

Abstract

Human immunodeficiency virus type-1 (HIV-1) protease is essential for viral propagation, and its inhibitors are key anti-HIV-1 drug candidates. In this study, we discovered a novel HIV-1 protease inhibitor (compound 16) with potent antiviral activity and oral bioavailability using a structure-based drug design approach via X-ray crystal structure analysis and improved metabolic stability, starting from hit macrocyclic peptides identified by mRNA display against HIV-1 protease. We found that the improvement of the proteolytic stability of macrocyclic peptides by introducing a methyl group to the α-position of amino acid is crucial to exhibit strong antiviral activity. In addition, macrocyclic peptides, which have moderate metabolic stability and solubility in solutions containing taurocholic acid, exhibited desirable plasma total clearance and oral bioavailability. These approaches may contribute to the successful discovery and development of orally bioavailable peptide drugs.
Keywords: HIV-1 protease inhibitor, Macrocyclic peptide, Antiviral activity, Oral bioavailability, Proteolytic stability, α-Methyl amino acid, mRNA display
Acquired immunodeficiency syndrome is a chronic, potentially life-threatening condition caused by the human immunodeficiency virus (HIV). HIV infects lymphocytes (mainly expressing CD4 proteins) and macrophages, which are vital to immunity, and aggravates the disease by progressively destroying the immune system.1,2 HIV-1 protease is one of the key enzymes responsible for the propagation of infectious virus particles3 and is an important target protein for anti-HIV drugs because it plays a key role in cleaving precursor polyproteins transcribed and translated from the HIV genome.4,5 After approval of the protease inhibitor saquinavir (Figure 1) in 1995, combination therapy using three antiretroviral agents of different anti-HIV-1 drugs was initiated as the standard of care.6,7 Darunavir, approved by the Food and Drug Administration in 2006, exhibits potent antiviral activity against drug-resistant HIV-1 clinical isolates.8−10 However, since darunavir contains a peptide-like core structure that mimics the protease-catalyzed transition state, this has resulted in poor oral bioavailability and metabolic stability. Moreover, it is essential to combine darunavir with ritonavir,11 a CYP3A4 inhibitor, as a pharmacokinetic (PK) booster in the prescription of darunavir; therefore, it is necessary to pay close attention to the drug–drug interaction risk.12
Figure 1.

Approved HIV-1 protease inhibitors.
Macrocyclic peptides have emerged as a new class of drug discovery modalities and attracted much attention.13,14 Typically, macrocyclic peptides are considered more likely to acquire strong interactions with a target protein owing to their restricted molecular motion and rigidity compared to linear peptides.15 Cyclosporine, a natural macrocyclic peptide approved as an oral immunosuppressive drug, penetrates the cell membrane by masking the hydrogen bond donor (HBD) of the amide groups via intramolecular hydrogen bonding.13 On the other hand, to the best of our knowledge, only a few structure–activity relationship (SAR) studies of macrocyclic peptides for inhibition of intracellular targets or oral bioavailable derivatives have been reported.16−24 Among them, a macrocyclic hexapeptide with G protein-coupled receptor CXCR7 modulating activity derived from template-fixed β-hairpin peptidomimetics led to a potent and orally macrocyclic peptide–peptoid hybrid CXCR7 modulator by incorporating an N-linked peptoid and reducing the polarity.23
Recently, mRNA display has been used to select peptides from large libraries on the basis of their binding affinity for a target protein and has demonstrated promising results against a variety of protein targets.25−27 However, large macrocyclic peptides (8–15 mer) tend to be identified by mRNA display and are considered less favorable with respect to drug-likeness, such as cell membrane permeability and oral bioavailability. Recently, MK-0616, a new cholesterol-lowering medicine in a class of drugs called PCSK9 inhibitors, has been studied in clinical trials conducted by Merck.31 MK-0616 identified by mRNA display is a macrocyclic peptide and has been optimized for the inhibitory potency and stabilization, resulting in tricyclic peptides with picomolar potency against PCSK9. MK-0616 is an orally bioavailable PCSK9 inhibitor with permeation enhancers. So far, it has not been reported that a macrocyclic peptide identified by mRNA display has been optimized to a lead macrocyclic peptide that possesses both intracellular activity and oral bioavailability without the permeation enhancers. Herein, we describe a SAR study of macrocyclic peptides originated from mRNA display for HIV-1 protease inhibitors and optimization to deliver highly potent and orally available macrocyclic peptides.
