

Abstract

In view of the worldwide antimicrobial resistance (AMR) threat, new bacterial targets and anti-infective agents are needed. Since important roles in bacterial pathogenesis have been demonstrated for the collagenase H and G (ColH and ColG) from Clostridium histolyticum, collagenase Q1 and A (ColQ1 and ColA) from Bacillus cereus represent attractive antivirulence targets. Furthermore, repurposing FDA-approved drugs may assist to tackle the AMR crisis and was addressed in this work. Here, we report on the discovery of two potent and chemically stable bacterial collagenase inhibitors: synthesized and FDA-approved diphosphonates and hydroxamates. Both classes showed high in vitro activity against the clostridial and bacillary collagenases. The potent diphosphonates reduced B. cereus-mediated detachment and death of cells and Galleria mellonella larvae. The hydroxamates were also tested in a similar manner; they did not have an effect in infection models. This might be due to their fast binding kinetics to bacterial collagenases.
Introduction
Bacterial resistance is on the rise, and a global economic and public health crisis might be on the horizon due to the high incidence of deaths caused by multidrug-resistant bacteria.1,2 This also comes together with the slow discovery of new antibiotics. If this trend continues, mild infections of today might become lethal in the future.1,2 To combat the rise in antibiotic resistance, alternative, nonantibiotic treatment approaches are urgently needed. Antivirulence agents (also called “pathoblockers”), which selectively inhibit pathogenicity factors of bacteria, and hence prevent or delay infection, are one potential strategy.3,4 This could—without exerting selective pressure—harness the host’s immune system to fight the infection.3,4 Finding new applications for existing FDA-approved drugs is gaining popularity to tackle the current antimicrobial-resistance crisis as it reduces the cost, time, and effort required for the regular discovery of new antibiotics.5 Pentamidine is an antiprotozoal medicine and an example of an FDA-approved drug that has been repurposed for antibacterial use.6 This approach holds promise as it may overcome the slow development of new antibiotics.
Clostridium histolyticum (C. histolyticum) and Bacillus cereus (B. cereus) are Gram-positive bacteria and the epitome of many serious opportunistic infections, including gas gangrene as well as wound, corneal, and gastro-intestinal infections.7,8 As these bacteria resist the activity of many antibiotics (such as penicillin), they are still susceptible to the activity of gentamycin, vancomycin, erythromycin, and other antibiotics.9,10 The pathogenicity of these microorganisms is linked to their secreted toxins and proteases, which assist them to elude defensive mechanisms, reach deep locations for nourishment, and consequently replicate and persist at the infection site.11 This might also enhance bacterial histotoxicity by promoting toxin diffusion.11 Bacterial collagenases are calcium- and zinc-dependent metalloproteases and the etiologic feature of the aforementioned pathogens; they destroy tissues by demolishing extracellular matrix (ECM) collagen.12,13 Fibrillar collagen is the most common protein in the ECM (up to 90%). It has a stable structure that resists proteolysis and can only be broken down by true bacterial and mammalian collagenases.12,13 Bacterial collagenases process triple helical collagen under physiological conditions into small peptides and amino acids by targeting multiple sites.12,13 Mammalian collagenases, on the other hand, such as the collagenolytic matrix metalloproteinases (MMPs), cleave collagen at a single site to generate the characteristic 3/4 and 1/4 fragments. After this initial cut, other enzymes assist in the further proteolysis of the collagen fragments.13 Collagen is involved in many vital physiological processes such as tissue regeneration and wound healing, in addition to its tissue-supporting functions.13 Therefore, any bacterial collagenase-induced imbalance in collagen structure or quantity will have detrimental effects on tissue regeneration and wound healing besides the formation of voids in the ECM, which allow the bacteria to invade and gain access to anaerobic areas.14 Consequently, protecting collagen from bacterial collagenase represents a promising approach for developing antivirulence agents that can be used to treat collagenase-positive infections.
In contrast to the homologous B. cereus collagenases, C. histolyticum collagenases have been well studied. They are composed of two main structural modules: the collagenase unit and collagen recruitment domains. The collagenase unit is composed of an activator and peptidase domains.15,16 In the latter, two histidine residues in a HEXXH motif and a downstream glutamate coordinate the catalytic zinc ion. The general-acid base glutamate and the zinc ion polarize the water molecule in the active site and activate it for the nucleophilic attack.15−18 The collagen recruitment domains are suggested to be involved in collagen swelling and binding to fibrillar and insoluble collagen.15−18 High-resolution crystal structures are available for C. histolyticum collagenases but not for B. cereus collagenases, which share a high sequence similarity (i.e., 70%).17,19−21
We focused in this study on C. histolyticum collagenases H and G (ColH and ColG), as well as B. cereus collagenases Q1 and A (ColQ1 and ColA). These bacterial collagenases are attractive candidates for the development of antivirulence drugs due to their role in bacterial pathogenicity as well as their extracellular localization.14 The penetration of the bacterial cell wall is generally a key challenge for the development of antibacterial agents; however, in this instance, it can be avoided.
Distinct collagenase inhibitors have been identified to date, the majority of which include a zinc-binding group (ZBG) that binds to the catalytic zinc ion, displacing the catalytic water molecule from the coordination sphere and rendering the enzyme inactive. Figure 1a shows examples of known ColH inhibitors.22−24 Their lack of selectivity over human MMPs is the primary drawback that prevents further development of these existing potent inhibitors.
Figure 1.

Examples of known bacterial collagenase H and Q1 (ColH and ColQ1) inhibitors. (a) Structures of bacterial ColH inhibitors.22−24 (b) Structures of the recently identified inhibitors of ColH and ColQ1.25−28 Zinc-binding groups are highlighted by dashed rectangles. n.d.: not determined.
Recently, we were able to develop selective inhibitors of bacterial collagenases. Compound 4a, a thiocarbamate, serves as a prodrug. By conversion into a free thiol 4b, it can bind to the zinc ion (Figure 1b).25 The rest of the molecule binds to the conserved clostridial nonprimed edge strand, explaining the selectivity for the bacterial collagenases over the human off-target MMPs.25 Succinimide 5 is a more rigid derivative of compound 4b (Figure 1b).26 Similarly to thiocarbamates, this class has been validated for its high selectivity for several bacterial collagenases, including ColH and ColG from C. histolyticum, ColT from C. tetani, and ColQ1 from B. cereus, over the unwanted inhibition of human MMPs. The main drawback of these two compound classes is their chemical instability due to the oxidation of the thiol group to the corresponding disulfide, which leads to the loss of their activity.
Replacing the thiol group with another stable ZBG to maintain the chemical stability of the inhibitor is necessary, in addition to maintaining the high selectivity toward bacterial metalloproteases. The first stable and selective inhibitor (i.e., compound 6) (Figure 1b) of ColH was recently reported.27,28 Among various ZBGs, a phosphonate group has high stability and selectivity but moderate activities on ColH and ColQ1.27,28
In this work, we aimed to find chemically stable, potent, and selective bacterial collagenase inhibitors. Furthermore, we set out to characterize a range of compounds bearing several different ZBGs. We identified two chemical classes, namely, diphosphonates (including FDA-approved drugs) and hydroxamates with excellent selectivity, low cytotoxicity, and remarkable micro- to submicromolar activity in vitro against B. cereus ColQ1, ColA, and C. histolyticum ColH and ColG. This finding demonstrates the potential of these compounds to exert a broad-spectrum activity toward a variety of disease-causing bacteria. In addition, we show their activity in increasingly complex whole-cell assays and efficacy in a simple Galleria mellonella infection model. Hydroxamates were tested analogously. Surprisingly, they did not demonstrate the same inhibitory impact in the infection models, despite similar potencies in enzyme-inhibition studies.
Results and Discussion
Screening of Compounds with Various ZBGs on ColQ1 and ColH
To discover new small-molecule inhibitors with a stable ZBG, a total of 38 compounds with distinct ZBGs were tested at 100 μM in an in vitro peptidolytic assay using a custom-made collagenase-specific quenched fluorescent substrate (Table S1 and Figure S1). Two compounds (13 and 27) showed strong inhibition against the collagenase unit of ColQ1 (COlQ1-CU) (98 ± 1% inhibition and 97 ± 2% inhibition) and the peptidase unit of ColH (ColH-PD) (83 ± 9% inhibition and 84 ± 2% inhibition), respectively (Figure 2). Both compounds were selected for further studies, and a small library based on 13 and 27 structures was designed and synthesized (i.e., 16 diphosphonates and 6 hydroxamates).
Figure 2.

Chemical structures of compounds 13 and 27 and their activities against the collagenase unit (CU) of ColQ1 and peptidase domain (PD) of ColH.
Synthesis of New Anticollagenase Pathoblocker Agents
Diphosphonate Synthesis
To get access to the different diphosphonate compounds, a synthetic route composed of four steps starting from the corresponding diamine was implemented. As depicted in Scheme 1, the synthesis of the diphosphonate compounds 7–12 started by refluxing the corresponding diamine with oxalic acid in 4 N hydrochloric acid to achieve the 1,4-dihydroquinoxaline-2,3-diones 7a–12a. These quinoxaline-dione intermediates were then reacted with POCl3 in DMF to provide the 2,3-dichloroquinoxalines 7b–12b, which were used in the following step without additional purification.29,30 The resulting dichloroquinoxalines 7b–12b were converted via an Arbuzov reaction to the corresponding diethyl phosphonate esters 7c–12 using triethyl phosphite and heated in a sealed tube at 150 °C. TMSBr was then utilized to cleave the diethyl phosphonate esters 7c–12c to their corresponding phosphonic acids 7–12 in good yields.
Scheme 1. Synthesis of Compounds 7–12.

Reagents and conditions: (a) oxalic acid, 4 N HCl, reflux, 6 h; (b) POCl3, DMF, 50 °C, 6 h; (c) triethyl phosphite, sealed tube, 150 °C, 18 h; and (d) bromotrimethylsilane, dry DCM, stirring, room temperature, 18 h.
Compound 13 was synthesized using the same general procedure for the synthesis of the diphosphonates (Scheme 2). To get further access to the monophosphonate compound 16, intermediate 13b was converted into 16a using LiOH in a dioxane/water mixture, as described by Yang et al. (Scheme 2).30 The latter was then subjected to the Arbuzov reaction followed by ester hydrolysis using TMSBr to afford the corresponding phosphonic acid 16.
Scheme 2. Synthesis of Compounds 13 and 16.

Reagents and conditions: (a) oxalic acid, 4 N HCl, reflux, 6 h; (b) POCl3, DMF, 50 °C, 6 h; (c) triethyl phosphite, sealed tube, 150 °C, 18 h; (d) bromotrimethylsilane, dry DCM, stirring, room temperature, 18 h; (e) dioxane/H2O (1:1), LiOH, 55 °C, 24 h; and (f) POCl3, DMF, 0 °C to room temperature, 5 min.
Refluxing of 3,4-diaminobenzoic acid and oxalic acid in 4 N hydrochloric acid yielded 14a, which reacted with POCl3 in DMF to afford the corresponding dichloro derivative 14b. Compound 14c was synthesized by reacting 2,3-dichloroquinoxaline-6-carboxylic acid 14b with 3,4-dichloroaniline in DCM at room temperature for 18 h using EDC·HCl as a coupling reagent. This was followed by the Arbuzov reaction and ester hydrolysis to achieve the desired compound 14 (Scheme 3).
Scheme 3. Synthesis of Compound 14.

Reagents and conditions: (a) oxalic acid, 4 N HCl, reflux, 6 h; (b) POCl3, DMF, 50 °C, 6 h; (c) 3,4-dichloroaniline, EDC.HCl, DCM, 18 h; (d) triethyl phosphite, sealed tube, 150 °C, 18 h; and (e) bromotrimethylsilane, dry DCM, stirring, room temperature, 18 h.
Compound 15a was obtained via a Suzuki cross-coupling reaction by reacting 4-bromobenzene-1,2-diamine and (4-chlorophenyl)boronic acid in the presence of 2 M sodium carbonate and (1,1′-bis(diphenylphosphino)ferrocene)palladium(II) dichloride [Pd(dppf)Cl2] as a catalyst in a mixture of dioxane/H2O (4:1) under microwave irradiation (150 °C, 150 W) for 20 min.31 This was followed by the general procedure for the diphosphonate synthesis to afford compound 15 (Scheme 4).
Scheme 4. Synthesis of Compound 15.

Reagents and conditions: (a) (4-chlorophenyl)boronic acid, dioxane/H2O (4:1), Na2CO3 (2 M), Pd(PPh3)4, microwave, 20 min; (b) oxalic acid, 4 N HCl, reflux, 6 h; (c) POCl3, DMF, 50 °C, 6 h; (d) triethyl phosphite, sealed tube, 150 °C, 18 h; and (e) bromotrimethylsilane, dry DCM, stirring, room temperature, 18 h.
Hydroxamate Synthesis
As described in Scheme 5, the hydroxamates 27–33 were synthesized in six steps. The monocarboxylic acids 22a and 22b were first obtained through a monosaponification using sodium hydroxide in a mixture of ethanol and water. These intermediates were then activated using EDC·HCl and HOBt, in dichloromethane with diisopropylethylamine, to form the desired amides 23a and 23b by reacting them with the free amine. Then, an addition of hydrochloric acid (4 N in dioxane) afforded the free amines 24a and 24b, and these were reacted with the diazo transfer reagent to form the azides 25a and 25b. The ethyl esters were engaged in a KCN-catalyzed aminolysis reaction, which led to the formation of the azido hydroxamic acids 26a and 26b. The final step was a copper-catalyzed Huisgen 1,3-dipolar cycloaddition to give the desired 1,4-disubstituted 1,2,3-triazoles 27–33, the needed alkynes 34a–34d being previously synthesized by the nucleophilic substitution of phenols or thiophenol on propargyl bromide.
Scheme 5. Synthesis of Hydroxamic Acid Compounds.

Reagents and conditions: (a) NaOH, EtOH/H2O (4:1), rt., 18 h; (b) tert-butyl N-(2-aminoethyl)carbamate, EDC.HCl, HOBt, DIPEA, CH2Cl2, rt., 18 h; (c) 4 N HCl, EtOH, 0 °C to rt., 18 h; (d) azide-N-diazoimidazole-1-sulfonamide hydrogen sulfate, K2CO3, ZnCl2, DIPEA, MeOH, rt., 18 h; (e) aq. hydroxylamine (50% in water w/w), KCN (cat.), MeOH, rt., 18 h; and (f) alkyne 34a–34d, prop-2-ynoxybenzene or prop-2-ynylsulfanylbenzene, CuSO4 (5H2O), NaAsc, N,N-dimethylformamide/H2O (1.2:1), rt., 18 h.
Activity on B. cereus ColQ1
Structure–Activity Relationships of the Synthesized and FDA-Approved Diphosphonate Compounds on ColQ1
The initial testing of the diphosphonate 13 and its synthesized derivatives using ColQ1-CU showed that the presence of both phosphonic acid groups is indispensable for inhibition (Table 1), indicating that the phosphonate groups act as ZBG to the catalytic zinc ion. The unsubstituted diphosphonic acid quinoxaline 7 showed lower activity than the 3,4-dichloro analogue 13 (Table 1). Introducing an electron-withdrawing group such as fluorine at position 6 of the quinoxaline moiety of 8 led to no significant change in activity. Interestingly, increasing the lipophilicity by adding chlorine or bromine at position 6 of the quinoxaline of 9, 10, and 13 led to a significant boost in activity in comparison with the unsubstituted 7, indicating the importance of the lipophilic groups at this part of the inhibitor (Table 1). This finding prompted us to further explore variable lipophilic moieties at this part of the molecule in an attempt to enhance the activity. Compounds 14 and 15 were synthesized and tested yielding less potent inhibitors, which could be an indication of inappropriate bulky groups at that part of the molecule. Adding an electron-donating group at position 6 of the quinoxaline moiety showed a slight improvement in the activity, as shown by the methyl derivative 11, while the methoxy derivative 12 resulted in a decrease in activity (Table 1).
Table 1. Percent Inhibition (at a Concentration of 100 μM) and IC50 Values of Diphosphonate Derivatives against ColQ1-CUa.

