

Abstract
In recent years, increasing attention has been paid to titin (TTN) and its mutations. Heterozygous TTN truncating variants (TTNtv) increase the risk of a cardiomyopathy. At the same time, TTNtv and few missense variants have been identified in patients with mainly recessive skeletal muscle diseases. The pathogenic mechanisms underlying titin‐related diseases are still partly unknown. Similarly, the titin mechanical and functional role in the muscle contraction are far from being exhaustively clarified. In the last few years, several animal models carrying variants in the titin gene have been developed and characterized to study the structural and mechanical properties of specific titin domains or to mimic patients' mutations. This review describes the main animal models so far characterized, including eight mice models and three fish models (Medaka and Zebrafish) and discusses the useful insights provided by a thorough characterization of the cell‐, tissue‐ and organism‐phenotypes in these models.
Keywords: animal models, congenital myopathy, dilated cardiomyopathy, mdm, medaka, mice, titin, zebrafish
1. INTRODUCTION
Titin is well‐known as the largest sarcomeric protein expressed in skeletal and cardiac muscle. 1 It plays a crucial structural and functional role in sarcomeres. 2 , 3 , 4
In the last few years, an increasing number of inherited myopathies and cardiac disorders associated with titin (TTN) variants have been identified. 5 These disorders differ with a very large spectrum regarding inheritance (dominant or recessive), age at onset, progression and pattern of affected muscles. 5
Heterozygous carriers of titin‐truncating variants (TTNtv) affecting exons constitutively expressed in heart have an increased risk of adult‐onset dilated cardiomyopathy (DCM). 6 , 7 , 8 A secondary genetic or non‐genetic hit is, however, usually required to develop the cardiac disease. 9 The pathomechanism underlying titin‐related cardiomyopathies is still unclear: a reduced expression of wild‐type protein and the stable expression of truncated titin protein have been recently reported analysing myocardial tissue samples from patients with titin‐related DCM. 10 , 11
Biallelic mutations, mainly TTNtv with few non‐truncating variants, cause skeletal muscle diseases with or without a cardiac involvement. 5 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 The increasing number of patients diagnosed with a recessive titinopathies is facilitating the delineation of genotype–phenotype correlations. 20 , 21 Finally, non‐truncating variants in two specific exons (344 and 364) cause distinct, well‐characterized, phenotypes (HMERF and TMD/LGMD2, respectively). 22 , 23 , 24
At the same time, spontaneous and genetically engineered animal models bearing variants in the titin gene have been characterized. This review aims to describe the main animal models so far studied, including eight mice models and three fish models (Medaka and Zebrafish) (Table 1).
TABLE 1.
Titin models described in this review
| Name | Animals | Mutation | Phenotype | References |
|---|---|---|---|---|
| mdm | Mice (spontaneus) | Deletion and a LINE‐1 element integration within I‐band region (between the N2A and the proximal PEVK regions) | Muscle dystrophy | Garvey et al. 42 ; Powers et al. 47 ; Tahir et al. 48 ; Hessel et al. 49 |
| Ttn Δ30‐38 (IG‐KO) | Mice (induced) | Deletion of 9 Ig‐like domains in the proximal I‐band segment | Cardiac hypertrophy; kyphosis and smaller soleus and diaphragm muscles. | Chung et al. 50 ; Buck et al. 52 |
| TtnΔ112‐158 | Mice (induced) | Deletion of 47 PEVK exons (corresponding to the human exons 112–158) | Longitudinal hypertrophy | Brynnel et al. 55 |
| Ttn Δ49 (N2B KO) | Mice (induced) | Deletion of the N2B specific region | Impaired diastolic function. | Radke et al. 56 ; Radke et al. 57 |
| Ttn Δ219‐225 (PEVK‐KO) | Mice (induced) | Deletion of 7 PEVK exons (corresponding to human exons 219–225 or 220–226 using the current numbering) | Impaired diastolic function. Changes in muscle contractility. | Van der Pijl et al. 60 |
| Ttn ΔIAjxn | Mice (induced) | Deletion of 14 Ig‐like and Fn3 domains (corresponding to human exons 252–270 in the current numbering) in the I‐band/A‐band (IA) junction | Exercise intolerance and a left ventricle hypertrophy. | Granzier et al. 58 |
| FINmaj | Mice (induced) | 11‐bp deletion/insertion mutation in the last exon | Mild to severe muscle dystrophy. Dilated cardiomyopathy in homozygous mice. | Charton et al. 64 |
| ΔMex5 | Mice (induced) | Deletion of second last exon (human exon 363 using the current numbering) | Muscle dystrophy and dilated cardiomyopathy. | Charton et al. 65 |
| Pickwick m171 | Zebrafish (induced) | Nonsense variant in the N2B region | Dilated cardiomyopathy | Xu et al. 71 |
| runzel | Zebrafish (induced) | Point mutation in the N2A region | Muscle dystrophy | Steffen et al. 72 |
| nsh | Medaka (spontaneus) | Point mutation in the Medaka exon 204 (ENSORLT00000022736), corresponding to human exon 349 (using the current numbering) | Hypertrophic cardiomyopathy | Higashikuse et al. 69 |
2. TITIN: ROLE AND IMPORTANCE
Titin is giant protein that resides within the sarcomere in the striated muscle and heart. 1 It spans the entire I‐and A‐bands of the sarcomere, connecting the Z‐disc end of the actin filament with the tip of the myosin filament, and running bound to the surface of the myosin thick filament, through the A‐band to the M‐line at the centre of the sarcomere. 2
The structural role of titin in the muscle fibres is well characterized (Figure 1). Given its high number of interactions with other sarcomeric proteins, titin is a central hub that controls many of the mechanical properties of the sarcomere at rest and during contraction. 2 , 25 In addition, Titin is also important for sarcomere formation, mechanosensing and signal transduction. 25 , 26 , 27 , 28
FIGURE 1.
