

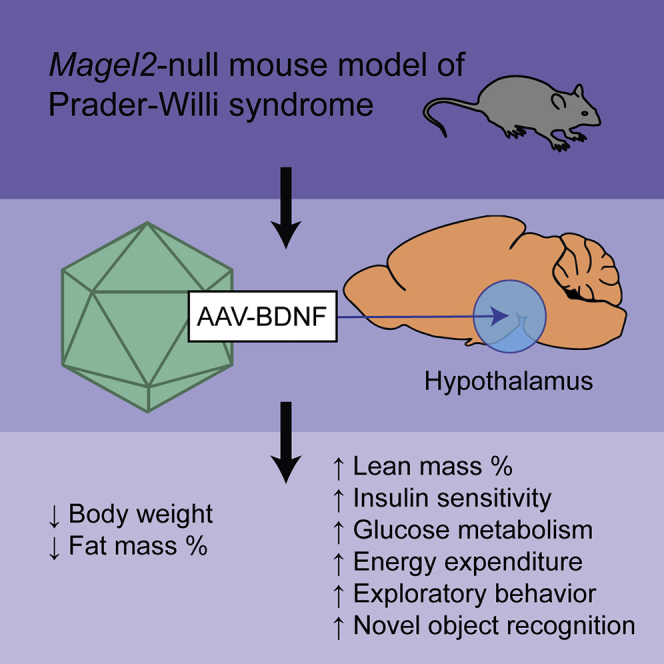
Abstract
Individuals with Prader-Willi syndrome (PWS) display developmental delays, cognitive impairment, excessive hunger, obesity, and various behavioral abnormalities. Current PWS treatments are limited to strict supervision of food intake and growth hormone therapy, highlighting the need for new therapeutic strategies. Brain-derived neurotrophic factor (BDNF) functions downstream of hypothalamic feeding circuitry and has roles in energy homeostasis and behavior. In this preclinical study, we assessed the translational potential of hypothalamic adeno-associated virus (AAV)-BDNF gene therapy as a therapeutic for metabolic dysfunction in the Magel2-null mouse model of PWS. To facilitate clinical translation, our BDNF vector included an autoregulatory element allowing for transgene titration in response to the host’s physiological needs. Hypothalamic BDNF gene transfer prevented weight gain, decreased fat mass, increased lean mass, and increased relative energy expenditure in female Magel2-null mice. Moreover, BDNF gene therapy improved glucose metabolism, insulin sensitivity, and circulating adipokine levels. Metabolic improvements were maintained through 23 weeks with no adverse behavioral effects, indicating high levels of efficacy and safety. Male Magel2-null mice also responded positively to BDNF gene therapy, displaying improved body composition, insulin sensitivity, and glucose metabolism. Together, these data suggest that regulating hypothalamic BDNF could be effective in the treatment of PWS-related metabolic abnormalities.
Keywords: Prader-Willi syndrome, BDNF, hypothalamus, hypothalamic, metabolic, metabolism, adeno-associated virus, AAV, gene therapy, molecular therapy
Graphical abstract
A brain-administered gene therapy ameliorates behavioral and metabolic deficits observed in a mouse model of Prader-Willi syndrome.
Introduction
Prader-Willi syndrome (PWS) is a contiguous gene syndrome that occurs in approximately 1 in 15,000 individuals.1 Individuals with PWS display developmental delays, cognitive impairment, excessive appetite, obesity, hypothalamic hypogonadism, obsessive compulsive behavior, anxiety, and temper tantrums.2, 3, 4 Current treatments to address metabolic dysfunction and behavioral abnormalities are limited to strict supervision of daily food intake and growth hormone (GH) therapy.5,6 While GH therapy remains the standard of care for PWS, it has several shortcomings, as (1) it requires daily administration and thus patient compliance, (2) it does not target the hypothalamic origins of PWS, (3) data about its efficacy in older adults are scant,7 and (4) exclusion criteria include common comorbidities such as severe obesity, uncontrolled diabetes, and active psychosis.8 Management of individuals with PWS is otherwise largely supportive and results in high levels of caregiver burden.9, 10, 11, 12
Loss of function of MAGEL2 (MAGE Family Member L2) is thought to contribute to several aspects of PWS pathophysiology,13 including alterations in the hypothalamic leptin-proopiomelanocortin (POMC) pathway. This pathway interprets peripheral signals of energy needs to drive feeding or fasting through opposing neuronal populations. In mice, Magel2 is required for leptin-mediated depolarization of hypothalamic anorexigenic POMC neurons.14 Furthermore, loss of Magel2 reduced anorexigenic α-melanocyte-stimulating hormone (α–MSH) axons,15,16 while orexigenic agouti-related peptide (AGRP) fibers remained unchanged.16,17 Thus, in the PWS-driven absence of Magel2, the response to peripheral hormones is blunted, resulting in an inability to maintain energy homeostasis.
Brain-derived neurotrophic factor (BDNF) serves as a potential therapeutic target, as it functions downstream in the leptin-POMC pathway and regulates energy homeostasis and behavior.18, 19, 20, 21, 22, 23, 24, 25 Furthermore, individuals with PWS display reductions in peripheral BDNF26 and PWS-related transcriptomic alterations in the leptin-POMC pathway may result from reduced BDNF expression.20 Administration of native or recombinant BDNF to the brain for therapeutic means proves difficult due to (1) the need for fine delivery localization, because, as a growth factor, BDNF has pleiotropic roles across various brain regions; (2) unfavorable pharmacokinetics; and (3) the need for repeated dosing.27, 28, 29
Previous work by our laboratory has shown that hypothalamic injection of a recombinant adeno-associated virus (rAAV)-BDNF vector in mice efficiently alleviates deficits in the leptin-POMC signaling pathway,30 thus mitigating obesity, hyperphagia, hyperglycemia, and hepatic steatosis. Moreover, adeno-associated virus (AAV)-BDNF gene therapy has been shown to reduce anxiety- and depression-like behavior and improve cognitive function.31, 32, 33 To facilitate clinical translation, we developed an autoregulatory AAV system to control therapeutic BDNF expression and mimic the body’s natural feedback systems. This autoregulatory approach leads to a sustainable plateau of body weight after substantial weight loss is achieved—minimizing the risk of cachexia—and has been validated in various physiologically related animal models.30,31,34, 35, 36 Given these data, we hypothesized that applying hypothalamic BDNF gene therapy to the Magel2-null murine model of PWS would ameliorate the metabolic dysregulation that results from deficiencies in the hypothalamic leptin-POMC signaling pathway. In this preclinical study, we assess the translational potential of hypothalamic AAV-BDNF gene therapy as a first-in-class therapeutic for PWS.
Results
Hypothalamic BDNF gene therapy improves metabolic function in female Magel2-null mice
Prior to vector administration, an EchoMRI was performed to assess body composition and allow for experimental group randomization. Consistent with genotype, female Magel2-null mice exhibited increased fat mass and decreased lean mass over wild-type controls at baseline (Figures S1A–S1D). Adult (16–20 weeks old) female wild-type and Magel2-null mice were injected with either AAV-yellow fluorescent protein (YFP) or AAV-BDNF vectors (Figure 1A) to the arcuate nucleus of the hypothalamus (ARC)/ventromedial hypothalamus (VMH), known sites of BDNF and POMC/AGRP action.18,37 In vivo monitoring continued over 23 weeks, beginning with metabolic measures (Figure 1B).
Figure 1.

Hypothalamic BDNF gene therapy improves metabolic function in female Magel2-null mice
(A) AAV constructs. (B) Experimental timeline of metabolic parameters, spanning from 0 to 12 weeks post AAV injection. (C) Body weight. (D) Percentage change body weight. (E) Average daily food intake. (F) Relative fat mass as measured by EchoMRI at 23 weeks post AAV injection. (G) Relative lean mass as measured by EchoMRI at 23 weeks post injection. (H) Insulin tolerance test at 6 weeks post AAV injection. (I) Area under the curve of the insulin tolerance test. (J) Glucose tolerance test at 8 weeks post AAV injection. (K) Area under the curve of the glucose tolerance test. Data are means ± SEM. Sample size: wild-type (WT) YFP n = 11, Magel2-null YFP n = 8, Magel2-null BDNF n = 8–9. ∗ or #p < 0.05, ∗∗ or ##p < 0.01, ∗∗∗ or ###p < 0.001, ∗∗∗∗p < 0.0001. For time course measurements, hash symbols (#) denote WT YFP versus Magel2-null YFP and asterisk symbols (∗) denote Magel2-null YFP versus Magel2-null BDNF.
As early as 3 weeks post injection, AAV-BDNF-treated female Magel2-null mice exhibited a significant decrease in body weight and prevention of excessive body weight gain over genotype-matched counterparts injected with AAV-YFP (Figures 1C and 1D). This reduction in body weight was maintained over the 23-week experiment. Magel2-null mice do not exhibit hyperphagia, despite manifestation of various other metabolic abnormalities.38,39 Consistent with previous reports, we observed reduced food intake in Magel2-null mice compared with wild-type counterparts (Figure 1E). AAV-BDNF-treated Magel2-null mice displayed a further reduction in food intake, consistent with BDNF’s anorexigenic actions.18,40,41
At 23 weeks post AAV injection, female Magel2-null mice treated with AAV-YFP displayed a significant increase in relative fat mass and decrease in relative lean mass over wild-type controls, whereas AAV-BDNF gene therapy rescued wild-type-like fat and lean mass composition (Figures 1F and 1G). AAV-BDNF-driven reductions in total body weight could be accounted for by losses in both absolute fat mass and lean mass (Figures S1E and S1F). Additional body composition measurement time points can be found in Figure S2.
At 6 weeks post AAV injection, an insulin tolerance test was performed. AAV-YFP-injected female Magel2-null mice exhibited no changes in sensitivity to an exogenous insulin bolus over wild-type counterparts, whereas AAV-BDNF-treated Magel2-null mice displayed improved insulin sensitivity (Figures 1H and 1I). At 8 weeks post AAV injection, a glucose tolerance test was performed to assess glycemic processing ability. AAV-YFP-treated Magel2-null mice exhibited worsened processing of an exogenous glucose bolus over wild-type controls; this deficit was rescued with AAV-BDNF administration (Figures 1J and 1K). Notably, compared with wild-type counterparts, AAV-YFP-injected Magel2-null mice exhibited elevated fasting blood glucose at baseline (t = 0) for both the insulin (Figure 1H) and glucose tolerance tests (Figure 1J), which was reversed by AAV-BDNF treatment.
