

Summary
Autophagy plays critical roles in neurodegeneration and development, but how this pathway is organized and regulated in neurons remains poorly understood. Here, we find that the dynein adaptor RILP is essential for retrograde transport of neuronal autophagosomes, and surprisingly, their biogenesis as well. We find that induction of autophagy by mTOR inhibition specifically up-regulates RILP expression and its localization to autophagosomes. RILP depletion or mutations in its LC3-binding LIR motifs strongly decrease autophagosome number suggesting an unexpected RILP role in autophagosome biogenesis. We find that RILP also interacts with ATG5 on isolation membranes, precluding premature dynein recruitment and autophagosome transport. RILP inhibition impedes autophagic turnover and causes p62/Sequestosome-1 aggregation. Together, our results identify an mTOR-responsive neuronal autophagy pathway, wherein RILP integrates the processes of autophagosome biogenesis and retrograde transport to control autophagic turnover. This pathway has important implications for understanding how autophagy contributes to neuronal function, development, and disease.
Keywords: Neuronal Autophagy, Sequestosome-1/p62, Autophagosome Biogenesis, Isolation Membranes, RILP, Dynein, Retrograde Transport, mTOR Regulation
Graphical Abstract

In Brief
Macroautophagy is required for neuronal homeostasis and is disregulated in several neurodevelopmental and degenerative disorders. Khobrekar et al. show that the Rab-Interacting Lysosomal Protein (RILP) controls the neuronal autophagy pathway through sequential interactions with ATG5, LC3, Rab7 and dynein to regulate autophagosome formation, transport and turnover in an mTOR-responsive mechanism.
Introduction
Macroautophagy (here referred to as “autophagy”) is a highly conserved cellular process used to degrade misfolded proteins and damaged organelles. It also serves to recycle amino acids and other metabolites during nutrient starvation, and to destroy invading pathogens (Kaushik and Cuervo, 2018, Dooley et al., 2014, Mizushima, 2018, Richter et al., 2016, Cecconi and Levine, 2008). Neurons exhibit high levels of basal autophagy, and several neurodevelopmental and degenerative disorders have been shown to arise from defects in this process (Narendra et al., 2008, Bordi et al., 2016, Gowrishankar et al., 2015).
Autophagosome biogenesis is initiated at PI3P-enriched nucleation sites within the ER and other membranous organelles (Derubeis et al., 2000, Axe et al., 2008, Uemura et al., 2014). Isolation membranes expand from these sites to engulf ubiquitinated cytoplasmic cargoes. Closure of the isolation membranes yields double-membraned LC3-positive autophagosomes (APs) (Carlsson and Simonsen, 2015, Dooley et al., 2014, Mizushima et al., 2011). In mammalian cells, the APs are transported retrogradely along microtubules, and fuse with Rab7-decorated late endosomes (LEs) and ultimately, lysosomes for Cathepsin-mediated degradation and recycling of the autophagosomal contents (Bright et al., 2016, Gao et al., 2018, Jia et al., 2017). mTOR kinase serves a critical role in sensing nutrient deprivation and other forms of stress. Inhibition of mTOR activates autophagy and prolongs cell survival. However, how mTOR regulates neuronal autophagy remains a critical, but poorly understood question (Maday and Holzbaur, 2016, Kim et al., 2011, Saxton and Sabatini, 2017).
Although the basic stages in autophagosome biogenesis and maturation have been extensively characterized in yeast and mammalian cells, they remain to be fully elucidated in neurons. Neuronal autophagosome behavior, including microtubule-based motility, is also of particular interest given the extreme asymmetry of these cells and the concentration of degradative organelles in the soma (Yap et al., 2018, Gowrishankar et al., 2015). Neuronal autophagosomes are transported along microtubules, though the stage of AP biogenesis at which motors are first acquired and the molecular mechanisms for regulation of transport, are incompletely understood (Maday and Holzbaur, 2016, Kim et al., 2011, Saxton and Sabatini, 2017, Takei and Nawa, 2014). Snapin and JIP1 have been implicated in microtubule motor protein recruitment to late autophagosomes (after fusion with Rab7-positive vesicles) in dorsal root ganglion (DRG) neurons. However, neither of these proteins was implicated in direct recruitment of the retrograde motor protein cytoplasmic dynein to autophagosomes (Cheng et al., 2015, Fu et al., 2014).
