

Abstract
Phagocytosis is the necessary first step to sense foreign microbes or particles and enables activation of innate immune pathways such as inflammasomes. However, the molecular mechanisms underlying how phagosomes modulate inflammasome activity are not fully understood. We show that in murine dendritic cells (DCs), the lysosomal histidine/peptide solute carrier transporter SLC15A4, associated with human inflammatory disorders, is recruited to phagosomes and is required for optimal inflammasome activity after infectious or sterile stimuli. Dextran sodium sulfate‐treated SLC15A4‐deficient mice exhibit decreased colon inflammation, reduced IL‐1β production by intestinal DCs, and increased autophagy. Similarly, SLC15A4‐deficient DCs infected with Salmonella typhimurium show reduced caspase‐1 cleavage and IL‐1β production. This correlates with peripheral NLRC4 inflammasome assembly and increased autophagy. Overexpression of constitutively active mTORC1 rescues decreased IL‐1β levels and caspase1 cleavage, and restores perinuclear inflammasome positioning. Our findings support that SLC15A4 couples phagocytosis with inflammasome perinuclear assembly and inhibition of autophagy through phagosomal content sensing. Our data also reveal the previously unappreciated importance of mTORC1 signaling pathways to promote and sustain inflammasome activity.
Keywords: dendritic cells, inflammasomes, mTORC1, phagocytosis, SLC15A4
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
SLC15A4 regulates inflammasome activity via the mTOR pathway and autophagy to promote inflammation in response to sterile and infectious stimuli.
Introduction
Microorganisms or sterile particles captured by phagocytosis are first sensed by pattern‐recognition receptors (PRRs) such as Toll‐like receptors (TLRs), which recognize pathogen‐ or danger‐associated molecular patterns (PAMPs or DAMPs) and are located at the cell surface and on phagosomes. Other PRRs localize to the cytosol and sense pathogen or phagocytosed cargo‐derived products that escape phagosomes. In turn, activation of cytosolic PRRs—such as the nucleotide‐binding domain leucine‐rich repeat‐containing proteins (NLRs) —may lead to assembly of a multi‐subunit complex known as the inflammasome, and production of the highly inflammatory cytokine IL‐1β, resulting in escalation of the immune response (Latz et al, 2013; Christgen & Kanneganti, 2020). Therefore, inflammasome activity must be tightly regulated to prevent chronic inflammation, which may cause excessive tissue damage and lead to disease. One negative regulator of inflammasomes is autophagy (Shi et al, 2012; Zhong et al, 2016; Brady et al, 2018; Takahama et al, 2018). We and others have shown that inflammasome components such as the adaptor apoptosis‐associated speck‐like protein containing a caspase‐recruitment domain (ASC) are targeted for autophagic sequestration (Shi et al, 2012; Mantegazza et al, 2017), leading to inflammasome silencing. However, the molecular mechanisms underlying the link between phagocytosis, inflammasome activity, and autophagy are not completely understood (Moretti & Blander, 2014).
Phagosomes in dendritic cells (DCs) mature through a series of interactions with the endolysosomal system, acquiring increasing degradative capacity further promoted by phagosomal autonomous TLR signaling (Blander & Medzhitov, 2006; Hoffmann et al, 2012; Lopez‐Haber et al, 2020). In addition to TLR recognition, phagolysosomal nutrient transporters may sense phagosomal degradation products and link nutrient sensing to the modulation of autophagy via the activation of mechanistic target of rapamycin complex 1 (mTORC1) kinase signaling on phagolysosomes. mTORC1 senses lysosomal homeostasis and links cellular nutrient status to cell growth through its interactions with the Rag A‐D GTPases and their regulators (Chantranupong et al, 2015; Perera & Zoncu, 2016). In nutrient‐sufficient conditions, Rag complexes recruit mTORC1 to the lysosomal membrane for activation (Bar‐Peled et al, 2012). Conversely, nutrient limitation inactivates Rag GTPases and consequently mTORC1. In turn, mTORC1 inactivation stimulates autophagy by relieving inhibition of autophagy initiation proteins such as unc‐51 like autophagy activating kinase 1 (ULK1) (Hosokawa et al, 2009; Kim et al, 2011) and/or of the master lysosomal/ autophagy gene transcriptional activators (Sardiello et al, 2009; Martina et al, 2012; Settembre et al, 2012; Raben & Puertollano, 2016). Importantly, dysregulation of nutrient sensing may lead to disease, as shown by the association of many solute transporters with human metabolic and inflammatory disorders such as gout, systemic lupus erythematosus (SLE) and inflammatory bowel disease (IBD) (Lin et al, 2015; Zhang et al, 2019).
Among phagolysosomal solute carrier (SLC) transporters is the histidine/peptide transporter SLC15A4. In plasmacytoid DCs (pDCs) and B cells—in which this transporter has been extensively studied—SLC15A4 is required for optimal endosomal TLR7/9 signaling and subsequent type I interferon and antibody production (Blasius et al, 2010; Sasai et al, 2010; Kobayashi et al, 2021a). In line with this, SLC15A4 is associated with SLE and IBD in genome‐wide association studies (Sasawatari et al, 2011; Baccala et al, 2013; Kobayashi et al, 2014, 2021a; Heinz et al, 2020). Moreover, increased SLC15A4 mRNA levels were detected in colon samples from a cohort of IBD patients (Lee et al, 2009). Interestingly, in conventional DCs—in which SLC15A4 has been much less studied—it allows bacterially derived peptidoglycan egress from endosomes to promote pro‐inflammatory signaling through the cytosolic sensors nucleotide‐binding oligomerization domain 1/2 (NOD1/2) (Nakamura et al, 2014), also associated with IBD (Hugot et al, 2001; Sasawatari et al, 2011). However, the potential role of SLC15A4 in inflammasome activity has not been explored. Of note, SLC15A4 bears a di‐leucine motif recognized by the endolysosomal adaptor protein‐3 (AP‐3), which promotes cargo sorting to lysosomes and lysosome‐related organelles (Blasius et al, 2010; Rimann et al, 2022). Our previous studies demonstrated that AP‐3 is required for optimal phagosomal TLR signaling in DCs and promotes inflammasome priming (Mantegazza et al, 2012, 2014, 2017; Lopez‐Haber et al, 2020). In addition, AP‐3 indirectly regulates NLRC4 inflammasome positioning to the perinuclear region to prevent autophagic sequestration and limits autophagy to sustain inflammation and control bacterial infection (Mantegazza et al, 2017). We hypothesized that the phenotype associated with AP‐3 deficiency might be partly attributed to its putative cargo SLC15A4. We now show that SLC15A4 is a novel link between phagocytosis and inflammasome activation by coupling phagosomal content sensing to mTORC1 signaling. We demonstrate that SLC15A4 promotes inflammasome activity after Salmonella enterica serovar Typhimurium (STm) infection or sterile particle stimulation by restraining autophagy both in vitro and in an in vivo model of dextran sodium sulfate (DSS)‐induced colitis. Additionally, our data unravel a heretofore unappreciated role for SLC15A4 and mTORC1 in sustaining NLRC4 inflammasome activity by ensuring proper complex assembly at the perinuclear region and limiting autophagy.
Results
SLC15A4 promotes inflammasome activity by inhibiting autophagy after phagocytosis in DCs, but not macrophages
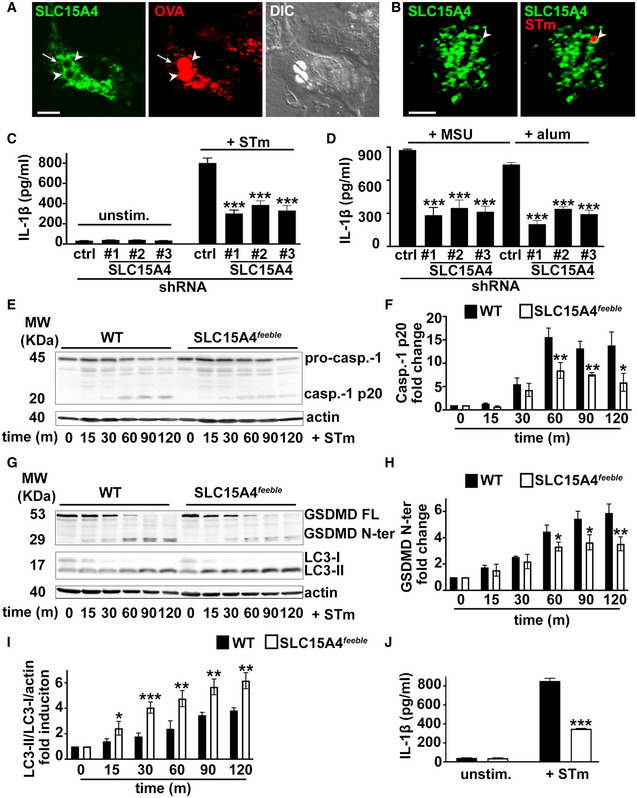
We previously showed that AP‐3 is required for optimal phagosomal maturation and formation of phagosomal tubules upon phagocytosis in DCs, which is dependent on the recruitment of lipid kinase phosphatidylinositol‐4‐kinase‐2alpha to phagosomes (Mantegazza et al, 2014; Lopez‐Haber et al, 2020). We also showed that AP‐3 indirectly sustains NLRP3 and NLRC4 inflammasome activity induced by phagosomal stimuli (Mantegazza et al, 2017). We propose that this function is likely due to AP‐3‐dependent recruitment of other unknown cargo to phagosomes. Given that SLC15A4 may bind AP‐3 (Blasius et al, 2010; Rimann et al, 2022), we first investigated the recruitment of SLC15A4 to DC phagosomes after phagocytosis of polystyrene beads coated with bacterial lipopolysaccharide (LPS) and Texas red (TxR)‐conjugated ovalbumin (OVA), as well as STm. We observed that SLC15A4‐GFP is indeed recruited to phagosomes and phagosomal tubules after LPS/OVA bead phagocytosis in wild‐type (WT) bone marrow‐derived DCs (BMDCs) and in the DC line DC2.4 (Figs 1A and EV1A and B; Movies EV1 and EV2), as well as to STm‐containing phagosomes (Fig 1B).
Figure 1. SLC15A4 promotes inflammasome activity by inhibiting autophagy.
-
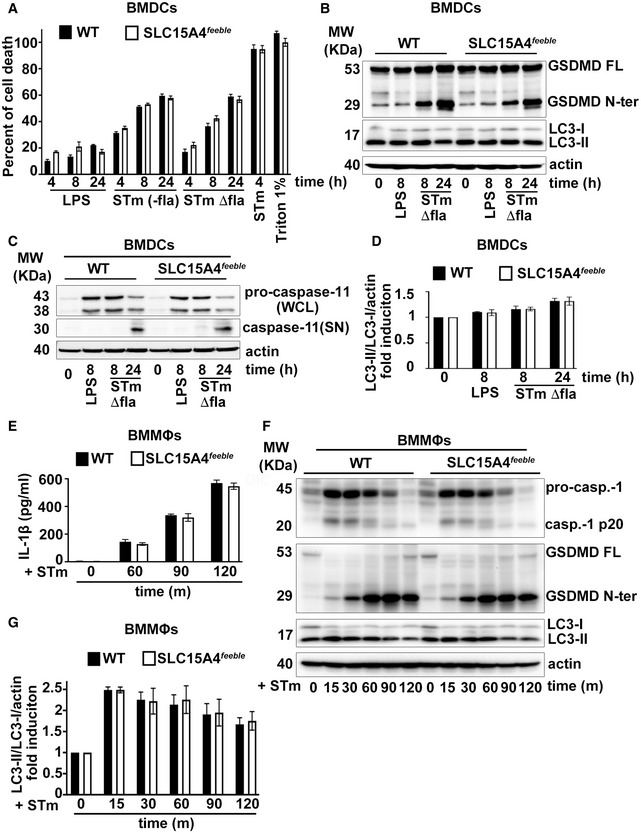
A–JWT BMDCs transduced with retrovirus encoding SLC15A4‐GFP (A, B), or lentiviruses encoding non‐target (ctrl) or any of three SLC15A4‐specific shRNAs (C, D) or WT or SLC15A4 feeble BMDCs (E–J) were treated with LPS/OVA‐TxR polystyrene beads (A) or STm‐mcherry (B), unstimulated or infected with flagellin‐expressing STm (C, E‐J) or primed for 3 h with LPS and stimulated with alum or MSU (D). (A, B). Cells were analyzed by live‐cell imaging 2 h after the pulse (A) or 1 h after infection (B). Representative images. Differential interference contrast (DIC) image shows cell shape and outline. Arrowheads, phagosomes; arrows, phagosomal tubules. Scale bars, 6 μm. (C–J). Cell supernatants collected 2 h (C, J) or 4 h (D) after treatment were assayed for IL‐1β by ELISA. (C, D, J). Representative plots of three independent experiments. (E–I). Cell pellets collected at the indicated time points after STm infection were lysed, fractionated by SDS–PAGE and immunoblotted for caspase‐1 (E, F), GSDMD or LC‐3 (G–I). (E, G). Representative immunoblots showing pro‐caspase‐1 (pro‐casp.‐1) and cleaved p20 (casp.‐1 p20) bands (E) or GSDMD full length (GSDMD FL), cleaved GSDMD N‐terminal fragment (GSDMD N‐ter), LC3‐II and LC3‐I bands in cell lysates (G). (F, H, I) Quantification of band intensities for caspase‐1 p20 normalized to pro‐caspase‐1 and actin (E), GSDMD N‐ter normalized to actin (G) and LC3‐II normalized to LC3‐I and actin (H) from three independent experiments are shown as fold change (F, H) or fold induction (I) relative to time 0.
