

Abstract

Essential plasmodial kinases PfGSK3 and PfPK6 are considered novel drug targets to combat rising resistance to traditional antimalarial therapy. Herein, we report the discovery of IKK16 as a dual PfGSK3/PfPK6 inhibitor active against blood stage Pf3D7 parasites. To establish structure–activity relationships for PfPK6 and PfGSK3, 52 analogues were synthesized and assessed for the inhibition of PfGSK3 and PfPK6, with potent inhibitors further assessed for activity against blood and liver stage parasites. This culminated in the discovery of dual PfGSK3/PfPK6 inhibitors 23d (PfGSK3/PfPK6 IC50 = 172/11 nM) and 23e (PfGSK3/PfPK6 IC50 = 97/8 nM) with antiplasmodial activity (23dPf3D7 EC50 = 552 ± 37 nM and 23ePf3D7 EC50 = 1400 ± 13 nM). However, both compounds exhibited significant promiscuity when tested in a panel of human kinase targets. Our results demonstrate that dual PfPK6/PfGSK3 inhibitors with antiplasmodial activity can be identified and can set the stage for further optimization efforts.
Introduction
Malaria is an infectious disease caused by the protozoan parasite known as Plasmodium, which is transmitted to humans through the bite of an infected female Anopheles mosquito. A human host is infected when a mosquito transfers the parasite form, termed sporozoites, into the blood stream during a blood meal. These sporozoites invade the liver where they multiply via asexual reproduction to form parasites capable of erythrocyte invasion, termed merozoites. The merozoites egress from liver cells to reach the blood stream, where they replicate within the red blood cells and rapidly multiply. Following that, a fraction of merozoites will mature into male and female gametocytes, which are ingested by the female mosquito where sexual reproduction results in a new cycle of infection.1 The World Health Organization (WHO) estimates that in 2020, there were 241 million malaria cases in 85 malaria endemic countries.2 Deaths from malaria have been estimated at 627,000 in 2020.2 There are five human-infective species of Plasmodium parasite, with Plasmodium falciparum (Pf) being responsible for the majority of deaths worldwide.3 Encouragingly, the mortality rate of malaria (as measured by deaths per 100,000 population at risk) has been reduced from 25 in the year 2000 to 10 in 2019. This is the result of the global effort to combat malaria where the total funding for malaria control and elimination in 2020 was estimated at $3.3 billion.2 Although much progress has been achieved in attenuating the debilitating effects of malaria, there is still a need to identify novel antimalarial agents due to the risk of the increase of drug-resistant parasites.
Antimalarial drug development studies usually target two stages of the parasite’s life cycle in the human host: (1) the erythrocytic stage, which is responsible for malaria pathogenesis and mortality, and (2) the hepatic stage, which is a clinically silent stage that is obligatory for the erythrocytic stage. Drugs that target the hepatic stage are considered prophylactic treatment. The parasites also have a transmission stage in the mosquito. Targeting this stage can block the disease from spreading.4 The WHO recommends artemisinin combination therapy (ACT) and vector control measures as the primary means to relieve the burden of malaria. However, artemisinin resistance has been detected in some parts of Asia, which has been linked to mutation in the PfKelch13 gene.5 Moreover, a recent study from Rwanda found that P. falciparum samples isolated from patients were resistant to artemisinin in vitro, the first report of resistance in Africa.5,6
Recently, plasmodial kinases have emerged as an attractive target for malaria treatment. Several reviews have highlighted the potential utility of plasmodial kinases as targets for antiplasmodial drug discovery.3,7−9 Recent studies have shown that many kinases of the P. falciparum kinome are indispensable for the survival of the parasite in humans and hence can be prioritized as drug targets.10,11 While the expression of kinases and their importance to viability can vary in the different life stages, several are essential throughout the life cycle.12 Thus, targeting the essential kinases using potent and selective inhibitors may provide a new avenue in the fight against malaria since none of the approved antiplasmodial agents have been shown to target plasmodial kinases. Additionally, if these inhibitors can be developed into drugs, they could complement the current treatment regimen and lower the selection pressure for parasite drug resistance. However, only a few members of the plasmodial kinase family have been pharmacologically validated as targets for antiplasmodial therapy. For instance, in 2017, Paquet et al. reported the discovery of a plasmodial phosphoinositol kinase inhibitor from a phenotypic screen, MMV390048 (Figure 1). MMV390048 targets Pf phosphatidylinositol 4-kinase (PfPI4K) and could block multiple malaria life cycle stages in the human host, which prompted its advancement to clinical validation.13
Figure 1.

Structures of reported Pf kinase inhibitors with antiplasmodial activity.
Kato et al. demonstrated that PfCDPK1 may be involved in regulating parasite motor-dependent processes that take place in the late schizont stage.14 Using an in vitro biochemical screen, they identified several structurally related 2,6,9-trisubstituted purines that were inhibitors of PfCDPK1. From this series, purfalcamine was the most potent inhibitor, showing an IC50 value of 17 nM against PfCDPK1 (Figure 1). Purfalcamine was also active against blood stage parasites, inhibiting the growth of five different strains with EC50 values in the nanomolar range. PfPKG has also emerged as a kinase that could be targeted for antiplasmodial activity. Recent reports described a series of imidazopyridine inhibitors of PfPKG, where the most potent compound of the series, ML10 (Figure 1), inhibited blood stage proliferation with an EC50 of 2.1 nM.15 This inhibitor not only targets merozoite invasion and egress but also prevents transmission of gametocytes to the mosquitos. PfCLK3 has been recently identified as a validated protein kinase drug target for antiplasmodial drug discovery. Studies have shown that PfCLK3 plays a role in the processing of parasite RNA, and thus, its inhibition could result in antiparasitic activity.16 This was confirmed with the identification of the potent PfCLK3 inhibitor TCMDC-135051 (Figure 1), which displayed nanomolar potency against the kinase, as well as showing 180 nM EC50 against asexual blood stage P. falciparum parasites.17,18 Further studies showed that targeting PfCLK3 can be used as a method for transmission blocking, as well as prophylaxis.18
Using an in vitro split luciferase assay,19 which utilizes the proprietary KinaseSeeker technology, Luceome Biotechnologies developed assays against 11 plasmodial kinases, most of which have been shown to be essential for parasite survival.10,20 A pilot screen of these 11 Pf kinases utilizing a 110-member kinase inhibitor library composed of research stage, clinical stage, and marketed inhibitors identified several compounds with sub-micromolar activity against one or more plasmodial kinases (Luceome internal data). IKK16, a 2,4-disubstituted pyrimidine compound, was one of the inhibitors identified from the 110-compound screen (Figure 2). IKK16 inhibited PfGSK3 and PfPK6, two of the 11 plasmodial kinases assayed, with IC50 values of 570 ± 90 and 460 ± 50 nM, respectively.21,22PfGSK3 is a serine/threonine kinase that shares 75/76% sequence similarity with the kinase domains of the human GSK3α/GSK3β. The exact functions of PfGSK3 are still unclear. However, one hypothesis implicates PfGSK3 in regulating the strong circadian rhythm of the parasite.23 Other reports outline the importance of PfGSK3 for parasite invasion into the host erythrocytes.24
Figure 2.
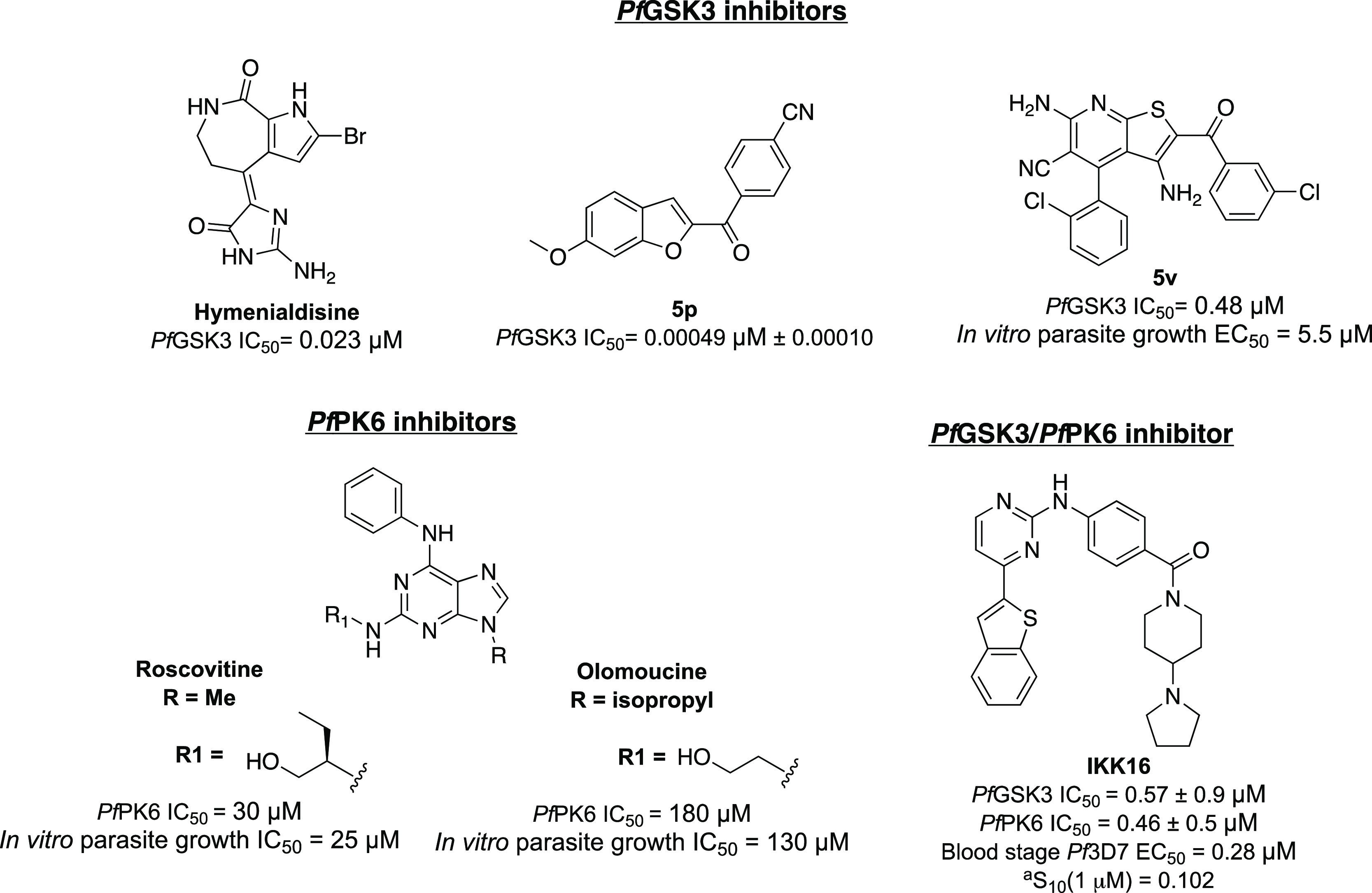
Exemplars of reported PfGSK3 and PfPK6 inhibitors25,27−29 and IKK16, a dual PfGSK3 and PfPK6 ligand identified as the starting point for this work. aSelectivity metric definition: S10 (1 μM) = number of kinases having an experimental value percent of control (PoC) ≤10% at 1 μM divided by the total number of distinct kinases tested. This metric quantifies the percentage of kinases in a panel that a compound binds to at a chosen threshold at a particular screening concentration.
PfPK6 is also a serine/threonine kinase that shares structural similarity with both human CDK2 and p38 mitogen-activated protein kinase (MAPK). It contains 14 of the 15 conserved amino acid residues that are usually associated with the kinase domains of eukaryotes.25PfPK6 is predominantly expressed during trophozoite and schizont stages of the intraerythrocytic cycle.9,25
In addition to the aforementioned findings, independent studies using reverse genetics and saturation mutagenesis have deemed these two kinases essential for P. falciparum asexual blood stage proliferation.10,20 Accordingly, PfGSK3 and PfPK6 may have utility as novel targets for antimalarial pharmacotherapy.
Several inhibitors of PfGSK3 and PfPK6 have been reported in the literature; however, most of them lack the optimum biological profile in terms of enzymatic IC50 or cellular EC50 to be considered as antiplasmodial agents (Figure 2). IKK16, on the other hand, possessed a favorable activity profile, displayed low toxicity in a resazurin-based cell viability assay, and is orally bioavailable in rats.21,22,26 For these reasons, IKK16 appeared attractive as a starting point for antimalarial drug discovery efforts.
Our enthusiasm for IKK16 stems from the fact that it inhibits not one but two plasmodial kinases thought to be essential for the survival of the malaria parasite. To our knowledge, no other compounds have been reported to target both kinases simultaneously with nanomolar potency. This multitargeted inhibition could be advantageous for antimalarial therapy, as it can reduce the likelihood of developing resistance. IKK16 was originally reported as a human IKK inhibitor; however, no data regarding its broad kinome selectivity has been reported to date.21 Accordingly, we decided to test it against a panel of 468 human kinase targets at 1 μM using the DiscoverX KINOMEscan platform. IKK16 showed an S10(1 μM) value of 0.10, hitting 41 kinase targets with >90% inhibition (Figure 2 and Table 7). Although this level of promiscuity against human kinases is not ideal, the literature report of low toxicity encouraged us to proceed to build an understanding of structure–activity relationships (SARs) for PfGSK3 and PfPK6.
Table 7. Evaluation of Kinase Selectivity Scores.
| compound | S10 (1 μM)a | no. of kinase hits |
|---|---|---|
| IKK16 | 0.10 | 41 |
| 9g | 0.08 | 32 |
| 18n | 0.25 | 100 |
| 18r | 0.22 | 88 |
| 23d | 0.26 | 103 |
| 23e | 0.16 | 64 |
Selectivity metric definition: S10 (1 μM) = number of kinases having an experimental value PoC ≤10% at 1 μM divided by the total number of distinct kinases tested. This metric quantifies the percentage of kinases in a panel that a compound binds to at a chosen threshold at a particular screening concentration.
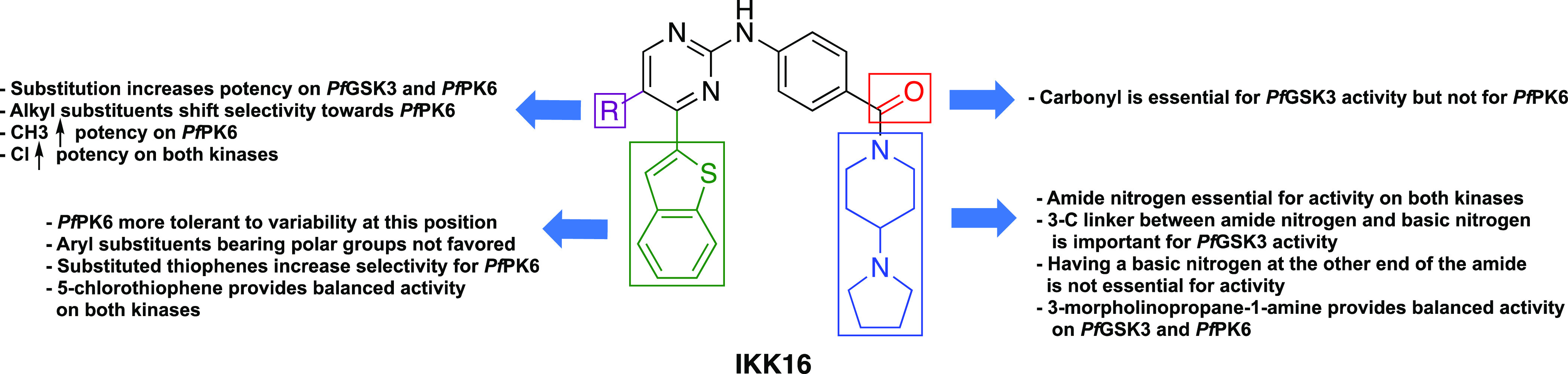
With this in mind, we pursued the 2-anilino-4-arylpyrimidine scaffold of IKK16 hoping to achieve three goals: (1) establish SARs for activity on PfPK6 and PfGSK3, (2) identify potent and selective dual inhibitors of plasmodial PfPK6 and PfGSK3, and (3) screen the most potent kinase inhibitors identified from aim 2 for multistage antiplasmodial activity. The long-term goal of our campaign is to identify compounds that could serve as lead molecules for the future discovery of novel antiplasmodial medicines working through kinase inhibition. Our SAR study and optimization plan are summarized in Figure 3.
Figure 3.
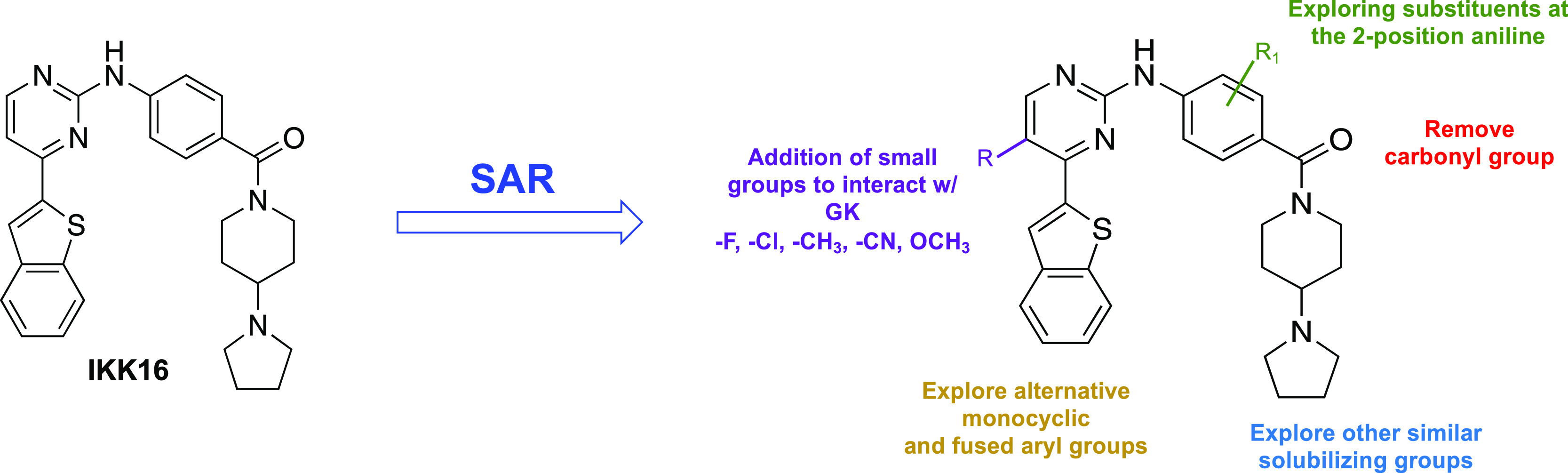
SAR study plan for the PfGSK3/PfPK6 dual inhibitor IKK16. (GK = gatekeeper residue of the kinase).
Results and Discussion
Inhibition of Plasmodial PfGSK3 and PfPK6
The synthesized compounds were tested for on target potency for inhibition of plasmodial PfPK6 and PfGSK3 using the proprietary KinaseSeeker split luciferase assay.19 Compounds were initially screened at 1 μM and only those showing ≥70% inhibition were further tested in a dose–response assay to determine their IC50 values.
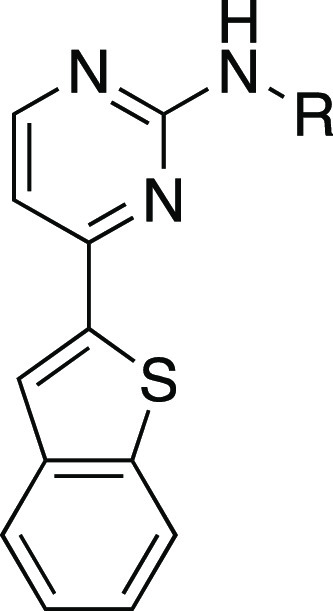
We began our SAR study by analyzing the role of the right-hand side of IKK16 (Table 1). Typically, for aminopyrimidine kinase scaffolds, this 2-amino group makes a key hydrogen bond with a backbone carbonyl in the hinge region of the kinase and the substituents on the phenyl ring project toward the solvent interface.30 Our first set of analogues investigated truncation of the 4-(pyrrolidinyl)piperidine moiety of IKK16 together with the amide group to observe how these alterations would affect activity at PfGSK3 and PfPK6. This modification proved to be detrimental for activity, as compound 4a, bearing only a simple aniline at the 2-position, failed to inhibit both kinases. We also evaluated various substituted anilines as 2-position substituents on the pyrimidine core. These anilines carried electron-donating as well as electron-withdrawing groups (4b–g). However, all these simplified analogues lacked any significant activity on both kinases. The benzylamine analogue 4h was also inactive. Reinstalling the carbonyl group through a methyl ester (5)21 or a carboxylic acid (6)21 resulted in some binding, but the activity did not reach the threshold we set for IC50 determination. Taken together, these results indicated the importance of an amide linker, as is present in IKK16, for activity on PfGSK3 and PfPK6.
Table 1. In Vitro Enzymatic Activity of Simplified Analogues 4a–h, 5, and 6 on PfGSK3 and PfPK6.
| compound | R | PfGSK3 PoC@1 μMa | PfPK6 PoC@1 μM |
|---|---|---|---|
| 4a | Ph | 100 | 100 |
| 4b | 4-Cl–Ph | 89 | 92 |
| 4c | 3-Cl–Ph | 89 | 100 |
| 4d | 2-Cl–Ph | 100 | 100 |
| 4e | 4-OMe–Ph | 97 | 88 |
| 4f | 3-OMe–Ph | 97 | 100 |
| 4g | 2-OMe–Ph | 100 | 100 |
| 4h | CH2–Ph | 100 | 100 |
| 5 | 4-CO2Me–Ph | 71 | 78 |
| 6 | 4-CO2H–Ph | 33 | 100 |
% enzymatic activity remaining after incubation with 1 μM of test compound.
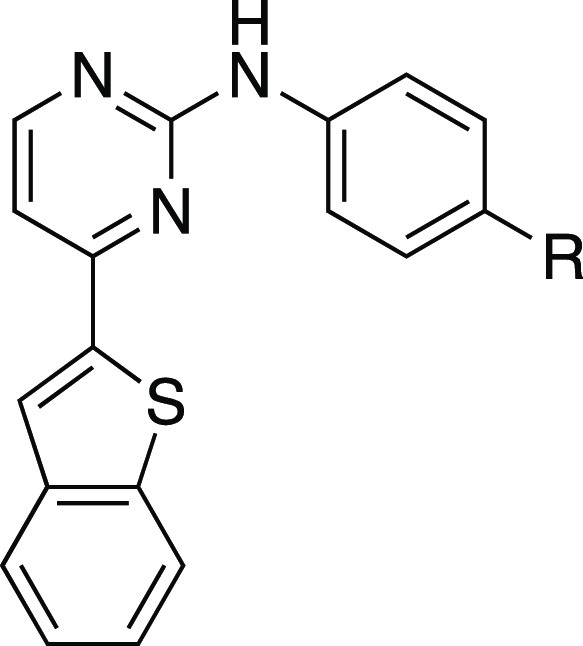
Next, we investigated the SAR around the amide group itself to assess the effect of various modifications on kinase activity (Table 2). We initially focused on the pyrrolidine ring of the 4-(pyrrolidinyl)piperidine side chain. The first modification we carried out was to open the pyrrolidine ring. Compound 8a lost significant activity on PfGSK3; however, it still retained activity at PfPK6 and in fact showed an improvement in potency relative to IKK16 (8aPfPK6 IC50 = 181 ± 4 nM). Other modifications to the pyrrolidine ring included replacing it with (R)-pyrrolidin-3-ol (8b), (S)-pyrrolidin-3-ol (8c), dimethyl amine (9a),21 as well as ring expansion to a six-membered ring (9b). The changes in 8b, 8c, and 9a were all well tolerated by PfPK6, with some improvement in potency and IC50 values ranging from 216 to 274 nM. The same compounds lacked significant activity on PfGSK3. Expanding the pyrrolidine ring to a six-membered piperidine ring (9b) provided roughly a 4-fold potency increase on PfPK6. This ring expansion was somewhat tolerated on PfGSK3, with 9b having a PfGSK3 IC50 of 695 ± 88 nM. These findings suggest that an unaltered pyrrolidinyl ring is important for activity on PfGSK3 since all modifications tried at this site, except for the subtle difference provided by ring expansion, resulted in significant loss of activity on PfGSK3.
Table 2. In Vitro Enzymatic Activity of Amide Analogues of IKK16 on PfGSK3 and PfPK6.

IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean values of two experiments performed in duplicate.
ND: not determined; the compound did not reach the threshold of inhibiting ≥70% kinase activity at 1 μM.