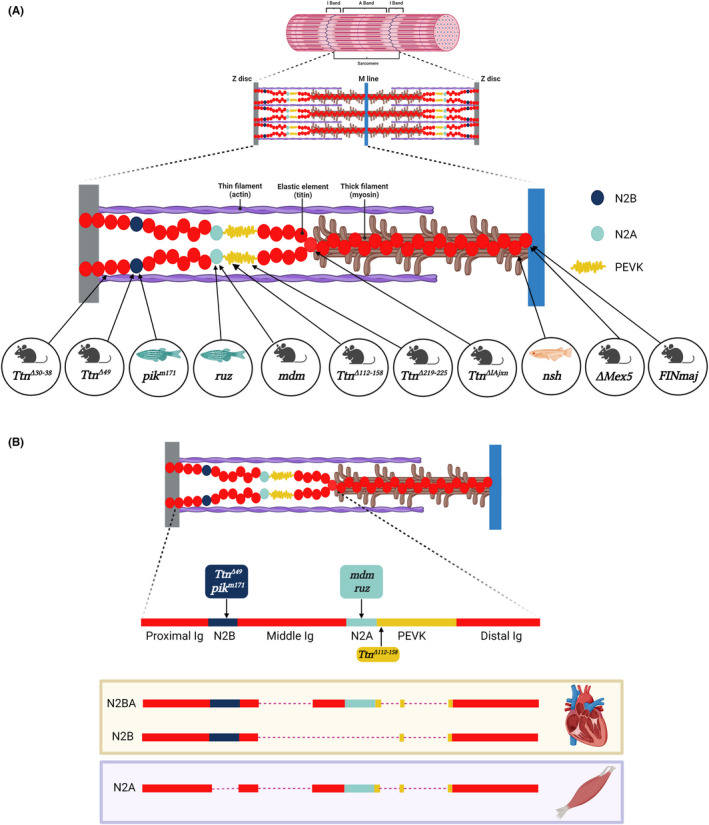
Schematic representation of titin localization into sarcomere and of titin cardiac and skeletal muscle main isoforms. (A) Two titin protein (red) spans each half‐sarcomeres, connecting the Z‐disc (grey) end of the actin thin filament (purple) with the tip of the myosin thick filament (brown), and running bound to the surface of this, up to the M‐line (blue) at the centre of the sarcomere. The Titin represented here is a theoretical isoform, in which all different domains fully expressed, to permit a correct localization in a unique figure of all the animal model mutation described in this review. (B) The main skeletal muscle isoform contains the N2A element (mutated in the mdm mice and in the ruz zebrafish model). The Titin cardiac isoforms include either the sole N2B element (mutated in the mice Ttn Δ49 and in the medaka pik m171 ), or both the N2A and N2B elements. The N2B isoforms also lack part of the Middle Ig domains and include a shorter PEVK (lacking those exons deleted in the Ttn Δ112–158 mice). The image is created with Biorender.
The N‐terminal portion of titin interacts with several structural and signalling proteins like nebulin, α‐actinin, telethonin, actin binding proteins and filamin C. 29 , 30 , 31 I‐band titin includes mainly repetitive immunoglobulin (Ig) domains and the so‐called PEVK region, which is named after the proline (P), glutamate (E), valine (V) and lysine (K) rich content. This region can extend when mechanical force is applied, providing the extensible or “spring‐like” function of titin. 1 , 4 , 32 A‐band titin region is a non‐extensible region that interacts with myosins. It is composed of repetitive Ig domains and fibronectin domains. The M‐band contain the serine/threonine kinase domain, a central hub for many signalling ligands, and interacts with myomesin and obscurin originating a scaffold that link thick filaments at the M‐line of the sarcomere. 33 , 34 , 35 , 36
3. TTN GENE AND TRANSCRIPTS
Human TTN gene includes 364 exons (363 coding and one additional non‐coding exon). 1 , 37 Titin transcripts undergo alternative splicing that can theoretically produce more than 1 million isoforms. 1 , 15 , 38 In particular, the I‐band region of Titin is prone to alternative splicing events, which regulate the inclusion or removal of Ig‐domains, the length of the elastic PEVK element 32 and also the expression of two important signalling hubs, the N2A and N2B elements. 1 , 2 , 4
The presence of these two elements (N2A and N2B) is likewise used to classify titin isoforms in three main categories. In particular, the N2A isoforms (containing only the N2A element) are expressed in the skeletal muscles. The two mainly cardiac isoforms are classified by the presence of only N2B element (N2B isoforms), or both the elements (N2BA isoforms). 3 , 39 The N2A and N2BA isoforms typically include a higher number of Ig‐domains and a longer PEVK than the N2B isoforms.