Data information: Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. Two‐tailed Student's t‐test. See also Figs EV1, EV2, EV3.
Source data are available online for this figure.
Figure EV1. SLC15A4 is recruited to phagosomes and phagosomal tubules and restrains autophagy induction in DCs.
-
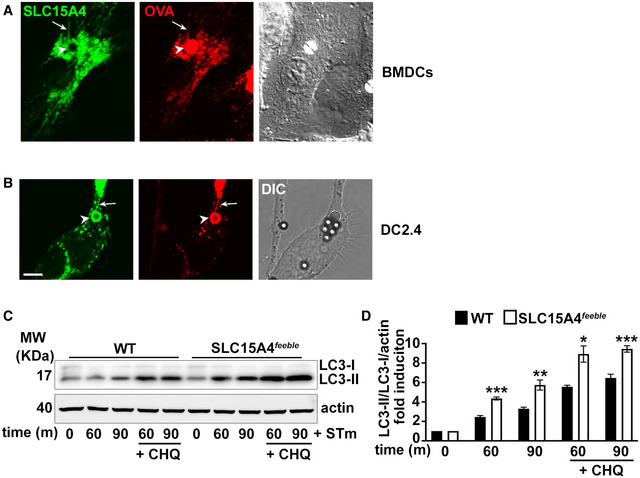
A, BWT BMDCs (A) or DC line DC2.4 (B) expressing SLC15A4‐GFP were pulsed with LPS/OVA‐TxR beads and analyzed by live‐cell imaging 2 h after the pulse. Representative images. DIC images show cell shape and outline. Arrowheads, phagosomes; arrows, phagosomal tubules. Scale bar, 6 μm.
-
C, DWT or SLC15A4 feeble BMDCs were infected with STm in the presence or absence of chloroquine (CHQ). Cell pellets collected at the indicated time points after STm infection were lysed, fractionated by SDS–PAGE, and immunoblotted for LC3 and actin. Representative immunoblots. (D) Quantification of band intensities for LC3‐II normalized to LC3‐I and actin from three independent experiments are shown as fold induction relative to time 0.
Data information: Data represent mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. Two‐tailed Student's t‐test.
Source data are available online for this figure.
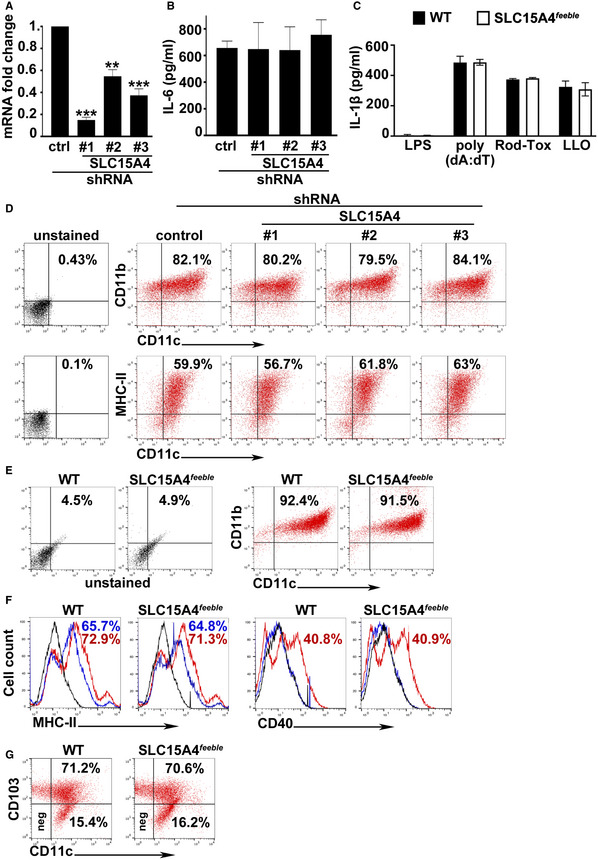
We then investigated whether SLC15A4 could regulate inflammasome activity after phagocytosis. To test this, we knocked down SLC15A4 in BMDCs using three different shRNA sequences. Knock‐down efficiency was between 50 and 80%, as assessed by quantitative real‐time PCR (Fig EV2A). SLC15A4 knockdown (KD) did not affect DC differentiation compared to shRNA non‐target control‐treated DCs (Fig EV2D). We then treated BMDCs with flagellin‐expressing STm—to induce NLRC4 activation (Wynosky‐Dolfi et al, 2014; Mantegazza et al, 2017)—and measured IL‐1β secretion as a read‐out for inflammasome activity by ELISA. IL‐1β secretion was reduced by 50% in SLC15A4 KD DCs relative to shRNA control‐treated DCs (Fig 1C). Similar results were observed when DCs were primed with LPS and subsequently stimulated with alum or monosodium urate crystals (MSU) to activate the NLRP3 inflammasome (Fig 1D). Conversely, secretion of IL‐6 after LPS treatment was not affected by SLC15A4 KD, suggesting that SLC15A4 does not play a role in the priming step of the inflammasome pathway (Fig EV2B). This agrees with previous observations indicating that SLC15A4 does not affect TLR signaling in conventional DCs (Blasius et al, 2010). To confirm our observations on SLC15A4 KD DCs, we differentiated DCs from WT and SLC15A4 feeble mice. The feeble mutation results in abnormal splicing of the Slc15a4 gene resulting in the transcription of two aberrant products and the absence of functional protein expression (Blasius et al, 2010). SLC15A4 feeble BMDCs differentiated and matured similarly in response to LPS compared to BMDCs (Fig EV2E and F). In agreement with our observations in SLC15A4 KD DCs, after STm stimulation, IL‐1β secretion was reduced by more than 50% in SLC15A4 feeble DCs compared to WT DCs (Fig 1J). Consistent with the decreased IL‐1β secretion, caspase 1 and its substrate gasdermin‐D (GSDMD) cleavage were also reduced in SLC15A4 feeble DCs between 60 and 120 min after STm stimulation (Fig 1E–H). In contrast, stimulation of NLRC4 or NLRP3 inflammasomes with soluble stimuli—STm type III secretion system inner rod protein fused to the N‐terminal domain of anthrax lethal factor and co‐administered with the protective antigen channel protein for cytosol delivery (Rod‐Tox) (Rauch et al, 2016; Reyes Ruiz et al, 2017), or Listeria monocytogenes pore‐forming listeriolysin O toxin (LLO)(Franchi et al, 2012), respectively—led to comparable levels of IL‐1β production between WT and SLC15A4 feeble DCs (Fig EV2C). Similarly, stimulation of absent in melanoma 2 (AIM2) inflammasome with double‐stranded DNA [poly(dA:dT)] (Rathinam et al, 2010) delivered to the cytosol by lipid complexes was independent of SLC15A4 (Fig EV2C). These observations indicate that SLC15A4 does not play a role in inflammasome activation upon plasma membrane damage or breaching, in agreement with its localization to lysosomes and phagosomes. Overall, our observations suggest that SLC15A4 is required for optimal inflammasome activity triggered by particulate stimuli.
Figure EV2. SLC15A4 knock‐down or feeble mutation does not significantly impair DC differentiation, maturation, LPS priming, or inflammasome stimulation with soluble ligands.
-
A–GWT BMDCs transduced with lentiviruses encoding non‐target (ctrl), or any of three SLC15A4 shRNAs (A, B, D), were untreated (A, D) or treated with LPS for 3 h (B), or WT and SLC15A4 feeble BMDCs were untreated (E) or treated with LPS for 3 h (C) or 18 h (F), or intestinal DCs isolated from colon of WT or SLC15A4 feeble mice were untreated (G). (A) cDNA generated from isolated RNA was analyzed by RT–qPCR. Data from three independent experiments were normalized to the average of two housekeeping genes, and the ΔΔCt values were calculated and represented as mean ± SD fold change of mRNA in SLC15A4 shRNA‐transduced cells relative to non‐target ctrl‐treated cells. (B) Cell supernatants collected 3 h after treatment were assayed for IL‐6 by ELISA. Representative plot of three independent experiments. (C) Cell supernatants collected 6 h after poly(dA:dT)/LyoVec treatment or 1 h after Rod‐Tox or LLO treatment were assayed for IL‐1β by ELISA. Representative plot of three independent experiments. (D, E). Representative dot plots with the percentages of CD11b+/CD11c+ BMDCs indicated as markers of DC differentiation after 7 days. (F) Representative histograms with the percentages of CD40+ or MHC‐II+ BMDCs as markers of DC maturation on day 7. Blue solid lines, untreated DCs; red solid lines, LPS‐treated DCs; black solid lines, unstained controls. (G) Representative dot plots with the percentages of CD11c+/CD103+ DCs indicated as markers of intestinal DCs.
Data information: **P < 0.01; ***P < 0.001. Two‐tailed Student's t‐test.
Source data are available online for this figure.
We previously showed that STm infection induces autophagy in BMDCs and that the absence of AP‐3 increases autophagy induction (Mantegazza et al, 2017). However, the mechanism by which AP‐3 modulates autophagy remains unknown. Considering the link between SLC15A4 and mTORC1—a known autophagy regulator (Martina et al, 2012; Settembre et al, 2012) —observed in other cell types (Kobayashi et al, 2014), we hypothesized that SLC15A4 might be required for optimal inflammasome activity in DCs by limiting autophagy. We detected induction of the lipidated form of the microtubule‐associated protein 1 light chain 3 α (LC3‐II) relative to the unlipidated LC3‐I (indicative of autophagy induction (Klionsky et al, 2021)), in cell lysates over time after STm stimulation in WT DCs (Fig 1G and I). Importantly, LC3‐II was significantly increased in SLC15A4 feeble DCs between 15 and 120 min after STm stimulation compared to WT DCs (Fig 1G and I). Notably, increased LC3‐II/LC3‐I ratio in SLC15A4 feeble DCs persisted after treatment with chloroquine, an alkalinizing agent that blocks autophagic flux by preventing autophagosome content degradation (Klionsky et al, 2021) (Fig EV1C and D), suggesting that the increase in LC3‐II/LC3‐I ratio reflected increased autophagy induction rather than decreased autophagosome clearance. These observations suggest that SLC15A4 promotes NLRC4 inflammasome activity by inhibiting autophagy induction after STm stimulation.
Furthermore, DC stimulation with mutant STm lacking flagellin (ΔfliCΔfljB) or STm grown to stationary phase to downregulate flagellin expression [STm (−fla)] —conditions that prevent NLRC4 activation (Wynosky‐Dolfi et al, 2014) —leads to similar induction of cell death [measured by lactate dehydrogenase (LDH) release; Fig EV3A]. Given that contrary to flagellin‐expressing STm, STm ΔfliCΔfljB or STm (−fla) induce an NLRC4‐independent, caspase‐11‐dependent delayed type of cell death (Kayagaki et al, 2011; Broz et al, 2012; Ross et al, 2018), our results suggest that SLC15A4 is not required for caspase‐11‐dependent non‐canonical inflammasome activity. Concordantly, caspase‐11 and its substrate GSDMD cleavage were not impaired in SLC15A4 feeble DCs (Fig EV3B and C). Interestingly, neither STm ΔfliCΔfljB nor STm (−fla) induced autophagy significantly, measured by LC3‐II induction (Fig EV3B and D), in contrast to flagellin‐expressing STm (Fig 1G and I). These observations may explain at least partly the lack of a SLC15A4 requirement in non‐canonical inflammasome function.
Figure EV3. SLC15A4 is not required for non‐canonical inflammasome function in DCs or NLRC4 inflammasome activity in MΦs.
-
A–DWT or SLC15A4 feeble BMDCs were treated with LPS (100 ng/ml) overnight and infected with non‐expressing flagellin STm [STm (−fla)] or STm lacking flagellin (ΔfliCΔfljB) (A–D) or flagellin‐expressing STm (STm; control) (A) at MOI 5:1 for 4–24 h. A. Cells were pelleted and LDH release into the supernatant was measured in three independent experiments. Percent of cell death was normalized to release from uninfected cells by 1% Triton X‐100 treatment (100% cell death). Representative experiment showing biological triplicates. (B, C) Cell pellets (B, C) or cell supernatants (C) collected at the indicated time points after STm infection were lysed, fractionated by SDS–PAGE, and immunoblotted for GSDMD, LC3, and actin (B), or pro‐caspase 11, caspase 11, and actin (C). Representative immunoblots. (D) Quantification of band intensities for LC3‐II normalized to LC3‐I and actin from three independent experiments are shown as fold induction relative to time 0. Data represent mean ± SD.
-
E–GWT or SLC15A4 feeble BMMΦs were unstimulated or infected with flagellin‐expressing STm for the indicated time points. (E) Cell supernatants collected after treatment were assayed for IL‐1β by ELISA. Representative plot of three independent experiments. (F) Cell pellets collected at the indicated time points after STm infection were lysed, fractionated by SDS–PAGE, and immunoblotted for caspase‐1, GSDMD, or LC‐3. Representative immunoblots. (G) Quantification of band intensities for LC3‐II normalized to LC3‐I and actin from three independent experiments are shown as fold change relative to time 0.