Next, we aimed to substitute the entire pyrrolidinyl–piperidine ring system of IKK16 with simpler basic and non-basic functionalities that may mimic important elements of the original moiety. Substitution with a piperazine ring in 9c saw a loss of activity on PfGSK3, but activity on PfPK6 was retained with an IC50 value of 398 ± 35 nM. Methylation of the piperazine-4-N atom resulted in 9d, which retained activity on PfPK6 but was less potent on PfGSK3. Compound 9e saw placing the amide nitrogen outside of the six-membered ring, and this modification restored activity on PfGSK3 (PfGSK3 IC50 = 308 ± 27 nM). This pointed to the potential importance of maintaining a three-carbon linker between the amide nitrogen and the basic ring nitrogen for PfGSK3 activity. In contrast, no difference in PfPK6 IC50 was observed for 9e compared to the N-methyl piperazine analogue, 9d.
Building upon this finding, we synthesized additional amides with substituents bearing a terminal basic nitrogen while maintaining the three-carbon linker between the amide nitrogen and the basic nitrogen, which was found to be important for PfGSK3 activity. Accordingly, we used 3-(4-methylpiperazin-1-yl)propan-1-amine (9f), 3-morpholinopropan-1-amine (9g), and N1,N1-dimethylpropane-1,3-diamine (9h) as amide substituents. The piperazine analogue 9f demonstrated an almost 3-fold increase in potency on PfPK6 and a 2-fold potency increase on PfGSK3 compared to IKK16 (9fPfPK6 IC50 = 153 ± 12 nM and PfGSK3 IC50 = 226 ± 14 nM). The morpholine analogue 9g exhibited comparable activity to 9f, indicating that the N-methyl of the piperazine ring is not an important element for the observed potency enhancement. The simplified dimethyl analogue 9h was less potent than the morpholine and piperazine analogues and demonstrated an IC50 value of 328 ± 40 nM on PfGSK3, while activity on PfPK6 remained comparable to that of IKK16.
We also explored several non-basic amide substituents. The 3-(methylsulfonyl)propan-1-amine substituent led to a small increase in potency of inhibition on both kinases (9iPfGSK3 IC50 = 223 ± 33 nM and PfPK6 IC50 = 215 ± 21 nM), indicating that the presence of a basic moiety on the solvent-exposed tail was not a strict requirement for binding to PfPK6 or PfGSK3. The cyclopropylamine analogue 9j showed a roughly 2-fold improvement in potency on PfGSK3, while activity on PfPK6 was largely unaffected in comparison to the parent, IKK16. Finally, compound 9k with the 1-methyl-1H-pyrazole substituent showed no activity against either Pf kinase, which implied that aromaticity may not be well tolerated at this position.
Removing the carbonyl functionality from the amide group of IKK16 resulted in the dibasic compound 12. Although this modification abolished activity on PfGSK3, the compound remained active on PfPK6, demonstrating an IC50 value of 173 ± 14 nM, a nearly 3-fold increase in potency from IKK16.
Results from the SAR analysis of the right-hand side of IKK16 demonstrate that several types of structural changes can be tolerated by PfPK6, while PfGSK3 appears to be more sensitive to changes, with several of the analogues losing PfGSK3 activity with subtle structural changes. We also learned that binding can be retained without having an ionizable basic functionality at the distal end of the solvent-exposed tail.
We designed the next set of analogues to explore the 4-position of the pyrimidine ring. In addition to utilizing the moieties of similar size as benzothiophene, we investigated replacement of the benzothiophene of IKK16 with smaller and less lipophilic aryl rings (Table 3). Incorporating the bioisosteric benzofuran to afford compound 18a led to the decrease of activity on PfGSK3, while activity on PfPK6 remained almost the same as IKK16 (18aPfPK6 IC50 = 317 ± 34 nM). A benzothiophene to naphthalene swap (18b) led to a significant loss of activity on PfGSK3, while PfPK6 activity remained comparable to that of the parent, IKK16. One possible explanation for this could be that the sulfur of the benzothiophene is somehow an important element for binding in the PfGSK3 catalytic site while being less important for PfPK6 binding. Next, we synthesized and tested a set of monocyclic and substituted monocyclic aromatic systems in the 4-position of the pyrimidine to probe ligand–kinase interactions at this position. Truncation of the fused phenyl ring of the benzothiophene of IKK16 afforded compound 18c, with an unsubstituted thiophene occupying the 4-position of the pyrimidine. Since this substitution restored a sulfur atom at this position, we were expecting to see some resurgence of PfGSK3 activity. However, this truncation led to inactivity on PfGSK3, but the IC50 on PfPK6 was found to be 236 ± 22 nM, a 2-fold increase in potency from IKK16. This indicated that the fused benzene ring could be essential for PfGSK3 activity. The other 4-position monocyclic aromatic rings included thiazole (18d), furan (18e), 1-methyl-1H-pyrazole (18f), and phenyl (18g). The thiazole analogue demonstrated a comparable activity profile to that of the thiophene counterpart, pointing to the minimal effect of the extra ring nitrogen on activity. Replacing the sulfur atom of the thiophene ring of 18c for an oxygen to afford the furan isostere resulted in more than 3-fold decrease in potency on PfPK6 (18ePfPK6 IC50 = 768 ± 98 nM) while remaining inactive on PfGSK3. The more polar 1-methyl-1H-pyrazole derivative 18f showed comparable activity to 18e on both kinases. Results from 18e and 18f indicate that having polar groups on the aryl moiety of the pyrimidine-4-position may not be favored by PfPK6. This finding was augmented by the observation that the more lipophilic phenyl analogue, 18g, also displayed diminished activity on PfGSK3 but showed a slightly better IC50 of 329 ± 64 nM on PfPK6.
Table 3. In Vitro Enzymatic Activity of 4-Substituted Pyrimidine Analogues on PfGSK3 and PfPK6.

IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean values of two experiments performed in duplicate.
ND: not determined; compound did not reach the threshold of inhibiting ≥70% kinase activity at 1 μM.
We attempted to mimic the benzothiophene by filling the space of the fused phenyl ring utilizing a 3,4-dichlorophenyl substituent. To our delight, compound 18h saw a reinstatement of PfGSK3 inhibitory activity (PfGSK3 IC50 = 698 ± 66 nM), while the compound remained equipotent to 18g on PfPK6.
So far, these results indicated that the benzothiophene was the optimum substituent on the pyrimidine 4-position for activity on PfGSK3, while PfPK6 tolerates some variability, such as thiophene 18c with PfPK6 IC50 = 236 ± 22 nM. Building on this, and starting from 18c, we attempted further structural exploration to restore activity on PfGSK3 and/or improve PfPK6 potency. We began by substituting the 5-position of the thiophene ring of 18c with different lipophilic groups such as a methyl (18i), chloro (18j), and phenyl (18k). While the 5-methylthiophene substituent (18i) resulted in some reinstatement of PfGSK3 activity (IC50 = 1141 ± 249 nM), its IC50 on PfPK6 remained roughly the same as that of the unsubstituted thiophene (18iPfPK6 IC50 = 222 ± 9 nM). Compound 18j, with a 5-chlorothiophene substituent, saw a 2-fold increase in PfGSK3 activity, showing an IC50 value of 550 ± 60 nM. Interestingly, 18j saw some increase in potency against PfPK6 also (18jPfPK6 IC50 = 130 ± 16 nM). The 5-phenylthiophene substituent (18k) afforded a compound that was inactive on PfGSK3 and showed comparable activity to IKK16 on PfPK6.
If this pyrimidine series binds to Pf kinases in the same binding mode as is typically observed for similar pyrimidine kinase inhibitors, then the aryl group at the pyrimidine-4-position is likely directed toward the catalytic lysine of the kinase active site.30 Consequently, we postulated that incorporation of a 4-position functional group capable of forming a hydrogen bond with this lysine residue could lead to better kinase inhibition. Previous studies have shown that such interaction is achievable (Lys208 in PfGSK3).27,28 Accordingly, we synthesized compound 18l with a (5-methylthiophen-2-yl)methanol group at the pyrimidine-4-position. This single analogue proved unsuccessful, as the activity on PfGSK3 remained unchanged compared to the unsubstituted thiophene while also leading to decreased activity on PfPK6. Further derivatives will need to be explored to see if engagement of the active site lysine is a viable strategy for potency enhancement. The trifluoroethyl-ether analogue 18m was obtained as a byproduct in the synthesis of 18l. Fortuitously, this ether analogue showed activity on PfGSK3 comparable to IKK16 (18mPfGSK3 IC50 = 490 ± 34 nM) while showing an ∼2-fold increase in potency on PfPK6 compared to its alcohol counterpart (18mPfPK6 IC50 = 292 ± 22 nM). Taken together, these results establish that a range of hydrophobic groups can be utilized in the 4-position of the pyrimidine core. Further exploration of additional polar functional groups will be needed to identify any polar moieties that can maintain or improve potency.
In conclusion to the SAR analysis performed on the aryl substituent of the pyrimidine-4-position, we have identified the 5-chlorothiophene substituent of 18j as a possible bioisostere of the benzothiophene with equipotent activity on PfGSK3 and with enhanced potency on PfPK6 in comparison to the parent, IKK16. Based on the latter result, 18j was considered a better lead compound, demonstrating enhanced PfPK6 ligand efficiency compared to IKK16 (18j LEPfPK6 = 0.30 and IKK16 LEPfPK6 = 0.26, Table S1).
The final position to be investigated in our SAR study was the 5-position of the pyrimidine (Table 4). Based on the putative binding mode of this pyrimidine inhibitor scaffold, inferred from observing different crystal structures (e.g., PDB: 2P33, 6VNE, and 2JKK), we expect this position to be proximal to the kinase gatekeeper residue. This prompted us to investigate the prospect of engaging the gatekeeper residues of PfGSK3 and PfPK6 (Met157 and Phe99, respectively) through various substituents while observing the effect on binding. We decided to add electron-donating, electron-withdrawing, as well as neutral substituents to this position. IKK16 analogues were synthesized bearing a methyl, cyclopropyl, methoxy, fluoro, chloro, bromo, and nitrile substituents at the 5-position of the pyrimidine core (18n–t, respectively). To our delight, all the synthesized analogues showed increased activity on PfPK6 with IC50 values between 19 and 294 nM. The most potent compounds were 18n, with a methyl substituent, and 18r, with a chloro substituent, showing a 9- and 20-fold increase in potency compared to the parent IKK16, respectively (18nPfPK6 IC50 = 45 ± 2 nM and 18rPfPK6 IC50 = 19 ± 3 nM). These 5-position substituents led to substantial variation in PfGSK3 activity. Whereas all the alkyl-containing substituents displayed low activity on PfGSK3, all the halogenated analogues showed considerable activity. The chloro-substituted 18r and the bromo-substituted 18s analogues were more active than the fluoro analogue 18q, with IC50 values of 174 ± 16 and 165 ± 18 nM for chloro and bromo, respectively, and 456 ± 43 nM for fluoro. One can speculate that this ∼3-fold enhancement arises from a putative halogen bonding interaction between the halogen atoms of these compounds and the sulfur atom of the methionine gatekeeper residue of PfGSK3, where several examples of this type of interaction have been reported in the literature.31−33
Table 4. In Vitro Enzymatic Activity of 5-Substituted Pyrimidine Analogues on PfGSK3 and PfPK6.
| compound | R | PfGSK3 IC50 (nM)a | PfPK6 IC50 (nM) |
|---|---|---|---|
| 18n | CH3 | 710 ± 97 | 45 ± 2 |
| 18o | cyclopropyl | bND | 114 ± 14 |
| 18p | OCH3 | ND | 294 ± 27 |
| 18q | F | 456 ± 43 | 66 ± 2 |
| 18r | Cl | 174 ± 16 | 19 ± 3 |
| 18s | Br | 165 ± 18 | 57 ± 2 |
| 18t | CN | ND | 77 ± 9 |
IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean values of two experiments performed in duplicate.
ND: not determined; compound did not reach the threshold of inhibiting ≥70% kinase activity at 1 μM.
Taken together, SAR analysis of the pyrimidine-5-position indicated that substitution in this region can be productive for modulating kinase activity and that there are differences in how PfGSK3 and PfPK6 respond to these changes. These results suggest that the PfGSK3 catalytic site poorly tolerates alkyl substituents and prefers halogens, especially chloro and bromo. On the other hand, both alkyl-based substituents and halogens enhanced the potency on PfPK6. The findings of our SAR study are summarized in Figure 4.
Figure 4.
Summary of SAR study on IKK16.
Evaluation of Inhibitors’ Binding Mode in PfPK6 and PfGSK3 Catalytic Sites Using Homology Modeling and Docking Studies
To gain insights into the binding mode of the developed series of compounds in PfPK6 and PfGSK3, we performed a molecular docking simulation into their ATP binding site. Starting with PfPK6, and since its crystal structure is not available, we built a homology model of the plasmodial enzyme based on the human cyclin-dependent kinase-2 (CDK2).34 The latter is an extensively studied kinase with several crystal structures in both active and inactive states. PfPK6 shows a high degree of sequence similarity to CDK2, with greatly conserved structural features including their ATP-binding sites (Table S5). The ATP binding site of PfPK6 is relatively narrower due to the presence of two tyrosine residues at its entry, which are not present in CDK2.34 The plasmodial kinase has a U-shaped mostly hydrophobic adenine binding site limited on both sides with polar residues.
We first docked the parent compound IKK16 into the ATP binding site of the PfPK6 model, followed by docking the most potent analogues from Tables 2–4, 9g, 18n, and 18r. All compounds bound in the expected orientation with their 2-anilino-pyrimidine group occupying the putative adenine binding site and making the canonical H-bond interactions with the backbone carbonyl and NH groups of Cys102 in the hinge region (Figure 5A–D). The pyrimidine core is anchored into this orientation by a hydrophobic clamp formed by Ile15 and Phe107. The benzothiophene group extends toward the hydrophobic back pocket and is near the polar residues involved in catalysis, Lys38 and Glu57. Extending from the pyrimidine 2-position of the molecules, the flexible pyrrolidino-piperidine or morpholino-propane chain protrude from the binding site toward the solvent-exposed region, making polar contacts with Asp105 and Asn108. The superior activities of 18n and 18r may be attributable to the presence of the 5-methyl or 5-chloro group on the pyrimidine nucleus, which is able to make favorable hydrophobic contacts with the gatekeeper residue Phe99 (referred to using a red dashed circle in Figure 5D).
Figure 5.

Homology model and docking studies of PfPK6 inhibitors. Proposed binding modes of IKK16 (A), 9g (B), 18n (C), and 18r (D) in the developed homology model of PfPK6. Protein backbone is shown as a gray cartoon and the important residues in the binding site are displayed as gray sticks. Ligands are shown as cyan sticks with important polar contacts displayed as red dashed lines. The hinge region is colored green, and the hydrophobic contact between the 4-chloropyrimidine of 18r and F99 is indicated by a red dashed circle (D).
Then, we turned our attention to studying the molecular interactions of these compounds in the catalytic site of PfGSK3. Again, the crystal structure of this enzyme is not available, so we constructed a homology model based on the human GSK-3β enzyme which shows a great degree of sequence similarity and a conserved ATP-binding site35 (Table S5). The model was built using multiple templates of the human enzyme to obtain a highly accurate sequence alignment. The parent compound, IKK16, was docked into the putative ATP-binding site of the developed model (Figure 6A). The 2-anilino-pyrimidine nucleus of IKK16 docks in the expected orientation of anilino-pyrimidines, forming the critical H-bonds with the backbone of Ile148 in the hinge region. It is also anchored in its place via hydrophobic interactions from both sides with Ala94 and Leu201. The benzothiazole ring fits into the hydrophobic back pocket stabilized by the interaction with Val81. Extending from the pyrimidine 2-position, the pyrrolidine ring extends toward the solvent, approaching Lys154 and Gln198 but does not appear to make significant interactions with these residues.
Figure 6.

Proposed binding modes of IKK16 (A) and 9g (B) in the developed homology model of PfGSK3. Protein backbone is shown as a gray cartoon and the important residues in the binding site are displayed as gray sticks. Ligands are shown as cyan sticks with important polar contacts displayed as red dashed lines.
We followed by docking 9g, which had also shown high activity against PfGSK3, into our homology model (Figure 6B). The 2-anilino-pyrimidine nucleus of 9g docks in the same orientation as that of IKK16, forming the critical H-bonds with the backbone of Ile148 in the hinge region. It is also anchored in its place via hydrophobic interactions with Ala94 and Leu201. Again, the benzothiazole ring fits into the hydrophobic back pocket stabilized by the interaction with Val81. Interestingly, the major difference from IKK16 is that the morpholine ring and its linker extend toward the solvent, making an extensive network of H-bonds with Ile73, Lys154, and Lys157, which appears to contribute to the binding pose stability. These observations could potentially explain the greater activity of this compound compared to the parent molecule IKK16 and warrants further SAR exploration.
Chemistry
The first step in the synthesis of IKK16 analogues involved a Suzuki reaction utilizing 2,4-dichloropyrimidine (1) and benzo[b]thiophen-2-ylboronic acid (2) to afford the aryl halide intermediate 3 in very good yield (79%) (Scheme 1). From 3, various analogues were synthesized using different anilines and amines as nucleophiles in a nucleophilic aromatic substitution (SNAr) reaction with catalytic amounts of 1 M HCl solution to afford the corresponding 2,4-disubstituted pyrimidines 4a–h in low to good yield (8–72%).
Scheme 1. Synthesis of 2-Anilino-4-aryl Pyrimidine Analogues of IKK16.
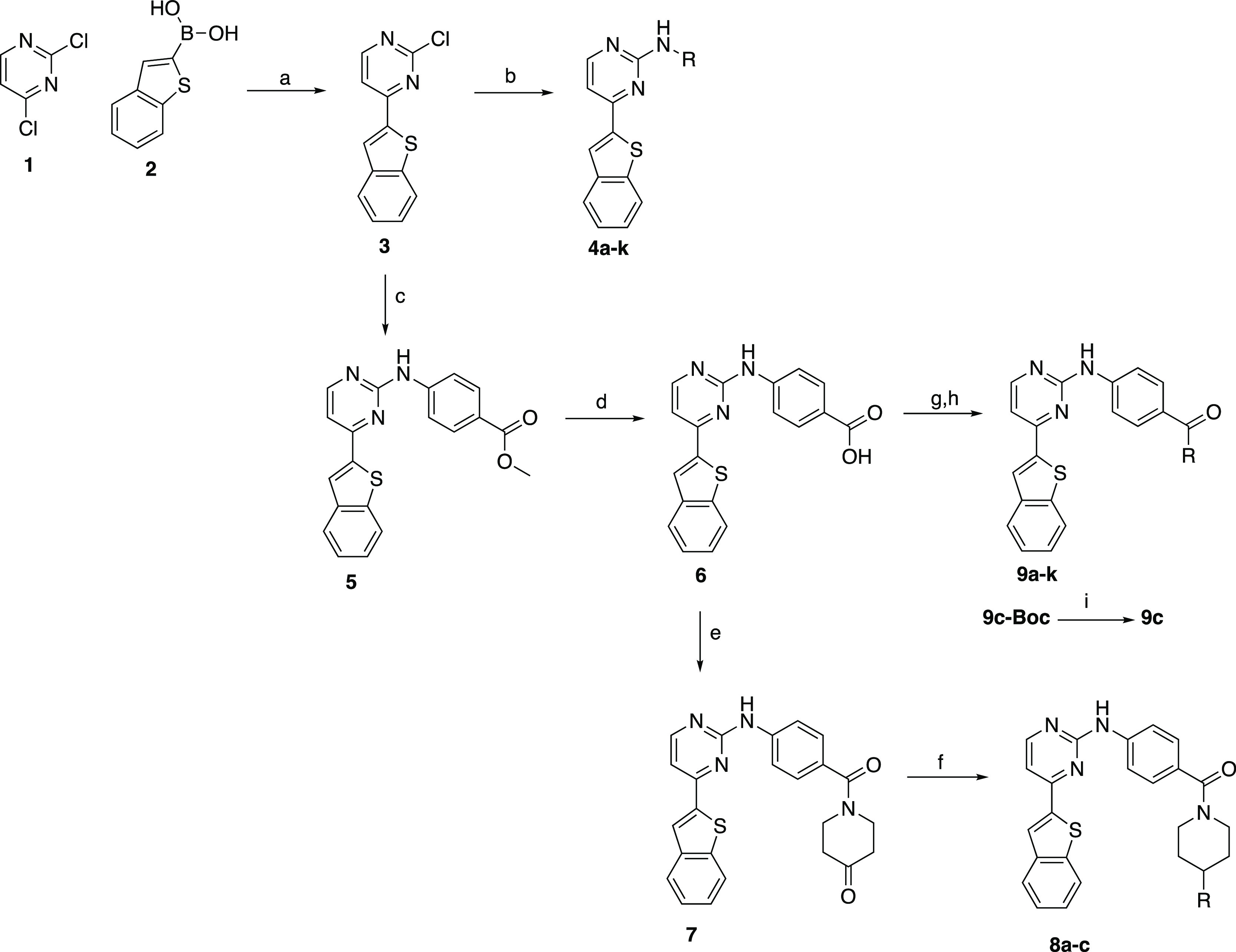
Reagents and conditions: (a) Pd(PPh3)4, Na2CO3, toluene/EtOH/water, 70 °C, and 79%; (b) aniline/amine, 1 N HCl, EtOH, μW, 160 °C, and 8–72%; (c) methyl-4-aminobenzoate, neat, 200 °C, and 40%; (d) 1 M LiOH, MeOH/THF, reflux, and 62%; (e) piperidine-4-one hydrochloride, EDCI, HOBt, DMF, TEA, rt, and 31%; (f) amine, NaBH(OAc)3, THF, rt, and 10–20%; (g) amines, EDCI, HOBt, DMF, TEA, rt (or 80 °C), and 5–58%; (h) HATU, TEA, DMF, 80 °C, and 4%; and (i) 4 M HCl/dioxane, DCM/MeOH, rt, and 42%.
Compounds 8a–c and 9a–k were prepared starting with the aryl halide 3, which was first subjected to neat SNAr conditions using methyl-4-aminobenzoate as the nucleophile at 200 °C to afford the corresponding ester 5 in 40% yield21 (Scheme 1). Saponification of 5 using aqueous LiOH in refluxing MeOH/THF as a solvent generated the carboxylic acid 6 in 62% yield. Carboxylic acid 6 was used as an intermediate to generate amide analogues of IKK16 in which the pyrrolidine ring was modified, or in which the 4-(pyrrolidin-1-yl)piperidine moiety was replaced all together. This was achieved using two different approaches. First, compounds 8a–c were generated by coupling 6 with piperidine-4-one to generate the ketone 7, followed by a reductive amination reaction using the desired amines with NaBH(OAc)3 as the reducing agent (10–20% yield). Compounds 9a–k were obtained via direct coupling of various cyclic, linear, aliphatic, or aromatic amines with the acid 6, utilizing either EDCI/HOBt or HATU as the coupling reagents. The corresponding amides were obtained in low to good yield (5–58%).
Compound 9c, bearing a piperazine ring at the amide position, was generated via deprotection of the Boc-protected precursor 9c-Boc under acidic conditions.
Compound 12, an analogue of IKK16 lacking the amide carbonyl moiety, was also synthesized from the common intermediate 3 (Scheme 2). A Buchwald–Hartwig amination between intermediate 3 and 4-aminophenylmethanol using Pd2(dba)3 and xantphos afforded the corresponding alcohol 10 in 26% yield.36 Afterward, SOCl2-mediated chlorination of the alcohol resulted in the alkyl halide 11, which was carried to the next step without isolation. Finally, addition of 4-(pyrrolidin-1-yl)piperidine to 11 in an SN2 reaction using K2CO3 as the base resulted in the desired diamine 12(37) (13% yield).
Scheme 2. Synthesis of 12.
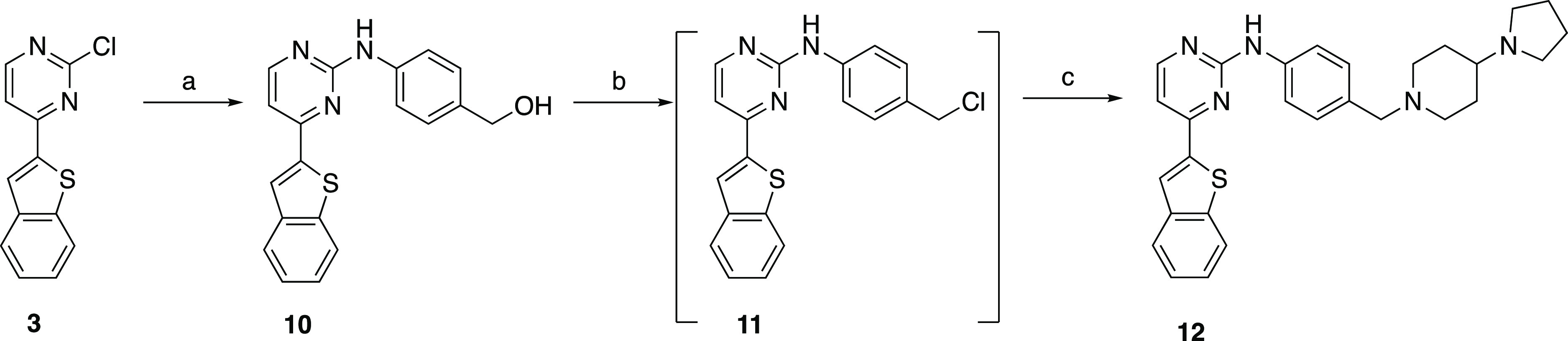
Reagents and conditions: (a) 4-aminophenylmethanol, Pd2(dba)3, xantphos, Cs2CO3, dioxane, 90 °C, and 26%; (b) SOCl2, DIPEA, THF, and rt; (c) 4-(pyrrolidin-1-yl)piperidine, K2CO3, DMF, rt, and 13%.