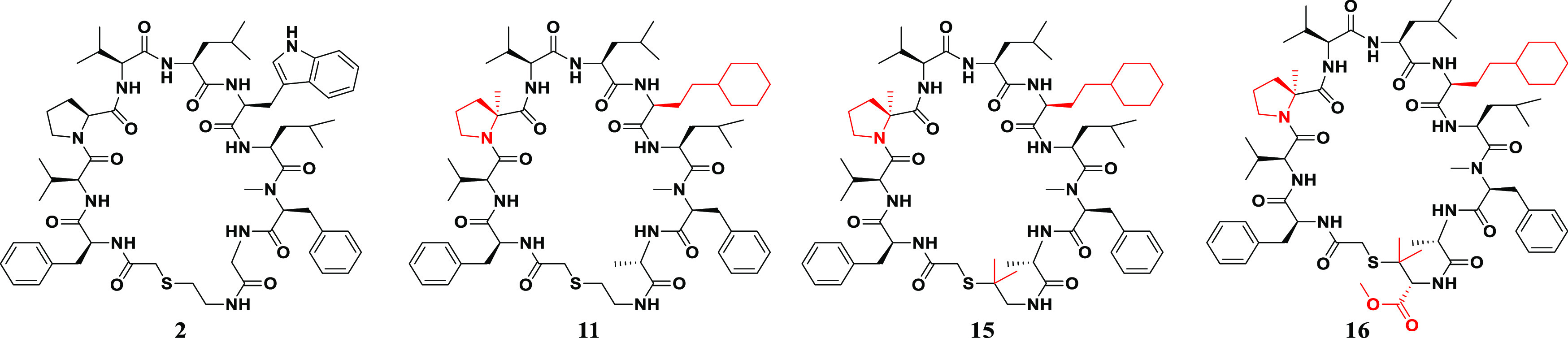
The HIV-1 protease mRNA display successfully identified hit compounds 1 and 2, which exhibited the desired enzyme inhibitory activity (IC50) (Figure 2). In terms of antiviral activity (EC50), compound 2 seemed to be unfavorable regarding membrane permeability owing to its additional HBD derived from an additional glycine unit, although the IC50 of compound 2 was better than that of compound 1. Therefore, we first selected compound 1 as the original compound and then applied the SAR findings to compound 2.
Figure 2.

Hit compounds identified by mRNA display against HIV-1 protease. aInhibitory activity of HIV-1 protease measured by fluorogenic peptide cleavage assay. bAnti-HIV-1 activity assay using the human T cell line (MT-4 cells).
Macrocyclic peptides consist of several amino acids and structural conversion sites. The X-ray crystal structure analysis of a protein–ligand complex is useful in the structure-based drug design to improve enzyme inhibitory activity. First, we used X-ray crystal structure analysis to improve the IC50 of compound 1. The cocrystal of compound 1 with a homodimer HIV-1 protease was successfully obtained by extensive screening of the conditions for cocrystallization. X-ray crystallographic analysis suggested that compound 1 forms a water-mediated interaction with aspartate residues (Asp25 and Asp25′) in the active center of the HIV-1 protease. In addition, hydrogen bonding between the backbone amide carbonyl of 1-Phe and the backbone amide (−NH) of Asp29′, the cation−π interaction between side chains of 1-Phe and Arg8, the hydrogen bonding between the backbone amide carbonyl of 5-Leu and the backbone amide (−NH) of Ile50, and CH−π interactions between side chains of 6-Trp and Ile54 and 8-MePhe and Pro81 were also confirmed. Besides, compound 1 formed four intramolecular hydrogen bonds (Figure 3).
Figure 3.

Interactions between HIV-1 protease (gray ribbon) and compound 1 (green sticks). The residues of HIV-1 protease interacting with compound 1 are highlighted as thin pink sticks and dark pink sticks. Yellow and orange dashed lines indicate hydrogen bonds and CH−π or cation−π interactions, respectively.