Data information: Data represent mean ± SD.
Source data are available online for this figure.
SLC15A4 is also expressed in macrophages (MΦs), where it promotes metabolic reprogramming upon stress (Kobayashi et al, 2021b). Given the differential phagolysosomal properties in terms of acidification, PRR signaling and inflammasome regulation between DCs and MΦs (Lukacs et al, 1991; Savina et al, 2006; Mantegazza et al, 2008, 2017; Yates & Russell, 2008; Zanoni et al, 2017), we investigated whether SLC15A4 could also play a role in inflammasome modulation in bone marrow‐derived MΦs (BMMΦs) upon stimulation with flagellin‐expressing STm. In contrast to our observations in BMDCs, neither IL‐1β production nor caspase‐1 or GSDMD cleavage was impaired by the absence of SLC15A4 (Fig EV3E and F). This is in agreement with our previous finding that the regulation of inflammasome activity upon particulate stimuli in BMMΦs is not dependent on AP‐3 (Mantegazza et al, 2017). Remarkably, LC3‐II/LC3‐I ratio is higher in WT BMMΦs compared to WT BMDCs 15 m after STm stimulation (Figs 1I and EV3G), similarly to what we previously showed upon alum stimulation (Mantegazza et al, 2017). These observations suggest that the role of SLC15A4 in the modulation of autophagy and inflammasome activity is cell‐type specific and may also be ligand‐dependent.
SLC15A4 feeble mice show faster recovery from acute dextran sodium sulfate‐induced colitis in mice
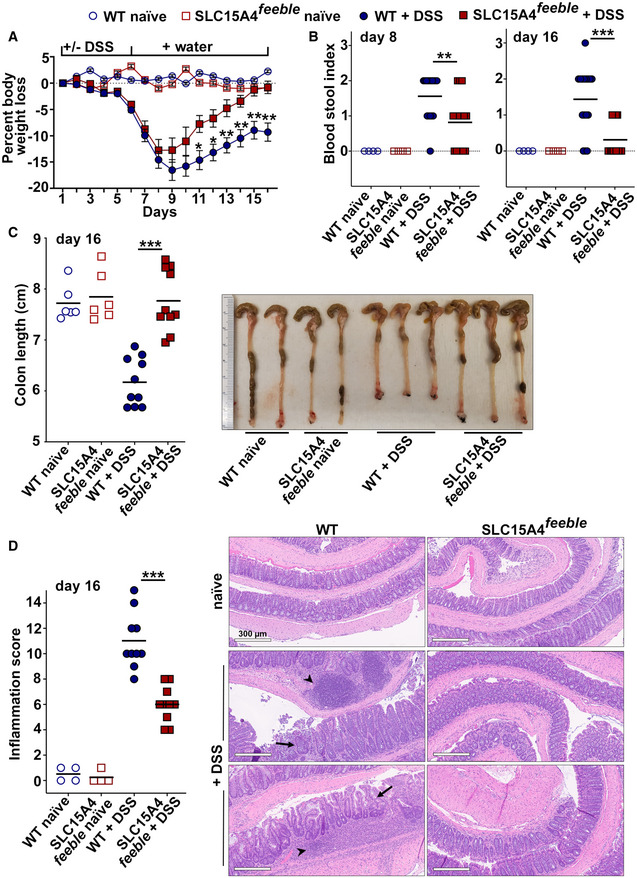
SLC15A4 expression was shown to be pro‐colitogenic in a mouse model of mild chronic DSS‐induced colitis due to its role in promoting TLR9 and NOD signaling (Sasawatari et al, 2011). However, the role played by SLC15A4 in inflammasome activity and the production of IL‐1β in DCs have not been investigated. To test whether SLC15A4 regulates inflammasome activity in vivo, we employed a model of acute DSS‐induced colitis. This model is proposed to cause intestinal injury associated with NLRP3 inflammasome activation and IL‐1β production (Bauer et al, 2010). WT and SLC15A4 feeble mice received either water (naïve mice) or 2.5% DSS in drinking water for 5 days, followed by water for 10 more days to allow recovery. Weight loss and stool appearance, consistency, and presence of blood were monitored daily after DSS administration. WT and SLC15A4 feeble mice showed a similar trend of body weight loss during the first 8 days after DSS administration—with an average of 15% and 13% body weight loss on day 7, respectively. However, after this time, SLC15A4 feeble mice recovered body weight significantly more efficiently than WT mice. Remarkably, on day 16 (endpoint), SLC15A4 feeble mice completely recovered their initial body weight, while WT mice's body weight remained significantly lower—0% and 10% of body weight loss, respectively (Fig 2A). Even though the percent of body weight loss was similar on day 8, the blood stool index was significantly different between WT and SLC15A4 feeble mice. Whereas most WT mice exhibited very soft stool consistency and visible traces of stool blood (score 2), most SLC15A4 feeble mice showed soft but formed stool consistency and positive hemoccult tests without visible traces of blood (score 1) (Fig 2B, left). On day 16, consistent with the faster recovery observed in body weight in SLC15A4 feeble mice, most of these mice exhibited normal stool appearance and negative hemoccult tests (score 0), whereas most WT mice remained showing higher blood stool index scores at the same time point (Fig 2B, right).
Figure 2. SLC15A4 feeble mice show faster recovery from acute dextran sodium sulfate‐induced colitis in mice.
-
AMice in three independent experiments were weighed daily. Percent of body weight loss overtime is represented relative to day 1.
-
BStool consistency and presence of blood were assessed. Blood stool index on days 8 (left panel) and 16 (right panel) was determined using the following score: 0, normal feces, negative hemoccult; 1, soft but formed feces, positive hemoccult; 2, very soft feces, visible traces of stool blood; 3, diarrhea, rectal bleeding. Data from three independent experiments are shown.
-
CLeft panel. Colon length was measured on day 16. Data from three independent experiments are shown. Right panel. Representative image of three independent experiments.
-
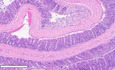
DColon swiss roll samples were formalin‐fixed, paraffin embedded, stained, and scored blinded by expert pathologist. Left panel. Inflammation score on day 16. Data from three independent experiments are shown. Right panels. Representative haematoxylin and eosin stained, formalin‐fixed paraffin embedded samples. Note disrupted crypt architecture (arrows) and inflammatory infiltrate (arrowheads) in DSS‐treated WT samples. Scale bar, 300 μm.
Data information: Data represent mean ± SD in panel A. Mean is indicated in panels B, C, and D. **P < 0.01; ***P < 0.001; Mann–Whitney non‐parametric statistical test.
Source data are available online for this figure.
Decreased colon length has been associated with increased inflammation (Chassaing et al, 2014). In agreement with this observation and with our previous results on day 16, while most DSS‐treated SLC15A4 feeble mice showed similar colon length compared to water‐treated mice (mean = 7.8 cm), all WT mice showed significantly reduced colon length compared to water‐treated mice and DSS‐treated SLC15A4 feeble mice (mean = 6.2 cm) (Fig 2C), suggesting that colon inflammation is reduced in the absence of SLC15A4. Consistent with these observations, histopathological evaluation of swiss roll samples prepared from mouse colon on day 16, revealed significant differences between WT and SLC15A4 feeble mice (Fig 2D). WT mouse samples displayed in most cases moderate to severe inflammation characterized by focally extensive ulceration of the mucosa with segmental involvement of the deep lamina propria and multifocal extension into the submucosa, while SLC15A4 feeble mouse samples showed mostly mild to moderate inflammation with modest infiltrate of mononuclear cells (Fig 2D, right). Accordingly, the inflammation score index—comprising mucosal/crypt loss, crypt inflammation, and inflammatory infiltrate—was remarkably higher in WT samples (mean = 11) compared to SLC15A4 feeble samples (mean = 6) (Fig 2D, left). These results show that the absence of SLC15A4 promotes faster recovery from acute DSS‐induced colitis.
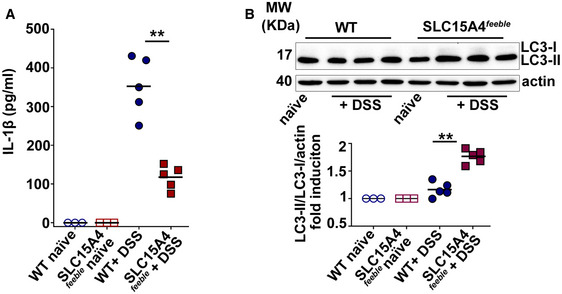
Given our observations that SLC15A4 favors IL‐1β production in BMDCs by restraining autophagy, we assessed the contribution of intestinal DCs to the DSS‐induced process. On day 16, DCs from WT and SLC15A4 feeble mice were isolated from colon. The percent of CD103+/CD11c+ DCs was similar between WT and SLC15A4 feeble mice (Fig EV2G). Isolated DCs were then cultured overnight without further stimulation. Remarkably, IL‐1β secretion was significantly higher in the cell culture supernatants from WT DCs relative to SLC15A4 feeble DCs as measured by ELISA (Fig 3A). In agreement with our in vitro results, cell lysates from isolated intestinal DCs showed increased LC3‐II induction in SLC15A4 feeble DCs compared to WT DCs (Fig 3B), suggesting that increased autophagy is limiting IL‐1β production in colonic DCs in vivo.
Figure 3. SLC15A4 feeble intestinal CD11c+ DCs show reduced IL‐1β secretion and increased autophagy.
-
AIL‐1β was measured from culture supernatants by ELISA in three independent experiments.
-
BCell pellets from three independent experiments were immunoblotted for LC3 and actin. Upper panel. Representative immunoblot. Lower panel. Quantification of band intensities for LC3‐II normalized to LC3‐I and actin are shown as fold induction relative to samples from untreated mice. Note that LC3‐I signal from ex vivo samples is almost undetectable.
Data information: Mean is indicated. **P < 0.01. Mann–Whitney non‐parametric statistical test. See also Fig EV2G.
Source data are available online for this figure.
SLC15A4 promotes inflammasome perinuclear positioning away from autophagic membranes
We and others showed that autophagy negatively regulates inflammasome activity and that the adaptor ASC aggregates or “specks” formed after inflammasome assembly appear to be at least partly sequestered by autophagic membranes, as measured by fluorescence microscopy and flow cytometry upon differential permeabilization (Shi et al, 2012; Mantegazza et al, 2017). To assess inflammasome assembly and ASC speck positioning, we transduced WT and SLC15A4 feeble DCs with the retroviral constructs ASC‐GFP and mcherry‐LC3 or probed endogenous ASC specks together with the autophagic adaptor p62/SQTM1 (p62), which links ubiquitylated substrates to LC3 (Clausen et al, 2010). ASC speck formation was monitored by fluorescence microscopy and flow cytometry. Neither the percentage of ASC‐GFP speck‐positive DCs nor the kinetics of ASC‐GFP speck formation after STm infection differed appreciably between WT and SLC15A4 feeble DCs as assessed by flow cytometry by analyzing GFP‐width and GFP‐height parameters as previously described (Sester et al, 2015; Hoss et al, 2018) (Fig EV4A–C).
Figure EV4. SLC15A4 does not affect the kinetics of ASC speck formation.
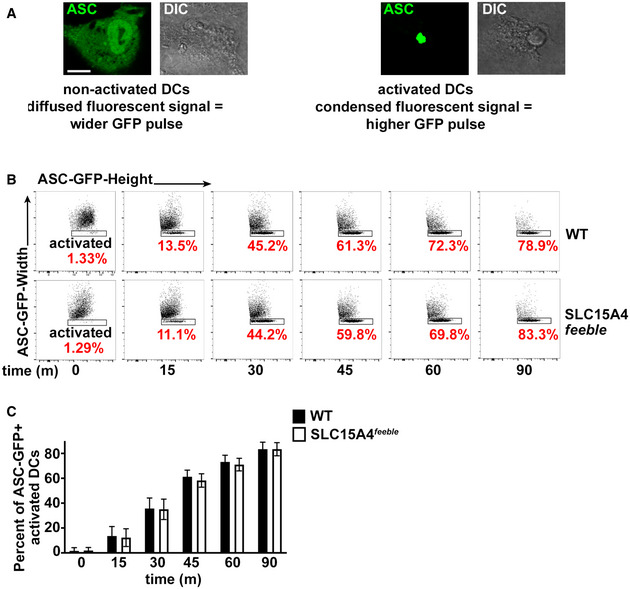
WT or SLC15A4 feeble BMDCs expressing ASC‐GFP were infected with flagellin‐expressing STm. Cells were fixed at the indicated time points after infection and analyzed by flow cytometry measuring GFP‐width and GFP‐height pulses.
-
ACytosolic‐diffused ASC‐GFP in non‐activated cells correlates to a wider GFP signal compared to condensed ASC‐GFP in activated cells. Representative images. DIC images show cell shape and outline. Scale bar, 6 μm.
-
BRepresentative dot plots, after gating on GFP+ cells. Note that the height of the GFP pulse is more prominent (rectangular shape) relative to the width of the GFP signal as the number of cells bearing ASC specks (activated DCs) increases over time. Percent of GFP+ activated DCs are indicated in red.
-
CPlot represents three independent experiments.
Data information: Data represent mean ± SD.
Source data are available online for this figure.