Means and SD of three independent experiments, n.i.: no inhibition (percent inhibition < 5%).
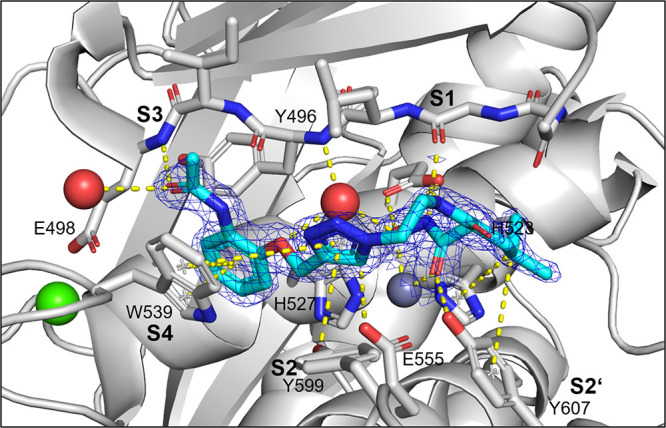
In the case of ColG-PD (a close homologue of ColQ1), the SAR could be rationalized by a crystal structure in complex with compound 13 determined at 1.95 Å resolution (Figure 3 and Table S2). In addition to a nonfunctional binding site at the rear of the peptidase domain, we could detect 13 in the primed binding pocket, albeit at relatively low occupancy/high mobility. The inhibitor could be modeled into the active site with the help of a polder map. One of the phosphonate groups acted as ZBG and simultaneously interacted with Glu555 and Tyr607, while the aromatic scaffold of 13 and the chlorides established hydrophobic interactions in the primed binding pockets with Phe515, His523, and Ile576.
Figure 3.
Crystal structure of ColG-PD in complex with 13 solved at a resolution of 1.95 Å. Close-up view of the active site in ball-and-stick representation. The inhibitor (cyan) is shown in sticks with a polder map contoured at 2.5 σ above the background. The catalytic zinc ion (dark gray) and the calcium ion (green) are shown as spheres (PDB code: 7ZBV).
As drug repurposing is gaining attraction these days, especially for finding new antimicrobial agents,5 we tested a number of FDA-approved diphosphonates regarding their effect on ColQ1-CU (Table 2). Tiludronate disodium and alendronate sodium resulted in a moderate inhibition of ColQ1-CU (63 ± 3 and 76 ± 1%, respectively) at a concentration of 100 μM. Diphosphonate FDA-approved drugs are routinely used in the treatment of bone diseases and were never reported to have antibacterial activity.32 This calls for testing more diphosphonate FDA-approved drugs for their activity on bacterial collagenases to find potent inhibitors with known data regarding their safety, efficacy, and pharmacokinetics.
Table 2. Percent Inhibition of ColQ1-CU by Selected FDA-Approved Diphosphonates at a Concentration of 100 μMa.

Means and SD of three independent experiments, n.i.: no inhibition (percent inhibition < 5%).
Structure–Activity Relationships of Hydroxamate Compounds on ColQ1
An initial screening led to the identification of hydroxamate 27; the SAR study was performed using COLQ1-CU, which is articulated around a 1,4-disubstituted 1,2,3-triazole ring (Figure S2 and Table 3). To explore the ortho-acetamide phenoxy group, four derivatives were synthesized (28–31). The move of the ortho-acetamide to meta- and para-positions as in 28 and 29, respectively, led to a slight decrease in activity, while its removal (compound 31) or its replacement by a 2-fluorine motif (compound 30) produced similar activities. The phenol replacement of 31 by thiophenol (compound 32) led to a lower ColQ1-CU inhibition. Interestingly, compound 33 demonstrated that bulky substituents in the α-position of the hydroxamate could be tolerated.
Table 3. Percent Inhibition (at a Concentration of 100 μM) or IC50 Values of Hydroxamates against ColQ1-CUa.


Means and SD of three independent experiments.
Next, we evaluated the potential of the most effective compounds 27 and 33 as broad-spectrum inhibitors of bacterial collagenases and determined the inhibition constants using ColG-CU, ColH-PD, ColA-CU (from B. cereus strain ATCC14579), and ColQ1-CU. The results revealed that both compounds inhibited clostridial and bacillary collagenases in the low micro- to submicromolar range (Table 4).
Table 4. Inhibition Constant (Ki) of 27 and 33 against Bacterial Collagenasesa.
| 27 | 33 | ||
|---|---|---|---|
| bacteria | protein | Ki (μM) | Ki (μM) |
| C. histolyticum | ColH-PD | 11.6 ± 0.4 | 1.7 ± 0.2 |
| ColG-CU | 31 ± 1 | 18.4 ± 0.6 | |
| B. cereus | ColQ1-CU | 0.10 ± 0.02 | 0.82 ± 0.07 |
| ColA-CU | 3.4 ± 4 | 4.7 ± 0.3 |
Means and SD of three independent experiments.
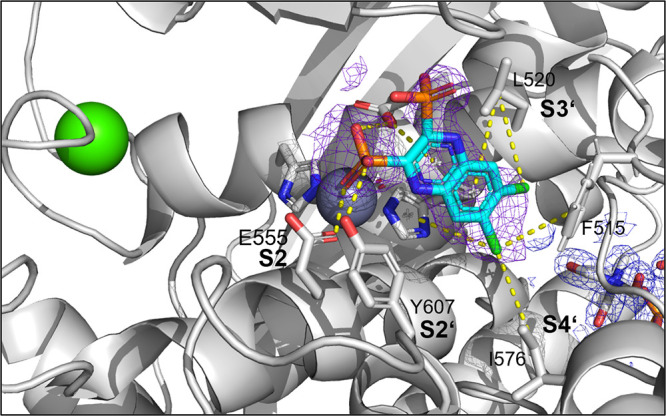
We rationalized the screening results based on the crystal structure of 27 in complex with the ColG-PD, solved at a 1.80 Å resolution (Figure 4 and Table S2). The bound inhibitor occupied the active site from the S3 pocket to the S2′ binding site. In the S3 binding pocket, the ortho-acetamide group directly interacted with the edge strand via a hydrogen bond to the backbone amide of Glu498, while the aromatic phenoxy ring established π–π stacking interactions with the side chain of Trp539. This explains the observed preference for the ortho-configuration of the acetamide group.
Figure 4.
Crystal structure of ColG-PD in complex with 27 solved at 1.80 Å resolution. Close-up view of the active site in ball-and-stick representation. The inhibitor (cyan) is shown in sticks with the maximum likelihood weighted 2Fo–Fc electron density map contoured at 1σ. The catalytic zinc ion (dark gray) and the calcium ion (green) and water molecules (red) are shown as spheres (PDB code: 7Z5U).
The central triazole ring hydrogen bonded with Glu555 and formed a π–hydrogen interaction with Tyr599 in the S2 binding pocket. The hydroxamate group coordinated, as expected, the catalytic zinc ion. The isobutyl group in the α-position formed hydrophobic interactions with Tyr607 and His523 in the S2′ pocket. The hydrophobic benzyl group of 33 can be expected to fit similarly into the S2′ pocket.
Selectivity over Human MMPs and Activity against Other Bacterial Collagenases
To assess the selectivity of the compounds over human MMPs and on other bacterial collagenases, selected compounds were tested against ColG-CU and ColH-PD from C. histolyticum and ColA-CU from B. cereus strain ATCC14579. In addition, the compounds were tested on catalytic domains of three human MMPs, which are characterized by different depths of the S1′ binding pocket (MMP-1 (shallow), −2 (intermediate), and −3 (deep)). They were also tested against other important human off-targets, which are involved in gene expression and the processing of TNF-α; these include HDAC-3, -8, TACE (ADAM-17),33,34 and COX-1.
Our data showed that the compounds have high activity against most tested bacterial collagenases, which confirms their broad-spectrum inhibitory potency against bacterial targets (Table 5). This broad activity is comparable to that previously observed for compounds carrying thiol or phosphonate ZBGs.25−27
Table 5. Percent Inhibition of ColA-CU, ColH-PD, and ColG-CU in the Presence of 100 μM of Compounds 13 14, 15, 27, Tiludronate Disodium, and Alendronate Sodiuma.
| class | cpd. | ColA-CU | ColH-PD | ColG-CU |
|---|---|---|---|---|
| synthesized diphosphonates | 13 | 71 ± 4 | 83 ± 9 | 68 ± 4 |
| 14 | 82 ± 2 | 96 ± 2 | 72 ± 3 | |
| 15 | 86 ± 8 | 97 ± 5 | 70 ± 8 | |
| FDA-approved diphosphonates | tiludronate disodium | 72 ± 2 | 91 ± 2 | 28 ± 7 |
| alendronate sodium | 18 ± 4 | 25 ± 5 | 28 ± 7 | |
| hydroxamate | 27 | 93 ± 2 | 84 ± 2 | 71 ± 7 |
Means and SD of at least two independent experiments.
On the other hand, the compounds possess a high selectivity over most of the tested human off-targets (except for 13 against the tested MMPs and tiludronate disodium against MMP-1) (Tables 6 and S3).
Table 6. Inhibition of Compounds 13 14, 15, 27, Tiludronate Disodium, and Alendronate Sodium against Three MMPsa.
| IC50 (μM) | ||||
|---|---|---|---|---|
| class | cpd. | MMP-1 | MMP-2 | MMP-3 |
| synthesized diphosphonates | 13 | 53 ± 3 | 79 ± 2 | 33 ± 11 |
| 14 | >100 | >100 | >100 | |
| 15 | >100 | >100 | >100 | |
| FDA-approved diphosphonates | tiludronate disodium | 50 ± 10 | >100 | >100 |
| alendronate sodium | >100 | >100 | >100 | |
| hydroxamate | 27 | >100 | >100 | >100 |
Means and SD of two independent experiments, >100: IC50 is higher than 100 μM.
Cytotoxicity against Mammalian Cell Lines
Besides the selectivity, cytotoxicity is also an important criterion, especially when it comes to a potential therapeutic application in humans. In this context, we evaluated the cytotoxicity of 13, 14, 15, and 27 against four mammalian cell lines, comprising HepG2 (hepatocellular carcinoma), HEK293 (embryonal kidney), NHDF (normal human dermal fibroblasts), and MDCK II (Madin–Darby canine kidney II) cells. Interestingly, the compounds did not show cytotoxic effects (IC50 values > 100 μM or 200 μM) against these cell lines (Table S4). This makes them suitable for further investigation to determine their ADMET profile.
Small-Molecule Collagenase Inhibitors Prevent Collagen I Cleavage
We examined collagen I (Col I) cleavage induced by the full-length ColQ1-FL with and without inhibitors to investigate whether the compounds have an anticollagenolytic effect on the collagenase’s natural substrate. After 4 h of co-incubation of ColQ1-FL with Col I, in the absence and presence of inhibitor, Col I breakdown was investigated. On reducing polyacrylamide gels, the hallmark bands of Col I (i.e., α1, α2, and β chains) were clearly visible. Compared to the negative control (no inhibitor), 13 and 27 displayed considerable anti-ColQ1 activity and maintained the chains of Col I, as shown at concentrations above 3 and 0.8 μM, respectively. Below these concentrations, substantial chain disintegration was detected (Figure 5). The diphosphonate derivatives 11, 10, 14, and 15 inhibited the collagenase activity at all concentrations tested (12.5–1.5 μM) (except for 10, which showed inhibition only at 12.5 μM) and protected Col I chains from cleavage (Figure 4). Similar findings were observed for other diphosphonate derivatives 7, 9, and 11 and the hydroxamate derivative 33 (Figure S3). In contrast, severe degradation was visible at all tested concentrations (12.5–1.5 μM) of diphosphonate16 and the hydroxamate derivatives 28, 29, 31, and 32 (Figure S3), indicating their low activity on ColQ1.
Figure 5.

Activity of ColQ1 inhibitors against the collagenolytic activity of the full-length (FL) ColQ1. Inhibitors prevented the cleavage of 1 mg/mL of Col I chains (i.e., β, α-1, and α-2). The Bacillus cereus ColQ1-FL (50 ng) was incubated with 1 mg/mL Col I for 3 h, and the degradation was then visualized by 12% SDS-PAGE. Col I: 1 mg/mL Col I without any protease. M (kDa): molecular weight standards, Col I: type I collagen, ColQ1: collagenase Q1.
These findings are corroborated by the previously determined inhibition data obtained with the peptidolytic assay. This highlights that several of our hydroxamate and diphosphonate compounds are capable in vitro of preventing cleavage of the large, structurally more complex physiological substrate of collagenases, i.e., Col I, which accounts for 80–90% of the collagen in the body.35 Based on these findings, we next investigated whether this protective effect would also prevail in a more complex cellular setting.
Small-Molecule Inhibitors Reduce Collagenase Activity and Preserve Fibroblast Cell Integrity
Collagenase Release during B. cereus Infection of NHDF Cells
To investigate the potential antivirulence activity of ColQ1 inhibitors, we developed an in vitro infection model using the common connective tissue cell line NHDF in the ECM. The fibroblasts have crucial functions in the ECM, as they synthesize their main components (such as collagen) and maintain their homeostasis.36 Previous reports revealed that bacillary ColQ1 and ColA have a prominent collagenolytic activity that is greater than or similar to the well-studied clostridial ColG and ColH.20 Therefore, B. cereus collagenases were used as model proteases to further evaluate the inhibitors’ effects in infection settings.
To establish the model, the release of B. cereus collagenases was investigated to determine the duration needed for the AH187 strain (expresses two collagenases: B7HV61 (sequence identical to ColQ1) and B7HZW5 (ColA)) to secrete considerable amounts of collagenase into the surrounding DMEM medium of NHDF cells. To inspect the release of collagenase, the DMEM medium was collected at various time points and investigated by gelatin zymography. The zymography analysis revealed that the bacteria required at least 4 h to release substantial amounts of collagenases, which increased over time, as seen in Figure S4b. In the zymograms, besides the bands of the full-length ColQ1 and ColA (109 kDa), also truncated isoforms (100–40 kDa) were detected (Figure S4b). This phenomenon has been previously reported for other bacterial collagenases.19 After 4 h of infection, we could also observe a massive reduction (>50%) in fibrillar collagen in the ECM quantified with the picrosirius red assay (Figure S4d).37,38 Concomitantly, NHDF cells started to detach and their morphology changed from spindle-shaped to round, as evidenced by light microscopy and SDS-PAGE analysis of the cell lysate (Figure S4a,c). In addition, we monitored the release of lactate dehydrogenase (LDH)39 into the DMEM to detect cells undergoing cell death. Significant amounts of LDH were excreted after 4 h, and the excretion increased over time (Figure S4e). Studies showed that B. cereus protein complexes hemolysin BL and nonhemolytic enterotoxin induced cytotoxicity and cell detachment, which means not only collagenases may induce cytotoxicity but other toxins also contribute.40,41 These results suggest that bacterial collagenases may play a role in inducing cellular necrosis. Based on these findings, we chose a time window of 4–6 h for testing the efficacy of collagenase inhibitors in the NHDF infection model.
Collagenase Inhibitors Suppress the Gelatinolytic Activity of B. cereus Collagenases Released in the NHDF Infection Model
We investigated the most potent compounds from our in vitro cleavage assays in the NHDF infection model at concentrations ranging from 0, 25, 50, 100–200 μM. We observed a dose-dependent inhibitory effect on the activity of the secreted collagenases into the supernatant of infected cells detected by gelatin zymography. At 200 μM, 13 completely suppressed the gelatinolytic activity, while for 14 a complete inhibition was already observed at a concentration of 100 μM (Figures 6 and S5). The other diphosphonate derivatives (10, 11, and 15) were also evaluated. Compound 15 also demonstrated a clear dose-dependent inhibitory effect on gelatin turnover (Figure S6), while this was less evident for 10 and 11, which inhibited the collagenase activity toward gelatin only marginally at 100 μM (Figure S6).
Figure 6.

