

Abstract
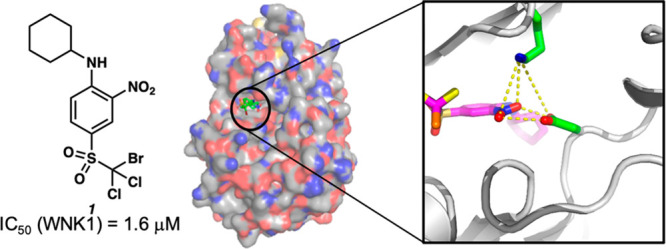
With No lysine (K) [WNK] kinases are structurally unique serine/threonine protein kinases that have therapeutic potential for blood pressure regulation and cancer. A novel class of trihalo-sulfone compounds was identified by high-throughput screening. Trihalo-sulfone 1 emerged as an effective inhibitor of WNK1 with an IC50 value of 1.6 μM. Herein, we define chemical features necessary for inhibition of WNK1 using chemical synthesis and X-ray crystallography. Analogues that probed the role of specific functional groups to the inhibitory activity were synthesized. X-ray structures of trihalo-sulfone 1 and a second trihalo-sulfone 23 bound to WNK1 revealed active site binding to two of the three previously defined canonical inhibitor binding pockets as well as a novel binding site for the trihalo-sulfone moiety. The elucidation of these novel interaction sites may allow for the strategic design of even more selective and potent WNK inhibitors.
Keywords: WNK1, kinase inhibitor, structure−activity-relationship, small-molecule, halogen bond, ATP binding pocket
With No lysine (K) [WNK] kinases are cytoplasmic serine/threonine protein kinases, named for their unique placement of a catalytic lysine residue in subdomain I rather than the more typical subdomain II (Figure 1).1 These enzymes are involved in transepithelial ion transport, cell volume control, and cell motility,2−5 making WNKs potential drug targets in several disease indications. Mice homozygous for WNK1 deletion are embryonically lethal, whereas mice with heterozygous deletion exhibit reduced blood pressure. Therefore, WNK1 has been identified as an essential kinase in hypertension and hypotension.6 WNK3 is also important in blood pressure regulation under certain conditions,7 thus establishing the WNK family as promising drug targets for hypertension. WNK kinases are also implicated in cancers of the breast, lung, ovary, and brain.8−12 A recent transposon insertional analysis identified WNK1 as a proto-oncogenic signature gene for triple negative breast cancer.13 WNK1 depletion suppresses the metastatic driver tyrosine kinase AXL.14 WNK3 knockout mice have reduced edema in a stroke model.15,16 Novartis previously reported on nanomolar pan-WNK and WNK1 selective inhibitors. WNK463, the pan-WNK inhibitor, is ATP competitive, whereas WNK476 and other analogues are allosteric, binding in a site adjacent to helix C.17,18 However, these compounds proved to be too toxic for use as antihypertensive agents. We hypothesized that the discovery of novel chemical scaffolds for the inhibition of WNK1 could provide additional opportunities to target this enzyme for hypertension as well as more acute clinical indications such as cancer.
Figure 1.

X-ray crystal structure of the mammalian serine/threonine protein kinase WNK1. (A) WNK1 (PDB 6CN9). Ribbon diagram mainly blue, with the activation loop in red. Closeup of active site highlighting the unique lysine in WNK1, the Mg2+ binding aspartic acid (oxygens red), and an inhibitory chloride ion in 6CN9 (magenta). (B) Key characteristics of WNK1. (C) Chemical structure of WNK463, a previously reported pan-inhibitor of WNK.
To identify new chemical scaffolds for WNK1 inhibition, we performed a high-throughput screen of >200 000 structurally diverse small molecules (unpublished results). Herein, we present the structural characterization and chemical interrogation of a novel trihalo-sulfone 1 that inhibits WNK1 with a IC50 value of 1.6 μM (Figure 2). X-ray crystallographic studies show that this compound adopts a binding mode in part overlapping with the reported pan-WNK inhibitor (WNK463).17,19 A small library of analogues was developed based on systematic functional modifications of parent trihalo-sulfone 1. Assays of the synthesized analogues reveal the importance of functional groups to the potency of inhibition. Interactions between 1 and a second trihalo-sulfone 23 with WNK1 revealed a unique binding mode for this class of inhibitors.
Figure 2.

Chemical structure and systematic functional group modification of WNK1 inhibitor, trihalo-sulfone 1.
