

Abstract
A series of potent, selective, and highly permeable human neuronal nitric oxide synthase inhibitors (hnNOS) based on the 2-aminopyridine scaffold with a shortened amino sidechain is reported. A rapid and simple protocol was developed to access these inhibitors in excellent yields. Neuronal nitric oxide synthase (nNOS) is a novel therapeutic target for the treatment of various neurological disorders. The major challenges in designing nNOS inhibitors in humans focus on potency, selectivity over other isoforms of nitric oxide synthases (NOSs), and blood-brain barrier permeability. In this context, we discovered a promising inhibitor, 6-(3-(4,4-difluoropiperidin-1-yl)propyl)-4-methylpyridin-2-amine dihydrochloride, that exhibits excellent potency for rat (Ki = 46 nM) and human nNOS (Ki = 48 nM), respectively, with 388-fold human eNOS and 135-fold human iNOS selectivity. It also displayed excellent permeability (Pe = 17.3 × 10−6 cm s−1) through a parallel artificial membrane permeability assay, a model for blood-brain permeability. We found that increasing lipophilicity by incorporation of fluorine atoms on the backbone of the inhibitors significantly increased potential blood-brain barrier permeability. In addition to measuring potency, isoform selectivity, and permeability of NOS inhibitors, we also explored structure-activity relationships via structures of key inhibitors complexed to various isoforms of nitric oxide synthases.
1. Introduction
The gaseous free radical nitric oxide (NO) has emerged as an important cell signaling molecule involved in numerous physiological and pathological processes.1–2 In human physiology, the role of NO is unique and regulates essential processes such as neurotransmission,3 vasodilation,4 smooth muscle relaxation,5 and immune response.6 In mammals, nitric oxide is produced endogenously through the oxidation of L-arginine to L-citrulline with the help of NADPH and molecular oxygen by three principal nitric oxide synthases (NOSs), namely, neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).7 Neuronal NOS (nNOS) catalyzes the oxidation of L-arginine in the central nervous system (CNS) to produce NO, a critical neurotransmitter for neuronal communication.8–10 Endothelial NOS (eNOS) is an isozyme of NOS that reduces blood pressure through relaxation of muscle.11 Inducible NOS (iNOS), on the other hand, produces cytotoxic NO and is responsible for activating the immune system to destroy pathogens and microorganisms.12
In the last decade, the excessive production of nitric oxide by nNOS has been recognized as a key player in the induction and progression of neurodegenerative diseases. Although the normal activity of producing physiological levels13 of NO by nNOS is important for neurotransmission, its overproduction is associated with the formation of the extremely reactive oxidant, peroxynitrite (ONOO−),14 in the presence of superoxide. nNOS has also been shown to be involved in several chronic neurodegenerative pathologies such as Alzheimer’s disease (AD),15 Parkinson’s disease (PD),16 Huntington’s disease (HD),17 amyotrophic lateral sclerosis (ALS),18 neuronal damage during stroke,19 as well as chronic headaches.20 Finally, at high concentrations, NO can cause excessive nitration and/or nitrosylation of proteins, leading to their degradation and misfolding.21
The three mammalian NOSs show high homology (>50% amino acid identity) and are active only as homodimers.7 Each monomer of NOS consists of a C-terminal reductase and an N-terminal oxygenase domain connected via a calmodulin-binding region. The C-terminal reductase domain contains the binding sites for reduced nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), and flavin mononucleotide (FMN) and is responsible for transporting electrons to the iron center of the heme during biosynthesis of NO.22 The N-terminal oxygenase domain contains the binding sites for noncatalytic zinc (Zn2+), tetrahydrobiopterin (H4B), and a heme-containing catalytic active site that binds the substrate, L-Arg.7 In the active site, NOSs catalyze the oxidation of L-arginine to L-citrulline and release NO in the presence of NADPH and molecular oxygen. This crucial cascade reaction is initiated at the reductase domain by the two-electron donor, NADPH, to the electron acceptor, the iron center of heme in the oxygenase domain, through FAD and FMN with a series of proton transfers in the process.23
The inhibition of nNOS has shown promise in treating or preventing neuronal damage in animal models.22 However, eNOS inhibition can cause cardiovascular problems and hypertension,24 and iNOS inhibition can result in immune defense failure.25 Therefore, inhibition of nNOS must be selective over the other isoforms to be a viable potential therapeutic approach for the treatment of neurodegenerative disorders.7,26–27 Small molecules that compete with the binding of L-Arg at the active site are one approach to inhibit nNOS. Design of such inhibitors can take advantage of slight but key differences in the active site structures of the three isoforms to build in nNOS selectivity.28–29
A major difficulty in treating central nervous system (CNS) diseases is ineffective drug delivery across the blood-brain barrier (BBB).30–33 The BBB is located at the interface of the blood vessels and brain tissues and is formed by brain capillary endothelial cells creating a tight junction to protect the brain from circulating xenobiotic molecules. Small molecules account for the vast majority of CNS drugs primarily because of their ability to penetrate the phospholipid membrane of the blood-brain barrier by passive or carrier-mediated mechanisms.34 In CNS drug development, factors such as: reduced molecular weight (MW < 500 Da; preferably nearer to 350–400 Da); increased lipophilicity (preferably ClogP <5); decreased topological polar surface area (TPSA < 76 Å2; preferably 25–60 Å2 ); the number of hydrogen bond donors, OH plus NH2 count (HBD = 5; preferably 3); the number of hydrogen bond acceptors, O plus N atoms (HBA = 10; preferably 7); a reduced number of rotatable bonds (RB < 8; preferably nearer to 4–5); reduced pKa of basic amino groups (pKa 7.5–10.5; preferably near to 8.4); and reduced P-glycoprotein (P-gp) efflux (ER < 2), are expected to significantly improve blood-brain barrier permeability based on properties of successful CNS drugs.32–33,35
In particular, the number of rotatable bonds (NRB) is a direct indication of flexibility of a chemical molecule, and studies have shown that increased rotatable bond count has a negative effect on the permeation rate.36 Reducing the number of rotatable bonds (NRB) not only improves the permeability rate but also oral bioavailability. Therefore, reducing molecular flexibility by lowering the number of rotatable bonds (NRB) is often an important consideration for improving BBB permeability.
Our laboratories have focused on the development of selective nNOS inhibitors for the treatment of neurodegenerative diseases for more than two decades, achieving big milestones in maintaining excellent potency while retaining high selectivity over other NOS isoforms and BBB permeability.22,37–40 Our efforts in improving the pharmacokinetic (PK) properties of nNOS inhibitors using various medicinal chemistry approaches such as the incorporation of intramolecular hydrogen bonds (1),41 suppressing amine basicity (2),42 converting to prodrugs (3),43 replacement of pharmacophores (4–5),44–45 introduction of a double-pyridine pharmacophore (6),46 incorporation of a pyridine linker (7),47 and exchanging the pyridine linker with fluoroaryl (8–9),37–38 have resulted in considerable improvements in the pharmacokinetic properties of NOS inhibitors (Figure 1).
Figure 1.

Structure of recent nNOS inhibitors.
We recently reported our first generation of nNOS inhibitors containing a 2-aminopyridine scaffold attached to a pyridine linker (7), which exhibits excellent potency with rat and human nNOS (Ki (rnNOS) = 16 nM; Ki (hnNOS) = 13 nM) as well as high selectivity over eNOS (hnNOS/heNOS = 1831). In addition, pharmacokinetic studies of 7 displayed very low cytochrome P450 inhibition, little human microsomal metabolism, and only 20% human plasma protein binding, all favorable properties. However, 7 displayed little or no Caco-2 permeability, predicting poor oral absorption and poor BBB permeability.47 However, exchanging the pyridine linker for a fluorobenzene ring (8-9) yielded significant improvement in membrane permeability while retaining high inhibitory activity. For example, 8 showed excellent potency and isoform selectivity toward human nNOS (Ki (hnNOS) = 30 nM; hnNOS/heNOS = 2799), but it still displayed a high efflux ratio (ER = 5.9) by a bidirectional Caco-2 permeability assay.37 Compound 9, with one additional fluorine atom on the fluorobenzene middle linker and an azetidine ring in the tail compared to 8, displayed excellent inhibition for human nNOS (Ki = 23 nM) and high selectivity over human eNOS (hn/he = 956) and human iNOS (hn/hi = 77) along with good brain penetration in PAMPA and Caco-2 permeability assays (Pe = 16.3 × 10−6 cm s−1; Papp = 17.0 × 10−6 cm s−1, respectively), and also had a lower efflux ratio (ER = 0.8).38
In continuation with these efforts, we report herein our development and optimization of a rapid and simple protocol for the synthesis of potent, selective, and highly permeable human neuronal nitric oxide synthase inhibitors based on the 2-aminopyridine scaffold, with the aim of improving cell-membrane permeability without sacrificing potency and isoform selectivity by lowering the number of rotatable bonds (NRB) in our molecules.
We performed several structural modifications to 9, based on recent X-ray crystallographic analysis, to improve the potency, isoform selectivity, and permeability of the inhibitors (Figure 2). In our earlier reports, we demonstrated that the 2-aminopyridine scaffold was crucial for the key anchoring interactions of nNOS inhibitors with Glu-592 and Glu-597 at the active sites of rat nNOS and human nNOS, respectively.37–38,40,47 Because of this, we retained the 2-aminopyridine head during further modification of our inhibitors. Structural modifications of this scaffold, especially at the tail amino functionality, was investigated to improve the bioavailability and selectivity of NOS inhibitors. Various strategies of structural modifications were employed to investigate their effect on biological activity. We found that the tail amino functionality is crucial for achieving high isoform selectivity after examining the inhibitory activity of 12. Therefore, the major modifications in this study focused on exploring the diversity of the tail amino functionality, including increasing lipophilicity, reducing the rigidity of the linker, and modulating the pKa of amine basicity.