As the X-ray crystal structure suggested that the indole NH of 6-Trp in compound 1 was not involved in the interaction with HIV-1 protease, we expected that the removal of the indole NH of 6-Trp would not affect the IC50. As expected, N-methylation of the indole moiety of 6-Trp to 1-methylindole was tolerable (compound 3, Table 1). Furthermore, the replacement with 2-naphthalene exhibited a 12-fold potent IC50 (compound 4). The reason for the enhanced activity may be that the 2-naphthalene ring filled the lipophilic pocket around the sixth position. Although compound 4 showed remarkable inhibitory activity, there was room for improvement regarding the antiviral activity, as a significant gap between IC50 and EC50 can be seen. Typically, peptides are susceptible to hydrolysis. Since compound 4 may be decomposed by intracellular proteolytic enzymes, as indicated by its moderate stability (46%) in human liver S9 without NADPH (to exclude oxidative metabolic pathways) (Figure 4), we attempted to enhance the EC50 of the human T cell line (MT-4 cells) by improving proteolytic enzyme stability. It is known that alkylation at the α-position of a constituent amino acid in a peptide improves protease tolerance owing to steric hindrance.28 Therefore, methylation at the α-position was carried out for each amino acid involved in compound 4. To our delight, methylation at the α-position improved stability against the S9 fraction and also demonstrated antiviral activities when Pro at the third position and Val at the fourth position were converted into α-methyl l-proline (αMePro, compound 5) and α-methyl l-valine (αMeVal, compound 6), respectively (Figure 4). In contrast, α-methyl amino acids at the other positions significantly decreased both IC50 and EC50 (data not shown). In order to further improve antiviral activity, we optimized the side chain at the sixth position of compound 5. As shown in Table 2, the nonaromatic substituents enhanced the EC50 along with their improved IC50 (compounds 8 and 9), and the IC50 and EC50 of compound 7 substituted by the phenethyl moiety was comparable with those of compound 5.
Table 1. Optimization of the 6th Position of Compound 1.


Inhibitory activity of HIV-1 protease measured by the fluorogenic peptide cleavage assay.
The anti-HIV-1 activity assay using the human T cell line (MT-4 cells).
Figure 4.

αMe introduction at the 3rd and 4th positions of compound 4. aInhibitory activity of HIV-1 protease measured by the fluorogenic peptide cleavage assay. bAnti-HIV-1 activity assay using the human T cell line (MT-4 cells). cMetabolic stability using human liver S9.
Table 2. Optimization of the 6th Position of Compound 5.


Inhibitory activity of HIV-1 protease measured by the fluorogenic peptide cleavage assay.
The anti-HIV-1 activity assay using the human T cell line (MT-4 cells).
Next, the SAR findings from the SAR exploration as described above were applied to the structural optimization of compound 2, which exhibited a more potent IC50 compared with that of compound 1. As expected, compound 10, which consisted of αMePro at the third position and l-homocyclohexylalanine at the sixth position, exhibited remarkable potency with a high stability to the human liver S9 fraction (Figure 5). In addition, we optimized the amino acid at the 10th position of compound 10 and found that Ala instead of Gly gave more potent compound 11 while Leu and Phe resulted in the diminished potencies (compounds 12 and 13) (Table 3). Since thioethers are typically susceptible to oxidative metabolism, the introduction of steric hindrance adjacent to the sulfur atom was conducted to improve liver metabolic stability (Table 4). A vicinal dimethyl group at the α-position of the sulfur atom improved liver microsomal metabolic stability (compounds 14 and 15). Interestingly, compound 16 bearing a methoxycarbonyl group, which is well-known as a metabolically labile functional group, still maintained the stability of compound 15 with a slight drop of potency.
Figure 5.

αMe introduction at the 3rd position and l-homocyclohexylalanine introduction at 6th position. aInhibitory activity of HIV-1 protease measured by the fluorogenic peptide cleavage assay. bAnti-HIV-1 activity assay using the human T cell line (MT-4 cells). cMetabolic stability using human liver S9.
Table 3. Optimization at the 10th Position of Compound 10.

| compound | 10-amino acid | IC50 (nM)a | EC50 (nM)b |
|---|---|---|---|
| 10 | Gly | 0.37 | 29 |
| 11 | Ala | 0.28 | 24 |
| 12 | Leu | 2.30 | 62 |
| 13 | Phe | 2.32 | 56 |
Inhibitory activity of HIV-1 protease measured by the fluorogenic peptide cleavage assay.