Moreover, three further isoforms (Novex‐1, Novex‐2, Novex‐3) have been reported. Novex‐1 and Novex‐2 are similar to the N2B isoforms, except for the inclusion of an isoform‐specific exon (exon 45 in Novex‐1 and exon 46 in Novex‐2). The Novex‐3 isoform exclusively contains the N‐terminal region of titin, because of an alternative stop codon in the exon 48. A recently discovered isoform, Cronos, lacks the N‐terminal and it is expressed in foetal and adult cardiac tissue. 40 , 41
An inferred transcript (metatranscript) including all the coding exons (except the exon 48) is also commonly used (NM_001267550.1). Several TTN exons from this metatranscript are not included in the aforementioned canonical post‐natal isoforms. These exons are referred to as metatranscript only exons and they are thought to be expressed during embryonic development, although some of them still have a low but detectable post‐natal expression. 14 , 38 Alternative splicing events (ASE) occurring in different developmental and physiological states or in anatomically different muscles result in longer or shorter titin isoforms with a variable expression. 42 , 43 , 44
4. MUSCULAR DYSTROPHY WITH MYOSITIS (MDM)
The muscular dystrophy with myositis mice (mdm) is by far the most studied titin animal model, widely used to characterize titin physiology in skeletal muscle. This spontaneous model contains a complex rearrangement (a deletion and a LINE‐1 element integration within titin I‐band region between the N2A and the proximal PEVK regions). The rearrangement leads to an aberrantly spliced transcript and, thereby, to a protein lacking 83 amino acids including the deletion of the N2A calpain‐3 binding region. 45 Homozygous mice (mdm/mdm) are smaller compared with their wild‐type siblings and show kyphosis, rigid and limited gait, associated with a severe muscle degeneration and necrotic fibres with phagocytosis, that had been initially mis‐interpreted as inflammation hallmarks (thereby, the name myositis). They develop an early onset progressive muscular dystrophy. Mechanical experiments have showed a significantly impaired muscle contractile function already at 6 weeks of age. 46 The mice die at 2 months of age, 45 due to the loss of diaphragmatic contractile function leading to respiratory failure.
The pathomechanism underlying the observed muscle phenotype has been delineated by several more recent studies. In 2016, Powers and colleagues suggested that, in psoas muscle, titin stiffness enhances the force by stabilizing the sarcomere during force development. This mechanism is lost in mdm mice in which the skipped domains in the N2A and in the proximal PEVK regions lead to a decrease in titin stiffness in an active sarcomere. 47 More recent studies on soleus confirmed that, during activation, titin is the only known sarcomeric non‐cross bridge viscoelastic element that interacts at high affinity with Ca2+ and actin. 48 , 49 This interaction is mediated by the regions deleted in mdm mice. 48 , 49
5. PROXIMAL IMMUNOGLOBULIN‐LIKE DOMAINS KNOCK‐OUT (IG‐KO OR Ttn Δ30 –38)
The immunoglobulin‐like domains knock‐out mouse model (IG‐KO) has been generated deleting nine Ig‐like domains in the proximal I‐band segment of titin. Overall, 783 amino acids encoded by exons corresponding to human exons 30–38, are deleted. This model, generated by Chung and colleagues in 2013, 50 partly recapitulates the phenotype observed in patients with heart failure with preserved ejection fraction (HFpEF). HFpEF patients and the IG‐KO model show a cardiac hypertrophy due to an increase of titin‐related diastolic stiffness. 51 Moreover, the IG‐KO mice also display kyphosis and smaller soleus and diaphragm muscles. 52
In these IG‐KO mice, the observed increased stiffness in heart is only due to the deletion of the nine Ig‐like domains in the proximal I‐band of titin since no compensatory changes in TTN cardiac isoform expression have been detected. 50 On the contrary, in the soleus of the IG‐KO mice, the I‐band deletion seems to alter the splicing pattern of the PEVK region resulting in smaller and much stiffer isoforms. Thereby, a significant increase of passive stress and a decrease of the developed active force is observed in the IG‐KO whole soleus muscle. 52 The noted high activity level of RBM20, a well‐known TTN splicing factor, could explain the presence of smaller isoforms due to the modified PEVK splicing pattern. 53 , 54
6. Ttn Δ112 –158 MICE
The Ttn Δ112–158 mouse model carries a deletion of 47 PEVK exons (corresponding to the human exons 112–158). The almost 75% reduction of the PEVK segment length observed in the soleus was expected to induce an increase of the passive stiffness at the levels of the sarcomere, single fibres and whole muscle. 55 However, a compensatory mechanism with a longitudinal hypertrophy and a consequently increased number of sarcomeres is observed. This readjustment process shortens the in vivo sarcomere length working range and thereby decreases the passive stiffness. The shortening of the thin filaments minimizes the negative effect of this readjustment on active force. The reduced weight of the TtnΔ112–158 mice is probably due to the high energetic cost of the aforementioned compensatory process that increases the number of sarcomeres by nearly 30%. 55
7. N2B KNOCK OUT MICE (N2B‐KO)
The N2B knock out mouse model (N2B‐KO) carries a deletion of the N2B specific exon (corresponding to the human exon 49). 56 This single exon deletion does not alter the reading frame and leads to normal inclusion of the mutant slightly truncated protein in the sarcomere. The N2B‐KO mice survive to adulthood and are also fertile. They show a smaller heart compared to the wild‐type controls with normal ejection volumes. Decreased levels were observed of the cardiac specific protein FHL2, a member of the LIM domain gene family. Although the mice show normal systolic function, diastolic wall stress is increased and accompanied with impaired diastolic function, due to slightly reduced sarcomere length and increased passive tension of the cardiomyocytes. 56 The increased passive tension is probably caused by the expression of the shortened titin isoform, without additional alterations in TTN expression or splicing profile. 57