Trihalo-sulfone 1 is a novel scaffold for the inhibition of WNK1 with unique structural features, including a bromodicholoro sulfone moiety and an ortho-nitro aniline. Systematic functional group modifications were designed to generate a focused library of analogues stemming from trihalo-sulfone 1, as shown in Figure 2. Modifications highlighted in blue include the use of aromatic, heterocyclic, and tertiary amines. The absence of the nitro group attached to the core scaffold was also explored. Highlighted in red are modifications encompassing the sulfone and halogen moieties. Chemical modifications to the structure were envisioned to probe the impact of each functional group on the inhibitory activity.
A synthetic pathway for trihalo-sulfone 1 was designed to include halogenated intermediates that could serve as branch points for diversification (Figure 3). The key intermediate 6 was prepared through the alkaline reduction of 2 using sodium sulfite to afford 3 in 71% yield. Sodium salt 3 was then converted into dichloromethyl-4-chlorophenyl sulfone 4 through a biphasic reaction with chloroform and water in an alkaline media in 41% yield. Sulfone 4 was identified as the first linchpin intermediate of the synthetic sequence, allowing access to brominated and nonbrominated analogues. Analogue 4 underwent nitration using a nitric acid and sulfuric acid mixture to obtain sulfone 12 in quantitative yield. A subsequent nucleophilic aromatic substitution with aliphatic amine 9 furnished product 20 in 73% yield. Alternatively, bromination of sulfone 4 was performed in the presence of LiHMDS and NBS to yield 5 in 58% yield. Bromodichloromethyl-4-chlorophenyl sulfone 5 was nitrated with a mixture of nitric acid and sulfuric acid to afford 6 as a white solid in 93% yield after trituration. Compound 6 served as a second lynchpin intermediate, allowing for the use of various aliphatic and aromatic amines in a nucleophilic aromatic substitution. Adduct 29 was synthesized through chlorination of intermediate 4 by employing N-chlorosuccinimide (NCS) as a chlorine source and LiHMDS as a base in THF to obtain 27 in 47% yield, followed by standard nitration conditions to afford 28 in 61% yield and last a nucleophilic aromatic substitution using cyclohexylamine to access 29 in 63% yield.
Figure 3.
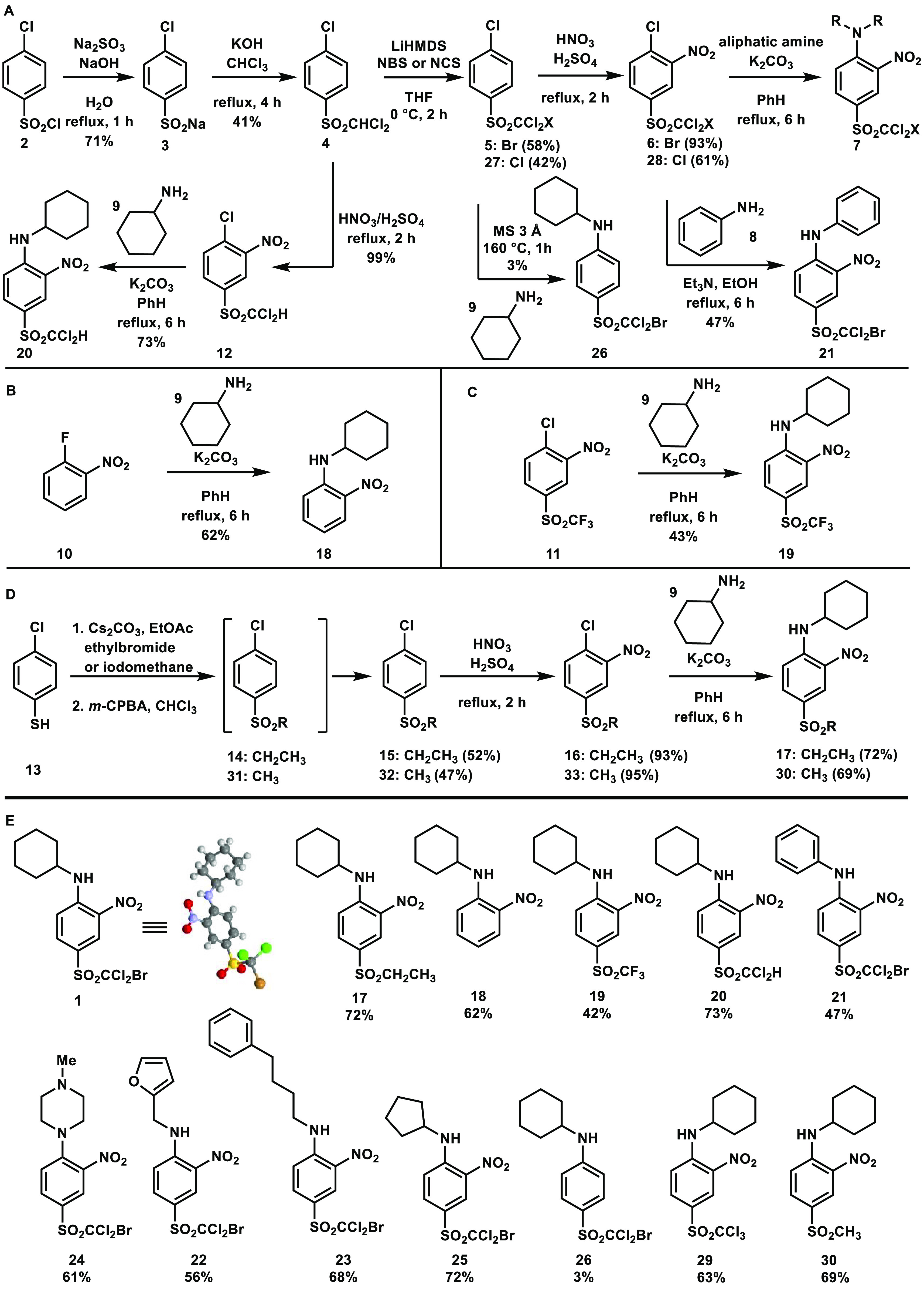
Synthesis of 1 and analogues 17–26, 29, 30.
