

Abstract
Post‐translational modifications by ubiquitin‐like proteins (UBLs) are essential for nearly all cellular processes. Ubiquitin‐related modifier 1 (Urm1) is a unique UBL, which plays a key role in tRNA anticodon thiolation as a sulfur carrier protein (SCP) and is linked to the noncanonical E1 enzyme Uba4 (ubiquitin‐like protein activator 4). While Urm1 has also been observed to conjugate to target proteins like other UBLs, the molecular mechanism of its attachment remains unknown. Here, we reconstitute the covalent attachment of thiocarboxylated Urm1 to various cellular target proteins in vitro, revealing that, unlike other known UBLs, this process is E2/E3‐independent and requires oxidative stress. Furthermore, we present the crystal structures of the peroxiredoxin Ahp1 before and after the covalent attachment of Urm1. Surprisingly, we show that urmylation is accompanied by the transfer of sulfur to cysteine residues in the target proteins, also known as cysteine persulfidation. Our results illustrate the role of the Uba4‐Urm1 system as a key evolutionary link between prokaryotic SCPs and the UBL modifications observed in modern eukaryotes.
Keywords: oxidative stress, persulfidation, sulfur transfer, ubiquitin‐like, Urm1
Subject Categories: Post-translational Modifications & Proteolysis, Structural Biology
Mechanistic analysis of conjugation of the ancient ubiquitin‐like protein Urm1 reveals that its attachment simultaneously leads to the persulfidation of cysteines in the target proteins.

Introduction
Ubiquitin‐related modifier 1 (Urm1) is an evolutionarily conserved member of the ubiquitin family that adopts the β‐grasp fold characteristic of all eukaryotic ubiquitin‐like proteins (UBLs; e.g., ubiquitin, SUMO, NEDD8, ATG12, UFM1; Bedford et al, 2011). Urm1 acts as a sulfur carrier protein (SCP) and is an essential member of the modification cascade that thiolates the C2 position of wobble uridine (s2U34) in eukaryotic tRNA anticodons via the Ncs2‐Ncs6 sulfur‐transferase complex (Nakai et al, 2008; Schlieker et al, 2008; Leidel et al, 2009; Noma et al, 2009). The attachment of a sulfur atom to the wobble base in the tRNA anticodon optimizes ribosomal dynamics during translational elongation (Nedialkova & Leidel, 2015; Ranjan & Rodnina, 2016) and promotes cellular responses to nutrient starvation (Laxman et al, 2013; Gupta et al, 2019; Bruch et al, 2020). The disruption of tRNA anticodon modifications can result in dramatic proteome perturbations and the onset of severe human diseases, including cancer and neurodegenerative pathologies (Rezgui et al, 2013; Nedialkova & Leidel, 2015; Schaffrath & Leidel, 2017; Close et al, 2018; Hawer et al, 2018). In addition to its role as SCP in tRNA thiolation, reports showed that Urm1 covalently attaches to target proteins in conditions of oxidative stress in different organisms, reminiscent of other UBLs (Furukawa et al, 2000; Goehring et al, 2003; Schlieker et al, 2008; Van der Veen et al, 2011; Jüdes et al, 2015; Khoshnood et al, 2017; Wang et al, 2019; Tan et al, 2022).
The conjugation reaction of a UBL to its target protein starts with adenylation of its C‐terminus by ATP‐dependent E1 ubiquitin‐activating enzymes. Next, the activated C‐terminus of the UBL is relayed via active site cysteines of an E1‐, E2‐, and E3‐enzyme cascade (Passmore & Barford, 2004). Finally, the UBL is attached to a specific target protein via a covalent isopeptide bond formed between a highly conserved diglycine motif at its C‐terminus and a lysine side chain of the target protein (Hochstrasser, 2000; McDowell & Philpott, 2013; Stewart et al, 2016; Cappadocia & Lima, 2018). Similarly, the initial activation steps for SCPs start with adenylation of the C‐terminus by specialized E1‐like proteins. Although SCPs typically do not form thioesters, our recent work revealed the necessity of a thioester intermediate for the activation of Urm1 by its dedicated E1 enzyme, Uba4 (Termathe & Leidel, 2018; Pabis et al, 2020). Following the formation of a thioester between Uba4 and Urm1, the rhodanese domain of Uba4 transfers a persulfide group to the activated C‐terminus of Urm1 (Kaduhr et al, 2021). The resulting thiocarboxylated C‐terminus (COSH) is present in all prokaryotic and archaeal SCPs (Kessler, 2006) and is required for the thiolation of nucleic acids as well as in the biosynthesis of Molybdenum cofactor (Moco) and thiamine (Leimkühler, 2017; Shigi, 2018). However, amongst eukaryotic UBLs only Urm1 is known to carry this unique terminal modification (Urm1‐SH). Hence, Urm1 and Uba4 are located at a crucial evolutionary branchpoint between prokaryotic SCPs and eukaryotic UBLs since they combine molecular features otherwise exclusively present in either SCPs or UBLs (Xu et al, 2006; Pedrioli et al, 2008; Jüdes et al, 2015).
As the role of Urm1‐SH for tRNA thiolation is well established and no E2 or E3 enzymes for Urm1 have been identified so far, the potential conjugation of Urm1 to proteins (also referred to as protein “urmylation”), analogous to other UBLs, has been questioned. Nonetheless, several mass spectrometry studies have identified proteins conjugated with Urm1 under specific environmental conditions (Schlieker et al, 2008; Van der Veen et al, 2011; Khoshnood et al, 2017). In vivo, the most robust evidence for conjugation of Urm1 was observed for yeast Ahp1 (Goehring et al, 2003; Van der Veen et al, 2011; Jüdes et al, 2015; Brachmann et al, 2020), a peroxiredoxin that acts as an antioxidant enzyme in scavenging reactive oxygen species (Lee et al, 1999). However, the biochemical details including which E2 and E3 enzymes are required for the conjugation reaction to Ahp1, or other targets remain undefined. Furthermore, as the unique COSH moiety at the C‐terminus of Urm1 is absent from all other eukaryotic UBLs, neither its functional role nor its fate after the conjugation reaction is clear. Finally, the necessity of oxidative stress and a redox‐active (also called peroxidatic) cysteine for the conjugation of Ahp1 (Brachmann et al, 2020) raised the question of whether a preferred site of conjugation exists in other proteins and whether the reaction requirements would be similar.
We sought to close these gaps by determining the mechanism of Urm1 conjugation to target proteins. We show that purified Urm1‐SH can be efficiently attached to Ahp1 and other target proteins in vitro using mild oxidative stress conditions in the absence of E2 enzymes or E3 ligases. We demonstrate that the conjugation depends on the thiocarboxylated C‐terminus of Urm1 and a redox‐active cysteine in the target protein, and that these covalent linkages can occur on a variety of lysine, serine, and threonine residues. Our high‐resolution crystal structures of Ahp1 before and after the conjugation reaction confirm our biochemical findings and provide in‐depth insights into the molecular mechanism of Urm1 attachment.
Strikingly, we observed that Urm1‐SH transfers its sulfur moiety to the urmylated target protein, resulting in directed persulfidation of specific redox‐active cysteines. Persulfidation of cysteines (also called protein S‐sulfhydration) has been directly linked to aging and may be regulated by intracellular H2S levels (Zivanovic et al, 2019). However, the pathway responsible for targeted cysteine persulfidation has remained elusive. Oxidation of specific cysteines triggers the process of Urm1 conjugation, providing a molecular mechanism that enables cells to protect vulnerable cysteine residues from reactive oxygen species (ROS) by a highly selective process. Finally, we used our mechanistic insights to engineer artificially thiocarboxylated proteins and recapitulate the conjugation reaction on a variety of model proteins, including other UBLs and GFP, providing a platform to covalently modify proteins of interest with proteinaceous tags through this selective reaction. Our findings demonstrate how Urm1 protects proteins during oxidative stress and reveal a critical evolutionary link between sulfur transfer and covalent modification by ubiquitin family proteins.
Results
Urm1‐SH directly conjugates to Ahp1 in vitro
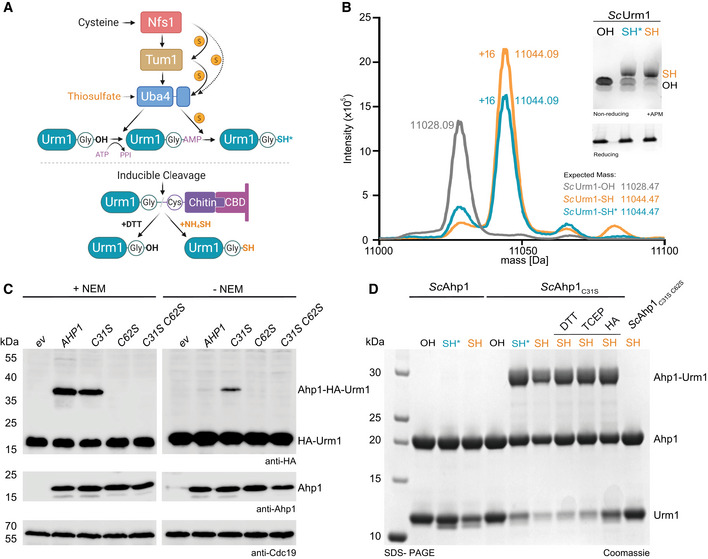
To study the function of Urm1 and understand the details of its protein conjugation reaction, we produced large quantities of purified Urm1‐SH using two methods. In vivo, the cysteine desulfurase Nfs1 and the sulfur‐transferase Tum1 relay sulfur to the rhodanese domain of Uba4, which transfers an activated sulfur group to the C‐terminus of Urm1 (Fig 1A; Noma et al, 2009). The use of thiosulfate as a sulfur source for Uba4 makes it possible to bypass Nfs1 and Tum1, thereby catalyzing the direct thiocarboxylation of Urm1 in vitro (Termathe & Leidel, 2018). We used this approach to produce Urm1‐SH from purified Saccharomyces cerevisiae Urm1 and Uba4 proteins (ScUrm1 and ScUba4). As an alternative strategy, we generated Urm1‐SH or Urm1‐OH from Homo sapiens and Chaetomium thermophilum, a thermophilic fungus harboring highly stable proteins (Bock et al, 2014), using a chitin‐based inducible cleavage (intein) system (Kinsland et al, 1998; Fig EV1A). We verified all purified proteins by mass spectrometry and independently confirmed the presence of a sulfur atom in Urm1‐SH by its increased gel retardation in PAGE supplemented with [(N‐acryloylamino)phenyl]mercuric chloride (APM) (Fig 1B; Igloi, 1988). Of note, the sequences of most eukaryotic Urm1 proteins do not contain cysteine residues, which would affect their behavior in the used APM gels. We removed the nonconserved surface cysteine (Cys55) in CtUrm1 to circumvent super‐shifting in the APM gels and facilitate the analyses. Positioning a cysteine at the C‐terminus of Urm1 (CtUrm1G111C) also led to a retardation of the purified protein in the APM gel (Fig EV1A).
Figure 1. Urm1‐SH forms an isopeptide bond in vitro with Ahp1 in response to thiol‐reactive species.
-
AScheme of different methodologies to generate Urm1‐SH in vitro. Sulfur from cysteine is passed from Nfs1 to Uba4 directly or via Tum1. Uba4 transfers sulfur to Urm1 by catalyzing sequential adenylation and thiocarboxylation reactions. The intein‐based inducible cleavage system can produce carboxylated Urm1 (OH) or thiocarboxylated Urm1 (SH) by using DTT or ammonium sulfide (NH4)2S, respectively. Intein‐produced SH is in orange and Uba4‐produced SH* is in blue.
-
BESI‐MS analyses of Urm1‐OH and Urm1‐SH produced via Uba4 (blue and asterisk) or the intein system (orange). The expected and assessed masses of the samples are listed. Inset: APM gel analyses of the same Urm1 samples. SDS–PAGE gel below for the same proteins using a sample buffer containing DTT. APM: [(N‐acryloylamino)phenyl]mercuric chloride. SDS–PAGE gels were stained with Coomassie.
-
CIn vivo conjugation analyses in the presence (left) or absence of NEM (N‐Ethylmaleimide) (right) with protein extracts obtained from the indicated yeast strains expressing HA‐URM1. Unconjugated Urm1 and Urm1‐conjugates were detected by anti‐HA (top panels) Western blots. Anti‐Ahp1 blots (middle) detect unconjugated Ahp1. Anti‐Cdc19 (bottom panels) served as a loading control.
-
DAnalysis of covalent adduct formation between ScAhp1 or ScAhp1C31S or ScAhp1C31S C62S mutant and carboxylated (OH) or thiocarboxylated ScUrm1 (SH) or Uba4 produced thiocarboxylated Urm1 (SH*) in the presence of TBH (tert‐Butyl hydroperoxide). Confirmation of the isopeptide bond was carried out by the addition of DTT, TCEP, or HA, respectively. Unconjugated Ahp1 and Urm1 as well as their formed conjugate (Ahp1‐Urm1) are indicated on the right. DTT: 1,4‐Dithiothreitol; TCEP: Tris(2‐carboxyethyl) phosphine; HA: Hydroxylamine.
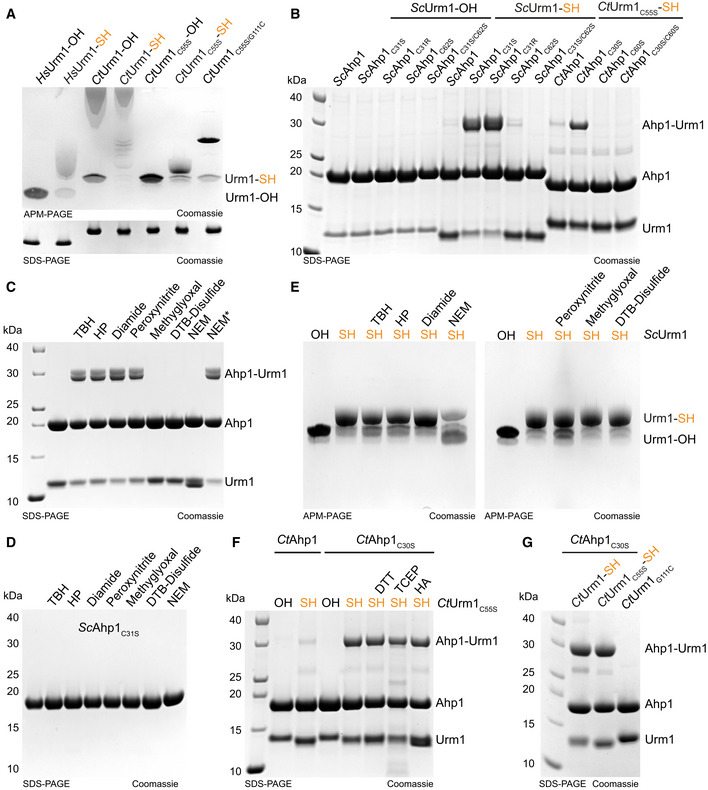
Figure EV1. Urm1 conjugation to Ahp1 in response to different reactive species.
-
AAnalysis of thiocarboxylated Urm1 generated using the intein system by APM gel electrophoresis and retardation. HsUrm1, CtUrm1, the cysteine mutant CtUrm1C55S, and CtUrm1C55S‐G111C are examined. Standard SDS page gel below for the same proteins with sample buffer containing DTT. Coomassie was used for staining.
-
BAnalysis of covalent adduct formation between ScAhp1 or ScAhp1C31S or ScAhp1C31R or ScAhp1C62S or ScAhp1C31S/C62S mutant and thiocarboxylated ScUrm1 (SH) or carboxylated ScUrm1 (OH) in the presence of TBH (tert‐Butyl hydroperoxide). Analysis of covalent adduct formation between CtAhp1 or CtAhp1C30S or CtAhp1C60S or CtAhp1C30S/60S mutant and thiocarboxylated CtUrm1C55S (SH) in the presence of TBH.
-
CAnalysis of isopeptide‐bond formation between ScAhp1C31S mutant and thiocarboxylated ScUrm1 in the absence or presence of different reactive species (RS) inducing chemicals such as TBH, HP, Diamide, Peroxynitrite, Methylglyoxal, and DTB‐Disulfide. NEM was added either before adding TBH or after (NEM*). HP: Hydrogen peroxide; DTB: Disulfide‐Di‐tert‐butyl disulfide; NEM: N‐Ethylmaleimide.
-
DAnalysis of ScAhp1C31S stability treated with TBH, HP, Diamide, Peroxynitrite, Methylglyoxal, DTB‐Disulfide, and NEM.
-
EAnalysis of the stability of the thiocarboxylated ScUrm1 subjected to various reactive species producing chemicals such as TBH, HP, Diamide, Peroxynitrite, Methylglyoxal, and DTB‐Disulfide. ScUrm1‐COSH subjected to NEM was also analyzed.
-
FAnalysis of covalent adduct formation between CtAhp1 or CtAhp1C30S mutant and thiocarboxylated CtUrm1C55S or carboxylated (OH) in the presence of TBH. Confirmation of an isopeptide bond was conducted by using DTT/TCEP/HA.
-
GAnalysis of covalent adduct formation between CtAhp1C30S and CtUrm1‐SH or CtUrm1C55S‐SH or CtUrm1G111C mutant in the presence of TBH.