We have now discovered an autophagy mechanism which mediates autophagosome biogenesis as well as microtubule-based transport in neurons. We find that the dynein adaptor protein RILP (Rab-Interacting Lysosomal Protein), controls these processes through direct sequential interactions with the core autophagy proteins ATG5 and LC3, as well as dynein. RILP is an α-helical coiled-coil-containing protein previously established to recruit cytoplasmic dynein to Rab7-positive late endosomes (LEs) in non-neuronal cells (Cantalupo et al., 2001, Jordens et al., 2001, Scherer et al., 2014). From our own prior work (Tan et al., 2011, Scherer et al., 2014) and that of others (Harrison et al., 2004, Starr et al., 2008, Wozniak et al., 2016, Seto et al., 2010), RILP has also been implicated in endo-lysosomal mobilization during the host cell response to adenoviral and bacterial infection. We speculated that RILP might specifically contribute to the autophagy pathway, a possibility so far unexplored.
We report that RILP recruitment to autophagic membranes is specifically upregulated upon induction of autophagy through mTOR inhibition. We find that, RILP directly binds to the well-known isolation membrane protein ATG5, and facilitates the maturation of isolation membranes into fully-formed autophagosomes (APs) through a dynein-independent mechanism. We find that RILP then associates with the APs via LC3, an interaction mediated by three RILP C-terminal LIR (LC3-binding) motifs we identify. Using RNAi and mutational analysis, we find all three LIR motifs to be both necessary and sufficient for RILP stimulation of autophagosome biogenesis, their retrograde transport, and the autophagic clearance of p62/Sequestosome-1. Together, our data lead to a model for RILP as an mTOR-sensitive master regulator of neuronal autophagy.
Results
RILP Associates with Autophagosomes through C-terminal LC3-Interacting Regions
Although RILP has been implicated in several host defense mechanisms, a RILP role in autophagy has not been investigated. To address this issue, we tested for co-distribution and co-behavior of RILP with autophagosomal membranes in neuronal and non-neuronal cells. We expressed GFP-tagged RILP in HeLaM and C6 glioma cells and co-immunostained for endogenous Rab7 or LC3. Endogenous Rab7 showed clear colocalization with a subset of RILP vesicles. Interestingly, LC3 also showed striking colocalization with RILP-positive structures in both cell lines (Sup Fig 1k–m). To test whether RILP also associated with LC3-positive APs in neurons, we monitored the relative behavior of GFP-RILP in DIV (days-in-vitro) 8 rat embryonic cortical neurons expressing RFP-LC3, using high temporal resolution dual-channel live imaging. We observed extensive punctate colocalization of RILP with LC3-positive autophagosomes (APs). The RILP-positive APs were highly motile, predominantly moving in the retrograde direction consistent with dynein-driven behavior (Fig 1, Video S1).
Figure 1. RILP Regulates Retrograde Axonal Transport of LC3-positive Autophagosomes in Neurons.