8. Ttn Δ219 –225 MICE OR PEVK‐KO
Seven PEVK exons (corresponding to human exons 219–225 or 220–226 using the current numbering) are deleted in the Ttn Δ219–225 mice (also known as PEVK‐KO). These exons are constitutively expressed in the main cardiac titin isoform N2B. 58
The PEVK region is a major source of titin elasticity in the range of the physiological sarcomere length. 59 As expected, excision of seven exons in this region leads to an increase of the titin‐based passive tension, causing diastolic dysfunction in the adult mice. Mutant mice show both longitudinal and cross‐sectional hypertrophy due to an increased number of sarcomeres in series and in parallel. PEVK‐KO mice have hypertrophied hearts with upregulated expression of FHL proteins, proving that the dysregulation of the FHL2 levels is associated with cardiac hypertrophy. 58
Further studies on skeletal muscle have revealed an unexpected increase (53% in soleus and 62% in EDL) of passive tension in TtnΔ219–225 mutants, in contrast to the predicted increment of 17%. Co‐expression of two titin isoforms: the TtnΔ219–225 N2A titin (larger isoform) and N2A2 (smaller isoform) is the likely explanation. The N2A2 isoform seems to be a splicing adaption leading to a reduced expression of elastic elements. Changes in muscle contractility have also been observed, implicating the PEVK region in the regulation of both passive and active muscle mechanics as well as of muscle plasticity. 60
9. Ttn ΔIAJXN
The Ttn ΔIAjxn mouse model has been generated to remove 14 immunoglobulin‐like and fibronectin type III domains (exons 251–269 corresponding to exons 252–270 in the current human numbering) located in the I‐band/A‐band (IA) junction. 61 This titin region is not a part of the extensible spring zone of titin and the deletion does not change thick filament length. However, it moves the attachment point of the titin Ig proximal region away from the Z disk (∼65 nm). Consequently, the mice heart shows the exercise intolerance and a left ventricle hypertrophy, mimicking the heart failure with preserved ejection fraction observed in some titin patients. 61 , 62
10. FINmaj MICE
The human FINmaj mutation is a 11‐bp deletion/insertion mutation located in the last exon of TTN. 23 The variant results in a 4‐amino acid exchange in the C‐terminal Ig domain M10 of the M‐band titin. FINmaj is a founder mutation in the Finnish population with a frequency close to 1/2000 individuals and causes the late onset distal myopathy named tibial muscular dystrophy (TMD). 23
Mice carrying the FINmaj mutation well recapitulate the phenotype observed in patients. 63 Heterozygous mice develop a late‐onset mild progressive myopathy with dystrophic changes visible from 9 months of age in three muscles, tibialis anterior, biceps femoris and quadriceps. No significant functional impairment is, however, detected. Homozygous mice show a more severe phenotype with an earlier onset (dystrophic features are visible in soleus already after 1 month of age). A dilated cardiomyopathy with heart muscle fibrosis and left ventricular dysfunction is only observed in the homozygous mice.
In patients as well as in the model mice, the mutation causes a pathological in cis cleavage of the last C‐terminal domains of the mutated titin protein. A calpain‐3 binding site is located in proximity in the cleaved off region and dysregulation of the proteolytic activity seems to play a crucial role in the pathology. 64
11. ΔMEX5 MICE
ΔMex5 mice carry a homozygous deletion of Ttn second last exon (exon 363), an alternatively splicing exon encoding the insertion sequence 7 (is7). 65 Interestingly, the exon 363 usage is highly variable in anatomically different muscles. Skeletal muscles undergoing aerobic exercises usually show a higher expression of is7+ isoforms. 38 , 66 Similarly, heart main isoform expresses the 363 exon. 7 The c‐terminal part of titin interacts with numerous partners and is7 is included in a binding site for calpain 3, an enzyme almost absent in the cardiac muscle. 67
The total absence of is7+ isoforms in the homozygous mice results in a dystrophic phenotype in those muscles where the exon is usually expressed (e.g. the soleus muscle). 65 The is7+ isoforms are highly expressed in heart, and the is7+ sequence, encoded by exon 363, is believed to be essential for proper cardiac function in mice, explaining the observed dilated cardiomyopathy in ΔMex5 mice. 68
Although the absence of is7 has a direct consequence only on the interaction with calpain‐3, which is not expressed in heart, it is highly probable that M‐line titin acts as a protein scaffold and, thereby, the absence of is7 may impact other interactions, partly explaining the observed pathology.
12. FISH MODELS (AN INNOVATIVE TOOL FOR THE INVESTIGATION OF TITIN MUTATION)
Zebrafish (Danio rerio) and Medaka (Oryzias latipes) are two fish models in which titin has a structure similar to the human one Table 2. These animal models are, therefore, emerging as alternative cheaper models to study titin function and to characterize the effect of large‐scale mutations on the cardiac and skeletal muscle phenotype. Teleost fish underwent a whole genome duplication event which led, in zebrafish, to the formation of two copies of titin, ttna and ttnb. In contrast, medaka has only one titin gene, which is thought to be composed of 219 exons and shows evolutionary highly conserved regions. 69 Because of the evolutionary distance between medaka and zebrafish (about 110 million years ago) and the consequent subfunctionalization of gene copies, the repertoire of the duplicated genes is different, and the two models are unlikely to show redundant phenotypes. Thus, the medaka fish model, due of its reduced genomic duplication and unique phenotypes attributable to specific mutations, is a promising model for future studies of titin mutations.
TABLE 2.