The well‐studied in vivo target Ahp1 is a 2‐Cys peroxiredoxin that consists of a resolving cysteine (CR) and a peroxidatic cysteine (CP; Lian et al, 2012). CP reacts with reactive oxygen species to form sulfenic acid and the CR from the other monomer reacts with the sulfenylated CP forming a disulfide bridge. In vivo, Urm1 conjugation requires the presence of oxidative stressors and peroxidatic cysteine (Cys62) in ScAhp1 (Van der Veen et al, 2011; Brachmann et al, 2020). Surprisingly, we observed that mutating the resolving cysteine in Ahp1 (C31S) not only permitted the attachment of Urm1 in vivo but even promoted the conjugation reaction in absence of N‐Ethylmaleimide (NEM), a compound that irreversibly alkylates cysteines and is generally required to detect ubiquitin‐like conjugates in cell lysates (Fig 1C). Hence, ScAhp1C31S represented a very promising tool to recapitulate the conjugation reaction in vitro. First, we tested purified ScUrm1‐OH and ScUrm1‐SH in combination with various purified variants of ScAhp1 (ScAhp1C31S, ScAhp1C31R, ScAhp1C62S, and ScAhp1C31S C62S; Fig 1C). We were not able to detect Urm1 conjugation to the Ahp1 substrate in vitro by incubating wild‐type ScAhp1 with ScUrm1‐OH in the presence of the oxidizing agent tert‐butyl hydroperoxide (TBH) previously shown to stimulate the reaction in vivo (Van der Veen et al, 2011; Brachmann et al, 2020). Using wild‐type ScAhp1 with ScUrm1‐SH led to the formation of Urm1 conjugates, but their levels remained close to the detection limit. However, ScAhp1C31S and ScAhp1C31R very efficiently formed specific conjugates with Urm1‐SH in the presence of TBH, resulting in an increase in the molecular weight of Ahp1 roughly by the mass of one Urm1 molecule (Figs 1D and EV1B). Of note, the conjugates formed in presence of Urm1‐SH prepared by either thiocarboxylation protocol but not with Urm1‐OH or in the absence of TBH (Fig 1D). To further characterize the conjugates, we treated the samples after the reaction with either Tris(2‐carboxyethyl)phosphine (TCEP) to reduce disulfide bonds, hydroxylamine (HA) to reduce thioester bonds, or DTT to reduce both (Termathe & Leidel, 2018). None of these reagents disrupted the conjugates, suggesting that a covalent isopeptide bond is formed between Urm1‐SH and ScAhp1C31S (Fig 1D). In agreement with previous in vivo data (Brachmann et al, 2020), the mutation of the peroxidatic cysteine (Cys62) in Ahp1 (ScAhp1C31S C62S) completely abolishes Urm1 attachment in vitro (Fig 1D). In addition, the Urm1‐conjugation efficiency of mutated monomeric Ahp1 is strongly reduced in vitro (Appendix Fig S1) and almost completely abolished in vivo (Brachmann et al, 2020). As different oxidative stressors can induce Urm1 conjugation in vivo (Van der Veen et al, 2011; Jüdes et al, 2015), we tested whether “urmylation” can similarly be triggered by different oxidative compounds in vitro. Indeed, conjugate formation likewise occurred in the presence of hydrogen peroxide (HP), peroxynitrite, and diamide. On the contrary, methylglyoxal, di‐tert‐butyl disulfide, and NEM did not promote the conjugation reaction but also did not influence the stability of Urm1 conjugation if added after the reaction (Fig EV1C). None of the reagents compromised ScAhp1 integrity (Fig EV1D), and only the alkylating agents NEM and peroxynitrite negatively affected the stability of the thiocarboxyl group at the C‐terminus of Urm1 (Fig EV1E). This observation suggests that NEM does not promote the urmylation of Ahp1 in vivo but rather facilitates its detection by blocking potential inhibitory factor(s) or deconjugating enzyme(s). In addition, we used CtUrm1 and variants of CtAhp1 to independently confirm the necessity of the C‐terminal thiocarboxyl group, oxidative stress conditions, and redox‐active cysteines for the reaction to occur (Fig EV1F). In summary, our results demonstrate that Urm1, like all other canonical eukaryotic UBLs, can form a covalent bond with a target protein. However, Urm1 conjugation does not depend on the canonical cascade of E2 ubiquitin‐conjugating enzymes and/or E3 ligases. Instead, the conjugation reaction requires a thiocarboxylated C‐terminus of Urm1 and a peroxidatic cysteine in the target protein, which in the case of Ahp1 appears to be promoted by site‐specific substitution of the resolving cysteine.
Structure of urmylated Ahp1 complex
To gain detailed molecular insights into the mechanisms of Urm1 conjugation, we purified and determined the structures of urmylated and unmodified CtAhp1 by macromolecular crystallography. The purified CtAhp1C30S‐Urm1C55S complex showed a clear shift in size in comparison to the individually purified Ahp1 and Urm1 proteins (Fig 2A). Of note, CtAhp1C30S also formed conjugates with wild‐type CtUrm1, showing that Cys55 is not required for the conjugation reaction in Chaetomium thermophilum (Fig EV1G). We independently crystallized CtAhp1 and CtAhp1C30S, which is equivalent to ScAhp1C31S, and collected complete datasets at 1.75 and 1.85 Å resolution, respectively (Table 1). We solved both structures by molecular replacement using ScAhp1 structure (PDB ID 4DSR; Lian et al, 2012) and refined the corresponding atomic models to R/Rfree values of 18.8%/20.2% and 15.7%/18.3%, respectively (Table 1, PDB ID 7Q68 and 7Q69). The structures confirm that CtAhp1, like ScAhp1, forms a homodimer, which is maintained by conserved hydrophobic residues (e.g., Phe56) at the center of the dimer interface (Fig 2B; Trivelli et al, 2003). Wild‐type CtAhp1 exists as a homodimer and forms a disulfide bond between Cys30 and Cys60 of opposite monomers, which indicates that in the absence of thioredoxins the dimer becomes trapped in the (post)oxidized state after the release of H2O. CtAhp1C30S cannot form the disulfide bond between the two redox‐active cysteines, resulting in a structural rearrangement of the entire loop region harboring the peroxidatic cysteine (Cys60). As the mutated residue (Ser30) remains in an almost identical position as in the wild‐type protein, CtAhp1C30S structurally mimics the reduced form that can accumulate oxidized/sulfenylated Cys60 (Fig 2B and Appendix Fig S2). Due to the presence of a reducing agent in the crystallization condition, there were no indications for the oxidation of the thiol group of Cys60.
Figure 2. Structure of the urmylated‐Ahp1 complex.
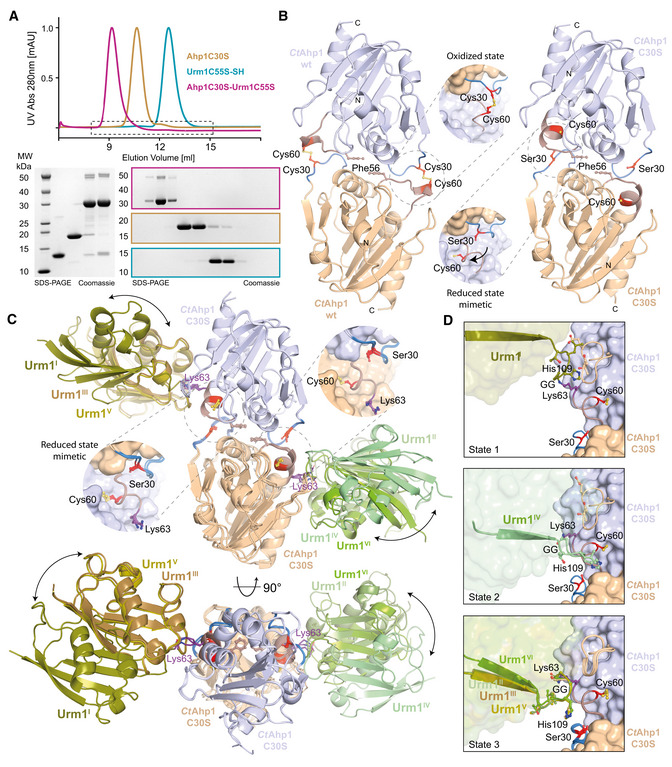
- Size‐exclusion chromatography profiles of CtAhp1C30S (yellow), CtUrm1C55S (cyan) and the CtAhp1C30S‐CtUrm1C55S complex (pink). SDS–PAGE analysis of the CtAhp1C30S‐CtUrm1C55S complex and individual proteins CtAhp1C30S and CtUrm1C55S‐SH. Equivalent fractions of each run were collected and analyzed separately. The expected molecular mass of the individual samples are as follows—CtUrm1C55S‐SH 12.5 kDa; CtAhp1C30S 18 kDa; CtAhp1C30S‐CtUrm1C55S complex 30.5 kDa.
- Crystal structures of dimeric CtAhp1 and CtAhp1C30S in cartoon representation. Insets: Close‐up of the active sites highlighting redox‐active cysteines (Cys60 and Cys/Ser30 in red).
- Front (up) and top (down) view of the CtAhp1C30S‐CtUrm1C55S complex structure in cartoon representation. All three entities in the asymmetric unit are superimposed on Ahp1 and the various Urm1 molecules are labeled. The position of Lys63 (violet) is highlighted and labeled. Insets: Close‐up of the active sites highlighting the position of the redox‐active cysteines (Cys60 and Ser30 in red).
- Comparison of the three observed states of Urm1 conjugation in relation to the linkage site of Ahp1. All 6 Ahp‐Urm1 entities in the asymmetric unit are superimposed on Ahp1. All relevant residues are labeled and highlighted in the ball and stick representation. The C‐terminal residues of the attached Urm1 are shown in cartoon representation and the remaining parts as a transparent surface. Cysteines are highlighted (Cys60 and Ser30 in red).
Table 1.
Data collection and refinement statistics (molecular replacement).
| CtAhp1 wt | CtAhp1C30S (Pre) | CtAhp1C30S (unreacted) | CtAhp1C30S‐Urm1 | CtAhp1C30S‐Urm1 | CtAhp1C30S‐Urm1 | |
|---|---|---|---|---|---|---|
| PDB ID 7Q68 | PDB ID 7Q69 | PDB ID 7Q6A | PDB ID 7Q5N | Zn peak | Zn HR | |
| Data collection | ESRF ID 23‐1 | BESSY 14.1 | BESSY 14.1 | BESSY 14.1 | BESSY 14.1 | BESSY 14.1 |
| Wavelength (Å) | 0.97625 | 0.9184 | 0.9184 | 0.9184 | 1.2823 | 1.3051 |
| Space group | P4322 (95) | P21 (4) | P1 (1) | C2221 (20) | C2221 (20) | C2221 (20) |
| Cell dimensions a, b, c (Å) | 75.25 75.25 110.26 | 60.14 101.74 69.42 | 35.85 41.10 60.83 | 185.43 197.22 139.18 | 186.51 197.46 138.85 | 186.31 197.05 138.57 |
| α, β, γ (°) | 90.0 90.0 90.0 | 90.0 107.33 90.0 | 77.09 75.12 67.27 | 90.0 90.0 90.0 | 90.0 90.0 90.0 | 90.0 90.0 90.0 |
| Resolution (Å) a | 50–1.75 (1.79–1.75) | 50–1.85 (1.9–1.85) | 50–1.10 (1.12–1.10) | 50–2.50 (2.57–2.50) | 50–2.85 (2.92–2.85) | 50–2.85 (2.92–2.85) |
| R meas (%) a , b | 7.1 (247.5) | 14.0 (99.1) | 6.8 (160.8) | 10.2 (423.6) | 12.7 (248.0) | 9.9 (129.8) |
| R pim a , c | 2.3 (80.7) | 7.7 (61.3) | 3.5 (84.7) | 3.9 (163.65) | 4.8 (95.1) | 3.8 (49.4) |
| I / σI a | 14.66 (0.82) | 11.24 (1.84) | 10.94 (0.97) | 12.18 (0.63) | 11.02 (0.96) | 13.40 (1.71) |
| CC1/2 a | 0.998 (0.516) | 0.994 (0.539) | 0.999 (0.464) | 0.999 (0.386) | 0.998 (0.423) | 0.998 (0.738) |
| Completeness (%) a | 99.8 (98.4) | 96.1 (80.1) | 89.3 (79.6) | 99.3 (97.8) | 99.3 (94.9) | 99.3 (94.5) |
| Redundancy a | 9.3 (9.4) | 3.34 (2.61) | 3.7 (3.6) | 6.74 (6.7) | 6.95 (6.8) | 6.94 (6.9) |
| Refinement | ||||||
| Resolution (Å) | 47.92–1.75 | 46.07–3.15 | 32.46–1.10 | 48.47–2.50 | ||
| No. reflections | 32,590 | 65,571 | 112,450 | 9,580 | ||
| R work/R free | 0.188/0.202 | 0.157/0.183 | 0.161/0.174 | 0.234/0.272 | ||
| No. of atoms | 1,387 | 6,003 | 3,022 | 12,664 | ||
| Protein | 1,254 | 5,038 | 2,519 | 2,877 | ||
| Ligand/ion | 32 | 68 | 54 | 1 | ||
| Water | 101 | 897 | 449 | 0 | ||
| B‐factors (Å2) | 55.6 | 26.4 | 19.1 | 109.0 | ||
| Protein | 54.8 | 24.7 | 16.4 | 156.76 | ||
| Ligand/ion | 91.5 | 47.1 | 46.6 | 110.05 | ||
| Water | 53.7 | 34.4 | 30.7 | n.a. | ||
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.005 | 0.007 | 0.005 | 0.006 | ||
| Bond angles (°) | 0.598 | 0.665 | 0.845 | 0.981 | ||
| Ramachandran | ||||||
| Outliers (%) | 0 | 0 | 0 | 0 | ||
| Allowed (%) | 1.8 | 2.4 | 1.8 | 4.8 | ||
| Favored (%) | 98.2 | 97.6 | 98.2 | 95.2 | ||
Values in parentheses are for the high‐resolution shell.
.
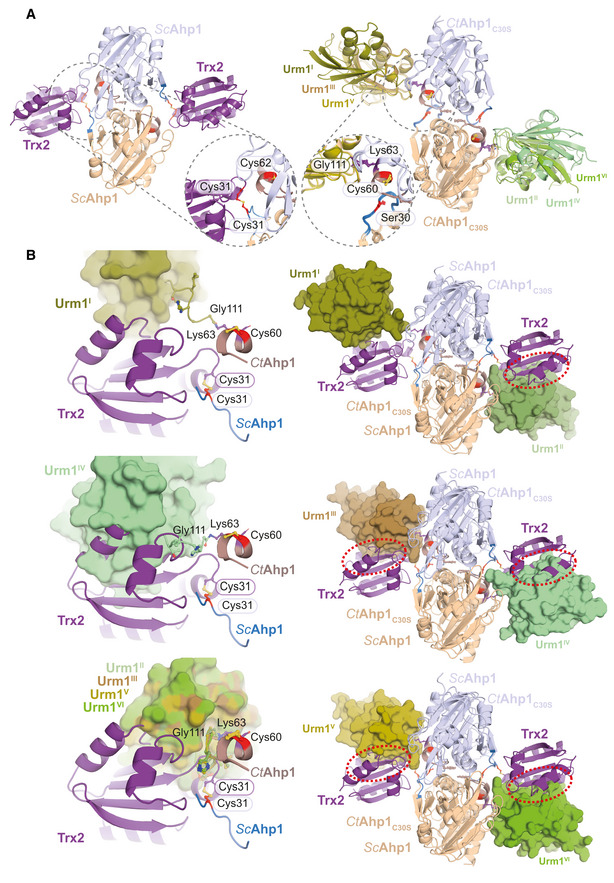
Next, we obtained crystals of the CtAhp1C30S‐Urm1C55S complex, which after several rounds of optimization, diffracted to an overall resolution of 2.5 Å (Table 1). We solved the structure by molecular replacement using the high‐resolution model of CtAhp1C30S in combination with the model of CtUrm1 taken from the Uba4C202K–Urm1 complex (PDB ID 6YUC; Pabis et al, 2020). The final model was refined to R/Rfree values of 23.4%/27.2% obeying perfect stereochemistry (Table 1, PDB ID 7Q5N). The structure shows an almost unchanged homodimer of CtAhp1C30S, where the Lys63 residue from each Ahp1 molecule forms a covalent isopeptide bond between the lysine side chain and the C‐terminus of Urm1 (Fig 2C). The overall dimeric structure of Ahp1 and the position of its peroxidatic cysteines remain almost identical in the presence or absence of Urm1 (Fig 2C). The asymmetric unit contains three individual CtAhp1C30S‐Urm1C55S dimers, which are all linked via the same peptide bond between Ahp1Lys63 and Urm1Gly111 (Appendix Fig S3A). The conformations of the three urmylated Ahp1 homodimers are superimposable, but the relative position of the respective Urm1 molecules, the quality of the density map, and the associated B‐factors differ between the individual copies (Fig 2C and Appendix Fig S3B). These variations arise most likely due to different packing environments in the crystal lattice and might result from the fact that Ahp1 and the attached Urm1 do not form an extensive interaction surface. However, there are no indications that a hexameric form of CtAhp1C30S‐Urm1C55S (Appendix Fig S3C) or dimerization of Urm1 between neighboring complexes (Appendix Fig S3D) occurs in solution or has any functional significance in vivo. Of note, the Urm1‐Urm1 packing interfaces are almost identical, and changes in relative positioning are not caused by structural changes of the individual Ahp1 and Urm1 proteins but are strictly related to variations in the flexible C‐terminal region of Urm1 (Appendix Fig S3E). Unexpectedly, we found three distinct types of Urm1 positioning relative to the attachment site around Ahp1Lys63 (Fig 2D). These differences are not only visible at the domain level but also the specific conformation of interacting residues in Urm1 and Ahp1 varies between the observed attachment sites. The structure of ScAhp1 bound to its recycling factor thioredoxin 2 (Trx2) has been determined in different reaction states (Lian et al, 2020, 2012), showing that Trx2 directly interacts with Cys30 of Ahp1 to resolve the disulfide bond formed between the redox‐active cysteines (Fig EV2A). By contrast, our urmylated Ahp1 structure places Urm1 close to the peroxidatic Cys62, indicating that certain conformations of Urm1 attachment are compatible with the binding of Trx2 while others are sterically impossible (Fig EV2B). Our crystallographic data confirm the formation of a covalent isopeptide bond between Ahp1 and Urm1. Furthermore, we identify Lys63 as one of the main attachment sites in CtAhp1 and reveal the structural variability of the conjugated complex, which is linked closely to the peroxidatic cysteine.