(A) RILP domain organization and interactions. RILP is an elongated α-helical coiled- coil (CC) – containing protein known to interact with cytoplasmic dynein and its regulator dynactin, as well as with the late endosomal marker Rab7. This study also shows RILP to interact with the autophagosomal membrane protein LC3. Three C-terminal LC3-interacting regions (LIRs) identified in this study are indicated. (B) LIR amino acid sequences, the second of which is a canonical (WxxL) motif, whereas the others are F-type (phenylalanine-containing). (C-E) GFP-RILP and RFP-LC3 were co-expressed in DIV (Day in vitro) 7–8 rat cortical neurons and monitored by dual-channel live time-lapse imaging. Single frames showing individual axons are presented at top for (C) wt; (D) LIR3- mutant; and (E) Rab7-binding mutant RILP vs. RFP-LC3. Shown below are kymographs indicating the change in AP position within the axon with time. (F-H) Quantification of AP behavior. (C) wt RILP colocalized and co-migrated with RFP-LC3-positive APs (n=31), predominantly in the retrograde direction. (D) In contrast, the LIR3-mutant RILP showed reduced localization and retrograde transport, and AP number was also markedly decreased (n=40). (E) Rab7-binding mutant RILP colocalized and co-migrated with APs (n=26). (F) Relative colocalization of wt and mutant RILP with APs. Rab7-binding site is dispensable for AP binding. (G) Decreased retrograde AP transport observed with LIR3- mutant RILP, but not with wt and Rab7-binding mutant RILP. (H) Decreased number of APs per unit length of axon in LIR3- mutant RILP expressing neurons suggests a role in AP formation. (I-K) GFP-RILP and mCh-Rab7 expressing DIV7–8 rat cortical neurons were monitored by live time-lapse imaging. Single frames showing individual axons are presented for (I) wt; (J) Rab7-binding mutant; and (K) LIR3-mutant RILP vs. mCh-Rab7. Shown below are kymographs and (L-N) quantification of LE behavior. (I) wt RILP colocalized and co-migrated with mCh-Rab7-positive LEs (n=31), predominantly in the retrograde direction. (n=30). (J) In contrast, the Rab7-binding mutant RILP showed reduced localization to LEs (n=16). (K) LIR3-mutant RILP colocalized and co-migrated with LEs (n=20). (L) Relative colocalization of wt and mutant RILP with LEs. LIRs are dispensable for Rab7-binding. (M) Decreased retrograde LE transport observed with Rab7-binding mutant RILP, but not wt and LIR3- mutant RILP. (N) No effect of Rab7-binding mutant RILP on LE number in axons. scale bars: x = 5 μm; y = 30 sec. P-values: *=0.05, **=0.01, ***=0.001.
To determine the molecular basis for RILP-LC3 colocalization, we first tested whether the Rab7-binding site in RILP was also responsible for its recruitment to autophagosomes. We introduced a single point mutation ((Met305→Ala) within the Rab7-binding site, which has been previously reported to disrupt RILP-Rab7 binding (Wu et al., 2005). As expected, dual-color live imaging in DIV8 cortical neurons showed a striking decrease in localization of the Rab7-binding mutant RILP with mCh-Rab7-positive LEs (Fig 1c, e–g, Video S4), though this mutant remained fully able to localize to and transport RFP-LC3-positive APs (Fig 1d, Video S3). Thus, the Rab7-binding site appears dispensable for RILP recruitment to APs. To further define the basis for RILP-AP binding, we examined the human RILP amino acid sequence for LC3- Interacting Regions (LIRs) (Alemu et al., 2012, Kalvari et al., 2014, Lamark et al., 2009), which have been identified in a number of ATG family proteins, and are required for their interaction with LC3. We found one canonical and two F-type (Phenylalanine-containing) LIRs in close proximity to, but not overlapping with the Rab7-GTP binding site within the C-terminal “cargo binding” domain of RILP (Fig 1a, b). To test the physiological significance of these motifs, we introduced missense mutations in the first aromatic amino acid residue to change each to valine. To determine the effects of the mutations in rat cortical neurons, we co-expressed single, double or triple LIR-mutant versions of GFP-tagged RILP, along with RFP-LC3. Relative to wt RILP, we observed a graded loss in RILP LIR mutant – AP colocalization, which was dependent on increase in the number of LIR mutations in RILP (Sup Fig 1 a–d). These results showed that all three LIR motifs were functional and, indeed, necessary for RILP recruitment to APs. The triple LIR-mutant RILP (Video S2) showed the most potent loss in AP localization, so for subsequent analysis we predominantly used this construct, here on referred to as “LIR3-mutant RILP”. This construct also caused a marked decrease in the frequency of retrograde AP transport (Fig 1g). Using biochemical analysis, we also observed an ~50% reduction in LC3-binding by the LIR3-mutant relative to wt RILP as judged by co-immunoprecipitation (Sup Fig 1e, f). Despite the decrease in localization to APs, the LIR3-mutant RILP still colocalized and co-migrated with Rab7-positive late endosomes (LEs) as efficiently as wt RILP (Fig 1 k, Video S4). The velocities and travel distances of residual APs and LEs that still exhibited retrograde motility remained unaltered relative to wt, suggesting normal dynein behavior for these vesicles (Sup Fig 1 g–h). These results show that RILP recruitment to APs occurs through discrete C-terminal LIR motifs, independent of the Rab7-binding site. Furthermore, the LIRs are necessary and sufficient for RILP recruitment to APs, independent of whether Rab7-binding is impaired. Together our results implicate RILP in dynein recruitment to both early (pre-LE fusion) as well as late (post-LE fusion) autophagosomal intermediates via RILP’s distinct LC3- and Rab7-binding sites.