The next series of compounds were synthesized to probe substitutions at the 4- and 5- positions of the core pyrimidine ring. We employed a convergent synthesis in which pyrimidines 17a–t, bearing the desired substituents, were prepared and then reacted with (4-aminophenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (15) in a microwave-assisted reaction (Scheme 3). First, 4-((tert-butoxycarbonyl)amino)benzoic acid (13) was coupled with 4-(pyrrolidin-1-yl)piperidine using HATU to afford the Boc-protected amide 14 in 80% yield (Scheme 3). Acid-catalyzed deprotection of the Boc group followed by basic work up afforded the aniline 15 in very good yield (67%). The substituted 2-chloropyrimidine intermediates 17a–t were prepared via a Suzuki reaction using either 5-substituted or unsubstituted 2,4-dichloropyrimidines with the appropriate boronic acids to afford the desired products in 8–66% yield. The thiazole intermediate, 17d, was prepared using Stille conditions utilizing 2-(tributylstannyl)thiazole and 2,4-dichloropyrimidine to afford the desired product in low yield (10%). Finally, reacting aniline 15 with 17a–t (except 17l and 17p) using TFA in 2,2,2-trifluoroethanol resulted in analogues 18a–t in 8–64% yield.38 For 18l and 18p, aniline 15 was reacted with 17l and 17p under Buchwald–Hartwig conditions using Pd(OAc)2 as the catalyst and rac-BINAP as the ligand to afford the desired analogues in 9–19% yield.
Scheme 3. Convergent Synthesis Leading to 4- and 5-Substituted Pyrimidine Analogues of IKK16.
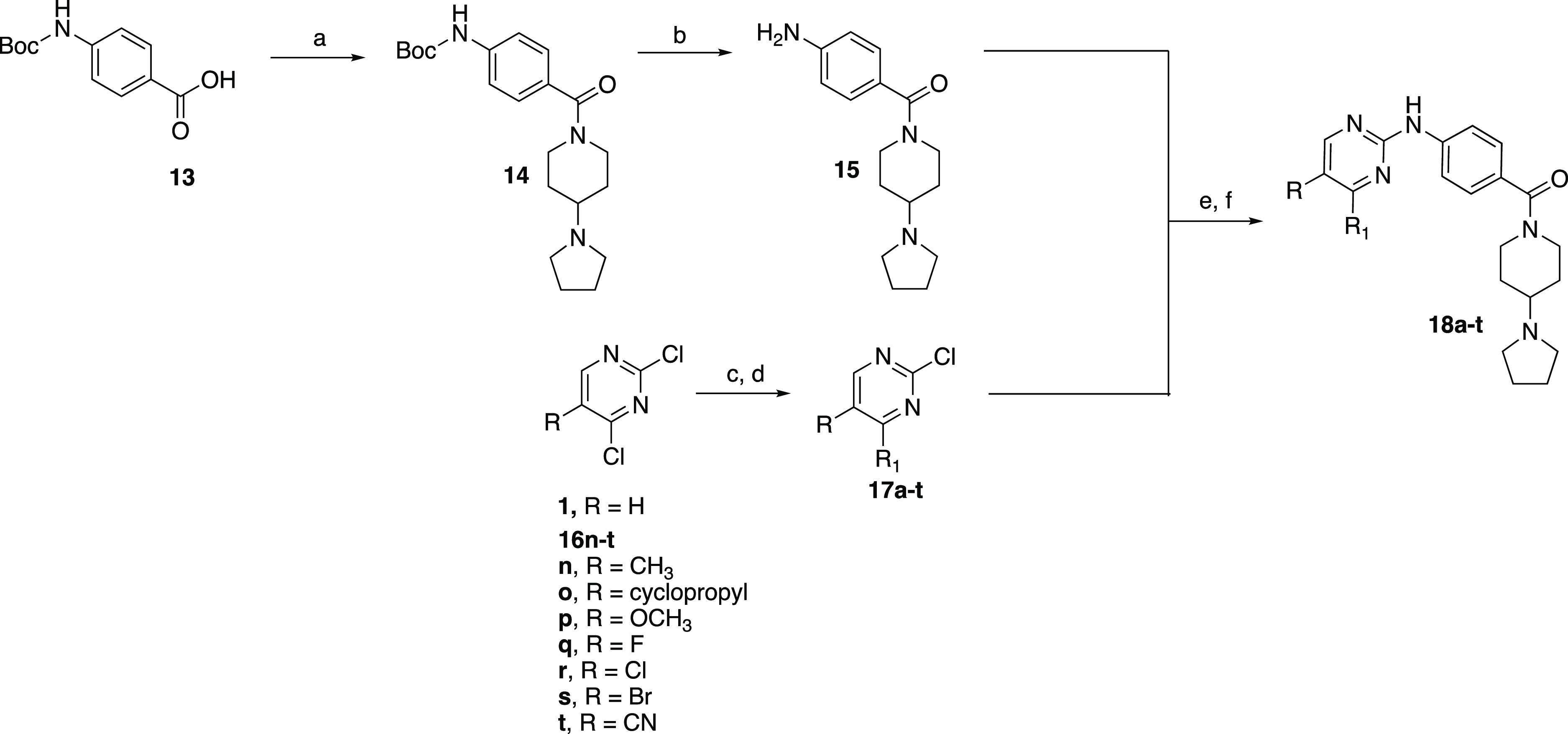
Reagents and conditions: (a) 4-(pyrrolidin-1-yl)piperidine, HATU, TEA, DMF, rt, and 80%; (b) 4 M HCl/dioxane, DCM, rt, and 67%; (c) Pd(PPh3)4, boronic acid, Na2CO3, toluene/EtOH/water (or dioxane/water), 70 °C, and 8–66%; (d) 2-(tributylstannyl)thiazole, Pd(Ph3P)4, LiCl, DMF, 70 °C, and 10%; (e) TFA, 2,2,2-trifluoroethanol, μW, 140 °C, and 8–64%; and (f) Pd(OAc)2, BINAP, Cs2CO3, dioxane, 80 °C, and 9–19%.
Compound 18t, bearing a nitrile group at the 5-position of the pyrimidine, was synthesized in a different manner from the rest of its congeners (Scheme 4). Briefly, the brominated analogue, 18s, was treated with Zn(CN)2 as a cyanide source, Pd(dppf)2Cl2 as a catalyst, and diisopropylethylamine as a base and then heated to 170 °C in DMF under microwave conditions. This procedure afforded 18t in 20% yield.39
Scheme 4. Conversion of 5-Bromo to 5-Nitrile.

Reagents and conditions: (a) Zn(CN)2, Pd(dppf)2Cl2, DIPEA, DMF, 170 °C, μW, and 20%.
Design of Hybrid Analogues
The initial SAR study on IKK16 (Tables 1–4) led to identification of analogues with enhanced enzymatic potency on both PfGSK3 and PfPK6. To drive the enzymatic potency even higher, we envisioned designing a second set of analogues based on combining the structural features of the best performing derivatives from Tables 1–4. Compounds chosen were 9g (Table 2), 18i and 18j (Table 3), and 18n and 18r (Table 4). The hybrid analogues would combine substitutions on the pyrimidine-4-position with those on the 5-position, as well as the amide region, in hope that this would lead to a synergistic effect, resulting in greater inhibition of the target kinases. The synthesized compounds were tested for in vitro enzymatic activity against PfGSK3 and PfPK6. In addition, we calculated the ligand efficiency values of these new hybrid analogues based on their PfPK6 potencies (Tables 5 and S1).
Table 5. In Vitro Enzymatic Activity of Hybrid Analogues on PfGSK3 and PfPK6 with Their Associated Ligand Efficiency Values.

IC50 values were determined using the KinaseSeeker assay with 5-fold dilutions, presented as mean values of two experiments performed in duplicate.
Ligand efficiency was calculated as follows: PfPK6 pIC50/number of heavy atoms in the molecule.
ND: not determined; compound did not reach the threshold of inhibiting ≥70% kinase activity at 1 μM.
We first combined the (5-methylthiopheno) substituent from the 4-position pyrimidine core with the 5-methyl and 5-chloro pyrimidine substituents, keeping the 4-(pyrrolidin-1-yl)piperidine tail from the pyrimidine 2-position as it is, culminating in 23a and 23b, respectively (Table 5). Consistent with previous findings, we observed that a methyl at the 5-position of the pyrimidine increases preference for PfPK6 over PfGSK3. This can be observed in 23a, which demonstrated 28 ± 3 nM IC50 at PfPK6, while activity at PfGSK3 in a screen at 1 μM did not reach our threshold for determining an IC50 value. On the other hand, the chloro analogue 23b was merely 9-fold selective for PfPK6. The activity of 23b on both kinases was comparable to that of most potent analogue from the first set, 18r.
Next, we combined the pyrimidine 5-position methyl and chloro substituents with (5-chlorothiophene) at the 4-position of the pyrimidine forming 23c and 23d (Table 5). Again, the same pattern of selectivity appears. Compound 23c showed micromolar activity at PfGSK3 while showing a 14 ± 1 nM IC50 at PfPK6. Compound 26d, on the other hand, was less selective, showing IC50 values of 172 ± 13 nM at PfGSK3 and 11 ± 1 nM at PfPK6.
Encouraged by these findings, we followed up by making the 3-morpholinopropan-1-amine analogues of 23b–d. We chose to make these because this side chain had shown enhancement of enzymatic activity on both kinases in our first analogue set (Table 2). The resulting new analogues 23e–g demonstrated excellent potency on PfPK6, with 23e displaying an IC50 value of 8 ± 1 nM. As for PfGSK3 activity, 23e and 23f are the first analogues reported in this study to show less than 100 nM IC50 against this kinase. Analogue 23g did not show significant activity on PfGSK3.
In addition to their high potency, all the compounds from Table 5 showed PfPK6 LE values greater than the recommended minimum of 0.3.40 Additionally, 23a–g also exhibited PfPK6 LE values greater than those of their parent congeners, as well as IKK16. This provided an indication (beyond potency alone) that our medicinal chemistry optimization campaign was heading in the appropriate direction.
Synthesis of Hybrid Analogues
The hybrid compounds 23a–g were synthesized using a similar method to the one used for the first set of analogues (Scheme 5). Briefly, commercially available pyrimidines 16n and 16r were subjected to Suzuki conditions with appropriate boronic acids to afford 19a–d in 38–50% yield. Concurrently, 4-nitrobenzoic acid (20) was coupled with 3-morpholinopropan-1-amine using HATU, giving amide 21 in 29% yield. Compound 21 was reduced to the aniline intermediate using Fe and NH4Cl under refluxing conditions. The resulting aniline 22 was carried to the next step without purification. The final analogues were obtained using microwave-assisted reactions, coupling 19a–d with either 15 or 22 to afford 23a–g in low yield (6–15%).
Scheme 5. Synthesis of Hybrid Analogues.
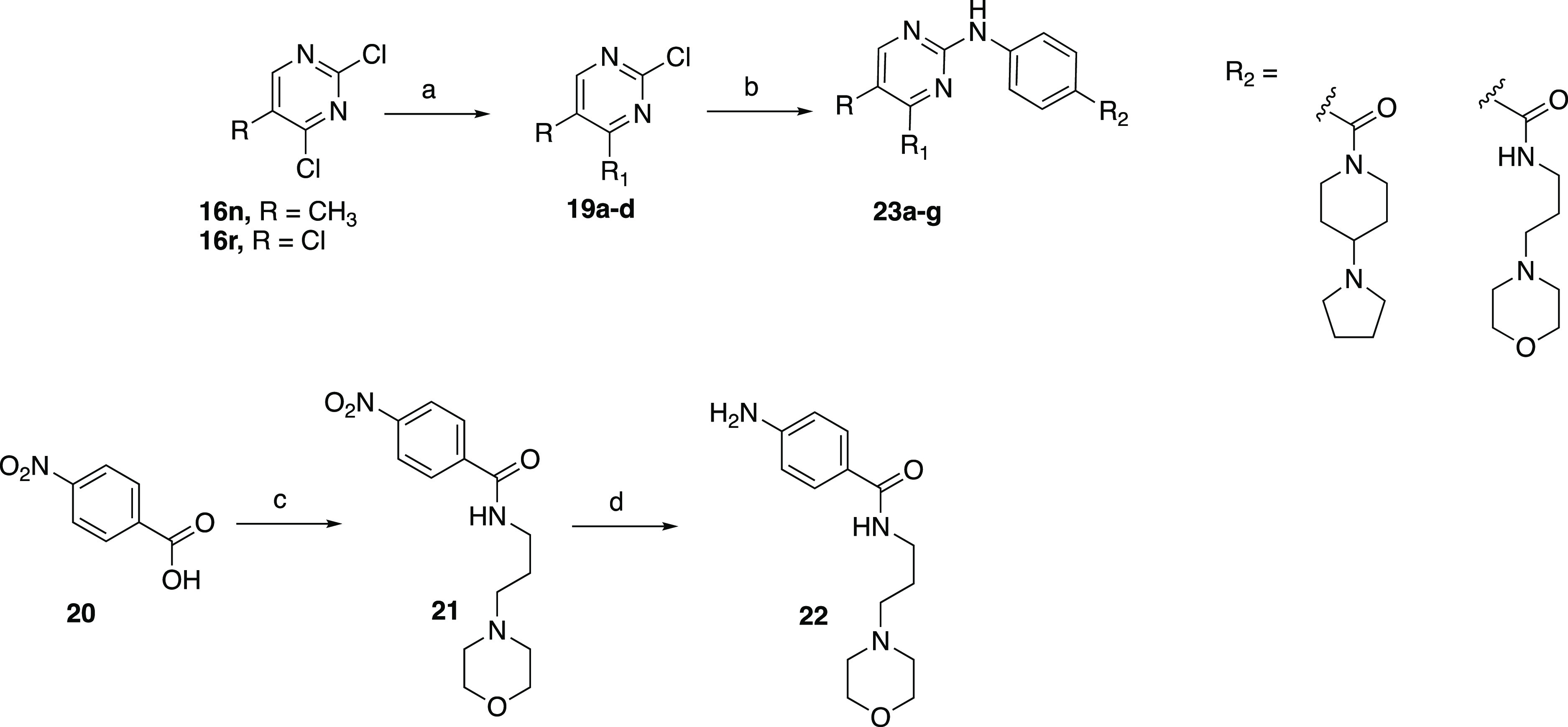
Reagents and conditions: (a) boronic acid, Pd(PPh3)4, Na2CO3, toluene/EtOH/water, 70 °C, and 38–50%; (b) 15 or 22, TFA, 2,2,2-trifluoroethanol, μW, 140 °C, and 6–15%; (c) 3-morpholinopropan-1-amine, HATU, DIPEA, 40 °C, and 29%; and (d) Fe, NH4Cl, EtOH/water, and reflux.
Activity on Blood Stage Parasites
All the synthesized compounds were evaluated for inhibition of P. falciparum 3D7 load in erythrocytes utilizing a SYBR green assay.41 Compounds of interest were initially screened at 1 μM concentration, and only compounds that inhibited >75% parasite load were considered active and subsequently evaluated in a dose–response study to determine their EC50 values (Table 6). Concurrently, we evaluated the analogues for cytotoxicity against HepG2 cells, a human hepatoma cell line, using a commercially available CellTiter-Glo assay (Promega). This assay is a commonly used proxy for evaluating cytotoxicity against human cells and has been previously used to evaluate cytotoxicity among the antimalarial hits from TCAMS.42 The cytotoxicity data is presented as % viability after incubation with 200 nM or 2 μM of the test compounds.
Table 6. Activity of IKK16 Analogues against P. falciparum Blood Stage Proliferation with the Associated Cytotoxicity against HepG2 Cells.
| compound | Pf3D7 EC50 (nM ± SEM)a | % viability@200 nMb | % viability@2 μMb |
|---|---|---|---|
| IKK16 | 280 ± 2 | 110 | 103 |
| 12 | 60 ± 5 | 100 | 97 |
| 18b | 541 ± 29 | 105 | 82 |
| 18m | 810 ± 19 | 110 | 87 |
| 18n | 760 ± 61 | 87 | 66 |
| 18o | 646 ± 10 | 83 | 55 |
| 18q | 607 ± 16 | 98 | 79 |
| 18r | 991 ± 65 | 94 | 61 |
| 18s | 300 ± 9 | 83 | 47 |
| 18t | 322 ± 7 | 95 | 84 |
| 23c | 576 ± 28 | 87 | 65 |
| 23d | 552 ± 37 | 113 | 34 |
| 23e | 1400 ± 13 | 102 | 65 |
| 23f | 2600 ± 35 | 99 | 54 |
EC50 values were determined using the SYBR green I-based assay with 2-fold dilutions, presented as mean values performed in triplicate.
% of viable cells after incubation with 200 nM and 2 μM of the test compounds, presented as an average of two experiments performed in quadruplicate.
Compounds which demonstrated >75% activity against P. falciparum blood stage parasites in the primary screen at 1 μM were 12, 18b, 18m–o, 18q–t, and 23c–f. Follow-up dose–response experiments for these compounds resulted in a wide range of EC50 values ranging from 60 to 2600 nM (Table 6). Compound 12 (EC50 = 60 ± 8 nM) showed the greatest antiparasitic effect, surpassing IKK16 (EC50 = 280 ± 2 nM). Analogues 18s and 18t demonstrated comparable activity to IKK16 despite showing improved kinase inhibition. However, these compounds also showed some cytotoxic effect at 2 μM concentration.
All the screened compounds did not show significant toxicity at 200 nM, with all of them resulting in >80% viability after incubation with the cells (Table 6). However, at 2 μM, we started to observe a trend where analogues which had a 5-position substituent on the pyrimidine ring tend to exhibit greater cytotoxicity (18n–s). This can be exemplified by observing the difference in cytotoxicity between IKK16 and 18n (which only differs by a methyl group at the pyrimidine-5-position), in which 18n shows ∼40% more cytotoxicity than IKK16. Compound 23d showed the greatest cytotoxic effect (34% viable cells) at 2 μM, indicating that it may have a small window between its antiplasmodial EC50 and CC50.
This cellular activity data indicated that the improvement in potency we observed in the in vitro enzymatic assay for these analogues did not necessarily translate into a significant improvement in the antiparasitic activity compared to the reference, IKK16. This could be attributed to multiple factors, such as differences related to kinase activity in an in vitro biochemical assay versus kinase activity in live parasite isolates, differing off-target profiles, as well as differences in physicochemical properties that may hamper access to parasites, and/or intracellular stability.
Given these results, we were interested in assessing some of the physicochemical properties of these compounds and how they compare to the parent IKK16, hoping that this could offer some insight. To do this, we chose our lead kinase inhibitors, 23b–f, and subjected them to a preliminary characterization of their kinetic solubility and effective permeability in the PAMPA assay (Table S2). The compounds showed a wide range of solubility values. Analogues 23b and 23d exhibited greater solubility than IKK16 with values of 56.3 and 55.3 μg/mL versus 19.4 μg/mL, respectively. Interestingly, 23e and 23d were almost completely insoluble using this assay format. This may be due to the lower basicity of the morpholine ring which reduces the ionized fraction of the compounds at physiological pH, thus lowering their overall aqueous solubility.
In the membrane permeability assay, all the analogues tested demonstrated much lower permeability than IKK16, which indicated that the physicochemical properties of these compounds need to be further optimized (Table S2).
Activity on Liver Stage Parasites
We next checked to see if any of our novel kinase inhibitors displayed activity against the liver stage parasites. We chose compounds that had shown <20 nM IC50 at either PfGSK3 or PfPK6 in hopes that this potency would translate to noticeable antiparasitic activity in liver cells. We utilized the common rodent malaria model Plasmodium berghei ANKA to infect human HepG2 liver cells. For this assay, the hepatocyte viability in the presence of inhibitors was evaluated in parallel with assessing their effect on the P. berghei parasite load (Table S4).
Based on their superior potency against PfGSK3/PfPK6, the compounds chosen for this assay were 23b–e. Unfortunately, none of the compounds exhibited a P. berghei EC50 that was 5-fold greater than the HepG2 CC50 (Table S4). Due to this low activity window, antiparasitic effects could not be distinguished from hepatotoxic action. Accordingly, the compounds were considered inactive against liver stage parasites.
Evaluation of Kinase Selectivity Scores
Compounds with the best kinase inhibitory profile from our first set of analogues, 9g, 18n, and 18r, together with the lead hybrid analogues 23d and 23e were screened against 468 human kinases using the DiscoverX KINOMEscan platform to determine the selectivity for their kinase targets (Table 7 and Supporting Information). Compounds were screened at 1 μM concentration, and their selectivity scores S10 (1 μM) were determined. Examining the selectivity scores, we noticed that substitution on the 5-position of the pyrimidine led to an increase in the number of human kinase targets inhibited by the analogues, compared to the parent molecule, IKK16. This can be observed in the high level of promiscuity of 18n and 18r, both of which have 5-position substituents and showing S10 (1 μM) values of 0.25 and 0.23, respectively, an almost 2-fold reduction in selectivity. Interestingly, replacing the pyrrolidino-piperidine tail group of IKK16 with 3-morpholinopropan-1-amine resulted in fewer kinase target hits and accordingly a smaller S10 (1 μM) value for 9g (S10 (1 μM) = 0.08). The hybrid compound 23d showed the highest level of promiscuity, hitting 103 kinase targets of the 468 kinases screened in the assay. On the other hand, replacing the pyrrolidino-piperidine tail group of 23d with 3-morpholinopropan-1-amine (23e) reduced the promiscuity by almost 40%, going from 103 to 64 kinase hits. The same pattern was observed when comparing IKK16 with 9g. Taken together, these results indicated that the substituent at the 5-position of the pyrimidine could be generating a common pharmacophore that is recognized by more human kinases than the unsubstituted pyrimidine, while the 3-morpholinopropan-1-amine tail leads to analogues with reduced promiscuity against human kinases screened in this panel. This underscores the need for further exploration and optimization of these analogues by utilizing the information we gained from the SAR study to increase the selectivity for their kinase targets.
Conclusions
With the noticeable rise in resistance to traditional first-line antimalarial therapy, we need new methods to combat the deadly malaria parasite. The emergence of plasmodial kinases as viable targets for antimalarial drugs has prompted the scientific community to pursue plasmodial kinase inhibition as an avenue to discover medicines with new modes of action for combating plasmodial infections. By screening a library of known kinase inhibitors against 11 plasmodial kinases, we identified IKK16, a sub-micromolar inhibitor of PfGSK3 and PfPK6, which was also active against blood stage parasites. Given the beneficial prospect of dual kinase inhibition, we conducted an extensive medicinal chemistry campaign focusing on establishing SAR around the IKK16 scaffold as well as identifying analogues with enhanced PfGSK3/PfPK6 inhibitor activity and reduced promiscuity. Our SAR study focused on exploring and modifying three different positions of the IKK16 scaffold. We observed that activity against PfGSK3 was typically more dramatically affected by these modifications, suggesting that, for this scaffold, its catalytic pocket is perhaps less tolerant of changes than that of PfPK6, which tolerated many of the structural modifications that were applied. In general, many of the analogues synthesized showed some degree of preference for PfPK6 over PfGSK3. We were able to identify the 5-chlorothiophene group as a suitable bioisostere of the bulky benzothiophene, having lower molecular weight and leading to better ligand efficiency. We noticed that the preference for PfPK6 may be modulated with certain substitutions, while the same could not be achieved for PfGSK3. This was most noticeable with the 5-position substituents, where a 5-CH3 resulted in almost a 16-fold preference for PfPK6, while the 5-chloro substituent was only 9-fold selective (18n vs 18r, respectively). The greatest enhancement of kinase inhibition potency was also achieved by substitution on the pyrimidine-5-position. Building upon this finding, we identified multiple potent inhibitors of PfGSK3 and PfPK6, most of which demonstrated greater kinase inhibition than the hit compound, IKK16. However, the 5-position substituent on the pyrimidine core also appears to be favored by many other kinase targets, as evidenced by the increased promiscuity of analogues having this substitution. In addition to their kinase activity, many of the analogues reported herein inhibited parasitemia, although with varying potencies ranging from micromolar to low nanomolar. Screening of the most potent kinase inhibitors identified, 23b–e, against P. berghei liver stage parasites, showed that their antiparasitic activity may not be Pf kinase dependent but rather a result of non-specific hepatotoxicity. By assessing some of the physicochemical properties of these compounds, we found that 23b–d showed acceptable kinetic solubility, while the morpholine analogues 23d and 23e were highly insoluble. On the other hand, all these analogues demonstrated poor apparent permeability compared to IKK16. This may, in part, contribute to poor compound accumulation in the erythrocytes and thus poor target engagement in the parasite. Analogues 23d and 23e were the most promising analogues identified in terms of dual kinase inhibition, ligand efficiency, and their modest cellular activity against blood stage parasites. To the best of our knowledge, 23d is among the few dual PfGSK3/PfPK6 inhibitors with nanomolar potency against both kinases as well as nanomolar cellular activity against blood stage parasites (23dPfGSK3 IC50 = 172 ± 13 nM, PfPK6 IC50 = 11 ± 1 nM, and blood stage EC50 = 552 ± 37 nM).