The anti-HIV-1 activity assay using the human T cell line (MT-4 cells).
Table 4. Optimization of the Thioether Linker of Compound 11.

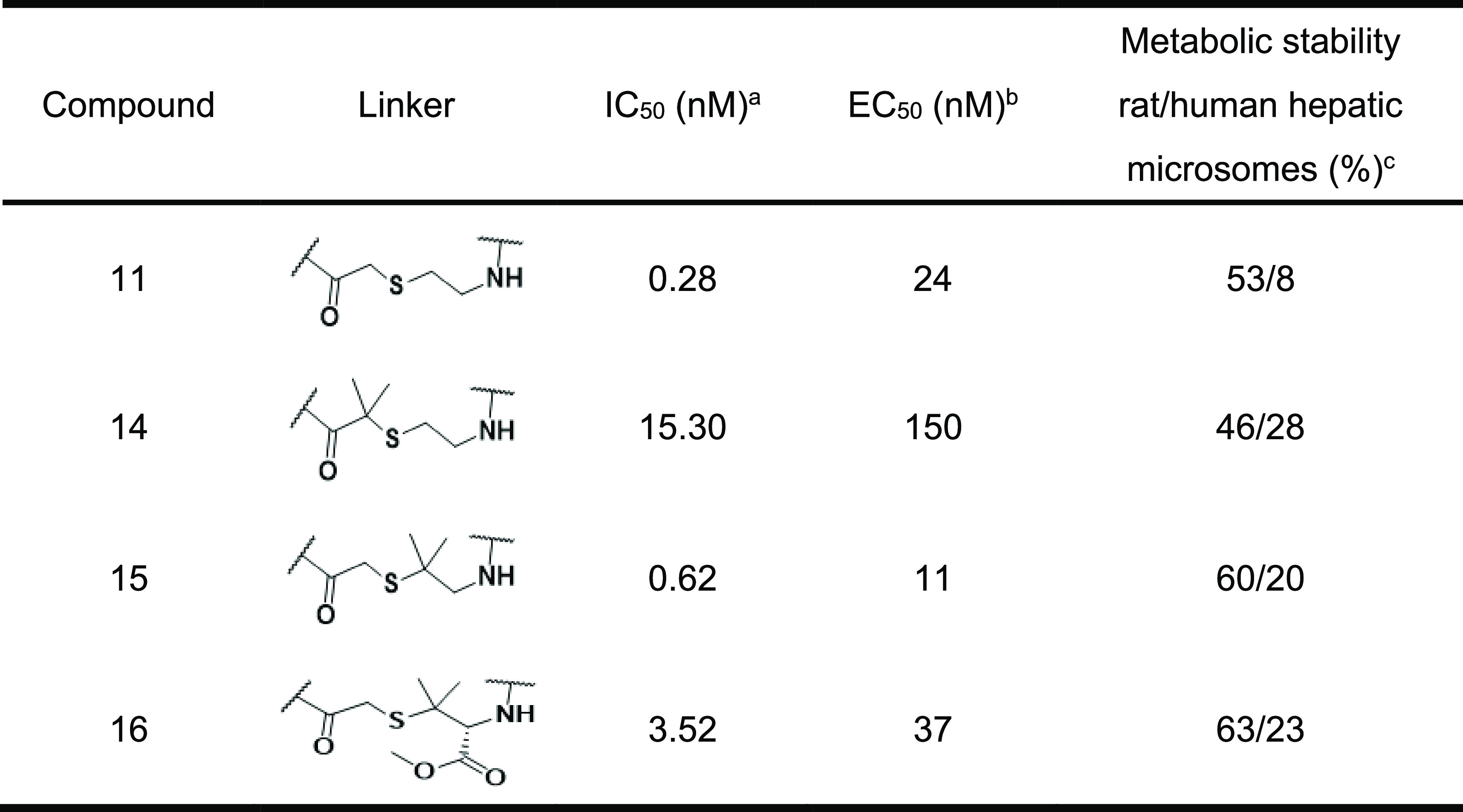
Measured by the fluorogenic peptide cleavage assay.
Anti-HIV-1 activity assay using MT-4 cells.
Metabolic stability using human and rat hepatic microsomes.