Figure 2.

Analogues from lead 9 with a reduced number of rotatable bonds
First, reduction of the number of rotatable bonds within the molecule was investigated through truncation (10–20), by disconnecting the middle fluorobenzene linker and reattaching the remaining aliphatic tail amine portion directly to the 2-aminopyridine head. Second, length variation through homologation (15–16) from three carbons to four carbons between the 2- aminopyridine and tail amino functionality was examined. Third, methylation of the secondary amine to tertiary amine (12) was carried out in a strategy to improve permeability by reducing the number of H-bond donors. Fourth, increasing the rigidity of the molecule by introducing an alkyne moiety (10, 13, 15, 17 and 19) between the 2-aminopyridine and the tail amino functionality was carried out to improve permeability. Finally, modulation of the basicity of the amino tail functionality and maximizing the lipophilicity within the molecule (17–20) was used to improve selectivity and permeability. All synthesized compounds were investigated for their nNOS inhibitory activity and selectivity over eNOS and iNOS. In addition, all inhibitors were examined in a parallel artificial membrane permeability assay for blood-brain barrier permeability (PAMPA−BBB).
2. Results and Discussion
2.1. Chemistry
The general synthetic route to compounds 10–14, and 17–20 is outlined in Scheme 1. Most of these compounds are accessed within two-three steps from the corresponding bromopyridines and alkyne precursors.
Scheme 1.

General synthetic approaches to compounds 10-14 and 17-20. (a) The appropriate propargyl amine, Pd(PPh3)4 (5 mol%), PPh3 (10 mol%), CuI (10 mol%), Et3N, 120 °C, CH3CN (0.23 M), 24 h, 28–93%; (b) Pd/C or Pd(OH)2/C (10–40 mol%), H2, CH3OH, 25–80 °C, 8–24 h, 27–89%.
The alkyne precursors (23, 25, and 27) were prepared from propargyl bromide and the corresponding amine or amine hydrochloride through SN2 substitution (Scheme 2).58 The generated alkyne precursors (23, 25, and 27) were then coupled with 6-bromo-4-methylpyridin-2-amine (21, Scheme 1) via Sonogashira cross-coupling48–49 to afford the desired alkynes (10, 13, 17, and 19) in good to excellent yields. The flexible saturated version of NOS inhibitors is accessed by hydrogenation of the corresponding alkyne precursors (10, 13, 17, and 19) using Pd/C45 or Pd(OH)2/C50 with H2.
Scheme 2.

Synthesis of propargyl amines 23, 25 and 27. (a) propargyl bromide, K2CO3, CH3CN, 25 °C, 8 h, 48–53%.
The synthetic approaches to compounds 15–16 are outlined in Scheme 3. The Sonogashira cross-coupling reaction51 between 6-bromo-4-methylpyridin-2-amine (21) and but-3-yn-1-ol was carried out at 120 °C under an inert atmosphere to obtain the corresponding alkynyl alcohol in 70% yield. The resulting primary alcohol is converted into the corresponding tosylate 28, which is subsequently reacted with N,N-dimethylamine to afford the desired alkyne 15 in good yield. Hydrogenation of alkyne 15 was performed using Pd/C with H2 to afford 16 in a 62% yield.
Scheme 3.

Synthesis of compounds 15-16. (a) But-3-yn-1-ol, Pd(PPh3)4 (5 mol%), CuI (10 mol%), Et3N, 120 °C, CH3CN (0.25 M), 18 h, 70%; (b) TsCl, DMAP, Et3N, DCM, 0 °C to 25 °C, 17 h, 53%; (c) (CH3)2NH, K2CO3, CH3CN, 80 °C, 9 h, 68%; (d) Pd/C (10%), H2, CH3OH, 25 °C, 24 h, 62%.
2.2. Biological activity
The inhibitory constant (Ki) of newly synthesized analogues (10–20) were measured using the NO-hemoglobin (Hb) capture assay.52 All compounds were assayed against purified rat nNOS, human nNOS, human iNOS, and human eNOS for the evaluation of potency as well as isoform selectivity. Inhibitory activities of the compounds are summarized in Table 1. The ratio of hnNOS/rnNOS is made by the ratio of the Ki values of these inhibitors with human and rat nNOS. The ratio hn/rn is important for evaluating the potential translation of these inhibitors from preclinical to clinical studies. The isoform selectivity for nNOS over iNOS (hn/hi) and eNOS (hn/he) was obtained by comparing the Ki values of human nNOS with human iNOS and human eNOS, respectively.
Table 1.
List of Ki values and selectivities of 10–20
| Compd | Ki (nM)a,b,c | Selectivity | |||||
|---|---|---|---|---|---|---|---|
| rat nNOS |
human nNOS |
human iNOS |
human eNOS |
hn/rn | hn/hi | hn/he | |
| 10 | 88 | 92 | 3575 | 1919 | 1.0 | 39 | 21 |
| 11 | 29 | 21 | 669 | 1469 | 0.7 | 32 | 70 |
| 12 | 26 | 29 | 2831 | 3662 | 1.1 | 98 | 126 |
| 13 | 947 | 2124 | 113733 | 43798 | 2.2 | 54 | 21 |
| 14 | 56 | 171 | 10160 | 12536 | 3.1 | 59 | 73 |
| 15 | 242 | 327 | 29835 | 70509 | 1.4 | 91 | 216 |
| 16 | 36 | 58 | 7671 | 7351 | 1.6 | 132 | 127 |
| 17 | 465 | 488 | 11089 | 5132 | 1.1 | 23 | 11 |
| 18 | 95 | 149 | 4358 | 6268 | 1.6 | 29 | 42 |
| 19 | 1591 | 1430 | 27369 | 18950 | 0.9 | 19 | 13 |
| 20 | 46 | 48 | 6480 | 18639 | 1.0 | 135 | 388 |
Ki values were calculated from the IC50 values of the corresponding dose−response curves using the Cheng−Prusoff equation. 8 to 11 concentrations were tested, and the IC50 value was calculated from an average of at least two duplicates. The standard errors were less than 10%. The ratio hn/rn was determined to evaluate the potential translation of these inhibitors from preclinical data to a clinical study. This ratio was aimed to be as close to 1.0 as possible so that there would be little to no significant difference in the amount of inhibitors used in rat and human dosage. Selectivity values are determined by calculating the ratios of respective Ki values.
The plate reader syringe lines are equilibrated with assay buffer (100mM HEPES, 10% glycerol, pH 7.4–7.5) before the assay.
The assay was performed using assay buffer (100 mM HEPES, 10% glycerol, pH 7.4–7.5), and stock buffer (100 mM HEPES, pH 7.4–7.5).
As shown in Figure 2, the simplified versions of NOS inhibitors (10–20) were accessed through truncation by disconnecting the middle difluorobenzene linker and reattaching the remaining aliphatic tail amine portion directly to the 2-aminopyridine head. The structure−activity studies of newly synthesized compounds (11–12) (Table 1) revealed that the steric bulk on the amino tail plays a crucial role in achieving good isoform selectivity. For example, compound 12, with a tertiary amine at the tail amino functionality, exhibits excellent potency for rat (Ki = 26 nM) and human nNOS (Ki = 29 nM), respectively, with 126-fold human eNOS and 98-fold human iNOS selectivity. In contrast, 11, with a secondary amine, shows a higher potency than that of 12 with hnNOS; however, a drop in selectivity was observed with heNOS, suggesting that the tertiary amine is important for achieving good isoform selectivity in this series. Disappointingly, 12 displayed poor membrane permeability (Pe = 2.0 × 10−6 cm s−1) due to the lack of the lipophilic difluoroaryl linker as in parent lead 9.
To improve the permeability of these inhibitors, we introduced C-F bonds at the backbone of the inhibitors. Interestingly, after installing a 4,4-difluoropiperidine at the tail amino functionality of 9, as in 19, we observed a dramatic increase in the permeability. In line with this result, we discovered the most promising inhibitor, i.e., compound 20, which has a fluorinated tertiary amine at the tail and exhibits excellent potency for rat (Ki = 46 nM) and human nNOS (Ki = 48 nM), with 388-fold human eNOS and 135-fold human iNOS selectivity. Compound 20 also displayed excellent permeability (Pe = 17.3 × 10−6 cm s−1) in the PAMPA-BBB assay (Table 2).
Table 2.
Effective permeability (Pe) of five commercial drugs and nNOS inhibitors in the PAMPA−BBB assay
| Compounds | Reported Pe (10−6 cm s−1)a |
Determined Pe (10−6 cm s−1)b–d |
Predictiona |
|---|---|---|---|
| (±)-Verapamil | 16 | 21.2 ± 0.43 | CNS (+) |
| Desipramine | 12 | 22.8 ± 0.93 | CNS (+) |
| Chlorpromazine | 6.5 | 10.8 ± 0.64 | CNS (+) |
| Dopamine | 0.2 | 0.13 ± 0.14 | CNS (−) |
| Theophylline | 0.12 | 0.1 ± 0.02 | CNS (−) |
| 10 | 4.7 ± 0.44 | CNS (+) | |
| 11 | 0.96 ± 0.03 | CNS (−) | |
| 12 | 2.0 ± 0.21 | CNS (+/−) | |
| 13 | 3.2 ± 0.12 | CNS (+/−) | |
| 14 | 2.2 ± 0.01 | CNS (+/−) | |
| 15 | 4.1 ± 0.12 | CNS (+) | |
| 16 | 2.3 ± 0.04 | CNS (+/−) | |
| 17 | 19.6 ± 0.86 | CNS (+) | |
| 18 | 17.5 ± 0.71 | CNS (+) | |
| 19 | 21.1 ± 0.15 | CNS (+) | |
| 20 | 17.3 ± 0.44 | CNS (+) |
The reported effective permeability (Pe) values from the literature.53
All assays were performed over 17 h at a concentration of 200 μM.