Although they have several favorable features as described above, 23d and 23e suffer from high levels of kinase promiscuity, originating in part from the 5-Cl substituent on the pyrimidine core. This promiscuity may also be a contributing factor to the modest cytotoxicity observed for these compounds. Our future work will include further modifications of the structure of these two hits that explore additional functional groups based on the knowledge from our SAR study, with the aim of reducing their kinase promiscuity while increasing the on-target potency. We also plan to work on improving their physicochemical properties, especially membrane permeability and solubility, to enhance the drug-likeness of these novel compounds and allow us to move them to more advanced malaria efficacy models.
Experimental Section
Chemistry
All reagents and solvents used were purchased from commercial sources and were used without further purification. NMR spectra were obtained using a Bruker 850 MHz or INOVA 400 MHz spectrometers at room temperature; chemical shifts are expressed in parts per million (ppm, δ units) and are referenced to the residual protons in the deuterated solvent used. Coupling constants are given in units of hertz (Hz). Splitting patterns describe apparent multiplicities and are designated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), and br s (broad singlet), dd (doublet of doublets), ddd (double double doublet), and tt (triplet of triplets). The purity of compounds submitted for biological screening was determined to be ≥95% as measured by high-performance liquid chromatography (HPLC). Analytical thin-layer chromatography (TLC) was performed on silica gel plates, 200 μm, with an F254 indicator. Column chromatography was performed using RediSep Rf preloaded silica gel cartridges on Isolera one Biotage automated purification systems. Samples for high-resolution mass spectrometry were analyzed with a Thermo Fisher Q Exactive HF-X (Thermo Fisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced via a heated electrospray source (HESI) at a flow rate of 0.3 mL/min. Electrospray source conditions were set as follows: spray voltage 3.0 kV, sheath gas (nitrogen) 60 arb, auxiliary gas (nitrogen) 20 arb, sweep gas (nitrogen) 0 arb, nebulizer temperature 375 °C, capillary temperature 380 °C, and RF funnel 45 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 120,000. Separations were conducted on a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm particle size). LC conditions were set at 95% water with 0.1% formic acid (A) ramped linearly over 5.0 min to 100% acetonitrile with 0.1% formic acid (B) and held until 6.0 min. At 7.0 min, the gradient was switched back to 95% (A) and allowed to re-equilibrate until 9.0 min. The injection volume for all samples was 3 μL. Analytical LC/MS data was obtained using a Waters Acquity ultrahigh-performance liquid chromatography (UPLC) system equipped with a photodiode array (PDA) detector using the following method: solvent A = water + 0.2% FA, solvent B = ACN + 0.1% FA, and flow rate = 1 mL/min. The gradient started at 95% A for 0.05 min. Afterward, it was ramped up to 100% B over 2 min and held for an additional minute at this concentration, before returning to the initial gradient. Compounds were purified on prep HPLC using an Agilent 1100 equipped with a Phenomenex column (phenyl–hexyl, 75 × 30 mm, 5 μm) using the following method: solvent A: water + 0.05% TFA; solvent B: MeOH; and flow rate: 70.00 mL/min. LC conditions were set at 90% (A) ramped linearly over 8.0 min to 100% (B) and held until 10.0 min at 100% B. At 10.0 min, the gradient was switched back to 90% (A).
General Procedure A: Suzuki Reaction
4-(Benzo[b]thiophen-2-yl)-2-chloropyrimidine (3)
2,4-Dichloropyrimidine (1) (1.30 g, 1.0 equiv, 8.40 mmol), benzo[b]thiophen-2-ylboronic acid (1.50 g, 1.0 equiv, 8.4 mmol) (2), and Na2CO3 (2.70 g, 3 equiv, 25.00 mmol) were dissolved in a mixture of toluene (37.00 mL), EtOH (9.250 mL), and water (9.250 mL). The solvent was degassed thoroughly by bubbling argon gas through it for 5 min. Pd(PPh3)4 (0.78 g, 0.08 equiv, 0.67 mmol) was added to the reaction mixture, and it was heated to 70 °C and stirred overnight. The reaction mixture was cooled to room temperature and then poured onto water. The layers were separated, and the aqueous layer was then extracted with ethyl acetate (×3). The combined organic extracts were washed with brine, dried with Na2SO4, and filtered. The solvent was removed in vacuo. The crude compound was purified using silica gel chromatography using a gradient of hexanes and DCM 100:0 to 50:50 to afford the title material as a white solid (1.65 g, 79%). 1H NMR (400 MHz, CDCl3): δ 8.54 (d, J = 5.2 Hz, 1H), 8.06 (s, 1H), 7.86–7.74 (m, 2H), 7.53 (d, J = 5.2 Hz, 1H), 7.42–7.26 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 162.32, 161.90, 159.69, 141.79, 140.11, 139.80, 126.93, 126.29, 125.30, 125.26, 122.95, 114.54. LCMS (ESI+) m/z: 247 [M + H]+.
General Procedure B: Microwave-Assisted SNAr
4-(Benzo[b]thiophen-2-yl)-N-phenylpyrimidin-2-amine (4a)
A mixture of 4-(benzo[b]thiophen-2-yl)-2-chloropyrimidine (0.05 g, 1 equiv, 0.20 mmol), aniline (0.02 g, 0.02 mL, 1.2 equiv, 0.24 mmol), and a catalytic amount of 1 N HCl solution was heated in EtOH (3.00 mL) for 1 h at 160 °C under microwave conditions. The solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a yellow solid (0.04 g, 60%). 1H NMR (400 MHz, DMSO): δ 9.74 (s, 1H), 8.56 (d, J = 5.1 Hz, 1H), 8.38 (s, 1H), 8.09–8.04 (m, 1H), 7.97–7.91 (m, 1H), 7.90–7.84 (m, 2H), 7.51 (d, J = 5.2 Hz, 1H), 7.49–7.42 (m, 2H), 7.38–7.31 (m, 2H), 6.99 (tt, J = 7.3, 1.1 Hz, 1H). 13C NMR (101 MHz, DMSO): δ 159.91, 158.89, 158.84, 142.77, 140.39, 140.24, 139.85, 128.53, 126.16, 125.06, 124.97, 124.81, 122.89, 121.54, 118.93, 107.01. HRMS–ESI+ (m/z): [M + H]+ calcd for C18H14N3S, 304.0903; found, 304.0892.
4-(Benzo[b]thiophen-2-yl)-N-(4-chlorophenyl)pyrimidin-2-amine (4b)
This compound was synthesized according to general procedure B starting from 3 and 4-chloroaniline (0.06 g, 0.486 mmol) to afford the title material as a yellowish-white solid (0.05 g, 36%). 1H NMR (400 MHz, DMSO): δ 9.91 (s, 1H), 8.58 (d, J = 5.2 Hz, 1H), 8.39 (s, 1H), 8.08–8.03 (m, 1H), 7.97–7.92 (m, 1H), 7.92–7.86 (m, 2H), 7.54 (d, J = 5.2 Hz, 1H), 7.50–7.42 (m, 2H), 7.41–7.36 (m, 2H). 13C NMR (101 MHz, DMSO): δ 159.66, 158.94, 158.91, 142.56, 140.25, 139.84, 139.39, 128.37, 126.23, 125.23, 125.00, 124.85, 122.87, 120.39, 107.38. HRMS–ESI+ (m/z): [M + H]+ calcd for C18H14ClN3S, 338.0513; found, 338.0502.
4-(Benzo[b]thiophen-2-yl)-N-(3-chlorophenyl)pyrimidin-2-amine (4c)
This compound was synthesized according to general procedure B starting from 3 and 3-chloroaniline (0.04 g, 0.290 mmol) to afford the title material as a yellowish-white solid (0.05 g, 55%). 1H NMR (400 MHz, DMSO): δ 9.98 (s, 1H), 8.60 (d, J = 5.2 Hz, 1H), 8.45–8.32 (m, 1H), 8.13 (t, J = 2.1 Hz, 1H), 8.08–8.02 (m, 1H), 7.98–7.90 (m, 1H), 7.75 (dd, J = 8.4, 1.2 Hz, 1H), 7.57 (d, J = 5.2 Hz, 1H), 7.51–7.42 (m, 2H), 7.35 (t, J = 8.1 Hz, 1H), 7.03 (dd, J = 8.0, 1.2 Hz, 1H). 13C NMR (101 MHz, DMSO): δ 159.63, 159.00, 158.94, 142.50, 141.99, 140.27, 139.85, 133.09, 130.16, 126.32, 125.40, 125.06, 124.91, 122.89, 121.02, 118.13, 117.19, 107.67. HRMS–ESI+ (m/z): [M + H]+ calcd for C18H14ClN3S, 338.0513; found, 338.0503.
4-(Benzo[b]thiophen-2-yl)-N-(2-chlorophenyl)pyrimidin-2-amine (4d)
4-(Benzo[b]thiophen-2-yl)-2-chloropyrimidine (0.10 g, 1 equiv, 0.405 mmol) (3), 2-chloroaniline (0.05 g, 0.043 mL, 1 equiv, 0.405 mmol), rac-BINAP (0.03 g, 0.1 equiv, 0.041 mmol), and Cs2CO3 (0.33 g, 2.5 equiv, 1.01 mmol) were stirred in dioxane (3.00 mL). The solvent was degassed by bubbling argon gas for 5 min. Pd(OAc)2 (0.005 g, 0.05 equiv, 0.020 mmol) was added, and the reaction mixture was heated to 90 °C and stirred for 24 h. Afterward, the reaction mixture was cooled to room temperature and then filtered through Celite. The crude compound was purified using silica chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a yellow solid (0.14 g, 61%). 1H NMR (850 MHz, DMSO): δ 8.91 (s, 1H), 8.52 (d, J = 5.1 Hz, 1H), 8.36 (s, 1H), 8.07–8.01 (m, 1H), 7.96–7.88 (m, 2H), 7.54 (dd, J = 8.0, 1.5 Hz, 1H), 7.52 (d, J = 5.1 Hz, 1H), 7.47–7.43 (m, 2H), 7.40 (td, J = 7.6, 1.5 Hz, 1H), 7.22–7.18 (m, 1H). 13C NMR (214 MHz, DMSO): δ 160.19, 159.09, 158.97, 142.50, 140.31, 139.78, 136.28, 129.45, 127.38, 127.09, 126.14, 125.62, 125.29, 125.13, 124.93, 124.79, 122.85, 107.46. HRMS–ESI+ (m/z): [M + H]+ calcd for C18H14ClN3S, 338.0513; found, 338.0501.
4-(Benzo[b]thiophen-2-yl)-N-(4-methoxyphenyl)pyrimidin-2-amine (4e)
This compound was synthesized according to general procedure B starting from 3 and 4-methoxyaniline (0.06 g, 0.486 mmol) to afford the title material as a yellow solid (0.10 g, 74%). 1H NMR (400 MHz, DMSO): δ 9.56 (s, 1H), 8.51 (d, J = 5.1 Hz, 1H), 8.35 (s, 1H), 8.09–8.00 (m, 1H), 7.96–7.88 (m, 1H), 7.75 (d, J = 9.0 Hz, 2H), 7.53–7.32 (m, 3H), 7.02–6.79 (m, 2H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO): δ 160.00, 158.84, 158.78, 154.29, 142.93, 140.20, 139.86, 133.51, 126.10, 124.93, 124.86, 124.77, 122.86, 120.61, 113.75, 106.42, 55.19. HRMS–ESI+ (m/z): [M + H]+ calcd for C19H16N3OS, 334.1009; found, 334.0999.
4-(Benzo[b]thiophen-2-yl)-N-(3-methoxyphenyl)pyrimidin-2-amine (4f)
This compound was synthesized according to general procedure B starting from 3 and 3-methoxyaniline (0.06 g, 0.486 mmol) to afford the title material as a yellow solid (0.02 g, 15%). 1H NMR (400 MHz, DMSO): δ 9.73 (s, 1H), 8.57 (d, J = 5.2 Hz, 1H), 8.39 (s, 1H), 8.12–8.02 (m, 1H), 8.00–7.89 (m, 1H), 7.61 (t, J = 2.2 Hz, 1H), 7.52 (d, J = 5.2 Hz, 1H), 7.49–7.37 (m, 3H), 7.23 (t, J = 8.2 Hz, 1H), 6.58 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 3.81 (s, 3H). 13C NMR (101 MHz, DMSO): δ 159.87, 159.60, 158.89, 158.83, 142.65, 141.59, 140.20, 139.83, 129.25, 126.18, 125.12, 125.00, 124.81, 122.90, 111.34, 107.12, 104.58, 55.07. HRMS–ESI+ (m/z): [M + H]+ calcd for C19H16N3OS, 334.1009; found, 334.0998.
4-(Benzo[b]thiophen-2-yl)-N-(2-methoxyphenyl)pyrimidin-2-amine (4g)
This compound was synthesized according to general procedure B starting from 3 and 2-methoxyaniline (0.06 g, 0.486 mmol) to afford the title material as a yellow solid (0.06 g, 41%). 1H NMR (400 MHz, DMSO): δ 8.54 (d, J = 5.2 Hz, 1H), 8.38 (d, J = 0.8 Hz, 1H), 8.27 (dd, J = 7.3, 1.7 Hz, 1H), 8.17 (s, 1H), 8.09–8.02 (m, 1H), 7.98–7.90 (m, 1H), 7.52 (d, J = 5.2 Hz, 1H), 7.50–7.40 (m, 2H), 7.12–6.98 (m, 3H), 3.88 (s, 3H). 13C NMR (101 MHz, DMSO): δ 159.78, 158.99, 158.98, 149.13, 142.53, 140.26, 139.83, 128.40, 126.19, 125.18, 124.97, 124.82, 122.91, 122.88, 120.43, 120.21, 110.90, 107.25, 55.80. HRMS–ESI+ (m/z): [M + H]+ calcd for C19H16N3OS, 334.1009; found, 334.0996.
4-(Benzo[b]thiophen-2-yl)-N-benzylpyrimidin-2-amine (4h)
A mixture of 3 (0.10 g, 1 equiv, 0.41 mmol), phenylmethanamine (0.07 g, 0.07 mL, 1.5 equiv, 0.61 mmol), and TEA (0.08 g, 0.11 mL, 2.0 equiv, 0.81 mmol) in EtOH (5.00 mL) was heated for 30 min at 120 °C under microwave conditions. The solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a white solid (0.01 g, 8%). 1H NMR (400 MHz, DMSO): δ 8.35 (d, J = 5.1 Hz, 1H), 8.27 (s, 1H), 8.04–7.98 (m, 1H), 7.94–7.85 (m, 2H), 7.49–7.34 (m, 4H), 7.34–7.28 (m, 2H), 7.25 (d, J = 5.2 Hz, 1H), 7.23–7.18 (m, 1H), 4.56 (d, J = 6.3 Hz, 2H). 13C NMR (101 MHz, DMSO): δ 162.19, 158.88, 140.41, 140.15, 139.81, 128.18, 127.41, 126.58, 125.91, 124.83, 124.65, 124.47, 122.81, 104.95, 44.04. HRMS–ESI+ (m/z): [M + H]+ calcd for C19H16N3S, 318.1059; found, 318.1049.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)benzoate (5)
A neat mixture of 3 (0.97 g, 1 equiv, 3.93 mmol) and methyl-4-aminobenzoate (0.71 g, 1.2 equiv, 4.72 mmol) was heated at 200 °C for 2 h. The resulting crude compound was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20 to afford the title material as a yellow solid (0.57 g, 40%). 1H NMR (400 MHz, DMSO): δ 10.23 (s, 1H), 8.64 (d, J = 5.2 Hz, 1H), 8.42 (s, 1H), 8.12–8.06 (m, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.98–7.89 (m, 3H), 7.62 (d, J = 5.2 Hz, 1H), 7.53–7.37 (m, 2H), 3.84 (s, 3H). 13C NMR (101 MHz, DMSO): δ 166.02, 159.50, 159.04, 158.96, 145.05, 142.42, 140.34, 139.85, 130.17, 126.27, 125.42, 125.02, 124.89, 122.96, 121.90, 117.90, 108.06, 51.74. HRMS–ESI+ (m/z): [M + H]+ calcd for C20H16N3O2S, 362.0958; found, 362.0945.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)benzoic acid (6)
To a solution of 5 (1.60 g, 1 equiv, 4.43 mmol) in THF/MeOH (1:1, 70 mL) was added 1 M LiOH solution (13.30 mL, 3 equiv, 13.30 mmol), and the reaction mixture was heated to reflux and stirred overnight. The solvent was evaporated, and the resulting solid was dissolved in ice water and stirred vigorously. After acidification with 2 N HCl solution until reaching a pH of 2, the precipitated solid was collected by filtration. The solid was washed with ice water and then dried in vacuo to afford the title material as a yellow solid (0.96 g, 62%). 1H NMR (400 MHz, DMSO): δ 10.15 (s, 1H), 8.63 (d, J = 5.2 Hz, 1H), 8.42 (s, 1H), 8.13–8.05 (m, 1H), 8.02–7.86 (m, 5H), 7.60 (d, J = 5.2 Hz, 1H), 7.51–7.37 (m, 2H). 13C NMR (101 MHz, DMSO): δ 167.26, 159.57, 159.02, 158.95, 144.47, 142.49, 140.33, 139.85, 130.25, 126.25, 125.37, 125.02, 124.88, 123.39, 122.96, 117.83, 107.90. HRMS–ESI+ (m/z): [M + H]+ calcd for C19H14N3O2S, 348.0801; found, 348.0792.
1-(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)benzoyl)piperidin-4-one (7)
To a stirring solution of 6 (0.44 g, 1 equiv, 1.28 mmol), 1-hydroxybenzotriazole-hydrate (0.328 g, 80% wt, 1.20 equiv, 1.53 mmol), and TEA (0.39 g, 0.53 mL, 3.00 equiv, 3.83 mmol) in DMF (8.00 mL) was added 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride (0.29 g, 1.20 equiv, 1.53 mmol). The reaction mixture was stirred at room temperature for 15 min, followed by addition of 4-oxopiperidin-1-ium chloride (0.21 g, 1.20 equiv, 1.53 mmol), and the reaction was left to stir at room temperature overnight. It was then poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water, brine, and dried with Na2SO4, and the solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a yellow solid (0.17 g, 31%). 1H NMR (850 MHz, DMSO): δ 8.60 (d, J = 5.1 Hz, 1H), 8.41 (d, J = 0.8 Hz, 1H), 8.09–8.03 (m, 1H), 7.98–7.93 (m, 3H), 7.57 (d, J = 5.1 Hz, 1H), 7.53–7.49 (m, 2H), 7.48–7.43 (m, 2H), 3.95–3.52 (m, 4H), 2.52–2.41 (m, 4H). LCMS (ESI+) m/z: 429 [M + H]+.
(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(methyl(propyl)amino)piperidin-1-yl)methanone (8a)
To a stirring solution of 7 (0.050 g, 1 equiv, 0.12 mmol) and two drops of glacial acetic acid in THF (1.50 mL) was added N-methylpropan-1-amine (0.01 g, 0.02 mL, 1.5 equiv, 0.18 mmol). The reaction mixture was stirred at room temperature for 15 min after which was added NaBH(OAc)3 (0.07 g, 3.0 equiv, 0.35 mmol), and then, it was left to stir at room temperature overnight. MeOH was carefully added to the reaction mixture, and the solvents were removed in vacuo. The crude compound was purified using silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20 to afford the title material as a white solid (0.008 g, 10%). 1H NMR (400 MHz, MeOD): δ 8.48 (d, J = 5.3 Hz, 1H), 8.17 (s, 1H), 7.97–7.86 (m, 4H), 7.48–7.36 (m, 5H), 4.78–3.78 (m, 2H), 3.25–2.81 (m, 2H), 2.79–2.67 (m, 1H), 2.52–2.42 (m, 2H), 2.30 (s, 3H), 2.10–1.69 (m, 2H), 1.63–1.45 (m, 4H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (214 MHz, MeOD): δ 172.64, 161.40, 161.28, 159.66, 144.16, 143.70, 142.48, 141.58, 129.59, 129.07, 127.22, 125.91, 125.86, 125.84, 123.60, 119.64, 108.59, 61.90, 56.75, 49.01, 43.14, 38.05, 28.94, 21.10, 12.16. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H32N5OS, 486.2322; found, 486.2310.
(R)-(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(3-hydroxypyrrolidin-1-yl)piperidin-1-yl)methanone (8b)
To a stirring solution of 7 (0.050 g, 1 equiv, 0.12 mmol) in THF (1.50 mL) was added (R)-pyrrolidin-3-ol (0.01 g, 0.01 mL, 1.20 equiv, 0.14 mmol), and the solution was stirred at room temperature for 1 h. Afterward, NaBH(OAc)3 (0.07 g, 3.0 equiv, 0.35 mmol) was added, and the reaction was stirred at room temperature overnight. The solvent was removed in vacuo, and then water and ethyl acetate were added. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were dried with Na2SO4, and the solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20 to afford the title material as a yellow solid (0.008 g, 10%). 1H NMR (400 MHz, MeOD): δ 8.47 (d, J = 5.2 Hz, 1H), 8.16 (s, 1H), 7.99–7.83 (m, 4H), 7.49–7.35 (m, 5H), 4.72–4.44 (m, 1H), 4.45–4.33 (m, 1H), 4.25–3.73 (m, 1H), 3.25–2.87 (m, 4H), 2.84–2.65 (m, 2H), 2.65–2.51 (m, 1H), 2.22–1.89 (m, 3H), 1.87–1.72 (m, 1H), 1.64–1.42 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.65, 161.37, 161.26, 159.66, 144.14, 143.74, 142.46, 141.57, 129.49, 129.06, 127.23, 125.91, 125.87, 125.85, 123.58, 119.64, 108.62, 70.84, 63.22, 60.99, 51.11, 42.31, 34.67, 31.84. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H30N5O2S, 500.2115; found, 500.2104.
(S)-(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(3-hydroxypyrrolidin-1-yl)piperidin-1-yl)methanone (8c)
This compound was synthesized using the same procedure for 8b, starting with 7 and (S)-pyrrolidin-3-ol (0.01 g, 0.11 mmol) to afford the title material as a yellow solid (0.008 g, 20%). 1H NMR (850 MHz, MeOD): δ 8.47 (d, J = 5.1 Hz, 1H), 8.17 (s, 1H), 7.95–7.91 (m, 3H), 7.91–7.85 (m, 1H), 7.46–7.37 (m, 5H), 4.66–4.46 (m, 1H), 4.41–4.32 (m, 1H), 4.08–3.81 (m, 1H), 3.25–3.04 (m, 1H), 2.93 (dd, J = 10.3, 6.2 Hz, 2H), 2.88–2.79 (m, 1H), 2.69–2.64 (m, 1H), 2.60 (dd, J = 10.4, 3.3 Hz, 1H), 2.46–2.40 (m, 1H), 2.15–2.09 (m, 1H), 2.09–1.81 (m, 2H), 1.77–1.71 (m, 1H), 1.59–1.37 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.63, 161.40, 161.27, 159.66, 144.16, 143.69, 142.48, 141.58, 129.62, 129.02, 127.23, 125.91, 125.86, 125.84, 123.60, 119.65, 108.60, 71.05, 63.11, 61.10, 51.05, 42.64, 34.84, 32.41. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H30N5O2S, 500.2115; found, 500.2101.
General Procedure C: Amide Coupling
(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(dimethylamino)piperidin-1-yl)methanone (9a)
To a stirring solution of 6 (0.15 g, 1 equiv, 0.43 mmol), N,N-dimethylpiperidin-4-amine (0.06 g, 1.2 equiv, 0.52 mmol), 1-hydroxybenzotriazole-hydrate (0.11 g, 80% wt, 1.2 equiv, 0.52 mmol), and TEA (0.13 g, 0.18 mL, 3 equiv, 1.30 mmol) in DMF (6 mL) was added 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride (0.10 g, 1.2 equiv, 0.52 mmol), and the reaction was stirred at room temperature for 24 h. The reaction mixture was poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and the solvent was removed in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 90:10 to afford the title material as a yellow solid (0.03 g, 13%). 1H NMR (400 MHz, DMSO): δ 9.98 (s, 1H), 8.59 (d, J = 5.1 Hz, 1H), 8.40 (s, 1H), 8.11–7.99 (m, 1H), 8.00–7.85 (m, 3H), 7.56 (d, J = 5.2 Hz, 1H), 7.49–7.42 (m, 2H), 7.42–7.37 (m, 2H), 3.14–2.72 (m, 4H), 2.49–2.40 (m, 1H), 2.25 (s, 6H), 1.88–1.71 (m, 2H), 1.37 (qd, J = 12.0, 4.1 Hz, 2H). 13C NMR (101 MHz, DMSO): δ 169.07, 159.75, 158.99, 142.59, 141.67, 140.30, 139.88, 128.75, 127.87, 126.28, 125.30, 125.05, 124.91, 122.93, 118.15, 107.57, 61.49, 41.16, 27.94. HRMS–ESI+ (m/z): [M + H]+ calcd for C26H28N5OS, 458.2009; found, 458.1995.