Figure EV2. Comparison between Ahp1‐Trx2 and Ahp1‐Urm1 complexes.
-
AStructural Comparison of Ahp1‐Trx2 vs Ahp1‐Urm1 and close‐up.
-
BSuperimposition of Ahp1‐Urm1 and Ahp1‐Trx2.
Urm1 can conjugate to various sites in Ahp1
Although the crystal structure of CtAhp1C30S‐Urm1C55S displays an exclusive linkage with unambiguous density between CtUrm1Gly111 and CtAhp1Lys63 (Fig 3A), we previously found that Urm1 can be conjugated to different lysine residues of ScAhp1 in vivo (Brachmann et al, 2020). Hence, we analyzed the products of the in vitro conjugation reactions by mass spectrometry to independently confirm the presence of HGG‐K linked peptides after digestion with chymotrypsin (Fig 3B). Since ubiquitin is known to form poly‐ubiquitin chains on target proteins, we asked whether we could detect this kind of polymerization for Urm1 conjugates. In our dataset, the discrete molecular weights of the conjugates indicated that internal conjugation events did not occur between Urm1 molecules under these conditions. In contrast to the exclusive Lys63‐mediated linkage in our crystals, we identified additional lysine residues of CtAhp1 (Lys44, Lys99, Lys141, Lys156, and Lys171) conjugated to the C‐terminus of CtUrm1. Furthermore, we used a CtAhp1C30S K63R mutant, lacking the primarily conjugated residue Lys63, to promote the occurrence of rare conjugation events and detected urmylation of Lys71, which is in close proximity to Cys60. Similarly, in ScAhp1, which harbors more lysine residues than CtAhp1 (Fig 3B), we detected various additional attachment sites by mass spectrometry (Lys32, Lys47/Lys48, Lys79, Lys81, Lys107, Lys124, and Lys156). Next, we mutated various lysine residues in CtAhp1 and ScAhp1 individually and performed in vitro conjugation reactions using these purified Ahp1 variants. Our results clearly demonstrated that even after simultaneously mutating the major attachment sites in CtAhp1 (CtAhp1K44R K63R K99R), residual conjugates were still detectable (Fig 3C). In ScAhp1, only the simultaneous mutation of several lysine residues (ScAhp1K32R,K47R,K48R,K124R,K156R) slightly reduced urmylation levels (Fig 3D). Last, we tested the effects of these lysine mutants in S. cerevisiae and did not detect any preferred modification site in vivo. After mutating different lysine residues, we neither observed any phenotypic consequences in the presence of TBH (Fig 3E) nor the complete inhibition of conjugation (Fig 3F).
Figure 3. Urmylation is an E2/E3‐independent, and primarily lysine‐directed protein conjugation.
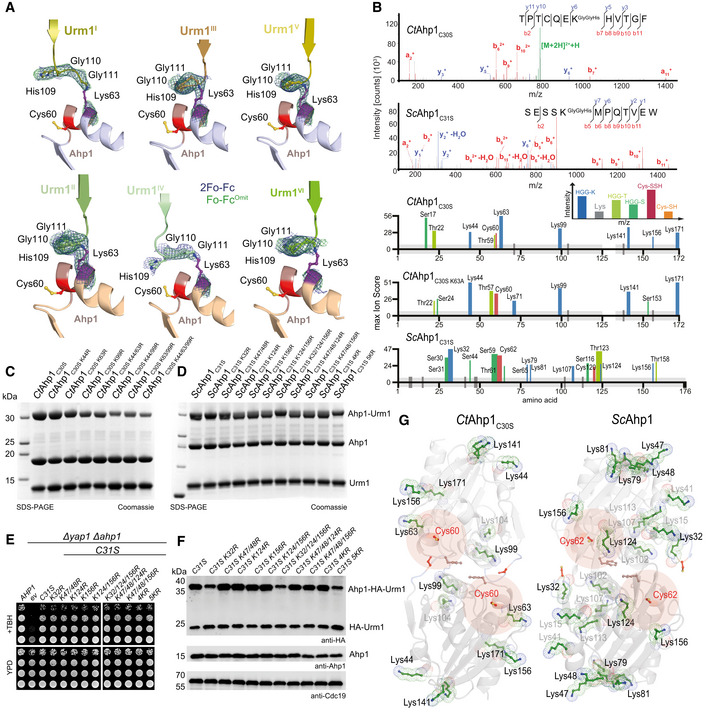
-
AComparison of the individual Ahp1‐Urm1 units in the asymmetric unit. Close‐up of the formed peptide bond with all nearby residues labeled and highlighted in a balls‐and‐sticks representation (Cys60 in red). The last strand of the attached Urm1 protein is shown in cartoon representation and the refined 2Fo‐Fc density (blue) and the Fo‐Fc omit map lacking the highlighted residues (green) is shown around Ahp1Lys63 and Urm1Gly111 at 1σ and 3σ, respectively.
-
BTop: MS/MS spectrum of the K63‐ε‐GGH containing peptide from CtAhp1C30S and K32‐ε‐GGH containing peptide from ScAhp1C31S. A representative annotated fragmentation spectrum is shown with the b‐ and y‐ions marked in red and blue, respectively. The precursor ions are labeled in green. The peptide sequence is shown at the top with the collision‐induced fragmentation pattern. m/z: mass to charge ratio. Bottom: Schematic summary of the detected conjugated peptides by mass spectrometry for CtAhp1C30S, CtAhp1C30S K63A, and ScAhp1C31S. The thickness of the line represents the number of replicates where the respective site was detected, and the height represents the associated maximum Mascot ion score (n ≥ 3). The urmylation was identified as HisGlyGly tag on lysines, serines, and threonines while persulfidation was searched as a mass shift: +32 Da (persulfidation) or +89 Da (persulfidation + carbamidomethylation) on cysteines.
-
CAnalysis of conjugation between CtAhp1C30S variants in combination with thiocarboxylated CtUrm1C55S in the presence of TBH. Unconjugated Ahp1 and Urm1 as well as the formed conjugates (Ahp1‐Urm1) are indicated on the right.
-
DAnalysis of conjugation between ScAhp1C31S variants in combination with thiocarboxylated ScUrm1 in the presence of TBH. Unconjugated Ahp1 and Urm1 as well as the formed conjugates (Ahp1‐Urm1) are indicated on the right. ScAhp14KR: ScAhp1C31S K47/48R K124R K156R; ScAhp15KR: ScAhp1C31S K32R K47/48R K124R K156R.
-
ETBH cytotoxicity assay in vivo of the ScAhp1 lysine mutants in yeast. Growth of yap1Δahp1Δ cells carrying empty vector (ev), wild‐type peroxiredoxin gene (AHP1), or indicated cysteine or lysine substitutions was monitored without or with 1 mM TBH. ScAhp14KR: ScAhp1C31S K47/48R K124R K156R; ScAhp15KR: ScAhp1C31S K32R K47/48R K124R K156R.
-
FIn vivo conjugation analyses of ScAhp1 lysine mutants with protein extracts obtained from the indicated yeast strains expressing HA‐URM1. Unconjugated Urm1 and Urm1‐Ahp1 conjugates were detected by anti‐HA (top panels) Western blots. Anti‐Ahp1 blots (middle) detect unmodified Ahp1. Anti‐Cdc19 (bottom panels) served as a loading control. ScAhp14KR: ScAhp1C31S K47/48R K124R K156R; ScAhp15KR: ScAhp1C31S K32R K47/48R K124R K156R.
-
GComparison of the crystal structures of the oxidized dimeric forms of Ahp1 from CtAhp1C30S (left) and ScAhp1 (right; PDB ID 4DSR; Lian et al, 2012). All lysine residues (green) are shown in the ball and sticks representation. Conjugated lysine residues detected by mass spectrometry (in B) are labeled and highlighted in black. Other lysine residues are labeled in gray and peroxidatic cysteines (Cys60 in CtAhp1 and Cys62 in ScAhp1) and their surroundings are labeled in red.
Of note, the linked lysine residues in CtAhp1 and ScAhp1 are not evolutionarily conserved, and their spatial distribution does not reveal a specific distribution pattern (Fig 3G). However, our complementary mass spectrometry and mutational analyses show that the attachment of Urm1 to lysine residues in Ahp1 exclusively depends on the presence of a peroxidatic cysteine in the target protein. Our results in ScAhp1 emphasize that in the absence of a particular lysine residue a different residue will be used as a target with similar efficiency, indicating low specificity for the particular residue, motif, or structural context at which urmylation can occur.
Urm1 conjugates to other residues in the absence of lysines
As noncanonical linkages to serine, threonine or cysteine residues have also been described for other UBLs (McDowell & Philpott, 2013), we analyzed our mass spectrometry data for any such possible Urm1 linkage on Ahp1 (ScAhp1C31S and CtAhp1C30S). Strikingly, we found Urm1 being conjugated to several serine and threonine residues in ScAhp1C31S and few in CtAhp1C30S (Fig 3B). To test whether lysine residues are essential for catalyzing the conjugation reaction, we simultaneously replaced all lysine residues in ScAhp1C31S and CtAhp1C30S with arginine residues (ScAhp1C31S‐KtoR and CtAhp1C30S‐KtoR). Nonetheless, we still observed the formation of Ahp1‐Urm1 conjugates with these lysine‐less Ahp1 variants in the presence of Urm1‐SH and mild oxidative stress in vitro (Fig 4A). We mutated semi‐conserved serine and threonine residues in the spatial proximity of the peroxidatic cysteine and other nearby residues in CtAhp1 (Arg151) or CtUrm1 (Leu108, His109; Fig 4B). Whereas none of the mutations affected the conjugation efficiency in the presence of lysine residues, the simultaneous depletion of all possible serine and threonine attachment sites that are in proximity to the peroxidatic cysteines in ScAhp1C31S‐KtoR and CtAhp1C30S‐KtoR strongly reduced the formation of Ahp1‐Urm1 conjugates in vitro (Fig 4C and Appendix Fig S4A). Therefore, these serine and threonine residues can act as alternative attachment sites but play no significant role during the canonical reaction when lysines are present. Since the lysine‐less variants still showed traces of conjugates, we used mass spectrometry to unambiguously test for other low‐frequency attachment sites. This approach allowed us to confirm noncanonical ester linkages between ScUrm1 and Ser59 and Thr61 sites on ScAhp1 and between CtUrm1 and Thr57 and Thr59 on CtAhp1 (Fig 4D). In summary, our detailed mutational analyses indicate that Urm1‐SH preferably conjugates to lysine residues but is also able to attach to other amino acid side chains as has been shown for other UBLs.
Figure 4. Urm1 conjugation directly mediates cysteine persulfidation in vitro .
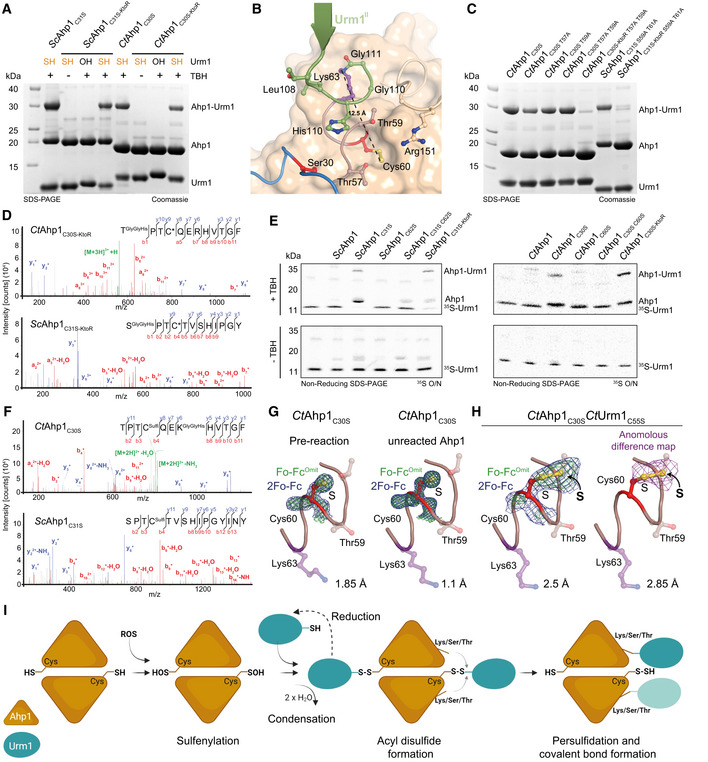
-
AAnalysis of conjugation between ScUrm1‐SH with ScAhp1C31S (left) and CtAhp1C30S (right) using lysine‐less variants (ScAhp1C31S‐KtoR and CtAhp1C30S‐KtoR). ScUrm1 or CtUrm1C55S were used. The control reactions with Urm1‐OH and without TBH are indicated.
-
BStructural close‐up of the Ahp1‐Urm1 conjugation site with all nearby residues labeled and highlighted in balls‐and‐sticks representation (Cys60 in red). The last strand of the attached Urm1 protein (green) is shown in the cartoon. The distance between the peptide bond that forms between Lys63 (violet)/Gly111 (green) and Cys60 (red) is indicated.
-
CAnalysis of conjugation between thiocarboxylated CtUrm1C55S and CtAhp1C30S variants, combining substitutions of serine and threonine residues in the active site and lysine‐less variants in the presence of TBH.
-
DMS/MS spectrum of the S‐ε‐GGH/T‐ε‐GGH containing peptide from CtAhp1C30S‐KtoR (top), and ScAhp1C31S‐KtoR (bottom) A representative annotated fragmentation spectrum is shown with the b‐ and y‐ions marked in red and blue, respectively. The peptide sequence is displayed at the top with the collision‐induced fragmentation pattern.
-
ERadioisotope assay for 35S‐labeled Urm1. Analysis of conjugation and sulfur transfer between Sc 35S‐Urm1 with ScAhp1 variants (left) and Ct 35S‐Urm1C55S with CtAhp1 variants in the presence (top) or absence of TBH (bottom). The samples were separated by nonreducing SDS–PAGE and dried gels were exposed overnight to storage phosphor screens. Uncropped images of the respective Coomassie‐stained gels before drying and the exposures themselves are provided in Appendix Fig S4D–G.
-
F(top) MS/MS spectrum of the persulfidated Cys60 and K63‐ε‐GGH containing peptide from CtAhp1C30S. (bottom) MS/MS spectrum of the persulfidated Cys62 peptide from ScAhp1C31S. Representative annotated fragmentation spectra are shown with the b‐ and y‐ions marked in red and blue, respectively. The peptide sequence is displayed at the top with the collision‐induced fragmentation pattern. Mass shift: +32 Da (persulfidation) or +89 Da (persulfidation + carbamidomethylation).
-
GStructural close‐up of the peroxidatic cysteine (Cys60) and Lys63 (violet) in CtAhp1C30S preconjugation (left) and unreacted Ahp1 from the conjugation reaction (right). The refined 2Fo‐Fc density (blue) and the Fo‐Fc omit map lacking the active site cysteine (green) is shown at comparable σ‐levels. For additional details see Appendix Fig S4.
-
HSame view as in G of the maps in the CtAhp1C30S‐CtUrm1C55S complex around Cys60 (red) at 3.5 σ (blue, left), 4 σ (green, left) and the anomalous difference Fourier map at 2 σ (violet, right). The respective resolution ranges of the maps in G and H are indicated.
-
IProposed mechanism of urmylation. The peroxidatic cysteine of Ahp1 gets oxidized and sulfenylated. Urm1‐SH recognizes the sulfenylated cysteine and condenses to form an acyl disulfide intermediate. The formation of this intermediate can be reverted by reduction. A lysine/serine/threonine residue at proximal distance from the cysteine of the target protein mediates a nucleophilic attack on Urm1 to resolve the acyl disulfide. This results in a persulfidated cysteine and the covalent conjugation of the C‐terminus of Urm1 with the respective lysine/serine/threonine acceptor residue.