Activities of FDA-approved tiludronate disodium and compound 14 on the fibroblast (NHDF) cells infected with Bacillus cereus. (a) The antigelatinolytic activities of compounds tiludronate disodium and 14 against B. cereus collagenases. The DMEM medium of the infected NHDF cells was applied to the zymograms. Clear regions against blue background indicate that gelatin in the gel has been cleaved. (b) The amount of fibrillar collagens maintained by tiludronate disodium and 14 in the infected NHDF cells (highlighted in the yellow background). (c) The cytotoxicity of B. cereus infection (highlighted in the yellow background) in NHDF cells treated with and without tiludronate disodium and 14. Ctrl represents the noninfected cells (gray column) and the infected cells and nontreated with inhibitors (red column). Statistical analysis was performed with one-way ANOVA, and statistical significance was analyzed by the Tukey test. Significance was calculated by comparing nontreated vs treated cells with compounds (mean ± SD, ****p < 0.0001, **p < 0.01, *p ≤ 0.05, ns: nonsignificant). Ctrl: control. M (kDa): molecular weight marker.
The most promising FDA-approved diphosphonate drugs from the in vitro enzyme assays were also tested. Tiludronate disodium and alendronate sodium could completely inhibit gelatin turnover in the zymography at a concentration of 100 μM and 200 μM, respectively (Figures 6 and S9). Interestingly, the hydroxamate compounds (27, 28, 29, 30, 31, 32, and 33) showed no inhibitory effect on the collagenase in the zymography at all tested concentrations (Figures S11 and S12).
Collagenase Inhibitors Prevent NHDF Cell Detachment and Cleavage of Fibrillar Collagens of the ECM
We further evaluated the effect of the diphosphonate and hydroxamate compounds on cell morphology, the fibrillar collagen content of the ECM, and cell viability in the infection model. As shown in Figures 6 and S5, the diphosphate compounds 13 and 14 showed a dose-dependent effect on the infected NHDF cells. Above a concentration of 25 μM, they were both able to sustain the cells during infection. The cells stayed attached and maintained their spindle shape (Figures S7 and S8), and above 50 μM, we observed a significant reduction in LDH release, indicating higher cell viability. The diphosphonate derivative 15 and the FDA-approved diphosphonate drugs tiludronate disodium and alendronate sodium also showed dose-dependent effects on NHDF morphology, fibrillar ECM collagen content, and LDH release, however, at a concentration of ≥100 μM (Figures 6 and S8–S10). In contrast, diphosphonates 10 and 11 showed a smaller effect at all concentrations tested (except for 10, which resulted in higher cell viability and attachment at 100 μM) (Figures S6 and S8).
We also examined the hydroxamate compounds 27, 28, 29, 31, 32, and 33. Interestingly, none of them showed the expected effects. They were neither able to rescue cell morphology nor able to maintain the collagen content of the ECM. Also, no inhibition of the collagenase in the gelatin zymography was detected at all concentrations tested (Figures S11 and S12). Intrigued by the results of the hydroxamate compounds, we investigated their stability with LC-MS in the conditions of the NHDF infection model to obtain a potential explanation for the observed findings. We used 27 as a representative example for this compound class. The LC-MS spectra (Figure S13) confirmed, however, the stability of 27 under the assay conditions.
To sum up, in the NHDF infection model, the potent diphosphonate compounds from the in vitro enzyme assays were shown to have anticollagenolytic activity by inactivating the bacterial collagenases. Furthermore, they reduced the cytotoxicity induced directly or indirectly by the released collagenases and maintained the spindle-shaped morphology of the cells. In contrast, the hydroxamate-based compounds displayed almost no anticollagenolytic activity in the infection model, despite their inhibitory activity in the in vitro enzyme assays.
Rapid, Slow, or Very Slow Reversibility of Diphosphonate Inhibitors Depends on Target Collagenase
Next, we compared the mechanism inhibition of the diphosphonate and the hydroxamate compounds to the bacterial collagenases. For this purpose, we performed rapid dilution assays to test the reversibility of compound inhibition with ColA-CU and ColQ1-CU from B. cereus and ColH-PD and ColG-CU from C. histolyticum.36 Upon rapid dilution, rapidly reversible inhibitors quickly dissociate from the enzyme and progress curves similar to the uninhibited control are observed, while irreversible or very slowly dissociating inhibitors remain bound to the enzyme, which only very gradually recovers activity.
Intriguingly, as demonstrated in Figure S14, we observed clear differences between the compound classes. While hydroxamates 27 and 33 behaved toward both bacillary and clostridial collagenases like rapidly reversible inhibitors—as expected from active-site directed competitive inhibitors—the diphosphonate compounds showed a more varied response. In the case of ColQ1-CU, both 13 and 15 displayed progress curves with approximately less than 9% residual activity, which is typical for irreversible or very slowly dissociating inhibitors.36 Therefore, we examined ColQ1-CU treated overnight with 13 by mass spectrometry of the intact protein and could confirm that the compound did not result in a covalent modification (Figure S15). This leads to the conclusion that 13 must be a very slowly dissociating reversible binder of ColQ1. In the case of ColA-CU, the rapid dilution assay revealed only a minimal inhibition in the presence of 13, but 15 displayed a progress curve typical for slowly dissociating inhibitors. In the case of ColH, the effect of 13 and 15 was reversed compared to ColA, and in the case of ColG, both diphosphonates behaved like rapidly reversible inhibitors (Figure S14).
Slowly and very slowly dissociating inhibition are interesting mechanisms for reducing enzyme activity, as they increase the lifetime of the enzyme–inhibitor complex. Its advantages appear in the latter phases of drug development when pharmacological properties (i.e., dosing interval and patient safety) need to be optimized.36,37 We speculate that the differential behavior observed in the infection models between the diphosphonate and hydroxamate compounds is caused by their different dissociation properties, which results in different enzyme–inhibitor complex lifetimes. The exact mechanism underlying the slow dissociation behavior, however, remains currently elusive, as we could not get a crystal structure of a diphosphonate inhibitor complex with ColA or ColQ1. Other reasons could also be related to the limited activity of the hydroxamate in the infection experiments. For instance, the accumulation of the hydroxamates inside the infected cells or binding to the large substrate of collagenase (i.e., collagen), which subsequently results in a reduced concentration available to bind with the extracellular bacterial collagenases.
Small-Molecule Inhibitors Reduce Collagenase Activity and Maintain Epithelial Cell Integrity
Collagenase Inhibitors Maintain the TEER of MDCK II Cells
Epithelial cells form intercellular tight junctions establishing a sealed epithelium to control the diffusion of membrane proteins, the uptake of small molecules, and protect the body from hazardous substances.42,43B. cereus-mediated cell detachment severely destroys the epithelial barrier function, allowing bacterial access to deeper areas of the tissue. To quantify the effect on the epithelial barrier function, we investigated selected compounds regarding their effect on the transepithelial electrical resistance (TEER) of polarized MDCK II cells. We treated MDCK II cells with our target compounds followed by the infection with the B. cereus AH187 or coincubation with the supernatant from AH187 strain culture, which contains ColQ1 and ColA. In comparison to the control without inhibitor, the TEER of the infected MDCK cells (Figures 7 and S16) was maintained with compounds 14, 15, tiludronate disodium, and alendronate sodium at 100 μM. On the other hand, compounds 13 and 10 failed to sustain the TEER of the infected cells at 200 μM (Figure S16) but they retain the TEER of the challenged cells with 50% (v/v) supernatant (Figure 7b).
Figure 7.

Change in the transepithelial electrical resistance (TEER) of the Madin–Darby canine kidney II (MDCK II) cells challenged with Bacillus cereus bacteria or 50% (v/v) culture supernatant and treated with or without collagenase inhibitors. (a) 14 and 15 preserve the TEER value of the MDCK II infected with B. cereus compared with the nontreated conditions with inhibitor. (b) Compounds 11, 13, 15, and tiludronate disodium maintained the TEER of MDCK II cells challenged with B. cereus supernatant. Each curve represents the average ± standard deviation of at least three independent experiments.
This discrepancy might be due to the lower amounts of collagenases secreted into the supernatant compared to the very high bacterial densities during infection that lead to a high collagenase secretion. This might have disrupted the optimum inhibitor/collagenase ratio and resulted in lower efficacy of some inhibitors. Thus, the effect of some inhibitors might be less in this case. Despite these variations in effect, our findings support the notion that collagenases are involved in attacking epithelial barriers and that our inhibitors can help to preserve the cellular junctions, thereby reducing bacterial invasion. These findings corroborate the theory that collagenases are one of the factors that might be involved in disturbing the TEER of epithelial cells. Our collagenase inhibitors proved their potential to preserve the cell attachment and the junction between them, subsequently reducing bacterial invasion.
Compounds Do Not Interfere with B. cereus Growth
The aim of this work was to develop an antivirulence agent that prevents bacteria from causing damage rather than killing them.3 Therefore, we tested the compounds on B. cereus growth to rule out antibacterial activity and ensure that the effects shown in the in vitro infection models were not caused by influencing bacterial viability. For this purpose, we selected the most potent compounds and tested them against the AH187 strain. As shown in Table S5, the compounds had no effect on AH187 growth and their minimum inhibitory concentration (MIC50) was >200 or >100 μM. These data support that the antivirulence activity, and not an antibacterial activity, was responsible for the observed effect in the infection experiments.
ColQ1 Inhibitors Maintain the Survival of G. mellonella Larvae
Finally, we evaluated selected compounds in an in vivo infection model. G. mellonella larvae are one of the most frequently used models to evaluate the effectiveness of newly discovered inhibitors and are well established for assessing B. cereus cytotoxicity.44 We reported previously that treating the G. mellonella larvae with ColQ1 caused death of the larvae.28 We established B. cereus infection of G. mellonella by injecting AH187 strain into the larvae in the presence or absence of our compounds. The larvae were incubated at 37 °C throughout the experiment, and their survival was monitored twice a day. Three synthesized inhibitors and two FDA-approved drugs were tested. Compounds 13, 14, 15, tiludronate disodium, and alendronate sodium ameliorated the survival of the larvae in a dose-dependent manner when compared to a control where no inhibitor was administered. At 100 μM, compounds 14 and 15 boosted the survival by around 50% and 35%, respectively. At lower concentration (i.e., 25 μM), both had a reduced effect (Figures 8 and S17). The FDA-approved tiludronate disodium and alendronate sodium increased the survival by 40% at 100 and 200 μM, respectively (Figures 8 and S17). A concentration of 200 μM of 13 showed the highest effect, increasing the survival by about 35%, while 50 μM showed the smallest effect and only improved survival by 5% (Figure S17). Overall, the data revealed the protective effect of the collagenase inhibition during B. cereus infection. As a result, these antivirulence compounds may be evaluated as a promising therapeutic agent in the future.
Figure 8.