On the contrary, ASC‐GFP specks in SLC15A4 feeble DCs were increasingly surrounded by LC3‐II puncta (within a radius of 1 μm), compared to WT DCs as observed by fluorescence microscopy 30 min after STm stimulation (Fig 4A and B). Similarly, endogenous ASC specks in SLC15A4 feeble DCs were also significantly more surrounded by p62 puncta at the same time point (Fig 4C). Additionally, ASC speck formation was visualized mostly in the perinuclear region (within a radius of 3 μm), in WT DCs (70 ± 10% of cells), while intriguingly, in most of SLC15A4 feeble DCs it was detected away from the nucleus (26 ± 9% of cells in perinuclear region; Fig 4A, C, and D). This behavior resembles our previous observations in AP‐3‐deficient DCs, in which ASC specks appear away from the nucleus and surrounded by autophagic membranes (Mantegazza et al, 2017).
Figure 4. SLC15A4 promotes inflammasome perinuclear positioning away from autophagic membranes.
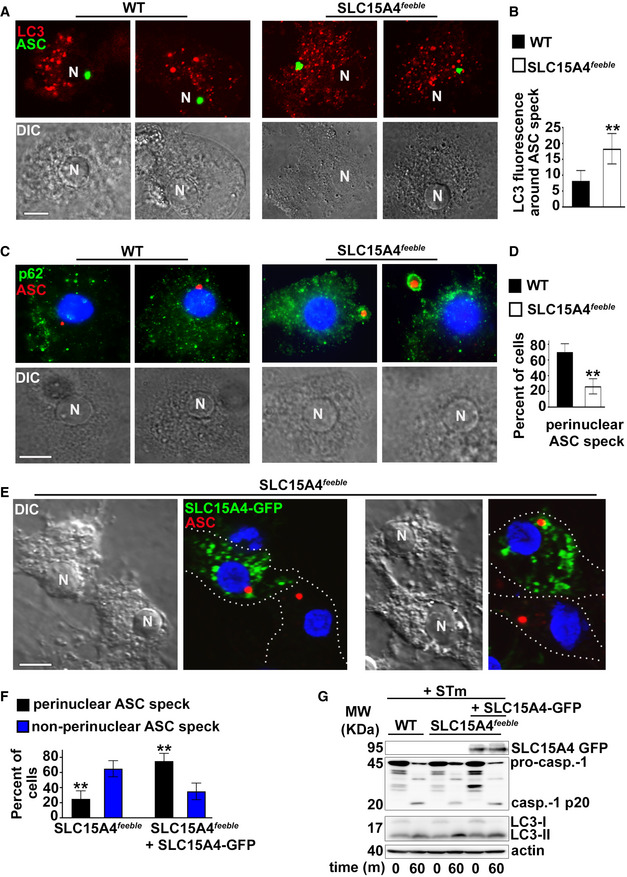
-
A–GWT or SLC15A4 feeble BMDCs expressing ASC‐GFP and mCherry‐LC3 (A, B, D) or non‐transduced (C) or SLC15A4 feeble BMDCs transduced with SLC15A4‐GFP (E, F, G) were infected with flagellin‐expressing STm and fixed (A–F) or lysed (G) 1 h after infection. (A) Cells were analyzed by fluorescence microscopy. Representative images showing ASC speck (green) relative to mCherry‐LC3 (red) in two infected WT and SLC15A4 feeble DCs each. (B) Quantification of LC3 fluorescence per unit area in a radius of 1 μm surrounding the ASC speck in 20 cells per cell type in each of three independent experiments. (C) Cells were stained for endogenous ASC and p62, labeled with DAPI and analyzed by fluorescence microscopy. Representative images showing ASC speck (red) relative to p62 (green) in two infected WT and SLC15A4 feeble DCs each. Note p62 staining surrounding ASC specks in SLC15A4 feeble DCs. (D) Quantification of perinuclear (within a radius of 3 μm from the nucleus) ASC specks in at least 30 cells per cell type in each of three independent experiments. (E, F) Cells were stained for endogenous ASC, labeled with DAPI and analyzed by fluorescence microscopy. Representative images showing ASC speck (red) and SLC15A4‐GFP (green) in SLC15A4 feeble DCs together with non‐transduced SLC15A4 feeble DCs. Note the perinuclear positioning of ASC specks in the transduced cells (E). Quantification of perinuclear (within a radius of 3 μm from the nucleus) and non‐perinuclear ASC specks in at least 30 cells per cell type in each of three independent experiments (F). (G) Representative immunoblots showing pro‐caspase‐1 (pro‐casp.‐1) and cleaved p20 (casp.‐1 p20), LC3‐II and LC3‐I, and actin bands. N, nucleus. Dotted white lines, cell outlines. Corresponding DIC images show nuclear position. Scale bar, 8 μm.
Data information: Data represent mean ± SD. **P < 0.01. Two‐tailed Student's t‐test. See also Fig EV4.
Source data are available online for this figure.
ASC speck positioning in SLC15A4 feeble DCs shifted from peripheral to perinuclear in cells transduced with human SLC15A4‐GFP (Fig 4E and F). This correlated with increased inflammasome activity as evidenced by augmented caspase‐1 cleavage in SLC15A4 feeble DCs expressing human SLC15A4‐GFP compared to non‐transduced counterparts (Fig 4G).
Altogether, these observations suggest that the kinetics of inflammasome formation is not affected by SLC15A4. However, the site of inflammasome assembly, inflammasome activity, and its targeting by autophagy are regulated by SLC15A4.
Constitutive mTORC1 signaling restores inflammasome activity and positioning in SLC15A4 feeble DCs
SLC15A4 was shown to regulate mTORC1 signaling in B cells and mast cells (Kobayashi et al, 2014, 2017). Given that mTORC1 activation inhibits autophagy (Martina et al, 2012; Settembre et al, 2012), we investigated whether mTORC1 signaling was dysregulated in SLC15A4 feeble DCs in our model of in vitro STm infection, in which SLC15A4 restrains autophagy (Fig 1G and I). WT and SLC15A4 feeble DCs non‐transduced or transduced with the control construct methionine aminopeptidase 2 (metap2; Gu et al, 2017) or GTP‐bound RagBQ99L—which lacks GTPase activity and renders mTORC1 constitutively active (Bar‐Peled et al, 2012)—were infected with STm for 1 h and assayed for mTORC1 activity. We observed ~50% reduced phosphorylation of mTOR on Ser 2481—the autophosphorylation site associated with mTORC1 catalytic activity (Kobayashi et al, 2014) —together with similar impaired phosphorylation of mTORC1 downstream substrates ULK1—required for autophagy initiation (Hosokawa et al, 2009)—, p70S6 kinase, and S6 ribosomal protein, in infected SLC15A4 feeble DCs compared to their WT counterparts (Fig 5A and B; non‐transduced and control lanes, and Fig EV5A). Reduced phosphorylation in SLC15A4 feeble DCs was rescued when DCs were transduced with GTP‐bound RagBQ99L (Fig 5A and B). Similar results were obtained after cell starvation in Earle's balanced salt solution (EBSS), which lacks essential amino acids (Sharifi et al, 2015) (Fig EV5B). Notably, reduced IL‐1β production together with decreased caspase‐1 and GSDMD cleavage in STm infected SLC15A4 feeble DCs were also rescued by overexpression of RagBQ99L (Fig 5C–E). Conversely, inhibition of mTOR signaling in WT BMDCs using torin, reduced IL‐1β production to the levels detected in SLC15A4 feeble DCs (Fig EV5C). These observations suggest that SLC15A4 positively regulates mTORC1 signaling in DCs, keeping autophagy at bay to promote inflammasome activity after STm infection. In agreement with this, knockdown of autophagy proteins ATG5 or ATG7 in WT and SLC15A4 feeble DCs increased inflammasome activity measured by IL‐1β production and caspase‐1 and GSDMD cleavage, and rescued defective inflammasome activity in SLC15A4 feeble DCs (Fig 5F and G).
Figure 5. Constitutive mTORC1 signaling restores inflammasome activity and positioning in SLC15A4 feeble DCs.
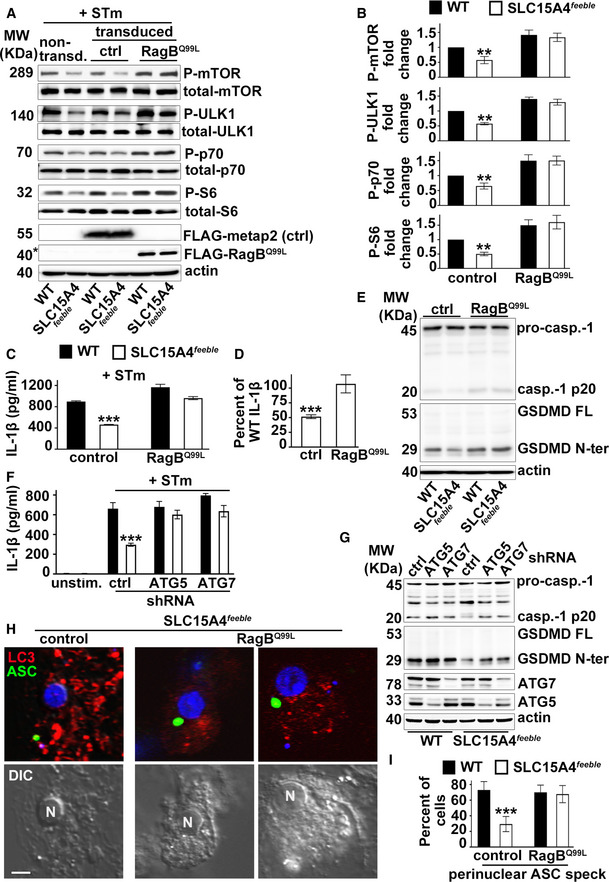
-
A–IWT or SLC15A4 feeble BMDCs transduced with constitutively active RagB (RagBQ99L) or metap2 (control) (A–E, H, I) and mcherry‐LC3 (H, I), or non‐target control, ATG5 or ATG7 shRNAs (F, G), were unstimulated or infected with flagellin‐expressing STm. (A, B). Cell pellets collected 1 h after STm infection were lysed, fractionated by SDS–PAGE, and immunoblotted for phospho (P) and total mTOR, ULK1 kinase, p70 kinase and S6, FLAG and actin. Representative immunoblots. Non‐specific band right above FLAG‐RagBQ99L is indicated with an asterisk (A). Quantification of band intensities for P‐mTOR normalized to total mTOR, P‐ULK1 normalized to total ULK1, P‐p70 normalized to total p70, and P‐S6 normalized to S6, in transduced cells from three independent experiments are shown as fold change relative to WT control (B). (C, D, F). Cell supernatants collected 1 h after infection were assayed for IL‐1β by ELISA. (C, F). Representative plots of three independent experiments. D. IL‐1β values of transduced SLC15A4 feeble BMDCs from three independent experiments are shown as percent of WT DC values to represent rescue of the defective phenotype. (E, G). Cell pellets collected 1 h after STm infection were probed for caspase‐1, GSDMD, actin (E, G), ATG5, and ATG 7 (G). Representative immunoblots showing pro‐caspase‐1 (pro‐casp.‐1) and cleaved p20 (casp.‐1 p20), GSDMD full length (GSDMD FL) and cleaved GSDMD N‐terminal fragment (GSDMD N‐ter) bands. (H, I). Cells were stained for endogenous ASC, labeled with DAPI, and analyzed by fluorescence microscopy. Representative images showing ASC speck (green) and LC3 (red) in SLC15A4 feeble DCs (H). Quantification of perinuclear (within a radius of 3 μm from the nucleus) in at least 30 cells per cell type in each of three independent experiments (I). N, nucleus. Corresponding DIC images show nuclear position. Scale bar, 8 μm.
Data information: Data represent mean ± SD. **P < 0.01; ***P < 0.001. Two‐tailed Student's t‐test. See also Fig EV5.
Source data are available online for this figure.
Figure EV5. Constitutively active mTOR rescues defective mTOR signaling, and mTOR inhibition in WT DCs phenocopies SLC15A4 feeble defects in inflammasome activity and positioning.
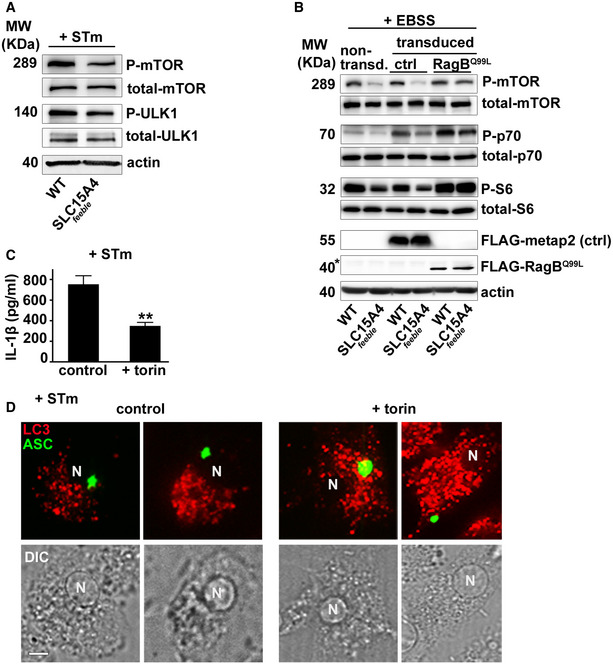
-
A–DWT or SLC15A4 feeble BMDCs were untreated (A–D) or pre‐treated with torin (C, D), non‐transduced (A), or transduced with FLAG‐metap2 (ctrl) or FLAG‐RagBQ99L (B) or ASC‐GFP and mcherry‐LC3 (D) and infected with STm (A, C, D) or starved in EBSS for 4 h (B). (A, B). Cell pellets were lysed, fractionated by SDS–PAGE, and immunoblotted for phospho (P) and total mTOR, ULK1, p70 kinase or S6, FLAG, or actin. Representative immunoblots of three independent experiments. (B) Non‐specific band right above FLAG‐RagBQ99L is indicated with an asterisk. (C) Cell supernatants collected 1 h after infection were assayed for IL‐1β by ELISA. Representative plot of three independent experiments. (D) Cells were fixed and analyzed by fluorescence microscopy 1 h after infection. Representative images showing ASC speck (green) relative to LC3 (red). Note peripheral ASC speck positioning in torin‐treated DCs. Corresponding DIC images show nuclear position. N, nucleus. Scale bar, 6 μm.