The synthetic pathway shown in Figure 3A served as an efficient route for the synthesis of most analogues. A few trihalo-sulfones required a different design due to starting material availability. Figure 3B depicts the synthesis of analogue 18. While 2-chloronitrobenzene was explored first, 2-fluoronitrobenzene 10 proved advantageous in the SNAr reaction with cyclohexyl amine 9. The ortho nitro group stabilized the resulting negative charge in the Meisenheimer complex as analogue 18 was accessed from commercially available 10 in one step in 62% yield. Figure 3C shows the synthesis of 19 from commercially available aryl chloride 11 in 43% yield. Last, Figure 3D illustrates analogue 17 and 30, which were designed to prove the effect of an ethyl or methyl group in place of the trihalogen functional group. Upon deprotonation of 4-chlorothiophenol 13 with a mild base, ethyl bromide served as the electrophile in a nucleophilic substitution followed by a tandem m-CPBA oxidation of the sulfide intermediate 14 to furnish the sulfone 15 in 52% yield. Standard nitration conditions provided access to intermediate 16 in 93% yield. Sulfone 16 was then transformed into desired analogue 17 (46% yield) via nucleophilic aromatic substitution. Analogue 30 was accessed through the exact same route of conditions but employing methyl iodide instead of ethyl bromide in the nucleophilic substitution step.
The commercial coupled ATP depletion assay Kinase-Glo (Promega) was used as an initial in vitro biochemical screen of WNK1 inhibition, and the results are summarized in Table 1. Kinase-Glo dose–response curves are given in the Supporting Information (SI), Figure S1. Data suggested that removal of the bromine atom (20) or replacement of the halogens for a saturated alkyl functionality (17, 30) are detrimental, resulting in diminished potency. It was hypothesized that the introduction of fluorine atoms could increase the potency of trihalo-sulfone 1. Fluorine often increases the activity of initial hits by causing changes in basicity, polarity, and lipophilicity.20,21 Nonetheless, incorporation of a new trifluoro or trichloro group (19, 29) proved pernicious. The bromodichloro moiety was confirmed as a key structural element when analogue 18 was designed with a complete deletion of it, rendering the analogue inactive. This modification suggested that both bromine and chlorine atoms are vital for an effective inhibitor–protein complex, either by participating in bonding interactions with the protein amino acid scaffold or by impacting conformational preferences of the inhibitor in the binding pocket.
Table 1. IC50 Characterization against Active WNK1 via Kinase-Glo and32P Radiometric Assaysa.

Each IC50 value averages three independent experiments.