All compounds were tested in a triplicate.
Effective permeability values obtained in our in-house conditions.
CNS (+) = high BBB permeation. CNS (−) = low BBB permeation. CNS (+/−) = BBB permeation uncertain.
It is interesting to note that the nNOS inhibitors with rigid alkynes such as 10, 13, 17, and 19 are less potent than their flexible saturated alkanes in potency and isoform selectivity. In contrast, the Pe values of those rigid alkynes (10, 13, 17, and 19) are slightly better than their flexible alkane nNOS inhibitors in this series.
Next, the length variation by homologation (15–16) from three carbon to four carbons between the 2-aminopyridine and the tail amino functionality was examined. Compound 16 maintained good potency (Ki (hnNOS) = 58 nM) and isoform selectivity (hn/hi = 132; hn/he = 127), although it suffered from poor permeability (Pe = 2.3 × 10−6 cm s−1; Table 2). Compound 15 maintained good isoform selectivity (hn/hi = 91; hn/he = 216) but lost its potency by six-fold as compared to its flexible alkane version. Compound 15 shares similar permeability as its rigid alkyne analogues (10, 13, 17, and 19) in the PAMPA-BBB assay.
2.3. Permeability
It is expected that reducing molecular flexibility through lowering the number of rotatable bonds (NRB) of an inhibitor should improve its membrane permeability (Table 2). To verify this hypothesis, lead 9 and its simplified versions 10–20 were examined in the PAMPA-BBB assay,37–38,53 and the results were compared based on their effective permeability (Pe) values. The results indicate that the absence of a fluoroaryl segment in the simplified version of inhibitors 10–14 negatively impacted their membrane permeability. However, we found that installing fluorine atoms at the substituted amine tail considerably enhanced the permeability of NOS inhibitors (17–20). It is important to note that 19, with an alkyne linker, showed the highest permeability (Pe = 21.1 × 10−6 cm s−1) in this series due to the presence of fluorine atoms in the amine tail as well as maintenance of the alkyne rigidity in the molecule. Because of the increased flexibility of 20 its permeability (Pe = 17.3 × 10−6 cm s−1) is considerably reduced relative to 19. These results also indicate that maintaining rigidity in the molecule could further improve permeability. For example, permeability values were considerably increased for rigid alkynes 10, 13, 15, 17, and 19, compared to their more flexible versions 11, 14, 16, 18, and 20, respectively. Based on the above experimental results, it is clear that maintaining rigidity and incorporating F atoms at the amine tail improve permeability without altering the potency and isoform selectivity of NOS inhibitors.
2.4. Structure-Activity Relationships
2.4.1. Truncation of lead 9
Reduction of the number of rotatable bonds within the molecule was investigated through truncation (10–20) by disconnecting the middle fluorobenzene linker and reattaching the remaining aliphatic tail amine portion directly to the 2-aminopyridine head. We showed in earlier reports37–38,40,47 that the 2-aminopyridine ring was crucial for the key interactions of nNOS inhibitors with Glu-592 and Glu-597 at the active sites of rat nNOS and human nNOS, respectively. Similarly, the terminal tail amine also plays a crucial role in the interactions with heme propionates for achieving higher potency. The linker between the 2-aminopyridine head and the amine tail provides additional variations for SAR studies. In this truncated series of compounds, we tested different tail amines, from simple tertiary amines to saturated cyclic amines, and further introduced fluorines into the cyclic amine to adjust its basicity. Other changes include different linker lengths and a rigid alkyne versus a flexible alkane linker. The collected crystallographic data reveal the following key features of SAR information: (1) A shorter linker provides better binding affinity; (2) The rigid alkyne linker leads to poor binding due to steric clashes, even through compounds with a rigid linker exhibit better permeability; (3) Compound 20, with a short flexible linker and a tail 4,4-difluoropiperidine ring, shows a unique binding mode resulting in both good potency and permeability.
First, we determined the crystal structures of rnNOS, hnNOS, and heNOS in complex with 12 that bears the N,N-dimethylaminopropyl side chain as the amine tail (Table 1 and Figure 3). The crystal structures of bound 12 revealed a unique inhibitor binding mode. In rnNOS (Figure 3A), in addition to the H-bonds from the 2-aminopyridine to Glu-592 and Trp-587, the tail tertiary amino group can approach the heme propionate A and form a H-bond directly. In contrast, the tail azetidine ring N atom in lead 9 forms a H-bond with the water sitting in between the H4B and propionate A, as seen in Figure S1. This is the main difference between the fluorophenyl ring-containing inhibitors and the current simplified versions. The removal of the difluorophenyl ring in lead 9 also removes the H-bonding interaction of Tyr-562 and Tyr-567 in rnNOS and hnNOS, respectively. These interactions were essential to achieve high potency and selectivity in the previous series. However, removal of this portion of lead 9, as depicted in the simplified NOS inhibitors, does not affect the potency significantly but had some unfavorable effects on isoform selectivity.
Figure 3.

Active site structures of 12 bound to rnNOS (A), hnNOS (B), and heNOS (C). In all structural figures, the following atomic color schemes are used: cyan, green, and yellow for carbon atoms in the rnNOS, hnNOS, and heNOS backbones, respectively; red, oxygen; blue, nitrogen; light cyan, fluorine. The omit Fo-Fc Polder electron density for the inhibitor is shown at the 3.0 σ contour level. Key H-bonds to the inhibitor are depicted with dashed lines.
Compound 12 maintained excellent potency towards rnNOS (Ki = 26 nM) and hnNOS (Ki = 29 nM), even though it loses the additional binding interactions that originate from the difluorophenyl group in lead 9 (Ki (rnNOS) = 26 nM and Ki (hnNOS) = 23 nM). Similarly, 12 binds to hnNOS (Figure 3B) with its 2-aminopyridine head to form H-bonds with Glu-597 and Trp-592 and its tail amino group to propionate A. Compared with the rnNOS structure of 12, the conformation of the tail amino linker in hnNOS is different: in rnNOS it is extended, while in hnNOS it is curled. The different conformation of the tail amino linker is due to the quite different orientation of the 2-aminopyridine ring in these structures. The reason for the difference is not clear; the side chain conformation of the anchoring Glu residue in these two structures is essentially the same.
Figure 3C shows the binding mode of 12 with human eNOS. Compound 12 binds well to human eNOS (Ki = 3362 nM). In addition to the H-bonds from the 2-aminopyridine to Glu-361 and Trp-356, the tail tertiary amino group also makes a H-bond directly with propionate A. Because of its better binding to heNOS, compound 12 displays an 8-fold drop in heNOS selectivity (hn/he = 126) as compared to lead 9 (hn/he = 956).38 The orientation of the 2-aminopyridine ring of 12 in the structure with rnNOS is significantly different from that observed with heNOS, much more tilted from the heme plane in rnNOS than in heNOS. However, in all these cases (rnNOS, hnNOS, and heNOS), the tail tertiary amino group can still approach propionate A for a H-bond.
Next, we examined the effect of steric bulk on the amine tail to improve isoform selectivity. Compound 11 is the secondary amine variant of the tail functionality similarly positioned as the tertiary amino group of 12. As shown in Figure 4, the binding interactions of 11 to hnNOS and heNOS are essentially the same. Compound 11 is capable of making a H-bond with propionate A in hnNOS, in addition to the other interactions such as the H-bonds to Glu-597 and Trp-592 with 2-aminopyridine. The advantage of the secondary amine in 11 is having fewer methyl groups compared to the tertiary amine in 12, thus having fewer steric clashes with propionate A while making the H-bond. This result agrees with the finding of slightly better potency (Ki (hnNOS) = 21 nM) of 11 relative to that of 12. Similarly, in heNOS, 11 makes a H-bond with propionate A, along with two other H-bonds to Glu-361 and Trp-356 with the 2-aminopyridine. However, a drop in selectivity was observed with heNOS (hn/he = 70) as compared to 12 (hn/he = 126), because the secondary amine exhibits better binding to heNOS.
Figure 4.
