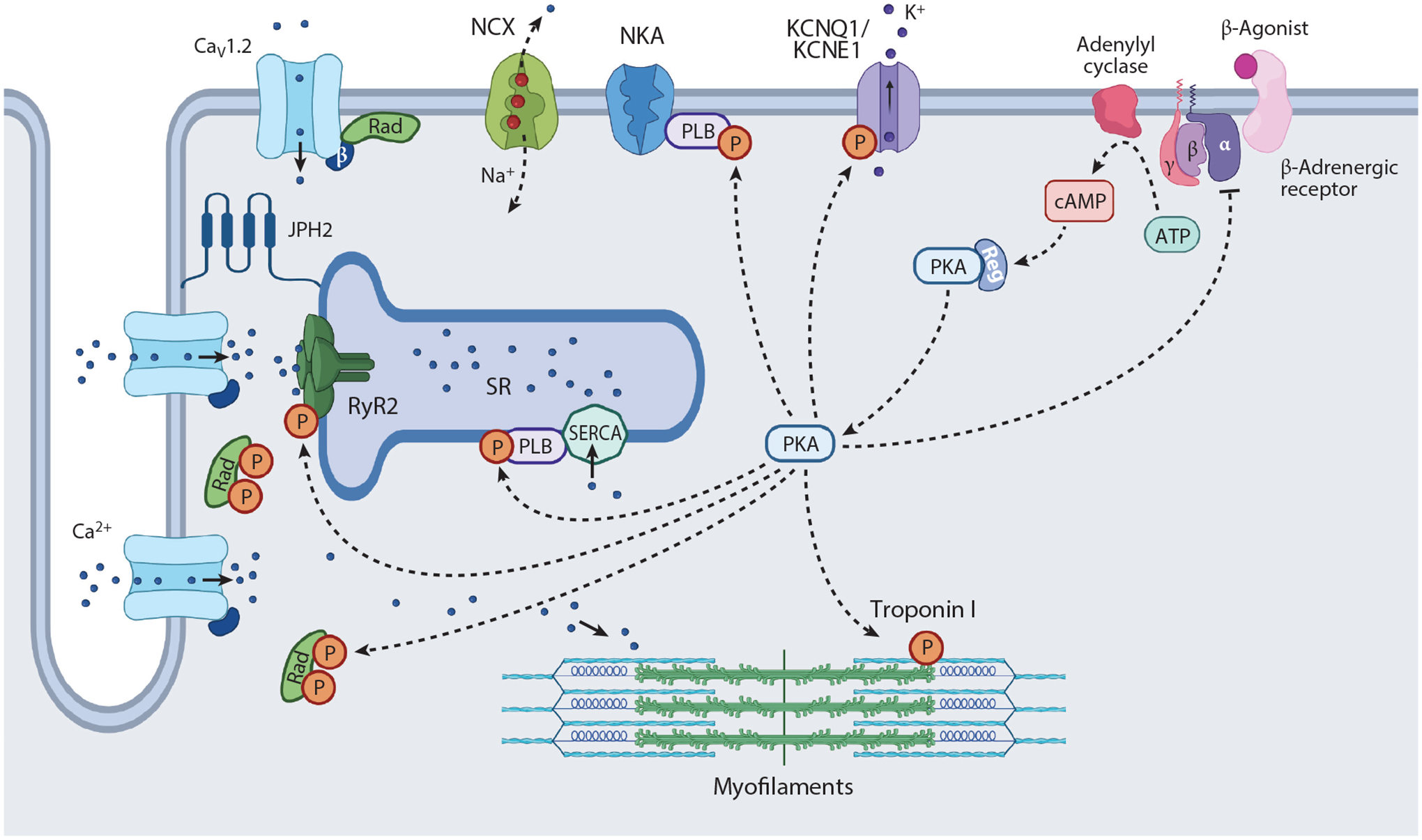
Schematic depicting β-adrenergic agonist stimulation in cardiomyocytes. Depolarization of membrane potential activates L-type Ca2+ channels, principally CaV1.2, in transverse tubules, which triggers RyR2 to open. Ca2+ stored in the SR is released, which binds troponin C, leading to myofilament cross-bridging. Thereupon, Ca2+ influx and Ca2+ release from the SR are terminated, and Ca2+ is either pumped back into the SR by the SERCA or transported out of the cell by the NCX. β-Adrenergic agonist binds to β-adrenergic receptor, which increases cAMP generation and activates PKA. PKA phosphorylates Rad, PLB, and RyR2, which causes increased Ca2+ influx, SR Ca2+ release, and SR Ca2+ reuptake, thereby augmenting inotropy and lusitropy. Phosphorylation of troponin I and KCNQ1 also contribute to enhanced lusitropy, the latter through increases in the IKS. Abbreviations: IKS, slowly activating delayed rectifier KCNQ1 channel; JPH2, junctophilin 2; NCX, Na+-Ca2+ exchanger; NKA, Na+-K+-ATPase; P, phosphorylation; PKA, protein kinase A; PLB, phospholamban; Rad, Ras associated with diabetes; Reg, regulatory; RyR2, ryanodine receptor 2; SERCA, sarcoplasmic-endoplasmic reticulum Ca2+-ATPase; SR, sarcoplasmic reticulum. Figure adapted from image created with BioRender.com.
T-tubules: transverse tubules
RyR2: ryanodine receptor 2
SR: sarcoplasmic reticulum
SERCA: sarcoplasmic-endoplasmic reticulum Ca2+-ATPase
PLB: phospholamban
NCX: Na+-Ca2+ exchanger
Cardiomyocyte contraction is augmented by β-adrenergic signaling and protein kinase A (PKA), the activation of which substantially increases the force of contraction, sarcomere shortening, and the rate of relaxation. PKA regulates several targets, including CaV1.2 channels, RyR2, and PLB, leading to increased Ca2+ entry, increased Ca2+ release from the SR, and increased Ca2+ reuptake by SERCA, respectively (5–10). Despite decades of investigations, the mechanisms responsible for adrenergic stimulation of Ca2+ channels in the heart were not well understood. Initially, the focus was on phosphorylation sites on the CaV1.2 pore-forming α1C subunit, which consists of four homologous transmembrane domains of six transmembrane segments and cytoplasmic N and C termini, and the channel’s β subunit, which interacts with an 18-residue sequence in the pore-forming subunit intracellular linker between domains I and II, termed the α-interacting domain (AID) (11–13). In this review, we offer an overview of the mechanisms responsible for adrenergic regulation of Ca2+ handling in the heart and focus on the investigations identifying the signaling responsible for PKA-mediated stimulation of Ca2+ influx in the heart.
PKA: protein kinase A
AID: α-interacting domain
SYMPATHETIC NERVOUS SYSTEM REGULATION OF CARDIAC FUNCTION
In 1895, George Oliver and Edward Albert Sharpey-Schäfer (14) showed that administering extracts of suprarenal or adrenal gland to anesthetized dogs augments both cardiac function and blood pressure and increases skeletal muscle perfusion at the expense of blood flow to nonessential vascular beds. By 1915, Walter Cannon (15, p. 108) described how the sympathetic nervous system rapidly coordinates the body’s adaptation to pain or excitement “…to meet by extra action the urgent demands of struggle or escape,” which he would later describe as the “fight or flight” response. In a series of experiments with collaborators, and drawing on the work of Max Lewandosky and T.R. Elliot, Cannon reproduced Oliver’s and Sharpey-Schäfer’s findings through electrical stimulation of the spine or splanchnic nerves, through purified adrenin and through washout release of tied-off adrenal veins (15, 16). He also found that both medical students before exams and felines subjected to either conscious confinement or anesthetized splanchnic nerve stimulation develop elevated blood sugar, adding to his theory of a multi-organ stress response that prioritizes the needs of the metabolically active immediately essential organs—the heart, lungs, brain, and skeletal muscle—at the expense of “the vegetative organs of the interior, which serve the routine needs of the body” (15, p. 108).
Ulf von Euler would win his share of a Nobel Prize for work identifying norepinephrine as the primary neurotransmitter of the sympathetic nervous system (16). Puzzled over the differences in potency of various synthetic and natural catecholamines on striated muscle as well as on vascular and airway constriction and relaxation, Raymond Ahlquist proposed the existence of separate excitatory and inhibitory α- and β-adrenergic receptors (17). This theory was lent credence in 1958, when C.E. Powell and I.H. Slater (18) published the physiologic effects of the first β-selective adrenergic-blocking drug, showing that it did not affect epinephrine- or norepinephrine-induced increases in blood pressure but did block isoproterenol-induced hypotension in cats. They additionally showed blockade of epinephrine-induced uterine relaxation and airway relaxation in different model systems. Neil Moran and Marjorie Perkins (17) blunted cardiac inotropic and chronotropic responses to catecholamines with a selective β-blocker in vagotomized and sympathectomized dogs, confirming the primacy of the β-adrenergic receptor in the cardiac fight or flight response. James Black would go on to win the Nobel Prize for his discovery of the first clinically useful beta-selective blocker, now known as propranolol (19). Other essential work that advanced the field and earned Nobel recognition includes Brian Kobilka’s and Robert Lefkowitz’s identification of the sequence and structure of the β2-adrenergic receptor as well as Earl Sutherland’s work identifying adenylyl cyclase activation and cAMP generation as key effectors of β-adrenergic stimulation (20, 21).
TARGETS OF THE SYMPATHETIC NERVOUS SYSTEM IN THE HEART
The sympathetic nervous system modulates the heart rate (chronotropy), the force of contraction (inotropy), and the rate of relaxation (lusitropy). The principal targets of PKA in ventricular and atrial cardiomyocytes are the L-type Ca2+ channels (principally CaV1.2), RyR2, and PLB, leading to increased Ca2+ entry, increased Ca2+ release from the SR, and increased Ca2+ reuptake by SERCA (5–10), respectively. This increased Ca2+ entry triggers yet more RyR2 openings, and because of greater Ca2+ reuptake and loading of the SR, increased fractional Ca2+ release (4, 22, 23). PKA also phosphorylates troponin I (TnI) and the cardiac myosin binding protein C (cMyBP-C), which reduces myofilament Ca2+ affinity, thereby promoting dissociation of Ca2+ from the myofilaments during diastole (24–26). PKA phosphorylation of cMyBP-C also accelerates cross-bridge detachment rates. The lusitropic effect of β-adrenergic stimulation, however, is predominantly mediated by the phosphorylation of PLB and the accelerated rate of SR Ca2+ reuptake (4).