Other modifications included the elimination of the nitro component (26) and changes to the N-substituent (21–25). The elimination of the nitro group shut down the inhibition potency completely, while modifications to the N-cyclohexyl moiety were better tolerated (see Table 1). Incorporation of a longer aliphatic chain with increased mobility and foldability (23) decreased the activity by ∼4-fold, and so did decreasing the ring size from a 6-membered ring to a 5-membered ring (24). Although a decrease in activity was evident, these modifications did not completely inactivate the inhibitor as observed with other structural changes. Analogue 21 introduced planarity and altered acid/base properties, with an aromatic ring instead of a cyclohexyl moiety, which resulted in reduced potency. Additionally, heterocyclic moieties such as a furan ring were investigated with analogue 22, which led to a decrease in potency by 3-fold. The decrease in activity of analogues 21–25 suggests the importance of the boat conformation of (1) in the active pocket (vide infra) because a reduced inhibition potency was observed with all analogues incapable of adapting this conformation. Furthermore, N-methyl piperazine was selected as a modification to replace the N-cyclohexyl group because this would examine the importance of the NH bond in 1. Nucleophilic displacement in the aromatic substitution with N-methyl piperazine as a nucleophilic source resulted in a tertiary amine (25) instead of a secondary amine, completely incapable of hydrogen bonding interactions. Unsurprisingly, the inhibitor lost all potency, confirming the deletion of the NH bond as an unfavorable modification.
Inhibitors showing the highest potency were selected for further evaluation in a radiometric assay with [γ-32P] ATP (Table 1, SI, Figure S2). This biochemical assay was necessary when using low enzyme concentrations and allowed for a more precise IC50 quantification. Trihalo-sulfone 1 showed the highest potency overall with a 1.6 μM IC50 value for WNK1. Trihalo-sulfones 22 and 23 exhibited higher potency when compared to their initial Kinase-Glo assay report, albeit not as a potent as 1. We performed cell-based assays on trihalo-sulfone 1 in the MDAMB231 breast cancer cell line. Activation of the endogenous substrate OSR1 was measured (SI, Figure S3C). In triplicated experiments, we found that trihalo-sulfone 1 inhibits endogenous OSR1 phosphorylation with IC50 of 4.3 μM. Cells were viable up to 12 μM.
Trihalo-sulfones 1 and 23 were tested for specificity against 50 kinases by Eurofins Inc. (France). The Eurofins screen contains kinases belonging to all five classes of protein kinases.25 In this screen, compound 1 exhibited ∼20% inhibition of WNK1 and WNK3 and similar inhibition of four kinases in diverse classes. Compound 23 exhibited ∼20% inhibition of WNK1 and WNK2 and showed even greater inhibition strength toward JNK2 and JNK3 (Jun N-terminal kinases 2 and 3) (SI, Figure S3). In our Kinase-Glo assay, trihalo-sulfone 1 is 10-fold more potent to WNK1 than WNK3 and trihalo-sulfone 23 is a better inhibitor of WNK3 (SI, Table S2).
X-ray crystallographic studies of WNK1 in complex with trihalo-sulfone 1 show that the inhibitor binds to the active site near the hinge region of the kinase to a pocket comprised of mixed charges and hydrophobic groups (Figure 4A). The binding site is aligned with pockets 1 and 2 defined by Gray and co-workers22 (Figure 4B).
Figure 4.
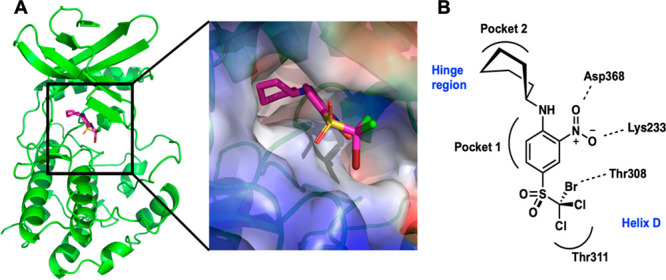
Location of the trihalo-sulfone 1 in WNK1/1. (A) The inhibitor binds in the active site to a complementary site. Drawn in PyMol. (B) Schematic of the site indicating the pockets defined by Gray22 and residues involved in inhibitor hydrogen bonding.