Other zebrafish titin models (described in Santiago et al. 70 )
| Name | Gene | Mutation Location |
|---|---|---|
| ttna_N2A, ttna_N2B, ttnb_N2A and ttnb_N2B morphants (MO) | ttna, ttnb | N2A, N2B |
| Herzschlag (Hel tg287 ) | ttna | I/A junction |
| n1, n2, n3 | ttna | Z‐disc, proximal I‐band, mid‐I band |
| c1, c2, c3 | ttna | Proximal, mid and distal A band |
| Ttn.2 xu064 | ttna | Z‐disc |
| Ttn.2 xu065 | ttna | A‐band |
| Ttn.1 xu066 | ttnb | Z‐disc |
| Ttn.1 xu067 | ttnb | A‐band |
| ttn xu068 | ttna | Z‐disc & A‐band |
| ttn xu069 | ttna ttnb | Z‐disc |
| ttn xu070 | ttna ttnb | A‐band |
| ttn xu071 | ttna ttnb | Z‐disc A‐band |
| ttna tv | ttna | A‐band |
A recent review published by Celine F. Santiago et al. 70 has discussed in depth fifteen different zebrafish titin mutated models created to characterize the cardiac structure and function and to study genetic and molecular mechanisms in muscle disease (Table 2). This was possible thanks to new tools of imaging and “‐omics” technologies for the cardiac phenotyping in small animal models. Conversely, to date just one medaka model has been reported with a titin mutation. Herein, we report two zebrafish models, which carry titin mutation linked to a cardiac (Pickwick m171 ) and a skeletal (runzel) myopathy, and the medaka cardiac model nsh.
13. ZEBRAFISH TITIN MODELS
13.1. Pickwickm171
Pickwick m171 (pik m171 ) is a recessive lethal mutation discovered in zebrafish during a large‐scale genetic screen. 71 The mutation found was a T > G transversion, located in the unique sequence of the cardiac N2B exon. The pik m171 heart develops normally during the first 3 days but is poorly contractile due to abnormal sarcomeres. 71 This mutation in the N2B exon causes the generation of a stop codon and a resulting truncated protein devoid of N2B and M‐line regions. The consequence is sarcomeric disassembly with thinner myofibrils and impaired systolic function. Using a morpholino approach to confirm the genotype–phenotype relation, Xu and colleagues disrupted the expression of the cardiac N2B exon in morphants, which led to the same phenotype as the pik m171 mutants. 71 These results indicate the requirement of titin for the correct assembly of the sarcomeres and resembles the cardiac phenotype of patients with dilated cardiomyopathy (DCM). 71
13.2. Runzel
Runzel (ruz) is a mutation in the N2A region, isolated after the n‐ethyl‐n‐nitrosourea (ENU) screening, and causing a recessive muscle dystrophy. 72 ruz zebrafish mutants show decreased muscle organization already at 3 days post fertilization (dpf) and low mobility with reduced swimming ability at 5–7 dpf, with a general sarcomeric disassembly and poorly organized myofibrils. 72 While heterozygotes did not show any abnormalities in mobility, in homozygosity this mutation appears to be lethal. Morpholino injections produced a phenocopy of the ruz mutant and, together with RNA and protein analyses in ruz mutants, confirmed that the ruz mutation is located in the skeletal titin locus. Also, ruz mutants displayed a progressive reduction of titin isoforms throughout developmental stages highlighting the lack of titin isoforms switching. 72
14. TITIN NSH MEDAKA
A point mutation causing an abnormal heart phenotype was reported after an n‐ethyl‐n‐nitrosourea (ENU)‐induced mutation screening in medaka Oryzyas latipes. 69 The mutant was named non‐spring heart (nsh). Positional cloning placed the point mutation in the exon 204 (ENSORLT00000022736, corresponding to human exon 349), that encodes an Ig‐domain near the MURF (muscle‐specific ring finger protein 1)‐binding region. The nsh‐homozygote embryos survived up to 8 days post fertilization (dpf) but, eventually, they did not hatch. 69 These embryos displayed hypertrophic myocardial walls, which caused slow or clogged blood flow. The cardiomyocytes showed loose arrangement of the contractile filaments, broken thick and thin filaments and non‐paralleled Z‐discs. Also, nsh homozygotes displayed a smaller body, in which the skeletal muscles had an abnormal sarcomeric structure with disrupted myofibrils. On the molecular level, these homozygote mutants exhibit differential expression of the cardiac stretch marker bnp (higher in the ventriculum and lower the atrium) compared with the wild‐type controls and an increased proportion of N2B isoforms probably causing less elasticity and increased passive stiffness of the cardiac myofibrils. Furthermore, the nsh adult heterozygotes showed M‐line disassembly in myofibrils linked to abnormal sarcomeric structures. Nevertheless, their size, movement, lifespan and fertility were not affected. Both these phenotypes are due to the Ig‐domain missense mutation located in the transition zone between the A‐band and the M‐line of titin, which leads to an increased binding of titin to MURF1 and an enhanced titin degradation by ubiquitination. Similar phenotypes with M‐line disassembly were found in hypertrophic cardiomyopathy (HCM) patients also associated with TTN mutations in the A‐band/M‐line transition zone. Thus, the impaired interaction of titin and MURF1 highlights a novel mechanism associated with the pathogenesis of HCM and suggests inhibition of MURF1 function as a possible therapy for HCM patients. 69
15. CONCLUSIONS AND PERSPECTIVES
To date, most TTN animal models have been developed to characterize the structural and mechanical properties of specific titin domains that are physiologically or clinically relevant. Except the FINmaj mouse (recapitulating the human TMD/LGMD R10 phenotypes) and the nsh Medaka models (carrying a missense variant also found in patients), 63 , 69 the genetic defects in the remaining animal models do not mirror the mutations identified in titinopathy patients so far. However, the thorough characterization of the cell‐, tissue‐ and organism‐phenotypes in these models has provided very useful insights:
Even large heterozygous but, probably, even homozygous in‐frame deletions of titin specific regions are relatively well tolerated, although still resulting in a cardiac and/or skeletal muscle phenotype somehow resembling the observed TTN‐related clinical entities;
These large deletions often activate a compensatory mechanism resulting in the expression of shorter and stiffer isoforms;
The main titin splicing factor, RBM20, could be a potential therapeutic target and its downregulation could lead to the expression of longer and more compliant titin isoforms (reverting the mechanism induced by the large deletions).