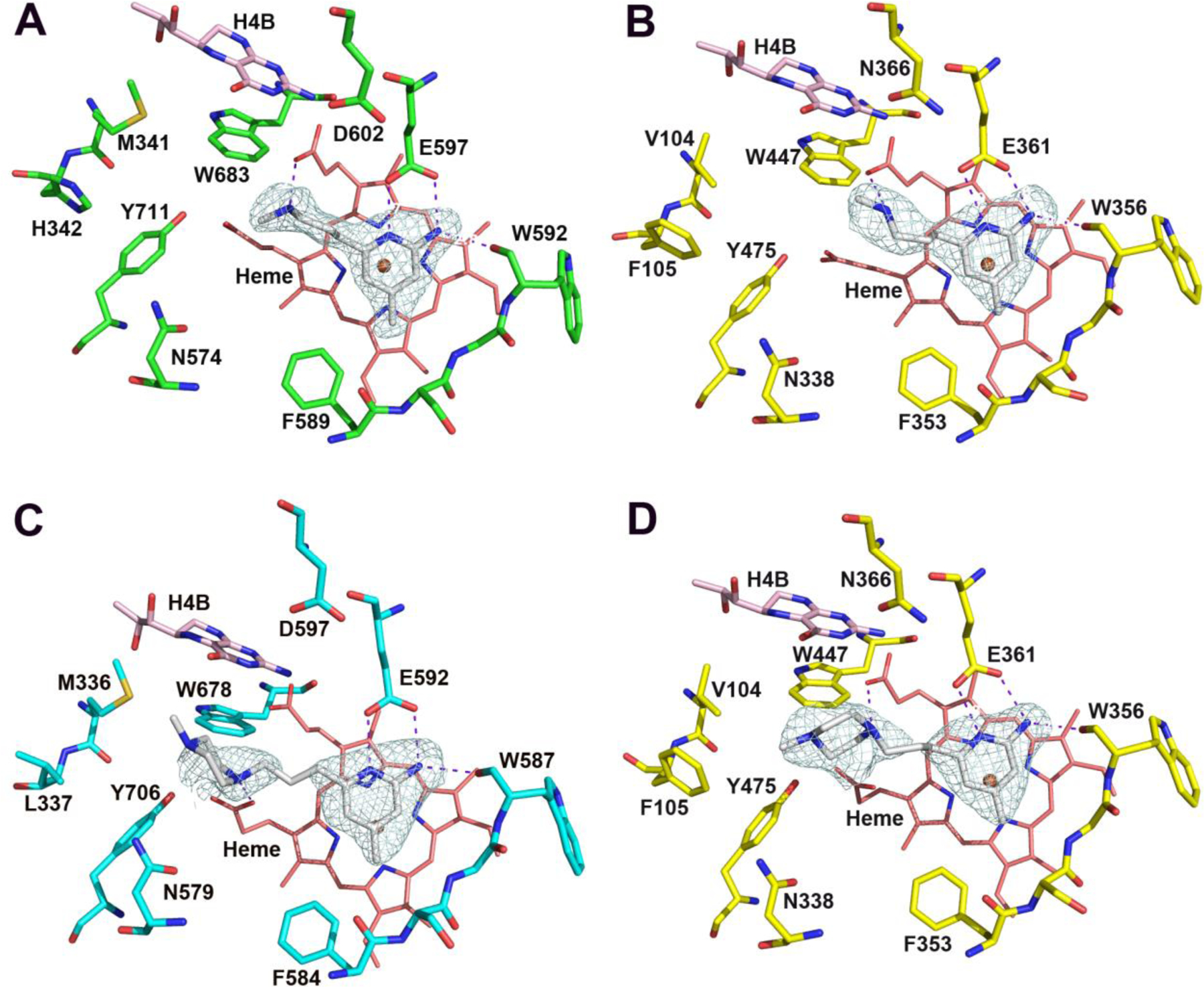
Active site structures of 11 bound to hnNOS (A) and heNOS (B); and 14 bound to rnNOS (C) and heNOS (D).
When the tail amino group is replaced by a piperazine ring, as in 14, the binding affinity drops two-fold in rnNOS (Ki (rnNOS) = 56 nM). The structure with this compound shows that one of the piperazine N atoms can approach for a H-bond to propionate D that has been bent over for the interaction (Figure 4 C). However, the flexibility of the piperazine ring of 14 is higher in the heNOS structure (Figure 4 D). A weaker H-bond is observed from the piperazine ring to propionate A, which is consistent with its much poorer affinity (Ki (heNOS) = 12536 nM) compared to 11 (Ki (heNOS) = 1469 nM).
2.4.2. Homologation
Based on the information accumulated from 11 and 12, we decided to incorporate a tertiary amine instead of a secondary amine as the tail amine functionality for future molecules in this truncated series. Further modification of 12 via homologation by increasing one-carbon (16) between the 2-aminopyridine and the tail amine functionality as represented in both the rigid alkyne (15) and its flexible saturated alkane (16) was investigated without further modification at the tertiary amine (N, N-dimethylamino group). The rigid alkyne (15) shows a slight improvement in heNOS selectivity (hn/he = 216) as well as permeability (Pe = 4.7 × 10−6 cm s−1); however, its potency towards hnNOS was reduced by 11-fold relative to 12. The available structure of hnNOS bound with 15 (Figure S2) indicates that the longer and rigid alkyne linker of 15 allows it to make a H-bond with propionate D rather than with propionate A as observed in 12. This creates a steric clash at 3.0 Å from propionate A to the rigid linker, which drives up the binding energy leading to high Ki value (327 nM). Compound 16 showed about a 2-fold drop in potency (Ki (hnNOS) = 58 nM), with a slight improvement in isoform selectivity (hn/hi = 132; hn/he = 127) compared to that of 12, and it suffered from poor permeability (Pe = 2.3 × 10−6 cm s−1; Table 2). The structure (Figure S2) shows that the flexible linker of 16 relaxes the clash between propionate A and the linker (3.1 – 3.2 Å); therefore, the affinity of 16 to hnNOS is significantly better than that of 15.
2.4.3. Introducing rigidity
Based on the above results, we attempted to improve the permeability of these inhibitors by increasing the rigidity at the linker section of 12 by incorporating an alkyne moiety between the 2-aminopyridine and the tail amino functionality. We designed and synthesized the appropriate NOS inhibitors and investigated their permeability through a PAMPA-BBB assay.
As shown in Table 2, the rigid alkynes 10, 13, 15, 17, and 19 exhibited moderate to excellent permeability values with increasing rigidity. Unfortunately, there was also a negative effect on the binding affinity to hnNOS: the approximately 3-fold (10), 73-fold (13), 17-fold (17), and 49-fold (19) drop in potency with hnNOS reflects their lower binding affinity relative to that of 12.
As shown in Figure 5 compound 10 retains its H-bonding interaction with propionate A in all three structures (rnNOS, hnNOS, and heNOS). While a 3-fold drop in affinity with rnNOS and hnNOS is not clearly explained, maintaining a rather high affinity to heNOS is agreeable with the good H-bond observed in the structure. When the tail secondary amine is replaced by a bulky piperazine ring as in 13 the binding affinity worsens significantly. To our surprise, in hnNOS one of the piperazine ring N atoms does form a H-bond with propionate D (Figure 6A). At a first glance, this seems to contradict the 73-fold affinity drop. By closely examining the enzyme-inhibitor interaction, the unfavorable clash (pink arrow) at about 3.0 Å observed between 13 and propionate A resulting from the rigidity of the alkyne linker could be responsible for the affinity loss. The poor affinity of 13 to heNOS does find support in the structure (Figure 6B). The tail piperazine ring is partially disordered and likely bends upward from propionates but close to Gln-247. In the heNOS structure, the clash (pink arrow) of 3.0 Å between 13 and propionate A is also present.
Figure 5.

Active site structures of 10 bound to rnNOS (A), hnNOS (B), and heNOS (C).
Figure 6.

Active site structures of 13 bound to hnNOS (A) and heNOS (B). The clashes observed between the rigid alkyne linker and propionate A are shown by the pink arrows.
2.4.4. Increasing lipophilicity
Various lipophilic amine moieties at the tail functionality were chosen to enhance lipophilicity of 12 by introduction of C-F bonds in the backbone of the saturated cyclic amine functionality (17–20). Modulation of basicity of the amino tail functionality was examined to increase the lipophilicity of the molecules (17–20) for improved selectivity and permeability. Both rigid alkynes (17 and 19) and the corresponding flexible saturated alkanes (18 and 20) with 3,3-difluoroazetidine and 4,4-difluoropiperidine rings at the tail, were investigated. Interestingly, these analogues were shown to exhibit a large improvement in permeability of about 9 to 11-fold over parent compound 12, based on their PAMPA-BBB permeability assay (Table 2). Rigid alkyne 19 was identified as the most permeable NOS inhibitor (Pe = 21.1 × 10−6 cm s−1) in this series, likely because of the presence of C-F bonds in the tertiary amine tail as well as the maintenance of alkyne rigidity in the molecule.
However, the addition of fluorine atoms to parent 12 also causes considerable changes in the binding affinity to NOS. The binding interaction of 17 is shown in Figure 7. In both rnNOS and hnNOS the difluoroazetidine ring bends toward propionate D, pushing it away. In this way, the 3,3-difluoroazetidine N atom can instead make a H-bond with propionate A. Also, in this bent conformation, the rigid linker of 17 can avoid clashes with propionate A. These observations are consistent with its better affinity to rnNOS and hnNOS than 13 (Table 1). In the new position, forced by inhibitor binding, the propionate D can make a H-bond with His341, which stabilizes the loop of residues 339–347, which are often disordered in hnNOS structures. However, the azetidine ring in heNOS bends upward away from the propionates (Figure 7C), and the fluorine atom makes van der Waals contacts with Gln-338. The clashes (pink arrow) from the alkyne linker to propionate A remain.
Figure 7.

Active site structures of 17 bound to rnNOS (A), hnNOS (B), and heNOS (C). A clash (pink arrow) was observed between the alkyne linker and propionate A in heNOS.
Compound 19 bears a 4,4-difluoropiperidine ring at the tail of a rigid alkyne linker. In all three structures (rnNOS, hnNOS, and heNOS), the 4,4-difluoropiperidine ring bends upward away from the heme propionates (Figure 8). In all cases the clashes (pink arrow) between the rigid alkyne linker and propionate A are inevitable. The combination of the lack of H-bonding with the piperidine ring and the presence of the unfavorable clashes account for the drop in potency by about 61- and 49-fold observed with rnNOS and hnNOS, respectively, compared with 12 (Table 1). The reduction in the potency of 19 is very likely the result of both the decrease in the pKa of the tail amino functionality as well as maintenance of alkyne rigidity, thereby diminishing electrostatic interactions with heme propionate A, which we also observed in an earlier study.37 Being able to maintain the H-bond with propionate A gives 17 a 3-fold better affinity than 19 with both rnNOS and hnNOS (Table 1). However, better binding of 17 to heNOS than 19 has no clear explanation.
Figure 8.

Active site structures of 19 bound to rnNOS (A), hnNOS (B), and heNOS (C). In all three cases, the clashes of the alkyne linker with propionate A were present (pink arrows).