The heart rate is principally controlled by the sinus node in the right atrium. Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels and L-type Ca2+ channels have been proposed to comprise the key mechanisms underlying the effects of catecholamines on heart rate (27). An increase in cAMP caused by adrenergic stimulation augments the open probability (Po) and shifts the activation curve of HCN channels, thereby increasing the rate of diastolic depolarization. Adrenergic agonists also control chronotropy by activating PKA that activates CaV1.3 channels (28–30) and RyR channels (31, 32) via phosphorylation.
As the heart rate increases, the ventricular action potential duration shortens, thereby increasing the diastolic filling time. The shortening of the action potential duration is detected as a reduction in the QT interval on an electrocardiogram. An important target for PKA-dependent phosphorylation in the regulation of the human cardiac action potential is the slowly activating delayed rectifier KCNQ1 channel (IKS) (Figure 1). PKA regulation of IKs requires a macromolecular complex consisting of the A-kinase anchoring protein (AKAP), also known as yotiao, which recruits PKA and protein phosphatase 1 (PP1) to the channel, and PKA phosphorylation of serine 27 (Ser27) on the N terminus (33).
AKAP: A-kinase anchoring protein
ADRENERGIC REGULATION OF VOLTAGE-GATED Ca2+ CHANNELS IN THE HEART
The mechanisms underlying β-adrenergic activation of Ca2+ influx in both atrial and ventricular cardiomyocytes have been studied for decades, with seminal studies demonstrating that activation of PKA is required (34–37). β-Adrenergic agonists increase Ca2+ current by 2–3 fold, principally by increasing the Po of the channel, shifting from inactive and low Po mode (modes 0 and 1, respectively) to mode 2, which is marked by high Po, with long and frequent openings (5, 34–37). Adrenergic agonists may also increase the mobilization of a subsarcolemmal pool of CaV1.2-cargo-carrying endosomes, leading to increased T-tubule sarcolemmal CaV1.2 abundance (38).
How PKA controls Ca2+ influx in the heart has been controversial. Moreover, heterologous expression systems proved to be unreliable in recapitulating the PKA-mediated activation of Ca2+ channels, making it challenging to establish mechanisms. Studies have focused on several possible approaches to establish regulatory mechanisms: (a) identifying PKA regulatory sites on the principal Ca2+ channel subunits in the heart, α1C and β2 (Figure 2a); (b) identifying a role for proteolytic cleavage of the C terminus in regulating PKA activation of CaV1.2; and (c) identifying a role for AKAPs in modulating adrenergic responsiveness. We review the major studies exploring these regulatory processes.
Figure 2.
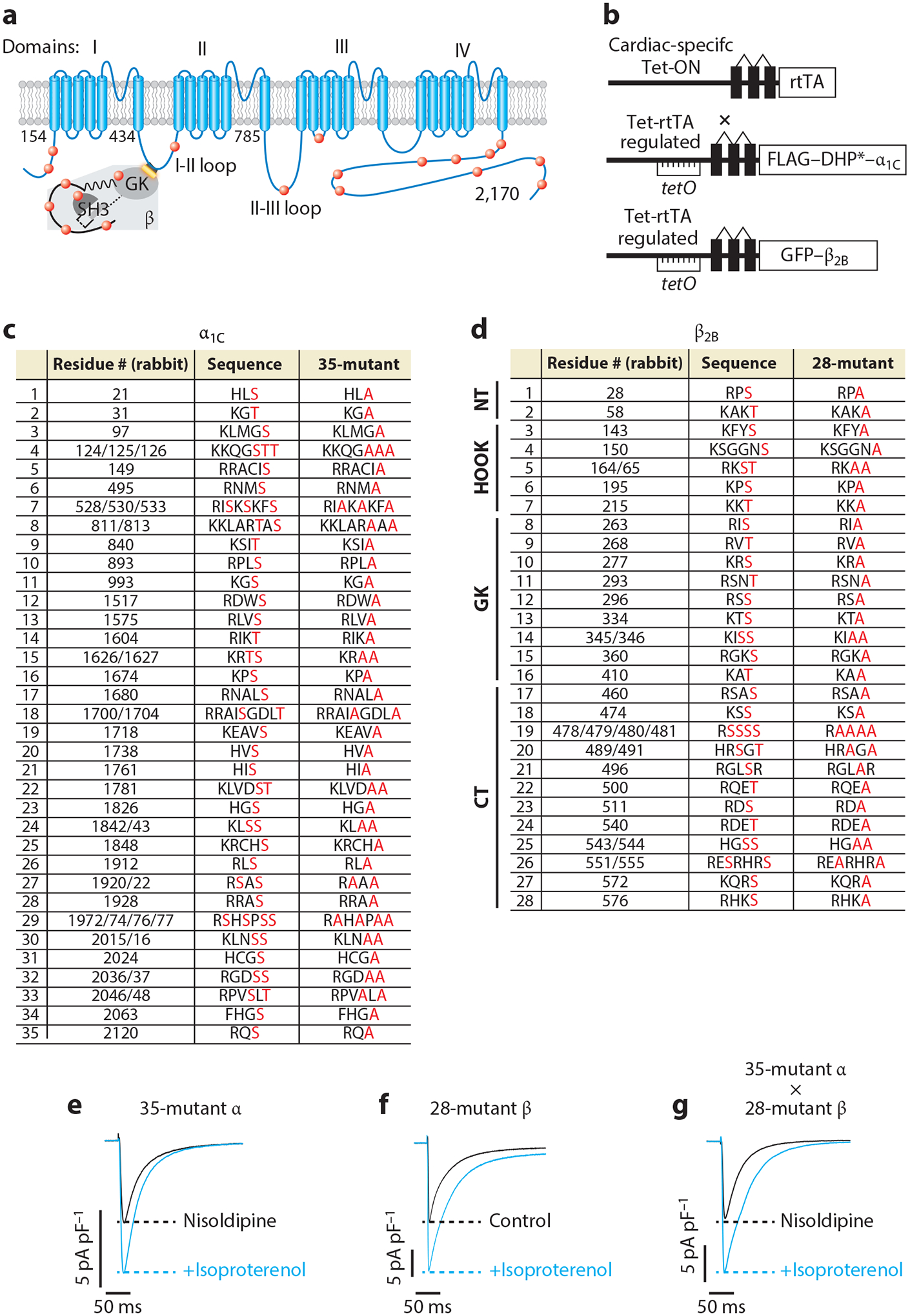
β-Adrenergic regulation of CaV1.2 does not require protein kinase A (PKA) phosphorylation of α1C and β subunits. (a) Schematic of rabbit cardiac α1C and β subunits. Red dots indicate some of the putative PKA phosphorylation sites. (b) Schematic of the transgene system (147) used for the creation of transgenic mice. Reverse tetracycline-controlled transactivator (rtTA) expression is driven by an α-myosin heavy chain (αMHC) promoter. The cDNAs for FLAG-DHP-resistant (DHP*) α1C or green fluorescent protein (GFP)-β2B were ligated behind tetO sequences. (c) The 35 putative PKA phosphorylation sites in rabbit α1C. The 51 residues in red, denoting predicted phosphorylation sites or within the immediate region of the predicted phosphorylation site, were replaced with alanine in the 35-mutant α1C transgenic mice. (d) The 37 residues within 28 sites are either predicted phosphorylation sites or within the immediate vicinity of predicted phosphorylation sites and were mutated to alanine in the 28-mutant β2B transgenic mice. The sites are distributed in the N-terminal (NT), HOOK, guanylate kinase–like (GK), and C-terminal (CT) domains of β2B. (e–g) Exemplar whole-cell CaV1.2 currents of 35-mutant α cardiomyocytes, 28-mutant β transgenic mice cardiomyocytes, and 35-mutant α X 28-mutant β transgenic mice cardiomyocytes. Figure adapted from Reference 68.
PHOSPHORYLATION OF α1C AND β2B
Full-length α1C contains a residue phosphorylated by PKA, Ser1928, that is absent from channels with cleavage of the distal 30-kDa C terminus (39–43). The phosphorylation of Ser1928 was demonstrated in dissociated rat cardiomyocytes exposed to β-adrenergic agonists using a phospho-epitope-specific antibody (44). Some studies showed that phosphorylation of Ser1928 was required for PKA-mediated upregulation of a heterologously expressed Ca2+ current (45, 46), although a subsequent study reported PKA-induced activation of Ca2+ channels with an α1C subunit truncated at residue 1905 (47). Several phosphorylation sites on the C terminus of the β subunit have also been identified (47, 48). The inability to reliably reconstitute PKA regulation using heterologous expression studies led the O’Rourke laboratory (49) to use adenovirus to express in cardiomyocytes a dihydropyridine (DHP)-resistant mutant α1C subunit harboring alanine substitution of the Ser1928 residue. DHP resistance enabled the pharmacological discrimination of endogenous DHP-sensitive CaV1.2 channels from the virally transduced DHP-resistant Ca2+ current. They found that phosphorylation of Ser1928 was not required for β-adrenergic stimulation of CaV1.2. Additionally, using adenoviral overexpression of mutant β2 subunits in cardiomyocytes, they demonstrated that phosphorylation of Ser478 and Ser479 of the β2 subunit was not required. Similarly, using adenoviral overexpression of β2 subunits, the Colecraft group (50) demonstrated that phosphorylation of Ser459 of the β2 subunit was also not required for β-adrenergic stimulation of Ca2+ currents in cardiomyocytes.
DHP: dihydropyridine
Experiments using knockin mice provided more definitive evidence that phosphorylation of neither Ser1928 on the pore-forming α1C subunit nor multiple sites on the C terminus of the auxiliary β2 subunit were necessary for β-adrenergic stimulation of Ca2+ currents in cardiomyocytes. Alanine substitution of Ser1928 had no effect on basal or β-adrenergic agonist stimulation of Ca2+ current in isolated ventricular cardiomyocytes (51). Insertion of a stop codon designed to delete the variable C terminus of the β2 subunit, which included all known PKA phosphorylation sites in β, did not prevent β-adrenergic regulation of CaV1.2 (52). Furthermore, mice with both alanine substitution of α1C Ser1928 and deletion of the C-terminal residues of the β2 subunit did not attenuate β-adrenergic stimulation of the CaV1.2 current (52), suggesting that regulation was not redundant at least for these residues. Thereafter, the Catterall group (53) identified two previously unrecognized phosphorylation sites in the C terminus of the skeletal muscle CaV1.1 channel by mass spectrometry (MS). These putative phosphorylation sites were conserved in CaV1.2 α1C subunits at Ser1700 and threonine 1704 (Thr1704) (53). Ser1700 was predicted to be a substrate for PKA and Ca2+/calmodulin-dependent protein kinase II (CaMKII), whereas Thr1704 was predicted to be a substrate of casein kinase II.