Survival analysis of Galleria mellonella larvae treated with Bacillus cereus AH187 with and without 14 and tiludronate disodium. Each curve represents results of three independent experiments; the statistical difference between groups treated with 100, 50, and 25 μM of compound 14 and B. cereus AH187 and with the group treated only with B. cereus AH187 is p < 0.0001, p = 0.0039, and p = 0.0173, respectively. The statistical difference between groups treated with 100 μM tiludronate disodium and with B. cereus AH187 is p = 0.0032 (log-rank test). The survival rate for the larvae treated with compound 14 and tiludronate disodium in PBS was 100%.
Conclusions
To tackle the AMR crisis and the slow discovery of new anti-infectives, non-traditional therapies and FDA-approved drugs repurposing are promising strategies. One potential non-traditional approach is the use of antivirulence agents, which inhibit the pathogenicity factors of bacteria and thus prevent or delay infection without exerting selective pressure. Bacterial collagenases are one of the antivirulence targets that are currently gaining wide attention. In this study, we identified two classes of inhibitors (synthesized and FDA-approved diphosphonates and hydroxamates) that target clostridial and bacillary collagenases. Among these, compounds, 13, 14, 15, tiludronate disodium, and alendronate sodium of the diphosphonate class and 27 and 33 of the hydroxamate class displayed high and broad-spectrum in vitro inhibition of clostridial collagenases ColH and ColG and on bacillary collagenases ColA and ColQ1. Furthermore, the majority of them demonstrated adequate selectivity over human MMPs and other off-targets. These compounds showed no cytotoxicity in four mammalian cell lines. We also studied the biological effects of these compounds in infection models using B. cereus as a representative bacterium-producing collagenase. In this context, we developed fibroblast- and epithelial cell infection models to characterize the effect of B. cereus collagenases and their inhibition. Our findings suggest that the inhibition of B. cereus collagenases by the most potent diphosphonate compounds maintained the fibroblast cell attachment, cell morphology, and cell viability, preserved the fibrillar collagen content, and sustained the TEER of epithelial cells. We also tested the compounds in vivo in the G. mellonella larvae infection model, where they enhanced the survival rate. The hydroxamates did not display any inhibition in the infection models; however, they showed similar potency as diphosphonates in the enzyme cleavage assays. This could be explained by their quick dissociation from the clostridial and bacillary collagenases. In contrast, the most active diphosphonate demonstrated high potency in both enzyme assays and infection models, which might be due to their slow to very slow dissociation from the bacillary collagenases. These findings offer insight into the role of bacterial collagenases in infections and the significance of their inhibition with small-molecule inhibitors and FDA-approved drugs, which might represent a potential treatment strategy in the future.
Materials and Methods
Chemistry
Chemical names follow the IUPAC nomenclature. Starting materials were purchased from Chempur, Sigma-Aldrich, Acros, Combi-Blocks, or Fluorochem and were used without purification. Column chromatography was performed using the automated flash chromatography system Combiflash Rf+ (Teledyne Isco) equipped with RediSepRf silica columns. The final products were dried in high vacuum. 1H NMR and 13C NMR spectra were measured on a Bruker AM500 spectrometer (at 500 and 125 MHz, respectively) at 300 K and on a Bruker Fourier 300 (at 300 and 75 MHz, respectively) at 300 K. Chemical shifts are reported in δ (parts per million: ppm) by reference to the hydrogenated residues of deuterated solvent as the internal standard: 2.05 ppm (1H NMR), 29.8, and 206.3 ppm (13C NMR) for acetone-d6, 2.50 ppm (1H NMR) and 39.52 ppm (13C NMR) for DMSO-d6. Signals are described as br (broad), s (singlet), d (doublet), t (triplet), dd (doublet of doublets), ddd (doublet of doublet of doublets), dt (doublet of triplets), and m (multiplet). All coupling constants (J) are given in Hertz (Hz). Mass spectrometry was performed on a TSQ Quantum (Thermo Fisher, Dreieich, Germany). The triple quadrupole mass spectrometer was equipped with an electrospray interface (ESI). Purity of compounds was determined by LC-MS using the area percentage method on the UV trace recorded at a wavelength of 254 nm and found to be >95%. The Surveyor-LC-system consisted of a pump, an auto sampler, and a PDA detector. The system was operated by the standard software Xcalibur. An RP C18 NUCLEODUR 100-5 (3 mm) column (Macherey-Nagel GmbH) was used as the stationary phase. All solvents were HPLC grade. In a gradient run, using acetonitrile and water, the percentage of acetonitrile (containing 0.1% trifluoroacetic acid) was increased from an initial concentration of 0% at 0 min to 100% at 13 min and kept at 100% for 2 min. The injection volume was 15 μL, and the flow rate was set to 800 μL/min. MS analysis was carried out at a needle voltage of 3000 V and a capillary temperature of 350 °C. Mass spectra were acquired in positive mode, using the electron spray ionization method, from 100 to 1000 m/z, and UV spectra were recorded at a wavelength of 254 nm and in some cases at 360 nm. High-resolution mass spectrometry (HRMS) measurements were recorded on a SpectraSystems-MSQ LC-MS system (Thermo Fisher).
Experimental Procedures of Diphosphonates
The following compounds were prepared according to previously described procedures: 7a–16a and 7b–14b.29−31
General Procedure A: Preparation of 1,4-Dihydro-2,3-quinoxaline-dione Derivatives 7a–14a and 15b
A mixture of 1,2-phenylenediamine derivative (1 equiv) and oxalic acid (1.2 equiv) was refluxed in 4 N HCl (20 mL) for 6 h, cooled to RT, poured over ice, and filtered. The product was washed with water and dried to give the title compound. The product was used in the next step without further purification.
General Procedure B: Preparation of 2,3-Dichloroquinoxaline Derivatives 7b–14b and 15c
A mixture of 1,4-dihydro-2,3-quinoxalinediones 7a–14a and 15b (2 mmol) and POCl3 (10 mL) was stirred at 50 °C in DMF (30 mL) for 2 h, cooled to RT, poured over ice, and filtered. The product was washed with water and dried to give the title compound. The product was used in the next step without further purification.
General Procedure C: Preparation of Diethyl Phosphonate Derivatives 7c–13c, 14d, 15d, and 16b
2,3-Dichloroquinoxaline derivatives 7b–14b, 15c, and 16a (1 equiv) were suspended in triethyl phosphite (10 equiv) and heated to 150 °C in a sealed tube for 18 h. Most of the unreacted triethyl phosphite was evaporated in vacuo, and the resultant oil was purified by column chromatography.
General Procedure D: Preparation of Phosphonic Acid Derivatives 7–16
To a solution of diethyl phosphonate (1 equiv) in dry DCM, bromotrimethylsilane (10 equiv) was added dropwise over a period of 15 min. The reaction mixture was stirred at RT overnight. Then, MeOH was added and stirred at RT for 30 min. Solvents were concentrated in vacuo, and the resultant oil was purified by preparative HPLC.
6-(4-Chlorophenyl)-1,4-dihydroquinoxaline-2,3-dione (15b)
Compound 15b was synthesized according to
general procedure A, using 4′-chloro-[1,1′-biphenyl]-3,4-diamine 15a (110 mg, 0.5 mmol) and oxalic acid (54 mg, 0.6 mmol).
The product was obtained as a yellow solid (90 mg, 66%). 1H NMR (500 MHz, DMSO-d6) δ: 11.96 (d, J = 10.7 Hz, 2H), 7.57 (d, J = 8.7 Hz, 2H), 7.53
(d, J = 8.7 Hz, 2H), 7.41 (dd, J = 8.5, 1.9 Hz, 1H), 7.37 (d, J = 1.9 Hz, 1H), 7.22
(d, J = 8.5 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ: 155.1, 155.5, 138.4, 133.9, 132.1, 129.0, 127.8,
126.6, 126.2, 122.0, 116.4, 113.1. MS (ESI+) m/z 273 [M + H]+.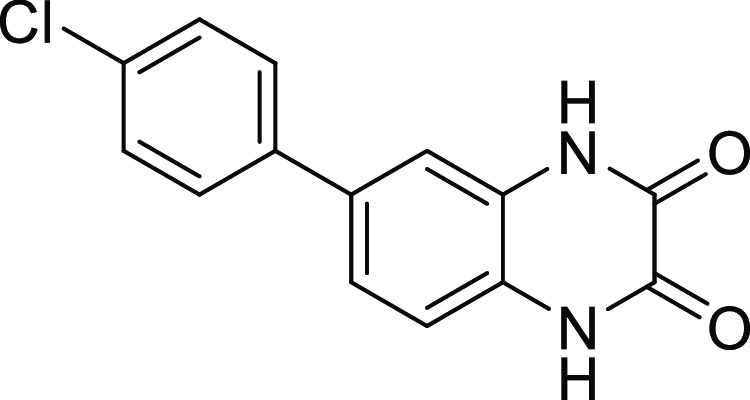
Tetraethyl Quinoxaline-2,3-diylbis(phosphonate) (7c)
Compound 7c was synthesized according to
general procedure C, using 2,3-dichloroquinoxaline 7b (90 mg, 0.45 mmol) and triethyl phosphite (819 μL, 4.5 mmol).
The residue was purified by automated column chromatography (Hex/EtOAc
= 1/1) to give the desired product (white solid, 129 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 8.12 (dd, J = 6.4, 3.5 Hz, 2H), 7.83 (dd, J = 6.4,
3.4 Hz, 2H), 4.56–3.98 (m, 8H), 1.37 (t, J = 7.1 Hz, 12H). 13C NMR (126 MHz, CDCl3) δ
149.9 (dd, JC-P = 226.4, 30.0 Hz),
140.9–140.1 (m), 132.4, 129.8, 63.9 (t, JC-P = 3.2 Hz), 19.3–9.9 (m). 31P NMR
(202 MHz, CDCl3) δ 6.83. MS (ESI+) m/z 403.05 [M + H]+.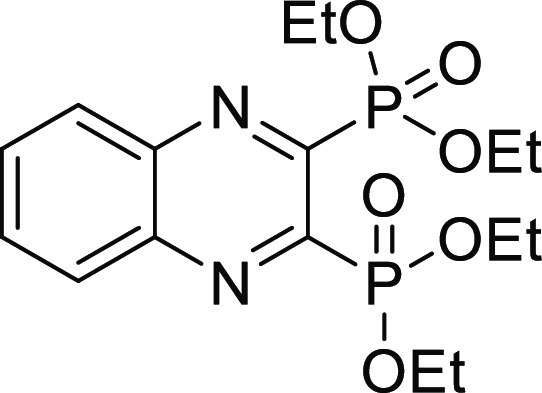
2,3-Dichloro-N-(3,4-dichlorophenyl)quinoxaline-6-carboxamide (14c)
2,3-Dichloroquinoxaline-6-carboxylic acid
(122 mg, 0.50 mmol) was dissolved in DCM (20 mL). EDC·HCl (191
mg, 1 mmol) was added, followed by 3,4-dichloroaniline (162 mg, 1
mmol). The reaction was stirred at room temperature for 24 h. The
crude product was purified using column chromatography DCM to DCM/MeOH
1%. The product was obtained as a white solid (107 mg, 55%). 1H NMR (500 MHz, DMSO) δ 10.87 (s, 1H), 8.71 (s, 1H),
8.39 (d, J = 6.7 Hz, 1H), 8.29–8.13 (m, 2H),
7.74 (dd, J = 70.6, 7.2 Hz, 2H). 13C NMR
(126 MHz, DMSO) δ 164.7, 147.0, 146.4, 141.8, 139.8, 139.4,
136.9, 131.4, 131.1, 130.6, 128.8, 127.9, 126.0, 122.0, 120.8. MS
(ESI+) m/z 387.96 [M + H]+.
2,3-Dichloro-6-(4-chlorophenyl)quinoxaline (15c)
Compound 15c was synthesized according to
general procedure B, using 6-(4-chlorophenyl)-1,4-dihydroquinoxaline-2,3-dione 15b (85 mg, 0.31 mmol) and POCl3 (5 mL). The product
was obtained as a white solid (69 mg, 72%). 1H NMR (500
MHz, DMSO) δ 8.38 (d, J = 1.9 Hz, 1H), 8.28
(dd, J = 8.8, 2.0 Hz, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H). 13C NMR (126 MHz, DMSO) δ
145.6, 145.0, 142.1, 140.8, 139.9, 137.2, 134.2, 131.0, 129.7, 129.6,
128.9, 125.3.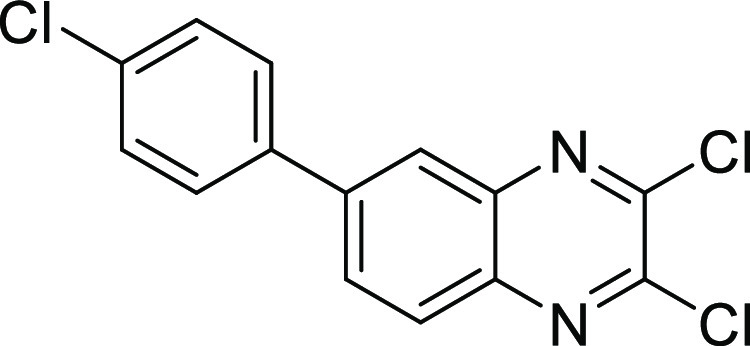
Quinoxaline-2,3-diylbis(phosphonic acid) (7)
Compound 7 was synthesized according to general procedure
D, using tetraethyl quinoxaline-2,3-diylbis(phosphonate) 7c (120 mg, 0.29 mmol), bromotrimethylsilane (444 μL, 2.9 mmol),
and DCM (15 mL). The reaction was stirred at RT overnight. The crude
product was purified using preparative HPLC (CH3CN (HCOOH
0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0). The product
was obtained as a white solid (53 mg, 61%). 1H NMR (500
MHz, DMSO) δ 8.23 (dd, J = 6.4, 3.4 Hz, 2H),
8.04 (dd, J = 6.4, 3.4 Hz, 2H). 13C NMR
(126 MHz, DMSO) δ 153.1 (dd, JC-P = 203.1, 30.4 Hz), 140.6 (dd, JC-P = 20.1, 4.5 Hz), 133.0, 129.7. 31P NMR (202 MHz, DMSO)
δ 5.24. Purity: 100%. HRMS (ESI–) calculated
for C8H7N2O6P2 [M – H]− 288.9784, found
288.9785.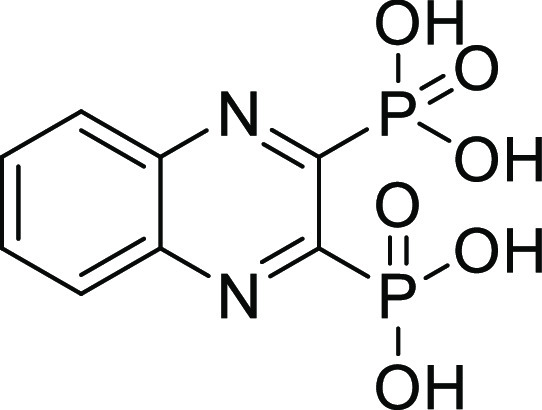
(6-Fluoroquinoxaline-2,3-diyl)bis(phosphonic acid) (8)
Compound 8 was synthesized according to general
procedure D, using tetraethyl (6-fluoroquinoxaline-2,3-diyl)bis(phosphonate) 8c (120 mg, 0.29 mmol), bromotrimethylsilane (444 μL,
2.9 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (63 mg, 72%). 1H NMR (500 MHz, MeOD) δ 8.31 (dd, J = 9.3,
5.7 Hz, 1H), 7.91 (dd, J = 8.9, 2.7 Hz, 1H), 7.85
(td, J = 8.8, 2.8 Hz, 1H). 13C NMR (126
MHz, MeOD) δ 165.6 (d, JC-F = 255.4 Hz), 153.5 (dd, JC-P,C-P = 208.9, 30.7 Hz), 151.9 (ddd, JC-P,C-P = 210.6, 30.4, 3.3 Hz), 143.2 (ddd, JC-P,C-F = 19.1, 13.8, 2.5 Hz), 139.7 (dd, JC-P,C-F = 18.8, 2.2 Hz), 133.4 (d, JC-F = 10.3 Hz), 124.3 (d, JC-F =
26.9 Hz), 113.8 (d, JC-F = 22.1
Hz). 31P NMR (202 MHz, MeOD) δ 6.89 (d, J = 7.1 Hz), 6.66 (d, J = 6.9 Hz). Purity: 96%. HRMS
(ESI–) calculated for C8H6FN2O6P2 [M–
H]− 306.9690, found 306.9690.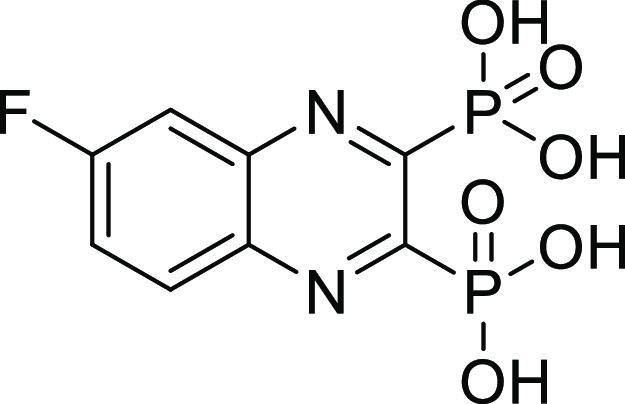
(6-Chloroquinoxaline-2,3-diyl)bis(phosphonic acid) (9)
Compound 9 was synthesized according to general
procedure D, using tetraethyl (6-chloroquinoxaline-2,3-diyl)bis(phosphonate) 9c (110 mg, 0.25 mmol), bromotrimethylsilane (326 μL,
2.5 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (48 mg, 59%). 1H NMR (500 MHz, DMSO) δ 8.28 (d, J = 2.3 Hz,
1H), 8.24 (d, J = 9.0 Hz, 1H), 8.04 (dd, J = 9.0, 2.3 Hz, 1H). 13C NMR (126 MHz, DMSO)
δ 154.3 (dd, JC-P = 201.5,
30.2 Hz), 153.6 (dd, JC-P = 202.3,
30.3 Hz), 140.9 (dd, JC-P = 18.4,
2.6 Hz), 139.3 (dd, JC-P = 18.3,
2.7 Hz), 137.3, 133.5, 131.6, 128.3. 31P NMR (202 MHz,
DMSO) δ 4.87 (d, J = 5.3 Hz), 4.72 (d, J = 6.1 Hz). Purity: 95%. HRMS (ESI–)
calculated for C8H6ClN2O6P2 [M – H]− 322.9395,
found 322.9395.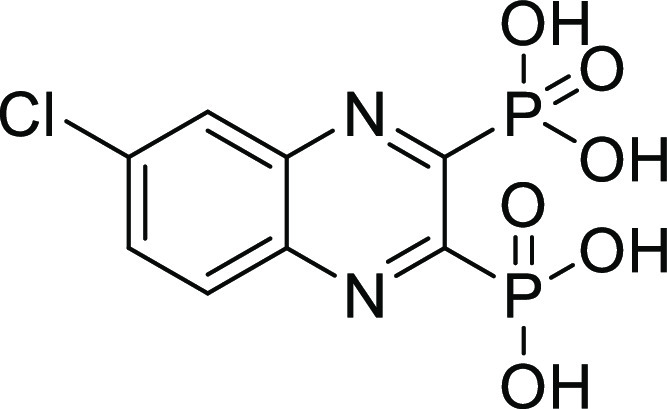
(6-Bromoquinoxaline-2,3-diyl)bis(phosphonic acid) (10)
Compound 10 was synthesized according to
general procedure D, using tetraethyl (6-bromoquinoxaline-2,3-diyl)bis(phosphonate) 10c (80 mg, 0.16 mmol), bromotrimethylsilane (209 μL,
1.6 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (46 mg, 75%). 1H NMR (500 MHz, DMSO) δ 8.42 (s, 1H), 8.14 (d, J = 0.8 Hz, 2H). 13C NMR (126 MHz, DMSO) δ 154.4
(dd, JC-P = 200.9, 30.1 Hz), 153.8
(dd, JC-P = 201.7, 30.0 Hz), 141.1
(dd, JC-P = 18.3, 2.4 Hz), 139.5