[1,4′-Bipiperidin]-1′-yl(4-((4-(benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)methanone (9b)
To a stirring solution of 6 (0.080 g, 1 equiv, 0.23 mmol), 1-hydroxybenzotriazole-hydrate (0.06 g, 80% wt, 1.2 equiv, 0.28 mmol), and TEA (0.070 g, 0.096 mL, 3.0 equiv, 0.69 mmol) in DMF (2.00 mL) was added 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride (0.05 g, 1.2 equiv, 0.28 mmol). The reaction was stirred at room temperature for 15 min, followed by addition of 1,4′-bipiperidine (0.05 g, 1.2 equiv, 0.28 mmol) after which the reaction was stirred at room temperature overnight. The reaction mixture was poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and the solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 90:10 to afford the title material as a yellow solid (0.04 g, 35%). 1H NMR (850 MHz, DMSO): δ 9.98 (s, 1H), 8.60 (d, J = 5.1 Hz, 1H), 8.40 (s, 1H), 8.11–8.04 (m, 1H), 7.96–7.94 (m, 1H), 7.94–7.90 (m, 2H), 7.56 (d, J = 5.1 Hz, 1H), 7.48–7.43 (m, 2H), 7.41–7.37 (m, 2H), 4.66–3.58 (m, 2H), 3.11–2.63 (m, 2H), 2.49–2.40 (m, 5H), 1.83–1.59 (m, 2H), 1.52–1.44 (m, 4H), 1.44–1.32 (m, 4H). 13C NMR (214 MHz, DMSO): δ 168.96, 159.72, 158.94, 142.56, 141.58, 140.27, 139.84, 128.82, 127.79, 126.21, 125.24, 124.99, 124.85, 122.90, 118.10, 107.51, 61.76, 49.66, 27.74, 26.03, 24.51. HRMS–ESI+ (m/z): [M + H]+ calcd for C29H32N5OS, 498.2322; found, 498.2308.
tert-Butyl 4-(4-((4-(benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)benzoyl)piperazine-1-carboxylate (9c-Boc)
This compound was synthesized according to general procedure C starting with 6 and tert-butylpiperazine-1-carboxylate (0.24 g, 1.29 mmol) to afford the title material as a yellow solid (0.26 g, 58%). 1H NMR (850 MHz, CDCl3): δ 8.47 (d, J = 5.1 Hz, 1H), 7.99 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.85 (dd, J = 7.3, 1.5 Hz, 1H), 7.83–7.78 (m, 2H), 7.50–7.46 (m, 3H), 7.44–7.37 (m, 2H), 7.22 (d, J = 5.1 Hz, 1H), 3.89–3.28 (m, 8H), 1.48 (s, 9H). MS ESI+ m/z: 516 [M + H]+.
(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(piperazin-1-yl)methanone (9c)
To a stirring solution of 9c-Boc (0.07 g, 1 equiv, 0.13 mmol) in DCM/MeOH (2:1, 0.5 mL) was added 4 M HCl in dioxane (16 equiv, 0.50 mL), and the reaction was left to stir at room temperature overnight. The solvent was removed in vacuo, and the residue was triturated with DCM and then filtered. The residue was washed with DCM and then purified using silica gel chromatography using a system of DCM/MeOH 100:0 to 85:15 to afford the title material as white solid (0.02 g, 42%). 1H NMR (400 MHz, DMSO): δ 10.04 (s, 1H), 8.61 (d, J = 5.1 Hz, 1H), 8.42 (s, 1H), 8.10–8.03 (m, 1H), 8.01–7.87 (m, 3H), 7.58 (d, J = 5.1 Hz, 1H), 7.52–7.39 (m, 4H), 3.79–3.64 (m, 4H), 3.19–3.03 (m, 4H). 13C NMR (214 MHz, DMSO): δ 169.47, 159.69, 158.99, 142.51, 142.21, 140.27, 139.86, 128.37, 127.20, 126.29, 125.35, 125.05, 124.91, 122.89, 118.09, 107.68, 42.66. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H22N5O2S, 416.1540; found, 416.1528.
(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-methylpiperazin-1-yl)methanone (9d)
This compound was synthesized according to general procedure C starting from 6 and 1-methylpiperazine (0.05 g, 0.06 mL, 0.52 mmol) to afford the title material as a yellow solid (0.06 g, 32%). 1H NMR (400 MHz, DMSO): δ 9.99 (s, 1H), 8.60 (d, J = 5.2 Hz, 1H), 8.41 (s, 1H), 8.10–8.04 (m, 1H), 7.98–7.88 (m, 3H), 7.57 (d, J = 5.1 Hz, 1H), 7.51–7.42 (m, 2H), 7.42–7.36 (m, 2H), 3.59–3.42 (m, 4H), 2.36–2.27 (m, 4H), 2.20 (s, 3H). 13C NMR (214 MHz, DMSO): δ 169.06, 159.69, 158.93, 142.53, 141.70, 140.26, 139.83, 128.40, 127.97, 126.20, 125.24, 124.98, 124.84, 122.88, 118.09, 107.53, 54.56, 45.62. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H24N5OS, 430.1696; found 430.1684.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(1-methylpiperidin-4-yl)benzamide (9e)
This compound was synthesized according to general procedure C starting from 6 and 1-methylpiperidin-4-amine (0.06 g, 0.48 mmol) to afford the title material as a yellow solid (0.02 g, 12%). 1H NMR (400 MHz, DMSO): δ 10.04 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.42 (s, 1H), 8.14 (d, J = 7.7 Hz, 1H), 8.11–8.05 (m, 1H), 7.99–7.93 (m, 3H), 7.91–7.85 (m, 2H), 7.59 (d, J = 5.2 Hz, 1H), 7.51–7.42 (m, 2H), 3.87–3.70 (m, 1H), 2.96–2.78 (m, 2H), 2.26 (s, 3H), 2.21–2.04 (m, 2H), 1.86–1.74 (m, 2H), 1.74–1.53 (m, 2H). 13C NMR (214 MHz, DMSO): δ 165.31, 159.66, 159.08, 158.86, 143.06, 142.59, 140.27, 139.90, 128.10, 127.21, 126.30, 125.31, 125.06, 124.91, 122.91, 117.63, 107.63, 54.20, 45.99, 45.38, 31.04. HRMS–ESI+ (m/z): [M + 2H]2+ calcd for C25H26N5OS, 222.5963; found, 222.5958.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-(4-methylpiperazin-1-yl)propyl)benzamide (9f)
This compound was synthesized according to general procedure C starting with 6 and 3-(4-methylpiperazin-1-yl)propan-1-amine (0.04 g, 0.27 mmol) to afford the title material as a yellow solid (0.05 g, 47%). 1H NMR (400 MHz, DMSO): δ 10.03 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.41 (s, 1H), 8.38 (t, J = 5.5 Hz, 1H), 8.09–8.05 (m, 1H), 7.98–7.93 (m, 3H), 7.88–7.82 (m, 2H), 7.58 (d, J = 5.2 Hz, 1H), 7.50–7.43 (m, 2H), 3.32–3.24 (m, 3H), 2.47–2.22 (m, 9H), 2.14 (s, 3H), 1.75–1.62 (m, 2H). 13C NMR (214 MHz, DMSO): δ 165.70, 159.66, 159.04, 158.87, 143.01, 142.57, 140.26, 139.88, 127.87, 127.26, 126.29, 125.30, 125.04, 124.89, 122.88, 117.74, 107.63, 55.93, 54.80, 52.74, 45.76, 38.02, 26.26. HRMS–ESI+ (m/z): [M + H]+ calcd for C27H31N6OS, 487.2275; found, 487.2263.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-morpholinopropyl)benzamide (9g)
To a stirring solution of 6 (0.08 g, 1 equiv, 0.23 mmol), 1-hydroxybenzotriazole-hydrate (0.059 g, 80% wt, 1.2 equiv, 0.28 mmol), and TEA (0.070 g, 0.10 mL, 3.0 equiv, 0.69 mmol) in DMF (2.00 mL) was added 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride (0.05 g, 1.2 equiv, 0.28 mmol). The reaction was stirred at room temperature for 15 min, then 3-morpholinopropan-1-amine (0.04 g, 0.04 mL, 1.2 equiv, 0.28 mmol) was added, and the reaction was heated to 80 °C and stirred for 18 h. Afterward, the reaction mixture was poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and the solvent was removed in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 90:10 to afford the title material as a white solid (0.04 g, 37%). 1H NMR (400 MHz, DMSO): δ 10.04 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.41 (s, 1H), 8.38 (t, J = 5.8 Hz, 1H), 8.09–8.04 (m, 1H), 7.99–7.93 (m, 3H), 7.88–7.83 (m, 2H), 7.58 (d, J = 5.2 Hz, 1H), 7.50–7.42 (m, 2H), 3.59 (t, J = 4.6 Hz, 4H), 3.32–3.27 (m, 2H), 2.43–2.29 (m, 6H), 1.70 (p, J = 7.0 Hz, 2H). 13C NMR (214 MHz, DMSO): δ 165.72, 159.65, 159.05, 158.86, 143.02, 142.60, 140.28, 139.88, 127.86, 127.25, 126.29, 125.30, 125.04, 124.88, 122.89, 117.74, 107.61, 66.23, 56.32, 53.38, 37.90, 25.91. HRMS–ESI+ (m/z): [M + H]+ calcd for C26H28N5O2S, 474.1958; found, 474.1944.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-(dimethylamino)propyl)benzamide (9h)
This compound was synthesized according to general procedure C starting from 6 and N1,N1-dimethylpropane-1,3-diamine (0.05 g, 0.07 mL, 0.52 mmol) to afford the title material as a yellow solid (0.02 g, 10%). 1H NMR (400 MHz, DMSO): δ 10.03 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.44–8.33 (m, 2H), 8.10–8.04 (m, 1H), 7.98–7.89 (m, 3H), 7.88–7.79 (m, 2H), 7.58 (d, J = 5.2 Hz, 1H), 7.51–7.41 (m, 2H), 3.31–3.24 (m, 2H), 2.26 (t, J = 7.1 Hz, 2H), 2.14 (s, 6H), 1.66 (p, J = 7.1 Hz, 2H). 13C NMR (214 MHz, DMSO): δ 166.07, 159.81, 159.24, 159.06, 143.18, 142.68, 140.42, 140.04, 128.05, 127.39, 126.53, 125.48, 125.26, 125.12, 123.05, 118.00, 107.84, 57.14, 45.33, 37.90, 27.32. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H26N5OS, 432.1853; found 432.1842.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-(methylsulfonyl)propyl)benzamide (9i)
This compound was synthesized according to general procedure C starting with 6 and 3-(methylsulfonyl)propan-1-aminium chloride (0.11 g, 0.61 mmol) to afford the title material as a yellow solid (0.05 g, 21%). 1H NMR (400 MHz, DMSO): δ 10.05 (s, 1H), 8.62 (d, J = 5.2 Hz, 1H), 8.44 (t, J = 5.8 Hz, 1H), 8.42 (s, 1H), 8.10–8.06 (m, 1H), 8.00–7.94 (m, 3H), 7.91–7.83 (m, 2H), 7.59 (d, J = 5.2 Hz, 1H), 7.51–7.41 (m, 2H), 3.44–3.35 (m, 2H), 3.21–3.15 (m, 2H), 2.99 (s, 3H), 2.02–1.89 (m, 2H). 13C NMR (214 MHz, DMSO): δ 166.01, 159.65, 159.06, 158.88, 143.16, 142.57, 140.27, 139.88, 128.00, 126.97, 126.30, 125.33, 125.06, 124.90, 122.90, 117.73, 107.67, 51.58, 40.13, 37.79, 22.42. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H23N4O3S2, 467.1206; found, 467.1194.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-cyclopropylbenzamide (9j)
This compound was synthesized according to general procedure C starting from 6 and cyclopropanamine to afford the title material as a yellow solid (0.01 g, 6%). The N.B. Product was purified using a gradient of hexanes and ethyl acetate. 1H NMR (400 MHz, DMSO): δ 10.03 (s, 1H), 8.61 (d, J = 5.2 Hz, 1H), 8.41 (s, 1H), 8.29 (d, J = 4.2 Hz, 1H), 8.10–8.04 (m, 1H), 7.99–7.90 (m, 3H), 7.87–7.78 (m, 2H), 7.58 (d, J = 5.2 Hz, 1H), 7.52–7.41 (m, 2H), 2.89–2.80 (m, 1H), 0.73–0.66 (m, 2H), 0.61–0.54 (m, 2H). 13C NMR (214 MHz, DMSO): δ 167.13, 159.66, 159.08, 158.88, 143.08, 142.59, 140.28, 139.90, 127.96, 127.03, 126.32, 125.33, 125.07, 124.92, 122.91, 117.69, 107.65, 23.03, 5.80. HRMS–ESI+ (m/z): [M + H]+ calcd for C22H19N5OS, 387.1274; found, 387.1264.
4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)-N-(1-methyl-1H-pyrazol-4-yl)benzamide (9k)
To a stirring solution of 6 (0.175 g, 1 equiv, 0.504 mmol), 1-methyl-1H-pyrazol-4-amine (0.06 g, 1.20 equiv, 0.61 mmol), and TEA (0.153 g, 0.21 mL, 3.00 equiv, 1.51 mmol) in DMF (4 mL) was added HATU (0.23 g, 1.20 equiv, 0.61 mmol). The reaction was heated to 80 °C and stirred overnight. The reaction mixture was poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and the solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a greenish-white solid (0.01 g, 4%). 1H NMR (400 MHz, DMSO): δ 10.27 (s, 1H), 10.11 (s, 1H), 8.63 (d, J = 5.2 Hz, 1H), 8.43 (s, 1H), 8.11–8.05 (m, 1H), 8.04–8.00 (m, 3H), 7.99–7.93 (m, 3H), 7.60 (d, J = 5.2 Hz, 1H), 7.58 (d, J = 0.8 Hz, 1H), 7.51–7.43 (m, 2H), 3.83 (s, 3H). 13C NMR (214 MHz, DMSO): δ 163.05, 159.64, 159.10, 158.89, 143.38, 142.55, 140.27, 139.89, 130.13, 128.19, 126.65, 126.32, 125.36, 125.07, 124.91, 122.90, 122.06, 121.52, 117.81, 107.74, 38.69. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H18N6OS, 427.1336; found, 427.1325.
(4-((4-(Benzo[b]thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)methanol (10)
A mixture of 3 (0.340 g, 1 equiv, 1.38 mmol), (4-aminophenyl)methanol (0.19 g, 1.10 equiv, 1.52 mmol), and Cs2CO3 (0.898 g, 2.00 equiv, 2.76 mmol) was stirred in dioxane (12 mL), and the solvent was degassed by bubbling argon gas for 5 minutes. Afterward, xantphos (0.08 g, 0.10 equiv, 0.14 mmol) and Pd2(dba)3 (0.13 g, 0.10 equiv, 0.14 mmol) were added to the reaction mixture, which was heated to 90 °C and stirred overnight. The mixture was cooled to room temperature and filtered through Celite. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel chromatography using a gradient of DCM/ethyl acetate to afford the title material as a yellow solid (0.12 g, 26%). 1H NMR (400 MHz, DMSO): δ 9.71 (s, 1H), 8.55 (d, J = 5.2 Hz, 1H), 8.37 (s, 1H), 8.11–8.04 (m, 1H), 7.97–7.90 (m, 1H), 7.84–7.76 (m, 2H), 7.49 (d, J = 5.2 Hz, 1H), 7.48–7.40 (m, 2H), 7.33–7.21 (m, 2H), 5.07 (t, J = 5.7 Hz, 1H), 4.47 (d, J = 5.8 Hz, 2H). LCMS (ESI+) m/z: 334 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-N-(4-(chloromethyl)phenyl)pyrimidin-2-amine (11)
To a stirring suspension of 10 (0.80 g, 1 equiv, 2.4 mmol) and DIPEA (0.62 g, 0.84 mL, 2 equiv, 4.8 mmol) in THF (23 mL) was added SOCl2 (0.86 g, 0.53 mL, 3 equiv, 7.2 mmol) dropwise, and the reaction mixture was stirred at room temperature overnight. Excess thionyl chloride and the reaction solvent were removed under reduced pressure. Afterward, water and DCM were added to the reaction mixture, and the layers were separated. The organic layer was washed with water and brine and then dried with Na2SO4. The solvent was removed in vacuo, and the crude product was used for the next step without further purification.
4-(Benzo[b]thiophen-2-yl)-N-(4-((4-(pyrrolidin-1-yl)piperidin-1-yl)methyl)phenyl)pyrimidin-2-amine (12)
To a stirring solution of 11 (0.10 g, 1 equiv, 0.28 mmol) and K2CO3 (0.12 g, 3.01 equiv, 0.85 mmol) in DMF (2.00 mL) was added 4-(pyrrolidin-1-yl)piperidine (0.09 g, 2.00 equiv, 0.57 mmol), and the reaction mixture was stirred at room temperature overnight. The reaction was quenched by adding water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with brine, dried with Na2SO4, and concentrated in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 40:60. The isolated product was re-purified using prep HPLC to afford the TFA salt of title material as a yellow solid (0.03 g, 13%). 1H NMR (400 MHz, MeOD): δ 8.48 (d, J = 5.3 Hz, 1H), 8.19 (s, 1H), 8.03–7.95 (m, 2H), 7.93–7.86 (m, 2H), 7.52–7.47 (m, 2H), 7.46–7.36 (m, 3H), 4.34 (s, 2H), 3.75–3.60 (m, 4H), 3.51–3.39 (m, 1H), 3.22–2.97 (m, 4H), 2.53–2.37 (m, 2H), 2.21–1.90 (m, 6H). 13C NMR (151 MHz, MeOD): δ 162.80, 162.34, 160.52, 145.26, 144.93, 143.77, 142.84, 134.25, 128.64, 127.43, 127.26, 127.23, 124.76, 121.81, 109.90, 62.60, 61.53, 54.55, 52.38, 28.71, 25.13. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H32N5S, 470.2373; found, 470.2360.
tert-Butyl (4-(4-(pyrrolidin-1-yl)piperidine-1-carbonyl)phenyl)carbamate (14)
To a stirring solution of 4-((tert-butoxycarbonyl)amino)benzoic acid 13 (0.50 g, 1.0 equiv, 2.11 mmol), 4-(pyrrolidin-1-yl)piperidine (0.39 g, 1.20 equiv, 2.53 mmol), and TEA (0.64 g, 0.88 mL, 3.00 equiv, 6.32 mmol) in DMF (10.00 mL) was added HATU (0.88 g, 1.10 equiv, 2.32 mmol), and the reaction was allowed to stir at room temperature for 24 h. Afterward, the reaction mixture was poured onto water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and the solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 90:10 to afford the title material as a white solid (0.63, 80%). 1H NMR (400 MHz, DMSO): δ 9.53 (s, 1H), 7.55–7.39 (m, 2H), 7.31–7.15 (m, 2H), 4.41–3.50 (m, 2H), 3.11–2.82 (m, 2H), 2.54–2.44 (m, 4H), 2.34–2.19 (m, 1H), 1.90–1.76 (m, 2H), 1.72–1.62 (m, 4H), 1.48 (s, 9H), 1.41–1.28 (m, 2H). 13C NMR (101 MHz, DMSO): δ 169.28, 153.12, 141.09, 129.94, 128.18, 117.86, 79.78, 61.04, 51.19, 46.12, 31.48, 28.51, 23.36. LCMS (ESI+) m/z: 374 [M + H]+.
(4-Aminophenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (15)
To a stirring solution of 14 (0.63 g, 1 equiv, 1.69 mmol) in DCM (2.00 mL) was added 4 M HCl in dioxane (5 equiv, 2.00 mL), and the reaction solution was stirred at room temperature for 48 h. The solvent was evaporated, and the residue was triturated with DCM and then filtered. The filtered residue was washed with DCM, collected, and then dried in vacuo. Afterward, it was dissolved in water, and the pH was adjusted to 12 using a 1 M NaOH solution. The aqueous layer was extracted with DCM (×3). The combined organic extracts were washed with brine and dried with Na2SO4, and the solvent was removed in vacuo to afford the title material as a white solid (0.31 g, 67%). 1H NMR (400 MHz, MeOD): δ 7.22–7.14 (m, 2H), 6.77–6.58 (m, 2H), 4.65–3.82 (m, 2H), 3.13–2.82 (m, 2H), 2.70–2.56 (m, 4H), 2.34 (tt, J = 11.0, 4.0 Hz, 1H), 2.08–1.93 (m, 2H), 1.88–1.74 (m, 4H), 1.53–1.38 (m, 2H). LCMS (ESI+) m/z: 274 [M + H]+.
4-(Benzofuran-2-yl)-2-chloropyrimidine (17a)
This compound was synthesized according to general procedure A starting from 1 and benzofuran-2-ylboronic acid (0.30 g, 1.85 mmol) to afford the title material as a white solid (0.03 g, 8%). 1H NMR (400 MHz, CDCl3): δ 8.68 (d, J = 5.1 Hz, 1H), 7.77 (d, J = 1.0 Hz, 1H), 7.74 (d, J = 5.2 Hz, 1H), 7.72–7.67 (m, 1H), 7.58 (dq, J = 8.4, 0.9 Hz, 1H), 7.44 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.32 (ddd, J = 8.0, 7.2, 1.0 Hz, 1H). LCMS (ESI+) m/z: 231 [M + H]+.
2-Chloro-4-(naphthalen-2-yl)pyrimidine (17b)
2,4-Dichloropyrimidine 1 (0.22 g, 1 equiv, 1.45 mmol) was dissolved in a mixture of dioxane (10.00 mL) and water (2.50 mL). The solution was degassed by bubbling nitrogen gas for 5 minutes. Naphthalen-2-ylboronic acid (0.25 g, 1 equiv, 1.45 mmol), Cs2CO3 (1.42 g, 3 equiv, 4.36 mmol), and Pd(PPh3)4 (0.13 g, 0.08 equiv, 0.116 mmol) were added to the reaction mixture, and the reaction mixture was heated to 70 °C and stirred for 20 h. Afterward, the reaction mixture was poured into water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine, dried with Na2SO4, filtered, and then concentrated. The crude compound was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a yellow solid (0.23 g, 66%). 1H NMR (400 MHz, DMSO): δ 8.86 (d, J = 5.3 Hz, 1H), 8.85–8.83 (m, 1H), 8.28 (d, J = 5.3 Hz, 1H), 8.27–8.21 (m, 1H), 8.15–8.11 (m, 1H), 8.09 (d, J = 8.7 Hz, 1H), 8.03–7.94 (m, 1H), 7.69–7.57 (m, 2H). LCMS (ESI+) m/z: 241 [M + H]+.
2-Chloro-4-(thiophen-2-yl)pyrimidine (17c)
This compound was synthesized according to general procedure A starting from 1 and thiophen-2-ylboronic acid (0.30 g, 2.34 mmol) to afford the title material as a white solid (0.27 g, 59%). 1H NMR (400 MHz, CDCl3): δ 8.54 (d, J = 5.3 Hz, 1H), 7.83 (dd, J = 3.8, 1.1 Hz, 1H), 7.60 (dd, J = 5.0, 1.1 Hz, 1H), 7.47 (d, J = 5.3 Hz, 1H), 7.18 (dd, J = 5.0, 3.8 Hz, 1H). LCMS (ESI+) m/z: 196 [M + H]+.
2-(2-Chloropyrimidin-4-yl)thiazole (17d)
To a solution of Pd(PPh3)4 (0.12 g, 0.05 equiv, 0.10 mmol) and LiCl (0.10 g, 1.20 equiv, 2.42 mmol) in anhydrous DMF (3.00 mL) were added 1 (0.30 g, 1 equiv, 2.01 mmol) and 2-(tributylstannyl)thiazole (0.90 g, 0.76 mL, 1.20 equiv, 2.42 mmol) under argon pressure, and the reaction mixture was stirred at 70 °C for 16 h. The reaction mixture was cooled to room temperature, followed by addition of a saturated KF solution. The formed precipitate was filtered through a Celite pad and washed with ethyl acetate. The filtrate was extracted with ethyl acetate, and the combined organic extracts were washed with brine and dried with Na2SO4. The solvent was removed in vacuo, and the crude product was purified by silica gel chromatography using a gradient of hexanes and ethyl acetate to afford the title material as a white solid (0.04 g, 10%). 1H NMR (400 MHz, CDCl3): δ 8.66 (d, J = 5.1 Hz, 1H), 7.99 (d, J = 5.1 Hz, 1H), 7.97 (d, J = 3.1 Hz, 1H), 7.57 (d, J = 3.1 Hz, 1H). LCMS (ESI+) m/z: 198 [M + H]+.