Finally, compounds with potent EC50 and good metabolic stability in rat hepatic microsomes were evaluated in rat in vivo PK studies (Table 5). Notably, compounds 15 and 16 achieved desirable plasma total clearance (5.7 and 8.2 mL/min/kg, respectively) and oral bioavailability (0.2% and 3.3%, respectively) in rats. Interestingly, there was a great difference in the oral bioavailability between compounds 15 and 16 although they have similar physicochemical properties and in vitro data. Using a preliminary Simcyp animal (version 21) simulation,33 we confirmed that a difference in in vivo distribution (Vdss and t1/2) between compounds 15 and 16 could affect the in vivo duration after oral administration to rats and improve oral bioavailability. In addition, low plasma concentration after oral administration to rats and the LLOQ (10 nM) of LC-MS/MS might overestimate the differences of the oral bioavailability. Although the oral bioavailability of compound 16 (3.3%) was less than that of the previous macrocyclic hexapeptide (compound 25,23 18%), the plasma total clearance of compound 16 (8.2 mL/min/kg) was superior to that of compound 25 (136 mL/min/kg); thus, the oral plasma exposure corrected by the dose of compound 16 was superior to that of the previous macrocyclic hexapeptide. The solubility of our macrocyclic peptides was improved by the presence of taurocholic acid, suggesting that macrocyclic peptides can be dissolved by micellization in the presence of bile acids, as reported previously.32 A previous macrocyclic hexapeptide (compound 25(23)) was administered in a self-emulsifying drug delivery system (SEDDS) formulation to enhance oral bioavailability, although compound 16 was administered in suspension. Therefore, the application of the SEDDS to compound 16 is expected to improve its oral bioavailability by enhancing the solubility. On the other hand, we consider that the disadvantage of the use of the self-microemulsifying drug delivery system (SMEDDS) is to address the issue of the chemical stability during the storage of the SMEDDS formulation because a marketed SMEDDS formulation such as Neoral (cyclosporine) is a capsule filled liquid and the chemical stability in the liquid should be required.34 In addition, the exposed polar surface area (EPSA) of compounds 15 and 16 was less than 100, although the EPSA of compound 2 was greater than 100. EPSA can be used as a filter with a cutoff value of 100 to predict acceptable permeability for the bRo5 chemical series.29,30 We consider that compound 2 does not have significant permeability. It is necessary to optimize the lipophilicity of the compound to achieve high passive membrane permeability, as reported previously.30 In the future, lead-to-candidate optimization to improve the solubility and permeability of macrocyclic peptides or the application of SEDDS will be required. Although further improvement in oral bioavailability is still required, we conducted SAR exploration and optimization of the strong binders from mRNA display and successfully obtained highly potent and orally available HIV-1 protease inhibitors as a new series.
Table 5. Pharmacokinetic Properties of Compounds in Ratsa.
| compound | metabolic stability rat/human (%)b | CLtot (mL/min/kg)c | Vdss (L/kg) | t1/2 (h) | F (%)c | serum fu (%) | solubility (μM) pH 1.2/pH 6.8/pH 6.8 with TCA | EPSAd |
|---|---|---|---|---|---|---|---|---|
| 2 | 29/3 | 6.8 | 0.1 | 0.7 | ND | 1.1 | ND/ND/26 | 113 |
| 11 | 53/18 | 11.4 | 0.8 | 1.6 | ND | 0.1 | ND/ND/NT | 88 |
| 15 | 60/20 | 5.7 | 0.7 | 2.0 | 0.2 | 0.1 | ND/ND/NT | 93 |
| 16 | 63/23 | 8.2 | 1.9 | 3.9 | 3.3 | 0.1 | ND/ND/10 | 92 |
ND: not detected; NT: not tested.
Metabolic stability using human and rat hepatic microsomes.
All compounds were administered to rats by intravenous administration at 1 μmol/kg solution and oral administration at 2 μmol/kg suspension.
Exposed polar surface area (EPSA) analysis by supercritical fluid chromatography.