Urm1‐SH transfers sulfur to the peroxidatic cysteine of Ahp1
To the best of our knowledge, Urm1 is the only eukaryotic UBL that carries a C‐terminal thiocarboxyl group, which we showed is essential for the conjugation reaction. Intriguingly, this unique sulfur atom was undetectable at the site of linkage in our mass spectrometry analyses above. Hence, we were curious about the fate of the attached sulfur moiety during and after isopeptide bond formation. To address this question, we generated Urm1‐SH carrying radioactive sulfur (35S) at its C‐terminus (see Materials and Methods and Appendix Fig S4B) for in vitro “urmylation” reactions (Appendix Fig S4C). We incubated 35S‐labeled thiocarboxylated ScUrm1 and CtUrm1C55S with various variants of ScAhp1 and CtAhp1 in the presence or absence of TBH (Fig 4E and Appendix Fig S4D–G). Strikingly, we detected a very efficient transfer of 35S from Urm1‐35SH to ScAhp1 and CtAhp1 variants (ScAhp1C31S, ScAhp1C31S‐KtoR, CtAhp1C30S and CtAhp1C30S‐KtoR). The mutation of the peroxidatic cysteine not only diminished the Urm1 conjugation reaction but also blocked the sulfur transfer between Urm1‐SH and Ahp1 (ScAhp1C62S, CtAhp1C60S, and CtAhp1C30S C60S). In the case of ScAhp1C31S C62S and ScAhp1C31S‐KtoR, we still observed a background of low‐level sulfur transfer, which appears to be conjugation‐independent and could be mediated by an additional cysteine residue in ScAhp1 (Cys120). Neither the unspecific reaction with Ahp1 nor the additional cysteine residue is present in CtAhp1. Interestingly, the 35S transfer to Ahp1 cannot only occur on the Urm1‐conjugated molecule of the Ahp1 homodimer but also on the other unconjugated chain of the homodimer.
Next, we searched our mass spectrometry datasets of the complexes for possible forms of peptide S‐sulfhydration. Strikingly, we identified persulfidation of the catalytically active peroxidatic cysteine in both ScAhp1 and CtAhp1 (Fig 4F), indicating that Urm1‐SH transfers its sulfur atom to the sulfenylated cysteine on its target protein. The archaeal TfuA protein, which is also C‐terminally thiocarboxylated, was recently shown to replace oxygen with sulfur in the peptide backbone of its target protein (Liu et al, 2021). We reanalyzed our crystallographic data to identify the precise spatial position of the post‐translational modification. A prominent density next to the sulfur atom of the cysteine side chain was identifiable in the crystal structure of the CtAhp1C30S‐Urm1C55S complex, which was absent from individual Ahp1 structures alone (Fig 4G and Appendix Fig S5). To confirm the position and identity of the additional sulfur moiety, we collected diffraction data for the CtAhp1C30S‐Urm1C55S crystals at suitable wavelengths and calculated anomalous difference Fourier maps (Fig 4H). Foremost, we did not observe an anomalous signal at the position of the backbone oxygen atom, excluding the possibility of a sulfur substitution reaction, like TfuA. Furthermore, we obtained a structure at 1.1 Å resolution of the nonreacted CtAhp1C30S, which we repurified after the conjugation reaction with Urm1‐SH (Table 1, PDB ID 7Q6A). Although this Ahp1 sample underwent an identical treatment, we did not observe any extra densities around Cys60 without Urm1 conjugation (unreacted Ahp1; Fig 4G). Our complementary biochemical, mass spectrometry, and crystallographic analyses show that the detected persulfide group is absent in the starting material and that the post‐translational thio‐modification of the cysteine side chain in Ahp1 occurred during the conjugation reaction with Urm1‐SH.
We propose a very simple reaction mechanism that directly couples urmylation with the observed sulfur transfer to the target protein (Fig 4I). In detail, the exposure of a redox‐active cysteine in Ahp1 to oxidative stress leads to the sulfenylation of its side chain (‐SOH). Urm1‐SH recognizes the sulfenylated cysteine and after condensation forms a short‐lived acyl disulfide intermediate (Kang et al, 2018), which we were not able to directly observe in our experiments due to its transient character. The reduction in the transient acyl disulfide intermediate can lead to the recycling of Urm1‐SH. Otherwise, a lysine, serine, or threonine residue at a proximal distance from the linked Cys‐Cys pair triggers a nucleophilic attack on the acyl disulfide. This reaction scheme simultaneously produces a persulfidated cysteine and a covalent conjugation site between the C‐terminus of Urm1 and respective lysine, serine, or threonine residue in the target protein (Fig 4I).
Our results show that the Urm1‐conjugation reaction is mechanistically and directly coupled to the process of cysteine persulfidation. Foremost, our results demonstrate that Urm1 can specifically transfer the sulfur atom of its thiocarboxyl group to redox‐active cysteine residues in Ahp1 if the target is exposed to oxidative conditions.
Cellular Urm1 targets are persulfidated in vitro
It is important to emphasize that Ahp1, an antioxidant enzyme, represents an unusual target of Urm1 under oxidative stress conditions. For instance, even under optimized experimental conditions, we detected only weak conjugation activity of wild‐type Ahp1 in vitro and we had to use ScAhp1C31S or CtAhp1C30S to detect substantial conjugation activity. To identify and characterize additional Urm1 targets in vitro, we used a list of 547 candidate proteins that had been identified in yeast by LC–MS/MS after Urm1 pull‐down. Twenty one of the hits were significantly enriched upon NEM treatment, recapitulating the behavior of Ahp1 (Fig 5A). We decided to focus on a shortlist of potential Urm1 targets from yeast and humans based on available purification protocols. Hence, we used Tdh3, Ses1, and Pyk1 (Weygand‐Durasevic et al, 1987; Jurica et al, 1998; Liu et al, 2012) as well as their human homologs (PRDX5 (human homolog of Ahp1), GAPDH (human homolog of Tdh3), SARS1 (human homolog of Ses1) and PKM2 (human homolog of Pyk1)). Strikingly, we were able to detect Urm1 conjugates for the entire set of yeast and human targets in vitro using either ScUrm1‐SH or HsUrm1‐SH, respectively (Fig 5B). Formation of these conjugates required the addition of an oxidizing agent (Fig 5B) and only occurred in the presence of Urm1‐SH but not Urm1‐OH (Appendix Fig S6A). Unlike Ahp1, these targets showed robust Urm1 conjugation without introducing additional mutations or removing potential resolving cysteines (Appendix Fig S6B). To confirm the formation of covalent peptide bonds, we performed exhaustive mass spectrometry analyses to identify numerous HGG‐K, HGG‐T and HGG‐S linked peptides and map the individually conjugated residues (Fig 5C, and Appendix Fig S6C and D). As the conjugation of Urm1 to Ahp1 requires at least one peroxidatic cysteine, we tested whether known catalytic cysteines are necessary for the conjugation reaction to the additional target proteins. We observed a complete loss of Urm1 conjugation to HsPRDX5, ScTdh3, and HsGAPDH when a single peroxidatic cysteine was mutated (Fig 5D). The mutation of individual cysteine residues in the other targets only partially reduced the Urm1 conjugation efficiency, indicating that several cysteines can support the Urm1 conjugation reaction in these targets (Appendix Fig S6B). Furthermore, we used Urm1‐35SH for in vitro conjugation of yeast targets and observed the direct transfer of sulfur during Urm1 conjugation onto ScTdh3, ScSes1, and ScPyk1 in the presence of diamide (Fig 5E). In addition, we used mass spectrometry to verify that the sulfur atom from Urm1‐SH was transferred, leading to persulfidation of individual cysteine residues in HsGAPDH, ScTdh3 and HsPRDX5 (Fig 5C). Consistent with the observation that mutation of individual cysteines did not abolish Urm1 conjugation, we found that indeed more than one peroxidatic cysteine was persulfidated in ScSes1, HsSARS1, ScPyk1 and HsPKM2 (Fig 5C). Our data thus uncover the previously unknown source of the sulfur donor for targeted persulfidation of eukaryotic proteins.
Figure 5. Cellular Urm1 targets are persulfidated upon conjugation in vitro .
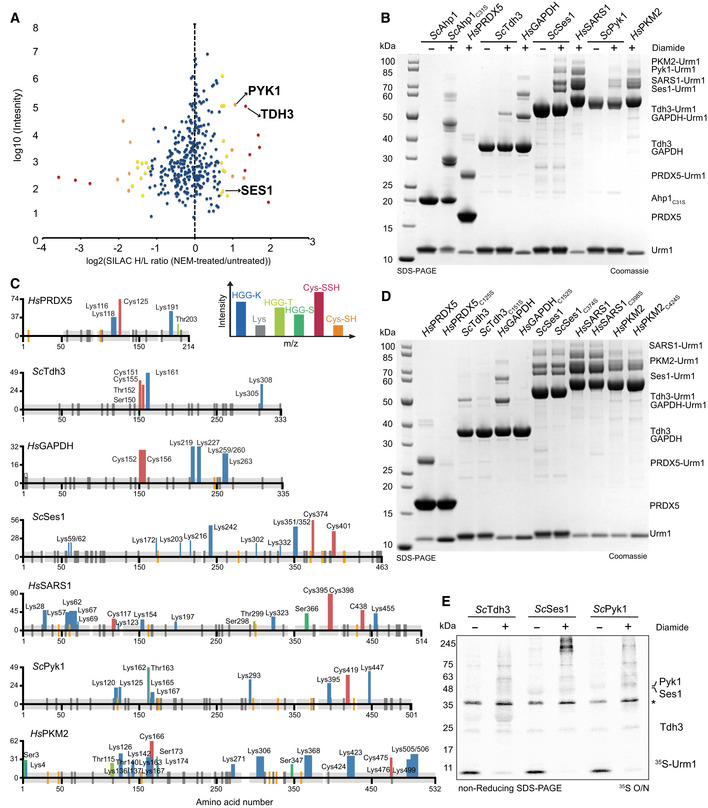
-
AScatter plot of normalized SILAC H/L ratios (NEM‐treated versus untreated) for 547 quantified proteins, plotted against the sum of the respective peptide intensities. Proteins are color‐coded according to their respective P‐values (red < 0.001, orange 0.001–0.01, yellow 0.01–0.05 and blue > 0.05).
-
BAnalysis of conjugation between thiocarboxylated ScUrm1 or HsUrm1 and various urmylation target proteins (ScAhp1C31S, HsPRDX5, ScTdh3, HsGAPDH, ScSes1, HsSARS1, ScPyk1, HsPKM2) in the presence of diamide. Unconjugated target proteins and Urm1 as well as their conjugates are indicated on the right.
-
CSchematic summary of the detected conjugated peptides by mass spectrometry for the various targets in (B). The thickness of the line represents the number of replicates where the respective site was detected, and the height represents the associated maximum Mascot ion score (n ≥ 3). The urmylation was identified as HisGlyGly tag on lysines, serines, and threonines, while persulfidation was searched as a mass shift: +32 Da (persulfidation) or +89 Da (persulfidation + carbamidomethylation) on cysteines.
-
DAnalysis of conjugation between thiocarboxylated ScUrm1 or HsUrm1 and various urmylation target proteins (ScAhp1C31S, HsPRDX5, ScTdh3, HsGAPDH, ScSes1, HsSARS1, ScPyk1, HsPKM2) and their cysteine mutants in the presence of diamide. Unconjugated target proteins and Urm1 as well as their conjugates are indicated on the right.
-
ERadioisotope assay for 35S‐labeled Urm1. Analysis of conjugation and sulfur transfer between Sc 35S‐Urm1 with ScTdh3, ScSes1, and ScPyk1 in the presence or absence of diamide. The samples were separated by nonreducing SDS–PAGE and visualized by Coomassie staining. The dry gels were exposed overnight to storage phosphor screens. *Uba4 background.
Any thiocarboxylated C‐terminus can conjugate to proteins
We established that Urm1 can be covalently conjugated to various target proteins in vitro without the need for specific E2 enzymes and E3 ligases. The intein system that we used to generate Urm1‐SH above enables us to produce virtually any protein with a thiocarboxylated C‐terminus. Therefore, we tested whether other UBLs that harbor the C‐terminal GlyGly‐motif can also be conjugated to the identified Urm1 target proteins in response to oxidative stress. Using purified ubiquitin‐SH or SUMO‐SH (Fig 6A), we were able to attach these synthetically thiocarboxylated UBLs to ScAhp1 and ScTdh3 under the same experimental conditions that worked for Urm1‐SH (Fig 6A). As expected, ubiquitin‐OH and SUMO‐OH that lack the thiocarboxyl group did not form conjugates with the target proteins. We used mass spectrometry to map all conjugation sites. Interestingly, the pattern of conjugation sites differs between the different thiocarboxylated UBLs (Fig 6B). Nonetheless, we confirmed the identity of the peptides linked to the C‐termini and found that the sulfur is similarly deposited as a persulfide on the same oxidizable cysteines in the target proteins (Fig 6B).
Figure 6. Thiocarboxylated Ubiquitin and SUMO conjugate to Ahp1 in vitro .
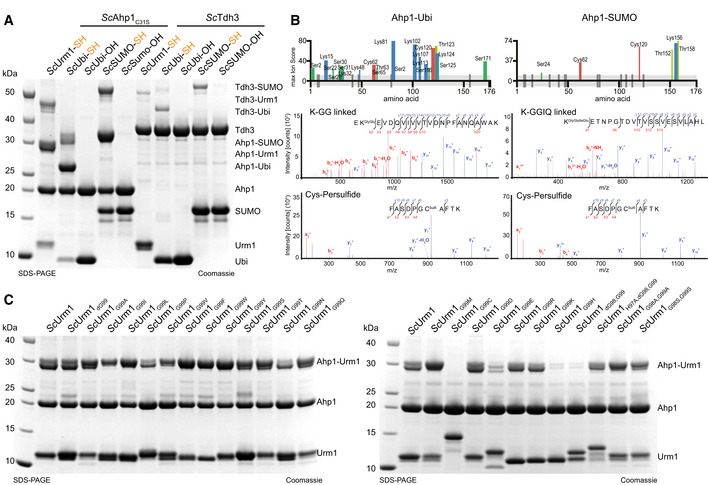
-
AAnalysis of covalent adduct formation by carboxylated (OH) or thiocarboxylated (SH) ScUbiquitin or ScSUMO on ScAhp1C31S or ScTdh3 in the presence of diamide. Thiocarboxylated ScUrm1 was used as a control. Unconjugated Ubls and target proteins as well as their conjugates are indicated on the right.
-
BSchematic summary of the conjugated peptides that were detected by mass spectrometry for ubiquitin or SUMO conjugation on Ahp1. The thickness of the line represents the number of replicates where the respective site was detected, and the height represents the associated ion score (n > 3). MS/MS spectrum of the ubiquitin (K‐ε‐GG/ S‐ε‐GG/ T‐ε‐GG) or SUMO (K‐ε‐GGIQ/ S‐ε‐GGIQ/ T‐ε‐GGIQ) conjugated peptide on ScAhp1C31S and the persulfidated cysteine spectrum by ubiquitin or SUMO. A representative annotated fragmentation spectrum is shown with b‐ and y‐ions marked in red and blue, respectively. The peptide sequence is shown at the top along with the collision‐induced fragmentation pattern. The ubiquitin or SUMO conjugation site was identified by detection of the GG and GGIQ remnant motif, respectively. m/z: mass to charge ratio. Persulfidation was searched as a mass shift: +32 Da (persulfidation) or +89 Da (persulfidation + carbamidomethylation) on cysteines.
-
CAnalysis of covalent adduct formation between ScAhp1C31S and various thiocarboxylated C‐terminal variants of ScUrm1 in the presence of TBH. Unconjugated Urm1, Ahp1, and the conjugates are indicated on the right.
Next, we tested whether the highly conserved C‐terminal GG motif of UBLs is required for the conjugation reaction. We replaced the terminal Gly99 of ScUrm1 with all other canonical amino acids and produced the thiocarboxylated versions via the intein system. Strikingly, the presence of nearly any amino acid at the C‐terminus permitted the conjugation reaction, as long as the protein remained thiocarboxylated (Fig 6C). ScUrm1G99C was not thiocarboxylated by the intein system, since it undergoes autocleavage before the final cleavage and elution step could be induced with ammonium sulfide. We also observed reduced conjugation efficiencies for ScUrm1G99E and ScUrm1G99H for reasons that remain unclear. When removing both C‐terminal glycine residues (ScUrm1HΔGG), histidine (His97) is located at the C‐terminus, which also led to strongly reduced conjugation efficiency. However, when histidine is replaced by alanine (ScUrm1H97AΔGG), or when both glycine were mutated to alanine or serine (ScUrm1G98A,G99A, ScUrm1G98S,G99S), Urm1 did conjugate even without the characteristic GG‐motif. In summary, our data show that Urm1 variants carrying any thiocarboxylated amino acid at their C‐termini can undergo a covalent attachment to defined target proteins in vitro. Next, we tested whether the attachment requires the typical β‐grasp fold of UBLs or whether an unrelated thiocarboxylated model protein could be attached to Urm1 targets in vitro. We generated thiocarboxylated versions of GFP (GFP‐SH) and a fusion protein between GFP and ScUrm1 (GFP‐Urm1‐SH). Both artificially thiocarboxylated proteins can be conjugated to ScAhp1C31S or ScPyk1 (Fig EV3A). As GFP‐SH contains a C‐terminal lysine residue, we verified the formation of a conjugated XK‐K peptide (Fig EV3B) by mass spectrometry.