Between 10 and 12 weeks post AAV injection, mice were subjected to indirect calorimetry. Body composition has known effects on energy expenditure (EE);42 larger animals typically have higher absolute rates of EE, due in part to increases in total metabolically active mass, whereas smaller animals typically have higher per-kilogram rates of EE.43 At the time of indirect calorimetry, AAV-YFP-treated female Magel2-null mice exhibited increased body weight over wild-type counterparts (Figure 2A), while AAV-BDNF-treated mice had reduced body weight. Accordingly, we observed lower hourly EE in the lighter AAV-BDNF-treated mice (Figures 2B and 2C), likely a byproduct of reductions in total metabolically active mass following gene therapy. Next, we compared the relationship between EE and body weight for each experimental group. Female AAV-BDNF-treated Magel2-null mice displayed increased EE per gram of body weight (Figure 2D), denoting increased energy efficiency over AAV-YFP genotype-matched counterparts (p = 0.049) and wild-type counterparts (p = 0.061). The respiratory exchange ratio (RER; calculated as VCO2/VO2) provides an indirect measurement of the predominant metabolic source (carbohydrate versus fat) for whole-body EE. Magel2-null AAV-YFP mice displayed reduced RER (Figures 2E and 2F) compared with wild-type counterparts, indicating fat as the main fuel source. This genotype-driven deficit was ameliorated by AAV-BDNF gene therapy; the RER indicated a shift toward carbohydrate use over fat use. Similar to previous reports,39,44 Magel2-null were hypoactive compared with wild-type counterparts (Figures 2G and 2H). Importantly, AAV-BDNF gene therapy did not ameliorate genotype-driven lethargy; increases in various metrics of metabolic function could not be explained by increased locomotor activity. Additional weight-normalized and absolute measures of VO2 and VCO2 can be found in Figure S3.
Figure 2.

Hypothalamic BDNF gene therapy increases EE per gram body weight in female Magel2-null mice
(A) Body weight at time of indirect calorimetry. (B) EE over time. (C) Average hourly EE. (D) Daily EE compared with body weight. (E) Respiratory exchange ratio (RER) over time. (F) Average hourly RER. (G) Ambulation over time. (H) Average hourly ambulation. Data are means ± SEM. Sample size: WT YFP n = 10, Magel2-null YFP n = 8, Magel2-null BDNF n = 9. Dark phases are denoted by shaded areas on indirect calorimetry time-course panels. Plus sign (+) indicates p < 0.10; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Hypothalamic BDNF gene therapy improves metabolic function in male Magel2-null mice
A similar experiment was initiated in male mice (between 16 and 22 weeks old at time of AAV injection). As seen in the female mice, metabolic measures were normalized in AAV-BDNF-treated male mice through 10 weeks post AAV injection. AAV-BDNF-treated male Magel2-null mice exhibited a significant decrease in body weight and prevention of excessive body weight gain over genotype-matched counterparts injected with AAV-YFP (Figures 3A and 3B). We observed a lack of hyperphagia in male AAV-YFP-injected Magel2-null mice compared with wild-type counterparts (Figure 3C). AAV-BDNF-treated Magel2-null mice displayed reduced food intake compared with genotype-matched controls (Figure 3C). At 4 weeks post AAV injection, male Magel2-null mice treated with AAV-YFP displayed a significant increase in relative fat mass and decrease in relative lean mass over wild-type controls, whereas AAV-BDNF gene therapy rescued wild-type-like fat and lean mass composition (Figures 3D and 3E). AAV-BDNF-driven reductions in total body weight could be accounted for by losses in both absolute fat mass and lean mass (Figures 3F and 3G). At 6 weeks post AAV injection, AAV-YFP-injected male Magel2-null mice exhibited no alterations in sensitivity to an exogenous insulin bolus over wild-type counterparts, whereas AAV-BDNF-treated Magel2-null mice displayed improved insulin sensitivity (Figures 3H and 3I). At 8 weeks post AAV injection, AAV-YFP-treated male Magel2-null mice exhibited a reduction in glycemic processing ability over wild-type controls; this deficit was rescued with AAV-BDNF administration (Figures 3J and 3K).
Figure 3.

Hypothalamic BDNF gene therapy improves metabolic function in male Magel2-null mice
(A) Body weight. (B) Percentage change body weight. (C) Average daily food intake. (D) Relative fat mass as measured by EchoMRI at 4 weeks post AAV injection. (E) Relative lean mass as measured by EchoMRI at 4 weeks post AAV injection. (F) Absolute fast mass as measured by EchoMRI at 4 weeks post AAV injection. (G) Absolute lean mass as measured by EchoMRI at 4 weeks post AAV injection. (H) Insulin tolerance test at 6 weeks post AAV injection. (I) Area under the curve of the insulin tolerance test. (J) Glucose tolerance test at 8 weeks post AAV injection. (K) Area under the curve of the glucose tolerance test. Data are means ± SEM. Sample size: WT YFP n = 19, Magel2-null YFP n = 14–15, Magel2-null BDNF n = 17. ∗ or #p < 0.05, ∗∗ or ##p < 0.01, ∗∗∗ or ###p < 0.001, ∗∗∗∗p < 0.0001. For time course measurements, hash symbols (#) denote WT YFP versus Magel2-null YFP and asterisk symbols (∗) denote Magel2-null YFP versus Magel2-null BDNF.
Hypothalamic BDNF gene therapy normalizes behavior in female Magel2-null mice
To assess safety and determine whether hypothalamic BDNF gene therapy alters behavior in female Magel2-null mice, comprehensive behavioral profiling was performed following metabolic assessments (Figure 4A). At 13 weeks post AAV injection, an open field test45 was performed, as Magel2-null mice exhibit alterations in exploratory activity and anxiety-like behavior.46,47 Magel2-null AAV-YFP-treated mice exhibited a trending—but not significant (p = 0.081)—decrease in exploratory activity over wild-type counterparts (Figure 4B). Magel2-null AAV-BDNF mice exhibited a significant increase in exploratory activity over AAV-YFP-treated Magel2-null counterparts Figure 4B). No changes were observed in percentage time spent in the periphery (Figure 4C) or center (Figure 4D) of the open field arena, indicating no changes in anxiety-like behavior.
Figure 4.

Hypothalamic BDNF gene therapy normalizes behavior in female Magel2-null mice
(A) Continued experimental timeline, spanning from 13 to 23 weeks post AAV injection. (B) Total distance traveled in the open field arena. (C) Percentage time spent in the periphery of the open field arena. (D) Percentage time spent in the center of the open field arena. (E) Novel object discrimination index (WT YFP n = 8, Magel2-null YFP n = 8, Magel2-null BDNF n = 9). (F) Marbles buried during the marble burying test. (G) Social preference index as measured during the three-chamber sociability test. (H) Social novelty index as measured during the three-chamber sociability test (WT YFP n = 10, Magel2-null YFP n = 8, Magel2-null BDNF n = 9). (I) Immobile time during the tail suspension test (WT YFP n = 8, Magel2-null YFP n = 8, Magel2-null BDNF n = 9). Data are means ± SEM. Sample size: WT YFP n = 11, Magel2-null YFP n = 8, Magel2-null BDNF n = 9 unless otherwise noted. Plus sign (+) indicates p < 0.10; ∗p < 0.05, ∗∗p < 0.01.
At 14 weeks post injection, the novel object recognition test was performed. Female Magel2-null mice have been shown to be averse to novel objects and environments.46 Consistent with the literature,46 we observed a reduced discrimination index in Magel2-null YFP-treated mice over wild-type controls (Figure 4E). This deficit was ameliorated in the Magel2-null mice receiving AAV-BDNF gene therapy treatment (Figure 4E). One interpretation of these results indicates that AAV-BDNF treatment resolves non-discrimination of novel objects previously reported in Magel2-null mice.46 The alternative interpretation of these data suggests that AAV-BDNF gene therapy improves short-term memory48,49 in Magel2-null mice.
At 16 weeks post injection, mice were subjected to the marble burying test to assess repetitive behaviors.50,51 Consistent with previous work,46 Magel2-null female mice buried fewer marbles than wild-type counterparts (Figure 4F). Wild-type-like burying behavior was observed in Magel2-null mice receiving AAV-BDNF gene therapy.
Previous reports suggest Magel2-deficient mice have altered social phenotypes.47,52 At 19 weeks post injection, mice were subjected to the three-chamber sociability test to assess social affiliation and social novelty preference.53 Mice were given the opportunity to wander three chambers while investigating social stimuli. In the first test phase—assessing social affiliation—mice were exposed to a novel confined peer and an opposing empty chamber. We observed no genotype- or AAV-BDNF-induced alterations in social preference (Figure 4G) during this first test phase. During the second test phase—assessing social novelty engagement—mice were exposed to both familiar and novel confined peers in opposing chambers. Similarly, we observed no genotype- or AAV-BDNF-induced alterations in social novelty-seeking behavior (Figure 4H).
At 21 week post injection, mice were subjected to the tail suspension test to assess depression-like behavior.54 AAV-YFP-treated Magel2-null mice exhibited increased levels of immobility compared with wild-type counterparts (Figure 4I). AAV-BDNF gene therapy yielded a trending, but not yet significant (p = 0.060), reduction in immobility of Magel2-null mice during this test (Figure 4I).
Hypothalamic BDNF gene therapy reduces white adipose tissue depot size and improves circulating markers of systemic metabolism in female Magel2-null mice
Female mice were euthanized at 23 weeks post AAV injection and tissues were collected. No changes were observed in relative brown adipose tissue (BAT) weight (Figure 5A). Consistent with EchoMRI observations, AAV-YFP-treated Magel2-null mice displayed increased relative mass of three white adipose tissue (WAT) depots—the inguinal white adipose tissue (iWAT), gonadal white adipose tissue (gWAT), and retroperitoneal adipose tissue (rWAT)—compared with wild-type counterparts (Figure 5A). Increases in Magel2-null mouse WAT depot size were ameliorated with AAV-BDNF gene therapy (Figure 5A) and relative liver weight was normalized.