In summary, we discovered the macrocyclic peptide series as a novel class of HIV-1 protease inhibitors through mRNA display technology. The structure-based drug design approach using the X-ray cocrystal structure allowed one to enhance the potency by removing one HBD. In the effort to enhance the antiviral activity, we found that proteolytic stability can be essential to fill the gap between cell-free inhibitory activity and cell-based antiviral activity. Methyl installation to the α-position of Pro delivered highly potent antiviral active compounds accordingly. In addition, the improvement of metabolic stability by modification of the linker part gave compounds 15 and 16 with desirable potencies, solubility in solutions containing taurocholic acid, and relatively lower EPSA values, and these compounds successfully demonstrated desirable plasma total clearance and oral bioavailability in the rat PK test. We strongly believe that this study would provide valuable insights to other peptide drug discovery projects aimed at intracellular targets and oral bioavailability.
Acknowledgments
We would like to thank to Motohiro Fujiu, Kouhei Matsui, Kazutaka Sekiguchi, Tomokazu Yoshinaga (Shionogi & Co., Ltd.), Akira Ueda, Masahiro Maeda, and Kazuya Matsuo (Shionogi TechnoAdvance Research Co., Ltd.) for their technical support and contributions.
Glossary
Abbreviations
- HIV-1
human immunodeficiency virus type-1
- SBDD
structure-based drug design
- CYP3A4
cytochrome P450 3A4
- PK
pharmacokinetic
- HBD
hydrogen bond donor
- SAR
structure–activity relationship
- CXCR7
C-X-C chemokine receptor type-7
- IC50
enzyme inhibitory activity
- EC50
antiviral activity
- Asp
l-aspartic acid
- Phe
l-phenylalanine
- Arg
l-arginine
- Leu
l-leucine
- Trp
L-tryptophan
- Ile
l-isoleucine
- MePhe
N-methyl l-phenylalanine
- Pro
l-proline
- Val
l-valine
- Gly
glycine
- Ala
l-alanine
- SEDDS
self-emulsifying drug delivery system
- EPSA
exposed polar surface area
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.2c00310.
Synthetic procedure for peptides, amino acids, and resins, characterization of peptides, protocols of biological assays, pharmacokinetic studies, and supporting figures of the X-ray structures (PDF)
This work was funded and conducted by Shionogi & Co., Ltd.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Medicinal Chemistry Letters virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Barré-Sinoussi F.; Chermann J. C.; Rey F.; Nugeyre M. T.; Chamaret S.; Gruest J.; Dauguet C.; Axler-Blin C.; Vézinet-Brun F.; Rouzioux C.; Rozenbaum W.; Montagnier L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immunedeficiency syndrome (AIDS). Science 1983, 220, 868–871. 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- Gallo R. C.; Sarin P. S.; Gelmann E. P.; Robert-Guroff M.; Richardson E.; Kalyanaraman V. S.; Mann D.; Sidhu G. D.; Stahl R. E.; Zolla-Pazner S.; Leibowitch J.; Popovic M. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983, 220, 865–867. 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- Brik A.; Wong C. H. HIV-1 protease: mechanism and drug discovery. Org. Biomol. Chem. 2003, 1, 5–14. 10.1039/b208248a. [DOI] [PubMed] [Google Scholar]
- Wlodawer A.; Miller M.; Jaskólski M.; Sathyanarayana B. K.; Baldwin E.; Weber I. T.; Selk L.; Clawson L.; Schneider J.; Kent S. B. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]
- Lapatto R.; Blundell T.; Hemmings A.; Overington J.; Wilderspin A.; Wood S.; Merson J. R.; Whittle P. J.; Danley D. E.; Geoghegan K. F.; Hawrylik S. J.; Lee S. E.; Scheld K. G.; Hobart P. M. X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature 1989, 342, 299–302. 10.1038/342299a0. [DOI] [PubMed] [Google Scholar]