The complex of WNK1/S382A (kinase domain) with 1 (WNK1/1) was solved by molecular replacement using the WNK1/SA in complex with WNK463 (PDB 5DRB(17)). Crystallographic data and refinement statistics are presented for data to 2.9 Å (SI, Table S1). The electron density contoured at 0.5σ (Figure 5A) is good over the entirety of 1. Trihalo-sulfone 23 also crystallized with WNK1 (WNK1/23). WNK1/23 crystals diffracted to 2.7 Å (SI, Table S1). The electron density for 23 is complete for the o-nitro-p-bromodichloro sulfone aniline and good for the phenyl ring of the γ-phenyl propyl moiety (Figure 5B). The density for the propyl moiety is weak. The aniline rings of 1 and 23 bind in pocket 1, occupying the nucleotide of the ATP binding pocket (Figure 5A,B) and making hydrophobic interactions with Phe256. The cyclohexyl ring of 1 and the 3-phenylpropyl group of 23 bind to pocket 2. Unexpectedly, the bromodichloro sulfone moieties bind to a site not previously identified, unveiling unexplored chemical space and a novel set of interactions for a kinase–inhibitor complex. The unconventional binding site consists of two threonine residues, Thr308 and Thr311, located in helix D (Figure 5C). Thr308 forms a halogen bond with the inhibitor’s bromine atom. Binding interactions via halogen bonds occur in protein kinases.23,24 These interactions tend to involve the backbone carbonyls in the hinge region; however, our data show that the trihalo-sulfone 1 halogen binds a side chain hydroxyl group.
Figure 5.
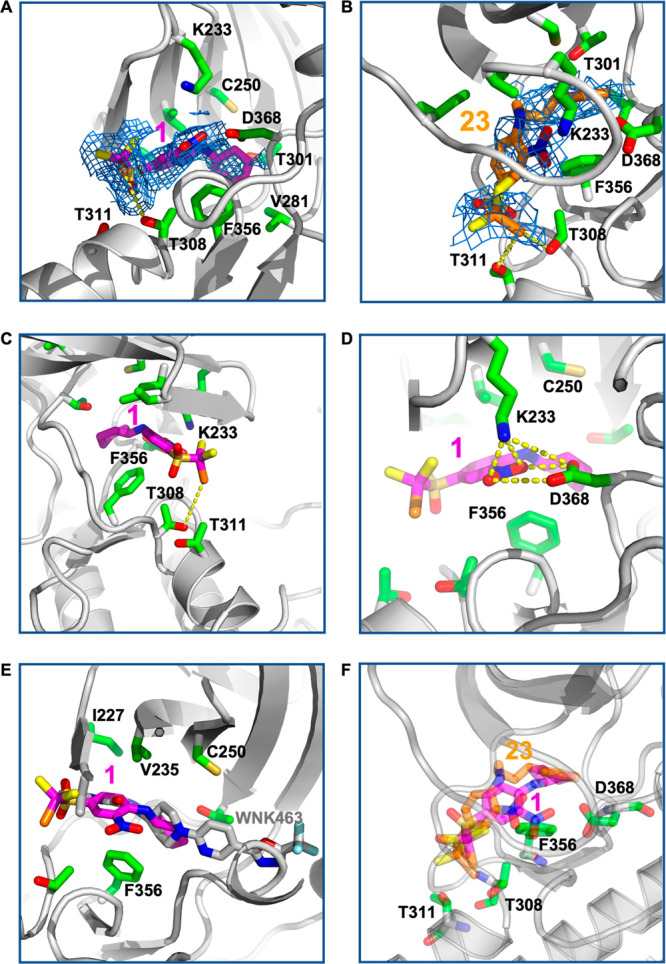
Interactions of 1 and 23 with WNK1. (A) Electron density for 1 contoured at 0.5σ. Electron density surrounding 1 is green, inhibitor carbon atoms are magenta, and protein carbon atoms are green. (B) Electron density for 23 contoured and colored similarly to (A). (C) Closeup of the trihalo-sulfone binding mode for trihalo-sulfone 1. (D) Closeup of the nitro-group interactions. (E) The WNK1/1 complex overlaid with the WNK1/WNK463 complex (PDB 5DRB(17)). (F) The WNK1/23 complex overlaid with the WNK1/1 complex.