Figure 5.

Hypothalamic BDNF gene therapy reduces WAT depot size and improves circulating markers of systemic metabolism in female Magel2-null mice
(A) Relative tissue weight at 23 weeks post AAV injection. (B) Serum leptin (WT YFP n = 9, Magel2-null YFP n = 8, Magel2-null BDNF n = 8). (C) Serum total adiponectin (WT YFP n = 10, Magel2-null YFP n = 7, Magel2-null BDNF n = 5). (D) Serum high-molecular-weight adiponectin (WT YFP n = 10, Magel2-null YFP n = 7, Magel2-null BDNF n = 5). (E) Ratio of high-molecular-weight adiponectin to total adiponectin within serum (WT YFP n = 10, Magel2-null YFP n = 7, Magel2-null BDNF n = 5). (F) Ratio of total adiponectin to leptin within serum (WT YFP n = 9, Magel2-null YFP n = 7, Magel2-null BDNF n = 5). (G) Serum glucose. (H) Serum insulin (WT YFP n = 8, Magel2-null YFP n = 8, Magel2-null BDNF n = 8). (I) Homeostatic model assessment for insulin resistance index (HOMA-IR) (WT YFP n = 8, Magel2-null YFP n = 7, Magel2-null BDNF n = 8). (J) Serum CCL2, also known as MCP-1. (K) Serum PAI-1. (L) Serum corticosterone. (M) Serum GH (WT YFP n = 10, Magel2-null YFP n = 7, Magel2-null BDNF n = 7). (N) Serum IGF-1 (WT YFP n = 9, Magel2-null YFP n = 8, Magel2-null BDNF n = 6). Data are means ± SEM. Sample size: WT YFP n = 11, Magel2-null YFP n = 8, Magel2-null BDNF n = 9 unless otherwise noted. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Serum was profiled to assess changes in systemic metabolism following AAV-BDNF gene therapy. Leptin serves as a central-peripheral messenger to maintain energy homeostasis and its production is positively correlated with adipose tissue mass.55 Consistent with gross adipose observations, AAV-YFP-treated Magel2-null mice exhibited increased serum leptin over wild-type counterparts (Figure 5B). Heightened serum leptin in Magel2-null mice was ameliorated with AAV-BDNF administration (Figure 5B). Adiponectin plays roles in insulin sensitivity, glucose homeostasis, and systemic metabolic function;56 circulating adiponectin levels are negatively correlated with adipose tissue mass and impaired glucose tolerance. Magel2-null mice exhibited no significant increase in total (Figure 5C) or high-molecular-weight (HMW) adiponectin (Figure 5D) compared with wild-type counterparts. AAV-BDNF gene therapy increased both total and HMW adiponectin in Magel2-null mice over wild-type AAV-YFP-treated mice (Figures 5C and 5D). We observed no genotype- or AAV-BDNF-induced alterations changes in the total/HMW adiponectin ratio (Figure 5E). Increased adiponectin/leptin ratios are correlated with reduced body mass index and cardiometabolic risk.57,58 AAV-YFP-treated Magel2-null mice exhibited reduced adiponectin/leptin ratios compared with wild-type controls; this deficit was ameliorated with AAV-BDNF gene therapy (Figure 5F). No genotype- or gene-therapy-induced changes were observed in circulating glucose levels (Figure 5G). Magel2-null AAV-BDNF-treated mice exhibited a significant decrease in fasting serum insulin over AAV-YFP-treated counterparts (Figure 5H). Similar results were observed in the HOMA-IR (homeostatic model assessment for insulin resistance) index; together, both are indicative of improved insulin sensitivity following AAV-BDNF gene therapy (Figure 5I). No changes were observed in two systemic inflammatory markers, chemokine (C-C motif) ligand 2 (CCL2, also known as MCP-1) and plasminogen activator inhibitor-1 (PAI-1) (Figures 5J and 5K). We observed a gene-therapy-induced increased in serum corticosterone (Figure 5L), consistent with previous reports that suggest BDNF activates the hypothalamic-pituitary-adrenal (HPA) axis.59, 60, 61 The GH/IGF-1 (insulin-like growth factor 1) axis is controlled in part by the hypothalamus and is dysfunctional in PWS patients and Magel2-null mice.62 Importantly, GH administration remains the current standard of care for PWS patients.5,6 AAV-YFP-treated wild-type and Magel2-null mice displayed no difference in GH levels, whereas BDNF gene therapy treatment resulted in a strong induction of circulating GH (Figure 5M). Serum IGF-1 was reduced in AAV-YFP-treated Magel2-null mice compared with wild-type counterparts; this genotype-driven decrease in IGF-1 was not ameliorated by AAV-BDNF gene therapy (Figure 5N).
Hypothalamic BDNF gene therapy normalizes adipocyte size in female Magel2-null mice
Previous work by our laboratory described a brain-adipose axis driven by hypothalamic BDNF expression.63 Environmental- or genetic-driven increases in hypothalamic BDNF confer a white-to-brown adipose phenotypic shift via increased sympathetic tone. Accordingly, we profiled expression of various metabolism-relevant genes in the largest visceral WAT depot, the gWAT (Figure 6A). Magel2-null mice showed a trend of Lep upregulation in gWAT (not reaching significance) compared with wild type, which appeared to be corrected by BDNF gene therapy. It is possible that the genotype-induced increase in circulating levels of leptin is not caused solely by transcription changes, but instead reflects increases in total fat mass. We observed no significant differences in Adipoq (encoding adiponectin), Hsl (encoding hormone-sensitive lipase), and Pten (encoding phosphatase and tensin homolog) expression. We observed a genotype-induced reduction in Adrb3 (encoding adrenoceptor β3). Magel2-null mice exhibited increased Ppargc1a (encoding peroxisome proliferator-activated receptor gamma coactivator 1-alpha) expression following gene therapy. Consistent with gross tissue observations, Magel2-null mice exhibited increased adipocyte size over wild-type counterparts; genotype-driven increases in adipocyte size were ameliorated with AAV-BDNF gene therapy (Figures 6B–6D).
Figure 6.

Hypothalamic BDNF gene therapy normalizes adipocyte size in female Magel2-null mice
(A) Relative gene expression of gWAT. (B) Representative images of gWAT. (C) Area distribution of all sampled adipocytes. (D) Mean adipocyte area per animal. Data are means ± SEM for (A) and (D). The violin plot in (C) depicts first quartiles, medians, and third quartiles. Sample size: WT YFP n = 6, Magel2-null YFP n = 6, Magel2-null BDNF n = 6 for (A) and (D). Plus sign (+) indicates p < 0.010; ∗p < 0.05, ∗∗∗∗p < 0.0001.
Target validation of a hypothalamic AAV-BDNF vector in female Magel2-null mice
To verify the expression of our vector, transgene expression was probed using various methods. Hypothalamic gene expression profiling revealed a 4.5-fold upregulation of Bdnf in AAV-BDNF-treated mice over AAV-YFP controls (Figure 7A). No changes were observed in BDNF receptor TrkB-FL (encoding tropomyosin receptor kinase B full length), Agrp, or Pomc gene expression. Consistent with obese states, we observed a baseline reduction in Mc4r (encoding melanocortin-4 receptor) expression and increase in Insr (encoding insulin receptor) expression in Magel2-null mice. Obrb (encoding leptin receptor long form) expression was increased in Magel2-null mice following BDNF gene therapy. We observed reduced expression of Crh (encoding corticotrophin-releasing hormone) in AAV-YFP-injected Magel2-null mice compared with wild-type counterparts; this deficit was ameliorated with AAV-BDNF gene therapy. Consistent with increased Bdnf expression,64 we observed a strong upregulation of Vgf (non-acronymic) in AAV-BDNF-treated mice over AAV-YFP-treated mice.
Figure 7.

Target validation of a hypothalamic AAV-BDNF vector in female Magel2-null mice
(A) Relative gene expression of hypothalamic tissue. (B) Immunoblotting of an HA tag as a proxy for BDNF expression; each lane depicts protein obtained from a unilateral hypothalamic dissection from an individual mouse. (C) Representative image of AAV-YFP fluorescence (ARC, arcuate nucleus of the hypothalamus; VMH, ventromedial hypothalamus; 3V, third ventricle). Data are means ± SEM. Sample size: WT YFP n = 6, Magel2-null YFP n = 6, Magel2-null BDNF n = 5–6. Plus sign (+) indicates p < 0.010; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Immunoblotting of a hemagglutinin (HA) tag was performed as a proxy for BDNF transgene expression. As expected, we observed no HA signal in AAV-YFP-treated controls and positive HA protein in AAV-BDNF-treated mice (Figure 7B). For spatial targeting validation, representative brain samples were sectioned and YFP fluorescence was observed in the ARC and VMH of the hypothalamus (Figure 7C).