- Hammer S. M.; Squires K. E.; Hughes M. D.; Grimes J. M.; Demeter L. M.; Currier J. S.; Eron J. J. Jr; Feinberg J. E.; Balfour H. H. Jr; Deyton L. R.; Chodakewitz J. A.; Fischl M. A.; Phair J. P.; Pedneault L.; Nguyen B.-Y.; Cook J. C. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med. 1997, 337, 725–733. 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- Gulick R. M.; Mellors J. W.; Havlir D.; Eron J. J.; Gonzalez C.; McMahon D.; Richman D. D.; Valentine F. T.; Jonas L.; Meibohm A.; Emini E. A.; Chodakewitz J. A.; Deutsch P.; Holder D.; Schleif W. A.; Condra J. H. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- Koh Y.; Nakata H.; Maeda K.; Ogata H.; Bilcer G.; Devasamudram T.; Kincaid J. F.; Boross P.; Wang Y. F.; Tie Y.; Volarath P.; Gaddis L.; Harrison R. W.; Weber I. T.; Ghosh A. K.; Mitsuya H. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2003, 47, 3123–3129. 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh Y.; Amano M.; Towata T.; Danish M.; Leshchenko-Yashchuk S.; Das D.; Nakayama M.; Tojo Y.; Ghosh A. K.; Mitsuya H. In vitro selection of highly darunavir-resistant and replicationcompetent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors. J. Virol. 2010, 84, 11961–11969. 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tie Y.; Boross P. I.; Wang Y. F.; Gaddis L.; Hussain A. K.; Leshchenko S.; Ghosh A. K.; Louis J. M.; Harrison R. W.; Weber I. T. High resolution crystal structures of HIV-1 protease with a potent nonpeptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol. 2004, 338, 341–352. 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- Kempf D J; Marsh K C; Denissen J F; McDonald E; Vasavanonda S; Flentge C A; Green B E; Fino L; Park C H; Kong X P ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 2484–2488. 10.1073/pnas.92.7.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar G. N.; Rodrigues A. D.; Buko A. M.; Denissen J. F. Cytochrome P450-mediated metabolism of the HIV-1 protease inhibitor ritonavir (ABT-538) in human liver microsomes. J. Pharmacol. Exp. Ther. 1996, 277, 423–431. [PubMed] [Google Scholar]
- Vinogradov A. A.; Yin Y.; Suga H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. 10.1021/jacs.8b13178. [DOI] [PubMed] [Google Scholar]
- Passioura T.; Katoh T.; Goto Y.; Suga H. Selection-based discovery of druglike macrocyclic peptides. Annu. Rev. Biochem. 2014, 83, 727–752. 10.1146/annurev-biochem-060713-035456. [DOI] [PubMed] [Google Scholar]
- Kim Y. K.; Arai M. A.; Arai T.; Lamenzo J. O.; Dean E. F. III; Patterson N.; Clemons P. A.; Schreiber S. L. Relationship of Stereochemical and Skeletal Diversity of Small Molecules to Cellular Measurement Space. J. Am. Chem. Soc. 2004, 126, 14740–14745. 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]
- White T. R.; Renzelman C. M.; Rand A. C.; Rezai T.; Mcewen C. M.; Gelev V. M.; Turner R. A.; Linington R. G.; Leung S. F.; Kalgutkar A. S.; Bauman J. N.; Zhang Y.; Liras S.; Price D. A.; Mathiowetz A. M.; Jacobson M. P.; Lokey R. S. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C. N.; Townsend C. E.; Jain A. N.; Naylor M. R.; Pye C. R.; Schwochert J.; Lokey R. S. Geometrically Diverse Lariat Peptide Scaffolds Reveal an Untapped Chemical Space of High Membrane Permeability. J. Am. Chem. Soc. 2021, 143, 705–714. 10.1021/jacs.0c06115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron E.; Chatterjee J.; Ovadia O.; Langenegger D.; Brueggen J.; Hoyer D.; Schmid H. A.; Jelinek R.; Gilon C.; Hoffman A.; Kessler H. Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem., Int. Ed. 2008, 47, 2595–2599. 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- Fouche M.; Schäfer M.; Berghausen J.; Desrayaud S.; Blatter M.; Piéchon P.; Dix I.; Garcia A. M.; Roth H. J. Design and Development of a Cyclic Decapeptide Scaffold with Suitable Properties for Bioavailability and Oral Exposure. ChemMedChem 2016, 11, 1048–1059. 10.1002/cmdc.201600082. [DOI] [PubMed] [Google Scholar]