Interactions between the nitro group of trihalo-sulfone 1 are also unusual. The nitro moiety binds to important catalytic residues, including Asp368, which acts as a Mg2+ binder. Moreover, the nitro group interacts with Lys233, a WNK-specific lysine residue, forming a buried electrostatic interaction. Even though the use of nitroarene scaffolds are often avoided due to their metabolic instability and mutagenic potential,25 this functionality has been employed in multiple FDA approved drugs, and it can be replaced with more stable isosteres. The cyclohexyl ring is not in its more stable chair conformation but has adopted a boat conformation and contacts Phe356 in b6. We hypothesize this type of spatial arrangement might render inactive all inhibitors that are unable to adopt a boat-shaped conformation. The aniline ring also makes hydrophobic contacts with Val235, Ile227, and Phe356. The binding site of trihalo-sulfone 1 partially overlaps with that of WNK463 (PDB 5DRB(17)) but is more exterior than WNK463 (Figure 5E).
Overlay of the WNK1/1 structure with WNK1/23 shows that 23 does not form as tight interactions as 1 with Asp368. This appears to be due to the larger size of the 3-phenylpropyl moiety compared with the boat-configured cyclohexyl moiety in 1 (Figure 5F). Rigidity at the back of the binding site, at residues Val281 and Thr301, may also contribute to the observed boat configuration.
Based on a hit compound from a high-throughput screening campaign of >200 000 compounds, a series of small-molecule WNK1 inhibitors was developed. This is the first report of an aniline scaffold bearing a trihalogen moiety in a WNK kinase inhibitor. Crystallographic data of WNK1/1 shows that 1 binds in the ATP binding pocket of WNK1. The binding interactions rely on the unique constellation of catalytic residues in WNK1. Furthermore, the trihalo-sulfone moiety binds to a surface pocket completely distinct from the three previously established kinase inhibitor pockets.22 Our strategic functional group modification in SAR studies revealed that all moieties in trihalo-sulfone 1 are essential for effective binding. We anticipate the elucidation of novel interaction sites may allow for the strategic design of even more selective and potent WNK inhibitors. Efforts to exploit novel binding modes in a structure-guided approach are underway.
Acknowledgments
Financial support was provided by the American Heart Association 16SA285300002 and 14GRNT20500035 (E.J.G.), Cancer Prevention and Research Institute of Texas (RP190421 to E.J.G. and U.K.T.), Welch Foundation (I-2100-20220331 to E.J.G., I-1748 to U.K.T.), W. W. Caruth, Jr. Endowed Scholarship (U.K.T.), Sloan Research Fellowship (U.K.T.), Bonnie Bell Harding Professorship in Biochemistry (U.K.T.), NIH-DK (DK110358 to E.J.G.), and NIH-NCI (T32CA124334 to M.R.). We thank Clinton Taylor and Melanie Cobb for the gOSR1 peptide, Shuguang Wu and Bruce Posner for help with HTS data collection, and Anwu Zhou and Prema Mallipeddi for HTS data analysis. Crystallographic studies were coordinated by Diana Tomchick in the UT Southwestern Structural Biology Laboratory. Results shown in this report were derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC), at the Advanced Photon Source. The SBC is operated by the U Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. We acknowledge Dr. Vincent Lynch (manager of the X-ray Diffraction Lab at UT Austin) for X-ray structural analysis of trihalo-sulfone 1. Finally, we thank our diverse group of laboratory members for creating an environment that supports our scientific endeavors.
Glossary
Abbreviations
- WNK
With No lysine
- ATP
adenosine 5′-triphosphate
- IC50
half-maximal inhibitory concentration
- LiHMDS
lithium bis(trimethylsilyl)amide
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- SNAr
nucleophilic aromatic substitution
- m-CPBA
meta-chloroperoxybenzoic acid
- JNK
Jun N-terminal kinase
- MOPS
3-morpholinopropane-1-sulfonic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00216.
Experimental details, characterization data, and spectral data (PDF)
Accession Codes
Coordinates for WNK1/1 and WNK1/23 have been deposited in the PDB (7UOS and 7UOU, respectively).