2-Chloro-4-(furan-2-yl)pyrimidine (17e)
This compound was synthesized according to general procedure A starting from 1 and (furan-2-ylboronic acid (0.19 g, 1.68 mmol)) to afford the title material as a white solid (0.20 g, 66%). 1H NMR (400 MHz, CDCl3): δ 8.58 (d, J = 5.2 Hz, 1H), 7.63 (dd, J = 1.8, 0.8 Hz, 1H), 7.52 (d, J = 5.2 Hz, 1H), 7.39 (dd, J = 3.6, 0.8 Hz, 1H), 6.61 (dd, J = 3.6, 1.7 Hz, 1H). LCMS (ESI+) m/z: 181 [M + H]+.
2-Chloro-4-(1-methyl-1H-pyrazol-4-yl)pyrimidine (17f)
This compound was synthesized according to general procedure A starting from 1 and (1-methyl-1H-pyrazol-4-yl)boronic acid (0.25 g, 1.99 mmol) to afford the title material as a pink solid (0.10 g, 26%). 1H NMR (400 MHz, CDCl3): δ 8.49 (d, J = 5.3 Hz, 1H), 8.10 (s, 1H), 8.01 (s, 1H), 7.29 (d, J = 5.3 Hz, 1H), 3.97 (s, 3H). LCMS (ESI+) m/z: 195 [M + H]+.
2-Chloro-4-phenylpyrimidine (17g)
This compound was synthesized according to general procedure A starting from 1 and phenylboronic acid (0.25 g, 2.01 mmol) to afford the title material as a white solid (0.25g, 65%). 1H NMR (400 MHz, CDCl3): δ 8.64 (d, J = 5.2 Hz, 1H), 8.13–8.03 (m, 2H), 7.65 (d, J = 5.2 Hz, 1H), 7.58–7.46 (m, 3H). LCMS (ESI+) m/z: 191 [M + H]+.
2-Bromo-4-(3,4-dichlorophenyl)pyrimidine (17h)
This compound was synthesized according to general procedure A starting from 2,4-dibromopyrimdine (0.30 g, 1.26 mmol) and (3,4-dichlorophenyl)boronic acid (0.24 g, 1.26 mmol) to afford the title material as a white solid (0.18 g, 47%). 1H NMR (400 MHz, CDCl3): δ 8.61 (d, J = 5.2 Hz, 1H), 8.21 (d, J = 2.2 Hz, 1H), 7.91 (dd, J = 8.4, 2.2 Hz, 1H), 7.65 (d, J = 5.2 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H). LCMS (ESI+) m/z: 303 [M + H]+.
2-Chloro-4-(5-methylthiophen-2-yl)pyrimidine (17i)
This compound was synthesized according to general procedure A starting from 1 and (5-methylthiophen-2-yl)boronic acid (0.29 g, 2.01 mmol) to afford the title material as a yellow solid (0.18 g, 41%). 1H NMR (400 MHz, CDCl3): δ 8.48 (d, J = 5.4 Hz, 1H), 7.65 (d, J = 3.7 Hz, 1H), 7.38 (d, J = 5.4 Hz, 1H), 6.88–6.75 (m, 1H), 2.56 (d, J = 0.5 Hz, 3H). LCMS (ESI+) m/z: 211 [M + H]+.
2-Chloro-4-(5-chlorothiophen-2-yl)pyrimidine (17j)
This compound was synthesized according to general procedure A starting from 1 and (5-chlorothiophen-2-yl)boronic acid (0.33 g, 2.01 mmol) to afford the title material as a green solid (0.26 g, 56%). 1H NMR (400 MHz, CDCl3): δ 8.54 (d, J = 5.3 Hz, 1H), 7.59 (d, J = 4.1 Hz, 1H), 7.39 (d, J = 5.3 Hz, 1H), 6.99 (d, J = 4.1 Hz, 1H). LCMS (ESI+) m/z: 230 [M + H]+.
2-Chloro-4-(5-phenylthiophen-2-yl)pyrimidine (17k)
This compound was synthesized according to general procedure A starting from 1 and (5-phenylthiophen-2-yl)boronic acid (0.34 g, 1.68 mmol) to afford the title material as a brown solid (0.25 g, 54%). 1H NMR (400 MHz, CDCl3): δ 8.51 (d, J = 5.5 Hz, 1H), 7.78 (d, J = 4.0 Hz, 1H), 7.69–7.61 (m, 2H), 7.47–7.39 (m, 3H), 7.39–7.33 (m, 2H). LCMS (ESI+) m/z: 272 [M + H]+.
(5-(2-Chloropyrimidin-4-yl)thiophen-2-yl)methanol (17l)
This compound was synthesized according to general procedure A starting from 1 and (5-(hydroxymethyl)thiophen-2-yl)boronic acid (0.25 g, 1.58 mmol) to afford the title material as a yellow solid (0.22 g, 60%). 1H NMR (400 MHz, CDCl3): δ 8.53 (d, J = 5.3 Hz, 1H), 7.72 (d, J = 3.8 Hz, 1H), 7.45–7.40 (m, 1H), 7.09–7.04 (m, 1H), 4.89 (br s, 2H). LCMS (ESI+) m/z: 227 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-2-chloro-5-methylpyrimidine (17n)
This compound was synthesized according to general procedure A starting from 2 and 4-dichloro-5-methylpyrimidine (16n) (0.23 g, 1.40 mmol) to afford the title material as a pink solid (0.09 g, 24%). 1H NMR (400 MHz, CDCl3): δ 8.47 (d, J = 1.0 Hz, 1H), 8.00 (d, J = 1.9 Hz, 1H), 7.89 (m, 2H), 7.48–7.35 (m, 2H), 2.64 (d, J = 1.7 Hz, 3H). LCMS (ESI+) m/z: 261 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-2-chloro-5-cyclopropylpyrimidine (17o)
This compound was synthesized according to general procedure A starting from 2 and 2,4-dichloro-5-cyclopropylpyrimidine (16o) (0.50 g, 2.81 mmol) to afford the title material as a white solid (0.20 g, 49%). 1H NMR (400 MHz, DMSO): δ 8.63 (d, J = 0.8 Hz, 1H), 8.56 (s, 1H), 8.08–7.97 (m, 2H), 7.53–7.40 (m, 2H), 2.32–2.19 (m, 1H), 1.27–1.13 (m, 2H), 0.96–0.80 (m, 2H). LCMS (ESI+) m/z: 287 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-2-chloro-5-methoxypyrimidine (17p)
This compound was synthesized according to general procedure A starting from 2 and 2,4-dichloro-5-methoxypyrimidine (16p) (0.25 g, 1.40 mmol) to afford the title material as a white solid (0.18 g, 49%). 1H NMR (400 MHz, DMSO): δ 8.71 (s, 1H), 8.50 (s, 1H), 8.10–7.94 (m, 2H), 7.53–7.35 (m, 2H), 4.16 (s, 3H). LCMS (ESI+) m/z: 277 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-2-chloro-5-fluoropyrimidine (17q)
This compound was synthesized according to general procedure A starting from 2 and 2,4-dichloro-5-fluoropyrimidine (16q) (0.23 g, 1.40 mmol) to afford the title material as a white solid (0.20 g, 55%). 1H NMR (400 MHz, CDCl3): δ 8.51 (d, J = 2.7 Hz, 1H), 8.29–8.23 (m, 1H), 7.95–7.84 (m, 2H), 7.50–7.37 (m, 2H). LCMS (ESI+) m/z: 264 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-2,5-dichloropyrimidine (17r)
This compound was synthesized according to general procedure A starting from 2 and 2,4,5-trichloropyrimidine (16r) (0.26 g, 1.40 mmol) to afford the title material as a white solid (0.20 g, 50%). 1H NMR (400 MHz, CDCl3): δ 8.63 (d, J = 0.9 Hz, 1H), 8.61 (s, 1H), 7.94–7.83 (m, 2H), 7.50–7.38 (m, 2H). LCMS (ESI+) m/z: 280 [M + H]+.
4-(Benzo[b]thiophen-2-yl)-5-bromo-2-chloropyrimidine (17s)
This compound was synthesized according to general procedure A starting from 2 and 5-bromo-2,4-dichloropyrimidine (16s) (0.30 g, 1.32 mmol) to afford the title material as a white solid (0.17 g, 39%). 1H NMR (400 MHz, CDCl3): δ 8.74 (s, 1H), 8.73 (s, 1H), 7.93–7.82 (m, 2H), 7.51–7.36 (m, 2H).
General Procedure D: SNAr Using TFA/TFE
4-(((4-(Benzofuran-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18a)
To a stirring suspension of 17a (0.03 g, 1 equiv, 0.12 mmol) and 15 (0.04 g, 1.20 equiv, 0.15 mmol) in 2,2,2-trifluoroethanol (1.15 mL) was added TFA (0.04 g, 0.02 mL, 2.52 equiv, 0.31 mmol) slowly under an argon atmosphere. The vial was then sealed and heated to 140 °C under microwave conditions for 1 h. The solvent was removed in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20 and then re-purified by prep HPLC to afford the tri-fluoroacetate salt of the title material as a yellow solid (0.02 g, 30%). 1H NMR (400 MHz, MeOD): δ 8.58 (d, J = 5.1 Hz, 1H), 7.99–7.88 (m, 2H), 7.74 (d, J = 7.8 Hz, 1H), 7.71 (s, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.49–7.41 (m, 3H), 7.38 (d, J = 5.1 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 4.81–3.90 (m, 2H), 3.75–3.60 (m, 2H), 3.55–3.40 (m, 1H), 3.26–2.80 (m, 4H), 2.32–2.13 (m, 4H), 2.10–1.94 (m, 2H), 1.76–1.57 (m, 2H). 13C NMR (214 MHz, MeOD): δ 174.12, 162.71, 161.66, 158.84, 158.39, 156.03, 145.37, 130.85, 130.53, 130.12, 128.92, 126.05, 124.60, 120.87, 113.84, 110.37, 109.98, 64.46, 54.16, 31.76, 25.12. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H30N5O2, 468.2394; found, 468.23935.
(4-((4-(Naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18b)
This compound was synthesized according to general procedure D from 15 and 17b (0.05 g, 0.21 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.02 g, 15%). 1H NMR (400 MHz, MeOD): δ 8.71 (s, 1H), 8.55 (d, J = 5.3 Hz, 1H), 8.28 (dd, J = 8.7, 1.8 Hz, 1H), 8.07–7.90 (m, 5H), 7.63–7.54 (m, 3H), 7.52–7.45 (m, 2H), 4.80–3.84 (m, 2H), 3.75–3.63 (m, 2H), 3.54–3.40 (m, 1H), 3.24–2.73 (m, 4H), 2.34–2.11 (m, 4H), 2.13–1.92 (m, 2H), 1.81–1.59 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.89, 166.27, 161.67, 159.84, 144.31, 136.17, 135.64, 134.71, 130.05, 129.60, 129.26, 128.78, 128.74, 128.55, 128.50, 127.72, 125.05, 119.68, 110.03, 63.20, 52.90, 30.06, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C30H32N5O, 478.2601; found, 478.2598.
(4-(Pyrrolidin-1-yl)piperidin-1-yl)(4-((4-(thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)methanone (18c)
This compound was synthesized according to general procedure D from 15 and 17c (0.04 g, 0.20 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (yield not determined). 1H NMR (400 MHz, MeOD): δ 8.41 (d, J = 5.5 Hz, 1H), 7.94 (dd, J = 3.8, 1.1 Hz, 1H), 7.92–7.86 (m, 2H), 7.72 (d, J = 4.8 Hz, 1H), 7.54–7.42 (m, 2H), 7.32 (d, J = 5.5 Hz, 1H), 7.22 (dd, J = 5.0, 3.8 Hz, 1H), 4.80–3.89 (m, 2H), 3.76–3.59 (m, 2H), 3.54–3.38 (m, 1H), 3.26–2.74 (m, 4H), 2.30–2.13 (m, 4H), 2.12–1.89 (m, 2H), 1.80–1.56 (m, 2H). 13C NMR (101 MHz, MeOD): δ 172.71, 162.06, 159.99, 157.93, 143.71, 143.50, 132.07, 129.86, 129.72, 129.40, 129.23, 120.11, 107.94, 63.18, 52.88, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H28N5OS, 434.2009; found, 434.2008.
(4-(Pyrrolidin-1-yl)piperidin-1-yl)(4-((4-(thiazol-2-yl)pyrimidin-2-yl)amino)phenyl)methanone (18d)
This compound was synthesized according to general procedure D from 15 and 17d (0.05 g, 0.18 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.01 g, 14%). 1H NMR (850 MHz, MeOD): δ 8.62 (d, J = 5.0 Hz, 1H), 8.03 (d, J = 3.1 Hz, 1H), 7.95–7.92 (m, 2H), 7.83 (d, J = 3.1 Hz, 1H), 7.57 (d, J = 5.0 Hz, 1H), 7.47–7.43 (m, 2H), 4.82–3.84 (m, 2H), 3.76–3.60 (m, 2H), 3.45 (tt, J = 11.8, 4.1 Hz, 1H), 3.28–2.74 (m, 4H), 2.38–2.11 (m, 4H), 2.10–1.93 (m, 2H), 1.82–1.53 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.78, 168.93, 161.39, 160.86, 159.34, 145.79, 143.86, 129.23, 129.10, 124.62, 119.76, 108.22, 63.20, 52.91, 30.09, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H27N6OS, 435.1962; found, 435.1950.
(4-((4-(Furan-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18e)
This compound was synthesized according to general procedure D from 15 and 17e (0.04 g, 0.19 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.05 g, 64%). 1H NMR (400 MHz, MeOD): δ 8.47 (d, J = 5.3 Hz, 1H), 7.96–7.88 (m, 2H), 7.77 (dd, J = 1.8, 0.8 Hz, 1H), 7.51–7.41 (m, 2H), 7.35 (dd, J = 3.6, 0.8 Hz, 1H), 7.20 (d, J = 5.4 Hz, 1H), 6.67 (dd, J = 3.5, 1.8 Hz, 1H), 4.80–3.86 (m, 2H), 3.80–3.60 (m, 2H), 3.51–3.39 (m, 1H), 3.27–2.72 (m, 4H), 2.33–2.10 (m, 4H), 2.08–1.90 (m, 2H), 1.78–1.49 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.86, 161.35, 159.85, 157.46, 153.38, 146.67, 144.19, 129.22, 128.72, 119.53, 113.46, 113.33, 107.64, 63.20, 52.89, 29.94, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H28N5O2, 418.2238; found, 418.2235.
(4-((4-(1-Methyl-1H-pyrazol-4-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18f)
To a stirred suspension of 17f (0.04 g, 1 equiv, 0.21 mmol) and 15 (0.06 g, 1.1 equiv, 0.23 mmol) in 2,2,2-trifluoroethanol (1.75 mL) was added TFA (0.06 g, 0.04 mL, 2.5 equiv, 0.52 mmol) slowly under argon pressure. The vial was sealed and heated to 140 °C under microwave conditions for 1 h. The solvent was removed in vacuo. The crude compound was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20. Afterward, the resulting product was re-purified using reversed-phase chromatography using a gradient of water (0.5% TFA) and MeOH to afford the trifluoroacetate salt of the title material as a yellow solid (0.02 g, 20%). 1H NMR (400 MHz, MeOD): δ 8.39–8.28 (m, 2H), 8.14 (d, J = 0.7 Hz, 1H), 7.90–7.79 (m, 2H), 7.52–7.43 (m, 2H), 7.17 (d, J = 5.7 Hz, 1H), 4.82–4.10 (m, 2H), 3.98 (s, 3H), 3.77–3.60 (m, 2H), 3.45 (tt, J = 11.8, 3.9 Hz, 1H), 3.28–2.79 (m, 4H), 2.33–2.11 (m, 4H), 2.10–1.97 (m, 2H), 1.75–1.59 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.88, 161.41, 161.13, 159.08, 144.26, 139.55, 132.38, 129.25, 128.65, 123.10, 119.57, 109.18, 63.20, 52.89, 39.29, 29.96, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H30N7O, 432.2506; found, 432.2504.
(4-((4-Phenylpyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18g)
This compound was synthesized according to general procedure D from 15 and 17g (0.06 g, 0.31 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.022 g, 16%). 1H NMR (850 MHz, MeOD): δ 8.51 (d, J = 5.3 Hz, 1H), 8.20–8.12 (m, 2H), 7.96–7.90 (m, 2H), 7.56–7.50 (m, 3H), 7.48–7.44 (m, 2H), 7.40 (d, J = 5.3 Hz, 1H), 4.85–3.88 (m, 2H), 3.74–3.59 (m, 2H), 3.45 (tt, J = 11.8, 4.0 Hz, 1H), 3.26–2.79 (m, 4H), 2.33–2.14 (m, 4H), 2.11–1.90 (m, 2H), 1.73–1.57 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.80, 166.87, 161.10, 159.06, 143.95, 138.16, 132.25, 129.97, 129.26, 129.07, 128.34, 119.91, 109.72, 63.20, 52.90, 30.06, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C26H30N5O, 428.2445; found, 428.2431.
(4-((4-(3,4-Dichlorophenyl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18h)
This compound was synthesized according to general procedure D from 15 and 17h (0.06 g, 0.20 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.025 g, 26%). 1H NMR (850 MHz, MeOD): δ 8.55 (d, J = 5.2 Hz, 1H), 8.41–8.34 (m, 1H), 8.12–8.03 (m, 1H), 7.94–7.86 (m, 2H), 7.69 (d, J = 8.3 Hz, 1H), 7.47–7.43 (m, 2H), 7.39 (dd, J = 5.2, 1.2 Hz, 1H), 4.81–3.94 (m, 2H), 3.73–3.59 (m, 2H), 3.45 (tt, J = 11.8, 4.1 Hz, 1H), 3.27–2.84 (m, 4H), 2.38–2.12 (m, 4H), 2.10–1.94 (m, 2H), 1.81–1.55 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.80, 163.76, 161.58, 160.30, 144.01, 138.70, 135.90, 134.13, 132.11, 130.13, 129.22, 129.01, 127.79, 119.84, 109.60, 63.20, 52.91, 30.15, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C26H28Cl2N5O, 496.1665; found, 496.1656.
(4-((4-(5-Methylthiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(yrrolidine-1-yl)piperidin-1-yl)methanone (18i)
This compound was synthesized according to general procedure D from 15 and 17i (0.04 g, 0.19 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.02 g, 24%). 1H NMR (400 MHz, MeOD): δ 8.35 (d, J = 5.6 Hz, 1H), 7.95–7.86 (m, 2H), 7.76 (d, J = 3.8 Hz, 1H), 7.50–7.43 (m, 2H), 7.25 (d, J = 5.6 Hz, 1H), 6.96–6.84 (m, 1H), 4.79–3.83 (m, 2H), 3.74–3.62 (m, 2H), 3.52–3.39 (m, 1H), 3.24–2.80 (m, 4H), 2.56 (s, 3H), 2.34–2.09 (m, 4H), 2.08–1.93 (m, 2H), 1.78–1.48 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.88, 161.32, 161.15, 159.15, 146.66, 144.17, 141.52, 129.45, 129.20, 128.68, 128.14, 119.55, 107.57, 63.20, 52.88, 29.95, 23.85, 15.64. HRMS–ESI+ (m/z): [M + H]+ calcd for C25H30N5OS, 448.2166; found, 448.2163.
(4-((4-(5-Chlorothiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18j)
This compound was synthesized according to general procedure D from 15 and 17j (0.04 g, 0.17 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.02 g, 27%). 1H NMR (400 MHz, MeOD): δ 8.44 (d, J = 5.3 Hz, 1H), 7.93–7.85 (m, 2H), 7.73 (d, J = 4.0 Hz, 1H), 7.50–7.41 (m, 2H), 7.24 (d, J = 5.3 Hz, 1H), 7.09 (d, J = 4.0 Hz, 1H), 4.78–3.90 (m, 2H), 3.76–3.59 (m, 2H), 3.45 (tt, J = 11.8, 4.0 Hz, 1H), 3.25–2.76 (m, 4H), 2.33–2.11 (m, 4H), 2.10–1.96 (m, 2H), 1.81–1.59 (m, 2H). 13C NMR (214 MHz, MeOD): δ 174.09, 162.47, 161.43, 161.10, 145.23, 144.29, 136.83, 130.49, 130.46, 130.18, 129.87, 120.95, 108.54, 64.45, 54.16, 31.18, 25.11. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H27ClN5OS, 468.1619; found, 468.1618.
(4-((4-(5-Phenylthiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18k)
This compound was synthesized according to general procedure D from 15 and 17k (0.05 g, 0.18 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.04 g, 43%). 1H NMR (400 MHz, MeOD): δ 8.41 (d, J = 5.4 Hz, 1H), 7.97–7.86 (m, 3H), 7.78–7.66 (m, 2H), 7.51–7.42 (m, 5H), 7.41–7.33 (m, 1H), 7.30 (d, J = 5.5 Hz, 1H), 4.83–3.83 (m, 2H), 3.78–3.59 (m, 2H), 3.53–3.38 (m, 1H), 3.26–2.81 (m, 4H), 2.33–2.13 (m, 4H), 2.09–1.96 (m, 2H), 1.80–1.55 (m, 2H). 13C NMR (214 MHz, MeOD): δ 174.15, 162.50, 162.24, 160.71, 151.12, 145.39, 144.24, 136.51, 131.47, 130.79, 130.49, 130.04, 128.20, 126.95, 120.91, 109.05, 64.46, 54.15, 31.48, 25.18. HRMS–ESI+ (m/z): [M + H]+ calcd for C30H32N5OS, 510.2322; found, 510.2324.
(4-((4-(5-(Hydroxymethyl)thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18l)
(5-(2-Chloropyrimidin-4-yl)thiophen-2-yl)methanol (17l) (0.07 g, 1 equiv, 0.31 mmol), 15 (0.10 g, 1.20 equiv, 0.37 mmol), and Cs2CO3 (0.30 g, 3 equiv, 0.93 mmol) were added to 1,4-dioxane (4.00 mL). The resulting suspension was degassed by bubbling argon gas through the solvent for 5 min. Afterward, BINAP (0.06 g, 0.30 equiv, 0.09 mmol) and Pd(OAc)2 (0.01 g, 0.14 equiv, 0.05 mmol) were added to the reaction mixture, which was heated to 90 °C and stirred for 3 h. The mixture was cooled to room temperature and filtered. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20. The isolated product was re-purified using prep HPLC to afford the trifluoroacetate salt of the title material as a yellow solid (0.03 g, 19%). 1H NMR (400 MHz, MeOD): δ 8.39 (d, J = 5.5 Hz, 1H), 7.94–7.88 (m, 2H), 7.81 (d, J = 3.8 Hz, 1H), 7.50–7.42 (m, 2H), 7.28 (d, J = 5.5 Hz, 1H), 7.12–7.04 (m, 1H), 4.81 (s, 2H), 4.75–3.76 (m, 2H), 3.76–3.58 (m, 2H), 3.53–3.39 (m, 1H), 3.25–2.77 (m, 4H), 2.33–2.13 (m, 4H), 2.10–1.95 (m, 2H), 1.78–1.53 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.88, 161.22, 159.40, 151.67, 144.15, 143.17, 129.21, 128.92, 128.72, 126.97, 119.61, 107.74, 63.20, 60.35, 52.90, 30.22, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C25H30N5O2S, 464.2115; found, 464.2102.
(4-(Pyrrolidin-1-yl)piperidin-1-yl)(4-((4-(5-((2,2,2-trifluoroethoxy)methyl)thiophen-2-yl)pyrimidin-2-yl)amino)phenyl)methanone (18m)
This compound was synthesized according to general procedure D from 15 and 17l (0.04 g, 0.18 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.02 g, 19%). 1H NMR (400 MHz, MeOD): δ 8.43 (d, J = 5.3 Hz, 1H), 7.95–7.88 (m, 2H), 7.80 (d, J = 3.8 Hz, 1H), 7.49–7.40 (m, 2H), 7.28 (d, J = 5.3 Hz, 1H), 7.19–7.13 (m, 1H), 4.89 (s, 2H), 4.82–4.10 (m, 2H), 4.00 (q, J = 8.9 Hz, 2H), 3.73–3.61 (m, 2H), 3.51–3.40 (m, 1H), 3.27–2.89 (m, 4H), 2.29–2.12 (m, 4H), 2.09–1.96 (m, 2H), 1.75–1.56 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.87, 161.28, 160.90, 159.65, 145.58, 144.88, 144.12, 129.48, 129.21, 128.75, 128.72, 125.69, 119.60, 107.86, 69.54, 67.86 (q, J = 34.7 Hz), 63.20, 52.90, 29.89, 23.84. HRMS–ESI+ (m/z): [M + H]+ calcd for C27H31F3N5O2S, 546.2145 [M + H]+; found, 546.2141.