- Fouché M.; Schäfer M.; Blatter M.; Berghausen J.; Desrayaud S.; Roth H. J. Pharmacokinetic Studies around the Mono- and Difunctionalization of a Bioavailable Cyclic Decapeptide Scaffold. ChemMedChem. 2016, 11, 1060–1068. 10.1002/cmdc.201600083. [DOI] [PubMed] [Google Scholar]
- Räder A. F. B.; Weinmüller M.; Reichart F.; Schumacher-Klinger A.; Merzbach S.; Gilon C.; Hoffman A.; Kessler H. Orally Active Peptides: Is There a Magic Bullet?. Angew. Chem., Int. Ed. 2018, 57, 14414–14438. 10.1002/anie.201807298. [DOI] [PubMed] [Google Scholar]
- Dougherty P. G.; Sahni A.; Pei D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. 10.1021/acs.chemrev.9b00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M.; Beaumont K.; Jones R.; Kalgutkar A. S.; Zhang L.; Atkinson K.; Bai G.; Brown J. A.; Eng H.; Goetz G. H.; Holder B. R.; Khunte B.; Lazzaro S.; Limberakis C.; Ryu S.; Shapiro M. J.; Tylaska L.; Yan J.; Turner R.; Leung S. S. F.; Ramaseshan M.; Price D. A.; Liras S.; Jacobson M. P.; Earp D. J.; Lokey R. S.; Mathiowetz A. M.; Menhaji-Klotz E. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide– Peptoid Hybrid CXCR7Modulators. J. Med. Chem. 2017, 60, 9653–9663. 10.1021/acs.jmedchem.7b01028. [DOI] [PubMed] [Google Scholar]
- Hayashi K.; Uehara S.; Yamamoto S.; Cary D. R.; Nishikawa J.; Ueda T.; Ozasa H.; Mihara K.; Yoshimura N.; Kawai T.; Ono T.; Yamamoto S.; Fumoto M.; Mikamiyama H. Macrocyclic Peptides as a Novel Class of NNMT Inhibitors: A SAR Study Aimed at Inhibitory Activity in the Cell. ACS Med. Chem. Lett. 2021, 12, 1093–1101. 10.1021/acsmedchemlett.1c00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary D. R.; Ohuchi M.; Reid P. C.; Masuya K. Constrained Peptides in Drug Discovery and Development. J. Synth. Org. Chem. Jpn. 2017, 75, 1171–1178. 10.5059/yukigoseikyokaishi.75.1171. [DOI] [Google Scholar]
- Murakami H.; Ohta A.; Ashigai H.; Suga H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- Goto Y.; Katoh T.; Suga H. Flexizymes for genetic code reprogramming. Nat. Protocols 2011, 6, 779–790. 10.1038/nprot.2011.331. [DOI] [PubMed] [Google Scholar]
- Oba M. Peptide Foldamers: Structural Control and Cell-penetrating Ability. Yakugaku Zasshi 2019, 139, 599–608. 10.1248/yakushi.18-00179-3. [DOI] [PubMed] [Google Scholar]
- Goetz G. H.; Philippe L.; Shapiro M. J. EPSA: a novel supercritical fluid chromatography technique enabling the design of permeable cyclic peptides. ACS Med. Chem. Lett. 2014, 5, 1167–1172. 10.1021/ml500239m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe A.; Uehara S.; Akazawa T.; Fujiu M. Optimization of Cyclic Peptide Property Using Chromatographic Capacity Factor on Permeability of Passive Cell Membrane and Human Induced Pluripotent Stem Cell-Derived Intestinal Membrane. J. Pharm. Sci. 2022, 111, 1879–1886. 10.1016/j.xphs.2022.03.019. [DOI] [PubMed] [Google Scholar]
- Mullard A. Merck readies oral, macrocyclic PCSK9 inhibitor for phase II test. Nat. Rev. Drug Discovery 2022, 21, 9. 10.1038/d41573-021-00195-4. [DOI] [PubMed] [Google Scholar]
- Sugano K.; Okazaki A.; Sugimoto S.; Tavornvipas S.; Omura A.; Mano T. Solubility and dissolution profile assessment in drug discovery. Drug Metab. Pharmacokinet. 2007, 22, 225–254. 10.2133/dmpk.22.225. [DOI] [PubMed] [Google Scholar]
- Certara Home Page; https://www.certara.com/software/simcyp-animal/ (accessed 2022-08-19).
- Dokania S.; Joshi A. K. Self-microemulsifying drug delivery system (SMEDDS)--challenges and road ahead. Drug Delivery 2015, 22 (6), 675–90. 10.3109/10717544.2014.896058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.