The X-ray crystal structures of 18 bound with all three NOSs (Figure 9) show a H-bond from the ring N atom of 3,3-difluoroazetidine to heme propionate A. Because of the more flexible linker in 18 than in 17, establishing the H-bonding interactions with propionate A by twisting the linker does not generate the clashes that were seen for 17. Keeping the lipophilicity but relaxing the rigidity of the linker proved crucial to finding the most promising NOS inhibitor in this series, compound 20, that not only shows excellent membrane permeability (Pe = 17.3 × 10−6 cm s−1) but also shows excellent potency for rat (Ki = 46 nM) and human nNOS (Ki = 48 nM), respectively, with 388-fold heNOS and 135-fold hiNOS selectivity (Table 1 and 2).
Figure 9.

Active site structures of 18 bound to rnNOS (A), hnNOS (B), and heNOS (C).
It is interesting to note that the binding interaction of 20 (Figure 10) is quite different from the other members of this series. The H-bond observed for 20 with propionate D in rat and human nNOS results because propionate D bends over to optimize the geometry for H-bonding with the ring N atom of the 4,4-difluoropiperidine ring. Propionate D in heNOS does not show this change of conformation when the tail amine of 20 approaches with its linker in a more extended conformation. Therefore, the H-bond between the 4,4-difluoropiperidine and propionate D is less optimized in heNOS. The potential nonbonded interaction between the bulky Phe-105 side chain and the fluorine atom of the 4,4-difluoropiperidine ring may play a role in this different behavior of H-bond formation in heNOS compared to that in hnNOS.
Figure 10.

Active site structures of 20 bound to rnNOS (A), hnNOS (B), and heNOS (C).
The structural observations for 20 are in good agreement with the observed higher potency and selectivity as compared to rigid analogue 19 because of its flexible nature (Table 1). A slight decrease in potency of 20 relative to that of 12 is very likely the result of its lower pKa due to the presence of the 4,4-difluoropiperidine tail group. However, 20 exhibits a much better membrane permeability, having a Pe value of 17.3 × 10−6 cm s−1 relative to that of 12 (Pe = 2.0 × 10−6 cm s−1). This suggests that achieving both higher potency and good permeability can be a delicate balance; i.e., a slight change in the skeleton of the inhibitor can alter its properties considerably. Overall, because of their flexible nature, 18 and 20 show better binding interactions than their rigid and linear linker analogues 17 and 19, thereby increasing their potency and selectivity.
3. Conclusion
In summary, a series of potent, selective, and highly permeable human nNOS inhibitors based on the 2-aminopyridine scaffold with a shortened amino sidechain was designed and evaluated. A rapid and simple protocol was developed to access these inhibitors with excellent yields. Compound 20, which has a reduced number of rotatable bonds, had outstanding properties that exhibit excellent potency for rat (Ki = 46 nM) and human nNOS (Ki = 48 nM), respectively, with 388-fold human eNOS and 135-fold human iNOS selectivity. Compound 20 also displayed excellent membrane permeability (Pe = 17.3 × 10−6 cm s−1) through a parallel artificial membrane permeability assay modeling the blood-brain barrier (PAMPA-BBB). We found that addition of fluorine atoms at the backbone of the amine tail of the inhibitors greatly increased membrane permeability, suggesting that lipophilicity will be a key component of clinically useful nNOS inhibitors. Increasing rigidity in the molecule also had an overall positive impact on permeability; however, it also decreased potency and isoform selectivity. Finally, we show that X-ray crystal structures of representative inhibitors, including 20, bound to NOS isoforms provide excellent interpretations for their properties in potency, isoform selectivity, and permeability.
4. Experimental section
Unless otherwise stated, all the described reactions were performed under an argon atmosphere. Anhydrous solvents such as THF, 1,4-dioxane, CH2Cl2, MeOH, Et3N, and DMF were purchased directly from commercial sources. N,N-dimethylpropargylamine and N-methylpropargylamine were purchased from Sigma-Aldrich. Metal catalysts, especially tetrakis(triphenylphosphine)palladium(0) was purchased from Strem Chemicals. Other metal catalysts such as bis(triphenylphosphine)palladium(II) dichloride, copper(I) iodide, and Pd/C (10 wt. % loading (dry basis), matrix carbon powder, wet support) were purchased from Sigma-Aldrich. Pd(OH)2/C (20% on carbon, 60% water, wet support) was acquired from Acros Organics. Sonogashira cross-coupling reactions were performed using Chemglass pressure vessels. All reactions were monitored by thin-layer chromatography (TLC) using Merck TLC silica gel 60 F254; 0.25 mm glass plates and components were visualized by ultraviolet light (254 nm) and/or KMnO4 or phosphomolybdic acid stain. 1H and 13C NMR spectra were recorded on a Bruker Avance III NMR spectrometer with an operating frequency of 500 and 126 MHz, respectively, in CDCl3, CD3OD, or DMSO. Chemical shifts are reported in parts per million, and multiplicities are indicated by s = singlet, d = doublet, t = triplet, q = quartet, sep = septet, dd = doublet of doublet, dt = doublet of triplet, m = multiplet, and br = broad resonance. Coupling constants “J” were reported in hertz. High-resolution mass spectral (HRMS) data were obtained on an Agilent 6210 LC-TOF spectrometer in the positive ion mode using electrospray ionization (ESI) with an Agilent G1312A high-performance liquid chromatography (HPLC) pump and an Agilent G1367B autoinjector at the Integrated Molecular Structure Education and Research Center (IMSERC), Northwestern University. Flash column chromatography was performed on an Agilent 971-FP automated flash purification system with a Varian column station and various SiliCycle cartridges (4−80 g, 40−63 μm, 60 Å). All compounds undergoing biological testing were >95% pure.
4.1. General procedure for the synthesis of NOS inhibitors
4.1.1. Synthesis of 6-(3-(4,4-difluoropiperidin-1-yl)propyl)-4-methylpyridin-2-amine dihydrochloride (20)
Synthesis of 6-(3-(4,4-difluoropiperidin-1-yl)propyl)-4-methylpyridin-2-amine dihydrochloride (20) is representative (Scheme 1).
4.1.2. Synthesis of propargyl amines 23, 25, and 27
Compounds 23, 25, and 27 were prepared following a reported procedure with slight modifications (Scheme 2).58 To a stirred suspension of 4,4-difluoropiperidine hydrochloride (0.500 mg, 3.170 mmol, 1.0 equiv) and K2CO3 (877 mg, 6.350 mmol, 2.0 equiv) in anhydrous CH3CN (10 mL) at 25 °C, a solution of propargyl bromide (0.420 mL, 3.810 mmol, 80% in toluene, 1.2 equiv) in anhydrous CH3CN (2 mL) was added. The mixture was allowed to stir at 25 °C for 8 h. After completion of the reaction as indicated by TLC, the reaction mixture was diluted with water and then extracted with DCM (3 × 15 mL). The combined organic layers were washed with 20 mL of brine, dried over Na2SO4, filtered, and concentrated in vacuo, provided propargyl amine 25 (260 mg, 1.650 mmol, 52%) as a yellow semisolid. The crude product was sufficiently pure to use in the next step without further purification. The same procedure was adopted for the synthesis of propargyl amines 23 (yellow oil, 243 mg, 1.850 mmol, 48%) and 27 (yellow solid, 367 mg, 2.660 mmol, 53%).
4.1.3. Typical procedure for Sonogashira cross-coupling reaction (compounds 10, 13, 17 and 19)
The synthesis of 6-(3-(4,4-difluoropiperidin-1-yl)prop-1-yn-1-yl)-4-methylpyridin-2-amine (19) is representative (Scheme 1). A flame-dried pressure vessel containing 6-bromo-4-methylpyridin-2-amine (21) (260 mg, 1.380 mmol, 1.0 equiv), Pd(PPh3)4 (24 mg, 0.02 mmol, 1.5 mol%), and CuI (26 mg, 0.14 mmol, 10 mol%) was filled with argon using three vacuum-argon flush cycles to remove oxygen. To this, CH3CN (4 mL, 0.25 M), Et3N (0.770 mL, 5.520 mmol, 4.0 equiv), and 4,4-difluoro-1-(prop-2-yn-1-yl)piperidine (25) (260 mg, 1.650 mmol, 1.2 equiv) in CH3CN (1.5 mL) were added, and the resulting mixture was stirred for 12 h at 120 °C. After completion of the reaction as indicated by TLC, the reaction mixture was cooled to 25 °C, filtered through Celite, and concentrated by rotary evaporation. Purification of the crude residue was performed by flash column chromatography eluting with hexane: EtOAc (20:80, v:v) for 10 min to remove any nonpolar residues and followed by the solvent mixture (DCM:CH3OH:Et3N, 80:10:10, v:v) for another 15 min to afford compound 19 (154 mg, 0.580 mmol, 42%) as a brown solid. 1H NMR (500 MHz, CDCl3): δ 6.67 (s, 1 H), 6.27 (s, 1 H), 4.47 (bs, 2 H), 3.54 (s, 2 H), 2.71 (t, J = 5.0 Hz, 4 H), 2.19 (s, 3 H), 2.13 – 1.95 (m, 4 H). 13C NMR (126 MHz, CDCl3): δ 158.4, 149.1, 140.5, 121.7 (t, J = 241.9 Hz), 119.4, 109.2, 85.4, 83.1, 49.3 (t, J = 5.7 Hz), 47.2 (t, J = 1.9 Hz), 34.1 (t, J = 22.7 Hz), 20.9. HRMS (ESI) Calcd for C14H18F2N3 [(M+H)+]: 266.1463, found: 266.1470.