Figure EV3. GFP‐Urm1 for tracing persulfidation in vivo .
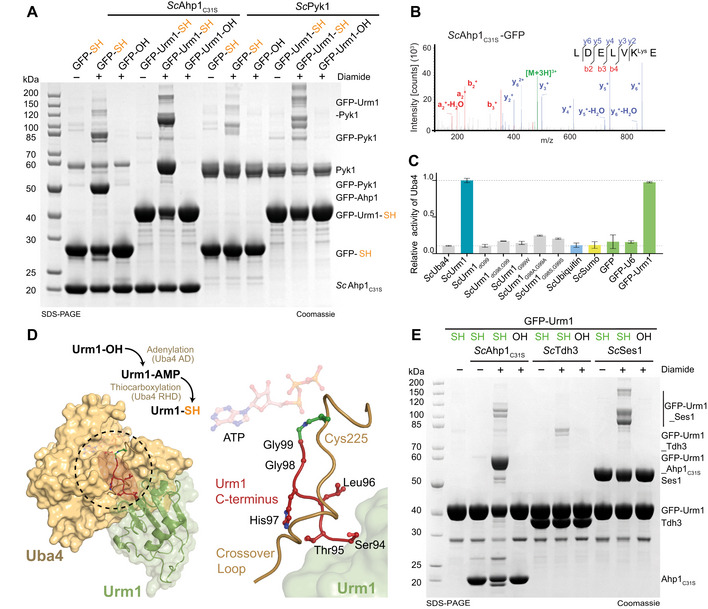
-
AAnalysis of in vitro conjugation between ScAhp1C31S or ScPyk1 and various thiocarboxylated variants of GFP in the presence of TBH. Unconjugated target proteins and the formed conjugates are indicated on the right.
-
BMS/MS Spectra of GFP conjugated to ScAhp1C31S via lysine. A representative annotated fragmentation spectrum is shown with the b‐ and y‐ions marked in red and blue, respectively. The peptide sequence is shown at the top with the collision‐induced fragmentation pattern. The GFP conjugation site was identified by the detection of conjugated lysine residues generated by chymotryptic digestion.
-
CATP hydrolysis by ScUba4 WT in the presence of ScUrm1 variants, ScUbiquitin, ScSumo, and GFP variants. Data are representative of three independent experiments (n > 3; technical replicates). The error bars represent the calculated standard error of the mean (SEM).
-
D(Left) Crystal structure of Uba4 in complex with Urm1 (PDB ID 6YUC). The C‐terminus of Urm1 highlighted in red is conjugated to the catalytic Uba4C202K that is a mimetic of the thioester bond usually formed. (Right) Structural close‐up view showing the Uba4 active site with the residues of the Urm1 C‐terminus (red), the Uba4 the crossover loop (brown), the thioester intermediate (green), and the ATP molecule (transparent) in the nucleotide‐binding site.
-
EAnalysis of in vitro conjugation of ScAhp1C31S, ScTdh3, and ScSes1 by carboxylated GFP‐Urm1 or Uba4‐thiocarboxylated GFP‐Urm1 in the presence or absence of diamide. Unconjugated proteins and the formed conjugates are indicated on the right.
The results of our in vitro studies suggest that the highly conserved C‐terminus of Urm1 is functionally not required for conjugation. However, it is important to highlight that by using the intein system to produce thiocarboxylated proteins, we can bypass the naturally occurring reaction catalyzed by the ATP‐dependent E1 activating enzyme Uba4. Our recent work showed that the C‐terminus of Urm1 is positioned deep inside the active site of Uba4 and that it most likely remains buried throughout the entire process of thiocarboxylation (Fig EV3D; Pabis et al, 2020). We performed an ATP hydrolysis assay to test whether different Urm1 variants and model proteins are able to activate Uba4 in vitro (Fig EV3C). Strikingly, only Urm1 and the GFP‐Urm1 fusion protein induced the ATPase activity of Uba4. Any variation in the C‐terminus of Urm1 (ScUrm1HGΔG, ScUrm1HΔGG, ScUrm1HG99W, ScUrm1HAA, ScUrm1HSS) failed to activate Uba4 (Fig EV3C). Furthermore, the use of ubiquitin, SUMO, GFP, or a GFP variant carrying the six most C‐terminal residues of Urm1 (GFP‐U6) did not induce a detectable Uba4 response (Fig EV3C). Finally, we performed Uba4‐mediated thiocarboxylation of GFP‐Urm1 in vitro and subsequently confirmed its direct conjugation to ScAhp1, ScTdh3, and ScSes1 (Fig EV3E).
In summary, our findings highlight the broad reactivity of various thiocarboxylated proteins with Urm1 target proteins in vitro under mild oxidative stress conditions. Even if all other UBLs are highly similar and carry the same C‐terminal GlyGly‐motif, the high specificity, and selectivity between Urm1 and Uba4 strongly suggest that Uba4 exclusively thiocarboxylates Urm1 in vivo, thereby ensuring that Urm1 is unique in its ability to covalently attach to target proteins independent of E2 or E3 enzymes and simultaneously catalyze persulfidation of its target.
Sulfur transfer by the Uba4‐Urm1 pathway to proteins is not linked to the H2S trans‐sulfuration pathway
Hitherto, the gaseous H2S molecule was thought to constitute the major cellular response signal to initiate the protection of cysteines by persulfidation upon oxidative stress (Kimura & Kimura, 2004; Mustafa et al, 2009; Zivanovic et al, 2019). Cys3, Cys4, and Tum1 are key components of the trans‐sulfuration pathway in yeast and were shown to regulate intracellular H2S levels. After obtaining experimental evidence for the existence of Urm1‐SH mediated persulfidation of cysteine residues in target proteins, we asked whether the previously known pathway is directly or indirectly linked to Urm1 and its role in tRNA and protein thiolation. We generated a panel of yeast strains carrying individual deletions of trans‐sulfuration or urmylation pathway components (ahp1Δ, urm1Δ, tum1Δ, ncs6Δ, cys3Δ, and cys4Δ) or double deletions of a trans‐sulfuration pathway component and URM1 (cys3Δurm1Δ and cys4Δurm1Δ). We found that only the deletion of AHP1 leads to a measurable growth defect in response to the presence of TBH (Fig 7A). The deletion of URM1 did not affect this response, confirming that Urm1 conjugation is not essential for the function of Ahp1 (Van der Veen et al, 2011; Brachmann et al, 2020). We also observed a modest growth defect for cys3Δ and cys4Δ mutants, in contrast to deletions of URM1 or known components of the tRNA thiolation pathway, which grew normally. To test whether the trans‐sulfuration pathway is linked to Urm1 activity and whether the Uba4/Urm1 pathway regulates H2S levels independently, we analyzed the generation of H2S in the different deletion strains. As expected, the cys3Δ and cys4Δ strains displayed dramatically altered H2S levels. However, the deletion of URM1, TUM1, or NCS6 had no detectable impact on intracellular H2S levels (Fig 7B). The cys3Δurm1Δ and cys4Δurm1Δ double mutants display phenotypes similar to cys3Δ and cys4Δ strains alone. Our data thus indicate that Urm1 does not regulate H2S directly or via Cys4/Cys3. Next, we analyzed in vivo conjugation rates of Urm1 to Ahp1 and tRNA thiolation levels in the various deletion strains. Importantly, loss of Tum1, but not Ncs6, strongly reduces Urm1 conjugation to Ahp1 in vivo (Fig 7C). Therefore, Urm1 conjugation and protein persulfidation do not require tRNA thiolation. Consistent with previous studies, the elimination of components of the tRNA thiolation cascade, like Urm1 or Ncs6, completely abolishes the formation of mcm5s2U34 in tRNAs from these strains (Fig 7D). By contrast, the inactivation of Cys3 or Cys4 leads to a reduction of tRNA modification levels, while the AHP1 knockout has no effect. The reduction in tRNA thiolation levels could be affected by the availability of intracellular sulfur sources that are regulated by Cys4 and Cys3 in the cysteine biosynthesis pathway. Nonetheless, the inactivation of Cys3 or Cys4 does not affect the Urm1 conjugation levels of Ahp1, indicating that Urm1‐SH is formed at similar levels. Foremost, we show that persulfidation of cysteines by Urm1 is independent of the previously established trans‐sulfuration pathway. Finally, our results indicate that protein conjugation and cysteine persulfidation by Urm1 are also independent of its role in tRNA thiolation (Fig 7E). Altogether, we reveal an unexpected function of Urm1, which appears to act as a universal SCP for the regulation of amino acid persulfidation and tRNA base thiolation.
Figure 7. Trans‐sulfuration and Uba4‐Urm1 are independent pathways for protein thiolation.
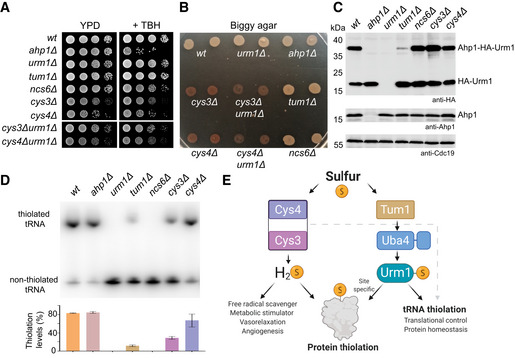
-
ATBH cytotoxicity assay in vivo. Basic growth and the synthetic effect in wild‐type ahp1Δ, urm1Δ, tum1Δ, ncs6Δ, cys3Δ, cys4Δ, cys3Δ urm1Δ, and cys4Δ urm1Δ strains was monitored without or with 1 mM TBH.
-
BH2S production using Biggy agar plates—Wild‐type, ahp1Δ, urm1Δ, tum1Δ, ncs6Δ, cys3Δ, cys4Δ, cys3Δ urm1Δ, and cys4Δ urm1Δ strains were grown on Biggy medium, and H2S production was recorded as indicated in the material and methods section.
-
CIn vivo Ahp1 conjugation analyses with protein extracts obtained from the indicated yeast strains expressing HA‐URM1. Unconjugated Urm1 and Urm1‐Ahp1 conjugates were detected by anti‐HA (top panels) Western blots. Anti‐Ahp1 blots (middle) detect unmodified Ahp1. Anti‐Cdc19 (bottom panels) served as a loading control.
-
DNorthern blot analysis for thiolation levels of in the genetic background of the indicated yeast strains. Total tRNA was resolved on denaturing PAGE supplemented with APM to retard the migration of thiolated . APM: ([N‐Acryloyl‐amino] phenyl) mercuric chloride. (bottom) Quantified thiolation levels from three independent biological replicates.
-
EScheme of sulfur source for thiolation. Sulfur released as H2S from trans‐sulfuration enzymes Cys4 and Cys3 is used for protein persulfidation, scavenging free radicals metabolic signaling/stimulation. The sulfur from the Uba4‐Urm1 pathway is directed to tRNA thiolation and to the persulfidation of specific cysteine of target proteins.
Discussion
The key importance of post‐translational modification by UBLs for molecular processes and cellular functions is undisputed. Numerous novel UBLs and noncanonical mechanisms of ubiquitination have recently been discovered (Bhogaraju et al, 2016, 2019; Qiu et al, 2016; Kalayil et al, 2018). While some bacterial and archaeal SCPs were shown to be conjugated to substrate proteins in vivo (Shigi, 2012; Hepowit et al, 2016; Xu et al, 2019), the mechanisms for conjugation remained unknown. Our results reveal a novel and surprising link between ubiquitin‐like conjugation of Urm1, a highly conserved eukaryotic SCP, to target proteins and post‐translational persulfidation of specific cysteine residues in these targets.
Several molecular characteristics indicate that the Urm1‐Uba4 system may represent an ancient precursor of ubiquitin‐like conjugation reactions in eukaryotes (Iyer et al, 2006; Burroughs et al, 2009; Schulman & Harper, 2009; Eme et al, 2017; Imachi et al, 2020). Urm1, with its thiocarboxylated C‐terminus and its bifunctional role as SCP and UBL, takes a unique position amongst all members of the eukaryotic UBL protein family. Our work establishes that Urm1‐SH is essential to catalyze the persulfidation of cysteines, while the lysine/serine/threonine‐directed attachment of UBLs in eukaryotes may have evolved as a side reaction. For instance, we found that the covalent attachment of Urm1 depends on the sulfur group that is simultaneously deposited at a redox‐active cysteine residue during the conjugation, highlighting the central role of the sulfur transfer during a highly coordinated reaction. Furthermore, the target motif only requires the modifiable cysteine residue and its preceding oxidation without the need for E2 enzymes or E3 ligases to direct Urm1 towards attachment sites and convey specificity. The observed high reactivity and broad specificity of the thiocarboxyl group might thus have been selected against the evolution of Urm1‐specific E2 and E3 enzymes and possibly, promoted the loss of the thiocarboxyl group in other UBLs. The identified Urm1 conjugation sites appear weakly conserved between homologs from different organisms (e.g., surface lysines in ScAhp1 versus CtAhp1) and removing all lysine residues still permits the sulfur transfer. Intriguingly, we did not detect functional consequences of deleting various lysine residues in Ahp1 in vivo, suggesting that the conjugation of Urm1 after the transfer of sulfur appears functionally dispensable. If this holds true, it would provide an optimal framework for the evolution of more complex E2/E3‐based conjugation systems around the formed GG‐K/GG‐S/GG‐T bond, potentially leading to functional specialization of the array of UBLs we see across eukaryotes today.
Furthermore, it is very likely that most of the Urm1 conjugates are removed by one or more unidentified Urm1‐specific deconjugating/deubiquitinating enzyme(s) (DUBs) that can be blocked by NEM. Therefore, the attachment of Urm1 might be transient and the cysteine persulfidation of the target protein would outlast the actual modification by attachment of a Urm1 molecule. The existence of a potential “deurmylase” might have led to a strong underestimation of the number of cellular targets and caused the inconsistencies between Urm1 target lists from various studies and organisms (Schlieker et al, 2008; Van der Veen et al, 2011; Khoshnood et al, 2016; Wang et al, 2019). As Urm1 can also attach to serines and threonines, the recently described class of ubiquitin esterases (de Cesare et al, 2021) could be highly relevant for the efficient removal of Urm1 from its target proteins in vivo. We suggest that the overall number of Urm1 targets and the comparison with the known list of persulfidated proteins need to be reassessed after experimentally eliminating the possibility of continuous removal of Urm1 from its targets by known DUBs, esterases, or a specific deurmylase in cells.
Urm1 conjugation is induced by oxidative stress in human cell lines, flies, plants, and yeast (Furukawa et al, 2000; Goehring et al, 2003; Van der Veen et al, 2011; Jüdes et al, 2015, 2016; Khoshnood et al, 2016; Wang et al, 2019). A recent study showed that the homolog of Urm1 in Toxoplasma gondii, an opportunistic parasite, also functions as a protein modifier in oxidative stress response. URM1 deletion strains of T. gondii display reduced proliferation, replication, invasion, and virulence in mice. Hence, TgURM1 plays a pivotal role in T. gondii survival and Urm1 could represent a novel target for the treatment of toxoplasmosis (Tan et al, 2022). Our data show that the transfer of the unique thiocarboxylated C‐terminal glycine of Urm1 requires the oxidation of catalytic or peroxidatic cysteines. Urm1‐SH condenses on the oxidized cysteine and may form a rather short‐lived acyl disulfide intermediate. Of note, we assume that diamide, which does not represent an oxidative stressor in vitro, is able to catalyze Urm1 conjugation by promoting the formation of the acyl disulfide intermediate. A nearby residue attacks the acyl disulfide, resulting in a persulfidated cysteine and the simultaneous formation of a covalent Urm1 conjugate. Hence, our in vitro observations provide a direct rationale for the dependency on cysteine oxidation. We further show that Urm1 conjugation and persulfidation are mechanistically coupled in all targets that we tested from yeast and human systems, providing substantial evidence that Urm1 is the elusive pathway that enables cysteine persulfidation throughout evolution. Recent studies revealed that motifs surrounding persulfidated cysteines are highly enriched with lysine residues (Longen et al, 2016; Fu et al, 2020). These observations provide further logical support for our conclusions on a proteome‐wide scale and support the concept that Urm1 may be directly responsible for the modification of a significant fraction of cysteines known to be persulfidated in cells. It was shown that cysteinyl‐tRNA synthetase can charge tRNACys with already persulfidated cysteine, which can be incorporated into proteins during ribosomal translation (Akaike et al, 2017). However, Urm1‐mediated persulfidation occurs post‐translationally, allowing for a direct molecular response to conditions of oxidative stress and damage.
Persulfides exhibit strong redox‐scavenging activities and contribute to redox signaling regulation, due to their nucleophilicity. However, the key role of persulfide modifications in proteins is to protect thiols in cysteine residues against irreversible oxidation (Nishida et al, 2012; Kasamatsu et al, 2016; Kimura, 2017). An overoxidized persulfidated cysteine can be reversed to a fully reduced cysteine, whereas an overoxidized cysteine can hardly be reduced under physiological conditions (Kasamatsu et al, 2016; Dóka et al, 2020). Selective persulfidation of cysteine residues constitutes an evolutionarily conserved defense mechanism against oxidative stress that is critical during aging (Zivanovic et al, 2019). The levels of persulfidation decrease throughout the lifetime of an organism, and likely correlate with a decline in the efficiency of the inducible protection mechanism. Furthermore, persulfidation can regulate the activity of proteins, and the modification of the catalytic cysteine (Cys152) in HsGAPDH represents one of the best‐studied examples (Mustafa et al, 2009)—and we confirmed in this study that Urm1 is responsible for Cys152 persulfidation in GAPDH. Our work shows that cysteine persulfidation can be installed post‐translationally by Urm1 in direct response to oxidative stress, providing potential new avenues for the development of Urm1‐based therapeutic strategies to protect the proteome against oxidative stress and cellular aging.