MS: mass spectrometry
TESTING COMBINATIONS OF PHOSPHORYLATION SITES IN α1C AND β2B SUBUNITS
Based on the lack of progress in identifying regulatory mechanisms, we felt that a combination of phosphorylation sites on the α1C and β2 subunits could explain the findings described above (Figure 2a). Overexpression of α1C or β subunits can attenuate the adrenergic stimulation of the Ca2+ channels in the heart (50, 54–56) and can induce cardiac dysfunction or apoptosis (57–60). Therefore, we developed an approach of using a doxycycline-inducible, tissue-specific, transgenic mouse–expressing FLAG-epitope-tagged, DHP-resistant α1C, which preserves hormonal regulation of CaV1.2 by limiting CaV1.2 overexpression (61) (Figure 2b). Similar to the work of O’Rourke and colleagues (49), we were able to distinguish between native DHP-sensitive CaV1.2 channels and transgenic DHP-resistant CaV1.2 using nisoldipine, a DHP antagonist. We have also used a similar approach to express mutant tetrodotoxin-sensitive or lidocaine-resistant NaV1.5 channels in the heart (62–64). Initially, we generated two transgenic mice with inducible cardiomyocyte-specific expression: (a) mice with N-terminal 3X FLAG-epitope–tagged, DHP-resistant α1C, designated pseudowild-type (pWT) α1C; and (b) mice with alanine substitutions of Ser1700 and Thr1704 as well as deletion of the presumed proteolytic cleavage site. We found that isoproterenol and forskolin regulation of the DHP-resistant channels in isolated pWT and S1700A/T1704A mice cardiomyocytes were equivalent, implying that phosphorylation of these two residues is not essential for β-adrenergic stimulation of Ca2+ currents (61).
Subsequently, S1700A and combined S1700A/T1704A knockin mice were generated by the Catterall group (65, 66) and were reported to have reduced basal and β-adrenergic-induced activation of Ca2+ currents. They based their conclusion, however, on an unconventional metric: the difference in absolute current amplitude rather than the fold increase after isoproterenol. Their metric is valid if the surface density of Ca2+ channels was unchanged, yet basal Ca2+ currents were substantially reduced (65, 66). Hofmann and colleagues (67) independently created S1700A/T1704A knockin mice, concluding that isoproterenol stimulated Ca2+ current in the control and mutant S1700A/T1704A cardiomyocytes to the same extent. Furthermore, Hofmann recalculated Catterall’s data and showed that in both groups’ knockin mice, the β-adrenergic stimulation for WT and mutant channels was equivalent. This confirmed our initial findings (61), which we have further substantiated with additional transgenic mice (68–70).
The use of DHP-resistant transgenic mice is an ideal approach to test the hypothesis that more than one phosphorylation site on α1C is required for adrenergic regulation. We identified conserved PKA consensus sequences, using bioinformatic methods, in the α1C subunit of five species: mouse, rat, rabbit, guinea pig, and human (Figure 2c). We then generated transgenic mice in which 17 conserved consensus PKA phosphorylation sites that were not previously studied and 5 conserved PKA/CaMKII phosphorylation sites known to be nonessential (including Ser1700 and Thr1704) were mutated to alanine (42, 49, 51, 52). Surprisingly, none were necessary (70). Could the functionally relevant PKA targets in α1C be different among these five species? To test this possibility, we generated transgenic mice with alanine mutations at additional potential PKA sites in the mouse channel that were not conserved in other species, with a total of 51 alanine substitutions at 35 sites (Figure 2c). DHP-resistant 35-α mutant Ca2+ channels were stimulated by isoproterenol or forskolin (Figure 2e), indicating that phosphorylation of α1C is not essential for adrenergic stimulation of CaV1.2 (68). The simplest explanation for this finding is that the β subunit contains previously unappreciated PKA phosphorylation sites (52). Similar to the approach used for testing putative phosphorylation sites in the α1C subunit, we generated transgenic mice expressing mutant human β2B subunits in which 37 alanine substitutions were made within 28 consensus PKA phosphorylation sites (68) (Figure 2d). Because there is no pharmacological approach to select for channels with mutant β subunits, we relied on the overexpression of mutant β subunits to competitively replace WT β2 subunits in the CaV1.2 complex, which was confirmed using coimmunoprecipitation studies. CaV1.2 channels with the mutant β subunit displayed a normal isoproterenol- or forskolin-induced increase in peak Ca2+ current (Figure 2f) and a hyperpolarizing shift in the V50 of activation. Furthermore, mice expressing the 35-α mutant and the 28-β mutant were crossed, and the DHP-resistant channels in these double-mutant (α1C and β2) progeny also displayed normal adrenergic regulation (68) (Figure 2g). These studies rule out a role for all consensus PKA phosphorylation sites in α1C and show that phospho-regulatory sites on α1C and β2B are not redundant and do not each fractionally contribute to the stimulatory effect of β-adrenergic agonists.
ROLE OF β SUBUNIT IN ADRENERGIC REGULATION OF CaV1.2
Global or cardiac-specific deletion of the dominant Cacnb2 gene is embryonic lethal due to abnormal heart development (71). Similarly, in cells heterologously expressing CaV1.2 channels, β is obligatory for α1C trafficking to the plasma membrane and for normalizing channel activation and inactivation gating properties (72–75). Unexpectedly, cardiomyocyte-specific, conditional deletion of the Cacnb2 gene in adult mice caused only a modest 29% reduction in Ca2+ current, with no obvious cardiac impairment (76). There were two possible interpretations for this unexpected result. First, in adult myocytes, β binding to α1C is not absolutely required for CaV1.2 surface expression. Alternatively, the remaining ~4% of β2 expression and the relatively low level of β3 expression could be sufficient for the trafficking and function of Ca2+ channels in the adult heart.
Although PKA phosphorylation of Ser or Thr residues in the β subunit is not required for adrenergic activation of CaV1.2 (68, 69), we suspected that β subunit binding to α1C may be required. We created transgenic mice with mutations in the AID of the α1-subunit I-II loop, thereby preventing the high-affinity binding of β and α1C subunits (11–13, 77). When expressed in cardiomyocytes, the FLAG-tagged AID-mutant α1C did not bind the β subunit. In contrast to heterologously expressed AID-mutant α1C channels, but consistent with the results obtained from the conditional β2 knockout mice (76), β-less Ca2+ channels in cardiomyocytes were capable of trafficking to the dyadic membrane (69). AID-mutant α1C channels exhibited rare sojourns to the high-activity mode and had a higher propensity for blank and low activity sweeps (78). Furthermore, the β-less Ca2+ channels were completely unresponsive to isoproterenol (69). Taken together, these findings suggest that β subunits are required for Ca2+ channels to enter a high Po state even though phosphorylation of β is not required.
The DHP-resistant AID-mutant transgenic mice also offered insights into the role of β-adrenergic stimulation of CaV1.2 in the fight or flight response (69): (a) In isolated AID-mutant α1C cardiomyocytes, isoproterenol, in the presence of nisoldipine, increased fractional shortening of myocytes by only 25% compared to 100% in pWT α1C cardiomyocytes; and (b) at the organ level, 200 nM isoproterenol increased cardiac contractility by 3.3 fold, whereas in hearts isolated from the AID-mutant mice, isoproterenol increased cardiac contractility by only 1.2 fold (69). This finding was predicted from modeling studies of rabbit (79) and mouse ventricular myocytes (80–82), both of which showed that specific removal of PKA stimulation of CaV1.2 decreased both the Ca2+ transient and force below basal levels. In contrast, acute removal of RyR2 phosphorylation or PLB phosphorylation had much smaller or negligible effects.
ROLE OF C-TERMINAL PROTEOLYTIC CLEAVAGE OF α1C IN ADRENERGIC REGULATION OF CaV1.2
Posttranslational proteolytic cleavage of α1C yields a distal C-terminal truncated α1C of ~210 kDa (42, 45, 83–85). The fraction of cleaved α1C subunits in cardiomyocytes is unresolved, with estimates ranging from none to full truncation, perhaps related to species, age, and experimental conditions (86). With optimized rapid extraction conditions, such as high concentrations of the calpain inhibitor, EGTA, and all solutions and instruments precooled to 0°C, immunoblotting showed that ~50% of the detectable α1C is in its long form in heart extracts (87). In skeletal muscle, the site of cleavage was determined by MS to be at Ala1664, which corresponds to Ala1800 in cardiac α1C. The distal C-terminal fragment is presumed to remain coupled by interactions with the proximal fragment, tonically inhibiting it (42, 84, 85, 88, 89). The distal C terminus is also partially localized to the nucleus where it may modify gene expression in neurons, smooth muscle, and cardiomyocytes (90–92).
Heterologous expression of an α1C cDNA with deletion of the distal C terminus demonstrated an increased current amplitude and Po and a hyperpolarizing shift in activation, which are similar to what is observed after adrenergic stimulation of CaV1.2 in the heart. Thus, it was speculated that PKA regulation of CaV1.2 requires the proteolytic cleavage of the distal C terminus and subsequent phosphorylation-dependent release of the inhibition imparted by the distal C terminus (53, 93, 94). Furthermore, Dascal’s group (95) demonstrated a small cAMP-/PKA-dependent increase in Ca2+ current in truncated α1C subunits but not full-length α1C subunits when heterologously expressed in Xenopus oocytes. Taken together, cleavage is proposed to set the basal CaV1.2 activity, which is then augmented by adrenergic stimulation (42, 53, 84, 85, 89, 96–98). One caveat is that a truncated α1C subunit is never expressed without its distal C terminus in vivo. Furthermore, knockin mice expressing a truncated α1C at either Gly1796 or Asp1904 die at birth due to low membrane expression of CaV1.2 (98, 99), which is inconsistent with the studies performed using heterologous expression.