(dd, JC-P = 18.1, 2.2 Hz), 136.0,
131.6, 131.5, 126.1. 31P NMR (202 MHz, DMSO) δ 4.90
(d, J = 7.1 Hz), 4.71 (d, J = 7.1
Hz). Purity: 100%. HRMS (ESI–) calculated for C8H6BrN2O6P2 [M – H]− 366.8890, found 366.8890.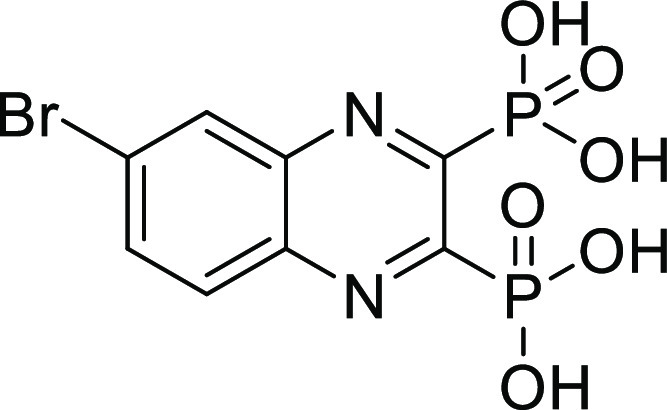
(6-Methylquinoxaline-2,3-diyl)bis(phosphonic acid) (11)
Compound 11 was synthesized according to
general procedure D, using tetraethyl (6-methylquinoxaline-2,3-diyl)bis(phosphonate) 11c (110 mg, 0.26 mmol), bromotrimethylsilane (340 μL,
2.6 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (64 mg, 80%). 1H NMR (500 MHz, DMSO) δ 8.10 (d, J = 8.6 Hz,
1H), 7.98 (s, 1H), 7.87 (dd, J = 8.6, 1.7 Hz, 1H),
2.63 (s, 3H). 13C NMR (126 MHz, DMSO) δ 152.9 (dd, JC-P = 203.4, 30.5 Hz), 151.9 (dd, JC-P = 204.2, 30.5 Hz), 143.6, 140.7 (dd, JC-P = 17.4, 1.4 Hz), 139.2 (dd, JC-P = 17.5, 1.4 Hz), 135.2, 129.2, 128.2,
21.9. 31P NMR (202 MHz, DMSO) δ 5.60 (d, J = 5.2 Hz), 5.48 (d, J = 7.0 Hz). Purity:
95%. HRMS (ESI–) calculated for C9H9N2O6P2 [M – H]− 302.9941, found 302.9941.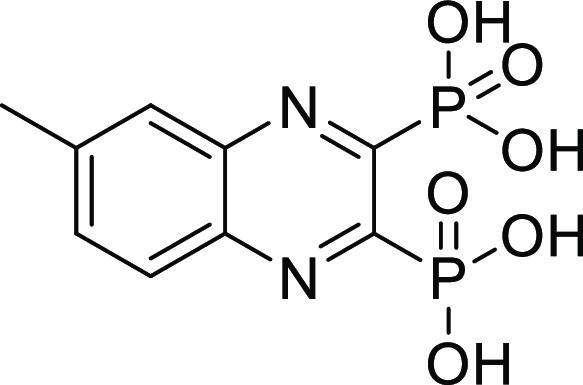
(6-Methoxyquinoxaline-2,3-diyl)bis(phosphonic acid) (12)
Compound 12 was synthesized according to
general procedure D, using tetraethyl (6-methoxyquinoxaline-2,3-diyl)bis(phosphonate) 12c (90 mg, 0.20 mmol), bromotrimethylsilane (260 μL,
2.0 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (46 mg, 70%). 1H NMR (500 MHz, DMSO) δ 8.11 (d, J = 9.2 Hz,
1H), 7.66 (dd, J = 9.2, 2.6 Hz, 1H), 7.52 (d, J = 2.4 Hz, 1H), 4.02 (s, 3H). 13C NMR (126 MHz,
MeOD) δ 163.1, 152.9 (dd, JC-P = 203.0, 30.3 Hz), 151.9 (dd, JC-P = 204.1, 30.3 Hz), 142.6 (dd, JC-P = 17.1, 1.4 Hz), 139.2 (dd, JC-P = 17.4, 1.4 Hz), 130.3, 126.1, 106.0, 55.3. 31P NMR (202
MHz, DMSO) δ 5.99 (d, J = 4.6 Hz), 5.68 (d, J = 6.0 Hz). Purity: 100%. HRMS (ESI–)
calculated for C9H9N2O7P2 [M – H]− 318.9890,
found 318.9890.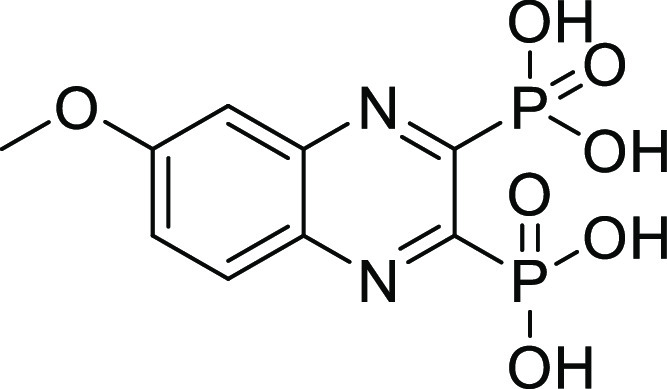
(6,7-Dichloroquinoxaline-2,3-diyl)bis(phosphonic acid) (13)
Compound 13 was synthesized according
to general procedure D, using tetraethyl (6,7-dichloroquinoxaline-2,3-diyl)bis(phosphonate) 13c (80 mg, 0.17 mmol), bromotrimethylsilane (222 μL,
1.7 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (39 mg, 65%). 1H NMR (500 MHz, DMSO) δ 8.51 (s, 2H). 13C NMR (126
MHz, DMSO) δ 155.1 (dd, JC-P = 199.4, 29.9 Hz), 139.6 (dd, JC-P = 19.9, 3.9 Hz), 135.58, 130.57. 31P NMR (202 MHz, DMSO)
δ 4.49. Purity: 98%. HRMS (ESI–) calculated
for C8H5Cl2N2O6P2 [M – H]− 356.9005,
found 356.9005.
(6-((3,4-Dichlorophenyl)carbamoyl)quinoxaline-2,3-diyl)bis(phosphonic acid) (14)
Compound 14 was synthesized
according to general procedure D, using tetraethyl (6-((3,4-dichlorophenyl)carbamoyl)quinoxaline-2,3-diyl)bis(phosphonate) 14d (100 mg, 0.17 mmol), bromotrimethylsilane (222 μL,
1.7 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (43 mg, 53%). 1H NMR (500 MHz, DMSO) δ 10.99 (s, 1H), 8.87 (d, J = 1.8 Hz, 1H), 8.45 (dd, J = 8.8, 1.8 Hz, 1H),
8.33 (d, J = 8.8 Hz, 1H), 8.25 (d, J = 2.4 Hz, 1H), 7.84 (dd, J = 8.8, 2.4 Hz, 1H),
7.68 (d, J = 8.8 Hz, 1H). 13C NMR (126
MHz, DMSO) δ 164.9, 155.6 (dd, JC-P = 70.9, 29.7 Hz), 154.0 (dd, JC-P = 71.9, 30.0 Hz), 141.8 (dd, JC-P = 18.3, 2.7 Hz), 139.9 (dd, JC-P = 18.4, 2.6 Hz), 139.5, 137.3, 131.4, 131.2, 131.1, 130.1, 129.3,
126.0, 122.0, 120.8. 31P NMR (202 MHz, DMSO) δ 4.95
(d, J = 4.8 Hz), 4.71 (d, J = 6.0
Hz). Purity: 98%. HRMS (ESI–) calculated for C15H10Cl2N3O7P2 [M – H]− 475.9376,
found 475.9376.
(6-(4-Chlorophenyl)quinoxaline-2,3-diyl)bis(phosphonic acid) (15)
Compound 15 was synthesized
according to general procedure D, using tetraethyl (6-(4-chlorophenyl)quinoxaline-2,3-diyl)bis(phosphonate) 15d (70 mg, 0.13 mmol), bromotrimethylsilane (170 μL,
1.3 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (24 mg, 44%). 1H NMR (500 MHz, DMSO) δ 8.45 (d, J = 1.9 Hz,
1H), 8.37 (dd, J = 8.8, 2.0 Hz, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.04–7.96 (m, 2H), 7.67–7.59
(m, 2H). 13C NMR (126 MHz, DMSO) δ 154.4 (dd, JC-P = 88.9, 30.6 Hz), 152.8 (dd, JC-P = 89.0, 29.9 Hz), 142.7, 140.9 (dd, JC-P = 20.3, 5.2 Hz), 140.0 (dd, JC-P = 20.4, 5.1 Hz), 137.4, 134.3, 131.8,
130.3, 129.8, 129.7, 126.5. 31P NMR (202 MHz, DMSO) δ
5.31, 5.27. Purity: 98%. HRMS (ESI–) calculated
for C14H10ClN2O6P2 [M – H]− 398.9708,
found 398.9707.
(6,7-Dichloro-3-oxo-3,4-dihydroquinoxalin-2-yl)phosphonic Acid (16)
Compound 16 was synthesized
according to general procedure D, using diethyl (6,7-dichloro-3-oxo-3,4-dihydroquinoxalin-2-yl)phosphonate 16b (90 mg, 0.25 mmol), bromotrimethylsilane (326 μL,
2.5 mmol), and DCM (15 mL). The reaction was stirred at RT overnight.
The crude product was purified using preparative HPLC (CH3CN (HCOOH 0.05%)/H2O (HCOOH 0.05%) = 0.2:9.8 to 10:0).
The product was obtained as a white solid (32 mg, 43%). 1H NMR (500 MHz, DMSO) δ 8.10 (s, 1H), 7.48 (s, 1H). 13C NMR (126 MHz, DMSO) δ 159.9 (d, JC-P = 213.5 Hz), 154.0 (d, JC-P =
28.6 Hz), 134.4, 132.9 (d, JC-P = 2.3 Hz), 131.2 (d, JC-P = 24.4
Hz), 130.7, 125.7, 117.2. 31P NMR (202 MHz, DMSO) δ
1.54. Purity: 95%. HRMS (ESI–) calculated for C8H4Cl2N2O4P [M – H]− 292.9291, found 292.9290.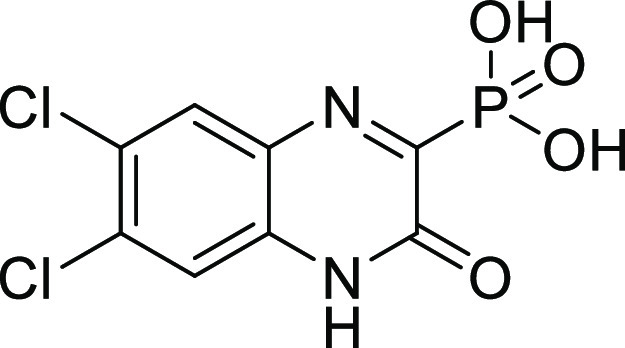
Experimental Procedures of Hydroxamates
General Procedure A2: Monosaponification
Malonate diester (1 equiv) was dissolved in a mixture of ethanol/water (4:1, 0.43–0.47 M), and sodium hydroxide (1.2 equiv) was added. The reaction mixture was stirred at RT overnight. Then, solvents were evaporated under reduced pressure, and the aqueous mixture remaining was diluted with sat. aq. NaHCO3 and washed with CH2Cl2. The aqueous layer was then carefully acidified (pH ∼ 1) with aq. HCl and extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure affording the desired product.
General Procedure B2: Amide Formation
Carboxylic acid (1 equiv) and amine (1.1 equiv) were dissolved in CH2Cl2 (0.21 M). HOBt (0.1 equiv), EDC.HCl (1.5 equiv), and diisopropylethylamine (3 equiv) were then added, and the mixture was stirred at RT overnight. The mixture was then washed with diluted aq. HCl (1 M), sat. aq. NaHCO3, and sat. aq. NaCl. The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure or the residue was finally purified by flash chromatography to give the desired amide.
General Procedure C2: Boc Deprotection
The Boc-protected intermediate was dissolved in a mixture of ethanol and dichloromethane (1:1, 0.08–0.14 M), and the reaction was cooled down to 0 °C before the addition of 4 N HCl in dioxane (0.17–0.28 M). The mixture was stirred at RT overnight. Then, solvents were evaporated to give the desired compound.
General Procedure D2: Diazo Transfer
A suspension of amine (1 equiv), ZnCl2 (0.06 equiv), and K2CO3 (1 equiv) in ethanol (0.8–0.93 M) under an inert atmosphere was cooled down to 0 °C with an ice bath. Separately, diisopropylethylamine (1.1–3.5 equiv) was slowly added to a solution of 1H-imidazole-1-sulfonyl azide; hydrogen chloride (1.2 equiv) was dissolved in ethanol (0.33–0.74 M) under an inert atmosphere (solution A). The azide-containing solution A was immediately added dropwise to the first mixture at 0 °C. Then, the cooling bath was removed, and the white mixture was stirred at RT overnight. The mixture was then cooled down to 0 °C, diluted with water, and carefully acidified to pH = 2 with 1 N aq. HCl. It was finally extracted with ethyl acetate. The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure or the residue was finally purified by flash chromatography to give the desired azide.
General Procedure E2: Aminolysis
To an ester solution (1 equiv) in methanol (0.15–0.19 M) were added aq. hydroxylamine (50% w/w in water, 0.15–0.19 M) and KCN (0.1 equiv). The mixture was stirred overnight. Then, the solvents were removed at RT under reduced pressure and the residue was purified by flash chromatography to afford the desired hydroxamate.
General Procedure F2: Copper-Catalyzed Click Reaction
Azide (1 equiv) and alkyne (1.0 equiv) were dissolved in N,N-dimethylformamide or dioxane (0.05–0.08 M) before the addition of copper sulfate pentahydrate (0.2 equiv) in water (0.1–0.15 M) and sodium ascorbate (0.5 equiv). The resulting mixture was stirred at room temperature overnight. The mixture was then diluted in water and extracted with ethyl acetate. The combined organic layers were dried over MgSO4, filtered, and concentrated under reduced pressure. The residue was finally purified by flash chromatography affording the desired 1,4-triazole.
General Procedure G2: Alkyne Formation
To a phenol solution (1 equiv) in N,N-dimethylformamide (0.2–0.5 M) were added K2CO3 (2 equiv) and propargyl bromide (1.1 equiv). The resulting solution was heated to 60 °C and stirred overnight. The mixture was then diluted in water and extracted three times with ethyl acetate. The organic layers were combined, and solvents were evaporated under reduced pressure affording the desired alkyne.
2-Ethoxycarbonyl-4-methyl-pentanoic Acid (22a)
Compound 22a was synthesized according to the general
procedure A2, using diethyl isopropylmalonate (4000 mg, 18.5 mmol)
and sodium hydroxide (888 mg, 22.2 mmol) in EtOH/H2O (40
mL, 32:8 v/v) overnight. Compound 22a was obtained as a colorless oil (2400 mg, 68%) and was
used in the next step without further purification. 1H
NMR (500 MHz, DMSO-d6) δ: 12.80
(br s, 1H), 4.10 (q, J = 7.1 Hz, 2H), 3.36–3.33
(m, 1H), 1.65–1.59 (m, 2H), 1.49 (sep, J =
6.7 Hz, 1H), 1.17 (t, J = 7.1 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ:
170.6, 169.6, 60.7, 49.8, 37.2, 25.7, 22.2, 22.1, 14.0. MS (ESI+): m/z = 189 [M + H]+.
2-Benzyl-3-ethoxy-3-oxo-propanoic Acid (22b)
Compound 22b was synthesized according to the general
procedure A2, using diethyl benzylmalonate (1070 mg, 4.3 mmol) and
sodium hydroxide (205 mg, 5.1 mmol) in EtOH/H2O (10 mL,
8:2 v/v) overnight. Compound 22b was
obtained as a colorless oil (694 mg, 72%) and was used in the next
step without further purification. 1H NMR (500 MHz, CDCl3) δ: 10.23 (br s, 1H), 7.36–7.31 (m, 2H), 7.30–7.26
(m, 3H), 4.22 (q, J = 7.1 Hz, 2H), 3.76 (t, J = 7.7 Hz, 1H), 3.30 (dd, J = 7.7 and
2.8 Hz, 2H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ: 174.3, 168.9, 137.4, 128.9
(2C), 128.7 (2C), 127.1, 62.0, 53.6, 34.9, 14.1. MS (ESI+): m/z = no ionization [M + H]+.
Ethyl 2-[2-(tert-Butoxycarbonylamino)ethylcarbamoyl]-4-methyl-pentanoate (23a)
Compound 23a was synthesized
according to the general procedure B2, using carboxylic acid 22a (2000 mg, 10.6 mmol), tert-butyl N-(2-aminoethyl)carbamate (1876 mg, 11.6 mmol), HOBt (162.8
mg, 1.0 mmol), EDC.HCl (3060 mg, 16.0 mmol), and diisopropylethylamine
(5560 μL, 31.8 mmol) in CH2Cl2 (50 mL)
overnight. The crude product was purified by flash chromatography
on silica gel (cHex to cHex/EtOAc 6:4) affording compound 23a as a yellowish solid (1868 mg, 53%). 1H NMR (DMSO-d6) δ: 8.13 (t, J = 5.3
Hz, 1H), 6.71 (t, J = 5.4 Hz, 1H), 4.10–4.01
(m, 2H), 3.29–3.26 (m, 1H), 3.15–3.01 (m, 2H), 2.98–2.95
(m, 2H), 1.64–1.53 (m, 2H), 1.45–1.40 (m, 1H), 1.37
(s, 9H), 1.15 (t, J = 7.1 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H), 0.84 (d, J = 6.6 Hz, 3H). 13C NMR (DMSO-d6) δ: 170.0,
168.4, 155.6, 77.7, 60.4, 50.2, 39.52, 38.8, 37.4, 28.2 (3C), 25.5,
22.6, 22.0, 14.0. MS (ESI+): m/z = 331 [M + H]+, 275 [M–tBu+H]+, 231 [M–Boc+H]+.
2-[(2-Ethoxycarbonyl-4-methyl-pentanoyl)amino]ethylammonium Chloride (24a)
Compound 24a was
synthesized according to the general procedure C2, using the Boc-protected
intermediate 23a (1855 mg, 5.6 mmol) and 4 N HCl in dioxane
(20 mL) in a mixture of CH2Cl2/EtOH (40 mL,
20:20 v/v) overnight. Compound 24a was
obtained as a colorless oil (1500 mg, quant. yield) and was used in
the next step without further purification. 1H NMR (300
MHz, DMSO-d6) δ: 8.53 (t, J = 5.5 Hz, 1H), 8.13 (br s, 3H), 4.09 (dd, J = 7.2 and 1.8 Hz, 1H), 4.04 (dd, J = 7.2 and 1.8
Hz, 1H), 3.35–3.28 (m, 3H), 2.89–2.77 (m, 2H), 1.65–1.59
(m, 2H), 1.44 (sep, J = 6.7 Hz, 1H), 1.16 (t, J = 7.1 Hz, 3H), 0.86 (d, J = 6.5 Hz, 3H),
0.85 (d, J = 6.5 Hz, 3H). 13C NMR (75
MHz, DMSO-d6) δ: 169.9, 168.8, 60.5,
50.2, 38.2, 37.3, 36.6, 25.6, 22.4, 22.2, 14.0. MS (ESI+): m/z = 231 [M + H]+.
Ethyl 2-(2-Azidoethylcarbamoyl)-4-methyl-pentanoate (25a)
Compound 25a was synthesized according to
the general procedure D2, using amine 24a (1500 mg, 5.6
mmol), ZnCl2 (46 mg, 0.34 mmol), K2CO3 (778 mg, 5.6 mmol), diisopropylethylamine (3420 μL, 19.6 mmol),
and diazo transfer reagent (1415 mg, 6.7 mmol) in EtOH (15 mL). The
mixture was stirred at room temperature overnight. The crude product
was purified by flash chromatography on silica gel (cHex/EtOAc 9:1
to 6:4) affording compound 25a as a colorless oil (1430
mg, quant. yield). 1H NMR (500 MHz, CDCl3) δ:
6.85–6.81 (m, 1H), 4.23–4.16 (m, 2H), 3.44–3.42
(m, 4H), 3.33–3.30 (m, 1H), 1.83–1.71 (m, 2H), 1.57
(sep, J = 6.7 Hz, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H), 0.92 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ: 172.6, 169.2, 61.7, 51.7, 50.9, 40.4, 39.3, 26.5, 22.6,
22.1, 14.2. MS (ESI+): m/z = 257 [M + H]+.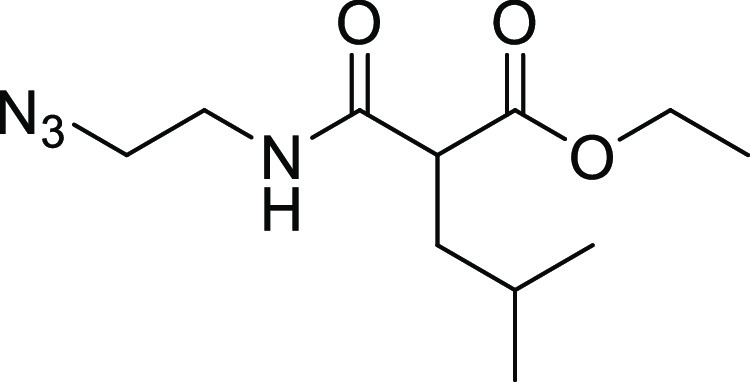