The essential role of Urm1 in tRNA thio‐modification (Termathe & Leidel, 2021) has long outshone its role in protein conjugation. Despite our unexpected findings, it remains undisputed that Urm1‐SH interacts with the Ncs2/Ncs6 thiolase and mediates the final transfer of sulfur to U34 of several tRNAs (Leidel et al, 2009; Yoshida et al, 2015). On the other hand, it remains to be shown whether other SCPs in prokaryotes that carry a thiocarboxylated C‐terminus are also able to catalyze cysteine persulfidation of specific key targets. Taken together, our work illustrates that Urm1‐SH can transfer its sulfur group to cysteine side chains of proteins. Foremost, this process is tightly linked to its E2/E3‐independent protein conjugation unknown among other eukaryotic UBLs.
Materials and Methods
Protein expression and purification
Wild‐type (Appendix Table S1), mutants, and truncated proteins (Appendix Table S2) were expressed in BL21(DE3) pRARE in TB media at 37°C for 6 h and overnight induction with 0.5 M IPTG at 20°C. Bacterial pellets were resuspended in lysis buffer (20 mM Tris–HCl pH 7.5, 200 mM NaCl, 10 mM imidazole, 0.15% TX‐100, 10 mM MgCl2, 1 mM β‐mercaptoethanol, 10 mg/ml DNase, 1 mg/ml lysozyme, 10% glycerol and a cocktail of protease inhibitors) and lysed to homogeneity using a high pressure homogenizer Emulsiflex C3 (Avestin). The proteins were purified with Ni‐NTA agarose (Qiagen) under standard conditions. Tags were cleaved with TEV protease and removed with a second Ni‐NTA purification step. Subsequently, the proteins were purified by size‐exclusion chromatography (SEC) on HiLoad 26/600 Superdex 200 and HiLoad 16/600 Superdex 75 prep grade columns (GE Healthcare) using ÄKTA™ start. Purified proteins were stored at −80°C in a storage buffer (20 mM Tris–HCl pH 7.5, 200 mM NaCl, and 1 mM DTT). Of note, ordering details of used chemicals and reagents are listed in Appendix Table S3.
Thiocarboxylated protein expression and purifications
To obtain thiocarboxylated HsUrm1, ScUrm1 CtUrm1, ScUbiquitin, ScSumo, and GFP, the sequences of the respective proteins were N‐terminally Intein‐CBD‐His6 fused and overexpressed in Escherichia coli and purified according to (Kinsland et al, 1998; Termathe & Leidel, 2018) with modifications. In brief, the bacterial pellet was resuspended in lysis buffer without a reducing agent (30 mM Tris–HCl pH 7.5, 300 mM NaCl, 30 mM imidazole, 0.15% TX‐100, 10 mM MgSO4, 10 mg/ml DNase, 1 mg/ml lysozyme, 10% glycerol and a cocktail of protease inhibitors) and lysed to homogeneity. The lysate was passed through a Ni‐NTA column, and, following washes, the fusion protein was eluted with elution buffer (30 mM Tris–HCl pH 7.5, 300 mM NaCl, 500 mM imidazole, and 10% glycerol). The eluates were dialyzed overnight to chitin‐column buffer (30 mM Tris–HCl pH 7.5 and 500 mM NaCl) and applied on a chitin column. The column was washed with chitin‐column buffer and the cleavage of the tag was induced through incubation with cleavage buffer (30 mM Tris–HCl pH 8.5, 500 mM NaCl, and 35 mM ammonium sulfide or 50 mM DTT) for 16 h at 4°C. This approach enabled us to purify proteins with C‐termini that were carboxylated (‐OH) by using DTT or thiocarboxylated (‐SH) by using ammonium sulfide and without additional residues at the N‐terminus (Kinsland et al, 1998). The eluted proteins were further purified by size‐exclusion chromatography on a HiLoad 16/600 Superdex 75 column on ™KTA™ start system and stored at −80°C in storage buffer (20 mM Tris pH 7.5 and 200 mM NaCl) for further use. The presence of thiocarboxylated C‐terminus was confirmed by running the protein on a polyacrylamide gel containing [(N‐acryloylamino)phenyl]mercuric chloride (APM). The Laemmli sample buffer without any reducing agent was used. For protein visualization, the gels were stained with Coomassie Brilliant Blue.
Thiocarboxylation of Urm1 by Uba4
Uba4‐mediated thiocarboxylation of Urm1 was performed as described (Termathe & Leidel, 2018). Briefly, 20 μM of Urm1 was mixed with 10 μM of Uba4 in the thiocarboxylation buffer (20 mM HEPES pH 8.5, 150 mM NaCl, and 2 mM MgCl2), supplemented with 5 mM ATP, 5 mM TCEP and 180 μM of sodium thiosulfate (Na2S2O3) was added to the reaction mix and incubated for 1 h at 30°C. For Urm1‐SH, samples were desalted using PD SpinTrapTM G‐25 (Cytiva) columns and either loaded on SDS–PAGE gels supplemented with 20 μM of APM to visualize the shift of the thiol group or buffer exchanged into 40 mM ammonium acetate and analyzed by ESI‐MS. Thiocarboxylation of Urm1 was scaled up and Urm1‐SH was purified by using Superdex 200 Increase 10/300 GL (Cytiva) on ÄKTA™ start system. The purified Urm1‐SH was snap frozen and stored at −80°C in storage buffer (20 mM Tris pH 7.5 and 200 mM NaCl) for further use.
Mass determination of thiocarboxylated proteins by mass spectrometry
Carboxylated (‐OH) and thiocarboxylated (‐SH) protein samples were analyzed using micrOTOF‐Q II mass spectrometer (Bruker Daltonics, Germany) equipped with an electrospray ionization source. The instrument was calibrated prior to measurements with ESI‐L Low Concentration Tuning Mix (Agilent Technologies). Samples were desalted on Amicon Ultra‐0.5 3 K (Millipore) using 0.05–0.1% formic acid (FA) as a washing solution. Samples at a protein concentration of about 0.2 mg/ml in formic acid (from 0.0125% up to 1%) and acetonitrile (from 25 to 50%) were directly infused into mass spectrometer with a syringe pump at a flow rate of 6 μl/min. Mass spectrometer was operated in positive mode with a spray voltage of 4,500 V and dry gas temperature of 180°C. MS scans were acquired over a mass range of m/z 500–3,000. The MS spectra were processed with the Maximum Entropy Deconvolution algorithm in Data Analysis 4.1 software (Bruker Daltonics, Germany).
In vitro Urm1 conjugation/urmylation assay
Twenty micromolar of Ahp1 and 10 μM of carboxylated or thiocarboxylated Urm1 were mixed in reaction buffer (20 mM Tris pH 7.5 and 200 mM NaCl). 0.5 mM tert‐Butyl hydroperoxide (TBH) was included and excluded as indicated. The reaction mix was incubated for 30 min at 37°C for Chaetomium thermophilum, and 30°C for Saccharomyces cerevisiae and Homo sapiens proteins. For the confirmation of the isopeptide bond TCEP, DTT and hydroxylamine were added at a final concentration of 5 mM after the initial 30‐min reaction and further incubated for 5 min at 37 or 30°C, respectively. The reactions were stopped by adding Laemmli sample buffer containing DTT and incubated for 5 min at 95°C. Subsequently, the samples were loaded on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). For protein visualization, the gels were stained with Coomassie Brilliant Blue.
In vitro Urm1 conjugation under different oxidative agents
Twenty micromolar of Ahp1 and 10 μM of thiocarboxylated Urm1 were mixed in reaction buffer (20 mM Tris pH 7.5 and 200 mM NaCl). Oxidative agents such as tert‐Butyl hydroperoxide (TBH), hydrogen peroxide (HP), diamide, peroxynitrite, methylglyoxal, and Di‐tert‐butyl disulfide (DTB‐disulfide) at a final concentration of 0.5 mM were included and excluded as indicated. The reaction mix was incubated for 30 min at 37°C for C. thermophilum, and 30°C for S. cerevisiae. The reactions were stopped by adding Laemmli sample buffer containing DTT and incubated for 5 min at 95°C. Subsequently, the samples were loaded on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). For protein visualization, the gels were stained with Coomassie Brilliant Blue.
Stability of Ahp1 and thiocarboxylated Urm1 to oxidative agents
Ten micromolar of Ahp1 or 10 μM of thiocarboxylated Urm1 were added to the reaction buffer (20 mM Tris pH 7.5 and 200 mM NaCl) and oxidative agents such as TBH, HP, diamide, peroxynitrite, methylglyoxal, and DTB‐disulfide at a final concentration of 0.5 mM were included or excluded as indicated. The reaction mix was incubated for 30 min at 30°C. The reactions were stopped by adding Laemmli sample buffer containing DTT for Ahp1 and Laemmli buffer without reducing agents for Urm1‐SH and incubated for 5 min at 95°C. Subsequently, the samples of Ahp1 were loaded on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). Samples of Urm1‐SH were loaded on SDS–PAGE gels supplemented with 20 μM of APM. For protein visualization, the gels were stained with Coomassie Brilliant Blue.
Urm1‐Ahp1 complex formation and purification for crystallization
Large‐scale urmylation of CtAhp1C30S and ScAhp1C31S was carried out for crystallization purposes. Two hundred and fifty micromolar of CtAhp1C30S and 125 μM of CtUrm1C55S‐SH were mixed in 4 ml reaction buffer (20 mM Tris pH 7.5 and 200 mM NaCl) and TBH was added for the final concentration of 0.5 mM. The reaction was carried out for 30 min at 37°C. The reaction mixtures were applied to an ion exchange column HiTrap Q FF GL column. The fractions were collected, and samples were run on SDS–PAGE to locate the fractions containing the CtAhp1C30S‐CtUrm1C55S complex. The fractions containing the complex were pooled and concentrated for further purification. The concentrated sample was loaded into HiLoad 16/600 Superdex 75 pg GL column on ÄKTA™ pure system that was equilibrated with 20 mM Tris pH 7.5, 200 mM NaCl, and 1 mM TCEP. The integrated peak areas corresponding to the complex CtAhp1C30S‐CtUrm1C55S and individual proteins CtAhp1C30S and CtUrm1C55S‐SH were calculated using the UNICORN 7.0 software. Subsequently, the samples from the fractions were loaded on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). For protein visualization, the gels were stained with Coomassie Brilliant Blue. The fractions containing the complex 30 kDa in size were collected and up concentrated to 25 mg/ml and proceeded with crystallization or snap frozen and stored at −80°C for further use. For urmylation of ScAhp1C31S and sample preparation of crystallization for the ScAhp1C31S‐ScUrm1 complex, similar procedures as CtAhp1C30S were used.
Crystallization, data collection, structure determination, and refinement
For crystallization, CtAhp1 and CtAhp1C30S were concentrated to 25 mg/ml in 20 mM Tris pH 7.5, 200 mM NaCl and 1 mM DTT. Crystals of the protein were grown at 21°C by vapor diffusion in sitting drops composed of equal volumes (1 μl each) of protein solution and crystallization buffer (2 M ammonium sulfate and 0.1 M sodium cacodylate pH 6.5). Crystals collected from reservoirs containing 2 M ammonium sulfate and 0.1 M sodium cacodylate‐pH 6.5 were cryoprotected by serial transfer into the cryoprotecting buffer (2 M ammonium sulfate and 0.1 M sodium cacodylate pH 6.5 and 50% glycerol). X‐ray diffraction data for CtAhp1 at 1.75 Å resolution were recorded at the ESRF beamline in Grenoble, France. X‐ray diffraction data for CtAhp1C30S at 1.85 Å resolution were recorded at BESSY in Berlin, Germany. Crystallization of unreacted CtAhp1C30S was carried out by pooling the unconjugated protein from the urmylation reaction and further loading into the HiLoad 16/600 Superdex 75 pg GL column on ÄKTA™ Pure system. The fractions of CtAhp1C30S were pooled and concentrated to 25 mg/ml 20 mM Tris pH 7.5, 200 mM NaCl and 1 mM TCEP. Crystals of the protein were grown at 21°C by vapor diffusion in sitting drops composed of equal volumes (1 μl each) of protein solution and crystallization buffer (2 M ammonium sulfate and 0.1 M sodium cacodylate pH 6.0). Crystals collected from reservoirs containing 2 M ammonium sulfate and 0.1 M sodium cacodylate pH 6.0 were cryoprotected by serial transfer into the cryoprotecting buffer (2 M ammonium sulfate and 0.1 M sodium cacodylate pH 6.0 and 30% glycerol). X‐ray diffraction data for CtAhp1C30S at 1.10 Å resolution were recorded at BESSY in Berlin, Germany. Crystals of CtAhp1C30S‐CtUrm1C55S complex were grown at 21°C using the hanging drop vapor diffusion technique. In each drop, 2 μl of protein sample was mixed with 2 μl of reservoir solution (0.05 M zinc acetate dihydrate, 0.1 M sodium cacodylate pH 6.5, 4% w/v polyethylene glycol 8000, and 30% w/v ethylene glycol). The crystals appeared after 48 h and grew to maximal size in 2 weeks. The crystals were cryoprotected by serial transfer into the cryoprotecting buffer (0.05 M zinc acetate dihydrate, 0.1 M sodium cacodylate pH 6.5, 4% w/v polyethylene glycol 8000, and 50% w/v ethylene glycol) and snap frozen in liquid nitrogen. X‐ray diffraction data at 2.40 Å resolution were collected at BESSY in Berlin, Germany. For data collection details see Table 1. The structures of CtAhp1, CtAhp1C30S and CtAhp1C30S‐CtUrm1C55S complex were determined by molecular replacement using Phaser (McCoy et al, 2007) with S. cerevisiae Ahp1 (PDB ID 4DSR; Lian et al, 2012) and Urm1 (PDB ID 2QJL; Yu & Zhou, 2008), respectively. Structures were refined using Phenix (Adams et al, 2010) and Refmac5 in CCP4 (Winn et al, 2011) programs. The comprehensive validation was done by MolProbity (Davis et al, 2007). Structural visualization was done with PyMOL (https://pymol.org/2/). For structure refinement statistics see Table 1. Anomalous density collection for Zn peak and below Zn peak data was collected for the CtAhp1C30S‐CtUrm1C55S complex at BESSY in Berlin, Germany. A complete list of all used software is listed in (Appendix Table S4).
In vitro Urm1 conjugation of in vivo targets
Twenty micromolar of target proteins such as HsPRDX5, ScTdh3, HsGAPDH, ScSes1, HsSARS1, ScPyk1, HsPKM2 and 20 μM of carboxylated or thiocarboxylated Urm1 were mixed in reaction buffer (20 mM Tris–HCl pH 7.5 and 200 mM NaCl). Diamide at a final concentration of 0.5 mM was included or excluded as indicated. The reaction mix was incubated for 30 min at 30°C. The reactions were stopped by adding Laemmli sample buffer containing DTT and incubated for 5 min at 95°C. Subsequently, the samples were loaded on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). For protein visualization, the gels were stained with Coomassie Brilliant Blue.
Protein identification from gel bands—Sample preparation and LC–MS/MS measurement
Samples were prepared and measured as described (Pabis et al, 2020) with minor changes. The flow rate during peptide separation on the analytical column was 250 nl/min. Moreover, different proteolytic enzymes were used depending on what modification was to be detected. Chymotrypsin was used to find urmylation and GFP conjugation, while trypsin to identify ubiquitination. To detect sumoylation, trypsin, chymotrypsin, and V8 protease were employed.
Protein identification from solution—Sample preparation and LC–MS/MS measurement
Protein samples were prepared in 20 mM Tris and 200 mM NaCl. A sample amount corresponding to about 10 μg of the protein of interest was used for digestion. Final volume of the digestion solution was 60 μl. Urea was added to the concentration of 0.5 M. Digestion solution was filled up to 60 μl with 50 mM ammonium bicarbonate. Chymotrypsin was used in the enzyme‐to‐protein ratio of 1:20. Digestion solution was supplemented with CaCl2 to the concentration of about 10 mM. Samples were incubated at 25°C overnight. The next day, digestion was stopped by adding trifluoroacetic acid to a concentration of 0.5%. Approximately 1% of digested samples were injected for LC–MS/MS analysis, which was performed the same as for samples from gel bands.