The functional relevance of proteolytic cleavage of α1C has not been demonstrated in cardiomyocytes. Indirect evidence, consisting of MS analysis of skeletal muscle α1S proteolytic peptides and sequence alignments of α1S and α1C, was the basis for the hypothesis that Ala1800 within the 1798NNAN motif is the α1C proteolytic site (85). The persistence of α1C cleavage in cardiomyocytes after deletion of the 1798NNAN motif could result from the presence of a nearby similar motif, 1794NANI1797. Thus, we created a transgenic mouse with cardiomyocyte expression of α1C with deletion of 1794NANINNANN1802, along with deletion of a nearby PEST sequence (1769DTESP) (70). Deletion of these sites, but not the PEST sequence alone, yielded a channel that was resistant to proteolytic cleavage. Further, we found that C-terminal proteolytic cleavage of α1C is not required for β-adrenergic stimulation of CaV1.2 in the heart. Although these experiments clearly rule out the necessity of C-terminal proteolytic cleavage for adrenergic agonist stimulation of Ca2+ channels in the heart, the methodology cannot address whether the C-terminal fragment is required for regulating transcription in the heart (91, 92).
ROLE OF A-KINASE-ANCHORING PROTEINS IN ADRENERGIC REGULATION OF CaV1.2
AKAPs may determine the specificity and the speed by which targets, such as ion channels, respond to sympathetic nervous system stimulation (100). During adrenergic stimulation, Ca2+ current amplitude increases at only half the rate of rising cAMP levels (101), implying that phosphorylation is rate limiting and that disrupting localization of PKA should be evident by the reduced rate and amplitude of stimulation. The importance of AKAPs in the adrenergic regulation of CaV1.2 is supported by considerable experimental evidence. Three groups have independently shown that β-adrenergic modulation of Ca2+ currents in neonatal rat, adult rat, and adult mouse cardiomyocytes is markedly blunted by the intracellular dialysis, via a patch pipette, of peptides designed to competitively disrupt the binding of PKA to an AKAP (45, 102–104). Although considerable evidence initially pointed to important roles for Akap5 and Akap7 (45, 53, 89, 104–107), mice in which Akap5, Akap7, or both are deleted retained full β-adrenergic upregulation of CaV1.2 channels in ventricular myocytes in response to isoproterenol (108, 109). Thus, it is likely that an AKAP other than Akap5 and Akap7 is required for β-adrenergic regulation of CaV1.2, or that several different AKAPs can permit PKA localization to the channel.
Cypher/Zasp, a member of the PDZ–LIM domain family that directly complexes with Z-line-associated proteins such as α-actinin-2, was proposed as an AKAP that regulates CaV1.2 function (110). In Cypher/Zasp null mice, the cell surface density of CaV1.2 channels and the basal Ca2+ current were significantly reduced despite a large increase in total CaV1.2 protein, indicating that the assembly of CaV1.2, insertion into the cell surface, and stability of CaV1.2 on the cell surface may be impaired. Isoproterenol stimulated Ca2+ currents by nearly twofold in Cypher/Zasp null mice, which was decreased by ~30% compared to control hearts (see 110, figure 3b). Taken together, these results suggest that Cypher/Zasp may contribute to β-adrenergic regulation, but there is likely redundancy.
USING PROXIMITY PROTEOMICS IN THE HEART TO ELUCIDATE ADRENERGIC SIGNALING
An alternative hypothesis to explain adrenergic regulation of CaV1.2 is that PKA phosphorylates an additional protein in the Ca2+ channel complex instead of direct phosphorylation of α1C or β. The PKA target could be either a channel activator, recruited to the CaV1.2 complex, or a channel inhibitor, released from either α1C or β subunits.
Important interacting proteins may associate transiently and as such are unlikely to be detected by coimmunoprecipitation. Moreover, standard methodologies to detect protein–protein interactions have limitations and do not always represent the protein interactions within the native intracellular milieu. Even among directly interacting proteins, many may have dissociation constants >10–100 μM and fast off-rate constants (111). We developed an in vivo platform to identify the interactome of CaV1.2 by adapting the ascorbate peroxidase 2 (APEX2) methodology (112–115). Unlike other proteome network maps in cardiomyocytes, this method enabled the identification of interacting and bystander proteins within ~20 nm in living cells (68). APEX2 is ideally suited for relatively rapid dynamic changes in subcellular neighborhoods because the labeling time is relatively short compared to biotin ligases such as TurboID or BioID (114–121).
APEX2: ascorbate peroxidase 2
We created transgenic mice with inducible, cardiomyocyte-specific expression of DHP-resistant α1C or β2B with APEX2 and a V5 epitope conjugated to the N termini (Figure 3a). Fusing APEX2 to α1C and β2B did not affect β-adrenergic agonist stimulation of CaV1.2 current (68) or the subcellular localization of CaV1.2 channels in the T-tubules (Figure 3b). Incubating isolated ventricular cardiomyocytes with biotin-phenol followed by exposure to H2O2 induced robust biotinylation of proteins in a striated z-disk pattern, which is consistent with localization of CaV1.2 channels at T-tubules (Figure 3b). Biotinylated proteins were affinity purified in denaturing conditions using streptavidin. Western blotting and tandem mass tag synchronous precursor selection triple-stage mass spectrometry (TMT SPS MS3) demonstrated the enrichment of known interacting proteins, such as calmodulin and junctophilin, as well as bystander proteins, such as RyR and NCX. Using proximity labeling in both isolated cardiomyocytes and Langendorff-perfused hearts and TMT SPS MS3 (Figure 3c), we found that isoproterenol induced a change in the extent of biotinylation of several proteins. Increased biotinylation of the PKA catalytic subunit likely indicated recruitment to the CaV1.2 channel neighborhood, whereas decreased biotinylation of Rad (Ras associated with diabetes) was indicative of reduced accessibility to labeling, likely due to less Rad near CaV1.2 channels (Figure 3d,e). In contrast, the amount of Rad did not change in cardiomyocytes isolated from nontransgenic mice (68).
Figure 3.
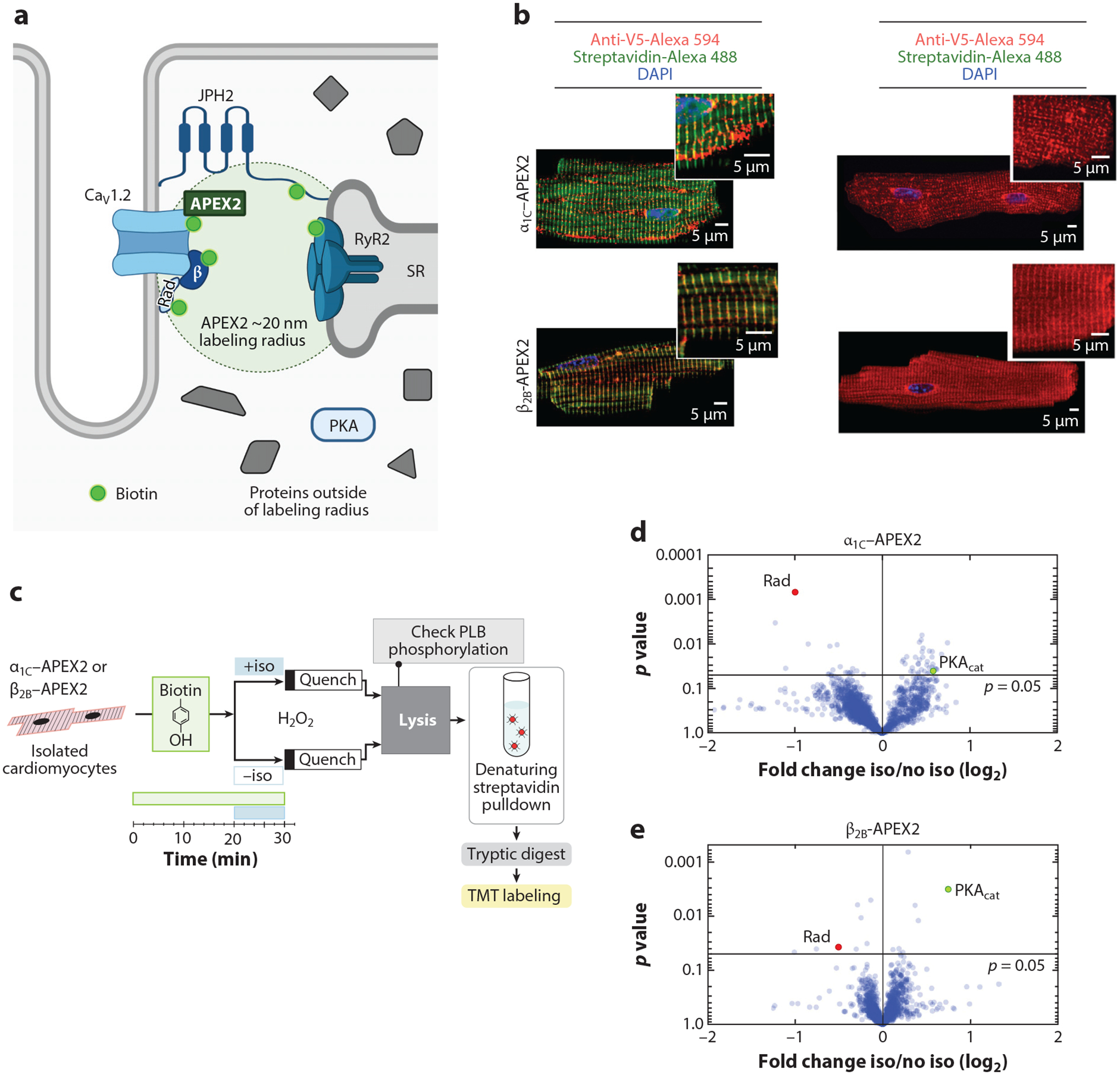
Proximity labeling using APEX2 to identify the mechanism underlying adrenergic agonist-induced augmentation of Ca2+ current. (a) Schematic depicting localization of APEX2-conjugated CaV1.2 channels in the dyadic space of cardiomyocytes. Panel a adapted from image created with BioRender.com. (b) Immunofluorescence of cardiomyocytes isolated from α1C-APEX2- and β2B-APEX2-expressing mice exposed to biotin-phenol and H2O2 or no H2O2. Nuclear labeling with DAPI stain. (c) Schematic of workflow for isolated cardiomyocytes. (d–e) Volcano plots of fold-change for relative protein quantification by tandem mass tag mass spectrometry of α1C-APEX2 and β2B-APEX2 samples. Non-adjusted unpaired two-tailed t-test. Rad (red dots) is reduced and PKA catalytic subunit (PKAcat; green dots) is increased. Panels b–e adapted from Reference 68. Abbreviations: APEX2, ascorbate peroxidase 2; JPH2, junctophilin 2; PLB, phospholamban; Rad, Ras associated with diabetes; RyR2, ryanodine receptor 2; SR, sarcoplasmic reticulum; TMT, tandem mass tag.