(4-((4-(Benzo[b]thiophen-2-yl)-5-methylpyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18n)
This compound was synthesized according to general procedure D from 15 and 17n (0.05 g, 0.19 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.01 g, 8%). 1H NMR (400 MHz, MeOD): δ 8.38 (s, 1H), 8.05 (s, 1H), 7.99–7.87 (m, 4H), 7.49–7.44 (m, 2H), 7.44–7.37 (m, 2H), 4.82–3.89 (m, 2H), 3.73–3.61 (m, 2H), 3.50–3.40 (m, 1H), 3.27–2.91 (m, 4H), 2.57 (s, 3H), 2.33–2.10 (m, 4H), 2.08–1.92 (m, 2H), 1.77–1.58 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.96, 161.96, 159.45, 158.67, 145.16, 144.35, 142.07, 141.83, 129.29, 128.40, 128.29, 127.24, 126.08, 125.74, 123.08, 119.43, 119.23, 63.21, 52.90, 30.01, 23.85, 17.97. HRMS–ESI+ (m/z): [M + H]+ calcd for C29H32N5OS, 498.2322; found, 498.23193.
(4-((4-(Benzo[b]thiophen-2-yl)-5-cyclopropylpyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18o)
To a stirred suspension of 4-(benzo[b]thiophen-2-yl)-2-chloro-5-cyclopropylpyrimidine (17o) (0.035 g, 1 equiv, 0.12 mmol) and 15 (0.04 g, 1.2 equiv, 0.15 mmol) in 2,2,2-trifluoroethanol (1.25 mL) was added TFA (0.04 g, 0.02 mL, 2.5 equiv, 0.31 mmol) slowly under argon pressure. The vial was sealed and heated to 140 °C under microwave conditions for 1 h. The solvent was removed in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 93:7 to afford the title material as a yellow solid (0.01 g, 19%). 1H NMR (400 MHz, MeOD): δ 8.42 (s, 1H), 8.38 (s, 1H), 7.98–7.83 (m, 4H), 7.47–7.43 (m, 2H), 7.43–7.33 (m, 2H), 4.75–3.71 (m, 2H), 3.58–3.31 (m, 3H), 3.22–2.77 (m, 4H), 2.34–1.98 (m, 7H), 1.74–1.56 (m, 2H), 1.22–1.06 (m, 2H), 0.86–0.64 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.92, 160.63, 160.35, 159.21, 144.67, 144.25, 142.10, 142.04, 129.37, 129.28, 128.55, 127.24, 126.12, 125.70, 124.21, 123.11, 119.32, 63.22, 52.88, 30.16, 23.87, 13.14, 7.87. HRMS–ESI+ (m/z): [M + H]+ calcd for C31H34N5OS, 524.2479; found, 524.2477.
(4-((4-(Benzo[b]thiophen-2-yl)-5-methoxypyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18p)
4-(Benzo[b]thiophen-2-yl)-2-chloro-5-methoxypyrimidine (17p) (0.060 g, 1 equiv, 0.22 mmol), 15 (0.065 g, 1.1 equiv, 0.24 mmol), and Cs2CO3 (0.21 g, 3 equiv, 0.65 mmol) were added to 1,4-dioxane (3.00 mL). The resulting suspension was degassed by bubbling argon gas for 5 min. Afterward, BINAP (0.041 g, 0.30 equiv, 0.066 mmol) and Pd(OAc)2 (0.007 g, 0.15 equiv, 0.03 mmol) were added to the reaction mixture, which was heated to 90 °C and stirred for 3 h. The mixture was cooled to room temperature and then filtered. The filtrate was concentrated in vacuo, and the crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20. The isolated product was re-purified using prep HPLC to afford the trifluoroacetate salt of the title material as a yellow solid (0.01 g, 9%). 1H NMR (400 MHz, MeOD): δ 8.42 (s, 2H), 7.96–7.82 (m, 4H), 7.49–7.42 (m, 2H), 7.44–7.35 (m, 2H), 4.80–4.16 (m, 2H), 4.11 (s, 3H), 3.75–3.60 (m, 2H), 3.53–3.39 (m, 1H), 3.26–2.67 (m, 4H), 2.36–2.11 (m, 4H), 2.11–1.91 (m, 2H), 1.81–1.55 (m, 2H). 13C NMR (151 MHz, MeOD): δ 173.05, 155.39, 149.11, 146.29, 144.81, 144.46, 142.11, 141.81, 130.00, 129.38, 127.86, 127.20, 126.04, 125.72, 123.13, 118.54, 63.24, 57.42, 52.92, 30.03, 23.85. HRMS–ESI+ (m/z): [M + H]+ calcd for C29H32N5O2S, 514.2271; found, 514.2269.
(4-((4-(Benzo[b]thiophen-2-yl)-5-fluoropyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18q)
This compound was synthesized according to general procedure D from 15 and 17p (0.04 g, 0.13 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.007 g, 10%). 1H NMR (400 MHz, MeOD): δ 8.45 (d, J = 3.4 Hz, 1H), 8.19 (dd, J = 1.6, 0.8 Hz, 1H), 7.96–7.84 (m, 4H), 7.50–7.37 (m, 4H), 4.78–3.80 (m, 2H), 3.77–3.60 (m, 2H), 3.52–3.39 (m, 1H), 3.26–2.75 (m, 4H), 2.33–2.11 (m, 4H), 2.10–1.92 (m, 2H), 1.79–1.55 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.86, 157.53 (d, JC–F = 2.7 Hz), 150.92 (d, JC–F = 256 Hz), 147.99 (d, JC–F = 25 Hz), 147.86 (d, JC–F = 10 Hz), 144.14, 142.12, 141.90, 139.41 (d, JC–F = 7.2 Hz), 129.79 (d, JC–F = 14 Hz), 129.29, 128.74, 127.72, 126.29, 126.05, 123.34, 119.12, 63.21, 52.90, 30.01, 23.86. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H29N5OSF, 502.207; found, 502.2069.
(4-((4-(Benzo[b]thiophen-2-yl)-5-chloropyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18r)
This compound was synthesized according to general procedure D from 15 and 17r (0.04 g, 0.12 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.01 g, 10%). 1H NMR (400 MHz, MeOD): δ 8.60 (d, J = 0.9 Hz, 1H), 8.50 (s, 1H), 7.99–7.79 (m, 4H), 7.53–7.32 (m, 4H), 4.80–3.92 (m, 2H), 3.67 (s, 2H), 3.57–3.41 (m, 1H), 3.28–2.84 (m, 4H), 2.34–2.11 (m, 4H), 2.10–1.91 (m, 2H), 1.83–1.52 (m, 2H). 13C NMR (101 MHz, MeOD): δ 171.34, 159.09, 157.56, 154.48, 142.23, 140.87, 140.82, 140.29, 128.71, 127.83, 127.74, 126.36, 125.00, 124.52, 121.69, 118.25, 116.51, 61.76, 51.47, 28.55, 22.43. HRMS–-ESI+ (m/z): [M + H]+ calcd for C28H29ClN5OS, 518.1776; found, 518.1775.
(4-((4-(Benzo[b]thiophen-2-yl)-5-bromopyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (18s)
This compound was synthesized according to general procedure D from 15 and 17s (0.06 g, 0.18 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.008 g, 8%). 1H NMR (400 MHz, MeOD): δ 8.73 (s, 1H), 8.63 (s, 1H), 8.01–7.88 (m, 4H), 7.54–7.34 (m, 4H), 4.79–3.85 (m, 2H), 3.74–3.59 (m, 2H), 3.53–3.37 (m, 1H), 3.26–2.75 (m, 4H), 2.32–2.11 (m, 4H), 2.09–1.94 (m, 2H), 1.74–1.54 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.77, 163.28, 159.33, 157.31, 143.60, 143.09, 142.45, 141.51, 129.89, 129.26, 129.24, 127.76, 126.41, 125.96, 123.13, 119.78, 105.82, 63.19, 52.90, 29.57, 23.86. HRMS–ESI+ (m/z): [M + H]+ calcd for C28H29N5OSBr, 562.127; found, 562.1268.
4-(Benzo[b]thiophen-2-yl)-2-((4-(4-(pyrrolidin-1-yl)piperidine-1-carbonyl)phenyl)amino)pyrimidine-5-carbonitrile (18t)
A mixture of 18s (0.025 g, 1 equiv, 0.037 mmol), Pd(dppf)2Cl2 (0.008 g, 0.30 equiv, 0.01 mmol), Zn(CN)2 (0.026 g, 0.014 mL, 6.0 equiv, 0.22 mmol), and DIPEA (0.019 g, 0.026 mL, 4.0 equiv, 0.15 mmol) in DMF (1.00 mL) was stirred at 170 °C under microwave conditions for 30 min. Water and NaHCO3 solution were added to the crude reaction mixture, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and dried with Na2SO4, and then the solvent was removed in vacuo. The crude product was purified by silica gel chromatography using a system of DCM/MeOH 100:0 to 80:20. The isolated product was re-purified by prep HPLC to afford the trifluoroacetate salt of the title material as a yellow solid (0.004 g, 20%). 1H NMR (850 MHz, MeOD): δ 8.77 (s, 1H), 8.65 (s, 1H), 7.98–7.87 (m, 4H), 7.53–7.50 (m, 2H), 7.49 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 7.44 (ddd, J = 8.0, 7.0, 1.0 Hz, 1H), 4.84–4.51 (m, 1H), 4.26–3.89 (m, 1H), 3.75–3.63 (m, 2H), 3.48 (tt, J = 11.7, 4.0 Hz, 1H), 3.29–2.74 (m, 4H), 2.39–2.12 (m, 4H), 2.08–1.99 (m, 2H), 1.81–1.66 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.43, 164.69, 161.69, 160.93, 142.86, 142.32, 141.58, 141.41, 130.83, 129.47, 129.18, 128.42, 126.71, 126.29, 123.41, 121.06, 118.54, 63.18, 52.87, 30.38, 23.89. HRMS–ESI+ (m/z): [M + H]+ calcd for C29H29N6OS, 509.2118; found, 509.2105.
2-Chloro-5-methyl-4-(5-methylthiophen-2-yl)pyrimidine (19a)
This compound was synthesized according to general procedure A starting from (5-methylthiophen-2-yl)boronic acid (0.26 g, 1.84mmol) and 2,4-dichloro-5-methylpyrimidine (16n) (0.30 g, 1.84 mmol) to afford the title material as a yellow solid (0.20 g, 50%). 1H NMR (400 MHz, CDCl3): δ 8.34 (d, J = 0.9 Hz, 1H), 7.59 (d, J = 3.8 Hz, 1H), 6.91–6.80 (m, 1H), 2.55 (d, J = 1.0 Hz, 3H), 2.49 (s, 3H). LCMS (ESI+) m/z: 225 [M + H]+.
2,5-Dichloro-4-(5-methylthiophen-2-yl)pyrimidine (19b)
This compound was synthesized according to general procedure A starting from (5-methylthiophen-2-yl)boronic acid (0.23 g, 1.64 mmol) and 2,4,5-trichloropyrimidine (16r) (0.30 g, 1.64 mmol) to afford the title material as a white solid (0.17 g, 44%). 1H NMR (400 MHz, DMSO): δ 8.85 (s, 1H), 8.18 (d, J = 3.9 Hz, 1H), 7.08–6.98 (m, 1H), 2.55 (d, J = 0.5 Hz, 3H). LCMS (ESI+) m/z: 245 [M + H]+.
2-Chloro-4-(5-chlorothiophen-2-yl)-5-methylpyrimidine (19c)
This compound was synthesized according to general procedure A starting from (5-chlorothiophen-2-yl)boronic acid (0.30 g, 1.84 mmol) and 2,4-dichloro-5-methylpyrimidine (16n) (0.30 g, 1.84 mmol) to afford the title material as a white solid (0.22 g, 48%). 1H NMR (400 MHz, CDCl3): δ 8.40 (q, J = 0.8 Hz, 1H), 7.54 (d, J = 4.2 Hz, 1H), 7.01 (d, J = 4.1 Hz, 1H), 2.50 (d, J = 0.8 Hz, 3H). LCMS (ESI+) m/z: 245 [M + H]+.
2,5-Dichloro-4-(5-chlorothiophen-2-yl)pyrimidine (19d)
This compound was synthesized according to general procedure A starting from (5-chlorothiophen-2-yl)boronic acid (0.27 g, 1.64 mmol) and 2,4,5-trichloropyrimidine (16r) (0.30 g, 1.64 mmol) to afford the title material as a white solid (0.16 g, 38%). 1H NMR (400 MHz, dmso): δ 8.93 (s, 1H), 8.21 (d, J = 4.3 Hz, 1H), 7.37 (d, J = 4.3 Hz, 1H). LCMS (ESI+) m/z: 265 [M + H]+.
N-(3-Morpholinopropyl)-4-nitrobenzamide (21)
To a stirring solution of 4-nitrobenzoic acid (20) (0.30 g, 1.00 equiv, 1.80 mmol) and DIPEA (0.46 g, 0.63 mL, 2.00 equiv, 3.59 mmol) in DMF (5.00 mL) was added HATU (1.37 g, 2.00 equiv, 3.59 mmol), and the reaction was allowed to stir at room temperature for 15 min. Afterward, 3-morpholinopropan-1-amine (0.29 g, 0.29 mL, 1.10 equiv, 1.98 mmol) was added, and the reaction was heated to 40 °C and stirred for 4 h. Upon completion of the reaction, the mixture was poured onto ice water, and the aqueous layer was extracted with ethyl acetate. The combined organic extracts were washed with water and brine and then dried with Na2SO4. After filtration, the solvent was removed in vacuo, and the residue was purified using silica gel chromatography using a system of DCM/MeOH–NH3 to afford the title material as a yellow solid (0.15 g, 29%). 1H NMR (400 MHz, DMSO): δ 8.82 (t, J = 5.6 Hz, 1H), 8.34–8.28 (m, 2H), 8.11–8.01 (m, 2H), 3.61–3.55 (m, 4H), 3.35–3.28 (m, 2H), 2.47–2.37 (m, 6H), 1.72 (p, J = 7.1 Hz, 2H). LCMS (ESI+) m/z: 294 [M + H]+.
4-Amino-N-(3-morpholinopropyl)benzamide (22)
To a stirring suspension of 21 (0.150 g, 1 equiv, 0.511 mmol) in EtOH/water (2.70 mL, 3:1) were added iron (0.09 g, 3.00 equiv, 1.53 mmol) and NH4Cl (0.14 g, 5.01 equiv, 2.56 mmol), and the reaction mixture was heated to reflux and stirred for 4 h. Upon completion, the reaction mixture was filtered while hot, and the filtering agent was rinsed with ethyl acetate. The filtrate was concentrated in vacuo, and the resulting crude product was used as such for the next step without further purification.
(4-((5-Methyl-4-(5-methylthiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (23a)
This compound was synthesized according to general procedure D from 15 and 19a (0.10 g, 0.45 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.037 g, 14%). 1H NMR (400 MHz, MeOD): δ 8.24 (s, 1H), 7.91–7.82 (m, 2H), 7.65 (d, J = 3.9 Hz, 1H), 7.48–7.40 (m, 2H), 6.97–6.86 (m, 1H), 4.82–3.91 (m, 2H), 3.71–3.57 (m, 2H), 3.44 (tt, J = 11.7, 7.8, 4.1 Hz, 1H), 3.25–2.80 (m, 4H), 2.56 (s, 3H), 2.44 (s, 3H), 2.28–2.11 (m, 4H), 2.07–1.92 (m, 2H), 1.77–1.54 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.78, 160.05, 158.94, 157.86, 147.48, 143.69, 142.31, 132.74, 129.26, 129.03, 128.53, 119.71, 118.13, 63.19, 52.88, 30.00, 23.85, 17.95, 15.49. HRMS–ESI+ (m/z): [M + H]+ calcd for C26H32N5OS, 462.2322; found, 462.2321.
(4-((5-Chloro-4-(5-methylthiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (23b)
This compound was synthesized according to general procedure D from 15 and 19b (0.10 g, 0.41 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.026 g, 11%). 1H NMR (400 MHz, MeOD): δ 8.42 (s, 1H), 8.14 (d, J = 3.9 Hz, 1H), 7.93–7.84 (m, 2H), 7.48–7.42 (m, 2H), 6.97–6.86 (m, 1H), 4.79–3.93 (m, 2H), 3.77–3.58 (m, 2H), 3.54–3.38 (m, 1H), 3.24–2.82 (m, 4H), 2.57 (s, 3H), 2.35–2.13 (m, 4H), 2.10–1.92 (m, 2H), 1.77–1.52 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.81, 160.08, 158.88, 155.95, 147.87, 143.84, 139.69, 133.60, 129.22, 128.96, 128.27, 119.53, 116.70, 63.19, 52.89, 29.91, 23.85, 15.47. HRMS–ESI+ (m/z): [M + H]+ calcd for C25H29ClN5OS, 482.1776; found, 482.1775.
(4-((4-(5-Chlorothiophen-2-yl)-5-methylpyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (23c)
This compound was synthesized according to general procedure D from 15 and 19c (0.10 g, 0.408 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.03 g, 12%). 1H NMR (400 MHz, MeOD): δ 8.33 (s, 1H), 7.91–7.81 (m, 2H), 7.60 (d, J = 4.2 Hz, 1H), 7.47–7.37 (m, 2H), 7.10 (d, J = 4.2 Hz, 1H), 4.79–3.98 (m, 2H), 3.74–3.61 (m, 2H), 3.51–3.40 (m, 1H), 3.25–2.92 (m, 4H), 2.45 (s, 3H), 2.30–2.09 (m, 4H), 2.07–1.93 (m, 2H), 1.75–1.59 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.90, 161.94, 159.26, 157.51, 144.41, 144.21, 135.58, 131.08, 129.25, 129.24, 128.54, 119.31, 118.18, 63.21, 52.90, 30.14, 23.85, 17.58. HRMS–ESI+ (m/z): [M + H]+ calcd for C25H29ClN5OS, 482.1776; found, 472.1775.
(4-((5-Chloro-4-(5-chlorothiophen-2-yl)pyrimidin-2-yl)amino)phenyl)(4-(pyrrolidin-1-yl)piperidin-1-yl)methanone (23d)
This compound was synthesized according to general procedure D from 15 and 19d (0.10 g, 0.37 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.022 g, 10%). 1H NMR (400 MHz, MeOD): δ 8.46 (s, 1H), 8.15 (d, J = 4.2 Hz, 1H), 7.93–7.74 (m, 2H), 7.53–7.33 (m, 2H), 7.12 (d, J = 4.2 Hz, 1H), 4.79–3.83 (m, 2H), 3.75–3.59 (m, 2H), 3.45 (tt, J = 11.7, 4.0 Hz, 1H), 3.26–2.82 (m, 4H), 2.34–2.11 (m, 4H), 2.06–1.93 (m, 2H), 1.74–1.54 (m, 2H). 13C NMR (214 MHz, MeOD): δ 172.72, 160.61, 158.89, 154.68, 143.56, 141.24, 136.97, 132.88, 129.31, 129.29, 129.21, 119.74, 116.70, 63.18, 52.90, 29.97, 23.86. HRMS–ESI+ (m/z): [M + H]+ calcd for C24H26Cl2N5OS, 502.1230; found, 502.1229.
4-((5-Chloro-4-(5-chlorothiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-morpholinopropyl)benzamide (23e)
This compound was synthesized according to general procedure D from 22 (0.04 g, 0.16 mmol) and 19d (0.05 g, 0.20 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.006 g, 6%). 1H NMR (850 MHz, MeOD): δ 8.50 (s, 1H), 8.17 (d, J = 4.1 Hz, 1H), 7.89–7.87 (m, 2H), 7.87–7.83 (m, 2H), 7.14 (d, J = 4.1 Hz, 1H), 4.13–4.07 (m, 2H), 3.85–3.76 (m, 2H), 3.55–3.49 (m, 4H), 3.26–3.21 (m, 2H), 3.20–3.13 (m, 2H), 2.12–2.05 (m, 2H). 13C NMR (214 MHz, MeOD): δ 170.72, 160.70, 158.82, 154.66, 145.03, 141.32, 136.99, 132.93, 129.37, 129.33, 127.66, 119.40, 116.88, 65.21, 55.95, 53.17, 37.44, 25.45. HRMS–ESI+ (m/z): [M + H]+ calcd for C22H24Cl2N5O2S, 492.1022; found, 492.1021.
4-((5-Chloro-4-(5-methylthiophen-2-yl)pyrimidin-2-yl)amino)-N-(3-morpholinopropyl)benzamide (23f)
This compound was synthesized according to general procedure D from 22 (0.04 g, 0.16 mmol) and 19b (0.04 g, 0.14 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.010 g, 10%). 1H NMR (850 MHz, MeOD): δ 8.44 (s, 1H), 8.16 (d, J = 3.8 Hz, 1H), 7.91–7.89 (m, 2H), 7.88–7.86 (m, 2H), 7.05–6.85 (m, 1H), 4.12–4.05 (m, 2H), 3.84–3.77 (m, 2H), 3.55–3.49 (m, 4H), 3.27–3.22 (m, 2H), 3.17 (td, J = 12.4, 3.7 Hz, 2H), 2.57 (d, J = 1.1 Hz, 3H), 2.11–2.05 (m, 2H). 13C NMR (214 MHz, MeOD): δ 170.80, 160.15, 158.79, 155.91, 147.88, 145.31, 139.76, 133.63, 129.32, 128.32, 127.33, 119.21, 116.86, 65.21, 55.93, 53.17, 37.43, 25.46, 15.46. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H27ClN5O2S, 472.1568; found, 472.1567.
4-((4-(5-Chlorothiophen-2-yl)-5-methylpyrimidin-2-yl)amino)-N-(3-morpholinopropyl)benzamide (23g)
This compound was synthesized according to general procedure D from 22 (0.04 g, 0.16 mmol) and 19c (0.04 g, 0.14 mmol) to afford the trifluoroacetate salt of the title material as a yellow solid (0.010 g, 15%). 1H NMR (850 MHz, MeOD): δ 8.35 (d, J = 0.8 Hz, 1H), 7.88–7.86 (m, 4H), 7.61 (d, J = 4.1 Hz, 1H), 7.11 (d, J = 4.1 Hz, 1H), 4.13–4.04 (m, 2H), 3.85–3.77 (m, 2H), 3.55–3.48 (m, 4H), 3.26–3.21 (m, 2H), 3.17 (td, J = 12.4, 3.7 Hz, 2H), 2.46 (s, 3H), 2.13–2.01 (m, 2H). 13C NMR (214 MHz, MeOD): δ 170.84, 161.84, 159.04, 157.56, 145.61, 144.43, 135.69, 131.20, 129.33, 129.31, 126.97, 119.02, 118.36, 65.21, 55.93, 53.16, 37.41, 25.46, 17.58. HRMS–ESI+ (m/z): [M + H]+ calcd for C23H27ClN5O2S, 472.1568; found, 472.1568.
Biology
KinaseSeeker Assay
Stock solutions (10 mM) of test compounds were serially diluted in DMSO to make assay stocks. Prior to initiating screening or IC50 determination, the test compounds were evaluated for false positive against split luciferase. The test compound was screened against Pf kinases at a minimum of eight different concentrations in duplicate. For kinase assays, a 24 mL aliquot of lysate containing Cfluc-kinase and Fos-Nfluc was incubated with either 1 μL of DMSO (for no-inhibitor control) or compound solution in DMSO for 2 h in the presence of a kinase-specific probe. Luciferin assay reagent (80 μL) was added to each solution, and luminescence was immediately measured on a luminometer. The % inhibition was calculated using the following equation: % inhibition = [ALU (control) – ALU (sample)]/ALU (control) × 100. For IC50 determinations, each compound was tested at a minimum of eight different concentrations. The % inhibition was plotted against compound concentration, and the IC50 value was determined for each compound using an eight-point curve.
P. falciparum Blood Stage Culture and Parasite Load Assays
P. falciparum 3D7 parasites were continuously cultured in vitro in complete medium [10.44 g/L RPMI 1640 (Thermo Fisher Scientific), 25 mM HEPES, pH 7.2 (Thermo Fisher Scientific), 0.37 mM hypoxanthine (Sigma), 24 mM sodium bicarbonate (Sigma), 0.5% (wt/vol) AlbuMAX II (Thermo Fisher Scientific), and 25 μg/mL gentamicin (Sigma)] supplemented with freshly washed human erythrocytes (Gulf Coast Regional Blood Center, Houston, TX) approximately every 48 h. The parasite cultures were maintained at 2–10% parasitemia with 1% hematocrit at 37 °C in a 3% O2, 5% CO2, and 92% N2 atmosphere. Highly synchronized cultures were generated by treatment with 25 volumes of 5% (wt/vol) D-sorbitol (Sigma) at 37 °C for 10 min during the early ring stage. Prior to the assays, P. falciparum 3D7 parasites were synchronized as described above and adjusted to 2% parasitemia and 2% hematocrit. Compounds were initially assayed for parasite inhibition at 1 μM. Dose–response curves were generated for select compounds by dispensing 100 μL of the culture into each well of a 96-well black plate (Corning), followed by administration of compounds (0–5 μM) in triplicate (HP D300 Digital Dispenser). Quinacrine at 500 nM was employed as the positive control and 0.5% DMSO as the negative control. Plates were incubated at 37 °C in a 3% O2, 5% CO2, and 92% N2 atmosphere after drug administration. At 34 h post-reinvasion (i.e., 72 h after drug administration), 40 μL of lysis solution [20 mM Tris–HCl, pH 7.5 (Fisher Chemical), 5 mM EDTA dipotassium salt dihydrate (Fisher Chemical), 0.16% (wt/vol) saponin (Sigma), and 1.6% (vol/vol) Triton X-100 (Fisher Chemical)] containing fresh 10x SYBR Green I (Thermo Fisher Scientific) was added to each well and incubated in the dark at room temperature for 24 h. The fluorescent signals were measured at 535 nm with excitation at 485 nm using an EnVision 2105 multimode plate reader (PerkinElmer). Data was normalized to the negative and positive controls to obtain the relative percent parasite load. EC50 values were determined by fitting data to a standard dose–response equation (GraphPad Prism). The Z-factor ranged from 0.5 to 0.9.
