

Abstract
Identification of targetable fusions as oncogenic drivers in non-small cell lung cancer has transformed its diagnostic and therapeutic paradigm. In a recent article in Nature, Izumi et al. report the discovery of CLIP1-LTK fusion as a novel oncogenic driver in lung cancer, targetable using the ALK tyrosine kinase inhibitor lorlatinib.
Chromosomal rearrangements resulting in gene fusions represent an important class of oncogenic driver across diverse tumor types. Tumors harboring these fusions often demonstrate oncogene addiction with remarkable sensitivity to small molecule inhibitors targeting the fusion oncoprotein. As prominent examples, in non-small cell lung cancer (NSCLC), ALK, ROS1, RET, and NTRK1–3 gene fusions have been established as actionable oncogenic drivers and numerous tyrosine kinase inhibitors (TKIs) targeting these fusions have transformed patient outcomes, demonstrating the therapeutic implications of oncogenic fusion detection (Thai et al., 2021). Yet, these fusions collectively account for less than 10% of all lung cancers, and in a substantial proportion of lung cancers (25–40% of adenocarcinomas), oncogenic drivers remain unknown (Chevallier et al., 2021). Discovery of novel oncogenic drivers in lung cancer deepens our understanding of tumorigenesis and further refines diagnostic and therapeutic stratification.
In their recent article in Nature, Izumi and colleagues (Izumi et al., 2021) make an important step forward with the identification of a novel oncogenic fusion driver. Through whole transcriptome sequencing of 75 NSCLC samples from the LC-SCRUM-Asia cohort known to be negative for described oncogenic drivers, they discovered in one patient a novel in-frame fusion transcript of CLIP1 (CAP-Gly domain containing linker protein 1, on chromosome 12q24) and LTK (leukocyte receptor tyrosine kinase, on chromosome 15q15). The presence of the CLIP1-LTK fusion in this index patient—predicted to harbor coiled-coil domains of CLIP1, a known fusion partner of other receptor tyrosine kinases (RTKs) such as ALK, ROS1, and RET, and the full kinase domain of LTK—was confirmed using reverse transcriptase-polymerase chain reaction (RT-PCR) and fluorescence in situ hybridization (FISH). The fusion was subsequently identified by RT-PCR in two additional patients out of 542 analyzed, yielding a prevalence of 0.4% in this Asian cohort. Of note, the tumors from all three patients were negative for other known oncogenic drivers, implying mutual exclusivity. In a series of elegant experiments, the authors went on to demonstrate that the LTK fusion protein is constitutively activated and drives transformation in vitro and in vivo.
LTK is a member of the anaplastic lymphoma kinase (ALK)/LTK subfamily within the insulin receptor superfamily of RTKs, sharing a near 80% identity in the kinase domain with ALK (Roll and Reuther, 2012). Knowledge about LTK is limited despite all that is known about its homolog ALK, a well-defined oncogenic driver in ~5% of NSCLC with multiple approved ALK TKIs (Lin et al., 2017). First described in 1988 (Ben-Neriah and Bauskin, 1988), LTK encodes an 864-amino-acid protein consisting of extracellular, transmembrane, and tyrosine kinase domains and a short carboxy terminus. Although LTK can activate the RAS/MAPK and PI3K/AKT pathways (Figure 1a) and is expressed in several organs including brain, bone marrow, lymphoid tissues, and placenta (Roll and Reuther, 2012), the precise biological role of LTK remains to be clarified. In malignancies, LTK has been found to be overexpressed in leukemia, and high expression of LTK in early-stage NSCLC has been associated with greater risk of metastasis (Muller-Tidow et al., 2005; Roll and Reuther, 2012). However, this work by Izumi et al. is the first to discover an in-frame LTK gene fusion as an oncogenic driver.
Figure 1. Schematic of LTK signaling and homology model.