TMT SPS MS3: tandem mass tag synchronous precursor selection triple-stage mass spectrometry
Rad: Ras associated with diabetes
RAD IS THE PKA TARGET
Rad is a member of the RGK (Rad, Rem, Rem2, Gem/Kir) Ras family of proteins. In a seminal study, Gem/Kir was discovered in a yeast two-hybrid screen of MIN6 cells as an interacting protein of CaVβ3 (122). All RGK proteins are intracellular inhibitors of high-voltage-activated Ca2+ channels (123–126). Sequence analysis and electrophysiological studies suggest that the interaction between RGK proteins and Ca2+ channels has been strictly conserved, originating prior to the deuterostome/protostome split (127). Rad, originally discovered as a protein overexpressed in skeletal muscle of patients with diabetes, is expressed in heart, placenta, lung, and skeletal muscle (128). Mice with deletion of Rad, either globally or specifically in heart, displayed increased basal Ca2+ currents with activation at lower voltages and reduced β-adrenergic stimulation of Ca2+ channels in isolated cardiomyocytes (129–131). Conditional Rad knockout mice also have elevated heart rates at baseline and during sleep (132).
RGK: Rad, Rem, Rem2, Gem/Kir
In patients with end-stage heart failure undergoing transplantation, Rad mRNA and protein levels were substantially reduced compared to controls (133). Could this be a natural compensatory mechanism to enhance Ca2+ entry and increase contractility, as was observed in Rad knockout mice (129–131, 134)? A reduction in Rad could also account, at least in part, for decreased adrenergic reserve. Furthermore, Rad may play an important role in cardiac hypertrophy. Within 24 h after transverse aortic constriction in rats, Rad protein levels decreased, which persisted for at least 14 days (133). In human embryonic stem cell (H9 cell line)-derived cardiomyocytes, deletion of Rad caused a hypertrophic phenotype, which was attenuated by blocking the increased Ca2+ current with verapamil (135).
The robust heterologous reconstitution of PKA regulation of CaV1.2 currents has been long pursued (136) but was unachievable with expression of only α1C and β subunits (Figure 4a). We found that Rad was the missing ingredient (68). Applying forskolin to HEK293T cells expressing WT α1C + β2B + Rad increased the maximal conductance (Gmax) (Figure 4b) and shifted the V50 for activation, similar to our observations in cardiomyocytes. Previous studies identified a single PKA phosphorylation site on the C terminus of Rad (137). Using MS, we identified an additional three phosphorylated residues in Rad after stimulation with forskolin (68). Alanine substitutions of these four Ser, two on the N terminus and two on the C terminus of Rad (4SA-mutant), prevented the forskolin-induced increase in Gmax and V50 shift. Phosphorylation of two Ser residues in the C-terminal polybasic membrane region of Rad is crucial to PKA regulation of Cav1.2. Alanine substitutions at Ser272 and Ser300 (2SA-mutant) prevented both the forskolin-induced increase in Gmax (Figure 4c) and V50 shift (68). Similar to cardiomyocytes, the forskolin-induced stimulation of Ca2+ channels was not dependent on phosphorylation of either the α1C or β2B subunits in HEK cells expressing Rad. The phosphorylation sites in Rad are conserved across species and across other members of the RGK GTPase family (68). Thus, at baseline, Rad, inhibits CaV1.2; on adrenergic activation, PKA phosphorylates Rad, which releases this inhibition. Disinhibition equals activation.
Figure 4.
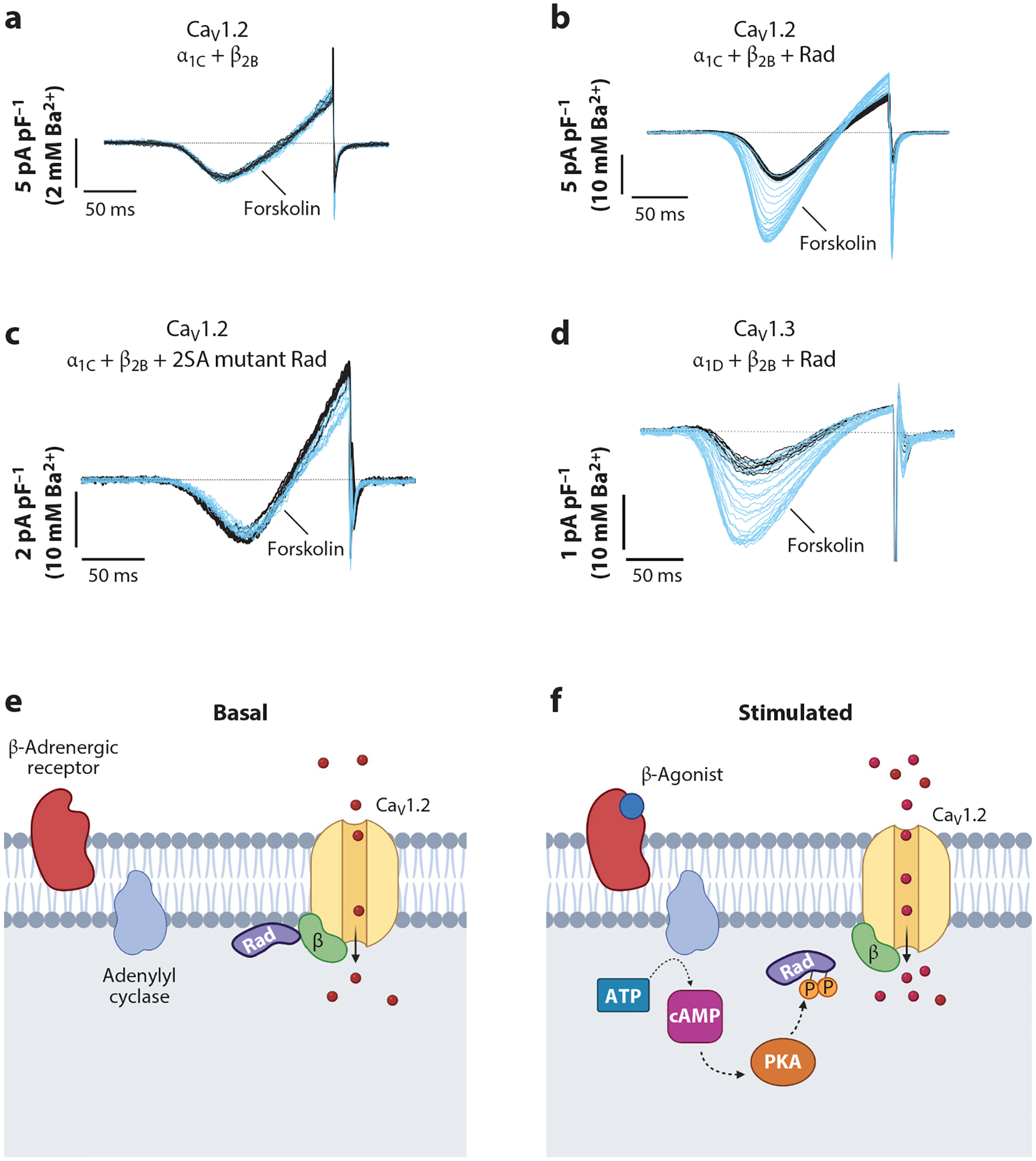
Phosphorylation of Rad is required for cAMP-PKA activation of voltage-gated Ca2+ channels. (a–c) Ba2+ current elicited by voltage ramp every 10 s, with black traces obtained before and blue traces obtained after forskolin. The α1C, β2B, and WT and mutant Rad were heterologously expressed in HEK293T cells. (d) CaV1.3 α1D, β2B, and Rad were heterologously expressed in HEK293T cells. Panels a–d adapted from Reference 68. (e–f) Proposed models of β-adrenergic regulation of CaV1.2 channels. PKA phosphorylates several residues on Rad, causing dissociation of Rad from the CaV1.2 complex and therefore increased Ca2+ influx. Panels e and f adapted from image created with BioRender.com. Abbreviations: P, phosphorylation; PKA, protein kinase A; Rad, Ras associated with diabetes; WT, wild-type.
ADRENERGIC CAV1.2 ACTIVATION VIA RAD PHOSPHORYLATION CONVERGES AT THE α1C I-II LOOP
Rad can inhibit CaV1.2 via binding to either the α1C or β subunits (138). We demonstrated that PKA phosphorylation of Rad markedly reduces Rad binding to the β subunit. Moreover, eliminating Rad binding to β, via mutation of the interaction site on either β or Rad, prevented both Rad inhibition and the adrenergic regulation of Ca2+ channels (68). The requirement of the β subunit is consistent with the prior findings that adrenergic regulation of CaV1.2 requires the interaction of α1C and β subunits (69).
To explore how distal conformational changes involving Rad interaction with the CaVβ subunit and phosphorylation-dependent signaling are ultimately conveyed to the channel pore-domain, we generated transgenic mice with expression of CaV1.2 α1C subunits with flexibility-inducing polyglycine substitutions in the I-II loop (GGG-α1C) (78). These mutations have been previously shown to disrupt coupling between the pore-domain and the AID of the α1C subunit (77, 139, 140). Introducing three glycine residues that disrupt a rigid IS6-AID helix reduced basal Po despite intact binding of CaVβ to the α1C I-II loop and eliminated β-adrenergic agonist stimulation of CaV1.2 current. Thus, we speculate that (a) CaVβ binding to α1C stabilizes an increased Po gating mode by a mechanism that requires an intact rigid linker between the β subunit binding site in the I-II loop and the channel pore; and (b) the release of Rad-mediated inhibition of Ca2+ channel activity by β-adrenergic agonists requires phosphorylation of the C terminus of Rad, which leads to decreased binding of Rad to the β subunit. With the dissociation of Rad, the β subunit can stabilize a high Po gating mode.