HepG2 CellTiter-Glo Cytotoxicity Assay
HepG2 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS, 1% NEAA, and 1% l-glutamine. No antibiotics were used. Cells were plated at 4000 cells/well in a 384-well plate (Costar) and incubated overnight (37 °C, 5% CO2) before adding compounds. Compounds were added in quadruplicate and incubated for 48 h. The DMSO percentage was constant across all concentrations of compound. The cell viability was measured using CellTiter-Glo2 (Promega), and the luminescence signal was read on a GloMax plate reader (Promega).
P. berghei Liver Stage Assays
HepG2 cells were maintained in DMEM with l-glutamine (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS) (v/v) (Sigma-Aldrich) and 1% antibiotic-antimycotic (Thermo Fisher Scientific) in a standard tissue culture incubator (37 °C, 5% CO2). P. berghei ANKA sporozoites used for liver stage experiments were isolated from freshly dissected salivary glands of infected mosquitoes (University of Georgia SporoCore). Dose–response curves were generated for select compounds by assessing the P. berghei parasite load in hepatocytes as previously described (PMID: 22586124). Briefly, HepG2 (8,000 cells/well) were seeded into 384-well white microplates (Corning). After 24 h, compounds (0–100 μM) were added (HP D300 Digital Dispenser) before infection with P. berghei ANKA sporozoites (4000 spz/well). DMSO (1% v/v) was added as the negative control. All samples were evaluated in triplicate and had a final DMSO concentration of 1%. After 44 h post-infection, the HepG2 cell viability and parasite load were assessed using CellTiter-Fluor (Promega) and Bright-Glo (Promega) reagents, respectively, according to manufacturer’s protocols. The relative fluorescence and luminescence signal was measured using an EnVision plate reader (PerkinElmer). The signal intensity of each well was normalized to the negative control (1% DMSO) to assess the relative viability. Dose–response analysis was performed with GraphPad Prism.
Molecular Modeling
The sequences of the target proteins, PfPK6 and PfGSK-3, were obtained from Uniprot (accession numbers: AF091845 and O77344, respectively). The homology models were generated using Modeller v10.1., where five different templates were used in building the target models of each protein. This technique helps in attaining the desired flexibility of the ATP-binding site of the target protein while, at the same time, providing a high degree of confidence about the sequence alignment. The X-ray crystal structures of the templates with their co-crystallized ligands were downloaded from RCSB in PDB format (sequence identity with the target proteins and resolution of each template are illustrated in Table S5, Supporting Information). The sequences of the target proteins were aligned with the sequences of the related templates using Clustal Omega online tool and, when needed, the adjustment was done by BioEdit. Modeller software was then used to build seven loop-refined models for each target protein, and the model with the lowest PDF violation was chosen for subsequent processing.43 The addition of hydrogen atoms and energy minimization of the crude models were performed using the Molecular Operating Environment (MOE) software, v2019. The minimization step was carried out to a gradient of 0.001 in three sequential steps, where the positional restraints on heavy atoms, backbone, and the entire protein were gradually reduced in each step, respectively. The final homology models were evaluated by different tools. The backbone RMSD of the model was measured relative to that of its template crystal structure individually, and fortunately, the deviation in all cases was less than 0.5 Å, suggesting an appropriate preservation of the template’s structural information. In addition, the overall stereochemical quality of the obtained models was also assessed using Procheck software. The analysis of the Ramachandran plot revealed that over 90% of the residues were found in the favorable regions, while more than 8% of the residues lie in the additional allowed region, with no steric clashes, distorted geometry, or outliers. The minimized proteins were then subjected to further refinement using molecular dynamics simulations to ensure better quality of the generated models. Initially, each protein was solvated in a rectangular TIP3P water box, with 10 Å edge distance, using CHARMM-GUI online tool. The protein–water system was then equilibrated in GROMACS software 2021 for 500 ps using the NVT ensemble, followed by an MD production stage for 10 ns.44 The final structure after simulation was first re-minimized using MOE (as per the protocol previously described) before being used in the molecular docking study. The Dock module in MOE software was utilized to dock the compounds into the putative binding site of the PfPK6 and PfGSK3 homology models. The active site was predicted using a site finder guided by the amino acids of the hinge region in both proteins, where dummy atoms were added to determine this binding pocket. Docking was performed using the induced fit protocol while other parameters were maintained as default. A maximum of 30 poses were generated for each ligand, and the best poses were selected based on the provided score as well as visual examination. The top scoring pose showing H-bonds with the hinge region was selected, minimized, and used for the analysis. Docking poses were analyzed using the PyMOL Molecular Graphics software 2.5.2 (Schrödinger, LLC., New York, NY, USA).
Acknowledgments
This work was supported in part by the National Institutes of Health under grant no. 1R44AI150237-01 and the National Science Foundation under grant nos. 1644868 and CHE-1726291. We thank the University of North Carolina’s Department of Chemistry Mass Spectrometry Core Laboratory, especially Diane Weatherspoon, for their assistance with mass spectrometry analysis. We are grateful for the support by the Structural Genomics Consortium (SGC), a registered charity (no. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and USA), Pfizer, and Takeda.
Glossary
Abbreviations used
- EDCI
N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- LE
ligand efficiency
- SAR
structure–activity relationship
- SN
nucleophilic aromatic substitution
- SN2
nucleophilic substitution (bimolecular)
- TCAMS
Tres Cantos antimalarial set
- TEA
triethylamine
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00996.
The authors declare no competing financial interest.
Supplementary Material
References
- Miller L. H.; Ackerman H. C.; Su X.; Wellems T. E. Malaria Biology and Disease Pathogenesis: Insights for New Treatments. Nat. Med. 2013, 19, 156–167. 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, World Health Organization . World Malaria Report 2021 Global Messaging, 2021.
- Cabrera D. G.; Horatscheck A.; Wilson C. R.; Basarab G.; Eyermann C. J.; Chibale K. Plasmodial Kinase Inhibitors: License to Cure?. J. Med. Chem. 2018, 61, 8061–8077. 10.1021/acs.jmedchem.8b00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biamonte M. A.; Wanner J.; Le Roch K. G. Recent Advances in Malaria Drug Discovery. Bioorg. Med. Chem. Lett. 2013, 23, 2829–2843. 10.1016/j.bmcl.2013.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwimana A.; Legrand E.; Stokes B. H.; Ndikumana J.-L. M.; Warsame M.; Umulisa N.; Ngamije D.; Munyaneza T.; Mazarati J.-B.; Munguti K.; Campagne P.; Criscuolo A.; Ariey F.; Murindahabi M.; Ringwald P.; Fidock D. A.; Mbituyumuremyi A.; Menard D. Emergence and Clonal Expansion of in Vitro Artemisinin-Resistant Plasmodium Falciparum Kelch13 R561H Mutant Parasites in Rwanda. Nat. Med. 2020, 26, 1602–1608. 10.1038/s41591-020-1005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balikagala B.; Fukuda N.; Ikeda M.; Katuro O. T.; Tachibana S.-I.; Yamauchi M.; Opio W.; Emoto S.; Anywar D. A.; Kimura E.; Palacpac N. M. Q.; Odongo-Aginya E. I.; Ogwang M.; Horii T.; Mita T. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. 10.1056/NEJMoa2101746. [DOI] [PubMed] [Google Scholar]
- Forte B.; Ottilie S.; Plater A.; Campo B.; Dechering K. J.; Gamo F. J.; Goldberg D. E.; Istvan E. S.; Lee M.; Lukens A. K.; McNamara C. W.; Niles J. C.; Okombo J.; Pasaje C. F. A.; Siegel M. G.; Wirth D.; Wyllie S.; Fidock D. A.; Baragaña B.; Winzeler E. A.; Gilbert I. H. Prioritization of Molecular Targets for Antimalarial Drug Discovery. ACS Infect. Dis. 2021, 7, 2764–2776. 10.1021/acsinfecdis.1c00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendse L. B.; Wyllie S.; Chibale K.; Gilbert I. H. Plasmodium Kinases as Potential Drug Targets for Malaria: Challenges and Opportunities. ACS Infect. Dis. 2021, 7, 518–534. 10.1021/acsinfecdis.0c00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang V. M.; Chavchich M.; Waters N. C. Targeting Protein Kinases in the Malaria Parasite: Update of an Antimalarial Drug Target. Curr. Top. Med. Chem. 2012, 12, 456–472. 10.2174/156802612799362922. [DOI] [PubMed] [Google Scholar]
- Solyakov L.; Halbert J.; Alam M. M.; Semblat J. P.; Dorin-Semblat D.; Reininger L.; Bottrill A. R.; Mistry S.; Abdi A.; Fennell C.; Holland Z.; Demarta C.; Bouza Y.; Sicard A.; Nivez M. P.; Eschenlauer S.; Lama T.; Thomas D. C.; Sharma P.; Agarwal S.; Kern S.; Pradel G.; Graciotti M.; Tobin A. B.; Doerig C. Global Kinomic and Phospho-Proteomic Analyses of the Human Malaria Parasite Plasmodium Falciparum. Nat. Commun. 2011, 2, 565. 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- Tewari R.; Straschil U.; Bateman A.; Böhme U.; Cherevach I.; Gong P.; Pain A.; Billker O. The Systematic Functional Analysis of Plasmodium Protein Kinases Identifies Essential Regulators of Mosquito Transmission. Cell Host Microbe 2010, 8, 377–387. 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho T. G.; Morahan B.; John von Freyend S.; Boeuf P.; Grau G.; Garcia-Bustos J.; Doerig C. The Ins and Outs of Phosphosignalling in Plasmodium: Parasite Regulation and Host Cell Manipulation. Mol. Biochem. Parasitol. 2016, 208, 2–15. 10.1016/j.molbiopara.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Paquet T.; Le Manach C.; Cabrera D. G.; Younis Y.; Henrich P. P.; Abraham T. S.; Lee M. C. S.; Basak R.; Ghidelli-Disse S.; Lafuente-Monasterio M. J.; Bantscheff M.; Ruecker A.; Blagborough A. M.; Zakutansky S. E.; Zeeman A. M.; White K. L.; Shackleford D. M.; Mannila J.; Morizzi J.; Scheurer C.; Angulo-Barturen I.; Martínez M.; Ferrer S.; Sanz L. M.; Gamo F. J.; Reader J.; Botha M.; Dechering K. J.; Sauerwein R. W.; Tungtaeng A.; Vanachayangkul P.; Lim C. S.; Burrows J.; Witty M. J.; Marsh K. C.; Bodenreider C.; Rochford R.; Solapure S. M.; Jiménez-Díaz M. B.; Wittlin S.; Charman S. A.; Donini C.; Campo B.; Birkholtz L. M.; Hanson K.; Drewes G.; Kocken C. M.; Delves M. J.; Leroy D.; Fidock D. A.; Waterson D.; Street L. J.; Chibale K. Antimalarial Efficacy of MMV390048, an Inhibitor of Plasmodium Phosphatidylinositol 4-Kinase. Sci. Transl. Med. 2017, 9, eaad9735 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N.; Sakata T.; Breton G.; Le Roch K. G.; Nagle A.; Andersen C.; Bursulaya B.; Henson K.; Johnson J.; Kumar K. A.; Marr F.; Mason D.; McNamara C.; Plouffe D.; Ramachandran V.; Spooner M.; Tuntland T.; Zhou Y.; Peters E. C.; Chatterjee A.; Schultz P. G.; Ward G. E.; Gray N.; Harper J.; Winzeler E. A. Gene Expression Signatures and Small-Molecule Compounds Link a Protein Kinase to Plasmodium Falciparum Motility. Nat. Chem. Biol. 2008, 4, 347–356. 10.1038/nchembio.87. [DOI] [PubMed] [Google Scholar]
- Baker D. A.; Stewart L. B.; Large J. M.; Bowyer P. W.; Ansell K. H.; Jiménez-Díaz M. B.; El Bakkouri M.; Birchall K.; Dechering K. J.; Bouloc N. S.; Coombs P. J.; Whalley D.; Harding D. J.; Smiljanic-Hurley E.; Wheldon M. C.; Walker E. M.; Dessens J. T.; Lafuente M. J.; Sanz L. M.; Gamo F.-J.; Ferrer S. B.; Hui R.; Bousema T.; Angulo-Barturén I.; Merritt A. T.; Croft S. L.; Gutteridge W. E.; Kettleborough C. A.; Osborne S. A. A Potent Series Targeting the Malarial CGMP-Dependent Protein Kinase Clears Infection and Blocks Transmission. Nat. Commun. 2017, 8, 430. 10.1038/s41467-017-00572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern S.; Agarwal S.; Huber K.; Gehring A. P.; Strödke B.; Wirth C. C.; Brügl T.; Abodo L. O.; Dandekar T.; Doerig C.; Fischer R.; Tobin A. B.; Alam M. M.; Bracher F.; Pradel G. Inhibition of the SR Protein-Phosphorylating CLK Kinases of Plasmodium Falciparum Impairs Blood Stage Replication and Malaria Transmission. PLoS One 2014, 9, e105732 10.1371/journal.pone.0105732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Sanchez-Azqueta A.; Janha O.; Flannery E. L.; Mahindra A.; Mapesa K.; Char A. B.; Sriranganadane D.; Brancucci N. M. B.; Antonova-Koch Y.; Crouch K.; Simwela N. V.; Millar S. B.; Akinwale J.; Mitcheson D.; Solyakov L.; Dudek K.; Jones C.; Zapatero C.; Doerig C.; Nwakanma D. C.; Vázquez M. J.; Colmenarejo G.; Lafuente-Monasterio M. J.; Leon M. L.; Godoi P. H. C.; Elkins J. M.; Waters A. P.; Jamieson A. G.; Álvaro E. F.; Ranford-Cartwright L. C.; Marti M.; Winzeler E. A.; Gamo F. J.; Tobin A. B. Validation of the Protein Kinase PfCLK3 as a Multistage Cross-Species Malarial Drug Target. Science 2019, 365, eaau1682 10.1126/science.aau1682. [DOI] [PubMed] [Google Scholar]
- Mahindra A.; Janha O.; Mapesa K.; Sanchez-Azqueta A.; Alam M. M.; Amambua-Ngwa A.; Nwakanma D. C.; Tobin A. B.; Jamieson A. G. Development of Potent PfCLK3 Inhibitors Based on TCMDC-135051 as a New Class of Antimalarials. J. Med. Chem. 2020, 63, 9300–9315. 10.1021/acs.jmedchem.0c00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jester B. W.; Cox K. J.; Gaj A.; Shomin C. D.; Porter J. R.; Ghosh I. A Coiled-Coil Enabled Split-Luciferase Three-Hybrid System: Applied Toward Profiling Inhibitors of Protein Kinases. J. Am. Chem. Soc. 2010, 132, 11727–11735. 10.1021/ja104491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Wang C.; Otto T. D.; Oberstaller J.; Liao X.; Adapa S. R.; Udenze K.; Bronner I. F.; Casandra D.; Mayho M.; Brown J.; Li S.; Swanson J.; Rayner J. C.; Jiang R. H. Y.; Adams J. H. Uncovering the Essential Genes of the Human Malaria Parasite Plasmodium Falciparum by Saturation Mutagenesis. Science 2018, 360, eaap7847 10.1126/science.aap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelchli R.; Bollbuck B.; Bruns C.; Buhl T.; Eder J.; Feifel R.; Hersperger R.; Janser P.; Revesz L.; Zerwes H. G.; Schlapbach A. Design and Preparation of 2-Benzamido-Pyrimidines as Inhibitors of IKK. Bioorg. Med. Chem. Lett. 2006, 16, 108–112. 10.1016/j.bmcl.2005.09.035. [DOI] [PubMed] [Google Scholar]
- Hermanson S. B.; Carlson C. B.; Riddle S. M.; Zhao J.; Vogel K. W.; Nichols R. J.; Bi K. Screening for Novel LRRK2 Inhibitors Using a High-Throughput TR-FRET Cellular Assay for LRRK2 Ser935 Phosphorylation. PLoS One 2012, 7, e43580 10.1371/journal.pone.0043580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droucheau E.; Primot A.; Thomas V.; Mattei D.; Knockaert M.; Richardson C.; Sallicandro P.; Alano P.; Jafarshad A.; Baratte B.; Kunick C.; Parzy D.; Pearl L.; Doerig C.; Meijer L. Plasmodium Falciparum Glycogen Synthase Kinase-3: Molecular Model, Expression, Intracellular Localisation and Selective Inhibitors. Biochim. Biophys. Acta, Proteins Proteomics 2004, 1697, 181–196. 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- Prinz B.; Harvey K. L.; Wilcke L.; Ruch U.; Engelberg K.; Biller L.; Lucet I.; Erkelenz S.; Heincke D.; Spielmann T.; Doerig C.; Kunick C.; Crabb B. S.; Gilson P. R.; Gilberger T. W. Hierarchical Phosphorylation of Apical Membrane Antigen 1 Is Required for Efficient Red Blood Cell Invasion by Malaria Parasites. Sci. Rep. 2016, 6, 34479. 10.1038/srep34479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracchi-ricard V.; Barik S.; Delvecchio C.; Doerig C.; Chakrabarti R.; Chakrabarti D. PfPK6, a Novel Cyclin-Dependent Kinase/Mitogen-Activated Protein Kinase-Related Protein Kinase from Plasmodium Falciparum. Biochem. J. 2000, 347, 255–263. 10.1042/0264-6021:3470255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coldewey S. M.; Rogazzo M.; Collino M.; Patel N. S. A.; Thiemermann C. Inhibition of IκB Kinase Reduces the Multiple Organ Dysfunction Caused by Sepsis in the Mouse. Dis. Models Mech. 2013, 6, 1031–1042. 10.1242/dmm.012435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masch A.; Kunick C. Selective Inhibitors of Plasmodium Falciparum Glycogen Synthase-3 (PfGSK-3): New Antimalarial Agents?. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1644–1649. 10.1016/j.bbapap.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Fugel W.; Oberholzer A. E.; Gschloessl B.; Dzikowski R.; Pressburger N.; Preu L.; Pearl L. H.; Baratte B.; Ratin M.; Okun I.; Doerig C.; Kruggel S.; Lemcke T.; Meijer L.; Kunick C. 3,6-Diamino-4-(2-Halophenyl)-2-Benzoylthieno[2,3- b ]Pyridine-5- Carbonitriles Are Selective Inhibitors of Plasmodium Falciparum Glycogen Synthase Kinase-3. J. Med. Chem. 2013, 56, 264–275. 10.1021/jm301575n. [DOI] [PubMed] [Google Scholar]
- Moolman C.; van der Sluis R.; Beteck R. M.; Legoabe L. J. Exploration of Benzofuran-Based Compounds as Potent and Selective Plasmodium Falciparum Glycogen Synthase Kinase-3 (PfGSK-3) Inhibitors. Bioorg. Chem. 2021, 112, 104839. 10.1016/j.bioorg.2021.104839. [DOI] [PubMed] [Google Scholar]
- van Linden O. P. J.; Kooistra A. J.; Leurs R.; de Esch I. J. P.; De Graaf C. A Knowledge-Based Structural Database to Navigate Kinase-Ligand Interaction Space. J. Med. Chem. 2014, 57, 249–277. 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- Lange A.; Günther M.; Büttner F. M.; Zimmermann M. O.; Heidrich J.; Hennig S.; Zahn S.; Schall C.; Sievers-Engler A.; Ansideri F.; Koch P.; Laemmerhofer M.; Stehle T.; Laufer S. A.; Boeckler F. M. Targeting the Gatekeeper MET146 of C-Jun N-Terminal Kinase 3 Induces a Bivalent Halogen/Chalcogen Bond. J. Am. Chem. Soc. 2015, 137, 14640–14652. 10.1021/jacs.5b07090. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Kirchner B.; Boeckler F. M. Addressing Methionine in Molecular Design through Directed Sulfur-Halogen Bonds. J. Chem. Theory Comput. 2011, 7, 2307–2315. 10.1021/ct200245e. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Joerger A. C.; Boeckler F. M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- Kontopidis G.; McInnes C.; Pandalaneni S. R.; McNae I.; Gibson D.; Mezna M.; Thomas M.; Wood G.; Wang S.; Walkinshaw M. D.; Fischer P. M. Differential Binding of Inhibitors to Active and Inactive CDK2 Provides Insights for Drug Design. Chem. Biol. 2006, 13, 201–211. 10.1016/j.chembiol.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Shin D.; Lee S.-C.; Heo Y.-S.; Lee W.-Y.; Cho Y.-S.; Kim Y. E.; Hyun Y.-L.; Cho J. M.; Lee Y. S.; Ro S. Design and Synthesis of 7-Hydroxy-1H-Benzoimidazole Derivatives as Novel Inhibitors of Glycogen Synthase Kinase-3β. Bioorg. Med. Chem. Lett. 2007, 17, 5686–5689. 10.1016/j.bmcl.2007.07.056. [DOI] [PubMed] [Google Scholar]
- Mollard A.; Warner S. L.; Call L. T.; Wade M. L.; Bearss J. J.; Verma A.; Sharma S.; Vankayalapati H.; Bearss D. J. Design, Synthesis, and Biological Evaluation of a Series of Novel AXL Kinase Inhibitors. ACS Med. Chem. Lett. 2011, 2, 907–912. 10.1021/ml200198x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M.; Wen J.; Tang F.; Tu C.; Shen H.; Zhao X. The Discovery of Novel N-(2-Pyrimidinylamino) Benzamide Derivatives as Potent Hedgehog Signaling Pathway Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6777–6783. 10.1016/j.bmcl.2013.10.022. [DOI] [PubMed] [Google Scholar]
- Carbain B.; Coxon C. R.; Lebraud H.; Elliott K. J.; Matheson C. J.; Meschini E.; Roberts A. R.; Turner D. M.; Wong C.; Cano C.; Griffin R. J.; Hardcastle I. R.; Golding B. T. Trifluoroacetic Acid in 2,2,2-Trifluoroethanol Facilitates SNAr Reactions of Heterocycles with Arylamines. Chem.—Eur. J. 2014, 20, 2311–2317. 10.1002/chem.201304336. [DOI] [PubMed] [Google Scholar]
- Jain R.; Lin X.; Ng S. C.; Pfister K. B.; Ramurthy S.; Rico A.; Subramanian S.; Wang X. M.. 2-arylaminoquinazolines for treating proliferative disease. WO 2009153313 A1, 2009.
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Kato N.; Comer E.; Sakata-Kato T.; Sharma A.; Sharma M.; Maetani M.; Bastien J.; Brancucci N. M.; Bittker J. A.; Corey V.; Clarke D.; Derbyshire E. R.; Dornan G. L.; Duffy S.; Eckley S.; Itoe M. A.; Koolen K. M. J.; Lewis T. A.; Lui P. S.; Lukens A. K.; Lund E.; March S.; Meibalan E.; Meier B. C.; McPhail J. A.; Mitasev B.; Moss E. L.; Sayes M.; Van Gessel Y.; Wawer M. J.; Yoshinaga T.; Zeeman A. M.; Avery V. M.; Bhatia S. N.; Burke J. E.; Catteruccia F.; Clardy J. C.; Clemons P. A.; Dechering K. J.; Duvall J. R.; Foley M. A.; Gusovsky F.; Kocken C. H. M.; Marti M.; Morningstar M. L.; Munoz B.; Neafsey D. E.; Sharma A.; Winzeler E. A.; Wirth D. F.; Scherer C. A.; Schreiber S. L. Diversity-Oriented Synthesis Yields Novel Multistage Antimalarial Inhibitors. Nature 2016, 538, 344–349. 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F.-J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J.-L.; Vanderwall D. E.; Green D. V. S.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of Chemical Starting Points for Antimalarial Lead Identification. Nature 2010, 465, 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Helal M. A.; Avery M. A. Combined Receptor-Based and Ligand-Based Approach to Delineate the Mode of Binding of Guaianolide–Endoperoxides to PfATP6. Bioorg. Med. Chem. Lett. 2012, 22, 5410–5414. 10.1016/j.bmcl.2012.07.053. [DOI] [PubMed] [Google Scholar]
- Saleh A. H.; Abdelwaly A.; Darwish K. M.; Eissa A. A. H. M.; Chittiboyina A.; Helal M. A. Deciphering the Molecular Basis of the Kappa Opioid Receptor Selectivity: A Molecular Dynamics Study. J. Mol. Graphics Modell. 2021, 106, 107940. 10.1016/j.jmgm.2021.107940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.