New animal models carrying mutations resembling those found in patients would be useful to characterize the pathomechanisms underlying human diseases.
At the same time, skinned fibres from patients´ muscle biopsies could be used for mechanical experiments although technical aspects (e.g. sample collection, amount and quality of patients‐derived material, the need for healthy sex‐ and aged‐matched control muscles) may hamper a relevant outcome of these experiments.
Finally, 3D culture models from human cells are emerging as a potential alternative approach other than vertebrate model organisms. 73 3D cultures mimic the structure and the functionality of human muscle tissues. Although currently the physiological properties of the 3D cultures are not yet comparable with those found in vivo in tissues, they may soon become a very useful tool for muscle disease modelling. 74 Since each of these approaches presents issues that can compromise its effectiveness, their synergistic use could be optimal to understand how a specific titin mutation leads to the development of a given clinical phenotype.
AUTHOR CONTRIBUTIONS
Matteo Marcello: Conceptualization (equal); data curation (equal); writing – original draft (lead); writing – review and editing (equal). Viviana Cetrangolo: Data curation (equal); writing – original draft (equal); writing – review and editing (supporting). Marco Savarese: Conceptualization (equal); data curation (equal); funding acquisition (equal); writing – original draft (supporting); writing – review and editing (lead). Bjarne Udd: Conceptualization (equal); funding acquisition (equal); writing – review and editing (equal).
FUNDING INFORMATION
M.S. work is supported by Academy of Finland (grant #339437 ´Improving the clinical interpretation of sequence variants´) and Sydäntutkimussäätiö (´Titin truncating variants and dilated cardiomyopathy´). B.U. work is supported by Academy of Finland and European Joint Programme on Rare Diseases (‘Improved diagnostic output in large sarcomeric genes IDOLS‐G’).
CONFLICT OF INTEREST
The authors declare no competing interests.
Marcello M, Cetrangolo V, Savarese M, Udd B. Use of animal models to understand titin physiology and pathology. J Cell Mol Med. 2022;26:5103‐5112. doi: 10.1111/jcmm.17533
Marco Savarese and Bjarne Udd equally contributed to this work
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Bang ML, Centner T, Fornoff F, et al. The complete gene sequence of titin, expression of an unusual approximately 700‐kDa titin isoform, and its interaction with obscurin identify a novel Z‐line to I‐band linking system. Circ Res. 2001;89(11):1065‐1072. [DOI] [PubMed] [Google Scholar]
- 2. Linke WA, Kulke M, Li H, et al. PEVK domain of titin: an entropic spring with actin‐binding properties. J Struct Biol. 2002;137(1–2):194‐205. [DOI] [PubMed] [Google Scholar]
- 3. Linke WA, Rudy DE, Centner T, et al. I‐band titin in cardiac muscle is a three‐element molecular spring and is critical for maintaining thin filament structure. J Cell Biol. 1999;146(3):631‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Linke WA, Stockmeier MR, Ivemeyer M, Hosser H, Mundel P. Characterizing titin's I‐band Ig domain region as an entropic spring. J Cell Sci. 1998;111(11):1567‐1574. [DOI] [PubMed] [Google Scholar]
- 5. Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3(3):293‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gerull B, Gramlich M, Atherton J, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30(2):201‐204. [DOI] [PubMed] [Google Scholar]
- 7. Roberts AM, Ware JS, Herman DS, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7(270):270‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ware JS, Li J, Mazaika E, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374(3):233‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cannata A, Merlo M, Dal Ferro M, et al. Association of titin variations with late‐onset dilated cardiomyopathy. JAMA Cardiol. 2022;7(4):371‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fomin A, Gartner A, Cyganek L, et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med. 2021;13(618):eabd3079. [DOI] [PubMed] [Google Scholar]
- 11. McAfee Q, Chen CY, Yang Y, et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13(618):eabd7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Savarese M, Maggi L, Vihola A, et al. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol. 2018;75(5):557‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Savarese M, Johari M, Johnson K, et al. Improved criteria for the classification of titin variants in inherited skeletal myopathies. J Neuromuscul Dis. 2020;7(2):153‐166. [DOI] [PubMed] [Google Scholar]
- 14. Oates EC, Jones KJ, Donkervoort S, et al. Congenital titinopathy: comprehensive characterization and pathogenic insights. Ann Neurol. 2018;83(6):1105‐1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bryen SJ, Ewans LJ, Pinner J, et al. Recurrent TTN metatranscript‐only c.39974‐11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. 2020;41(2):403‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23(4):980‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evila A, Palmio J, Vihola A, et al. Targeted next‐generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol Neurobiol. 2017;54(9):7212‐7223. [DOI] [PubMed] [Google Scholar]
- 18. Salih MA, Hamad MH, Savarese M, et al. Exome sequencing reveals novel TTN variants in Saudi patients with congenital titinopathies. Genet Test Mol Biomarkers. 2021;25(12):757‐764. [DOI] [PubMed] [Google Scholar]