RGK GTPase REGULATION OF CaV1.3 AND CaV2.2
The family of RGK GTPases is known to inhibit other voltage-gated Ca2+ channels. We speculated that phosphorylation of RGK GTPases could be a common mechanism to regulate voltage-gated Ca2+ channels that bind β subunits. CaV1.3 channels contribute to pacemaker activity in the sinus node and atrial cells (141) and are expressed in adrenal chromaffin cells (142). Similar to CaV1.2 channels, expression and PKA phosphorylation of Rad are also required for PKA-dependent activation of heterologously expressed CaV1.3 channels (68) (Figure 4d). CaV2.2 channels are expressed in presynaptic terminals in the brain. Coexpression of Rad or Rem enabled PKA-dependent activation of CaV2.2 channels in HEK cells (68). Thus, it is apparent that this mechanism of regulation of voltage-gated Ca2+ channels is modular.
CONCLUSIONS AND FUTURE DIRECTIONS
Despite the importance of adrenergic regulation of cardiac function, elucidating the underlying mechanisms for β-adrenergic stimulation of Ca2+ influx has been difficult. Similar to the inhibition of SERCA by PLB and the release of inhibition by PKA and CaMKII phosphorylation of PLB, the adrenergic regulation of Ca2+ channels involves a phosphorylation-dependent disinhibition mediated by RGK GTPases (Figure 4e,f). Phosphorylation of α1C or β2B subunits is not required in the heart (68). The relevant C-terminal phosphorylation sites of Rad are conserved through evolution and in all RGK proteins, implying the importance of this signaling pathway in other tissues. We speculate that this mechanism of regulation is advantageous by permitting tissue-specific modulation because CaV1.2 is expressed ubiquitously, whereas Rad is not, and the extent of adrenergic regulation varies substantially in different organs, with the heart being the greatest. Although phosphorylation of Ser1928 in the C terminus of α1C is not required for adrenergic regulation of CaV1.2 in cardiomyocytes (49, 51, 68, 70) or for adrenergic agonist stimulation of heterologously expressed CaV1.2 channels in HEK cells (68) and Xenopus oocytes (143), it has been shown to modulate Ca2+ currents in hippocampal neurons and vascular smooth muscle (106, 144, 145). The mechanisms that impart a stimulatory effect on Ser1928 phosphorylation in hippocampal neurons and vascular smooth muscle, but not cardiomyocytes, are not known. The experimental conditions may be important, as Ser1928-dependent stimulation in hippocampal neurons was recorded in the presence of Bay K 8644, an activator of CaV1.2 (145). As opposed to the physiological role for β-adrenergic agonist stimulation of Ca2+ currents in the heart, β-adrenergic agonists promote vasorelaxation in the vasculature, dependent on activation of K+ channel–induced hyperpolarization and a subsequent reduction in Ca2+ influx in vascular smooth muscle. In vascular smooth muscle, the role of Ser1928 phosphorylation may be linked to hyperglycemia-mediated activation of purinergic receptors, which promotes Ca2+ influx and increased vasoreactivity (106, 146).
Identification of the mechanisms responsible for adrenergic regulation of CaV1.2 and CaV1.3 enables substantial additional investigations. The next steps will be to create genetically altered mice with mutations of the phosphorylation sites of Rad or the interacting sites of β2B or Rad. Conditions such as heart failure or hypertrophy alter the global expression of Rad in animals and humans (133), but we do not yet know whether heart failure causes changes in the CaV1.2 macromolecular complex or whether signaling that affects Rad phosphorylation is altered. Harnessing the mechanism by which the sympathetic nervous system regulates Ca2+ influx could be an innovative approach for the treatment of heart failure and arrhythmias.
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants NIH T32 HL120826 and F31 HL158232 and National Science Foundation grant 1644869 to A.P.; NIH K08 grant HL151969, American Heart Association Career Development Award 35320208, and the Louis V. Gerstner, Jr. Scholars Program to J.K.; and NIH R01 HL121253, R01 HL140934, R01 HL155377, and R01 HL146149 to S.O.M.
Footnotes
DISCLOSURE STATEMENT
The authors are not of aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Moore B 1911. In memory of Sidney Ringer [1835–1910]: some account of the fundamental discoveries of the great pioneer of the bio-chemistry of crystallo-colloids in living cells. Biochem. J 5:ib3–xix [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabiato A, Fabiato F. 1975. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J. Physiol 249:469–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabiato A, Fabiato F. 1979. Calcium and cardiac excitation-contraction coupling. Annu. Rev. Physiol 41:473–84 [DOI] [PubMed] [Google Scholar]
- 4.Bers DM. 2002. Cardiac excitation-contraction coupling. Nature 415:198–205 [DOI] [PubMed] [Google Scholar]
- 5.Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC. 1986. Mechanisms of calcium channel modulation by β-adrenergic agents and dihydropyridine calcium agonists. J. Mol. Cell. Cardiol 18:691–710 [DOI] [PubMed] [Google Scholar]
- 6.Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. 1983. β-Adrenergic stimulation of phospholamban phosphorylation and Ca2+-ATPase activity in guinea pig ventricles. J. Biol. Chem 258:464–71 [PubMed] [Google Scholar]
- 7.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, et al. 2000. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101:365–76 [DOI] [PubMed] [Google Scholar]
- 8.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, et al. 2003. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113:829–40 [DOI] [PubMed] [Google Scholar]
- 9.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, et al. 2010. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Investig 120:4388–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Potenza DM, Janicek R, Fernandez-Tenorio M, Camors E, Ramos-Mondragon R, et al. 2019. Phosphorylation of the ryanodine receptor 2 at serine 2030 is required for a complete β-adrenergic response. J. Gen. Physiol 151:131–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen YH, Li MH, Zhang Y, He LL, Yamada Y, et al. 2004. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature 429:675–80 [DOI] [PubMed] [Google Scholar]
- 12.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. 2004. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron 42:387–99 [DOI] [PubMed] [Google Scholar]
- 13.Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr. 2004. Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429:671–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliver G, Schäfer EA. 1895. The physiological effects of extracts of the suprarenal capsules. J. Physiol 18:230–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannon WB. 1915. Bodily Changes in Pain, Hunger, Fear and Rage: An Account of Recent Researches into the Function of Emotional Excitement. New York: D Appleton & Co. [Google Scholar]
- 16.Snyder SH. 2006. Turning off neurotransmitters. Cell 125:13–15 [DOI] [PubMed] [Google Scholar]
- 17.Moran NC, Perkins ME. 1958. Adrenergic blockade of the mammalian heart by a dichloro analogue of isoproterenol. J. Pharmacol. Exp. Ther 124:223–37 [PubMed] [Google Scholar]
- 18.Powell CE, Slater IH. 1958. Blocking of inhibitory adrenergic receptors by a dichloro analog of isoproterenol. J. Pharmacol. Exp. Ther 122:480–88 [PubMed] [Google Scholar]
- 19.Black JW, Crowther AF, Shanks RG, Smith LH, Dornhorst AC. 1964. A new adrenergic β-receptor antagonist. Lancet 1:1080–81 [DOI] [PubMed] [Google Scholar]
- 20.Sutherland EW. 1972. Studies on the mechanism of hormone action. Science 177:401–8 [DOI] [PubMed] [Google Scholar]
- 21.Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. 1988. Chimeric α2-, β2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240:1310–16 [DOI] [PubMed] [Google Scholar]
- 22.Shannon TR, Ginsburg KS, Bers DM. 2002. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ. Res 91:594–600 [DOI] [PubMed] [Google Scholar]
- 23.Hunter DR, Haworth RA, Berkoff HA. 1983. Modulation of cellular calcium stores in the perfused rat heart by isoproterenol and ryanodine. Circ. Res 53:703–12 [DOI] [PubMed] [Google Scholar]
- 24.Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, et al. 2015. Phosphorylation of cardiac myosin-binding protein-C is a critical mediator of diastolic function. Circ. Heart Fail 8:582–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li L, Desantiago J, Chu G, Kranias EG, Bers DM. 2000. Phosphorylation of phospholamban and troponin I in β-adrenergic-induced acceleration of cardiac relaxation. Am. J. Physiol. Heart Circ. Physiol 278:H769–79 [DOI] [PubMed] [Google Scholar]
- 26.Tong CW, Wu X, Liu Y, Rosas PC, Sadayappan S, et al. 2015. Phosphoregulation of cardiac inotropy via myosin binding protein-C during increased pacing frequency or β1-adrenergic stimulation. Circ. Heart Fail 8:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mangoni ME, Nargeot J. 2008. Genesis and regulation of the heart automaticity. Physiol. Rev 88:919–82 [DOI] [PubMed] [Google Scholar]
- 28.Zaza A, Robinson RB, DiFrancesco D. 1996. Basal responses of the L-type Ca2+ and hyperpolarization-activated currents to autonomic agonists in the rabbit sino-atrial node. J. Physiol 491(Part 2):347–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choate JK, Feldman R. 2003. Neuronal control of heart rate in isolated mouse atria. Am. J. Physiol. Heart Circ. Physiol 285:H1340–46 [DOI] [PubMed] [Google Scholar]
- 30.Matthes J, Huber I, Haaf O, Antepohl W, Striessnig J, Herzig S. 2000. Pharmacodynamic interaction between mibefradil and other calcium channel blockers. Naunyn-Schmiedebergs Arch. Pharmacol 361:578–83 [DOI] [PubMed] [Google Scholar]
- 31.Rigg L, Heath BM, Cui Y, Terrar DA. 2000. Localisation and functional significance of ryanodine receptors during β-adrenoceptor stimulation in the guinea-pig sino-atrial node. Cardiovasc. Res 48:254–64 [DOI] [PubMed] [Google Scholar]
- 32.Vinogradova TM, Bogdanov KY, Lakatta EG. 2002. β-Adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res 90:73–79 [DOI] [PubMed] [Google Scholar]
- 33.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, et al. 2002. Requirement of a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295:496–99 [DOI] [PubMed] [Google Scholar]
- 34.Yue DT, Herzig S, Marban E. 1990. β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. PNAS 87:753–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirano Y, Suzuki K, Yamawake N, Hiraoka M. 1994. Multiple kinetic effects of β-adrenergic stimulation on single cardiac L-type Ca channels. Am. J. Physiol 266:C1714–21 [DOI] [PubMed] [Google Scholar]
- 36.Herzig S, Patil P, Neumann J, Staschen CM, Yue DT. 1993. Mechanisms of β-adrenergic stimulation of cardiac Ca2+ channels revealed by discrete-time Markov analysis of slow gating. Biophys. J 65:1599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hess P, Lansman JB, Tsien RW. 1984. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311:538–44 [DOI] [PubMed] [Google Scholar]