Discussion
For the past two decades, GH therapy and strict supervision of food intake have remained the standard of care for PWS. Unfortunately, caregiver burden remains high, and, in some estimates, exceeds that of caregivers for persons with dementia, Alzheimer’s, and traumatic brain injury.11,12 Recent work has focused on therapeutics that can regulate food intake and EE through mechanisms targeting the hypothalamic leptin-POMC pathway. Melanotan II (MT-II) and setmelanotide are MC4R agonists that have known roles in food intake reduction. Although Magel2-null mice are sensitive to both compounds,14,44 setmelanotide translation into the clinic proved less than fruitful (NCT02311673). More recently, intranasal carbetocin was proposed for treatment of hyperphagia, anxiousness, and distress associated with PWS (CARE-PWS; NCT03649477). Study trial difficulties stemming from early 2020 COVID-19 pandemic lockdowns limited efficacy data and ultimately led the US Food and Drug Administration (FDA) to deem the phase 3 study’s data to be insufficient for approval, although the drug was regarded as generally safe and well tolerated. To date, no FDA-approved therapies exist to target the day-to-day challenges PWS patients and families face surrounding excessive hyperphagia and emotional reactivity.65
Notably, our work represents the first attempt at an AAV-based, PWS-targeted gene therapy. The gene-therapy field has grown dramatically over the last decade as researchers and clinicians have focused efforts on treating genetic diseases such as PWS. With all gene therapies, the risk/benefit ratio must be considered. While general obesity can be better managed with interventions targeting exercise and nutrition, certain severe genetic forms of obesity may indicate the use of CNS-targeted gene therapies. Importantly, typical obesity interventions—such as self-monitoring of food intake—remain difficult in the PWS population due to incessant food-seeking behavior and associated cognitive rigidity. Patients are highly dependent upon caregivers for maintenance of “food security; restricting of food access and control of food intake occurs through drastic measures such as locks on refrigerators and pantries.66 To some, surgical methods may seem to be a better alternative to the lock-and-key method. While sleeve gastrectomy was well tolerated in Magel2-null mice,67 bariatric procedures in PWS patients result in far greater levels of complications than those observed in non-PWS obese patients68 and furthermore require patient or caregiver post-surgical compliance to guarantee adequate weight loss.69 Exercise and behavioral interventions have proved successful70, 71, 72 to treat symptoms but remain limited in their ability to address the root causes of satiety imbalance. In sum, therapies targeting the hypothalamic and genetic roots of PWS are sorely needed.
Energy homeostasis requires dynamic feedback systems; peripheral tissues are in constant communication with the brain to convey the need for fasting or feeding. Our autoregulatory AAV-BDNF vector provides significant advantages over other pharmacological interventions, as transgene expression is inherently tied to central-peripheral feedback systems and thus reflects the host’s physiological needs. In contrast to repeated dosing of MC4R agonists, our therapeutic vector would need to be administered only once for long-term efficacy and would require limited patient/caregiver dosing compliance. Notably, AAV vectors have shown reasonable safety and tolerability when administered to the human CNS, although administration methods vary.73, 74, 75, 76 Development and optimization of minimally invasive administration methods continues to be an area of focus in the field.77 Importantly, intracranial injection of hypothalamic tissue in humans—as in this proof-of-concept experiment—remains highly challenging due to the region’s deep location within the brain. Alternative administration methods (e.g., delivery of AAV to the hypothalamus via endoscopic endonasal procedures) must be considered on the path to clinical development.
As with any preclinical AAV work, future experiments will need to validate the vector in larger animal models, determine the optimal serotype and administration method, and assess toxicity and immune response. Furthermore, determination of the proper dose will be necessary to observe a functional response, but not one that is clinically dangerous. Special care must be taken to avoid hypoglycemic states, as individuals with PWS remain insulin sensitive.78 While cachexia is unlikely due to the autoregulatory nature of the vector, it remains unclear whether body weight and food intake reduction below that of healthy individuals would be clinically indicated, as was observed in our AAV-BDNF-treated Magel2-null mice.
Our data demonstrate that hypothalamic BDNF gene therapy results in sustained metabolic improvement in Magel2-null mice. Several caveats and limitations of the current study remain. MAGEL2 is just one of many genes located within the PWS deletion region of chromosome 15q11-q13. Mouse models with large deletions at the PWS locus were found to display high levels of lethality, leading researchers to favor single-gene knockout models targeting various PWS genes including Magel2, Snord116, and Ndn.65,79 As such, current preclinical models remain limited, as they recapitulate some, but not all, PWS phenotypes.65,79 Furthermore, animal models cannot fully mirror nuanced human emotionality and behavioral aspects of food-seeking behavior. Regarding specific limitations of the model used herein, Magel2-null mice do not mirror human hyperphagia,38,39 despite broad systemic metabolic dysfunction and impaired sensitivity to peripheral hormones. Notably, Schaaf-Yang syndrome is driven by loss of function of MAGEL2; individuals with Schaaf-Yang syndrome display many PWS-like phenotypes, albeit with a notable lack of hyperphagia and obesity.80, 81, 82 Despite this modeling issue, we observed an anorexigenic effect of AAV-BDNF administration that may prove useful in PWS-relevant translation efforts. Moreover, this BDNF gene therapy reversed hyperphagia in Mc4r-deficient mice, a model for the most common monogenic form of human obesity.30 On a different note, while AAV-BDNF gene therapy ameliorated metabolic deficits in both female and male mice, sex differences may exist for other outcomes. Notably, BDNF has known interactions with estrogen that may influence metabolic outcomes,83, 84, 85 and previous work suggests female and male Magel2-null mice display some variations in endocrine responses.62 Further investigation is warranted prior to translational efforts.
The hypothalamic leptin-POMC pathway remains a prime target for PWS therapeutics. In Magel2-null mice, leptin administration fails to (1) reduce food intake, (2) depolarize anorexigenic POMC+ neurons in the ARC, and (3) induce STAT3 phosphorylation.14 Additional work suggests that MAGEL2 regulates leptin receptor cell surface abundance, subcellular localization, and lysosomal degradation.86 In tandem with unchanged levels of orexigenic AGRP fibers,16 alterations in leptin sensitivity14,15,86 and POMC activity17 underlie metabolic dysregulation in the Magel2-null mouse model. Here, we did not observe changes in POMC expression as measured by qPCR. It is unclear whether normalization occurred over time or an alternative explanation exists. Future work by our laboratory will provide an unbiased screening of transcriptomic-wide changes in the hypothalamus of Magel2-null mice following short-term AAV-BDNF gene therapy.
On a different note, BDNF works downstream18 of aberrant leptin-POMC signaling to regulate food intake and energy homeostasis. Previous work by our group describes how environmental or genetic-driven increases in hypothalamic BDNF yield elevated β-adrenergic signaling, thermogenesis, and browning in adipose tissue alongside reductions in fat depot size and circulating leptin levels.63 We observed reduced circulating leptin levels and increased hypothalamic Obrb expression in Magel2-null mice following BDNF gene therapy. Notably, reduced leptin levels restore leptin sensitivity in POMC neurons,87 providing one feasible mechanism for observations presented herein. Additional work may delineate whether BDNF gene therapy alters central leptin sensitivity through direct or indirect means, thus normalizing central-peripheral feedback systems in the Magel2-null mouse model. Such work may provide additional mechanistic insights relevant to PWS etiology and potential druggable targets.
In this preclinical study, we conducted several behavior tests as safety assessments and observed no adverse behavioral changes in female Magel2-null mice following AAV-BDNF treatment. Unexpectedly, AAV-BDNF gene therapy rescued genotype-associated behavioral alterations in female Magel2-null mice, improving novel object recognition and increasing exploratory activity. Improved cognition in the novel object recognition test is of particular interest, as it is thought to be hippocampus dependent. It remains to be seen whether systemic metabolic improvements induced by hypothalamic BDNF gene transfer reflect back to extrahypothalamic regions of brain, thereby ameliorating cognitive and affective deficits of PWS models. Regarding the open field and marble burying tests, we observed genotype-driven behavioral deficits were ameliorated by AAV-BDNF gene therapy. While baseline reductions in total distance traveled and marbles buried (Figures 4B and 4F) can feasibly be explained by the hypoactivity observed in the Magel2-null model, the rescue of these behavioral outcomes following BDNF gene therapy cannot be explained by a mere amelioration of hypoactivity; AAV-BDNF-treated Magel2-null mice displayed no change in locomotion over AAV-YFP-injected Magel2-null counterparts (Figures 2G and 2H). Another interesting finding is that Magel2-null mice displayed increased immobility in tail suspension test—a commonly used test for depression-like behavior—while BDNF gene therapy rescued this deficit. Notably, scarce data regarding depression-like behavior have been reported. It is possible that our observations reflect differences in lean muscle mass and overall weight, known confounds of the tail suspension test.88 Nevertheless, these behavioral data encourage further investigation of potential therapeutic benefits beyond metabolic improvement.
In summary, we present preclinical data that suggest hypothalamic AAV-BDNF gene therapy is efficacious and safe for treatment of metabolic dysfunction in female and male Magel2-null mice. Furthermore, we observed BDNF gene therapy has no adverse effects on behavior in female Magel2-null mice. These proof-of-concept data indicate BDNF as a promising molecular target for PWS and other genetic forms of obesity.
Materials and methods
Animals
Magel2-null mice harbor a maternally inherited imprinted/silenced wild-type allele and a paternally inherited Magel2-lacZ knockin allele that abolishes endogenous Magel2 gene function. Male mice containing the Magel2-lacZ allele (Jackson Labs #009062) were bred with female C57BL/6 mice to produce both wild-type mice and Magel2-null littermates. Mice were genotyped from ear notch biopsies. Identification of mutant offspring was performed by polymerase chain reaction genotyping with Magel2 and LacZ oligonucleotide primers (common forward, 5′-ATGGCTCCATCAGGAGAAC; Magel2 reverse, 5′-GATGGAAAGACCCTTGAGGT; and LacZ reverse, RW4237, 5′-GGGATAGGTCACGTTGGTGT). Magel2-null mice develop metabolic deficiencies over time15,16; systemic manifestation occurs by 16 weeks of age.38 Therefore, all mice were between 16 and 22 weeks of age at experiment start date. All mice had ad libitum access to food (normal chow diet, 11% fat, caloric density 3.4 kcal/g, Teklad) and water. Mice were housed in standard laboratory cages (19.4 × 18.1 × 39.8 cm) within temperature (22°C–23°C) and humidity (30%–70%) controlled rooms under a 12:12 light:dark cycle. All animal experiments were in accordance with the regulations of The Ohio State University’s Institutional Animal Care and Use Committee (IACUC).