4.1.4. Typical procedure for hydrogenation (compounds 11–12, 14, 18, and 20)50
Compound 19 (154 mg, 0.580 mmol, 1.0 equiv) was diluted with CH3OH (10 mL) and flushed with nitrogen before adding a catalytic amount of Pd(OH)2/C (62 mg; 40% of the weight of the substrate). The inert atmosphere of this heterogeneous mixture was replaced with a hydrogen balloon at room temperature and stirring was continued at 80 °C for 24 h. The crude mixture was then filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with hexane: EtOAc (20:80 v:v) for 10 min to remove any nonpolar residues and followed by the solvent mixture (DCM:CH3OH:Et3N, 80:10:10, v:v) for 15 another min to afford 20 (42 mg, 0.16 mmol, 27%) as an off-white solid. 1H NMR (500 MHz, CDCl3): δ 6.34 (s, 1 H), 6.15 (s, 1 H), 4.32 (s, NH2, 2 H), 2.58 – 2.49 (m, 6 H), 2.45 – 2.38 (m, 2 H), 2.18 (s, 3 H), 2.01 – 1.92 (m, 4 H), 1.89 – 1.79 (m, 2 H). 13C NMR (126 MHz, CDCl3): δ 160.0, 158.2, 149.2, 122.3 (t, J = 241.9 Hz), 114.4, 106.5, 57.3, 50.1 (t, J = 5.0 Hz), 35.8, 34.1 (t, J = 23.3 Hz), 27.3, 21.1. HRMS (ESI) Calcd for C14H22F2N3 [(M+H)+]: 270.1776, found: 270.1786.
4.1.5. Synthesis of 4-methyl-6-(3-(methylamino)prop-1-yn-1-yl)pyridin-2-amine dihydrochloride (10)
Compound 10 was obtained in a similar manner to that described for the synthesis of compound 19 in Scheme 1. Yield: 286 mg (1.630 mmol, 61%); brown solid. 1H NMR (500 MHz, CD3OD): δ 6.72 (s, 2 H), 3.28 – 3.20 (m, 2 H), 2.93 (s, 6 H), 2.84 (t, J = 7.8 Hz, 2 H), 2.38 (s, 3 H), 2.22 – 2.16 (m, 2 H). 13C NMR (126 MHz, CD3OD): δ 159.1, 155.9, 148.5, 114.9, 111.3, 57.7 (2xC), 43.6, 30.3, 24.7, 22.0. HRMS (ESI) Calcd for C10H14N3 [(M+H)+]: 176.1182, found: 176.1177.
4.1.6. Synthesis of 4-methyl-6-(3-(methylamino)propyl)pyridin-2-amine dihydrochloride (11)
Compound 11 was obtained in a similar manner to that described for the synthesis of compound 20 in Scheme 1. The hydrogenation was performed using 10% Pd/C at 25 °C for 12 h. Yield: 202 mg (1.130 mmol, 89%); pale yellow solid. 1H NMR (500 MHz, CD3OD): δ 6.69 (s, 1H), 6.68 (s, 1H), 3.13 – 3.04 (m, 2H), 2.92 (s, NH, 1H), 2.86 – 2.82 (m, 2H), 2.73 (s, 3H), 2.38 (s, 3H), 2.14 – 2.07 (m, 2H). 13C NMR (126 MHz, CD3OD): δ 159.2, 156.0, 148.7, 114.8, 111.3, 49.2, 33.6, 30.4, 26.0, 22.0. HRMS (ESI) Calcd for C10H18N3 [(M+H)+]: 180.1495, found: 180.1469.
4.1.7. Synthesis of 6-(3-(dimethylamino)propyl)-4-methylpyridin-2-amine dihydrochloride (12)
Compound 12 was obtained in a similar manner to that described for the synthesis of compound 20 in Scheme 1. The hydrogenation was performed using 10% Pd/C at 60 °C for 24 h. Yield: 172 mg (0.890 mmol, 84%); brown solid. 1H NMR (500 MHz, CD3OD): δ 6.72 (s, 2 H), 3.28 – 3.20 (m, 2 H), 2.93 (s, 6 H), 2.84 (t, J = 7.8 Hz, 2 H), 2.38 (s, 3 H), 2.22 – 2.16 (m, 2 H). 13C NMR (126 MHz, CD3OD): δ 159.1, 155.9, 148.5, 114.9, 111.3, 57.7 (2xC), 43.6, 30.3, 24.7, 22.0. HRMS (ESI) Calcd for C11H20N3 [(M+H)+]: 194.1652, found: 194.1631.
4.1.8. Synthesis of 4-methyl-6-(3-(4-methylpiperazin-1-yl)prop-1-yn-1-yl)pyridin-2-amine dihydrochloride (13)
Compound 13 was obtained in a similar manner to that described for the synthesis of compound 19 in Scheme 1. Yield: 600 mg (2.460 mmol, 92%); yellow solid. 1H NMR (500 MHz, CDCl3): δ 6.62 (s, 1 H), 6.22 (d, J = 1.5 Hz, 1 H), 3.50 (s, 2 H), 2.82 – 2.32 (m, 8 H), 2.27 (s, 3 H), 2.15 (s, 3 H). 13C NMR (126 MHz, CDCl3): δ 158.4, 148.9, 140.6, 119.1, 108.8, 85.4, 83.1, 55.1, 52.0, 47.6, 46.1, 20.8. HRMS (ESI) Calcd for C14H21N4 [(M+H)+]: 245.1761, found: 245.1766.
4.1.9. Synthesis of 4-methyl-6-(3-(4-methylpiperazin-1-yl)propyl)pyridin-2-amine dihydrochloride (14)
Compound 14 was obtained in a similar manner to that described for the synthesis of compound 20 in Scheme 1. The hydrogenation was performed using 10% Pd/C at 25 °C for 24 h. Yield: 472 mg (1.900 mmol, 80%); pale yellow solid. 1H NMR (500 MHz, CDCl3): δ 6.34 (s, 1 H), 6.14 (s, 1 H), 4.27 (s, 2 H), 2.73 – 2.33 (m, 11 H), 2.27 (s, 3 H), 2.18 (s, 4 H), 1.88 – 1.82 (m, 2 H). 13C NMR (126 MHz, CDCl3): δ 160.3, 158.2, 149.1, 114.4, 106.4, 58.3, 55.3, 53.3, 46.2, 36.0, 27.1, 21.0. HRMS (ESI) Calcd for C14H25N4 [(M+H)+]: 249.2074, found: 249.2078.
4.1.10. Synthesis of 6-(3-(3,3-difluoroazetidin-1-yl)prop-1-yn-1-yl)-4-methylpyridin-2-amine dihydrochloride (17)
Compound 17 was obtained in a similar manner to that described for the synthesis of compound 19 in Scheme 1. Yield: 257 mg (1.080 mmol, 28%); brown solid. 1H NMR (500 MHz, CDCl3): δ 6.69 (s, 1 H), 6.29 (s, 1 H), 4.46 (bs, 2 H), 3.77 (t, J = 12.5 Hz, 4 H), 3.62 (s, 2 H), 2.20 (s, 3 H). 13C NMR (126 MHz, CDCl3): δ 158.5, 149.2, 140.2, 119.5, 116.4 (t, J = 274.7 Hz), 109.3, 85.9, 81.3, 62.5 (t, J = 23.3 Hz), 45.1, 20.9. HRMS (ESI) Calcd for C12H14F2N3 [(M+H)+]: 238.1150, found: 238.1147.
4.1.11. Synthesis of 6-(3-(3,3-difluoroazetidin-1-yl)propyl)-4-methylpyridin-2-amine dihydrochloride (18)
Compound 18 was obtained in a similar manner to that described for the synthesis of compound 20 in Scheme 1. The hydrogenation was performed using 10% Pd/C at 25 °C for 8 h. Yield: 123 mg (0.510 mmol, 62%); brown solid. 1H NMR (500 MHz, (CD3)2SO, shows mixture of rotamers): δ (6.68 + 6.67 + 6.63 + 6.60 (s, 2 H)), 4.41 – 4.32 (m, 3 H), 3.71 (t, J = 12.5 Hz, 1 H), 3.10 – 2.99 (m, 2 H), 2.78 – 2.73 (m, 2 H), (2.28 +2.27 (s, 3 H)), 2.16 – 2.09 (m, 1 H), 1.93 – 1.87 (m, 1 H). 13C NMR (126 MHz, (CD3)2SO, shows mixture of rotamers): 156.1 (s, 1xC merged), 154.2 (s, 1xC merged, (147.7 + 147.5, s, 1xC), (118.4 (t, J = 243.8 Hz) + 114.8 (t, J = 269.4 Hz), 1xCF2), (112.8 +112.7 (s, 1xCH)), (109.4 + 109.3 (s, 1xCH)), (63.5 (t, J = 26.9 Hz, 1xCH2 merged), 54.3 (s, 1xCH2), 47.5 (t, J = 25.8 Hz, 1xCH2), 46.8 (s, 1xCH2), 43.2 (t, J = 27.1 Hz, 1xCH2), (29.0 + 28.9 (s, 1xCH2)), (24.1 + 23.7 (s, 1xCH2)), (21.6 + 21.5 (s, 1xCH3)). HRMS (ESI) Calcd for C12H18F2N3 [(M+H)+]: 242.1463, found: 242.1462.