Data information: Data represent mean ± SD. **P < 0.01. Two‐tailed Student's t‐test.
Source data are available online for this figure.
Remarkably, overexpression of RagBQ99L, but not metap2 (control), in SLC15A4 feeble DCs induced ASC speck formation in the perinuclear region after NLRC4 stimulation (Fig 5H and I), a location that appears to be protected from autophagic membranes (Fig 4A–D), and our previous reports (Mantegazza et al, 2017). In line with this observation, WT BMDCs treated with torin showed ASC speck formation away from the nucleus, similar to SLC15A4 feeble DCs (Fig EV5D). Altogether, our data indicate that mTORC1 signaling ensures optimal IL‐1β secretion by limiting autophagy and promoting the assembly of the NLRC4 inflammasome at the perinuclear region where it appears excluded from autophagic membranes.
Discussion
The molecular mechanisms underlying the regulation of inflammasome activity by phagosomal signaling are not completely elucidated (Moretti & Blander, 2014), nor are the pathways that bring together the processes of phagocytosis, autophagy, and downmodulation of inflammasomes. Autophagy has been increasingly recognized as an anti‐inflammatory process (Shi et al, 2012; Zhong et al, 2016; Deretic & Levine, 2018). However, the molecular players that prompt autophagy after phagocytosis are only beginning to be unraveled. PRR stimulation by PAMPs/DAMPs on phagosomes is one mechanism proposed to trigger lysosomal and autophagy pathways by promoting expression of the coordinated lysosomal expression and regulation gene network (Gray et al, 2016; Pastore et al, 2016). However, PRR stimulation also promotes inflammasome priming (Latz et al, 2013), leading to a pro‐inflammatory response. Therefore, pro‐ and anti‐inflammatory forces triggered after phagocytosis must be finely tuned according to the nature of the phagocytic cargo.
Here we show that the lysosomal histidine/peptide transporter SLC15A4 is recruited to phagosomes and phagosomal tubules in DCs, as also observed for some PRRs (Blander, 2007; Mantegazza et al, 2012), and promotes inflammasome activity. An extensive recruitment surface may be required to ensure proper ligand or nutrient sensing and consequent cytosolic signaling, as observed in lysosomes (Nakamura et al, 2014). The role played by SLC15A4 in promoting type I interferon production and TLR9 signaling in pDCs and B cells has been extensively studied and supports the importance of this transporter in the pathogenesis of inflammatory disorders, including IBD (Blasius et al, 2010; Sasawatari et al, 2011; Baccala et al, 2013; Kobayashi et al, 2014, 2017, 2021a,b). However, the contribution of SLC15A4 to conventional DC function has been less studied (Nakamura et al, 2014). We now describe a previously unappreciated mechanism explaining the pro‐inflammatory role of SLC15A4 in vitro and in an in vivo model of DSS‐induced colitis. Even though we do not rule out the contribution of other cell types to colon inflammation, we show that SLC15A4 promotes inflammasome activity and supports IL‐1β production in conventional intestinal DCs by restraining autophagy, a process that genome‐wide association studies have correlated with IBD (Larabi et al, 2020).
We demonstrate that SLC15A4 promotes inflammasome activity triggered by particulate sterile and infectious stimuli—but not by ligands directly delivered into the cytosol through the plasma membrane—in BMDCs, but not BMMΦs. The absence of SLC15A4 in DCs led to decreased caspase‐1 and GSDMD cleavage and decreased IL‐1β production, which correlated with increased autophagy induction. In contrast, inflammasome activity and autophagy in BMMΦs do not appear to be regulated by SLC15A4. We speculate that intrinsic differences in phagosomal properties such as increased vacuolar H+ ATPase activity, a known autophagy stimulator (Zoncu et al, 2011a; Chung et al, 2019), in MΦs compared to DCs (Lukacs et al, 1991; Mantegazza et al, 2008), may hinder the effect of SLC15A4 in BMMΦ phagosomes. Regarding the lack of a SLC15A4 requirement for the regulation of inflammasomes triggered by soluble toxins or ligands delivered to the cytosol by lipid complexes, this resembles our previous observations on the role of the lysosomal adaptor AP‐3—which recognizes a dileucine motif on SLC15A4 for phagolysosomal targeting. One plausible explanation is that the plasma membrane is not on the AP‐3/ SLC15A4 trafficking route (Peden et al, 2004; Rimann et al, 2022). Alternatively, differences in inflammasome regulation may be ligand‐dependent, and associated with the differential triggering of regulatory responses, such as autophagy. In this regard, DC stimulation with mutant STm or STm grown in conditions that prevent NLRC4 stimulation to favor caspase‐11‐dependent non‐canonical inflammasome activity (Kayagaki et al, 2011; Broz et al, 2012; Wynosky‐Dolfi et al, 2014; Ross et al, 2018) did not induce autophagy appreciably. In this context, SLC15A4 was not required for inflammasome function, suggesting that the role played by SLC15A4 in autophagy regulation may also be dictated by the nature of the phagocytic cargo. Similar cell‐intrinsic and ligand‐dependent responses were observed upon inflammasome stimulation for the secretion of IL‐1β from hyperactive living DCs, in both mouse and human cells (Chen et al, 2014; Zanoni et al, 2016; Hatscher et al, 2021). Whether autophagic pathways and/or SLC15A4 modulate DC hyperactivation remains to be addressed.
How does SLC15A4 inhibit autophagy upon STm stimulation in DCs? We hypothesized that autophagy inhibition was dependent on mTORC1, the master regulator of cell growth, which inhibits autophagy in nutrient‐sufficient conditions (Zoncu et al, 2011b). We speculated that loss of histidine sensing caused by the absence of SLC15A4 would repress mTORC1 activation and trigger autophagy, similarly to the lysosomal arginine transporter SLC38A9 (Wang et al, 2015; Rebsamen & Superti‐Furga, 2016). In agreement with this, phosphorylation of mTOR and downstream effectors, including ULK1, was impaired in the absence of SLC15A4. Notably, decreased inflammasome activity in SLC15A4 feeble DCs was restored by the expression of constitutively active Rag B, which keeps mTORC1 active and is therefore predicted to inhibit autophagy. In line with this, knockdown of autophagy proteins ATG5 or ATG7 also restored defective inflammasome activity in SLC15A4 feeble DCs. Conversely, inhibiting mTORC1 decreased inflammasome activity in WT cells. Recent studies show that the Ragulator‐Rag‐mTORC1 axis is required for GSDMD oligomerization—post‐cleavage—via ROS production (Evavold et al, 2021). Of note, these studies focus on late inflammasome events. Our observations, on the other hand, unravel a novel role for Rag B and mTORC1 in the regulation of inflammasome activity in the initial steps of inflammasome activation.
Inflammasome activity is also suggested to depend on the site of inflammasome assembly (Martin et al, 2014; Magupalli et al, 2020). We previously observed that NLRC4 inflammasome formation at the perinuclear region appears to protect specks from sequestration by autophagic membranes (Mantegazza et al, 2017). We now show that the absence of SLC15A4 drives ASC speck formation away from the perinuclear region, and that remarkably, constitutively active mTORC1 rescues this aberrant phenotype. NLRP3 and pyrin inflammasome assembly occur at the microtubule‐organizing center (MTOC), and perinuclear positioning is dependent on histone deacetylase 6, an adaptor of the motor protein dynein (Magupalli et al, 2020). In contrast, NLRC4 inflammasome specks do not appear to localize at the MTOC and do not require microtubule transport for their activation ((Magupalli et al, 2020) and our unpublished observations). However, other cellular cytoskeletal filaments may be required for NLRC4 positioning. Similar to our observations in SLC15A4 feeble BMDCs, peripheral ASC speck positioning is observed in the absence of AP‐3 (Mantegazza et al, 2017). Interestingly, AP‐3 associates with the intermediate filament protein vimentin (Styers et al, 2004, 2005), which was shown to interact with NLRP3 (dos Santos et al, 2015). Intermediate filaments also interact with motor proteins such as kinesins and dyneins (Helfand et al, 2004) and may be required to recruit these motors to sites of inflammasome assembly. Whether any of these associations are impaired in the absence of SLC15A4 and are relevant for NLRC4 assembly remains to be addressed. Furthermore, raptor, a component of mTORC1, is shown to associate with kinesins, dyneins, and other molecules involved in cytoskeletal‐filament assembly or function (Rabanal‐Ruiz et al, 2021). Therefore, it is conceivable that reduced mTORC1 signaling caused by the absence of SLC15A4 impairs binding of ASC specks to cytoskeletal filaments or associated motor proteins that remain to be characterized. Regardless, our current observations support a novel and unexpected function for SLC15A4 and mTORC1 in ensuring NLRC4 inflammasome positioning at the perinuclear region, a location proposed to be required for proper function (Martin et al, 2014) and seemingly protected from autophagy, after phagocytosis (Mantegazza et al, 2017).
We propose that the balance between inflammasome activation and deactivation by autophagy is regulated by recruitment of not only PRRs but also certain SLCs such as SLC15A4 to DC phagosomes. In turn, PAMPs/DAMPs and nutrient sensing performed by PRRs and SLCs link phagocytosis to mTORC1 signaling, the regulation of autophagy and the ultimate control of the immune response. In homeostatic conditions, lysosomal mTORC1 signaling is turned on, keeping autophagy at bay. In the case of pathogenic microorganisms, if the integrity of phagosomes is compromised, inflammasomes are triggered and autophagy is induced in response to PRR signaling, organelle damage, or the pathogen itself (Deretic & Levine, 2018). In this scenario, SLC15A4 restrains autophagy to promote anti‐microbial responses. Thus, SLC15A4 contributes to microbial sensing in phagosomes and allies with conventional PRRs to optimize immune responses against certain bacterial pathogens. In contrast, in the case of sterile inflammation such as IBD, SLC15A4 would play a detrimental role by sustaining inflammation and subsequent tissue damage. We speculate that strategies aimed at downmodulating SLC15A4 function in this scenario would be beneficial to mitigate inflammation. Whether this is a generalized mechanism and other phagosomal SLCs also contribute to the regulation of the phagosome‐inflammasome‐autophagy axis, certainly warrants further investigation.
Materials and Methods
Mice
C57BL/6 wild‐type (WT) mice and SLC15A4 feeble (C57BL/6J‐Slc15a4 m1Btlr ) mice were originally purchased from The Jackson Laboratories (Bar Harbor, ME). SLC15A4 feeble mice were previously described (https://mutagenetix.utsouthwestern.edu/phenotypic/phenotypic_rec.cfm?pk=426). Sex‐ and age‐matched mice between 6 and 12 weeks of age were used in all experiments.
Ethics statement
Mice were bred under pathogen‐free conditions in the Department of Veterinary Resources at the Children's Hospital of Philadelphia or at Thomas Jefferson University, and were euthanized by carbon dioxide narcosis according to guidelines of the American Veterinary Medical Association Guidelines on Euthanasia. All animal studies were performed in compliance with the federal regulations set forth in the recommendations in the Public Health Service Policy on the Humane Care and Use of Laboratory Animals, the National Research Council's Guide for the Care and Use of Laboratory Animals, the National Institutes of Health Office of Laboratory Animal Welfare, the American Veterinary Medical Association Guidelines on Euthanasia, and the guidelines of the Institutional Animal Care and Use Committees of Children's Hospital of Philadelphia and Thomas Jefferson University. All protocols used in this study were approved by the Institutional Animal Care and Use Committee at the Children's Hospital of Philadelphia (protocols #14–001064 and #16–001064) and Thomas Jefferson University (protocol #21‐04‐368).