- 19. Perrin A, Juntas Morales R, Chapon F, et al. Novel dominant distal titinopathy phenotype associated with copy number variation. Ann Clin Transl Neurol. 2021;8(9):1906‐1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Savarese M, Vihola A, Oates EC, et al. Genotype‐phenotype correlations in recessive titinopathies. Genet Med. 2020;22(12):2029‐2040. [DOI] [PubMed] [Google Scholar]
- 21. Perrin A, Juntas Morales R, Rivier F, et al. The importance of an integrated genotype‐phenotype strategy to unravel the molecular bases of titinopathies. Neuromuscul Disord. 2020;30(11):877‐887. [DOI] [PubMed] [Google Scholar]
- 22. Palmio J, Leonard‐Louis S, Sacconi S, et al. Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol. 2019;266(3):680‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal‐muscle protein titin. Am J Hum Genet. 2002;71(3):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C‐terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord. 2008;18(12):922‐928. [DOI] [PubMed] [Google Scholar]
- 25. Gerull B. The rapidly evolving role of titin in cardiac physiology and cardiomyopathy. Can J Cardiol. 2015;31(11):1351‐1359. [DOI] [PubMed] [Google Scholar]
- 26. Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L. The giant protein titin's role in cardiomyopathy: genetic, transcriptional, and post‐translational modifications of TTN and their contribution to cardiac disease. Front Physiol. 2019;10:1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kruger M, Linke WA. The giant protein titin: a regulatory node that integrates myocyte signaling pathways. J Biol Chem. 2011;286(12):9905‐9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kontrogianni‐Konstantopoulos A, Ackermann MA, Bowman AL, Yap SV, Bloch RJ. Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol Rev. 2009;89(4):1217‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z‐disk: two types of titin interactions lead to an asymmetrical sorting of alpha‐actinin. EMBO J. 1998;17(6):1614‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gregorio CC, Trombitas K, Centner T, et al. The NH2 terminus of titin spans the Z‐disc: its interaction with a novel 19‐kD ligand (T‐cap) is required for sarcomeric integrity. J Cell Biol. 1998;143(4):1013‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Witt CC, Burkart C, Labeit D, et al. Nebulin regulates thin filament length, contractility, and Z‐disk structure in vivo. EMBO J. 2006;25(16):3843‐3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gautel M, Lehtonen E, Pietruschka F. Assembly of the cardiac I‐band region of titin/connectin: expression of the cardiac‐specific regions and their structural relation to the elastic segments. J Muscle Res Cell Motil. 1996;17(4):449‐461. [DOI] [PubMed] [Google Scholar]
- 33. Fukuzawa A, Lange S, Holt M, et al. Interactions with titin and myomesin target obscurin and obscurin‐like 1 to the M‐band: implications for hereditary myopathies. J Cell Sci. 2008;121(11):1841‐1851. [DOI] [PubMed] [Google Scholar]
- 34. Sarparanta J, Blandin G, Charton K, et al. Interactions with M‐band titin and calpain 3 link myospryn (CMYA5) to tibial and limb‐girdle muscular dystrophies. J Biol Chem. 2010;285(39):30304‐30315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lange S, Xiang F, Yakovenko A, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308(5728):1599‐1603. [DOI] [PubMed] [Google Scholar]
- 36. Gautel M, Castiglione Morelli MA, Pfuhl M, Motta A, Pastore A. A calmodulin‐binding sequence in the C‐terminus of human cardiac titin kinase. Eur J Biochem. 1995;230(2):752‐759. [PubMed] [Google Scholar]
- 37. Savarese M, Valipakka S, Johari M, Hackman P, Udd B. Is Gene‐Size an Issue for the Diagnosis of Skeletal Muscle Disorders? J Neuromuscul Dis. 2020;7(3):203‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Savarese M, Jonson PH, Huovinen S, et al. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018;8(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Labeit S, Lahmers S, Burkart C, et al. Expression of distinct classes of titin isoforms in striated and smooth muscles by alternative splicing, and their conserved interaction with filamins. J Mol Biol. 2006;362(4):664‐681. [DOI] [PubMed] [Google Scholar]
- 40. Zou J, Tran D, Baalbaki M, et al. An internal promoter underlies the difference in disease severity between N‐ and C‐terminal truncation mutations of titin in zebrafish. Elife. 2015;4:e09406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zaunbrecher RJ, Abel AN, Beussman K, et al. Cronos titin is expressed in human cardiomyocytes and necessary for normal sarcomere function. Circulation. 2019;140(20):1647‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greaser ML, Guo W, Bharmal SJ, Esbona K. Titin diversityalternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity‐‐alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garvey SM, Rajan C, Lerner AP, Frankel WN, Cox GA. The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle‐specific domain of titin. Genomics. 2002;79(2):146‐149. [DOI] [PubMed] [Google Scholar]
- 46. Lopez MA, Pardo PS, Cox GA, Boriek AM. Early mechanical dysfunction of the diaphragm in the muscular dystrophy with myositis (Ttnmdm) model. Am J Physiol Cell Physiol. 2008;295(5):C1092‐C1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Powers K, Nishikawa K, Joumaa V, Herzog W. Decreased force enhancement in skeletal muscle sarcomeres with a deletion in titin. J Exp Biol. 2016;219(Pt 9):1311‐1316. [DOI] [PubMed] [Google Scholar]