4.2. Synthesis of 4-(6-amino-4-methylpyridin-2-yl)but-3-yn-1-yl 4-methylbenzenesulfonate (28)
Compound 28 was prepared following a reported procedure with slight modifications (Scheme 3).51 A flame-dried pressure vessel containing 6-bromo-4-methylpyridin-2-amine (21) (1.000 g, 5.350 mmol), Pd(PPh3)4 (123 mg, 0.107 mmol) and CuI (102 mg, 0.535 mmol) was filled with argon using three vacuum-argon flush cycles to remove oxygen. To this, CH3CN (18 mL, 0.25 M), Et3N (2.98 mL, 21.400 mmol), and But-3-yn-1-ol (0.506 mL, 10.700 mmol) in CH3CN (3.4 mL) were added and the resulting mixture was stirred for18 h at 120 °C. After completion of the reaction as indicated by TLC, the reaction mixture was cooled to 25 °C, filtered through Celite, and concentrated in a rotary evaporator. Purification of the crude residue was performed by flash column chromatography eluting with hexane: EtOAc (20:80, v:v) for 10 min to remove any nonpolar residues and followed by the solvent mixture (DCM:CH3OH:Et3N, 80:10:10, v:v) for another 15 min to afford 4-(6-amino-4-methylpyridin-2-yl)but-3-yn-1-ol (660 mg, 3.750 mmol, 70%) as a brown solid. 1H NMR (500 MHz, CDCl3): 6.62 (s, 1 H), 6.28 (s, 1 H), 3.80 (t, J = 6.3 Hz, 2 H), 2.66 (t, J = 6.3 Hz, 2H), 2.18 (s, 3 H). 13C NMR (126 MHz, CDCl3): 158.3, 149.3, 140.7, 119.0, 109.0, 86.3, 82.0, 60.9, 23.9, 20.9. LRMS (ESI) Calcd for C10H13N2O [(M+H)+]: 177.10, found: 178.54. To a stirred solution of 4-(6-amino-4-methylpyridin-2-yl)but-3-yn-1-ol (650 mg, 3.690 mmol) in dry DCM (20 mL) at 0 °C was added TsCl (740 mg, 3.870 mmol), 4-(Dimethylamino)pyridine (45 mg, 0.37 mmol) and Et3N (0.620 mL, 4.420 mmol). After the addition, the reaction mixture was slowly warmed to room temperature and stirred for 8 h. After completion of the reaction as indicated by TLC, the reaction mixture was diluted with 1% aqueous K2CO3 (50 mL) and extracted with DCM (2 × 20 mL). The combined organic layer was then dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography eluting with hexane: EtOAc (20:80, v:v) to EtOAc (100) to afford 28 (650 mg, 1.970 mmol, 53%) as yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.80 (d, J = 8.3 Hz, 2 H), 7.35 – 7.28 (m, 2 H), 6.63 – 6.54 (m, 1 H), 6.27 (t, J = 1.1 Hz, 1 H), 4.39 (s, 2 H), 4.16 (t, J = 7.3 Hz, 2 H), 2.77 (t, J = 7.3 Hz, 2 H), 2.43 (s, 3 H), 2.19 (s, 3 H). 13C NMR (126 MHz, CDCl3): 158.3, 149.1, 145.1, 140.5, 132.8, 130.1, 128.2, 119.2, 109.1, 82.6 (2xC), 67.5, 21.8, 21.0, 20.4. LRMS (ESI) Calcd for C17H19N2O3 [(M+H)+]: 331.11, found: 331.25.
4.2.1. Synthesis of 6-(4-(dimethylamino)but-1-yn-1-yl)-4-methylpyridin-2-amine dihydrochloride (15)
Compound 15 was prepared following a reported procedure with slight modifications (Scheme 3).51 To a stirred solution of 28 (500 mg, 1.510 mmol) and K2CO3 (1.050 g, 7.570 mmol) in anhydrous CH3CN (10 mL) was added dimethylamine (0.910 mL, 1.820 mmol, 2 M in THF) at 25 °C. The resulting heterogeneous mixture was then heated to 80 °C for 9 h. After completion of reaction as indicated by TLC, the reaction mixture was cooled to room temperature, diluted with water, and the organic layer was extracted with ether (2 × 20 mL), followed by DCM:iPrOH (9:1 v:v; 1 × 20 mL). The combined organic layer was then dried over Na2SO4 and concentrated in vacuo. Purification of the crude residue was performed by flash column chromatography eluting with hexane: EtOAc (10:90, v:v) for 10 min to remove any nonpolar residues and followed by the solvent mixture (DCM:CH3OH:Et3N, 80:10:10, v:v) for another 15 min to afford 15 (210 mg, 1.030 mmol, 68%) as a brown oil. 1H NMR (500 MHz, CDCl3): δ 6.61 (s, 1 H), 6.23 (d, 1 H), 4.42 (bs, NH2, 2 H), 2.67 – 2.50 (m, 4 H), 2.26 (s, 6 H), 2.17 (s, 3 H). 13C NMR (126 MHz, CDCl3): δ 158.3, 149.0, 141.2, 118.9, 108.6, 87.3, 81.2, 58.1, 45.3, 20.9, 18.3. HRMS (ESI) Calcd for C12H18N3 [(M+H)+]: 204.1495, found: 204.1498.
4.2.2. Synthesis of 6-(4-(dimethylamino)butyl)-4-methylpyridin-2-amine dihydrochloride (16)
Compound 16 was prepared following a reported procedure with slight modifications (Scheme 3).45 Compound 15 (130 mg, 0.640 mmol) was diluted with dry CH3OH (15 mL; degassed) and flushed with nitrogen before adding a catalytic amount of Pd/C (13 mg; 10% of the weight of the substrate). The inert atmosphere of this heterogeneous mixture was replaced with a hydrogen balloon at room temperature and continued the stirring at the same temperature for 24 h. The crude mixture was then filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The crude product was purified by reverse phase flash column chromatography eluting with CH3CN:H2O (10:90, v:v) to CH3CN:H2O (30:70, v:v) to afford 16 (82 mg, 0.40 mmol, 62%) as a yellow oil. 1H NMR (500 MHz, CDCl3): δ 6.35 (s, 1 H), 6.14 (s, 1 H), 4.30 (bs, NH2, 2 H), 2.63 – 2.50 (m, 2 H), 2.33 – 2.23 (m, 2 H), 2.20 (s, 6 H), 2.18 (s, 3 H), 1.72 – 1.58 (m, 2 H), 1.58 – 1.43 (m, 2 H). 13C NMR (126 MHz, CDCl3): δ 160.5, 158.2, 149.2, 114.4, 106.4, 59.8, 45.7, 38.1, 27.8, 27.7, 21.1. HRMS (ESI) Calcd for C12H22N3 [(M+H)+]: 208.1808, found: 208.1808.
Supplementary Material
Acknowledgments
We are grateful for the generous support from the National Institutes of Health (R35GM131788 to R.B.S., GM057353 and GM131920 to T.L.P.). We thank Radoslaw T. Chrzanowski for assistance in the MOE docking studies. This work made use of the IMSERC at Northwestern University, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633); the State of Illinois and International Institute for Nanotechnology (IIN). We also thank the beamline staff at SSRL and ALS for their assistance during the remote X-ray diffraction data collections.
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2022XXXXX
References:
- (1).Kerwin JF Jr.; Lancaster JR; Feldman PL Nitric oxide: a new paradigm for second messengers. J. Med. Chem 1995, 38, 4343–4362. [DOI] [PubMed] [Google Scholar]
- (2).Vallance P, Leiper J Blocking NO synthesis: how, where and why? Nat. Rev. Drug Discovery 2002, 1, 939–950. [DOI] [PubMed] [Google Scholar]
- (3).Vincent SR Nitric oxide: a radical neurotransmitter in the central nervous system. Prog. Neurobiol 1994, 42, 129–160. [DOI] [PubMed] [Google Scholar]
- (4).Palmer RM; Ferrige AG; Moncada S Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [DOI] [PubMed] [Google Scholar]
- (5).Ignarro LJ; Lippton H; Edwards JC; Baricos WH; Hyman AL; Kadowitz PJ; Gruetter CA Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: evidence for the involvement of S-nitrosothiols as active intermediates. J. Pharmacol. Exp. Ther 1981, 218, 739–749. [PubMed] [Google Scholar]
- (6).Bogdan C Nitric oxide and the immune response. Nat. Immunol 2001, 2, 907–916. [DOI] [PubMed] [Google Scholar]
- (7).Alderton WK, Cooper CE, Knowles RG Nitric oxide synthases: structure, function and inhibition. Biochem. J 2001, 357, 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Barañano DE; Snyder SH Neural roles for heme oxygenase: contrasts to nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 10996–11002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Esplugues JV NO as a signalling molecule in the nervous system. Br J Pharmacol 2002, 135, 1079–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang Y; Newton DC; Marsden PA Neuronal NOS: Gene Structure, mRNA Diversity, and Functional Relevance. Crit. Rev. Neurobiol 1999, 13, 21–43. [DOI] [PubMed] [Google Scholar]
- (11).Roe ND; Ren J Nitric oxide synthase uncoupling: a therapeutic target in cardiovascular diseases. Vasc. Pharmacol 2012, 57, 168–172. [DOI] [PubMed] [Google Scholar]
- (12).Karpuzoglu E; Ahmed SA Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide 2006, 15, 177–186. [DOI] [PubMed] [Google Scholar]
- (13).Hall CN; Garthwaite J What is the real physiological NO concentration in vivo? Nitric Oxide 2009, 21, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Torreilles F; Salman-Tabcheh S; Guérin M; Torreilles J Neurodegenerative disorders: the role of peroxynitrite. Brain Res. Rev 1999, 30, 153–163. [DOI] [PubMed] [Google Scholar]
- (15).Dorheim M-A; Tracey WR; Pollock JS; Grammas P Nitric oxide synthase activity is elevated in brain microvessels in Alzheimer′ s disease. Biochem. Biophys. Res. Commun 1994, 205, 659–665. [DOI] [PubMed] [Google Scholar]
- (16).Zhang L; Dawson VL; Dawson TM Role of nitric oxide in Parkinson’s disease. Pharmacol. Ther 2006, 109, 33–41. [DOI] [PubMed] [Google Scholar]
- (17).Norris PJ; Waldvogel HJ; Faull RLM; Love DR; Emson PC Decreased neuronal nitric oxide synthase messenger RNA and somatostatin messenger RNA in the striatum of Huntington’s disease. Neuronsicence 1996, 72, 1037–1047. [DOI] [PubMed] [Google Scholar]