- 38.Del Villar SG, Voelker TL, Westhoff M, Reddy GR, Spooner HC, et al. 2021. β-Adrenergic control of sarcolemmal CaV1.2 abundance by small GTPase Rab proteins. PNAS 118:e2017937118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang FC, Hosey MM. 1988. Dihydropyridine and phenylalkylamine receptors associated with cardiac and skeletal muscle calcium channels are structurally different. J. Biol. Chem 263:18929–37 [PubMed] [Google Scholar]
- 40.Yoshida A, Takahashi M, Fujimoto Y, Takisawa H, Nakamura T. 1990. Molecular characterization of 1,4-dihydropyridine-sensitive calcium channels of chick heart and skeletal muscle. J. Biochem 107:608–12 [DOI] [PubMed] [Google Scholar]
- 41.Yoshida A, Takahashi M, Nishimura S, Takeshima H, Kokubun S. 1992. Cyclic AMP-dependent phosphorylation and regulation of the cardiac dihydropyridine-sensitive Ca channel. FEBS Lett 309:343–49 [DOI] [PubMed] [Google Scholar]
- 42.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. 1996. Specific phosphorylation of a site in the full-length form of the α1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochemistry 35:10392–402 [DOI] [PubMed] [Google Scholar]
- 43.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Striessnig J. 1996. Identification of PK-A phosphorylation sites in the carboxyl terminus of L-type calcium channel α1 subunits. Biochemistry 35:9400–6 [DOI] [PubMed] [Google Scholar]
- 44.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. 2006. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during β1-adrenergic regulation. PNAS 103:16574–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao T, Yatani A, Dell’Acqua ML, Sako H, Green SA, et al. 1997. cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19:185–96 [DOI] [PubMed] [Google Scholar]
- 46.Naguro I, Nagao T, Adachi-Akahane S. 2001. Ser1901 of α1C subunit is required for the PKA-mediated enhancement of L-type Ca2+ channel currents but not for the negative shift of activation. FEBS Lett 489:87–91 [DOI] [PubMed] [Google Scholar]
- 47.Bunemann M, Gerhardstein BL, Gao T, Hosey MM. 1999. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J. Biol. Chem 274:33851–54 [DOI] [PubMed] [Google Scholar]
- 48.Haase H, Bartel S, Karczewski P, Morano I, Krause EG. 1996. In-vivo phosphorylation of the cardiac L-type calcium channel β-subunit in response to catecholamines. Mol. Cell. Biochem 163–164:99–106 [DOI] [PubMed] [Google Scholar]
- 49.Ganesan AN, Maack C, Johns DC, Sidor A, O’Rourke B. 2006. β-Adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ. Res 98:e11–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miriyala J, Nguyen T, Yue DT, Colecraft HM. 2008. Role of CaVβ subunits, and lack of functional reserve, in protein kinase A modulation of cardiac CaV1.2 channels. Circ. Res 102:e54–64 [DOI] [PubMed] [Google Scholar]
- 51.Lemke T, Welling A, Christel CJ, Blaich A, Bernhard D, et al. 2008. Unchanged β-adrenergic stimulation of cardiac L-type calcium channels in Cav1.2 phosphorylation site S1928A mutant mice. J. Biol. Chem 283:34738–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brandmayr J, Poomvanicha M, Domes K, Ding J, Blaich A, et al. 2012. Deletion of the C-terminal phosphorylation sites in the cardiac β-subunit does not affect the basic β-adrenergic response of the heart and the Cav1.2 channel. J. Biol. Chem 287:22584–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuller MD, Emrick MA, Sadilek M, Scheuer T, Catterall WA. 2010. Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci. Signal 3:ra70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beetz N, Hein L, Meszaros J, Gilsbach R, Barreto F, et al. 2009. Transgenic simulation of human heart failure-like L-type Ca2+-channels: implications for fibrosis and heart rate in mice. Cardiovasc. Res 84:396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muth JN, Yamaguchi H, Mikala G, Grupp IL, Lewis W, et al. 1999. Cardiac-specific overexpression of the α1 subunit of the L-type voltage-dependent Ca2+ channel in transgenic mice. Loss of isoproterenol-induced contraction. J. Biol. Chem 274:21503–6 [DOI] [PubMed] [Google Scholar]
- 56.Groner F, Rubio M, Schulte-Euler P, Matthes J, Khan IF, et al. 2004. Single-channel gating and regulation of human L-type calcium channels in cardiomyocytes of transgenic mice. Biochem. Biophys. Res. Commun 314:878–84 [DOI] [PubMed] [Google Scholar]
- 57.Tang M, Zhang X, Li Y, Guan Y, Ai X, et al. 2010. Enhanced basal contractility but reduced excitation-contraction coupling efficiency and β-adrenergic reserve of hearts with increased Cav1.2 activity. Am. J. Physiol. Heart Circ. Physiol 299:H519–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen X, Zhang X, Kubo H, Harris DM, Mills GD, et al. 2005. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res 97:1009–17 [DOI] [PubMed] [Google Scholar]
- 59.Chen X, Nakayama H, Zhang X, Ai X, Harris DM, et al. 2011. Calcium influx through Cav1.2 is a proximal signal for pathological cardiomyocyte hypertrophy. J. Mol. Cell. Cardiol 50:460–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Ziman B, Bodi I, Rubio M, Zhou YY, et al. 2009. Dilated cardiomyopathy with increased SR Ca2+ loading preceded by a hypercontractile state and diastolic failure in the α1CTG mouse. PLOS ONE 4:e4133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang L, Katchman A, Samad T, Morrow J, Weinberg R, Marx SO. 2013. β-Adrenergic regulation of the L-type Ca2+ channel does not require phosphorylation of α1C Ser1700. Circ. Res 113:871–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, et al. 2016. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J. Clin. Investig 126:112–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avula UMR, Abrams J, Katchman A, Zakharov S, Mironov S, et al. 2019. Heterogeneity of the action potential duration is required for sustained atrial fibrillation. JCI Insight 4:e128765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abrams J, Roybal D, Chakouri N, Katchman AN, Weinberg R, et al. 2020. Fibroblast growth factor homologous factors tune arrhythmogenic late NaV1.5 current in calmodulin binding-deficient channels. JCI Insight 5:e141736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fu Y, Westenbroek RE, Scheuer T, Catterall WA. 2014. Basal and β-adrenergic regulation of the cardiac calcium channel CaV1.2 requires phosphorylation of serine 1700. PNAS 111:16598–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fu Y, Westenbroek RE, Scheuer T, Catterall WA. 2013. Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. PNAS 110:19621–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poomvanicha M, Matthes J, Domes K, Patrucco E, Angermeier E, et al. 2017. β-Adrenergic regulation of the heart expressing the Ser1700A/Thr1704A mutated Cav1.2 channel. J. Mol. Cell. Cardiol 111:10–16 [DOI] [PubMed] [Google Scholar]
- 68.Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, et al. 2020. Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, et al. 2019. Cardiac CaV1.2 channels require β subunits for β-adrenergic-mediated modulation but not trafficking. J. Clin. Investig 129:647–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katchman A, Yang L, Zakharov SI, Kushner J, Abrams J, et al. 2017. Proteolytic cleavage and PKA phosphorylation of α1C subunit are not required for adrenergic regulation of CaV1.2 in the heart. PNAS 114:9194–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weissgerber P, Held B, Bloch W, Kaestner L, Chien KR, et al. 2006. Reduced cardiac L-type Ca2+ current in CaVβ2−/− embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ. Res 99:749–57 [DOI] [PubMed] [Google Scholar]
- 72.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, et al. 1992. Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J. Biol. Chem 267:1792–97 [PubMed] [Google Scholar]
- 73.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. 1991. The roles of the subunits in the function of the calcium channel. Science 253:1553–57 [DOI] [PubMed] [Google Scholar]
- 74.Buraei Z, Yang J. 2010. The β subunit of voltage-gated Ca2+ channels. Physiol. Rev 90:1461–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takahashi SX, Miriyala J, Colecraft HM. 2004. Membrane-associated guanylate kinase-like properties of β-subunits required for modulation of voltage-dependent Ca2+ channels. PNAS 101:7193–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meissner M, Weissgerber P, Londono JE, Prenen J, Link S, et al. 2011. Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J. Biol. Chem 286:15875–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Van Petegem F, Duderstadt KE, Clark KA, Wang M, Minor DL Jr. 2008. Alanine-scanning mutagenesis defines a conserved energetic hotspot in the CaVα1 AID-CaVβ interaction site that is critical for channel modulation. Structure 16:280–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Papa A, Kushner J, Hennessey JA, Katchman AN, Zakharov SI, et al. 2021. Adrenergic CaV1.2 activation via rad phosphorylation converges at α1C I-II loop. Circ. Res 128:76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Negroni JA, Morotti S, Lascano EC, Gomes AV, Grandi E, et al. 2015. β-Adrenergic effects on cardiac myofilaments and contraction in an integrated rabbit ventricular myocyte model. J. Mol. Cell. Cardiol 81:162–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bondarenko VE. 2014. A compartmentalized mathematical model of the β1-adrenergic signaling system in mouse ventricular myocytes. PLOS ONE 9:e89113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mullins PD, Bondarenko VE. 2020. Mathematical model for β1-adrenergic regulation of the mouse ventricular myocyte contraction. Am. J. Physiol. Heart Circ. Physiol 318:H264–82 [DOI] [PubMed] [Google Scholar]
- 82.Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. 2014. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J. Physiol 592:1181–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, et al. 1993. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J. Cell Biol 123:949–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.De Jongh KS, Warner C, Colvin AA, Catterall WA. 1991. Characterization of the two size forms of the α1 subunit of skeletal muscle L-type calcium channels. PNAS 88:10778–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG 2nd, et al. 2005. Sites of proteolytic processing and noncovalent association of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. PNAS 102:5274–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weiss S, Oz S, Benmocha A, Dascal N. 2013. Regulation of cardiac L-type Ca2+ channel CaV1.2 via the β-adrenergic-cAMP-protein kinase A pathway: old dogmas, advances, and new uncertainties. Circ. Res 113:617–31 [DOI] [PubMed] [Google Scholar]
- 87.Dai S, Hall DD, Hell JW. 2009. Supramolecular assemblies and localized regulation of voltage-gated ion channels. Physiol. Rev 89:411–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, et al. 2001. C-terminal fragments of the α1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated α1C subunits. J. Biol. Chem 276:21089–97 [DOI] [PubMed] [Google Scholar]