- 48. Tahir U, Monroy JA, Rice NA, Nishikawa KC. Effects of a titin mutation on force enhancement and force depression in mouse soleus muscles. J Exp Biol. 2020;223(2):jeb197038. [DOI] [PubMed] [Google Scholar]
- 49. Hessel AL, Monroy JA, Nishikawa KC. Non‐cross bridge viscoelastic elements contribute to muscle force and work during stretch‐shortening cycles: evidence from whole muscles and permeabilized fibers. Front Physiol. 2021;12:648019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chung CS, Hutchinson KR, Methawasin M, et al. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128(1):19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Borbely A, Falcao‐Pires I, van Heerebeek L, et al. Hypophosphorylation of the stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104(6):780‐786. [DOI] [PubMed] [Google Scholar]
- 52. Buck D, Smith JE 3rd, Chung CS, et al. Removal of immunoglobulin‐like domains from titin's spring segment alters titin splicing in mouse skeletal muscle and causes myopathy. J Gen Physiol. 2014;143(2):215‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guo W, Schafer S, Greaser ML, et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18(5):766‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li S, Guo W, Dewey CN, Greaser ML. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013;41(4):2659‐2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brynnel A, Hernandez Y, Kiss B, et al. Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. Elife. 2018;7:e40532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Radke MH, Peng J, Wu Y, et al. Targeted deletion of titin N2B region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci U S A. 2007;104(9):3444‐3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Radke MH, Badillo‐Lisakowski V, Britto‐Borges T, et al. Therapeutic inhibition of RBM20 improves diastolic function in a murine heart failure model and human engineered heart tissue. Sci Transl Med. 2021;13(622):eabe8952. [DOI] [PubMed] [Google Scholar]
- 58. Granzier HL, Radke MH, Peng J, et al. Truncation of titin's elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105(6):557‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trombitas K, Freiburg A, Centner T, Labeit S, Granzier H. Molecular dissection of N2B cardiac titin's extensibility. Biophys J. 1999;77(6):3189‐3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van der Pijl RJ, Hudson B, Granzier‐Nakajima T, et al. Deleting titin's C‐terminal PEVK exons increases passive stiffness, alters splicing, and induces cross‐sectional and longitudinal hypertrophy in skeletal muscle. Front Physiol. 2020;11:494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Granzier HL, Hutchinson KR, Tonino P, et al. Deleting titin's I‐band/A‐band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A. 2014;111(40):14589‐14594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Butler J, Fonarow GC, Zile MR, et al. Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail. 2014;2(2):97‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Charton K, Daniele N, Vihola A, et al. Removal of the calpain 3 protease reverses the myopathology in a mouse model for titinopathies. Hum Mol Genet. 2010;19(23):4608‐4624. [DOI] [PubMed] [Google Scholar]
- 64. Charton K, Sarparanta J, Vihola A, et al. CAPN3‐mediated processing of C‐terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet. 2015;24(13):3718‐3731. [DOI] [PubMed] [Google Scholar]
- 65. Charton K, Suel L, Henriques SF, et al. Exploiting the CRISPR/Cas9 system to study alternative splicing in vivo: application to titin. Hum Mol Genet. 2016;25(20):4518‐4532. [DOI] [PubMed] [Google Scholar]
- 66. Kolmerer B, Olivieri N, Witt CC, Herrmann BG, Labeit S. Genomic organization of M line titin and its tissue‐specific expression in two distinct isoforms. J Mol Biol. 1996;256(3):556‐563. [DOI] [PubMed] [Google Scholar]
- 67. Hu LY, Ackermann MA, Kontrogianni‐Konstantopoulos A. The sarcomeric M‐region: a molecular command center for diverse cellular processes. Biomed Res Int. 2015;2015:714197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Biquand A, Spinozzi S, Tonino P, et al. Titin M‐line insertion sequence 7 is required for proper cardiac function in mice. J Cell Sci. 2021;134(18):jcs258684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Higashikuse Y, Mittal N, Arimura T, et al. Perturbation of the titin/MURF1 signaling complex is associated with hypertrophic cardiomyopathy in a fish model and in human patients. Dis Model Mech. 2019;12(11):dmm041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Santiago CF, Huttner IG, Fatkin D. Mechanisms of TTNtv‐related dilated cardiomyopathy: insights from zebrafish models. J Cardiovasc Dev Dis. 2021;8(2):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xu X, Meiler SE, Zhong TP, et al. Cardiomyopathy in zebrafish due to mutation in an alternatively spliced exon of titin. Nat Genet. 2002;30(2):205‐209. [DOI] [PubMed] [Google Scholar]
- 72. Steffen LS, Guyon JR, Vogel ED, et al. The zebrafish runzel muscular dystrophy is linked to the titin gene. Dev Biol. 2007;309(2):180‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Romagnoli C, Iantomasi T, Brandi ML. Available in vitro models for human satellite cells from skeletal muscle. Int J Mol Sci. 2021;22(24):13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dessauge F, Schleder C, Perruchot MH, Rouger K. 3D in vitro models of skeletal muscle: myopshere, myobundle and bioprinted muscle construct. Vet Res. 2021;52(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.