DSS‐induced mouse model of colitis, colon sample preparation and analysis
WT and SLC15A4 feeble male mice were randomized into control and experimental groups and co‐housed at weaning across multiple cages. Experiments were performed three times with a total of 15 mice per genotype per experimental group and 12 male mice per genotype per control group. In experimental groups, 8 weeks‐old mice were given 2.5% DSS (40,000–50,000 KDa molecular weight; Alfa Aesar J14489, Tewksbury, MA) in drinking water for 5 days, followed by normal drinking water for 10 more days. Age‐matched control mice received water only. Body weight and stool appearance, consistency, and presence of blood were recorded daily. Fecal occult blood was detected using Hemoccult single slides (Beckman Coulter Inc., Brea, CA). Blood stool index was determined using the following score: 0, normal feces, negative hemoccult; 1, soft but formed feces, positive hemoccult; 2, very soft feces, visible traces of stool blood; 3, diarrhea, rectal bleeding (Wirtz et al, 2007; Perse & Cerar, 2012). On day 16, mice were sacrificed, colon was dissected, length was measured, and Swiss rolls were prepared from PBS rinsed colon, fixed in 10% neutral buffered formalin (Polysciences) at room temperature for 48 h, rinsed in PBS, and stored in 50% ethanol for histological staining and analysis. Haematoxilin and eosin staining, paraffin‐embedding, and histopathological analysis were performed at the Comparative Pathology Core, University of Pennsylvania, School of Veterinary Medicine. Histopathological analysis and scoring were performed blinded by expert veterinary pathologist Dr. Enrico Radaelli. Histopathological analysis was focused on assessing the mucosal changes (inflammatory and proliferative) of the mid and distal colorectal tract present in each sample (Perse & Cerar, 2012; Gadaleta et al, 2017). Briefly, scoring comprised the evaluation of mucosal/ crypt loss, crypt inflammation, mononuclear cells, neutrophils, epithelial hyperplasia, and oedema/ fibrosis.
Reagents
LPS, MSU, poly (dA:dT)/Lyovec and type III secretion system inner rod protein fused to B. anthracis lethal factor (LFn‐Rod) were purchased from InvivoGen (San Diego, CA) and alum was from ThermoFisher Scientific (Rockford, IL). TxR‐conjugated OVA and EBSS were from Invitrogen (ThermoFisher Scientific). Mouse monoclonal anti‐caspase‐1 p20 (Casper‐1) and rabbit polyclonal anti‐ASC (AL177) were from Adipogen (San Diego, CA); recombinant LLO protein, rabbit polyclonal anti‐LC3B (ab48394), mouse monoclonal anti‐p62 (ab56416), rabbit monoclonal anti‐ATG7 (ab133528), and rabbit monoclonal anti‐ATG5 (ab108327) were from Abcam (Cambridge, MA); chloroquine, anthrax protective antigen from B. anthracis (PA), mouse monoclonal anti‐β actin (clone AC‐15), mouse monoclonal anti‐FLAG (clone M2), and rat monoclonal anti‐caspase 11 (clone 17D9) were from Sigma; rat monoclonal anti‐CD40 (3/23), anti I‐Ab (AF‐120.1), anti‐CD11c (HL3), anti‐CD11b (M1/70), anti‐CD86 (GL1), and anti‐CD103 (M290) were from BD Biosciences (San Jose, CA); mouse monoclonal anti‐GFP was from Roche (Indianapolis, IN); rabbit anti‐mTOR (7C10), anti‐phospho‐mTOR (Ser2481), anti‐ULK1 (D8H5), anti‐phospho‐ULK1 (Ser757), anti‐p70 S6 kinase (49D7), anti‐phospho‐p70 S6 kinase (Thr389), anti‐phospho‐S6 ribosomal protein (Ser235/236), and mouse anti‐S6 ribosomal protein (54D2) were from Cell Signaling Technology (Danvers, MA). ELISA Ready‐SET‐Go! kits for mouse interleukin‐6, was from eBioscience (San Diego, CA); the anti‐mouse‐IL‐1β ELISA set was from R&D (Minneapolis, MN). All secondary antibodies were from Jackson Immunoresearch (West Grove, PA). All restriction enzymes were from New England Biolabs (Ipswich, MA). Polymerase chain reaction (PCR) was performed using GoTaq kit (Promega, Madison, WI).
Cell culture
Bone marrow cells were isolated and cultured for 7–9 days in RPMI‐1640 medium (Gibco, ThermoFisher Scientific, Waltham, MA) supplemented with 10% low endotoxin‐FBS (Hyclone, Logan, UT), 2 mM L‐Gln, 50 μM 2‐mercaptoethanol (Invitrogen), and either 30% granulocyte‐macrophage colony stimulating factor (GM‐CSF)‐containing conditioned medium from J558L cells (kindly provided by R. Steinman former laboratory, Rockefeller University, NY) for differentiation to DCs as described (Winzler et al, 1997; Mantegazza & Marks, 2015), or 30% M‐CSF‐containing L929 conditioned medium for differentiation to MΦs (Mantegazza et al, 2017). DC2.4 mouse dendritic cell line (Shen et al, 1997; Chen et al, 2020) (kindly provided by Dr. Janis Burkhardt, Children's Hospital of Philadelphia) was cultured in RPMI‐1640 medium (Gibco, ThermoFisher Scientific) supplemented with 10% low endotoxin‐FBS (Hyclone), 2 mM L‐Gln and 50 μM 2‐mercaptoethanol (Invitrogen).
Intestinal lamina propria was isolated after dissecting colon and dissociating the intestinal epithelium in Ca2+/Mg2+‐free HBSS (Invitrogen) supplemented with 5% FBS and 2 mM EDTA for 20 min at 37°. Lamina propria was then digested by treatment with 1.5 mg/ml type VII collagenase (Sigma) and 40 μg/ml DNase I (Sigma) for 15 min at 37° to obtain single‐cell suspensions (as shown in http://www.jove.com/video/4040/; Geem et al, 2012). Colonic DCs were then purified with anti‐CD11c (N418) microbeads (Miltenyi Biotec Inc., Auburn, CA) (Geem et al, 2012).
DNA retroviral constructs, retroviral production, and transduction of dendritic cells
pMSCV‐ASC‐GFP, pLZRS‐mCherry‐LC3B, and MigR1 retroviral constructs were kindly provided by Teresa Fernandes‐Alnemri (Thomas Jefferson University, Philadelphia, PA), Erika Holzbaur (University of Pennsylvania, Philadelphia, PA), and Warren Pear (University of Pennsylvania), respectively. GFP was amplified using the forward primer 5′‐ATCTCTCGAGATGGTGAGCAAG GGCGAG‐3′ (XhoI restriction site in bold) and reverse primer 5′‐ ATCTGAATTCTTACTTGTAC AGCTCGTC (EcoRI restriction site in bold) and sucloned between XhoI and EcoRI restriction sites of the MigR1‐NotI vector (previously described (Lopez‐Haber et al, 2020)) resulting in MigR1‐NotI‐GFP. hSLC15A4 was then cloned between BglII and XhoI restriction sites of the MigR1‐NotI‐GFP vector by digesting pLenti‐hSLC15A4 (RC215932L2, NM_145648, OriGene, Rockville, MD) with BamHI and XhoI restriction enzymes resulting in MigR1‐hSLC15A4‐GFP.
Retrovirus was produced by transfection of packaging cell line Platinum‐E (Plat‐E) (Morita et al, 2000) (a generous gift of Mitchell Weiss, St. Jude Children's Research Hospital, Memphis, TN) using Lipofectamine 2000 (Invitrogen, ThermoFisher Scientific) and harvested from cell supernatants 2 days later. 3 × 106 BM cells were seeded on 6‐well non‐tissue culture treated plates per well for transduction 2 days after isolation, and transduced by spinoculation with 3 ml of transfected Plat‐E cell supernatant in the presence of 8 μg/ml polybrene and 20 mM HEPES for 2 h at 37°C. Retrovirus‐containing media were then replaced with DC culture media, and cells were collected for experiments 6 days later.
shRNAs, lentiviral production, and transduction of dendritic cells
pLKO.1‐puromycin derived lentiviral vectors (Stewart et al, 2003) for small hairpin RNAs (shRNAs) against SLC15A4, ATG5, ATG7 and non‐target shRNAs were obtained from the High‐throughput Screening Core of the University of Pennsylvania. SLC15A4 #1 sense sequence: CCACCTGCATTACTACTTCTT; SLC15A4 #2 sense sequence: CCTCATTGTGTCTGTGAAGT A; SLC15A4 #3 sense sequence: CCAGAGTGTCTTCATCACCAA. ATG5 sense sequence: GCAGAACCATACTATTTGCTT; ATG7 sense sequence: GCCTGGCATTTGATAAATGTA. Non‐target sense sequence: GCGCGATAGCGCTAATAATTT. pLJM1‐FLAG‐RagBQ99L and pLVX‐FLAG‐metap2 (control) were kindly provided by Dr. Roberto Zoncu (University of California, Berkeley, CA).
Lentivirus was produced by co‐transfection of 293T cells (obtained from American Type Culture Collection, Mannassas, VA) with packaging vectors pDM2.G and pSPAx2 using calcium phosphate precipitation (Marks et al, 1995) and harvested from cell supernatants 2 d later. 3 × 106 BM cells were seeded on 6‐well non‐tissue culture treated plates per well for transduction two d after isolation and transduced by spinoculation with 3 ml of transfected 293T cell supernatant in the presence of 8 μg/ml polybrene and 20 mM HEPES for 2 h at 37°C (Savina et al, 2009). Lentivirus‐containing media were then replaced with DC culture media. Puromycin (2 μg/ml) was added 3 days after infection, and cells were collected for experiments 3 days later.
For BM cell transduction with both retroviral constructs and lentiviral shRNAs, cells were first transduced with the indicated retroviral constructs, washed, and subsequently transduced with the indicated lentiviral shRNAs. Lentivirus‐containing media were then replaced with DC culture media. Puromycin (2 μg/ml) was added 3 days after transduction, and cells were collected for experiments 3 days later. Only lentiviral shRNAs are puromycin resistant.
Transfection of DC2.4 mouse dendritic cell line
DC2.4 were transfected with pEGFP‐N1‐hSLC15A4 (Nakamura et al, 2014) (kindly provided by Drs. Ira Mellman and Gerry Strasser, Genentech, South San Francisco, CA, upon material transfer agreement) using Neon transfection system (ThermoFisher Scientific) according to manufacturer's instructions. Briefly, 2 × 106 cells were washed in Ca2+/ Mg2+ free PBS and resuspended in 100 μl resuspension buffer R. Cells were then pulsed once with 5 μg of plasmid during 5 ms and 1,680 voltage and incubated overnight at 37°C.
Bacterial strains and infections
STm strains SL1344 and ΔfliCΔfljB SL1344 were kindly provided by Dr. Igor Brodsky, University of Pennsylvania, Philadelphia, PA. For in vitro infections, STm were grown overnight in streptomycin containing LB medium at 37°C with aeration, diluted into fresh LB containing 300 mM NaCl, and grown standing at 37°C for 3 h to induce flagellin expression (Wynosky‐Dolfi et al, 2014) unless otherwise indicated. Bacteria were washed with prewarmed Dulbecco's Modified Eagle Medium (DMEM, Gibco), added to cells at an MOI of 5:1, and spun onto cells at 200 × g for 5 min. Cells were incubated at 37°C in a tissue culture incubator with 5% CO2. Gentamycin (100 μg/ml) was added 1 h after infection and cells were harvested or analyzed by immunofluorescence microscopy or live‐cell imaging at the indicated time points. In some experiments, 50 μM chloroquine was added at the time of STm infection.
Real‐time quantitative PCR (RT–qPCR)
RNA was isolated from shRNA‐transduced BMDCs using RNeasy kit (Qiagen, Germantown, MD). 1 μg RNA was reverse transcribed to cDNA using TaqMan™ Reverse Transcription Kit (Invitrogen, ThermoFisher Scientific). qPCR was performed in a StepOnePlus RT–PCR System (Applied Biosystems, ThermoFisher Scientific) using TaqMan™ Fast Advanced master mix and TaqMan™ FAM probes (Applied Biosystems) according to manufacturer's instructions. TaqMan™ FAM probes specific for Slc15a4 and the housekeeping genes β2‐microglobulin (β2‐m) and glyceraldehyde‐3‐phosphate‐dehydrogenase (Gapdh) were Mm00505709_m1 (Slc15a4), Mm00437762_m1 (β2‐m), and Mm99999915_g1 (Gapdh). PCR product formation was continuously monitored using the StepOnePlus™ Real‐Time PCR System. Relative levels of Slc15a4 mRNA were calculated with the ΔΔCt method (Caino et al, 2011), normalized to the average of housekeeping genes and represented as mRNA fold change compared to control shRNA‐treated cells. qPCR reactions were performed in triplicate. Experiments were independently performed three times.
Inflammasome activation and measurement of cytokine production
200,000 BMDCs or BMMΦs were seeded in triplicate in RPMI medium on 96‐well round‐bottom plates, primed where indicated with 30 ng/ml LPS for 3 h, and then incubated with the indicated inflammasome stimuli (200 μg/ml alum; 200 μg/ml MSU crystals; 1 μg/ml LLO; 2 μg/ml poly (dA:dT)/LyoVec; 1 μg/ml PA and 10 ng/ml LFn‐Rod or infected with STm at MOI of 5:1, for 1 to 6 h; Rathinam et al, 2010; Gross, 2012; Reyes Ruiz et al, 2017). For non‐canonical inflammasome stimulation, BMDCs were primed with 100 ng/ml LPS overnight to induce pro‐caspase‐11 expression (Moretti et al, 2022) and then infected with STm ΔfliCΔfljB or non‐flagellin expressing STm for 4–24 h. Cells were pelleted at 200 × g at the indicated time points and supernatants were collected to measure cytokines using commercial ELISA kits (Gross, 2012), or for caspase‐11 p30 or LDH detection (see below).
For cytokine detection after in vivo DSS‐treatment, isolated colonic DCs were cultured overnight, supernatants were collected to measure IL‐1β by ELISA, and cell pellets were lysed with Laemmli sample buffer with 2‐mercapto‐ethanol for immunoblotting analysis of LC3.