LC–MS/MS data analysis for protein identification from gel bands and solution
The LC–MS/MS data were processed using the Proteome Discoverer platform (v.1.4; Thermo Scientific) and searched using an in‐house MASCOT server (v.2.5.1; Matrix Science, London, UK) against cRAP database (https://www.thegpm.org/crap/) supplemented with the sequences of the proteins of interest. The following modifications were included in search parameters depending on the sample: urmylation (HisGlyGly tag after chymotrypsin, ∆ mass = 251.101839); ubiquitination (GlyGly tag after trypsin, ∆ mass = 114.042927); sumoylation (GlyGlyIleGlnGlu tag after trypsin, ∆ mass = 503.246552; GlyGlyIleGln tag after chymotrypsin or V8 protease, ∆ mass = 374.203959); GFP conjugation (Lys tag after chymotrypsin; ∆ mass = 128.094963); cysteine carbamidomethylation (∆ mass = 57.021464); cysteine persulfidation (∆ mass = 31.972071); carbamidomethylated cysteine persulfidation (∆ mass = 88.993534); methionine oxidation (∆ mass = 15.994915).
Thiocarboxylation of Urm1 using L‐cysteine as sulfur source
To produce free sulfide from L‐Cys, 10 μM of E. coli desulfurase IscS was incubated at 25°C overnight with 1 mM L‐Cys and 20 μM PLP cofactor in a desulfurase buffer (20 mM Hepes pH 8.5, 150 mM NaCl, 2 mM MgCl2). To examine the fate of sulfur in the yeast system, 10 μM of ScUba4 and 40 μM of ScUrm1 were added, with 2.5 mM ATP and 1 mM TCEP, and incubated for 1 h at 30°C. Analogously, for Ct., 10 μM of CtUba4 and 40 μM of CtUrm1C55S were added with 2.5 mM ATP and 1 mM TCEP, and incubated for 1 h at 37°C. Following incubation, the samples were desalted using PD SpinTrapTM G‐25 (Cytiva) columns, buffer exchanged into 40 mM ammonium acetate, and analyzed by ESI‐MS as previously described.
Radioactive sulfur transfer assay
To generate thiocarboxylated Urm1 labeled with 35S, we purchased 35S‐containing L‐cysteine (Perkin Elmer, NEG022T001MC) and used it as a sulfur source that simulates one of the plausible sulfur sources in vivo. Approximately 0.1 mCi of 35S L‐cysteine was desulfurated using a 20 μM recombinant IscS desulfurase from E. coli for 1 h at room temperature in a desulfuration buffer containing 20 mM HEPES pH 8.5, 150 mM NaCl, 2 mM MgCl2, and 80 μM pyridoxal phosphate (PLP) necessary for IscS activity. Next, 10 μM Uba4 and 70 μM Urm1 proteins together with 2 mM ATP and 1 mM TCEP were added to the tube containing desulfurated cysteine. The Urm1 thiocarboxylation reaction was executed for 1 h at 30°C. To ensure that most of the 35S atoms were transferred to Urm1, 180 μM of sodium thiosulfate was added to the reaction mix and incubated for 15 min. Samples were desalted using Amersham WB MiniTrap kit (Cytiva) following the manufacturer's protocol to get rid of free cysteine, 35S, and reducing agent. Fresh 35S‐thiocarboxylated Urm1 was used for urmylation reactions in combination with 20 μM corresponding target proteins in presence of 0.5 mM oxidizing agents (TBH or diamide). The urmylation reaction was performed at 30°C for 1 h after which samples were denatured at 95°C in sample buffer without any reducing agents. Proteins were separated using SDS–PAGE, and gels were stained with Coomassie and dried for 2 h in the Gel Dryer Model 583 (BioRad). Dry gels were exposed overnight to storage phosphor screens (Cytiva) and the accumulated signal was visualized using the Personal Molecular Imager system (BioRad). All assays shown herein were repeated at least three times.
Malachite green ATPase assay
To examine the activatory potential of different UBLs on the ATPase activity of Uba4, we used a commercially available Malachite Green Kit (Sigma‐Aldrich). Assembled reactions contained 40 μM Uba4 and 60 μM of the UBL of interest, resuspended in 100 mM MES pH 6.0, 100 mM NaCl, 2 mM MgCl2 and 160 μM ATP. The addition of 5 mU/reaction of inorganic pyrophosphatase (Thermo Fischer Scientific) allowed us to convert all inorganic pyrophosphate crated by Uba4 to phosphate molecules that can be quantified using Malachite Green. The reaction took place for 90 min at 37°C. The reaction product was diluted 20× in water and developed according to the manufacturer's instructions. Absorbance was measured at 620 nm using SpectraMax 190 Microplate Reader (Molecular Devices). Data were acquired in three independent experiments with two technical replicates each.
Thiocarboxylation of GFP‐ScUrm1
ScUba4‐mediated thiocarboxylation of GFP‐ScUrm1 was done as described for ScUrm1. Briefly, 20 μM of GFP‐ScUrm1 was mixed with 10 μM of ScUba4 in the thiocarboxylation buffer (20 mM Hepes pH 8.5, 150 mM NaCl, 2 mM MgCl2), supplemented with 5 mM ATP and 5 mM TCEP and incubated for 1 h at 30°C. The GFP‐ScUrm1‐SH sample was loaded on Superdex 200 Increase 10/300 GL (Cytiva) to isolate GFP‐ScUrm1‐SH.
Conjugation reaction of GFP‐ScUrm1‐SH and target proteins
Conjugation reactions between 20 μM of GFP‐ScUrm1‐SH and 20 μM of target proteins ScAhp1, ScPyk1, and ScSes1 were done in the presence of 0.5 mM diamide for 30 min at 30°C. Carboxylated GFP‐ScUrm1‐OH was used as a negative control. The protein samples were denatured at 95°C in the presence of Laemmli sample buffer and analyzed on Bolt™ 4–12% Bis‐Tris Plus Gels (Thermo Fisher Scientific). For protein visualization, the gels were stained with Coomassie Brilliant Blue.
Generation of yeast strains and plasmid constructions
Saccharomyces cerevisiae strains used and generated in this study (Appendix Table S5) were grown on complete (YPD) or synthetic (SC) media (Sherman, 1991) at 30°C unless otherwise indicated. 80 μg/ml L‐cysteine hydrochloride was supplemented for the growth of auxotrophic strains when required. Yeast gene deletions were generated by PCR using the pUG plasmid system (Gueldener et al, 2002) and gene‐specific oligonucleotides (Appendix Table S6). Correct gene replacements were confirmed by PCR using primer pairs located outside of the target loci (Appendix Table S6). For the construction of yeast expression vectors (Gietz & Akio, 1988) carrying Ahp1 lysine to arginine mutants, we used the FastCloning technique (Li et al, 2011). In brief, the mutated AHP1 gene sequence was amplified from the corresponding pETM‐30 vector (Appendix Table S2) using primer pairs (Appendix Table S6) adding overlapping ends homologous to the linearized pAJ31 target plasmid. All mutations were verified by Sanger‐based DNA sequencing. The yeast expression plasmid pHA‐URM1 (Appendix Table S7) was used as a template to N‐terminally insert a polyhistidine affinity tag (8xHis) by PCR‐based site‐directed mutagenesis (Wang & Malcolm, 1999) with the appropriate oligonucleotide primers (Appendix Table S7) resulting in pLK20 (Appendix Table S7) that was used for in vivo urmylation assays. Transformation of yeast cells with PCR products or plasmids (Appendix Table S7) was performed as previously published (Gietz & Woods, 2002).
In vivo urmylation
In vivo urmylation studies were performed as previously described (Jüdes et al, 2015). In brief, yeast cells grown to an OD600nm of 1.0 were harvested and lysed mechanically with glass beads in a buffer (10 mM K‐HEPES pH 7.0, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM PMSF, and 2 mM benzamidine) containing complete protease inhibitors (Roche) and 2.5 mM N‐ethylmaleimide (NEM). Protein concentrations were determined according to Bradford assay (Bradford, 1976) and lysates were mixed with SDS sample buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 0.002% bromophenol blue, and 5% β‐mercaptoethanol) according to Laemmli (Laemmli, 1970). For Western blot analyses, proteins were transferred to PVDF membranes and incubated with primary anti‐HA antibodies (F7, Santa Cruz Biotechnology or 2–2.214, Invitrogen). Unconjugated Ahp1 was detected using anti‐Ahp1 serum (Iwai et al, 2010) kindly provided by Dr Kuge (Tohoku Pharmaceutical University, Japan). Equal protein loading was verified with anti‐Cdc19 antibodies donated by Dr Thorner (University of California‐Berkeley, USA). Detection of target proteins involved horseradish peroxidase‐conjugated secondary goat anti‐mouse or anti‐rabbit IgGs (Jackson ImmunoResearch) and the WesternBright ECL Spray (Advansta Inc.) according to manufacturers' instructions. For visualization of target proteins, the Odyssey® Fc Imaging System (LI‐COR, Inc.) was used.
Phenotypical analyses and TBH toxicity assay
Overnight yeast cultures were diluted to an OD600nm of 1.0. Ten‐fold serial dilutions were prepared ranging from 10−1 to 10−3 and transferred to YPD medium using a pin tool. For toxicity assays, the medium was supplemented with the organic peroxide TBH as indicated. Plates were incubated at 30°C unless otherwise stated and monitored after 36–48 h.
RNA isolation and northern blot analysis
Total tRNA was extracted from yeast as previously described (Krutyhołowa et al, 2019), using NucleoZOL reagent according to the manufacturers' instructions. The pelleted tRNA was washed once with 75% ethanol and stored in 100% ethanol for subsequent northern blot analysis. 0.4 μg of total tRNA was separated by electrophoresis on 12% denaturing polyacrylamide gels (7 M Urea, 0.5× TBE buffer), visualized by SYBR Gold (Invitrogen), and transferred to a nylon membrane (Immobilon‐Ny+) at 400 mA for 45 min using a Trans‐Blot® SD Semi‐Dry Transfer Cell (BioRad). To analyze the tRNA thiolation levels, the gels were supplemented with [(N‐acryloylamino)phenyl]mercuric chloride (APM) at a final concentration of 60 μg/ml (APM stock solution: 3 μg/μl in formamide). For APM+ gels the transfer was performed for 1 h and the transfer buffer was supplemented with 10 mM DTT to improve the transfer of thiolated tRNA. Membranes were hybridized at 42°C to a 32P‐5′‐end‐labeled DNA probe 5′‐tggctccgatacggggagtcgaac‐3′, which is complementary to a 3′ part of . Labeling of the probe with [γ‐32P]‐ATP, hybridization, and subsequent steps of the Northern blotting procedure were performed as described in (Leidel et al, 2009). The quantitative analysis was performed using a GelAnalyzer system. Thiolation levels (%) were calculated for three biological replicates. Aliquots from the spotting experiment were thawed on ice and total RNA was extracted by using hot phenol/chloroform extraction. One microgram of total RNA was resolved on an 8% PAGE containing 0.5 × TBE, 7 M Urea, and 50 μg/ml APM (Igloi, 1988). Northern blot analysis was performed as described previously by using the probe against (5′‐tggctccgatacggggagtcgaac‐3′; Leidel et al, 2009).
Protein isolation and Western blot analysis
Aliquots (3 OD600 units) from spotting experiments were thawed on ice and total proteins were extracted as previously described (von der Haar, 2007). Total protein extracts were resolved by SDS–PAGE and transferred by semi‐dry blotting onto a PVDF membrane. Membranes were probed using a monoclonal anti‐HA antibody (Covance MMS‐101R).
Qualitative identification of H2S production
The yeast strain used in this experiment was BY4742. The wild‐type strain together with the single knockout strains ahp1Δ, urm1Δ, tum1Δ, ncs6Δ, cys3Δ, and cys4Δ and the double knockout deletion strains cys3Δurm1Δ and cys4Δurm1Δ were maintained and grown on yeast extract‐peptone‐dextrose medium (YPD). For the Bismuth Glucose Glycine Yeast agar (BiGGY) screening, strains were grown overnight in a YPD medium and diluted to an OD600nm = 0.8 and OD600nm = 0.4. Four microliters of each strain were spotted on BiGGY agar plates and incubated for 72 h at 27°C. The production of H2S was evaluated using a color scale dependent on the production of sulfide, the more precipitation of bismuth sulfide, the darker the colony. Taking this into consideration, the scale comprises the following colors: white, cream, light brown, and dark brown (Mezzetti et al, 2014; Cirigliano et al, 2016).
Author contributions
Keerthiraju E Ravichandran: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Lars Kaduhr: Formal analysis; investigation; visualization; methodology; writing – review and editing. Bozena Skupien‐Rabian: Data curation; investigation; visualization; methodology. Ekaterina Shvetsova: Investigation; methodology. Mikołaj Sokołowski: Data curation; formal analysis; investigation; visualization; methodology; writing – review and editing. Ros´cisław Krutyhołowa: Data curation; formal analysis; investigation; visualization; methodology; writing – review and editing. Dominika Kwasna: Investigation; writing – review and editing. cindy Brachmann: Data curation; investigation; methodology. Sean Lin: Data curation; investigation; methodology. Sebastian Guzman Perez: Data curation; formal analysis; validation; investigation; visualization; methodology. Piotr Wilk: Data curation; formal analysis; validation; investigation; methodology. Manuel Kösters: Data curation; investigation. Przemyslaw Grudnik: Data curation; formal analysis; supervision; validation; investigation; visualization; methodology. Urszula Jankowska: Data curation; formal analysis; supervision; validation; investigation; visualization; methodology; writing – review and editing. Sebastian A Leidel: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; writing – original draft; project administration; writing – review and editing. Raffael Schaffrath: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; writing – original draft; project administration; writing – review and editing. Sebastian Glatt: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
KER and SG are inventors on a filed patent application (EP 22461521.1) relating to certain aspects of the presented work in this article.
Supporting information
Appendix
Expanded View Figures PDF
PDF+
Acknowledgements
We thank the members of the Glatt, Schaffrath, and Leidel laboratories for vivid discussion and suggestions. In particular, Martin Termathe, Marta Pabis, and Soumyananda Chakraborti for materials and support during the early stages of the project. In addition, we would like to thank Tobias Dick and Claudio Alfieri for their feedback on the manuscript. We would like to express our gratitude to the support from Stefan Jentsch (MPI Martinsried). We thank Drs Kuge (Tohoku Pharmaceutical University, Japan) and Thorner (University of California, USA) for providing anti‐Ahp1 and anti‐Cdc19 antibodies, respectively, and Dr Ohsumi (Tokyo Institute of Technology, Japan) for the kind gift of the HA‐Urm1 expression plasmid. Furthermore, we thank the beamline staff at beamlines 14.1 (BESSY) Helmholtz‐Zentrum Berlin, ESRF beamline in Grenoble, France, Swiss Light Source (SLS) Paul Scherrer Institut, Switzerland, XDR2 (Elettra Sincrotrone Trieste), and P11 (PETRAIII) for support during data collection and the Heddle, Yamada, Dubin and Kantyka labs at the MCB for the access to specialized equipment. This work was supported by the First Team Grant (FirstTEAM/2016‐1/2; KER, MS, SG) from the Foundation for Polish Science and the OPUS16 grant (2018/31/B/NZ1/03559; RK and SG) from the National Science Centre. This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement No 101001394, KER, MS, and SG). We acknowledge Urm1 project support by Deutsche Forschungsgemeinschaft, Germany, to RS (SCHA750/15‐2) and their SPP1784 program Chemical Biology of Native Nucleic Acid Modifications to RS (SCHA750/20‐2) and SAL (LE 3260/2‐2), the framework of the European Union Cost Action (EPITRAN CA16120) to RS and the Swiss National Science Foundation (310030_184947) to SAL. LK has been awarded a PhD scholarship by the Otto Braun‐Fonds (B. Braun, Melsungen AG, Germany) and is supported by ZFF‐PILOT grant #2620 (University of Kassel, Germany) to RS. In addition, we thank the MCB structural biology core facility (supported by the TEAM TECH CORE FACILITY/2017‐4/6 grant from Foundation for Polish Science) for providing instruments and support. This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 730872. The open‐access publication of this article was funded by the Priority Research Area BioS under the program “Initiative of Excellence—Research University” at the Jagiellonian University in Krakow.
The EMBO Journal (2022) 41: e111318.
Data availability
The atomic coordinates and respective structure factors for CtAhp1 (PDB ID 7Q68; https://www.ebi.ac.uk/pdbe/entry/pdb/7q68), CtAhp1C30S (PDB ID 7Q69; https://www.ebi.ac.uk/pdbe/entry/pdb/7q69), CtAhp1C30S‐Urm1C55S complex (PDB ID 7Q5N; https://www.ebi.ac.uk/pdbe/entry/pdb/7q5n), and unreacted CtAhp1C30S (PDB ID 7Q6A; https://www.ebi.ac.uk/pdbe/entry/pdb/7q6a) have been validated and deposited at the European Protein Data Bank. The mass spectrometry data have been deposited to the MassIVE repository with the dataset identifier MSV000088390 (https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=83d1e72af2da4f4abc609e91c5f92c1a).