rAAV design
The rAAV vector (Figure 1A) was designed to be self-regulating in nature to allow for sustainable, safe weight loss. HA-tagged human BDNF (HA-BDNF, referred to as BDNF) or destabilized YFP control (dsYFP, referred to as YFP) was inserted in a multiple cloning site following a cytomegalovirus enhancer and a chicken β-actin promoter. Woodchuck posttranscriptional regulatory element (WPRE) and a bovine GH polyadenosine (BGH poly(A)) tail followed BDNF or YFP transgenes. In the BDNF vector, a second regulatory cassette included an AGRP promoter driving a microRNA targeting BDNF (miR-Bdnf) and WPRE/polyA elements. As body weight decreases and AGRP is physiologically induced, microRNA expression is activated to inhibit BDNF transgene expression; this leads to a sustainable plateau of body weight after a substantial weight loss is achieved, thus limiting the risk of cachexia.34 All vectors were packaged into serotype 1 capsids and purified by iodixanol gradient centrifugation as previously described.34
Hypothalamic injections of AAV
Mice received either AAV-BDNF-miR-Bdnf (denoted as AAV-BDNF throughout the text) or AAV-YFP to the hypothalamus, 1 × 1010 viral genomes per site, bilaterally. Prior to surgery, mice were treated with Ethiqua XR (3.25 mg/kg body weight), an extended-release buprenorphine solution, for pain management. Mice were anesthetized with a single intraperitoneal dose of ketamine/xylazine (80 mg/kg and 5 mg/kg) and secured via ear and incisor bars on a stereotaxic frame (Kopf, Tujunga, CA). Anesthesia was further maintained with 2.5% isoflurane at 1 L/min during the injection procedure. A single midline incision was made through the scalp to expose the skull, and two small holes were made with a dental drill above the injection sites. rAAV vectors were administered bilaterally (0.5 μL per site) by a 10-μL Hamilton syringe (Reno, NV) and a Micro4 Micro Syringe Pump Controller (World Precision Instruments, Sarasota, FL) at 200 nL/min to the arcuate/ventromedial hypothalamus (AP, −1.1 mm; ML, ±0.50 mm; DV, −6.20). When the infusion was finished, the syringe was slowly retracted from the brain and the scalp was sutured. Animals were returned to clean cages resting atop a heating pad. Mice were provided with supplemental care—HydroGel (ClearH2O, Westbrook, ME) and mash—and were carefully monitored post surgery until fully recovered.
Food intake measurement
Food was measured at the cage level on a weekly basis throughout the experiment. Data were excluded if they were collected in weeks during which invasive profiling occurred that might disrupt normal feeding patterns (e.g., removal from home cages for indirect calorimetry monitoring and fasting periods for glucose tolerance tests).
Body composition assessment
An EchoMRI was utilized to measure fat and lean mass in live mice without anesthesia. At baseline, 4 weeks, between 14 and 15 weeks, and 23 weeks post AAV injection, body composition analysis was performed with a 3-in-1 Analyzer (EchoMRI LLC, Houston, TX) according to manufacturer instructions. Mice were subjected to a 5-Gauss magnetic field and whole-body masses of fat, lean, free water, and total water were determined during separate cycles by manufacturer software comparison with a canola oil standard.
Insulin tolerance test
At 6 weeks post AAV injection, mice were injected intraperitoneally with an insulin solution (0.5 U of insulin per kilogram body weight) following a 2-h fast. Tails were cut (<0.5 mm) with surgical scissors to elicit blood flow. Blood was obtained from the tail at baseline, 15, 30, 60, 90, and 120 min after insulin injection. Blood glucose concentrations were measured with a portable glucose meter using default manufacturer settings (Bayer Contour Next). Following blood collection, styptic powder was used to stop bleeding.
Glucose tolerance test
At 8 weeks post AAV injection, mice were injected intraperitoneally with glucose solution (2.0 g glucose per kilogram body weight) after a 16-h overnight fast. Tails were cut (<0.5 mm) with surgical scissors to elicit blood flow. Blood was obtained from the tail at 0, 15, 30, 60, 90, and 120 min after glucose injection. Blood glucose concentrations were measured with a portable glucometer using default manufacturer settings (Bayer Contour Next). Following blood collection, styptic powder was used to stop bleeding.
Indirect calorimetry
Between 10 and 12 weeks post AAV injection, mice underwent indirect calorimetry using the Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH). In short, the indirect calorimetry system pushes a flow of fresh air through gas-tight animal housing cages. The system collects and mixes expired air, measures the flow rate, and determines the incoming/outgoing O2 and CO2 concentrations.42 Mice were placed in calorimetry chambers and allowed to habituate to the testing environment for 16-18 h. Various physiological and behavioral parameters (VO2, VCO2, RER, EE, and ambulation) were recorded for 24 h. Mice were returned to their home cages after indirect calorimetry was performed.
Open field test
At 13 weeks post AAV injection, mice were individually placed into the center of an open square arena (60 × 60 cm, enclosed by walls of 48 cm). Each mouse was allowed to explore the arena for 10 min, during which time and locomotion—in the center and the periphery of the open field—was recorded and analyzed via TopScan (Clever Sys) software. Between each trial, the arena was cleaned with 70% ethanol to remove odor cues.
Novel object recognition test
At 14 weeks post AAV injection, mice were subjected to the novel object recognition test.49 For the test familiarization period, mice were placed in an open arena (60 × 60 cm, enclosed by walls of 48 cm) with two identical objects (either two Falcon tubes filled with water or two stacks of large Legos; habituation objects were varied between mice). Mice were allowed to explore the identical objects until 20 s of cumulative exploration time was achieved. Mice were returned to their home cage while the arena/objects were cleaned with 70% ethanol. During the novel object recognition test session, the two training objects were replaced with one matched item from the training session and a novel item. Mice were placed in the arena and allowed to explore both the novel and learned object until 20 s of cumulative exploration time was achieved, with a maximal time of 10 min to reach the criteria; those that did not meet the criteria were excluded from statistical analyses. Time spent exploring each respective object was recorded. Mice were returned to their home cages and the arena/objects were cleaned with 70% ethanol. As previously described, exploration activity was defined as “directing the nose toward the object at a distance less than or equal to 2 cm” and time spent climbing on or chewing on objects was not deemed exploration activity.49 The discrimination index was calculated by dividing the novel object exploration time by the total amount of object exploration during the test.
Marble burying test
At 16 weeks post AAV injection, mice were individually placed into cages (19.4 × 18.1 × 39.8 cm) with evenly spaced glass marbles arranged in a three-by-four grid on the surface of clean aspen bedding (5 cm in depth). An experimenter who was unaware of genotype/treatment scored the number of marbles buried after 30 min. Marbles were washed with mild detergent and water between each trial.
Three-chamber sociability test
At 19 weeks post AAV injection, mice were placed in an apparatus consisting of three connected plexiglass chambers (18 × 41 × 20 cm each) with removable dividers between each chamber. Each test subject was individually placed in the center plexiglass chamber for 5 min of habituation. In the first phase—testing social affiliation—another, unfamiliar mouse was placed in either the right or left chamber in a small wire cage, while another wire cage remained empty in the opposite chamber. The wire cage restricted social or aggressive interactions between the two mice beyond nose contact. Chamber dividers were lifted after the habituation period to allow the test subject to move freely about all three chambers for a 10-min observation period. A second 10-min test—assessing novel social engagement—was performed immediately afterward, using the conspecific from the first test (now denoted as a familiar mouse) and a novel unfamiliar mouse in the opposite chamber. Between each trial, the arena was cleaned with Opticide to remove odor cues. Trials were video recorded. An observer unaware of genotype/treatment used open-source event-logging software, BORIS,89 to create a user-scored ethogram with timestamps and length calculations for behavioral activities (i.e., chamber entry, mouse investigation). Once the behavioral coding process was completed, observation data were exported and analyzed. The social preference index was calculated by dividing the amount of time spent with the novel mouse by the amount of time spent in the empty chamber during the first test phase. The social novelty index was calculated by dividing the amount of time spent with the novel unfamiliar mouse by the amount of time spent with the familiar mouse during the second test phase.
Tail suspension test
At 21 week post AAV injection, a small plastic cylinder generated from a cut syringe (Becton, Dickinson and Company, Franklin Lakes, NJ) was slipped over each mouse’s tail to prevent climbing motion and escape from the test; mice that circumvented this anti-climb apparatus were excluded. Mice were suspended in air individually by tape attached to a shelf (64-cm height with bubble wrap laid beneath for safety) for 6 min. Trials were video recorded An observer unaware of genotype/treatment used the open-source event-logging software, BORIS,89 to create a user-scored ethogram with timestamps and length calculations for behavioral activities (i.e., immobility, mobility). Once the behavioral coding process was completed, observation data were exported and analyzed.
Tissue collection
At 23 weeks post AAV injection, mice were euthanized following a 4-h fast. Mice were anesthetized with 2.5% isoflurane (1.0 L/min) and then decapitated to collect trunk blood. Tissues to be used for mRNA and protein analyses were flash frozen on dry ice and stored at −80°C until further analysis. Hypothalamus was collected under a dissection microscope at sacrifice, with the left and right sides being collected separately to allow for RNA and protein isolation from each mouse.
Serum harvest and analysis
Trunk blood was collected at euthanasia, clotted on ice, and centrifuged at 10,000 rpm for 10 min at 4°C. The serum component was collected and stored at −20°C until further analysis. R&D Systems ELISA kits were used to assay serum leptin (#DY498), insulin-like growth factor (#DY791), PAI-1 (#DY3828), and CCL2 (#DY479). Additional ELISAs were performed for insulin (ALPCO #80-INSMSU-E01), corticosterone (Enzo #ADI-900-097), GH (EMD Millipore #EZRMGH-45K), and adiponectin (ALPCO #47-ADPMS-E01). Caymen Chemical colorimetric assay kits were used to assay triglycerides (#10010303) and glucose (#10009582). Homeostatic model assessment for insulin resistance (HOMA-IR) index was calculated as [fasting serum glucose (mmol/L) × fasting serum insulin (pmol/L)/22.5] as described elsewhere.90
qRT-PCR
Following tissue sonication, RNA was isolated using the RNeasy Mini kit (QIAGEN #74804) with RNase-free DNase treatment. cDNA was reverse transcribed using Taqman Reverse Transcription Reagents (Applied Biosystems #N8080234). qRT-PCR was completed on the StepOnePlus Real-Time PCR System using Power SYBR Green (Applied Biosystems #A25742) PCR Master Mix. Primer sequences are available in Table S1. We calibrated data to endogenous controls—Hprt1 for hypothalamus, Actinb for gWAT—and quantified the relative gene expression using the 2−ΔΔCT method.91
Immunoblotting
Tissue samples were homogenized in RIPA buffer (Pierce #89901) containing 1× PhosSTOP (Roche #4906845001) and protease inhibitor cocktail III (Calbiochem #539134). Tissue lysates were separated by gradient gel (4%–20%, Mini-PROTEAN TGX, Bio-Rad #4561096) and then transferred to a nitrocellulose membrane (Bio-Rad #1620115). Blots were incubated overnight at 4°C with the following Cell Signaling Technology primary antibodies: beta actin #3700, 1:500; HA tag #3724, 1:500. Blots were rinsed and incubated with HRP-conjugated secondary antibodies (Bio-Rad, 1:3,000). Chemiluminescence signal was detected and visualized by Odyssey Fc imaging (LI-COR Biotechnology, Lincoln, NE).