Cell death assay
Cytotoxicity was detected using the LDH Cytotoxicity Detection Kit (Clontech Laboratories, Inc., Mountain View, CA). 3 × 106 BMDCs per well were seeded into 6‐well plates in RPMI medium in triplicates. Cells were infected at MOI 5:1 with STm ΔfliCΔfljB or STm grown to stationary phase to prevent flagellin expression (Wynosky‐Dolfi et al, 2014), or flagellin‐expressing STm as control. Gentamycin (100 μg/ml) was added 1 h after infection, and supernatants were harvested 4, 8, and 24 h after infection. LDH release was quantified according to the manufacturer's instructions. Cytotoxicity was normalized to cell samples treated with 1% (v/v) Triton X‐100, and LDH release from uninfected cells was used as negative controls.
Immunoblotting
For LC3, GSDMD, caspase‐1 and caspase‐11 detection, BMDCs were infected with STm, harvested by centrifugation at 200 g at the indicated time points, and lysed in Laemmli sample buffer with 2‐mercaptoethanol. Samples were then fractionated by SDS–PAGE on 10% (for caspase‐11), 12% (for GSDM, caspase‐1 or LC3) or 15% polyacrylamide gels (for LC3), transferred to PVDF membranes (Immobilon‐P, Millipore) and analyzed using horseradish peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch), enhanced chemiluminescence (GE Healthcare, Pittsburgh, PA), and iBright imaging system (ThermoFisher Scientific) or FluorChem R imaging system (ProteinSimple, Biotechne, San Jose, CA). For the detection of cleaved caspase‐11, cell supernatants were precipitated with 10% TCA (vol/vol) for 1 h on ice. Precipitated proteins were pelleted at 20,000 g for 30 min at 4°C, air‐dried, resuspended in Laemmli sample buffer 2× with 2‐mercaptoethanol, and heated at 95°C for 5 min (Broz et al, 2012; Koontz, 2014). Precipitated supernatants from 3x106 BMDCs were loaded per well on polyacrylamide gels. For mTOR, ULK1, p70 S6 kinase, and S6 detection, samples were first lysed in lysis buffer containing 1% Triton X‐100, 10 mM sodium pyrophosphate, 10 mM sodium β‐glycerophosphate, 4 mM EDTA, 40 mM HEPES, and EDTA‐free protease inhibitors (Roche) at pH 7.4 (Chung et al, 2019) and then resuspended in Laemmli sample buffer with 2‐mercaptoethanol. Samples were then fractionated on 6% (mTOR and ULK1) or 12% (p70S6 kinase and S6) polyacrylamide gels, transferred to PVDF membranes (Immobilon‐P, Millipore) and analyzed using horseradish peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch), enhanced chemiluminescence (GE Healthcare, Pittsburgh, PA), and FluorChem R imaging system. For actin detection, Alexa Fluor 680 secondary antibodies or horseradish peroxidase‐conjugated secondary antibodies (Jackson ImmunoResearch) were used and developed using FluorChem R imaging system. Densitometric analyses of band intensity was performed using NIH Image J software, normalizing to control protein levels (Gross, 2012). For LC‐II/ LC‐I, cleaved fragment/full‐length protein and phospho‐protein/total protein assessment, fold induction, or fold change values were calculated upon treatment and/or over time, to normalize between different cell types, culture conditions and equipment, as recommended (Klionsky et al, 2021) and indicated in the Figure legends.
Immunofluorescence microscopy and flow cytometry
Non‐transduced BMDCs or BMDCs, expressing SLC15A4‐GFP or ASC‐GFP and/or mcherry‐LC3 and/or RagBQ99L and metap2 (control), were infected with STm for the indicated time points, fixed with 3% formaldehyde in PBS, permeabilized with Permwash (BD Biosciences, San Jose, CA), and labeled with primary antibodies and Alexa Fluor‐conjugated secondary antibodies (Jackson ImmunoResearch). Cells were analyzed by fluorescence microscopy using a Leica DMI6000 B inverted microscope, a Hamamatsu ORCA‐flash 4.0 camera, and Leica Microsystems Application Suite X software at the Department of Pathology at Children's Hospital of Philadelphia or a Nikon A1R laser scanning confocal microscope with spectral detectors and Nikon NIS‐Elements acquisition software at the Sidney Kimmel Cancer Center (SKCC) Bioimaging facility at Thomas Jefferson University. Images were analyzed using ImageJ (National Institutes of Health). LC3 fluorescence surrounding ASC specks was measured in a radio of 1 μm and normalized per unit area using ImageJ. ASC speck distance from nuclei was quantified using Analyze/Measure tools as detailed in ImageJ tutorial (https://imagej.nih.gov/ij/docs/pdfs/ImageJ.pdf). Magnification: 100×.
BMDC phenotype was characterized by flow cytometry using an LSR‐II and FloJo software (BD Biosciences). To assess ASC speck formation by flow cytometry, BMDCs were infected with STm for the indicated time points, fixed with 2% PFA in PBS for 10 m. Cells were finally washed in PBS with 0.5% BSA and 2 mM EDTA and analyzed by flow cytometry using a CytoFLEX (Beckman Coulter, Indianapolis, IN) and FlowJo software (BD Biosciences). Selection of activated and non‐activated cell gates was performed using GFP‐width and GFP‐height parameters based on the differential shape of the fluorescence pulse depending on the fluorophore distribution within the cell, as previously described (Sester et al, 2015; Hoss et al, 2018) and shown in Fig EV4.
Live‐cell imaging
BMDCs or DC2.4 cells expressing SLC15A4‐GFP were seeded on poly‐L‐lysine–coated glass‐bottom 35‐mm culture dishes (MatTek, Ashland, MA) on day 6 of culture. On day 7, cells were pulsed for 15 min with TxR‐conjugated OVA (Invitrogen, ThermoFisher scientific) and LPS (100 μg/ml) coupled to 3‐μm amino polystyrene beads (Polysciences Inc., Warrington, PA) as described previously (Savina et al, 2010; Mantegazza & Marks, 2015). DCs were then washed with RPMI, chased for 0–2.5 h, and visualized using a Nikon A1R laser scanning confocal microscope with spectral detectors and equipped with a Tokai‐Hit temperature and CO2‐controlled chamber at the SKCC Bioimaging facility at Thomas Jefferson University. Images or videos were obtained with Nikon NIS‐Elements acquisition software and analyzed using ImageJ (National Institutes of Health). Magnification: 100×.
Statistical analyses
Statistical analyses and data plots were performed using Microsoft Excel (Redmond, WA) and GraphPad Prism software (San Diego, CA). Statistical significance for in vitro experimental samples relative to untreated or control cells (as indicated) was determined using the unpaired Student's t‐test and ANOVA after normality assessment using GraphPad. Statistical significance for mouse analyses and mouse samples in DSS‐treated SLC15A4 feeble mice relative to WT mice was determined using the non‐parametric Mann–Whitney test (Fay & Proschan, 2010). All experiments were performed independently at least three times.
Author contributions
Cynthia López‐Haber: Formal analysis; investigation; methodology; writing—review and editing. Daniel J Netting: Investigation; methodology; writing—review and editing. Zachary Hutchins: Investigation; methodology; writing—review and editing. Xianghui Ma: Methodology; writing—review and editing. Kathryn E Hamilton: Supervision; funding acquisition; investigation; methodology; writing—review and editing. Adriana R Mantegazza: Conceptualization; supervision; funding acquisition; investigation; visualization; methodology; writing—original draft; project administration; writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization: ARM; Methodology: CLH, XM, DJN, KEH, ARM; Investigation: CLH, DJN, ZH, ARM; Visualization: CLH, DJN, ZH, ARM; Funding acquisition: KEH, ARM; Project administration: ARM; Supervision: KEH, ARM; Writing—original draft: ARM; Writing—review & editing: CLH, DJN, ZH, XM, KEH, ARM.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Movie EV1
Movie EV2
PDF+
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Julie Brill (The Hospital for Sick Children Research Institute, Toronto, Canada) and Ira Mellman for critical reading of the manuscript; Roberto Zoncu, Igor Brodsky and James Grayczyk, Ira Mellman and Gerry Strasser, Erika Holzbaur, Teresa Fernandes‐Alnemri and Emad Alnemri, Warren Pear, and Janis Burkhardt for the generous gifts of reagents; Michael S. Marks (Department of Pathology and Laboratory Medicine, University of Pennsylvania), the Department of Pathology at Children's Hospital of Philadelphia and the Department of Microbiology and Immunology at Thomas Jefferson University for reagents, equipment and helpful discussions; María Yolanda Covarrubias at the Sidney Kimmel Cancer Center (SKCC) Bioimaging facility at Thomas Jefferson University and David Schultz and the High‐throughput Screening core at the University of Pennsylvania for expert technical assistance, and the Flow Cytometry cores and Offices of Animal Resources at the Children's Hospital of Philadelphia and Thomas Jefferson University. This work was supported by NIH Grant R01 AI137173 (to CL‐H and ARM) and R01 DK124369 (to KEH). The veterinary pathologists of the Penn Vet Comparative Pathology Core are partially supported by the Abramson Cancer Center Support Grant (P30 CA016520). The Aperio Versa 200 scanner used for whole slide imaging and the image analysis software was supported by an NIH Shared Instrumentation Grant (S10 OD023465‐01A1). Thomas Jefferson University SKCC flow cytometry core is supported by NCI Core grant (P30 CA056036). The summary figure was created with BioRender.com, through Thomas Jefferson University library portal.
The EMBO Journal (2022) 41: e111161.
Data availability
This study includes no data deposited in external repositories. Source data has been provided upon manuscript submission.