- 89.Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, Catterall WA. 2006. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J. Physiol 576:87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bannister JP, Leo MD, Narayanan D, Jangsangthong W, Nair A, et al. 2013. The voltage-dependent L-type Ca2+ (CaV1.2) channel C-terminus fragment is a bi-modal vasodilator. J. Physiol 591:2987–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. 2006. The C terminus of the L-type voltage-gated calcium channel CaV1.2 encodes a transcription factor. Cell 127:591–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schroder E, Byse M, Satin J. 2009. L-type calcium channel C terminus autoregulates transcription. Circ. Res 104:1373–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, et al. 2000. Proteolytic processing of the C terminus of the α1C subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J. Biol. Chem 275:8556–63 [DOI] [PubMed] [Google Scholar]
- 94.Catterall WA. 2015. Regulation of cardiac calcium channels in the fight-or-flight response. Curr. Mol. Pharmacol 8:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oz S, Pankonien I, Belkacemi A, Flockerzi V, Klussmann E, et al. 2017. Protein kinase A regulates C-terminally truncated CaV1.2 in Xenopus oocytes: roles of N- and C-termini of the α1C subunit. J. Physiol 595:3181–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Jongh KS, Merrick DK, Catterall WA. 1989. Subunits of purified calcium channels: a 212-kDa form of α1 and partial amino acid sequence of a phosphorylation site of an independent β subunit. PNAS 86:8585–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao T, Puri TS, Gerhardstein BL, Chien AJ, Green RD, Hosey MM. 1997. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J. Biol. Chem 272:19401–7 [DOI] [PubMed] [Google Scholar]
- 98.Fu Y, Westenbroek RE, Yu FH, Clark JP 3rd, Marshall MR, et al. 2011. Deletion of the distal C terminus of CaV1.2 channels leads to loss of β-adrenergic regulation and heart failure in vivo. J. Biol. Chem 286:12617–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Domes K, Ding J, Lemke T, Blaich A, Wegener JW, et al. 2011. Truncation of murine CaV1.2 at Asp1904 results in heart failure after birth. J. Biol. Chem 286:33863–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pawson T, Scott JD. 1997. Signaling through scaffold, anchoring, and adaptor proteins. Science 278:2075–80 [DOI] [PubMed] [Google Scholar]
- 101.Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, et al. 2008. Spatiotemporal dynamics of β-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ. Res 102:1091–100 [DOI] [PubMed] [Google Scholar]
- 102.Hundsrucker C, Rosenthal W, Klussmann E. 2006. Peptides for disruption of PKA anchoring. Biochem. Soc. Trans 34:472–73 [DOI] [PubMed] [Google Scholar]
- 103.Hundsrucker C, Krause G, Beyermann M, Prinz A, Zimmermann B, et al. 2006. High-affinity AKAP7δ-protein kinase A interaction yields novel protein kinase A-anchoring disruptor peptides. Biochem. J 396:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. 2003. β-Adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. PNAS 100:13093–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, et al. 2007. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochemistry 46:1635–46 [DOI] [PubMed] [Google Scholar]
- 106.Nystoriak MA, Nieves-Cintron M, Patriarchi T, Buonarati OR, Prada MP, et al. 2017. Ser1928 phosphorylation by PKA stimulates the L-type Ca2+ channel CaV1.2 and vasoconstriction during acute hyperglycemia and diabetes. Sci. Signal 10:eaaf9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, et al. 2014. AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. 7:1577–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, et al. 2010. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ. Res 107:747–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jones BW, Brunet S, Gilbert ML, Nichols CB, Su T, et al. 2012. Cardiomyocytes from AKAP7 knockout mice respond normally to adrenergic stimulation. PNAS 109:17099–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu H, Yuan C, Westenbroek RE, Catterall WA. 2018. The AKAP Cypher/Zasp contributes to β-adrenergic/PKA stimulation of cardiac CaV1.2 calcium channels. J. Gen. Physiol 150:883–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rees JS, Li XW, Perrett S, Lilley KS, Jackson AP. 2015. Protein neighbors and proximity proteomics. Mol. Cell. Proteom 14:2848–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, et al. 2014. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol. Cell 55:332–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, et al. 2016. Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat. Protoc 11:456–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, et al. 2013. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339:1328–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Paek J, Kalocsay M, Staus DP, Wingler L, Pascolutti R, et al. 2017. Multidimensional tracking of GPCR signaling via peroxidase-catalyzed proximity labeling. Cell 169:338–49 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cho KF, Branon TC, Udeshi ND, Myers SA, Carr SA, Ting AY. 2020. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc 15:3971–99 [DOI] [PubMed] [Google Scholar]
- 117.Cho KF, Branon TC, Rajeev S, Svinkina T, Udeshi ND, et al. 2020. Split-TurboID enables contact-dependent proximity labeling in cells. PNAS 117:12143–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, et al. 2018. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol 36:880–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han S, Li J, Ting AY. 2018. Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr. Opin. Neurobiol 50:17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, et al. 2015. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12:51–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, et al. 2012. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol 30:1143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, et al. 2001. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 411:701–6 [DOI] [PubMed] [Google Scholar]
- 123.Colecraft HM. 2020. Designer genetically encoded voltage-dependent calcium channel inhibitors inspired by RGK GTPases. J. Physiol 598:1683–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Finlin BS, Crump SM, Satin J, Andres DA. 2003. Regulation of voltage-gated calcium channel activity by the Rem and Rad GTPases. PNAS 100:14469–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Flynn R, Zamponi GW. 2010. Regulation of calcium channels by RGK proteins. Channels 4:434–39 [DOI] [PubMed] [Google Scholar]
- 126.Buraei Z, Yang J. 2015. Inhibition of voltage-gated calcium channels by RGK proteins. Curr. Mol. Pharmacol 8:180–87 [DOI] [PubMed] [Google Scholar]
- 127.Puhl HL 3rd, Lu VB, Won YJ, Sasson Y, Hirsch JA, et al. 2014. Ancient origins of RGK protein function: modulation of voltage-gated calcium channels preceded the protostome and deuterostome split. PLOS ONE 9:e100694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Reynet C, Kahn CR. 1993. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 262:1441–44 [DOI] [PubMed] [Google Scholar]
- 129.Manning JR, Yin G, Kaminski CN, Magyar J, Feng HZ, et al. 2013. Rad GTPase deletion increases L-type calcium channel current leading to increased cardiac contraction. J. Am. Heart Assoc 2:e000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Levitan BM, Manning JR, Withers CN, Smith JD, Shaw RM, et al. 2016. Rad-deletion phenocopies tonic sympathetic stimulation of the heart. J. Cardiovasc. Transl. Res 9:432–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ahern BM, Levitan BM, Veeranki S, Shah M, Ali N, et al. 2019. Myocardial-restricted ablation of the GTPase RAD results in a pro-adaptive heart response in mice. J. Biol. Chem 294:10913–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Levitan BM, Ahern BM, Aloysius A, Brown L, Wen Y, et al. 2021. Rad-GTPase contributes to heart rate via L-type calcium channel regulation. J. Mol. Cell. Cardiol 154:60–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chang L, Zhang J, Tseng YH, Xie CQ, Ilany J, et al. 2007. Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 116:2976–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Manning JR, Withers CN, Levitan B, Smith JD, Andres DA, Satin J. 2015. Loss of Rad-GTPase produces a novel adaptive cardiac phenotype resistant to systolic decline with aging. Am. J. Physiol. Heart Circ. Physiol 309:H1336–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Y, Chang Y, Li X, Li X, Gao J, et al. 2020. RAD-deficient human cardiomyocytes develop hypertrophic cardiomyopathy phenotypes due to calcium dysregulation. Front. Cell Dev. Biol 8:585879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang X, Tsien RW. 2020. Suspect that modulates the heartbeat is ensnared. Nature 577:624–26 [DOI] [PubMed] [Google Scholar]
- 137.Moyers JS, Zhu J, Kahn CR. 1998. Effects of phosphorylation on function of the Rad GTPase. Biochem. J 333(Part 3):609–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yang T, Puckerin A, Colecraft HM. 2012. Distinct RGK GTPases differentially use α1- and auxiliary β-binding-dependent mechanisms to inhibit CaV1.2/CaV2.2 channels. PLOS ONE 7:e37079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arias JM, Murbartian J, Vitko I, Lee JH, Perez-Reyes E. 2005. Transfer of β subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett 579:3907–12 [DOI] [PubMed] [Google Scholar]
- 140.Findeisen F, Minor DL Jr. 2009. Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J. Gen. Physiol 133:327–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, et al. 2003. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. PNAS 100:5543–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mahapatra S, Marcantoni A, Zuccotti A, Carabelli V, Carbone E. 2012. Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J. Physiol 590:5053–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Katz M, Subramaniam S, Chomsky-Hecht O, Tsemakhovich V, Flockerzi V, et al. 2021. Reconstitution of β-adrenergic regulation of CaV1.2: Rad-dependent and Rad-independent protein kinase A mechanisms. PNAS 118:e2100021118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Man KNM, Bartels P, Horne MC, Hell JW. 2020. Tissue-specific adrenergic regulation of the L-type Ca2+ channel CaV1.2. Sci. Signal 13:eabc6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qian H, Patriarchi T, Price JL, Matt L, Lee B, et al. 2017. Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the β2-adrenergic receptor in neurons. Sci. Signal 10:eaaf9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, et al. 2019. A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. eLife 8:e42214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hambleton M, York A, Sargent MA, Kaiser RA, Lorenz JN, et al. 2007. Inducible and myocyte-specific inhibition of PKCα enhances cardiac contractility and protects against infarction-induced heart failure. Am. J. Physiol. Heart Circ. Physiol 293:H3768–71 [DOI] [PMC free article] [PubMed] [Google Scholar]