Adipocyte size analysis
Portions of fat depots were fixed in 10% neutral buffered formalin for 72 h and dehydrated in 70% ethanol prior to processing. Paraffin sections (6 μm) were created and an H&E stain was performed by the Comparative Pathology & Digital Imaging Shared Resource of The Ohio State University’s College of Veterinary Medicine. Adipose tissue sections were imaged at 20× magnification using an Olympus BX43 microscope with an Olympus SC30 color camera attachment and Olympus cellSens software. Downstream analysis was performed as previously described.92
For each field, adipocyte area was measured in FIJI using a semi-automated custom method based on the Adiposoft algorithm.93, 94, 95 Briefly, each image was split into H&E color channels by Color Deconvolution, then made binary by the Otsu thresholding algorithm.96 The images were then filtered with opening and median operators with the Morphological Filters plugin to sharpen cell boundaries.97 Automated particle subtraction and manual removal of extraneous particles were performed to reduce noise. A cutoff of 100 μm2 was used as the minimum size for an adipocyte. Six images were analyzed per animal. Adipocyte distribution curves show all sampled adipocytes within a given group. Average adipocyte sizes were calculated on a per-animal basis and then compared.
Perfusion and YFP fluorescence imaging
A subset of mice was anesthetized and transcardially perfused with phosphate-buffered saline (PBS), followed by 4% paraformaldehyde (PFA) (Sigma, St. Louis, MO) in PBS. Fixed brains were extracted and incubated overnight in 4% PFA on a rocker at 4°C. Brains were rinsed three times with PBS for 30 min before being submerged overnight in 30% sucrose with 0.03% sodium azide on a rocker at 4°C. The sucrose solution was replaced and left at 4°C for at least 3 days. Brains were embedded in optimal cutting temperature (O.C.T.) compound (Sakura Finetek, Torrance, CA), sectioned into 30-μm slices on a Thermo Scientific HM525NX cryostat (Waltham, MA), and placed on glass microscope slides. Fluorescence microscopy was performed on a Zeiss microscope (Thornwood, NY), and images were captured with Zeiss Zen software.
Statistical analysis
Data are expressed as means ± SEM. Microsoft Excel, IBM SPSS v.25, GraphPad Prism 9, and R v.4.1.3. software were used to analyze data. One-way ANOVAs with Tukey’s post hoc test were utilized for comparisons between three groups. Time course data (BWs, GTT, ITT, indirect calorimetry) were analyzed using a mixed ANOVA and area-under-the-curve calculations were performed where applicable. Normality was tested using the Shapiro-Wilk method. Outliers were determined and removed using the robust regression and outlier removal (ROUT) method.
Data availability
The authors confirm that data supporting the findings and conclusion of this study are presented within the article and supplemental information.
Acknowledgments
This work was supported by a grant from the Foundation for Prader-Willi Research (FPWR), NIH grant CA166590, and internal funding from The Ohio State University Comprehensive Cancer Center. Additional support was provided by the National Institute of Neurological Diseases and Stroke Institutional Center Core Grant P30 NS104177. We thank Julie Fitzgerald and the Rodent Behavior Core at The Ohio State University for materials used during the three-chamber sociability test.
Author contributions
Conceptualization, N.J.Q. and L.C.; methodology, N.J.Q., R.W., and L.C.; validation, N.J.Q. and L.C.; formal analysis, N.J.Q., J.M.A., and L.C.; investigation, N.J.Q., X.Z., J.M.A, W.H., B.A., S.K., and L.C.; resources, N.J.Q., W.H., R.W., and L.C.; data curation, N.J.Q., J.M.A., and L.C.; writing – original draft, N.J.Q. and L.C.; writing – review and editing, N.J.Q., R.W., and L.C.; visualization, N.J.Q.; supervision, W.H. and L.C.; project administration, L.C.; funding acquisition, L.C.
Declaration of interests
L.C. is an inventor of US patent 9,265,843 B2 on the autoregulatory BDNF vector.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.09.012.
Supplemental information
References
- 1.Resnick J.L., Nicholls R.D., Wevrick R., Prader-Willi Syndrome animal models working, G. Recommendations for the investigation of animal models of Prader-Willi syndrome. Mamm. Genome. 2013;24:165–178. doi: 10.1007/s00335-013-9454-2. [DOI] [PubMed] [Google Scholar]
- 2.Elena G., Bruna C., Benedetta M., Stefania D.C., Giuseppe C. Prader-Willi syndrome: clinical aspects. J. Obes. 2012;2012:473941. doi: 10.1155/2012/473941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verhoeven W.M., Curfs L.M., Tuinier S. Prader-Willi syndrome and cycloid psychoses. J. Intellect. Disabil. Res. 1998;42:455–462. doi: 10.1046/j.1365-2788.1998.4260455.x. [DOI] [PubMed] [Google Scholar]
- 4.Verhoeven W.M.A., Tuinier S. In: Catatonia in Autism Spectrum Disorders. Dhossche D.M., Wing L., Ohta M., Neumärker K.J., editors. Academic Press; 2006. Prader–willi syndrome: a typical psychoses and motor dysfunctions; pp. 119–130. [DOI] [Google Scholar]
- 5.Carrel A.L., Myers S.E., Whitman B.Y., Allen D.B. Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study. J. Clin. Endocrinol. Metab. 2002;87:1581–1585. doi: 10.1210/jcem.87.4.8414. [DOI] [PubMed] [Google Scholar]
- 6.Burman P., Ritzen E.M., Lindgren A.C. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocr. Rev. 2001;22:787–799. doi: 10.1210/edrv.22.6.0447. [DOI] [PubMed] [Google Scholar]
- 7.Grugni G., Sartorio A., Crino A. Growth hormone therapy for Prader-willi syndrome: challenges and solutions. Ther. Clin. Risk Manag. 2016;12:873–881. doi: 10.2147/TCRM.S70068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deal C.L., Tony M., Hoybye C., Allen D.B., Tauber M., Christiansen J.S., Growth hormone in prader-willi syndrome clinical care Guidelines Workshop, P. Growth hormone research society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J. Clin. Endocrinol. Metab. 2013;98:E1072–E1087. doi: 10.1210/jc.2012-3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassidy S.B., Schwartz S., Miller J.L., Driscoll D.J. Prader-Willi syndrome. Genet. Med. 2012;14:10–26. doi: 10.1038/gim.0b013e31822bead0. [DOI] [PubMed] [Google Scholar]
- 10.Shoffstall A.J., Gaebler J.A., Kreher N.C., Niecko T., Douglas D., Strong T.V., Miller J.L., Stafford D.E., Butler M.G. The high direct medical costs of Prader-Willi syndrome. J. Pediatr. 2016;175:137–143. doi: 10.1016/j.jpeds.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kayadjanian N., Schwartz L., Farrar E., Comtois K.A., Strong T.V. High levels of caregiver burden in Prader-Willi syndrome. PLoS One. 2018;13:e0194655. doi: 10.1371/journal.pone.0194655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kayadjanian N., Vrana-Diaz C., Bohonowych J., Strong T.V., Morin J., Potvin D., Schwartz L. Characteristics and relationship between hyperphagia, anxiety, behavioral challenges and caregiver burden in Prader-Willi syndrome. PLoS One. 2021;16:e0248739. doi: 10.1371/journal.pone.0248739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaaf C.P., Gonzalez-Garay M.L., Xia F., Potocki L., Gripp K.W., Zhang B., Peters B.A., McElwain M.A., Drmanac R., Beaudet A.L., et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat. Genet. 2013;45:1405–1408. doi: 10.1038/ng.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mercer R.E., Michaelson S.D., Chee M.J., Atallah T.A., Wevrick R., Colmers W.F. Magel2 is required for leptin-mediated depolarization of POMC neurons in the hypothalamic arcuate nucleus in mice. PLoS Genet. 2013;9:e1003207. doi: 10.1371/journal.pgen.1003207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pravdivyi I., Ballanyi K., Colmers W.F., Wevrick R. Progressive postnatal decline in leptin sensitivity of arcuate hypothalamic neurons in the Magel2-null mouse model of Prader-Willi syndrome. Hum. Mol. Genet. 2015;24:4276–4283. doi: 10.1093/hmg/ddv159. [DOI] [PubMed] [Google Scholar]
- 16.Maillard J., Park S., Croizier S., Vanacker C., Cook J.H., Prevot V., Tauber M., Bouret S.G. Loss of Magel2 impairs the development of hypothalamic Anorexigenic circuits. Hum. Mol. Genet. 2016;25:3208–3215. doi: 10.1093/hmg/ddw169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oncul M., Dilsiz P., Ates Oz E., Ates T., Aklan I., Celik E., Sayar Atasoy N., Atasoy D. Impaired melanocortin pathway function in Prader-Willi syndrome gene-Magel2 deficient mice. Hum. Mol. Genet. 2018;27:3129–3136. doi: 10.1093/hmg/ddy216. [DOI] [PubMed] [Google Scholar]
- 18.Xu B., Goulding E.H., Zang K., Cepoi D., Cone R.D., Jones K.R., Tecott L.H., Reichardt L.F. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 2003;6:736–742. doi: 10.1038/nn1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholson J.R., Peter J.C., Lecourt A.C., Barde Y.A., Hofbauer K.G. Melanocortin-4 receptor activation stimulates hypothalamic brain-derived neurotrophic factor release to regulate food intake, body temperature and cardiovascular function. J. Neuroendocrinol. 2007;19:974–982. doi: 10.1111/j.1365-2826.2007.01610.x. [DOI] [PubMed] [Google Scholar]