References
- Baccala R, Gonzalez‐Quintial R, Blasius AL, Rimann I, Ozato K, Kono DH, Beutler B, Theofilopoulos AN (2013) Essential requirement for IRF8 and SLC15A4 implicates plasmacytoid dendritic cells in the pathogenesis of lupus. Proc Natl Acad Sci USA 110: 2940–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M (2010) Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59: 1192–1199 [DOI] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R (2006) Toll‐dependent selection of microbial antigens for presentation by dendritic cells. Nature 440: 808–812 [DOI] [PubMed] [Google Scholar]
- Blander JM (2007) Signalling and phagocytosis in the orchestration of host defence. Cell Microbiol 9: 290–299 [DOI] [PubMed] [Google Scholar]
- Blasius AL, Arnold CN, Georgel P, Rutschmann S, Xia Y, Lin P, Ross C, Li X, Smart NG, Beutler B (2010) Slc15a4, AP‐3, and Hermansky‐Pudlak syndrome proteins are required for toll‐like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci USA 107: 19973–19978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Martina JA, Puertollano R (2018) Emerging roles for TFEB in the immune response and inflammation. Autophagy 14: 181–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM (2012) Caspase‐11 increases susceptibility to salmonella infection in the absence of caspase‐1. Nature 490: 288–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caino MC, von Burstin VA, Lopez‐Haber C, Kazanietz MG (2011) Differential regulation of gene expression by protein kinase C isozymes as determined by genome‐wide expression analysis. J Biol Chem 286: 11254–11264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Sabatini DM (2015) Nutrient‐sensing mechanisms across evolution. Cell 161: 67–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, Vijay‐Kumar M (2014) Dextran sulfate sodium (DSS)‐induced colitis in mice. Curr Protoc Immunol 104: 15.25.11–15.25.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K (2014) The neutrophil NLRC4 inflammasome selectively promotes IL‐1beta maturation without pyroptosis during acute salmonella challenge. Cell Rep 8: 570–582 [DOI] [PubMed] [Google Scholar]
- Chen M, Sun H, Boot M, Shao L, Chang SJ, Wang W, Lam TT, Lara‐Tejero M, Rego EH, Galan JE (2020) Itaconate is an effector of a Rab GTPase cell‐autonomous host defense pathway against salmonella. Science 369: 450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christgen S, Kanneganti TD (2020) Inflammasomes and the fine line between defense and disease. Curr Opin Immunol 62: 39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Shin HR, Berdan CA, Ford B, Ward CC, Olzmann JA, Zoncu R, Nomura DK (2019) Covalent targeting of the vacuolar H(+)‐ATPase activates autophagy via mTORC1 inhibition. Nat Chem Biol 15: 776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A et al (2010) p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6: 330–344 [DOI] [PubMed] [Google Scholar]
- Deretic V, Levine B (2018) Autophagy balances inflammation in innate immunity. Autophagy 14: 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales‐Nebreda L, Sennello JA, Kutuzov MA, Sitikov A, Davis JM et al (2015) Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 6: 6574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Hafner‐Bratkovic I, Devant P, D'Andrea JM, Ngwa EM, Borsic E, Doench JG, LaFleur MW, Sharpe AH, Thiagarajah JR et al (2021) Control of gasdermin D oligomerization and pyroptosis by the Ragulator‐rag‐mTORC1 pathway. Cell 184: 4495–4511.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay MP, Proschan MA (2010) Wilcoxon‐Mann‐Whitney or t‐test? On assumptions for hypothesis tests and multiple interpretations of decision rules. Stat Surv 4: 1–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L, Munoz‐Planillo R, Nunez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta RM, Garcia‐Irigoyen O, Moschetta A (2017) Exploration of inflammatory bowel disease in mice: chemically induced murine models of inflammatory bowel disease (IBD). Curr Protoc Mouse Biol 7: 13–28 [DOI] [PubMed] [Google Scholar]
- Geem D, Medina‐Contreras O, Kim W, Huang CS, Denning TL (2012) Isolation and characterization of dendritic cells and macrophages from the mouse intestine. J Vis Exp 63: e4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MA, Choy CH, Dayam RM, Ospina‐Escobar E, Somerville A, Xiao X, Ferguson SM, Botelho RJ (2016) Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB. Curr Biol 26: 1955–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O (2012) Measuring the inflammasome. Methods Mol Biol 844: 199–222 [DOI] [PubMed] [Google Scholar]
- Gu X, Orozco JM, Saxton RA, Condon KJ, Liu GY, Krawczyk PA, Scaria SM, Harper JW, Gygi SP, Sabatini DM (2017) SAMTOR is an S‐adenosylmethionine sensor for the mTORC1 pathway. Science 358: 813–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatscher L, Lehmann CHK, Purbojo A, Onderka C, Liang C, Hartmann A, Cesnjevar R, Bruns H, Gross O, Nimmerjahn F et al (2021) Select hyperactivating NLRP3 ligands enhance the TH1‐ and TH17‐inducing potential of human type 2 conventional dendritic cells. Sci Signal 14: eabe1757 [DOI] [PubMed] [Google Scholar]
- Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, Cesar‐Razquin A, Essletzbichler P, Goldmann U, Stefanovic A et al (2020) TASL is the SLC15A4‐associated adaptor for IRF5 activation by TLR7‐9. Nature 581: 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand BT, Chang L, Goldman RD (2004) Intermediate filaments are dynamic and motile elements of cellular architecture. J Cell Sci 117: 133–141 [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S (2012) Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc Natl Acad Sci USA 109: 14556–14561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N et al (2009) Nutrient‐dependent mTORC1 association with the ULK1‐Atg13‐FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoss F, Rolfes V, Davanso MR, Braga TT, Franklin BS (2018) Detection of ASC speck formation by flow cytometry and chemical cross‐linking. Methods Mol Biol 1714: 149–165 [DOI] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M et al (2001) Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 411: 599–603 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Abdel‐Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, Abeliovich H, Abildgaard MH, Abudu YP, Acevedo‐Arozena A et al (2021) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17: 1–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Shimabukuro‐Demoto S, Yoshida‐Sugitani R, Furuyama‐Tanaka K, Karyu H, Sugiura Y, Shimizu Y, Hosaka T, Goto M, Kato N et al (2014) The histidine transporter SLC15A4 coordinates mTOR‐dependent inflammatory responses and pathogenic antibody production. Immunity 41: 375–388 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Tsutsui H, Shimabukuro‐Demoto S, Yoshida‐Sugitani R, Karyu H, Furuyama‐Tanaka K, Ohshima D, Kato N, Okamura T, Toyama‐Sorimachi N (2017) Lysosome biogenesis regulated by the amino‐acid transporter SLC15A4 is critical for functional integrity of mast cells. Int Immunol 29: 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Nguyen‐Tien D, Ohshima D, Karyu H, Shimabukuro‐Demoto S, Yoshida‐Sugitani R, Toyama‐Sorimachi N (2021a) Human SLC15A4 is crucial for TLR‐mediated type I interferon production and mitochondrial integrity. Int Immunol 33: 399–406 [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Nguyen‐Tien D, Sorimachi Y, Sugiura Y, Suzuki T, Karyu H, Shimabukuro‐Demoto S, Uemura T, Okamura T, Taguchi T et al (2021b) SLC15A4 mediates M1‐prone metabolic shifts in macrophages and guards immune cells from metabolic stress. Proc Natl Acad Sci USA 118: e2100295118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz L (2014) TCA precipitation. Methods Enzymol 541: 3–10 [DOI] [PubMed] [Google Scholar]
- Larabi A, Barnich N, Nguyen HTT (2020) New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 16: 38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE (2009) pH‐dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem 284: 23818–23829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Yee SW, Kim RB, Giacomini KM (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov 14: 543–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Haber C, Levin‐Konigsberg R, Zhu Y, Bi‐Karchin J, Balla T, Grinstein S, Marks MS, Mantegazza AR (2020) Phosphatidylinositol‐4‐kinase IIalpha licenses phagosomes for TLR4 signaling and MHC‐II presentation in dendritic cells. Proc Natl Acad Sci USA 117: 28251–28262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs GL, Rotstein OD, Grinstein S (1991) Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)‐ATPase activity, counterion conductance, and H+ "leak". J Biol Chem 266: 24540–24548 [PubMed] [Google Scholar]
- Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z et al (2020) HDAC6 mediates an aggresome‐like mechanism for NLRP3 and pyrin inflammasome activation. Science 369: eaas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, Amigorena S (2008) NADPH oxidase controls phagosomal pH and antigen cross‐presentation in human dendritic cells. Blood 112: 4712–4722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Guttentag SH, El‐Benna J, Sasai M, Iwasaki A, Shen H, Laufer TM, Marks MS (2012) Adaptor protein‐3 in dendritic cells facilitates phagosomal toll‐like receptor signaling and antigen presentation to CD4(+) T cells. Immunity 36: 782–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Zajac AL, Twelvetrees A, Holzbaur EL, Amigorena S, Marks MS (2014) TLR‐dependent phagosome tubulation in dendritic cells promotes phagosome cross‐talk to optimize MHC‐II antigen presentation. Proc Natl Acad Sci USA 111: 15508–15513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Marks MS (2015) Visualizing toll‐like receptor‐dependent phagosomal dynamics in murine dendritic cells using live cell microscopy. Methods Mol Biol 1270: 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza AR, Wynosky‐Dolfi MA, Casson CN, Lefkovith AJ, Shin S, Brodsky IE, Marks MS (2017) Increased autophagic sequestration in adaptor protein‐3 deficient dendritic cells limits inflammasome activity and impairs antibacterial immunity. PLoS Pathog 13: e1006785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MS, Roche PA, van Donselaar E, Woodruff L, Peters PJ, Bonifacino JS (1995) A lysosomal targeting signal in the cytoplasmic tail of the beta chain directs HLA‐DM to MHC class II compartments. J Cell Biol 131: 351–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BN, Wang C, Willette‐Brown J, Herjan T, Gulen MF, Zhou H, Bulek K, Franchi L, Sato T, Alnemri ES et al (2014) IKKalpha negatively regulates ASC‐dependent inflammasome activation. Nat Commun 5: 4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti J, Blander JM (2014) Insights into phagocytosis‐coupled activation of pattern recognition receptors and inflammasomes. Curr Opin Immunol 26: 100–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti J, Jia B, Hutchins Z, Roy S, Yip H, Wu J, Shan M, Jaffrey SR, Coers J, Blander JM (2022) Caspase‐11 interaction with NLRP3 potentiates the noncanonical activation of the NLRP3 inflammasome. Nat Immunol 23: 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, Kitamura T (2000) Plat‐E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7: 1063–1066 [DOI] [PubMed] [Google Scholar]
- Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Maziere A, Klumperman J, Schlatter M, Delamarre L, Mellman I (2014) Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 509: 240–244 [DOI] [PubMed] [Google Scholar]
- Pastore N, Brady OA, Diab HI, Martina JA, Sun L, Huynh T, Lim JA, Zare H, Raben N, Ballabio A et al (2016) TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12: 1240–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J (2004) Localization of the AP‐3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol 164: 1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Zoncu R (2016) The lysosome as a regulatory hub. Annu Rev Cell Dev Biol 32: 223–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perse M, Cerar A (2012) Dextran sodium sulphate colitis mouse model: traps and tricks. J Biomed Biotechnol 2012: 718617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal‐Ruiz Y, Byron A, Wirth A, Madsen R, Sedlackova L, Hewitt G, Nelson G, Stingele J, Wills JC, Zhang T et al (2021) mTORC1 activity is supported by spatial association with focal adhesions. J Cell Biol 220: e202004010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Puertollano R (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 32: 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E et al (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11: 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch I, Tenthorey JL, Nichols RD, Al Moussawi K, Kang JJ, Kang C, Kazmierczak BI, Vance RE (2016) NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo . J Exp Med 213: 657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsamen M, Superti‐Furga G (2016) SLC38A9: a lysosomal amino acid transporter at the core of the amino acid‐sensing machinery that controls MTORC1. Autophagy 12: 1061–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes Ruiz VM, Ramirez J, Naseer N, Palacio NM, Siddarthan IJ, Yan BM, Boyer MA, Pensinger DA, Sauer JD, Shin S (2017) Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc Natl Acad Sci USA 114: 13242–13247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimann I, Gonzalez‐Quintial R, Baccala R, Kiosses WB, Teijaro JR, Parker CG, Li X, Beutler B, Kono DH, Theofilopoulos AN (2022) The solute carrier SLC15A4 is required for optimal trafficking of nucleic acid‐sensing TLRs and ligands to endolysosomes. Proc Natl Acad Sci USA 119: e2200544119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C, Chan AH, Von Pein J, Boucher D, Schroder K (2018) Dimerization and auto‐processing induce caspase‐11 protease activation within the non‐canonical inflammasome. Life Sci Alliance 1: e201800237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS et al (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, Iwasaki A (2010) Bifurcation of toll‐like receptor 9 signaling by adaptor protein 3. Science 329: 1530–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasawatari S, Okamura T, Kasumi E, Tanaka‐Furuyama K, Yanobu‐Takanashi R, Shirasawa S, Kato N, Toyama‐Sorimachi N (2011) The solute carrier family 15A4 regulates TLR9 and NOD1 functions in the innate immune system and promotes colitis in mice. Gastroenterology 140: 1513–1525 [DOI] [PubMed] [Google Scholar]
- Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon‐Duménil A‐M, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218 [DOI] [PubMed] [Google Scholar]
- Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, Moita LF, Amigorena S (2009) The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity 30: 544–555 [DOI] [PubMed] [Google Scholar]
- Savina A, Vargas P, Guermonprez P, Lennon AM, Amigorena S (2010) Measuring pH, ROS production, maturation, and degradation in dendritic cell phagosomes using cytofluorometry‐based assays. Methods Mol Biol 595: 383–402 [DOI] [PubMed] [Google Scholar]
- Sester DP, Thygesen SJ, Sagulenko V, Vajjhala PR, Cridland JA, Vitak N, Chen KW, Osborne GW, Schroder K, Stacey KJ (2015) A novel flow cytometric method to assess inflammasome formation. J Immunol 194: 455–462 [DOI] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC et al (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi MN, Mowers EE, Drake LE, Macleod KF (2015) Measuring autophagy in stressed cells. Methods Mol Biol 1292: 129–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Z, Reznikoff G, Dranoff G, Rock KL (1997) Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol 158: 2723–2730 [PubMed] [Google Scholar]
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu‐Asab M, Fitzgerald KA, Sher A, Kehrl JH (2012) Activation of autophagy by inflammatory signals limits IL‐1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 13: 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA et al (2003) Lentivirus‐delivered stable gene silencing by RNAi in primary cells. RNA 9: 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styers ML, Salazar G, Love R, Peden AA, Kowalczyk AP, Faundez V (2004) The endo‐lysosomal sorting machinery interacts with the intermediate filament cytoskeleton. Mol Biol Cell 15: 5369–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styers ML, Kowalczyk AP, Faundez V (2005) Intermediate filaments and vesicular membrane traffic: the odd couple's first dance? Traffic 6: 359–365 [DOI] [PubMed] [Google Scholar]
- Takahama M, Akira S, Saitoh T (2018) Autophagy limits activation of the inflammasomes. Immunol Rev 281: 62–73 [DOI] [PubMed] [Google Scholar]
- Wang S, Tsun ZY, Wolfson RL, Shen K, Wyant GA, Plovanich ME, Yuan ED, Jones TD, Chantranupong L, Comb W et al (2015) Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347: 188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzler C, Rovere P, Rescigno M, Granucci F, Penna G, Adorini L, Zimmermann VS, Davoust J, Ricciardi‐Castagnoli P (1997) Maturation stages of mouse dendritic cells in growth factor‐dependent long‐term cultures. J Exp Med 185: 317–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz S, Neufert C, Weigmann B, Neurath MF (2007) Chemically induced mouse models of intestinal inflammation. Nat Protoc 2: 541–546 [DOI] [PubMed] [Google Scholar]
- Wynosky‐Dolfi MA, Snyder AG, Philip NH, Doonan PJ, Poffenberger MC, Avizonis D, Zwack EE, Riblett AM, Hu B, Strowig T et al (2014) Oxidative metabolism enables salmonella evasion of the NLRP3 inflammasome. J Exp Med 211: 653–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates RM, Russell DG (2008) Real‐time spectrofluorometric assays for the lumenal environment of the maturing phagosome. Methods Mol Biol 445: 311–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR et al (2016) An endogenous caspase‐11 ligand elicits interleukin‐1 release from living dendritic cells. Science 352: 1232–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Springstead JR, Kagan JC (2017) By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome‐dependent phagocyte hyperactivation. Immunity 47: 697–709.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Sun K, Meng Z, Chen L (2019) The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J Mol Cell Biol 11: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Sanchez‐Lopez E, Karin M (2016) Autophagy, NLRP3 inflammasome and auto‐inflammatory/immune diseases. Clin Exp Rheumatol 34: 12–16 [PubMed] [Google Scholar]
- Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011a) mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science 334: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011b) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]