N-(2-Azidoethyl)-2-(hydroxycarbamoyl)-4-methyl-pentanamide (26a)
Compound 26a was synthesized
according to the general procedure E2, using ester 25a (600 mg, 2.3 mmol), KCN (30.5 mg, 0.47 mmol), and aq. NH2OH (12 mL, 50% w/w in water) in
MeOH (12 mL) overnight. The crude product was purified by flash chromatography
on silica gel (CH2Cl2 to CH2Cl2/MeOH 9:1) affording compound 26a as a white
solid (366 mg, 65%). 1H NMR (500 MHz, DMSO-d6) δ: 10.50 (s, 1H), 8.93 (s, 1H), 7.85 (t, J = 5.1 Hz, 1H), 3.36–3.34 (m, 2H), 3.25–3.22
(m, 2H), 3.00–2.97 (m, 1H), 1.64–1.58 (m, 1H), 1.56–1.51
(m, 1H), 1.43 (sep, J = 6.6 Hz, 1H), 0.84 (d, J =
6.4 Hz, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 169.4, 166.6, 49.9, 49.0, 38.6, 38.4, 25.6, 22.6,
22.1. MS (ESI+): m/z =
243 [M + H]+.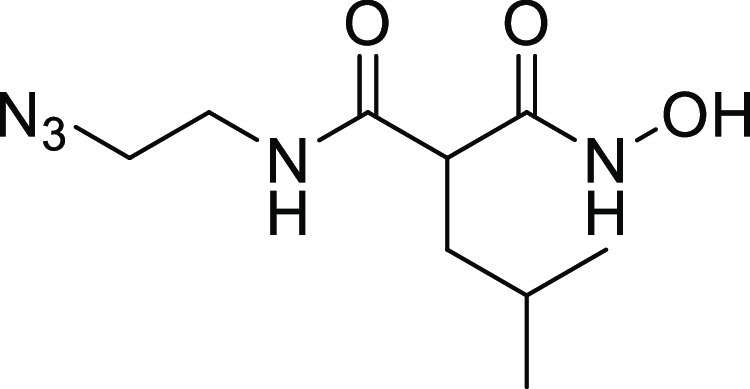
N-(2-Prop-2-ynoxyphenyl)acetamide (34a)
Compound 34a was synthesized according to
the general procedure G2, using N-(2-hydroxyphenyl)acetamide
(150 mg, 0.99 mmol), K2CO3 (274 mg, 1.98 mmol),
and propargyl bromide (103 μL, 1.09 mmol) in DMF (5 mL). The
resulting solution was heated to 60 °C and stirred overnight.
The mixture was then diluted in water and extracted three times with
ethyl acetate. Organic layers were combined, and solvents were evaporated
under reduced pressure affording compound 34a as a brown
solid (182 mg, 96%). 1H NMR (500 MHz, DMSO-d6) δ: 9.13 (s, 1H), 7.91 (d, J =
7.5 Hz, 1H), 7.12–7.11 (m, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.93 (t, J = 7.5 Hz, 1H), 4.86 (d, J = 2.0 Hz, 2H), 3.60 (t, J = 2.2 Hz, 1H),
2.08 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 168.5, 147.6, 127.9, 124.1, 122.6, 121.0,
112.9, 79.2, 78.6, 56.0, 23.8. MS (ESI+): m/z = 190 [M + H]+.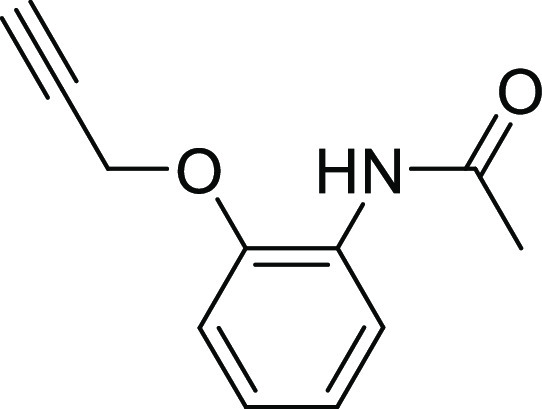
N-[2-[4-[(2-Acetamidophenoxy)methyl]triazol-1-yl]ethyl]-2-(hydroxycarbamoyl)-4-methyl-pentanamide (27)
Compound 27 was synthesized
according to the general procedure F2, using azide 26a (88 mg, 0.36 mmol), alkyne 34a (69 mg, 0.36 mmol),
copper (II) sulfate pentahydrate (18.1 mg, 0.07 mmol), and sodium
ascorbate (35.8 mg, 0.18 mmol) in DMF (5.5 mL) and H2O
(4.5 mL). The mixture was stirred at room temperature overnight. The
crude product was purified by preparative HPLC (H2O + 0.05%
FA/CAN + 0.05% FA 95:5 to 5:95) affording 27 as a white
solid after lyophilization (82 mg, 51%). 1H NMR (500 MHz,
DMSO-d6) δ: 10.52 (s, 1H), 9.01
(s, 1H), 8.95 (s, 1H), 8.15 (s, 1H), 7.91–7.90 (m, 1H), 7.84
(t, J = 5.7 Hz, 1H), 7.24–7.23 (m, 1H), 7.07–7.04
(m, 1H), 6.92–6.89 (m, 1H), 5.20 (s, 2H), 4.44 (t, J = 6.1 Hz, 2H), 3.52–3.50 (m, 2H), 2.94 (t, J = 7.7 Hz, 1H), 2.07 (s, 3H), 1.57–1.46 (m, 2H),
1.35 (sep, J = 6.6 Hz, 1H), 0.80 (d, J = 6.6 Hz, 3H), 0.79 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 169.4,
168.4, 166.6, 148.5, 142.7, 127.9, 124.8, 124.2, 122.3, 120.8, 113.2,
62.3, 49.0, 48.6, 39.0, 38.4, 25.5, 23.9, 22.4, 22.1. Purity: 100%.
HRMS–ESI+ (m/z): calculated for C20H29N6O5 [M + H]+ 433.2199, found 433.2190.
N-[2-[4-[(3-Acetamidophenoxy)methyl]triazol-1-yl]ethyl]-2-(hydroxycarbamoyl)-4-methyl-pentanamide (28)
Compound 28 was synthesized
according to the general procedure F2, using azide 26a (55 mg, 0.23 mmol), alkyne 34b (43 mg, 0.23 mmol),
copper (II) sulfate pentahydrate (11.3 mg, 0.05 mmol), and sodium
ascorbate (22.4 mg, 0.11 mmol) in DMF (3 mL) and H2O (2
mL). The mixture was stirred at RT overnight. The crude product was
purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH 9:1) affording 28 as a white solid after lyophilization (22 mg, 22%). 1H NMR (500 MHz, DMSO-d6) δ:
10.50 (s, 1H), 9.91 (s, 1H), 8.94 (s, 1H), 8.13 (s, 1H), 7.85 (t, J = 5.3 Hz, 1H), 7.34 (s, 1H), 7.21–7.18 (m, 1H),
7.12–7.10 (m, 1H), 6.73–6.72 (m, 1H), 5.07 (s, 2H),
4.43 (t, J = 5.2 Hz, 2H), 3.53–3.49 (m, 2H),
2.94 (t, J = 7.6 Hz, 1H), 2.03 (s, 3H), 1.57–1.47
(m, 2H), 1.38–1.30 (m, 1H), 0.81 (d, J = 6.2
Hz, 3H), 0.80 (d, J = 6.2 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 169.4, 168.3, 166.6, 158.3, 142.5,
140.5, 129.5, 124.7, 111.7, 108.9, 105.8, 61.0, 48.9, 48.6, 39.0,
38.3, 25.5, 24.1, 22.4, 22.2. Purity: 97%. HRMS–ESI+ (m/z): calculated for C20H29N6O5 [M + H]+ 433.2199, found 433.2190.
N-[2-[4-[(4-Acetamidophenoxy)methyl]triazol-1-yl]ethyl]-2-(hydroxycarbamoyl)-4-methyl-pentanamide (29)
Compound 29 was synthesized
according to the general procedure F2, using azide 26a (55 mg, 0.23 mmol), alkyne 34c (43 mg, 0.23 mmol),
copper (II) sulfate pentahydrate (11.3 mg, 0.05 mmol), and sodium
ascorbate (22.4 mg, 0.11 mmol) in DMF (3 mL) and H2O (2
mL). The mixture was stirred at RT overnight. The crude product was
purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH 9:1) affording 29 as a white solid after lyophilization (43 mg, 43%). 1H NMR (500 MHz, DMSO-d6) δ:
10.52 (s, 1H), 9.81 (s, 1H), 8.97 (s, 1H), 8.13 (s, 1H), 7.87 (t, J = 5.5 Hz, 1H), 7.48 (d, J = 8.9 Hz, 2H),
6.96 (d, J = 8.9 Hz, 2H), 5.06 (s, 2H), 4.43 (t, J = 5.7 Hz, 2H), 3.52–3.48 (m, 2H), 2.94 (t, J = 7.6 Hz, 1H), 2.00 (s, 3H), 1.56–1.46 (m, 2H),
1.38–1.31 (m, 1H), 0.81 (d, J = 6.3 Hz, 3H),
0.80 (d, J = 6.3 Hz, 3H). 13C NMR (126
MHz, DMSO-d6) δ: 169.5, 167.8, 166.7,
153.9, 142.7, 132.9, 124.8, 120.5 (2C), 114.7 (2C), 61.3, 48.9, 48.7,
39.0, 38.4, 25.5, 23.9, 22.4, 22.2. Purity: 98%. HRMS–ESI+ (m/z): calculated for C20H29N6O5 [M + H]+ 433.2199, found 433.2166.
N-[2-[4-[(2-Fluorophenoxy)methyl]triazol-1-yl]ethyl]-2-(hydroxycarbamoyl)-4-methyl-pentanamide (30)
Compound 30 was synthesized
according to the general procedure F2, using azide 26a (70 mg, 0.29 mmol), alkyne 34d (37 μL, 0.29 mmol),
copper (II) sulfate pentahydrate (14.4 mg, 0.06 mmol), and sodium
ascorbate (28.5 mg, 0.14 mmol) in dioxane (4 mL) and H2O (3 mL). The mixture was stirred at RT overnight. The crude product
was purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH 9:1) affording 30 as a white solid after lyophilization (74 mg, 65%). 1H NMR (500 MHz, DMSO-d6) δ:
10.52 (s, 1H), 8.97 (s, 1H), 8.17 (s, 1H), 7.86 (t, J = 5.5 Hz, 1H), 7.38–7.35 (m, 1H), 7.23–7.19 (m, 1H),
7.16–7.13 (m, 1H), 6.97–6.93 (m, 1H), 5.19 (s, 2H),
4.44 (t, J = 5.2 Hz, 2H), 3.53–3.49 (m, 2H),
2.93 (t, J = 7.6 Hz, 1H), 1.56–1.45 (m, 2H),
1.37–1.29 (m, 1H), 0.80 (d, J = 6.4 Hz, 3H),
0.79 (d, J = 6.4 Hz, 3H). 13C NMR (126
MHz, DMSO-d6) δ: 169.5, 166.6, 151.8
(d, J = 243.5 Hz), 146.0 (d, J =
10.6 Hz), 142.1, 125.2, 124.9 (d, J = 3.6 Hz), 121.4
(d, J = 6.0 Hz), 116.1 (d, J = 17.5
Hz), 115.4, 61.9, 48.9, 48.7, 39.0, 38.4, 25.5, 22.4, 22.2. 19F NMR (471 MHz, DMSO-d6) δ: −134.7.
Purity: 100%. HRMS–ESI+ (m/z): calculated for C18H25FN5O4 [M + H]+ 394.1890, found
394.1862.
2-(Hydroxycarbamoyl)-4-methyl-N-[2-[4-(phenoxymethyl)triazol-1-yl]ethyl]pentanamide (31)
Compound 31 was synthesized
according to the general procedure F2, using azide 26a (70 mg, 0.29 mmol), prop-2-ynoxybenzene (37 μL, 0.29 mmol),
copper (II) sulfate pentahydrate (14.4 mg, 0.06 mmol), and sodium
ascorbate (28.5 mg, 0.14 mmol) in dioxane (4 mL) and H2O (3 mL). The mixture was stirred at RT overnight. The crude product
was purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH 9:1) affording 31 as a white solid after lyophilization (56 mg, 51%). 1H NMR (500 MHz, DMSO-d6) δ:
10.51 (br s, 1H), 8.97 (s, 1H), 8.14 (s, 1H), 7.87 (t, J = 5.5 Hz, 1H), 7.32–7.28 (m, 2H), 7.04–7.02 (m, 2H),
6.96–6.93 (m, 2H), 5.10 (s, 2H), 4.43 (t, J = 5.1 Hz, 2H), 3.52–3.49 (m, 2H), 2.94 (t, J = 7.6 Hz, 1H), 1.57–1.46 (m, 2H), 1.38–1.30 (m, 1H),
0.81 (d, J = 6.4 Hz, 3H), 0.80 (d, J = 6.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ: 169.5, 166.6, 158.1, 142.6, 129.6 (2C), 124.9,
120.9, 114.6 (2C), 61.0, 48.9, 48.7, 39.0, 38.4, 25.5, 22.4, 22.2.
Purity: 100%. HRMS–ESI+ (m/z): calculated for C18H26N5O4 [M + H]+ 376.1985, found
376.1956.
2-(Hydroxycarbamoyl)-4-methyl-N-[2-[4-(phenylsulfanylmethyl)triazol-1 yl]ethyl]pentanamide (32)
Compound 32 was synthesized according to the general procedure F2, using azide 26a (70 mg, 0.29 mmol), prop-2-ynylsulfanylbenzene (43 mg,
0.29 mmol), copper (II) sulfate pentahydrate (14.4 mg, 0.06 mmol),
and sodium ascorbate (28.5 mg, 0.14 mmol) in dioxane (4 mL) and H2O (3 mL). The mixture was stirred at RT overnight. The crude
product was purified by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH 95:5)
affording 27 as a light-green solid after lyophilization
(64 mg, 56%). 1H NMR (500 MHz, DMSO-d6) δ: 10.49 (s, 1H), 8.94 (s, 1H), 7.89 (s, 1H), 7.80
(t, J = 5.9 Hz, 1H), 7.38–7.36 (m, 2H), 7.33–7.30
(m, 2H), 7.20–7.17 (m, 1H), 4.38–4.36 (m, 2H), 4.26
(s, 2H), 3.48–3.44 (m, 2H), 2.93 (t, J = 7.6
Hz, 1H), 1.57–1.45 (m, 2H), 1.40–1.32 (m, 1H), 0.82–0.81
(m, 6H). 13C NMR (126 MHz, DMSO-d6) δ: 169.4, 166.6, 143.2, 136.1, 129.0 (2C), 128.0 (2C),
125.8, 123.5, 48.9, 48.5, 38.9, 38.4, 27.2, 25.5, 22.4, 22.1. Purity:
100%. HRMS–ESI+ (m/z): calculated for C18H26N5O3S [M + H]+ 392.1756, found 392.1727.
N-[2-[4-[(2-Acetamidophenoxy)methyl]triazol-1-yl]ethyl]-2-benzyl-3-(hydroxyamino)-3-oxo-propanamide (33)
Compound 33 was synthesized
according to the general procedure F2, using azide 21b (45 mg, 0.1 mmol), alkyne 29a (43 mg, 0.1 mmol), copper
(II) sulfate pentahydrate (5 mg, 0.02 mmol), and sodium ascorbate
(9.9 mg, 0.05 mmol) in dioxane (2 mL) and H2O (1 mL). The
mixture was stirred at RT overnight. The crude product was purified
by flash chromatography on silica gel (CH2Cl2 to CH2Cl2/MeOH: 9/1) affording 28 as a white solid after lyophilization (7 mg, 15%). 1H
NMR (500 MHz, DMSO-d6) δ: 10.45
(s, 1H), 9.00–8.93 (m, 2H), 8.04–7.91 (m, 3H), 7.24–6.91
(m, 8H), 5.19 (s, 2H), 4.39 (br s, 2H), 3.49 (br s, 2H), 3.21–3.17
(m, 1H), 2.99–2.96 (br s, 2H), 2.06 (s, 3H). 13C
NMR (126 MHz, DMSO-d6) δ: 168.7,
168.4, 165.6, 148.5, 142.7, 138.8, 128.7 (2C), 128.2 (2C), 127.9,
126.2, 124.9, 124.2, 122.3, 120.8, 113.2, 62.2, 52.2, 48.6, 39.0,
34.6. Purity: 96%. HRMS–ESI+ (m/z): calculated for C23H27N6O5 [M + H]+ 467.2047,
found 467.2013.
Expression and Purification of ColQ1
ColQ1 was produced and purified in its complete length and collagenase unit from B. cereus strain Q1 (Uniprot: B9J3S4; Tyr94-Gly765), as previously described.20
In Vitro FRET-Based Proteolytic Assay (ColQ1, ColA, ColG, and ColH)
For all targets, the percent of inhibition and IC50 measurements were carried out as previously described.20,25,26 Experiments were performed in triplicate, and the results are provided as means ± standard deviation. For the determination of the inhibition constant (Ki), similar assay conditions were chosen. However, nominal final enzyme concentrations of 1 nM ColQ1-CU, 10 nM ColH-PD, 35 nM ColA, and 60 nM ColG were used and the reactions were monitored for 2 min 24 s. Regression analysis was performed using GraphPad Prism v 9.0.0 (GraphPad Software, San Diego, CA). The experiments were performed under first-order conditions ([S0] ≪ KM), which resulted in an approximation of the Kiapp to the true inhibition constant (Ki); therefore, the results are reported as Ki values.
Reversibility Assays by Rapid Dilution
The recovery of enzymatic activity after a rapid large dilution was performed following Copeland, 2013.32 In short, ColA, ColG, ColH, and ColQ1 were incubated for 30 min at 100-fold the concentration required for the activity assay (i.e., 4.375, 7.500, 1.250, and 0.125 μM, respectively) with a concentration of inhibitor equivalent to 10-fold the IC50. The mixture was then diluted 100-fold into the reaction buffer. The reaction was immediately initiated by the addition of the quenched-fluorescent substrate FS1-1 at a final concentration of 2 μM. The reaction was monitored for 2 min (excitation: 328 nm, emission: 392 nm) at 25 °C in an Infinite M200 plate reader (Tecan, Grödig, Austria).
Mass Spectrometric Analysis of Collagenase–Ligand Interactions
The collagenase subunit of ColQ1 incubated with either Ilomastat, 13, or no inhibitor was investigated by high-performance liquid chromatography coupled to mass spectrometry (HPLC-MS). Prior to HPLC-MS measurements, ColQ1-CU samples were buffer-exchanged to 150 mmol·L–1 ammonium acetate on 3 kDa molecular weight cutoff centrifugal filters (Amicon Ultra 0.5 mL, Merck, Darmstadt, Germany) and reconstituted to a concentration of 0.5 mg·mL–1. ColQ1-CU samples were separated on a capillary HPLC instrument (UltiMate U3000 RSLC, Thermo Fisher Scientific, Germering, Germany) equipped with a Supelco Discovery C18 column (150 × 2.1 mm2 i.d., 3 μm particle size, 300 Å pore size; Sigma-Aldrich, Vienna, Austria). The column was operated at a flow rate of 150 μL·min–1 and a column oven temperature of 70 °C. Using in-line split-loop mode, 7 microliters of sample [0.5 mg mL–1] was injected in triplicate. The separation was carried out employing a gradient of mobile phase A (H2O + 0.10% formic acid (98–100%; Sigma-Aldrich)) and B (acetonitrile (HiPerSolv; VWR chemicals, Radnor, PA) + 0.10% formic acid) as follows: 25.0% B for 5.0 min, 25.0–90.0% B in 20 min, 95.0% B for 5.0 min, and 25.0% B for 10 min. Water (H2O) was purified by a MilliQ Integral 3 system (Merck Millipore, Burlington, MA). Mass spectrometric data was acquired on a Thermo Scientific QExactive benchtop quadrupole-Orbitrap mass spectrometer employing an Ion Max source with a heated electrospray ionization probe (both from Thermo Fisher Scientific, Bremen, Germany) and an MXT715-000—MX Series II Switching Valve (IDEX Health & Science, LLC, Oak Harbor, WA), as described earlier.45 MS settings were optimized for the ColQ1-CU protein: the resolution was set to 17,500 at m/z 200 in a mass range of m/z 500–2700, the source heater temperature was lowered to 200 °C, and the in-source collision-induced dissociation was increased to 30.0 eV. Data was acquired between minutes 5 and 35. Mass spectra were deconvoluted employing the ReSpect algorithm implemented in the Chromeleon Chromatography Data System, version 7.2.10 (Thermo Fisher Scientific, Waltham, MA).