- 20.Bochukova E.G., Lawler K., Croizier S., Keogh J.M., Patel N., Strohbehn G., Lo K.K., Humphrey J., Hokken-Koelega A., Damen L., et al. A transcriptomic signature of the hypothalamic response to fasting and BDNF deficiency in Prader-Willi syndrome. Cell Rep. 2018;22:3401–3408. doi: 10.1016/j.celrep.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kernie S.G., Liebl D.J., Parada L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt H.D., Duman R.S. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology. 2010;35:2378–2391. doi: 10.1038/npp.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang H.J., Kim J.M., Lee J.Y., Kim S.Y., Bae K.Y., Kim S.W., Shin I.S., Kim H.R., Shin M.G., Yoon J.S. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord. 2013;151:679–685. doi: 10.1016/j.jad.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Marais L., Stein D.J., Daniels W.M. Exercise increases BDNF levels in the striatum and decreases depressive-like behavior in chronically stressed rats. Metab. Brain Dis. 2009;24:587–597. doi: 10.1007/s11011-009-9157-2. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z.Y., Jing D., Bath K.G., Ieraci A., Khan T., Siao C.J., Herrera D.G., Toth M., Yang C., McEwen B.S., et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han J.C., Muehlbauer M.J., Cui H.N., Newgard C.B., Haqq A.M. Lower brain-derived neurotrophic factor in patients with Prader-Willi syndrome compared to obese and lean control subjects. J. Clin. Endocrinol. Metab. 2010;95:3532–3536. doi: 10.1210/jc.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagahara A.H., Tuszynski M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011;10:209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- 28.Kishino A., Katayama N., Ishige Y., Yamamoto Y., Ogo H., Tatsuno T., Mine T., Noguchi H., Nakayama C. Analysis of effects and pharmacokinetics of subcutaneously administered BDNF. Neuroreport. 2001;12:1067–1072. doi: 10.1097/00001756-200104170-00040. [DOI] [PubMed] [Google Scholar]
- 29.Poduslo J.F., Curran G.L. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Mol. Brain Res. 1996;36:280–286. doi: 10.1016/0169-328x(95)00250-v. [DOI] [PubMed] [Google Scholar]
- 30.Siu J.J., Queen N.J., Liu X., Huang W., McMurphy T., Cao L. Molecular therapy of melanocortin-4-receptor obesity by an autoregulatory BDNF vector. Molecular therapy. Methods Clin. Dev. 2017;7:83–95. doi: 10.1016/j.omtm.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMurphy T., Huang W., Liu X., Siu J.J., Queen N.J., Xiao R., Cao L. Hypothalamic gene transfer of BDNF promotes healthy aging in mice. Aging Cell. 2019;18:e12846. doi: 10.1111/acel.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Y.Y., Ruan C.S., Yang C.R., Li J.Y., Kang Z.L., Zhou L., Liu D., Zeng Y.Q., Wang T.H., Tian C.F., et al. ProBDNF signaling regulates depression-like behaviors in Rodents under Chronic stress. Neuropsychopharmacology. 2016;41:2882–2892. doi: 10.1038/npp.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Connor B., Sun Y., von Hieber D., Tang S.K., Jones K.S., Maucksch C. AAV1/2-mediated BDNF gene therapy in a transgenic rat model of Huntington's disease. Gene Ther. 2016;23:283–295. doi: 10.1038/gt.2015.113. [DOI] [PubMed] [Google Scholar]
- 34.Cao L., Lin E.J., Cahill M.C., Wang C., Liu X., During M.J. Molecular therapy of obesity and diabetes by a physiological autoregulatory approach. Nat. Med. 2009;15:447–454. doi: 10.1038/nm.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cao L., Liu X., Lin E.J., Wang C., Choi E.Y., Riban V., Lin B., During M.J. Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition. Cell. 2010;142:52–64. doi: 10.1016/j.cell.2010.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X., McMurphy T., Xiao R., Slater A., Huang W., Cao L. Hypothalamic gene transfer of BDNF inhibits breast cancer progression and metastasis in middle age obese mice. Mol. Ther. 2014;22:1275–1284. doi: 10.1038/mt.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cone R.D., Cowley M.A., Butler A.A., Fan W., Marks D.L., Low M.J. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. Obes. 2001;25:S63–S67. doi: 10.1038/sj.ijo.0801913. [DOI] [PubMed] [Google Scholar]
- 38.Bischof J.M., Stewart C.L., Wevrick R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Genet. 2007;16:2713–2719. doi: 10.1093/hmg/ddm225. [DOI] [PubMed] [Google Scholar]
- 39.Kozlov S.V., Bogenpohl J.W., Howell M.P., Wevrick R., Panda S., Hogenesch J.B., Muglia L.J., Van Gelder R.N., Herzog E.D., Stewart C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007;39:1266–1272. doi: 10.1038/ng2114. [DOI] [PubMed] [Google Scholar]
- 40.Bariohay B., Lebrun B., Moyse E., Jean A. Brain-derived neurotrophic factor plays a role as an anorexigenic factor in the dorsal vagal complex. Endocrinology. 2005;146:5612–5620. doi: 10.1210/en.2005-0419. [DOI] [PubMed] [Google Scholar]
- 41.Pelleymounter M.A., Cullen M.J., Wellman C.L. Characteristics of BDNF-induced weight loss. Exp. Neurol. 1995;131:229–238. doi: 10.1016/0014-4886(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 42.Tschöp M.H., Speakman J.R., Arch J.R.S., Auwerx J., Brüning J.C., Chan L., Eckel R.H., Farese R.V., Galgani J.E., Hambly C. A guide to analysis of mouse energy metabolism. Nat. Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaiyala K.J., Schwartz M.W. Toward a more complete (and less controversial) understanding of energy expenditure and its role in obesity Pathogenesis. Diabetes. 2011;60:17–23. doi: 10.2337/db10-0909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bischof J.M., Van Der Ploeg L.H., Colmers W.F., Wevrick R. Magel2-null mice are hyper-responsive to setmelanotide, a melanocortin 4 receptor agonist. Br. J. Pharmacol. 2016;173:2614–2621. doi: 10.1111/bph.13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gould T.D., Dao D.T., Kovacsics C.E. Mood and Anxiety Related Phenotypes in Mice. Springer; 2009. The open field test; pp. 1–20. [DOI] [Google Scholar]
- 46.Mercer R.E., Kwolek E.M., Bischof J.M., van Eede M., Henkelman R.M., Wevrick R. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009;150B:1085–1099. doi: 10.1002/ajmg.b.30934. [DOI] [PubMed] [Google Scholar]
- 47.Fountain M.D., Tao H., Chen C.A., Yin J., Schaaf C.P. Magel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty. Gene Brain Behav. 2017;16:592–600. doi: 10.1111/gbb.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antunes M., Biala G. The novel object recognition memory: neurobiology, test procedure, and its modifications. Cognit. Process. 2012;13:93–110. doi: 10.1007/s10339-011-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leger M., Quiedeville A., Bouet V., Haelewyn B., Boulouard M., Schumann-Bard P., Freret T. Object recognition test in mice. Nat. Protoc. 2013;8:2531–2537. doi: 10.1038/nprot.2013.155. [DOI] [PubMed] [Google Scholar]
- 50.Thomas A., Burant A., Bui N., Graham D., Yuva-Paylor L.A., Paylor R. Marble burying reflects a repetitive and perseverative behavior more than novelty-induced anxiety. Psychopharmacology (Berl) 2009;204:361–373. doi: 10.1007/s00213-009-1466-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angoa-Perez M., Kane M.J., Briggs D.I., Francescutti D.M., Kuhn D.M. Marble burying and nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J. Vis. Exp. 2013:50978. doi: 10.3791/50978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meziane H., Schaller F., Bauer S., Villard C., Matarazzo V., Riet F., Guillon G., Lafitte D., Desarmenien M.G., Tauber M., Muscatelli F. An early postnatal oxytocin treatment prevents social and learning deficits in adult mice deficient for Magel2, a gene involved in Prader-Willi syndrome and autism. Biol. Psychiatr. 2015;78:85–94. doi: 10.1016/j.biopsych.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 53.Kaidanovich-Beilin O., Lipina T., Vukobradovic I., Roder J., Woodgett J.R. Assessment of social interaction behaviors. J. Vis. Exp. 2011 doi: 10.3791/2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steru L., Chermat R., Thierry B., Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- 55.Abella V., Scotece M., Conde J., Pino J., Gonzalez-Gay M.A., Gomez-Reino J.J., Mera A., Lago F., Gomez R., Gualillo O. Leptin in the interplay of inflammation, metabolism and immune system disorders. Nat. Rev. Rheumatol. 2017;13:100–109. doi: 10.1038/nrrheum.2016.209. [DOI] [PubMed] [Google Scholar]