Author Contributions
§ M.R. and A.K. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Xu B.; English J. M.; Wilsbacher J. L.; Stippec S.; Goldsmith E. J.; Cobb M. H. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J. Biol. Chem. 2000, 275 (22), 16795–16801. 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- Kahle K. T.; Rinehart J.; Lifton R. P. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta 2010, 1802 (12), 1150–1158. 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Rangel S.; Gamba G.; Ramos-Mandujano G.; Pasantes-Morales H. Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch 2012, 464 (3), 317–330. 10.1007/s00424-012-1137-4. [DOI] [PubMed] [Google Scholar]
- Choe K. P.; Strange K. Evolutionarily conserved WNK and Ste20 kinases are essential for acute volume recovery and survival after hypertonic shrinkage in Caenorhabditis elegans. Am. J. Physiol Cell Physiol 2007, 293 (3), C915–C927. 10.1152/ajpcell.00126.2007. [DOI] [PubMed] [Google Scholar]
- Tu S. W.; Bugde A.; Luby-Phelps K.; Cobb M. H. WNK1 is required for mitosis and abscission. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (4), 1385–1390. 10.1073/pnas.1018567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz B. P.; Abuin A.; Ramirez-Solis R.; Richter L. J.; Piggott J.; BeltrandelRio H.; Buxton E. C.; Edwards J.; Finch R. A.; Friddle C. J.; Gupta A.; Hansen G.; Hu Y.; Huang W.; Jaing C.; Key B. W.; Kipp P.; Kohlhauff B.; Ma Z.-Q.; Markesich D.; Payne R.; Potter D. G.; Qian N.; Shaw J.; Schrick J.; Shi Z.-Z.; Sparks M. J.; Van Sligtenhorst I.; Vogel P.; Walke W.; Xu N.; Zhu Q.; Person C.; Sands A. T. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention. Proc. Natl. Acad. Sci. U. S. A. 2003, 100 (24), 14109–14114. 10.1073/pnas.2336103100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oi K.; Sohara E.; Rai T.; Misawa M.; Chiga M.; Alessi D. R.; Sasaki S.; Uchida S. A minor role of WNK3 in regulating phosphorylation of renal NKCC2 and NCC co-transporters in vivo. Biol. Open 2012, 1 (2), 120–127. 10.1242/bio.2011048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H.; Hunter C.; Smith R.; Stephens P.; Greenman C.; Bignell G.; Teague J.; Butler A.; Edkins S.; Stevens C.; et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer research 2005, 65 (17), 7591–7595. 10.1158/0008-5472.CAN-05-1855. [DOI] [PubMed] [Google Scholar]
- Jinawath N.; Vasoontara C.; Jinawath A.; Fang X.; Zhao K.; Yap K.-L.; Guo T.; Lee C. S.; Wang W.; Balgley B. M.; et al. Oncoproteomic analysis reveals co-upregulation of RELA and STAT5 in carboplatin resistant ovarian carcinoma. PloS one 2010, 5 (6), e11198. 10.1371/journal.pone.0011198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W.; Begum G.; Pointer K.; Clark P. A.; Yang S.-S.; Lin S.-H.; Kahle K. T.; Kuo J. S.; Sun D. WNK1-OSR1 kinase-mediated phospho-activation of Na+-K+-2Cl-cotransporter facilitates glioma migration. Molecular cancer 2014, 13, 31. 10.1186/1476-4598-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T.; d’Ario G.; Lamb J. R.; Martin E.; Wang K.; Tejpar S.; Delorenzi M.; Bosman F. T.; Roth A. D.; Yan P.; et al. A comprehensive characterization of genome-wide copy number aberrations in colorectal cancer reveals novel oncogenes and patterns of alterations. PloS one 2012, 7 (7), e42001. 10.1371/journal.pone.0042001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens P.; Edkins S.; Davies H.; Greenman C.; Cox C.; Hunter C.; Bignell G.; Teague J.; Smith R.; Stevens C.; et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nature genetics 2005, 37 (6), 590–592. 10.1038/ng1571. [DOI] [PubMed] [Google Scholar]