Selectivity toward Human MMPs
The MMP inhibition assay (Sigma-Aldrich, Saint Louise, MO) was performed as previously reported and in accordance with the manufacturer’s instructions. The fluorescence signals were measured in a CLARIOstar plate reader (BMG LABTECH, Ortenberg, Germany) at a concentration of 100 μM.
Compound Toxicity
Cytotoxicity assays on HepG2, HEK293, NHDF, and MDCK II cells were carried out as described previously.25
In Vitro Collagen Cleavage Assay
The experiments were done as described before.20,28 Briefly, in a buffer containing 250 mM HEPES, 150 mM NaCl, 5 mM CaCl2, 5 μM ZnCl2, pH 7.5, 1 mg/mL acid-soluble type I collagen from bovine tail (Thermo Fischer Scientific) was incubated with 50 ng full-length ColQ1. Compounds were evaluated at various concentrations and incubated together with collagen and ColQ1 at 25 °C for 3 h. After stopping the reaction with 50 mM EDTA, the mixture was loaded on 12% SDS-PAGE gel and stained with colloidal coomassie G-250 dye.46 Two separate experiments were carried out for each compound.
In Vitro NHDF Infection Model
NHDF cells (1 × 105, Promo Cell C-12302) per well were seeded in 24-well plates (Greiner) with DMEM medium (Gibco) containing 10% (v/v) fetal calf serum (FCS, Gibco) and 1% (v/v) glutamine (Gibco). Prior to the treatment, the cells were cultured at 37 °C for 24 h with 5% CO2. In brain heart infusion (BHI) medium, B. cereus AH187 bacteria were cultivated to a mid-exponential phase. The culture was centrifuged at 4000g for 7 min at RT before being rinsed and diluted in PBS. Then, B. cereus suspension was added to the cells to give an MOI (multiplicity of infection) of 0.03. Before the infection, cells were starved for 1 h with DMEM containing only 1% (v/v) glutamine and no FCS. The cells were infected with B. cereus for 1, 2, 4, and 6 h to examine the kinetics of collagenase release. The DMEM supernatant was collected and harvested cleared by centrifugation at 4000g at 4 °C for 10 min. After the cells were washed, they were lysed using a lysis buffer (20 mM tris pH 7.5, 1 mM EDTA, 100 mM NaCl, 1% Triton 100, 0.5% Na-deoxycholate, 0.1% SDS, 1 × PIT, 1 mM Na3VO4, 1 mM Na-molybdate, 20 mM NaF, 20 mM β-glycerophosphate). Cell debris was removed by centrifugation with 12,000g at 4 °C for 10 min. The DMEM supernatants and cell lysates were stored at −80 °C for further investigation. The cell morphology was monitored with a light microscope (Olympus) using a 20X objective. The kinetic study was performed in three independent experiments. To study the behavior of ColQ1 inhibitors in this model, the experiment was done as stated above with a few changes; the compounds were added to the NHDF cells along with the bacterial suspension, and all were incubated at 37 °C for 5 h and 5% CO2. Two controls were considered in each experiment, uninfected cells, and infected cells without inhibitor. Each experiment was repeated three times in total.
Nonreducing Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Zymography
To perform the zymography, we collected supernatants from the NHDF infection experiments and mixed them with 1% nonreducing loading buffer. They were then electrophoretically separated after loading onto 10% SDS-PAGE gels containing 0.1% gelatin (Roth, Karlsruhe, Germany). Following separation, the gel was incubated at 4 °C for 2 × 30 min with gentle agitation in a renaturation buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10 mM CaCl2, 10 μM ZnCl2, 2.5% Triton X-100). The gel was then treated in a developing buffer (50 mM HEPES pH 7.5, 200 mM NaCl, 10 mM CaCl2, 10 μM ZnCl2, 0.02% Brij-35) at 37 °C overnight. By staining the gel with 0.1% Coomassie brilliant blue R-250 dye overnight, transparent bands of gelatinolytic activity could be seen. The ChemiDoc XRS+ imaging system (Biorad) was used to scan the gels, and image analysis was performed with Image lab software (Li-Cor Biosciences).
Reducing Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis
The cell lysate was placed onto a 12% SDS-PAGE gel with similar total protein content and stained overnight with Coomassie brilliant blue G-250. The gel was then visualized with ChemiDoc XRS+ imaging system (Biorad), and the signal analysis was exerted with Image lab software.
Picrosirius Red Assay
After infection, the NHDF cells were washed 3 × with PBS and then incubated with Bouin solution (Sigma-Aldrich) at RT for 20 min. The cells were incubated with 0.1% Picrosirius red dye (ab150681) at RT for 2 h. Then, they were washed 1 × with 0.01 N HCl and the matrix was dissolved in 0.01 N NaOH. The absorption was measured at 570 nm using a Tecan Infinite M200 plate reader (Tecan, Grödig, Austria). By dividing the absorbance of each sample by the absorbance of the healthy sample, the relative collagen quantity was determined. For each condition, the experiment was performed three times.
Lactate Dehydrogenase (LDH) Release Assay
The manufacturer’s procedure was followed to measure the released LDH amount in the supernatant of NHDF cells. Briefly, in a 96-well plate (Grenier), 50 μL of the supernatant was combined with 50 μL of substrate. The plate was incubated at RT for 30 min in the dark, and then the reaction was stopped with 50 μL of the stop solution. The absorbance was measured at 490 nm with a Tecan Infinite M200 plate reader (Tecan, Grödig, Austria). The cytotoxicity was calculated relative to the control (no inhibitor).
Stability of 27 with LC-MS
A concentration of 200 μM of compound 27 was incubated with DMEM medium at 37 °C for 5 h and 5% CO2. After the incubation, 2 μL of the compound was transferred to LC-MS vials containing 200 μL of acetonitrile and LC-MS spectra were measured. Three controls were included: (i) 27 in DMEO, (ii) 27 in DMEM without incubation, and (iii) DMEM medium.
Transepithelial Electric Resistance (TEER) Experiment
MDCK II cells were seeded at a density of 3 × 104 cells/mL onto a Millipore hanging cell culture insert at 37 °C for 12 days with 5% CO2. On day 5, the medium was changed. Prior to treatment, the cells were starved for 16 h in FCS-free RPMI medium (Gibco). The bacteria or bacterial-free supernatant was prepared; the bacteria was used at an MOI of 0.03, while 50% (v/v) of supernatant was added. The ColQ1 inhibitors were added into the inner compartment. The TEER of the cells was measured with Millicell ERS-2 (Electrical Resistance System) over time. Three readings were recorded for each well, and the unit area resistance (UAR) was calculated using the mean values of the TEER following the equation
Changes in TEER were normalized to the initial UAR (t = 0), which was set to 100%.
B. cereus Supernatant Production
B. cereus AH187 strain was cultured in FCS-free DMEM medium at 37 °C. The supernatant was harvested by centrifugation and kept at −80 °C until needed. The supernatant was sterile-filtered with a 0.22 μm filter (Greiner).
B. cereus Growth Inhibition Assay
The effect of the compounds on B. cereus growth was carried out by growing the bacteria in BHI medium until the mid-growth phase. Next, the bacterial suspension was diluted until OD600 nm was 0.2 and combined with compounds in a range of 200–3 μM in a 96-well plate. The plates were subsequently incubated at 37 °C for 48 h in a Tecan Infinite M200 plate reader (Tecan, Grödig, Austria). The MIC values presented are the average of at least two independent determinations.
In vivo Galleria Mellonella Infection Model
G. mellonella larvae were purchased from a fishing store. Injections were carried out using an LA120 syringe pump (Landgraf Laborsysteme, Langenhagen, Germany) equipped with 1 mL Injekt-F tuberculin syringes (B. Braun, Melsungen, Germany) and Sterican 0.30 × 12 mm2, 30 G × 1.5 needles (B. Braun). The larvae were divided into five groups depending on their treatment: (i) sterile PBS, (ii) no injection, (iii) only compound, (iv) BC AH187 bacterial suspension, and (v) BC AH187 bacterial suspension with compound. Larvae were incubated at 37 °C for 3 days and inspected twice daily. The total larvae used in all three experiments were 40 larvae per group. When the larvae became black and did not move when simulated with a tweezer, they were deemed dead.
Crystallization, X-ray Data Collection, and Analysis
Crystals of ColG-PD were grown in 0.1 M tris-Bicine pH 8.5, 0.04 M pentaethylene glycol, 0.04 M diethylene glycol, 0.04 M triethylene glycol, 0.04 M tetraethylene glycol, 10% (w/v) poly(ethylene glycol) 20,000, and 20% (v/v) poly(ethylene glycol) 550 monomethyl ether in sitting-vapor diffusion plates. Crystals were soaked with 10 mM 27 and 13 for 2 weeks. The crystals were cryoprotected with MiTeGen LV Cryo-oil (MiTeGen, Ithaca, NY) and immediately flash-frozen in liquid nitrogen. X-ray diffraction data was collected on beamline ID30 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. The data sets were indexed, integrated, and scaled using XDS47 and AIMLESS.48 Molecular replacement was performed with PHASER49 using as search model PDB entry 2y6i (ligand and activator domain deleted). Ligand coordinates and restraints were generated using the Grade Web Server.50 Final structures were obtained using PHENIX51 together with model building in WinCoot.52 PyMOL v 4.0.0 was used for figure generation (The PyMOL Molecular Graphics System, Version 4.0.0 Schrödinger, LLC). The final refined structures were deposited in the Protein Data Bank (PDB) as entries 7Z5U and 7ZBV. Data collection and refinement statistics are listed in Table S2.
Statistical Analysis
Graphical data in the manuscript is presented as the means ± SDs. Statistical comparisons are performed by Tukey one-way ANOVA test, which shows significant differences between conditions. Parametric/nonparametric statistical analysis used in the study was based on normality and homogeneity of variance. A value of p ≤ 0.05 was considered statistically significant, while p > 0.05 was considered nonsignificant. The normalized measurements were statistically compared between treated and nontreated groups using the generalized estimating equation model to account for correlated data arising from repeated measures. The survival of G. mellonella was computed using the Kaplan–Meier method, and the log-rank test was applied to calculate the significance of differences between conditions.
Acknowledgments
The authors are grateful to Jeannine Jung, Selina Wolter, Isabella Widlroither, Martina Wiesbauer, and Melanie Anja Schwarz for their technical support. The authors acknowledge financial support from the Austrian Science Fund (FWF) grant P 31843 (E.S.), grant W 1213 (C.B. and C.G.H.), and grant I 4360 (S.W.), the European Research Council (ERC starting grant 757913, A.K.H.H.), and by the Helmholtz Association’s Initiative and Networking Fund (A.K.H.H.).
Glossary
Abbreviations Used
- BHI
brain heart infusion medium
- Col I
collagen I
- ColQ1 and ColA
collagenases from Bacillus cereus AH187 and Q1 strains
- ColH
ColG Clostridium histolyticum collagenases
- CU
collagenase unit
- DMEM
Dulbecco’s modified Eagle’s medium
- ECM
extracellular matrix
- FCS
fetal calf serum
- FRET
fluorescence resonance energy transfer
- HEK293
embryonal kidney cell
- HepG2
hepatocellular carcinoma cell
- IC50
the half-maximal inhibitory concentration
- LDH
lactate dehydrogenase
- M (kDa)
molecular weight standards in kilo Dalton
- MDCK II
Madin–Darby canine kidney II
- MIC
minimum inhibitory concentration
- MMPs
human matrix metalloproteases
- NHDF
normal human dermal fibroblast cell
- PD
peptidase unit
- RPMI
Roswell Park Memorial Institute Medium
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide
- TEER
transepithelial electrical resistance
- ZBG
zinc-binding group
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00785.
Molecular Formula Strings (CSV)
Screening results for ColH and ColQ1, inhibition of MMPs and other off-targets by selected compounds, and cytotoxicity and antibacterial activity of selected compounds (Tables S1–S5); inhibition of the screened compounds on ColQ1 and ColH, activity of the compounds on Col I cleavage, on NHDF infected cells, on TEER of MDCK II cells, and on G. mellonella infection model; 1H NMR and 13C NMR and LC-MS spectra for final compounds and data collection and refinement statistics (Figures S4–S17) (PDF)