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: A comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM, Ihara H, Sawa T, Matsunaga T, Kasamatsu S et al (2017) Cysteinyl‐tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun 8: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE (2011) Ubiquitin‐like protein conjugation and the ubiquiting‐proteasome system as drug targets. Nat Rev Drug Discov 10: 29–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Bonn F, Mukherjee R, Adams M, Pfleiderer MM, Galej WP, Matkovic V, Lopez‐Mosqueda J, Kalayil S, Shin D et al (2019) Inhibition of bacterial ubiquitin ligases by SidJ–calmodulin catalysed glutamylation. Nature 572: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhogaraju S, Kalayil S, Liu Y, Bonn F, Colby T, Matic I, Dikic I (2016) Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 167: 1636–1649 [DOI] [PubMed] [Google Scholar]
- Bock T, Chen W‐H, Ori A, Malik N, Silva‐Martin N, Huerta‐Cepas J, Powell ST, Kastritis PL, Smyshlyaev G, Vonkova I et al (2014) An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res 42: 13525–13533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann C, Kaduhr L, Jüdes A, Ravichandran KE, West JD, Glatt S, Schaffrath R (2020) Redox requirements for ubiquitin‐like urmylation of Ahp1, a 2‐Cys peroxiredoxin from yeast. Redox Biol 30: 101438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72: 248–254 [DOI] [PubMed] [Google Scholar]
- Bruch A, Laguna T, Butter F, Schaffrath R, Klassen R (2020) Misactivation of multiple starvation responses in yeast by loss of tRNA modifications. Nucleic Acids Res 48: 7307–7320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Iyer LM, Aravind L (2009) Natural history of the E1‐like superfamily: Implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 75: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappadocia L, Lima CD (2018) Ubiquitin‐like protein conjugation: Structures, chemistry, and mechanism. Chem Rev 118: 889–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cesare V, Lopez DC, Mabbitt PD, Fletcher AJ, Soetens M, Antico O, Wood NT, Virdee S (2021) Deubiquitinating enzyme amino acid profiling reveals a class of ubiquitin esterases. Proc Natl Acad Sci USA 118: e2006947118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirigliano A, Stirpe A, Menta S, Mori M, Dell'Edera D, Pick E, Negri R, Botta B, Rinaldi T (2016) Yeast as a tool to select inhibitors of the cullin deneddylating enzyme Csn5. J Enzyme Inhib Med Chem 31: 1632–1637 [DOI] [PubMed] [Google Scholar]
- Close P, Bose D, Chariot A, Leidel SA (2018) Dynamic regulation of tRNA modifications in cancer. In Cancer and Noncoding RNAs, Chakrabarti J, Mitra S (eds), pp 163–186. Amsterdam: Elsevier Inc; [Google Scholar]
- Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS et al (2007) MolProbity: All‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dóka IT, Dagnell M, Abiko Y, Luong NC, Balog N, Takata T, Espinosa B, Nishimura A, Cheng Q, Funato Y et al (2020) Control of protein function through oxidation and reduction of persulfidated states. Sci Adv 6: eaax8358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eme L, Spang A, Lombard J, Stairs CW, Ettema TJG (2017) Archaea and the origin of eukaryotes. Nat Rev Microbiol 15: 711–723 [DOI] [PubMed] [Google Scholar]
- Fu L, Liu K, He J, Tian C, Yu X, Yang J (2020) Direct proteomic mapping of cysteine Persulfidation. Antioxid Redox Signal 33: 1061–1076 [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mizushima N, Noda T, Ohsumi Y (2000) A protein conjugation system in yeast with homology to biosynthetic enzyme reaction of prokaryotes. J Biol Chem 275: 7462–7465 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Akio S (1988) New yeast‐Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six‐base pair restriction sites. Gene 74: 527–534 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA (2002) Transformation of yeast by lithium acetate/single‐stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350: 87–96 [DOI] [PubMed] [Google Scholar]
- Goehring AS, Rivers DM, Sprague GF (2003) Attachment of the ubiquitin‐related protein Urm1p to the antioxidant protein Ahp1p. Eukaryot Cell 2: 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre‐mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Walvekar AS, Liang S, Rashida Z, Shah P, Laxman S (2019) A tRNA modification balances carbon and nitrogen metabolism by regulating phosphate homeostasis. Elife 8: e44795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Haar T (2007) Optimized protein extraction for quantitative proteomics of yeasts. PLoS ONE 2: e1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawer H, Hammermeister A, Ravichandran KE, Glatt S, Schaffrath R, Klassen R (2018) Roles of elongator dependent tRNA modification pathways in neurodegeneration and cancer. Genes (Basel) 10: 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepowit NL, de Vera IMS, Cao S, Fu X, Wu Y, Uthandi S, Chavarria NE, Englert M, Su D, Sӧll D et al (2016) Mechanistic insight into protein modification and sulfur mobilization activities of noncanonical E1 and associated ubiquitin‐like proteins of archaea. FEBS J 283: 3567–3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M (2000) Evolution and function of ubiquitin‐like protein‐conjugation systems. Nat Cell Biol 2: E153–E157 [DOI] [PubMed] [Google Scholar]
- Igloi GL (1988) Interaction of tRNAs and of phosphorothioate‐substituted nucleic acids with an organomercurial. Probing the chemical environment of thiolated residues by affinity electrophoresis. Biochemistry 27: 3842–3849 [DOI] [PubMed] [Google Scholar]
- Imachi H, Nobu MK, Nakahara N, Morono Y, Ogawara M, Takaki Y, Takano Y, Uematsu K, Ikuta T, Ito M et al (2020) Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 577: 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai K, Naganuma A, Kuge S (2010) Peroxiredoxin Ahp1 acts as a receptor for alkylhydroperoxides to induce disulfide bond formation in the Cad1 transcription factor. J Biol Chem 285: 10597–10604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer LM, Burroughs AM, Aravind L (2006) The prokaryotic antecedents of the ubiquitin‐signaling system and the early evolution of ubiquitin‐like β‐grasp domains. Genome Biol 7: 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüdes A, Bruch A, Klassen R, Helm M, Schaffrath R (2016) Sulfur transfer and activation by ubiquitin‐like modifier system Uba4•Urm1 link protein urmylation and tRNA thiolation in yeast. Microb Cell 3: 554–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüdes A, Ebert F, Bär C, Thüring KL, Harrer A, Klassen R, Helm M, Stark MJR, Schaffrath R (2015) Urmylation and tRNA thiolation functions of ubiquitin‐like Uba4.Urm1 systems are conserved from yeast to man. FEBS Lett 589: 904–909 [DOI] [PubMed] [Google Scholar]
- Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL (1998) The allosteric regulation of pyruvate kinase by fructose‐1,6‐bisphosphate. Structure 6: 195–210 [DOI] [PubMed] [Google Scholar]
- Kaduhr L, Brachmann C, Ravichandran KE, West JD, Glatt S, Schaffrath R (2021) Urm1, not quite a ubiquitin‐like modifier? Microb Cell 8: 256–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayil S, Bhogaraju S, Bonn F, Shin D, Liu Y, Gan N, Basquin J, Grumati P, Luo ZQ, Dikic I (2018) Insights into catalysis and function of phosphoribosyl‐linked serine ubiquitination. Nature 557: 734–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Ferrell AJ, Chen W, Wang D, Xian M (2018) Cyclic acyl disulfides and acyl selenylsulfides as the precursors for Persulfides (RSSH), selenylsulfides (RSeSH), and hydrogen sulfide (H2S). Org Lett 20: 852–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasamatsu S, Nishimura A, Morita M, Matsunaga T, Hamid HA, Akaike T (2016) Redox signaling regulated by cysteine persulfide and protein polysulfidation. Molecules 21: 6–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler D (2006) Enzymatic activation of sulfur for incorporation into biomolecules in prokaryotes. FEMS Microbiol Rev 30: 825–840 [DOI] [PubMed] [Google Scholar]
- Khoshnood B, Dacklin I, Grabbe C (2016) Urm1: an essential regulator of JNK signaling and oxidative stress in Drosophila melanogaster . Cell Mol Life Sci 73: 1939–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnood B, Dacklin I, Grabbe C (2017) A proteomics approach to identify targets of the ubiquitin‐like molecule Urm1 in Drosophila melanogaster . PLoS ONE 12: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H (2017) Hydrogen sulfide and polysulfide signaling. Antioxid Redox Signal 27: 619–621 [DOI] [PubMed] [Google Scholar]
- Kimura Y, Kimura H (2004) Hydrogen sulfide protects neurons from oxidative stress. FASEB J 18: 1165–1167 [DOI] [PubMed] [Google Scholar]
- Kinsland C, Taylor SV, Kelleher NL, McLafferty FW, Begley TP (1998) Overexpression of recombinant proteins with a C‐terminal thiocarboxylate: implications for protein semisynthesis and thiamin biosynthesis. Protein Sci 7: 1839–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutyhołowa R, Hammermeister A, Zabel R, Abdel‐Fattah W, Reinhardt‐Tews A, Helm M, Stark MJR, Breunig KD, Schaffrath R, Glatt S (2019) Kti12, a PSTK‐like tRNA dependent ATPase essential for tRNA modification by elongator. Nucleic Acids Res 47: 4814–4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- Laxman S, Sutter BM, Wu X, Kumar S, Guo X, Trudgian DC, Mirzaei H, Tu BP (2013) Sulfur amino acids regulate translational capacity and metabolic homeostasis through modulation of tRNA thiolation. Cell 154: 416–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Spector D, Godon C, Labarre J, Toledano MB (1999) A new antioxidant with alkyl hydroperoxide defense properties in yeast. J Biol Chem 274: 4537–4544 [DOI] [PubMed] [Google Scholar]
- Leidel S, Pedrioli PG, Bucher T, Brost R, Costanzo M, Schmidt A, Aebersold R, Boone C, Hofmann K, Peter M (2009) Ubiquitin‐related modifier Urm1 acts as a Sulphur carrier in thiolation of eukaryotic transfer RNA. Nature 458: 228–232 [DOI] [PubMed] [Google Scholar]
- Leimkühler S (2017) Shared function and moonlighting proteins in molybdenum cofactor biosynthesis. Biol Chem 398: 1009–1026 [DOI] [PubMed] [Google Scholar]
- Li C, Wen A, Shen B, Lu J, Huang Y, Chang Y (2011) FastCloning: a highly simplified, purification‐free, sequence‐ and ligation‐independent PCR cloning method. BMC Biotechnol 11: 92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian FM, Jiang YL, Yang W, Yang X (2020) Crystal structure of sulfonic peroxiredoxin Ahp1 in complex with thioredoxin Trx2 mimics a conformational intermediate during the catalytic cycle. Int J Biol Macromol 161: 1055–1060 [DOI] [PubMed] [Google Scholar]
- Lian FM, Yu J, Xiao‐Xiao M, Yu XJ, Chen Y, Zhou CZ (2012) Structural snapshots of yeast alkyl hydroperoxide reductase Ahp1 peroxiredoxin reveal a novel two‐cysteine mechanism of electron transfer to eliminate reactive oxygen species. J Biol Chem 287: 17077–17087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Si Y, Dong SH, Mahanta N, Penkala HN, Nair SK, Mitchell DA (2021) Functional elucidation of TfuA in peptide backbone thioamidation. Nat Chem Biol 17: 585–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Wang H, Liu H, Teng M, Li X (2012) Preliminary crystallographic analysis of glyceraldehyde‐3‐phosphate dehydrogenase 3 from Saccharomyces cerevisiae. Acta Crystallogr Sect F Struct Biol Cryst Commun 68: 978–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longen S, Richter F, Köhler Y, Wittig I, Beck KF, Pfeilschifter J (2016) Quantitative Persulfide site identification (qPerS‐SID) reveals protein targets of H2S releasing donors in mammalian cells. Sci Rep 6: 29808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, IUCr (2007) Phaser crystallographic software. J Appl Cryst 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell GS, Philpott A (2013) Non‐canonical ubiquitylation: mechanisms and consequences. Int J Biochem Cell Biol 45: 1833–1842 [DOI] [PubMed] [Google Scholar]
- Mezzetti F, de Vero L, Giudici P (2014) Evolved Saccharomyces cerevisiae wine strains with enhanced glutathione production obtained by an evolution‐based strategy. FEMS Yeast Res 14: 977–987 [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH (2009) HS signals through protein S‐Sulfhydration. Sci Signal 2: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y, Nakai M, Hayashi H (2008) Thio‐modification of yeast cytosolic tRNA requires a ubiquitin‐related system that resembles bacterial sulfur transfer systems. J Biol Chem 283: 27469–27476 [DOI] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA (2015) Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, Ihara H, Motohashi H, Yamamoto M, Suematsu M, Kurose H et al (2012) Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat Chem Biol 8: 714–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma A, Sakaguchi Y, Suzuki T (2009) Mechanistic characterization of the sulfur‐relay system for eukaryotic 2‐thiouridine biogenesis at tRNA wobble positions. Nucleic Acids Res 37: 1335–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabis M, Termathe M, Ravichandran KE, Kienast SD, Krutyhołowa R, Sokołowski M, Jankowska U, Grudnik P, Leidel SA, Glatt S (2020) Molecular basis for the bifunctional Uba4–Urm1 sulfur‐relay system in tRNA thiolation and ubiquitin‐like conjugation. EMBO J 39: e105087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore LA, Barford D (2004) Getting into position: the catalytic mechanisms of protein of protein ubiquitylation. Biochem J 379: 513–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrioli PGA, Leidel S, Hofmann K (2008) Urm1 at the crossroad of modifications. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep 9: 1196–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Sheedlo MJ, Yu K, Tan Y, Nakayasu ES, Das C, Liu X, Luo ZQ (2016) Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 533: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjan N, Rodnina MV (2016) tRNA wobble modifications and protein homeostasis. Translation (Austin) 4: e1143076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezgui VA, Tyagi K, Ranjan N, Konevega AL, Mittelstaet J, Rodnina MV, Peter M, Pedrioli PG (2013) tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine‐tune protein translation by promoting ribosome A‐site binding. Proc Natl Acad Sci USA 110: 12289–12294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffrath R, Leidel SA (2017) Wobble uridine modifications–a reason to live, a reason to die?! RNA Biol 14: 1209–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlieker CD, Van der Veen AG, Damon JR, Spooner E, Ploegh HL (2008) A functional proteomics approach links the ubiquitin‐related modifier Urm1 to a tRNA modification pathway. Proc Natl Acad Sci USA 105: 18255–18260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman BA, Harper JW (2009) Ubiquitin‐like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat Rev Mol Cell Biol 10: 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21 [DOI] [PubMed] [Google Scholar]
- Shigi N (2012) Posttranslational modification of cellular proteins by a ubiquitin‐like protein in bacteria. J Biol Chem 287: 17568–17577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigi N (2018) Recent advances in our understanding of the biosynthesis of sulfur modifications in tRNAs. Front Microbiol 9: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS (2016) E2 enzymes: More than just middle men. Cell Res 26: 423–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Wang J, Chen J, Liu X, Chen X, Xiao Q, Li J, Li H, Zhao X, Zhang X (2022) Involvement of Urm1, a ubiquitin‐like protein, in the regulation of oxidative stress response of toxoplasma gondii. Microbiol Spectr 10: e02394‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termathe M, Leidel SA (2018) The Uba4 domain interplay is mediated via a thioester that is critical for tRNA thiolation through Urm1 thiocarboxylation. Nucleic Acids Res 46: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Termathe M, Leidel SA (2021) Urm1: a non‐canonical ubl. Biomolecules 11: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivelli X, Krimm I, Ebel C, Verdoucq L, Prouzet‐Mauléon V, Chartier Y, Tsan P, Lauquin G, Meyer Y, Lancelin JM (2003) Characterization of the yeast peroxiredoxin Ahp1 in its reduced active and overoxidized inactive forms using NMR. Biochemistry 42: 14139–14149 [DOI] [PubMed] [Google Scholar]
- Van der Veen AG, Schorpp K, Schlieker C, Buti L, Damon JR, Spooner E, Ploegh HL, Jentsch S (2011) Role of the ubiquitin‐like protein Urm1 as a noncanonical lysine‐directed protein modifier. Proc Natl Acad Sci USA 108: 1763–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Cai X, Xing J, Liu C, Hendy A, Chen XL (2019) URM1‐mediated ubiquitin‐like modification is required for oxidative stress adaptation during infection of the Rice blast fungus. Front Microbiol 10: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Malcolm BA (1999) Two‐stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange(TM) site‐directed mutagenesis. Biotechniques 26: 680–682 [DOI] [PubMed] [Google Scholar]
- Weygand‐Durasevic I, Johnson‐Burke D, Söll D (1987) Cloning and characterization of the gene coding for cytoplasmic seryl‐tRNA synthetase from Saccharomyces cerevisiae. Nucleic Acids Res 15: 1887–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang J, Wang L, Zhou J, Huang H, Wu J, Zhong Y, Shi Y (2006) Solution structure of Urm1 and its implications for the origin of protein modifiers. Proc Natl Acad Sci USA 103: 11625–11630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang T, Niu Y, Liang K, Yang Y (2019) The ubiquitin‐like modification by ThiS and ThiF in Escherichia coli. Int J Biol Macromol 141: 351–357 [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kataoka N, Miyauchi K, Ohe K, Iida K, Yoshida S, Nojima T, Okuno Y, Onogi H, Usui T et al (2015) Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci USA 112: 2764–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhou CZ (2008) Crystal structure of the dimeric Urm1 from the yeast Saccharomyces cerevisiae . Proteins 71: 1050–1055 [DOI] [PubMed] [Google Scholar]
- Zivanovic J, Kouroussis E, Kohl JB, Adhikari B, Bursac B, Schott‐Roux S, Petrovic D, Miljkovic JL, Thomas‐Lopez D, Jung Y et al (2019) Selective Persulfide detection reveals evolutionarily conserved antiaging effects of S‐Sulfhydration. Cell Metab 30: 1152–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]