- (18).Drechsel DA; Estévez AG; Barbeito L; Beckman JS Nitric oxide-mediated oxidative damage and the progressive demise of motor neurons in ALS. Neurotoxic. Res 2012, 22, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sims NR; Anderson MF Mitochondrial contributions to tissue damage in stroke. Neurochem. Int 2002, 40, 511–526. [DOI] [PubMed] [Google Scholar]
- (20).Ashina M Nitric oxide synthase inhibitors for the treatment of chronic tension-type headache Exp. Opin. Pharmacother 2002, 3, 395–399. [DOI] [PubMed] [Google Scholar]
- (21).Uehara T; Nakamura T; Yao D; Shi Z-Q; Gu Z; Ma Y; Masliah E; Nomura Y; Lipton SA S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [DOI] [PubMed] [Google Scholar]
- (22).Mukherjee P; Cinelli MA; Kang S; Silverman RB Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain. Chem. Soc. Rev 2014, 43, 6814–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Feng C Mechanism of Nitric Oxide Synthase regulation: electron transfer and interdomain interactions. Coord. Chem. Rev 2012, 256, 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kobayashi Y; Ikeda K; Shinozuka K; Nara Y; Yamori Y; Hattori K L-nitroarginine increases blood pressure in the rat. Clin. Exp. Pharmacol. Physiol 1991, 18, 397–399. [DOI] [PubMed] [Google Scholar]
- (25).Cinelli MA; Do HT; Miley GP; Silverman RB Inducible nitric oxide synthase: Regulation, structure, and inhibition, Med. Res. Rev 2020, 40, 158–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Southan GJ; Szabo C Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem. Pharmacol 1996, 51, 383–394. [DOI] [PubMed] [Google Scholar]
- (27).Babu BR; Griffith OW Design of isoform-selective inhibitors of nitric oxide synthase. Curr. Opin. Chem. Biol 1998, 2, 491–500. [DOI] [PubMed] [Google Scholar]
- (28).Silverman RB Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res 2009, 42, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Poulos TL; Li H Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc. Chem. Res 2013, 46, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Di L; Rong H; Feng B Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem 2013, 56, 2–12. [DOI] [PubMed] [Google Scholar]
- (31).Misra A; Ganesh S; Shahiwala A; Shah SP Drug delivery to the central nervous system: a review, J Pharm Pharm Sci 2003, 6, 252–73. [PubMed] [Google Scholar]
- (32).Rankovic Z CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem 2015, 58, 2584–2608. [DOI] [PubMed] [Google Scholar]
- (33).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mikitsh JL; Chacko A-M Pathways for Small Molecule Delivery to the Central Nervous System across the Blood-Brain Barrier. Perspect Medicin Chem, 2014, 6, 11–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wager TT; Chandrasekaran RY; Hou X; Troutman MD; Verhoest PR; Villalobos A; Will Y Defining Desirable Central Nervous System Drug Space through the Alignment of Molecular Properties, in Vitro ADME, and Safety Attributes. ACS Chem. Neurosci, 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Veber DF; Johnson SR; Cheng H-Y; Smith BR; Ward KW; Kopple KD Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem 2002, 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- (37).Do HT; Wang H-Y; Li H; Chreifi G; Poulos TL; Silverman RB Improvement of Cell Permeability of Human Neuronal Nitric Oxide Synthase Inhibitors Using Potent and Selective 2-Aminopyridine-Based Scaffolds with a Fluorobenzene Linker. J. Med. Chem 2017, 60, 9360–9375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38) <j/>.Do HT; Li H; Chreifi G; Poulos TL; Silverman RB Optimization of Blood−Brain Barrier Permeability with Potent and Selective Human Neuronal Nitric Oxide Synthase Inhibitors Having a 2‑Aminopyridine Scaffold. J. Med. Chem 2019, 62, 2690–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ji H; Stanton BZ; Igarashi J; Li H; Martásek P; Roman LJ; Poulos TL; Silverman RB Minimal pharmacophoric elements and fragment hopping, an approach directed at molecular diversity and isozyme selectivity. Design of selective neuronal nitric oxide synthase inhibitors. J. Am. Chem. Soc 2008, 130, 3900–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kang S; Li H; Tang W; Martásek P; Roman LJ; Poulos TL; Silverman RB 2-Aminopyridines with a truncated side chain to improve human neuronal nitric oxide synthase inhibitory potency and selectivity. J. Med. Chem 2015, 58, 5548–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Labby KJ; Xue F; Kraus JM; Ji H; Mataka J; Li H; Martásek P; Roman LJ; Poulos TL; Silverman RB Intramolecular hydrogen bonding: a potential strategy for more bioavailable inhibitors of neuronal nitric oxide synthase. Bioorg. Med. Chem 2012, 20, 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Xue F; Li H; Delker SL; Fang J; Martásek P; Roman LJ; Poulos TL; Silverman RB Potent, highly selective, and orally bioavailable gem-difluorinated monocationic inhibitors of neuronal nitric oxide synthase. J. Am. Chem. Soc 2010, 132, 14229–14238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Silverman RB; Lawton GR; Ranaivo HR; Chico LK; Seo J; Watterson DM Effect of potential amine prodrugs of selective neuronal nitric oxide synthase inhibitors on blood−brain barrier penetration. Bioorg. Med. Chem 2009, 17, 7593–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Mukherjee P; Li H; Sevrioukova I; Chreifi G; Martásek P; Roman LJ; Poulos TL; Silverman RB Novel 2,4-disubstituted pyrimidines as potent, selective, and cell-permeable inhibitors of neuronal nitric oxide synthase. J. Med. Chem 2015, 58, 1067–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Cinelli MA; Li H; Chreifi G; Poulos TL; Silverman RB Nitrile in the hole: discovery of a small auxiliary pocket in neuronal nitric oxide synthase leading to the development of potent and selective 2-aminoquinoline inhibitors. J. Med. Chem 2017, 60, 3958–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Nitric Oxide Synthase Inhibitors That Interact with Both Heme Propionate and Tetrahydrobiopterin Show High Isoform Selectivity. Kang S; Tang W; Li H; Chreifi G; Martásek P; Roman LJ; Poulos TL; Silverman RB J. Med. Chem 2014, 57, 4382–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang H-Y; Qin Y; Li H; Roman LJ; Martásek P; Poulos TL; Silverman RB Potent and selective human neuronal nitric oxide synthase inhibition by optimization of the 2-aminopyridine based scaffold with a pyridine linker. J. Med. Chem 2016, 59, 4913–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sonogashira K; Tohda Y; Hagihara N A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett 1975, 16, 4467–4470. [Google Scholar]
- (49).Feuerstein M; Doucet H; Santelli M Sonogashira reaction of heteroaryl halides with alkynes catalysed by a palladium-tetraphosphine complex. J. Mol. Catal. Chem 2006, 256, 75–84. [Google Scholar]
- (50).Angle SR; Henry RM Synthesis of enantiopure Indolizidine alkaloids from a-amino acids: Total synthesis of (−)-Indolizidine 167B, J. Org. Chem 1997, 62, 8549–8552. [DOI] [PubMed] [Google Scholar]
- (51).Wagner M; Schepmann D; Ametamey SM; Wünsch B Modification of the 4-phenylbutyl side chain of potent 3-benzazepine-based GluN2B receptor antagonists, Bioorg. Med. Chem 2019, 27, 3559–3567. [DOI] [PubMed] [Google Scholar]
- (52).Hevel JM; Marletta MA [25] Nitric-oxide synthase assays. Methods Enzymol 1994, 233, 250–258. [DOI] [PubMed] [Google Scholar]
- (53).Di L; Kerns EH; Fan K; McConnell OJ; Carter GT High throughput artificial membrane permeability assay for blood− brain barrier. Eur. J. Med. Chem 2003, 38, 223–232. [DOI] [PubMed] [Google Scholar]
- (54).Gürdere MB; Budak Y; Ceylan M Synthesis of 2,3,4,5-tetraphenylfuran, -thiophene and -pyrrole from toluene, Asian J. Chem 2008, 20, 1425–1430. [Google Scholar]
- (55).Das S; Ko N; Lee E; Kim SE; Lee BC. Stereoselective three-component cascade synthesis of α-substituted 2,4-dienamides from gem-difluorochloro ethanes, Chem. Commun, 2019, 55, 14355–14358. [DOI] [PubMed] [Google Scholar]
- (56).McPherson CG, Caldwell N, Jamieson C, Simpson I, Watson AJB Amidation of unactivated ester derivatives mediated by trifluoroethanol, Org. Biomol. Chem, 2017, 15, 3507–3518. [DOI] [PubMed] [Google Scholar]
- (57).Terry CA; Fernández M-J, Gude L; Lorente A; Grant KB Physiologically relevant concentrations of NaCl and KCl increase DNA photocleavage by an N-substituted 9-aminomethylanthracene dye, Biochemistry, 2011, 50, 10375–10389. [DOI] [PubMed] [Google Scholar]
- (58).Di Porzio A; Galli U; Amato J; Zizza P; Iachettini S; Iaccarino N; Marzano S; Santoro F; Brancaccio D; Carotenuto A; De Tito S; Biroccio A; Pagano B; Tron GC; Randazzo A Synthesis and Characterization of Bis-Triazolyl-Pyridine Derivatives as Noncanonical DNA-Interacting Compounds. Int. J. Mol. Sci 2021, 22, 11959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.