Author Contributions
⊥ A.A. and A.S.A. contributed equally to this work.
Helmholtz Association’s Initiative and Networking Fund European Research Council (ERC): ERC starting grant 757913. Austrian Science Fund (FWF): P 31843, W 1213, and I 4360. Open Access is funded by the Austrian Science Fund (FWF).
The authors declare the following competing financial interest(s): The authors declare the following financial interest(s): A.S.A, V.C., J.H., and A.K.H.H are co-inventors on an international patent application WO2022/043322 that incorporates compounds outlined in the library screening in the supplementary information.
Notes
PDB codes: compound 13 (7ZBV) and compound 27 (7Z5U). The authors will release the atomic coordinates upon article publication.
Supplementary Material
References
- Antibiotic resistance. https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance. (accessed Feb 13, 2022).
- No Time to Wait: Securing the Future from Drug-Resistant Infections. https://www.who.int/publications/i/item/no-time-to-wait-securing-the-future-from-drug-resistant-infections. (accessed Feb 13, 2022).
- Vale P. F.; Fenton A.; Brown S. P. Limiting Damage during Infection: Lessons from Infection Tolerance for Novel Therapeutics. PLoS Biol. 2014, 12, e1001769 10.1371/journal.pbio.1001769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen R. C.; Popat R.; Diggle S. P.; Brown S. P. Targeting Virulence: Can We Make Evolution-Proof Drugs?. Nat. Rev. Microbiol. 2014, 12, 300–308. 10.1038/nrmicro3232. [DOI] [PubMed] [Google Scholar]
- Farha M. A.; Brown E. D. Drug Repurposing for Antimicrobial Discovery. Nat. Microbiol. 2019, 4, 565–577. 10.1038/s41564-019-0357-1. [DOI] [PubMed] [Google Scholar]
- Bean D. C.; Wareham D. W. Pentamidine: A Drug to Consider Re-Purposing in the Targeted Treatment of Multi-Drug Resistant Bacterial Infections?. J. Lab. Precis. Med. 2017, 2, 49. 10.21037/jlpm.2017.06.18. [DOI] [Google Scholar]
- Bottone E. J. Bacillus Cereus, a Volatile Human Pathogen. Clin. Microbiol. Rev. 2010, 23, 382–398. 10.1128/CMR.00073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclennan J. D. The Histotoxic Clostridial Infections of Man. Bacteriol. Rev. 1962, 26, 177–276. 10.1128/mmbr.26.2_pt_1-2.177-274.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sárvári K. P.; Schoblocher D. The Antibiotic Susceptibility Pattern of Gas Gangrene-Forming Clostridium Spp. Clinical Isolates from South-Eastern Hungary. Infect. Dis. 2020, 52, 196–201. 10.1080/23744235.2019.1696472. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhang J.; Zhan L.; Chen H.; Zhang Z.; Huang C.; Yue M.. Prevalence and Antimicrobial-Resistant Characterization of Bacillus Cereus Isolated from Ready-to-Eat Rice Products in Eastern China. Front. Microbiol. 2022, 13. 10.3389/fmicb.2022.964823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. W.Bacterial Pathogenesis. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston (TX), 1996. [PubMed] [Google Scholar]
- Zhang Y. Z.; Ran L. Y.; Li C. Y.; Chen X. L. Diversity, Structures, and Collagen-Degrading Mechanisms of Bacterial Collagenolytic Proteases. Appl. Environ. Microbiol. 2015, 81, 6098–6107. 10.1128/AEM.00883-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte A. S.; Correia A.; Esteves A. C. Bacterial Collagenases - A Review. Crit. Rev. Microbiol. 2016, 42, 106–126. 10.3109/1040841X.2014.904270. [DOI] [PubMed] [Google Scholar]
- Harrington D. J. Bacterial Collagenases and Collagen-Degrading Enzymes and Their Potential Role in Human Disease. Infect. Immun. 1996, 64, 1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R.; Wilson J. J.; Philominathan S. T. L.; Davis D.; Matsushita O.; Sakona J. Structural Comparison of ColH and ColG Collagen-Binding Domains from Clostridium Histolyticum. J. Bacteriol. 2013, 195, 318–327. 10.1128/JB.00010-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhard U.; Schönauer E.; Ducka P.; Briza P.; Nüss D.; Brandstetter H. Biochemical Characterization of the Catalytic Domains of Three Different Clostridial Collagenases. Biol. Chem. 2009, 390, 11–18. 10.1515/BC.2009.004. [DOI] [PubMed] [Google Scholar]
- Eckhard U.; Schönauer E.; Nüss D.; Brandstetter H. Structure of Collagenase G Reveals a Chew-and-Digest Mechanism of Bacterial Collagenolysis. Nat. Struct. Mol. Biol. 2010, 18, 1109–1114. 10.1038/nsmb.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita O.; Koide T.; Kobayashi R.; Nagata K.; Okabe A. Substrate Recognition by the Collagen-Binding Domain of Clostridium Histolyticum Class I Collagenase. J. Biol. Chem. 2001, 276, 8761–8770. 10.1074/jbc.M003450200. [DOI] [PubMed] [Google Scholar]
- Abfalter C. M.; Schönauer E.; Ponnuraj K.; Huemer M.; Gadermaier G.; Regl C.; Briza P.; Ferreira F.; Huber C. G.; Brandstetter H.; Posselt G.; Wessler S. Cloning, Purification and Characterization of the Collagenase ColA Expressed by Bacillus Cereus ATCC 14579. PLoS One 2016, 11, 1–19. 10.1371/journal.pone.0162433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe I. J.; Brandstetter H.; Schönauer E. Biochemical Characterisation of a Collagenase from Bacillus Cereus Strain Q1. Sci. Rep. 2021, 11, 4187 10.1038/s41598-021-83744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhard U.; Schönauer E.; Brandstetter H. Structural Basis for Activity Regulation and Substrate Preference of Clostridial Collagenases G, H, and T. J. Biol. Chem. 2013, 288, 20184–20194. 10.1074/jbc.M112.448548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima N.; Narukawa Y.; Takeda T.; Kiuchi F. Collagenase Inhibitors from Viola Yedoensis. J. Nat. Med. 2013, 67, 240–245. 10.1007/s11418-012-0665-8. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Protease Inhibitors: Synthesis of Matrix Metalloproteinase and Bacterial Collagenase Inhibitors Incorporating 5-Amino-2-Mercapto-1,3,4-Thiadiazole Zinc Binding Functions. Bioorganic Med. Chem. Lett. 2002, 12, 2667–2672. 10.1016/S0960-894X(02)00564-4. [DOI] [PubMed] [Google Scholar]
- Scozzafava A.; Supuran C. T. Protease Inhibitors: Synthesis of Matrix Metalloproteinase and Bacterial Collagenase Inhibitors Incorporating 5-Amino-2-Mercapto-1,3,4-Thiadiazole Zinc Binding Functions. Bioorganic Med. Chem. Lett. 2000, 12, 2667–2672. 10.1016/S0960-894X(02)00564-4. [DOI] [PubMed] [Google Scholar]
- Schönauer E.; Kany A. M.; Haupenthal J.; Hüsecken K.; Hoppe I. J.; Voos K.; Yahiaoui S.; Elsässer B.; Ducho C.; Brandstetter H.; Hartmann R. W. Discovery of a Potent Inhibitor Class with High Selectivity toward Clostridial Collagenases. J. Am. Chem. Soc. 2017, 139, 12696–12703. 10.1021/jacs.7b06935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinović J.; Yahiaoui S.; Alhayek A.; Haupenthal J.; Schönauer E.; Andreas A.; Kany A. M.; Müller R.; Koehnke J.; Berger F. K.; Bischoff M.; Hartmann R. W.; Brandstetter H.; Hirsch A. K. H. N-Aryl-3-Mercaptosuccinimides as Antivirulence Agents Targeting Pseudomonas Aeruginosa Elastase and Clostridium Collagenases. J. Med. Chem. 2020, 63, 8359–8368. 10.1021/acs.jmedchem.0c00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voos K.; Schönauer E.; Alhayek A.; Haupenthal J.; Andreas A.; Müller R.; Hartmann R. W.; Brandstetter H.; Hirsch A. K. H.; Ducho C. Phosphonate as a Stable Zinc-Binding Group for “Pathoblocker” Inhibitors of Clostridial Collagenase H (ColH). ChemMedChem 2021, 1–12. 10.1002/cmdc.202000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhayek A.; Khan E. S.; Schönauer E.; Däinghaus T.; Shafiei R.; Voos K.; Han M. K. L.; Ducho C.; Posselt G.; Wessler S.; Brandstetter H.; Haupenthal J.; del Campo A.; Hirsch A. K. H. Inhibition of Collagenase Q1 of Bacillus Cereus as a Novel Antivirulence Strategy for the Treatment of Skin-Wound Infections. Adv. Ther. 2022, 2100222 10.1002/adtp.202100222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.-M.; Li Y.-W.; Zhou J.; Gan L.-L.; Chen Y.-J.; Gan Z.-J.; Yu Y. A One-Pot Facile Synthesis of 2,3-Dihydroxyquinoxaline and 2,3-Dichloroquinoxaline Derivatives Using Silica Gel as an Efficient Catalyst. J. Heterocycl. Chem. 2018, 55, 1809–1814. 10.1002/jhet.3224. [DOI] [Google Scholar]
- Yang Y.; Zhang S.; Wu B.; Ma M.; Chen X.; Qin X.; He M.; Hussain S.; Jing C.; Ma B.; Zhu C. An Efficient Synthesis of Quinoxalinone Derivatives as Potent Inhibitors of Aldose Reductase. ChemMedChem 2012, 7, 823–835. 10.1002/cmdc.201200054. [DOI] [PubMed] [Google Scholar]
- Rangarajan M.; Kim J. S.; Sim S.-P.; Liu A.; Liu L. F.; LaVoie E. J. Topoisomerase I Inhibition and Cytotoxicity of 5-Bromo- and 5-Phenylterbenzimidazoles. Bioorg. Med. Chem. 2000, 8, 2591–2600. 10.1016/S0968-0896(00)00188-7. [DOI] [PubMed] [Google Scholar]
- Lozano-Calderon S. A.; Colman M. W.; Raskin K. A.; Hornicek F. J.; Gebhardt M. Use of Bisphosphonates in Orthopedic Surgery. Orthop. Clin. North Am. 2014, 45, 403–416. 10.1016/j.ocl.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Gooz M. ADAM-17: The Enzyme That Does It All. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 146–169. 10.3109/10409231003628015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropero S.; Esteller M. The Role of Histone Deacetylases (HDACs) in Human Cancer. Mol. Oncol. 2007, 1, 19–25. 10.1016/j.molonc.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelse K.; Pöschl E.; Aigner T. Collagens - Structure, Function, and Biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. 10.1016/j.addr.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Healing W. The Fibroblast. Proc. R. Soc. Med. 1967, 60, 778–782. 10.1177/003591576706000829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattouf R.; Younes R.; Lutomski D.; Naaman N.; Godeau G.; Senni K.; Changotade S. Picrosirius Red Staining: A Useful Tool to Appraise Collagen Networks in Normal and Pathological Tissues. J. Histochem. Cytochem. 2014, 62, 751–758. 10.1369/0022155414545787. [DOI] [PubMed] [Google Scholar]
- Junqueira L. C. U.; Cossermelli W.; Brentani R. Differential Staining of Collagens Type I, II and III by Sirius Red and Polarization Microscopy. Arch. Histol. Jpn. 1978, 41, 267–274. 10.1679/aohc1950.41.267. [DOI] [PubMed] [Google Scholar]
- Das C.; Lucia MS H. K.; T J. Quantification of Lactate Dehydrogenase for Cell Viability Testing Using Cell Lines and Primary Cultured Astrocytes. Physiol. Behav. 2017, 176, 139–148. 10.1002/cptx.21.Quantification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindbäck T.; Hardy S. P.; Dietrich R.; Sødring M.; Didier A.; Moravek M.; Fagerlund A.; Bock S.; Nielsen C.; Casteel M.; Granum P. E.; Märtlbauer E. Cytotoxicity of the Bacillus Cereus Nhe Enterotoxin Requires Specific Binding Order of Its Three Exoprotein Components. Infect. Immun. 2010, 78, 3813–3821. 10.1128/IAI.00247-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tausch F.; Dietrich R.; Schauer K.; Janowski R.; Niessing D.; Märtlbauer E.; Jessberger N. Evidence for Complex Formation of the Bacillus Cereus Haemolysin BL Components in Solution. Toxins 2017, 9, 1–18. 10.3390/toxins9090288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Einspanier R.; Schoen J. Transepithelial Electrical Resistance (TEER): A Functional Parameter to Monitor the Quality of Oviduct Epithelial Cells Cultured on Filter Supports. Histochem. Cell Biol. 2015, 144, 509–515. 10.1007/s00418-015-1351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan B.; Shuler L.; Hickman J. J.; et al. TEER Measurement Techniques for in Vitro Barrier Model Systems. SLAS Technol. 2015, 20, 107–126. 10.1177/2211068214561025.TEER. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramarao N.; Nielsen-Leroux C.; Lereclus D. The Insect Galleria Mellonella as a Powerful Infection Model to Investigate Bacterial Pathogenesis. J. Vis. Exp. 2012, 70, e4392. 10.3791/4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blöchl C.; Regl C.; Huber C. G.; Winter P.; Weiss R.; Wohlschlager T. Towards Middle-up Analysis of Polyclonal Antibodies: Subclass-Specific N-Glycosylation Profiling of Murine Immunoglobulin G (IgG) by Means of HPLC-MS. Sci. Rep. 2020, 10, 18080 10.1038/s41598-020-75045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyballa N.; Metzger S. Fast and Sensitive Colloidal Coomassie G-250 Staining for Proteins in Polyacrylamide Gels. J. Vis. Exp. 2009, 30, 2–5. 10.3791/1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. How Good Are My Data and What Is the Resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. Solving Structures of Protein Complexes by Molecular Replacement with Phaser. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 32–41. 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart O. S.; Womack T. O.; Flensburg C.; Keller P.; Paciorek W.; Sharff A.; Vonrhein C.; Bricogne G. Exploiting Structure Similarity in Refinement: Automated NCS and Target-Structure Restraints in BUSTER. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 368–380. 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkóczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L.-W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.