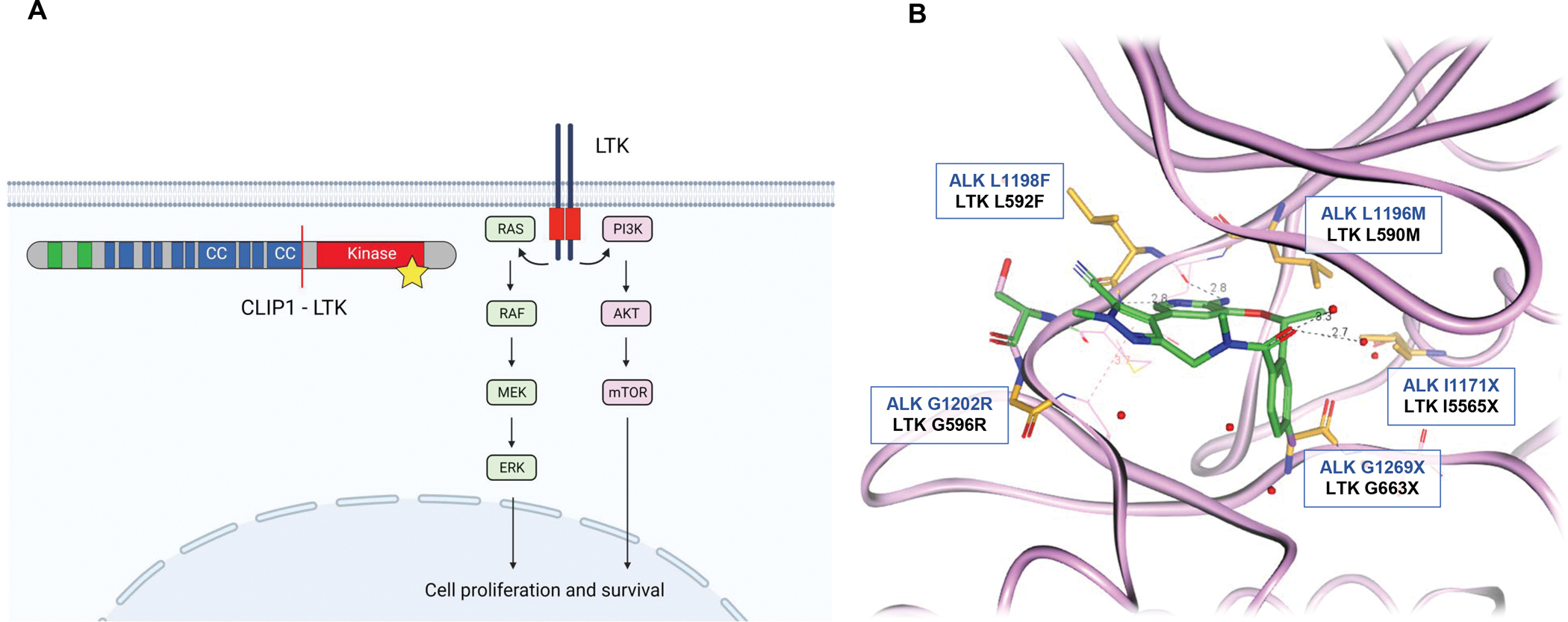
(A) The wild-type LTK receptor (right; red box indicates the kinase domain) promotes signaling through the RAS/MAPK and PI3K-AKT pathways upon activation. The CLIP1-LTK fusion (left; adapted from Izumi et al., Nature 2021) results in constitutive activation of LTK (indicated by star) and activation of AKT and ERK. In the fusion protein schematic, green represents cytoskeleton-associated protein glycine-rich (CAP-Gly) domain; blue represents coiled-coil domain (CC). Note: Schematic is not drawn to scale. (B) LTK homology model (colored pink) built from lorlatinib co-crystal structure (colored green) in the ALK kinase domain. LTK and ALK share 95% homology in residues within 6 Å of lorlatinib, which is the most critical for binding. The single difference within this distance corresponds to ALK A1200 and LTK S594. LTK residues mapped onto the ALK structure were energy minimized using the OPLS3 forcefield with the remainder of the protein fixed. Known clinical ALK resistance mutations (against various generations of ALK inhibitors and involved as part of compound mutations against lorlatinib) are colored orange with corresponding ALK (blue text) and LTK (black text) residues shown.
Importantly, the findings by Izumi et al. demonstrate the CLIP1-LTK fusion to be a therapeutically actionable oncogenic driver. Given the homology between LTK and ALK, the authors hypothesized that ALK-targeted TKIs may be potent against the LTK fusion. Indeed, the five FDA-approved ALK inhibitors (crizotinib, ceritinib, alectinib, brigatinib, and lorlatinib), and entrectinib and repotrectinib (multikinase inhibitors targeting ROS1/TRK/ALK), demonstrated variable degrees of preclinical activity against CLIP1-LTK, with lorlatinib—a third-generation ALK/ROS1 TKI—being the most potent (cellular IC50 of 1.1 nM in Ba/F3 cells expressing CLIP1-LTK). Based on the preclinical evidence that lorlatinib successfully inhibits CLIP1-LTK, the index patient received lorlatinib off-label and had a dramatic clinical response, albeit of unknown duration.
The discovery and validation of LTK as a therapeutic target in NSCLC has several important, immediate implications. First, the findings show that LTK fusions define a unique molecular subset of NSCLC, and their actionability supports the consideration of screening for LTK fusions in the diagnostic flow for advanced NSCLC. Prior studies have illuminated the benefits of RNA-based next-generation sequencing (NGS) in enabling multiplex testing for gene fusions in lung cancer (Benayed et al., 2019). It is worth noting that LTK fusions may not yet be identifiable using some of the currently available NGS gene panels. FISH could alternatively be developed to probe for LTK fusion in cases where NGS testing is not feasible.