- 56.Ye R., Scherer P.E. Adiponectin, driver or passenger on the road to insulin sensitivity? Mol. Metabol. 2013;2:133–141. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frühbeck G., Catalán V., Rodríguez A., Gómez-Ambrosi J. Adiponectin-leptin ratio: a promising index to estimate adipose tissue dysfunction. Relation with obesity-associated cardiometabolic risk. Adipocyte. 2018;7:57–62. doi: 10.1080/21623945.2017.1402151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao S., Kusminski C.M., Scherer P.E. Adiponectin, leptin and cardiovascular disorders. Circ. Res. 2021;128:136–149. doi: 10.1161/CIRCRESAHA.120.314458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeanneteau F.D., Lambert W.M., Ismaili N., Bath K.G., Lee F.S., Garabedian M.J., Chao M.V. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc. Natl. Acad. Sci. USA. 2012;109:1305–1310. doi: 10.1073/pnas.1114122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao R., Bergin S.M., Huang W., Mansour A.G., Liu X., Judd R.T., Widstrom K.J., Queen N.J., Wilkins R.K., Siu J.J., et al. Enriched environment regulates thymocyte development and alleviates experimental autoimmune encephalomyelitis in mice. Brain Behav. Immun. 2019;75:137–148. doi: 10.1016/j.bbi.2018.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiao R., Bergin S.M., Huang W., Slater A.M., Liu X., Judd R.T., Lin E.D., Widstrom K.J., Scoville S.D., Yu J., et al. Environmental and genetic activation of hypothalamic BDNF modulates T-cell immunity to exert an anticancer phenotype. Cancer Immunol. Res. 2016;4:488–497. doi: 10.1158/2326-6066.CIR-15-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tennese A.A., Wevrick R. Impaired hypothalamic regulation of endocrine function and delayed counterregulatory response to hypoglycemia in Magel2-null mice. Endocrinology. 2011;152:967–978. doi: 10.1210/en.2010-0709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cao L., Choi E.Y., Liu X., Martin A., Wang C., Xu X., During M.J. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metabol. 2011;14:324–338. doi: 10.1016/j.cmet.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Foglesong G.D., Huang W., Liu X., Slater A.M., Siu J., Yildiz V., Salton S.R.J., Cao L. Role of hypothalamic VGF in energy balance and metabolic adaption to environmental enrichment in mice. Endocrinology. 2016;2016:34–46. doi: 10.1210/en.2015-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carias K.V., Wevrick R. Preclinical testing in translational animal models of Prader-Willi syndrome: overview and gap analysis. Mol. Ther. Methods Clin. Dev. 2019;13:344–358. doi: 10.1016/j.omtm.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwartz L., Caixàs A., Dimitropoulos A., Dykens E., Duis J., Einfeld S., Gallagher L., Holland A., Rice L., Roof E., et al. Behavioral features in Prader-Willi syndrome (PWS): consensus paper from the International PWS clinical trial Consortium. J. Neurodev. Disord. 2021;13:25. doi: 10.1186/s11689-021-09373-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arble D.M., Pressler J.W., Sorrell J., Wevrick R., Sandoval D.A. Sleeve gastrectomy leads to weight loss in the Magel2 knockout mouse. Surg. Obes. Relat. Dis. 2016;12:1795–1802. doi: 10.1016/j.soard.2016.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scheimann A.O., Butler M.G., Gourash L., Cuffari C., Klish W. Critical analysis of bariatric procedures in Prader-Willi syndrome. J. Pediatr. Gastroenterol. Nutr. 2008;46:80. doi: 10.1097/01.mpg.0000304458.30294.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Muscogiuri G., Barrea L., Faggiano F., Maiorino M.I., Parrillo M., Pugliese G., Ruggeri R.M., Scarano E., Savastano S., Colao A., Restare Obesity in Prader–Willi syndrome: physiopathological mechanisms, nutritional and pharmacological approaches. J. Endocrinol. Invest. 2021;44:2057–2070. doi: 10.1007/s40618-021-01574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reus L., van Vlimmeren L.A., Staal J.B., Otten B.J., Nijhuis-van der Sanden M.W.G. The effect of growth hormone treatment or physical training on motor performance in Prader–Willi syndrome: a systematic review. Neurosci. Biobehav. Rev. 2012;36:1817–1838. doi: 10.1016/j.neubiorev.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 71.Morales J.S., Valenzuela P.L., Pareja-Galeano H., Rincón-Castanedo C., Rubin D.A., Lucia A. Physical exercise and Prader-Willi syndrome: a systematic review. Clin. Endocrinol. 2019;90:649–661. doi: 10.1111/cen.13953. [DOI] [PubMed] [Google Scholar]
- 72.Singh N.N., Lancioni G.E., Singh A.N., Winton A.S.W., Singh J., McAleavey K.M., Adkins A.D. A mindfulness-based health wellness program for an adolescent with Prader-Willi syndrome. Behav. Modif. 2008;32:167–181. doi: 10.1177/0145445507308582. [DOI] [PubMed] [Google Scholar]
- 73.McPhee S.W.J., Janson C.G., Li C., Samulski R.J., Camp A.S., Francis J., Shera D., Lioutermann L., Feely M., Freese A. Immune responses to AAV in a phase I study for Canavan disease. J. Gene Med. 2006;8:577–588. doi: 10.1002/jgm.885. [DOI] [PubMed] [Google Scholar]
- 74.Worgall S., Sondhi D., Hackett N.R., Kosofsky B., Kekatpure M.V., Neyzi N., Dyke J.P., Ballon D., Heier L., Greenwald B.M. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum. Gene Ther. 2008;19:463–474. doi: 10.1089/hum.2008.022. [DOI] [PubMed] [Google Scholar]
- 75.Marks W.J., Ostrem J.L., Verhagen L., Starr P.A., Larson P.S., Bakay R.A.E., Taylor R., Cahn-Weiner D.A., Stoessl A.J., Olanow C.W., Bartus R.T. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2–neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol. 2008;7:400–408. doi: 10.1016/S1474-4422(08)70065-6. [DOI] [PubMed] [Google Scholar]
- 76.Kaplitt M.G., Feigin A., Tang C., Fitzsimons H.L., Mattis P., Lawlor P.A., Bland R.J., Young D., Strybing K., Eidelberg D., During M.J. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- 77.Taghian T., Marosfoi M.G., Puri A.S., Cataltepe O.I., King R.M., Diffie E.B., Maguire A.S., Martin D.R., Fernau D., Batista A.R., et al. A safe and reliable technique for CNS delivery of AAV vectors in the cisterna magna. Mol. Ther. 2020;28:411–421. doi: 10.1016/j.ymthe.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haqq A.M., Muehlbauer M.J., Newgard C.B., Grambow S., Freemark M. The metabolic phenotype of Prader-Willi syndrome (PWS) in childhood: heightened insulin sensitivity relative to body mass index. J. Clin. Endocrinol. Metab. 2011;96:E225–E232. doi: 10.1210/jc.2010-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bervini S., Herzog H. Mouse models of Prader-Willi Syndrome: a systematic review. Front. Neuroendocrinol. 2013;34:107–119. doi: 10.1016/j.yfrne.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 80.Fountain M.D., Aten E., Cho M.T., Juusola J., Walkiewicz M.A., Ray J.W., Xia F., Yang Y., Graham B.H., Bacino C.A. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet. Med. 2017;19:45–52. doi: 10.1038/gim.2016.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCarthy J., Lupo P.J., Kovar E., Rech M., Bostwick B., Scott D., Kraft K., Roscioli T., Charrow J., Schrier Vergano S.A. Schaaf-Yang syndrome overview: Report of 78 individuals. Am. J. Med. Genet. 2018;176:2564–2574. doi: 10.1002/ajmg.a.40650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fountain M.D., Schaaf C.P. Prader-Willi syndrome and Schaaf-Yang syndrome: neurodevelopmental diseases intersecting at the MAGEL2 gene. Diseases. 2016;4:2. doi: 10.3390/diseases4010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Solum D.T., Handa R.J. Estrogen regulates the development of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus. J. Neurosci. 2002;22:2650–2659. doi: 10.1523/JNEUROSCI.22-07-02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sohrabji F., Miranda R.C., Toran-Allerand C.D. Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA. 1995;92:11110–11114. doi: 10.1073/pnas.92.24.11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pluchino N., Russo M., Santoro A.N., Litta P., Cela V., Genazzani A.R. Steroid hormones and BDNF. Neuroscience. 2013;239:271–279. doi: 10.1016/j.neuroscience.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 86.Wijesuriya T.M., De Ceuninck L., Masschaele D., Sanderson M.R., Carias K.V., Tavernier J., Wevrick R. The Prader-Willi syndrome proteins MAGEL2 and necdin regulate leptin receptor cell surface abundance through ubiquitination pathways. Hum. Mol. Genet. 2017;26:4215–4230. doi: 10.1093/hmg/ddx311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao S., Zhu Y.I., Schultz R.D., Li N., He Z., Zhang Z., Caron A., Zhu Q., Sun K., Xiong W. Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metabol. 2019;30:706–719. doi: 10.1016/j.cmet.2019.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Can A., Dao D.T., Terrillion C.E., Piantadosi S.C., Bhat S., Gould T.D. The tail suspension test. JoVE. 2012:e3769. doi: 10.3791/3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Friard O., Gamba M. BORIS: a free, versatile open-source event-logging software for video/audio coding and live observations. Methods Ecol. Evol. 2016;7:1325–1330. doi: 10.1111/2041-210X.12584. [DOI] [Google Scholar]
- 90.Fraulob J.C., Ogg-Diamantino R., Fernandes-Santos C., Aguila M.B., Mandarim-de-Lacerda C.A. A mouse model of metabolic syndrome: insulin resistance, fatty liver and non-Alcoholic fatty Pancreas disease (NAFPD) in C57BL/6 mice fed a high fat diet. J. Clin. Biochem. Nutr. 2010;46:212–223. doi: 10.3164/jcbn.09-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif.) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 92.Huang W., Queen N.J., McMurphy T.B., Ali S., Cao L. Adipose PTEN regulates adult adipose tissue homeostasis and redistribution via a PTEN-leptin-sympathetic loop. Mol. Metabol. 2019;30:48–60. doi: 10.1016/j.molmet.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rueden C.T., Schindelin J., Hiner M.C., DeZonia B.E., Walter A.E., Arena E.T., Eliceiri K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinf. 2017;18:529. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Galarraga M., Campion J., Munoz-Barrutia A., Boque N., Moreno H., Martinez J.A., Milagro F., Ortiz-de-Solorzano C. Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J. Lipid Res. 2012;53:2791–2796. doi: 10.1194/jlr.D023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Otsu N. A threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979;9:62–66. doi: 10.1109/TSMC.1979.4310076. [DOI] [Google Scholar]
- 97.Legland D., Arganda-Carreras I., Andrey P. MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics. 2016;32:3532–3534. doi: 10.1093/bioinformatics/btw413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that data supporting the findings and conclusion of this study are presented within the article and supplemental information.