- Chen L.; Jenjaroenpun P.; Pillai A. M.; Ivshina A. V.; Ow G. S.; Efthimios M.; Zhiqun T.; Tan T. Z.; Lee S. C.; Rogers K.; Ward J. M.; Mori S.; Adams D. J.; Jenkins N. A.; Copeland N. G.; Ban K. H.; Kuznetsov V. A.; Thiery J. P. Transposon insertional mutagenesis in mice identifies human breast cancer susceptibility genes and signatures for stratification. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E2215–E2224. 10.1073/pnas.1701512114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaykumar A. B.; Jung J. U.; Parida P. K.; Dang T. T.; Wichaidit C.; Kannangara A. R.; Earnest S.; Goldsmith E. J.; Pearson G. W.; Malladi S.; Cobb M. H. WNK1 Enhances Migration and Invasion in Breast Cancer Models. Mol. Cancer Ther 2021, 20 (10), 1800–1808. 10.1158/1535-7163.MCT-21-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G.; Yuan H.; Kahle K. T.; Li L.; Wang S.; Shi Y.; Shmukler B. E.; Yang S.-S.; Lin S.-H.; Alper S. L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46 (7), 1956–1965. 10.1161/STROKEAHA.115.008939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum G.; Song S.; Wang S.; Zhao H.; Bhuiyan M. I. H.; Li E.; Nepomuceno R.; Ye Q.; Sun M.; Calderon M. J.; Stolz D. B.; St. Croix C.; Watkins S. C.; Chen Y.; He P.; Shull G. E.; Sun D. Selective knockout of astrocytic Na(+) /H(+) exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018, 66, 126–144. 10.1002/glia.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Park H. M.; Rigel D. F.; DiPetrillo K.; Whalen E. J.; Anisowicz A.; Beil M.; Berstler J.; Brocklehurst C. E.; Burdick D. A.; Caplan S. L.; Capparelli M. P.; Chen G.; Chen W.; Dale B.; Deng L.; Fu F.; Hamamatsu N.; Harasaki K.; Herr T.; Hoffmann P.; Hu Q. Y.; Huang W. J.; Idamakanti N.; Imase H.; Iwaki Y.; Jain M.; Jeyaseelan J.; Kato M.; Kaushik V. K.; Kohls D.; Kunjathoor V.; LaSala D.; Lee J.; Liu J.; Luo Y.; Ma F.; Mo R.; Mowbray S.; Mogi M.; Ossola F.; Pandey P.; Patel S. J.; Raghavan S.; Salem B.; Shanado Y. H.; Trakshel G. M.; Turner G.; Wakai H.; Wang C.; Weldon S.; Wielicki J. B.; Xie X.; Xu L.; Yagi Y. I.; Yasoshima K.; Yin J.; Yowe D.; Zhang J. H.; Zheng G.; Monovich L. Small-molecule WNK inhibition regulates cardiovascular and renal function. Nat. Chem. Biol. 2016, 12 (11), 896–898. 10.1038/nchembio.2168. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Zhang J. H.; Xie X.; Reinhardt J.; Xie A. Q.; LaSala D.; Kohls D.; Yowe D.; Burdick D.; Yoshisue H.; Wakai H.; Schmidt I.; Gunawan J.; Yasoshima K.; Yue Q. K.; Kato M.; Mogi M.; Idamakanti N.; Kreder N.; Drueckes P.; Pandey P.; Kawanami T.; Huang W.; Yagi Y. I.; Deng Z.; Park H. M. Discovery and Characterization of Allosteric WNK Kinase Inhibitors. ACS Chem. Biol. 2016, 11 (12), 3338–3346. 10.1021/acschembio.6b00511. [DOI] [PubMed] [Google Scholar]
- Yamada K.; Levell J.; Yoon T.; Kohls D.; Yowe D.; Rigel D. F.; Imase H.; Yuan J.; Yasoshima K.; DiPetrillo K.; Monovich L.; Xu L.; Zhu M.; Kato M.; Jain M.; Idamakanti N.; Taslimi P.; Kawanami T.; Argikar U. A.; Kunjathoor V.; Xie X.; Yagi Y. I.; Iwaki Y.; Robinson Z.; Park H. M. Optimization of Allosteric With-No-Lysine (WNK) Kinase Inhibitors and Efficacy in Rodent Hypertension Models. J. Med. Chem. 2017, 60 (16), 7099–7107. 10.1021/acs.jmedchem.7b00708. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sumii Y.; Shibata N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5 (19), 10633–10640. 10.1021/acsomega.0c00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni C.; Hu M.; Hu J. Good partnership between sulfur and fluorine: sulfur-based fluorination and fluoroalkylation reagents for organic synthesis. Chem. Rev. 2015, 115 (2), 765–825. 10.1021/cr5002386. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9 (1), 28–39. 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Poznanski J.; Winiewska M.; Czapinska H.; Poznanska A.; Shugar D. Halogen bonds involved in binding of halogenated ligands by protein kinases. Acta Biochim Pol 2016, 63, 203–214. 10.18388/abp.2015_1106. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Canagarajah B. J.; Boehm J. C.; Kassisa S.; Cobb M. H.; Young P. R.; Abdel-Meguid S.; Adams J. L.; Goldsmith E. J. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998, 6 (9), 1117–1128. 10.1016/S0969-2126(98)00113-0. [DOI] [PubMed] [Google Scholar]
- Nepali K.; Lee H. Y.; Liou J. P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019, 62 (6), 2851–2893. 10.1021/acs.jmedchem.8b00147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.