Systematic assessment for the presence of LTK fusions in lung cancers will, in turn, shed more light on the molecular biology and clinical characteristics associated with these fusions. In NSCLC, other oncogenic fusions such as ALK, ROS1, and RET fusions are enriched in patients with adenocarcinoma and never or minimal smoking history (Lin et al., 2017). Furthermore, certain fusions (i.e., ALK) have been associated with higher incidence of metastases to the central nervous system (CNS) (Gainor et al., 2017), underscoring the importance of developing brain-penetrant drugs. In the work by Izumi et al., the three patients whose tumors harbored CLIP1-LTK had variable smoking histories, and one patient had baseline CNS metastasis. Investigation of a larger cohort of patients with LTK fusion-positive lung cancers will help define the true prevalence of this novel driver and the subset of patients in whom it may be enriched. In parallel, these efforts will enable the identification of other fusion partner genes of LTK aside from CLIP1 and inform studies into the mechanism of downstream pathway activation by the chimeric gene fusions.
Second, the efficacy achieved with lorlatinib highlights the potential value of repurposing available drugs with multiple RTK targets. Even though lorlatinib is highly selective for ALK and ROS1 (which share 77% homology in the ATP-binding site) (Johnson et al., 2014), it is also able to inhibit LTK, probably due to high degree of structural similarity between these closely related RTKs. Indeed, the homology for amino acid residues surrounding the ATP-binding pocket between LTK and ALK is near 100%, and the amino acid differences would not be predicted to contact lorlatinib (Figure 1b). At the same time, the efficacy of lorlatinib including duration of response in a larger cohort of patients with LTK fusion-positive NSCLC remains to be determined. The availability of TKI(s) that may be repurposed to treat LTK fusion-positive lung cancers should not deter the development of selective, potent LTK inhibitors.
Third, our experiences with targeted therapies across lung cancers have indicated that drug resistance is an inevitable clinical problem leading to disease relapse. In ALK fusion-positive NSCLC, both on-target resistance (due to acquired ALK kinase domain resistance mutations including compound ALK mutations; Figure 1b) and off-target resistance mechanisms (due to non-ALK alterations such as bypass signaling or histologic transformation) can result in ALK TKI resistance, with the distribution and spectrum of resistance mechanisms varying depending on the specific ALK inhibitor (Lin et al., 2017). As more knowledge is acquired regarding LTK fusion-positive cancers and as LTK inhibitors are developed, studies elucidating mechanisms of resistance to LTK inhibition will be warranted in parallel to continue to advance treatment options.
Finally, the oncogenic driver remains undetermined in a significant proportion of patients with NSCLC. The discovery of the LTK fusion represents a significant step forward, yet it also serves to underscore that much remains to be learned and much progress remains to be made in lung cancer. Continued efforts to identify actionable drivers and to develop broadly accessible biomarker testing and targeted therapies will be critical.
Acknowledgments
Declaration of Interests:
AJC declares no competing interests.
LVS has received consulting fees from AstraZeneca, Genentech, Pfizer, Takeda, and Janssen, and has received institutional research support from Boehringer-Ingelheim, Novartis, AstraZeneca, and Delfi.
TWJ is an employee of Pfizer.
JJL has served as a compensated consultant for Genentech, C4 Therapeutics, Blueprint Medicines, Nuvalent, and Turning Point Therapeutics; received honorarium and travel support from Pfizer; received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Bayer, Elevation Oncology, Roche, Linnaeus, and Novartis; received CME funding from OncLive, MedStar Health, and Northwell Health.
References:
- Ben-Neriah Y, and Bauskin AR (1988). Leukocytes express a novel gene encoding a putative transmembrane protein-kinase devoid of an extracellular domain. Nature 333, 672–676. [DOI] [PubMed] [Google Scholar]
- Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, Ptashkin R, Won H, Chang J, Halpenny D, et al. (2019). High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res 25, 4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevallier M, Borgeaud M, Addeo A, and Friedlaender A (2021). Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J Clin Oncol 12, 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor JF, Tseng D, Yoda S, Dagogo-Jack I, Friboulet L, Lin JJ, Hubbeling HG, Dardaei L, Farago AF, Schultz KR, et al. (2017). Patterns of Metastatic Spread and Mechanisms of Resistance to Crizotinib in ROS1-Positive Non-Small-Cell Lung Cancer. JCO Precis Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi H, Matsumoto S, Liu J, Tanaka K, Mori S, Hayashi K, Kumagai S, Shibata Y, Hayashida T, Watanabe K, et al. (2021). The CLIP1-LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, et al. (2014). Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem 57, 4720–4744. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Riely GJ, and Shaw AT (2017). Targeting ALK: Precision Medicine Takes on Drug Resistance. Cancer Discov 7, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Tidow C, Diederichs S, Bulk E, Pohle T, Steffen B, Schwable J, Plewka S, Thomas M, Metzger R, Schneider PM, et al. (2005). Identification of metastasis-associated receptor tyrosine kinases in non-small cell lung cancer. Cancer Res 65, 1778–1782. [DOI] [PubMed] [Google Scholar]
- Roll JD, and Reuther GW (2012). ALK-activating homologous mutations in LTK induce cellular transformation. PLoS One 7, e31733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thai AA, Solomon BJ, Sequist LV, Gainor JF, and Heist RS (2021). Lung cancer. The Lancet 398, 535–554. [DOI] [PubMed] [Google Scholar]