Intriguingly, expression of the LIR3-mutant RILP resulted in a striking decrease in the total number of APs per unit length of axon compared to wt RILP (Fig 1h). In these neurons, RFP-LC3 exhibited a predominantly diffuse cytoplasmic distribution. The Rab7-binding mutant RILP, in contrast, had no effect on the abundance of LEs or APs in neurons (Fig 1n). These results suggest that, in addition to playing a key role in the retrograde transport of autophagosomes in neurons, the RILP-LC3 interaction might be involved in their biogenesis as well.
RILP Depletion and LIR Mutations Inhibit Retrograde Autophagosome Transport, and Prevent Autophagic Clearance
To test whether RILP plays an essential role in neuronal autophagy, we performed RNAi to deplete endogenous RILP in cortical neurons (Fig 2a, b; Sup Fig 2 g). This resulted in a marked decrease in the number of retrogradely transported APs and an increase in stationary APs along the axon (Fig 2 c–d, Video S5, Sup Fig 2 d–e). There was also a modest but detectable decrease in anterogradely transported APs, which might reflect functional interactions between retrograde and anterograde motor proteins (Yi et al., 2011, Ally et al., 2009). In agreement with our observations with the LIR3-mutant RILP, RILP RNAi also reduced axonal AP number (Fig 2 g), further supporting an unexpected and completely novel role for RILP in AP biogenesis. In keeping with the previously reported RILP-Rab7 interaction (Wu et al., 2005), RILP RNAi also resulted in a decreased retrograde axonal transport of Rab7-positive late endosomes in cortical neurons (Fig 2e–f, Video S5, Sup Fig 2a, b). Finally, distances travelled by residual APs and LEs still exhibiting retrograde transport after RILP depletion appeared unaltered (Sup Fig 2 c, f), confirming that dynein activity in these neurons remained normal.
Figure 2. RILP is Essential for Axonal Autophagosome Transport.

RILP was knocked down in DIV3 rat cortical neurons using RNAi and its effects on AP and LE behavior were monitored by time-lapse imaging of (C) RFP-LC3-positive APs, or (E) mCh-Rab7-positive LEs (n >20). Kymographs indicate the change in axonal AP and LE position over time in control (top) and RILP RNAi (bottom) neurons (scale bars: x = 5 μm; y = 30 sec). (A) Western blot showing depletion of RILP protein upon 72 hr RILP RNAi in rat C6 glioma cells, quantified in (B). Kymographs show processive retrograde transport of APs (C) and LEs (E) in control axons, which is inhibited upon RILP RNAi. (D) Quantitative analysis reveals specific RILP RNAi-mediated inhibition of retrograde AP and (F) LE transport. (G) Decreased number of APs upon RILP RNAi indicates a RILP role in AP formation. P-values: *=0.05, **=0.01 and ***=0.001.
To examine whether the RILP role that we identified in neuronal autophagy has a functional consequence in clearance of autophagic cargoes, we monitored the behavior of myc-tagged Sequestosome 1 (SQSTM1/p62) (Zaffagnini et al., 2018a, Zaffagnini et al., 2018b) in RILP-depleted C6 glioma cells. We observed few myc-p62 puncta in the cytoplasm of control cells, but a striking increase in the number of p62 aggregates following RILP knockdown (Fig 3 a–b, e). These results provide evidence for a functional RILP role in turnover of autophagic substrates. We also performed this assay in C6 glioma cells transiently expressing LIR3-mutant RILP vs. wt RILP or GFP alone. We found no detectable difference in the number of p62 aggregates in GFP vs. wt RILP expressing cells; however, there was a substantial increase in the number of p62 aggregates in LIR3-mutant RILP expressing cells (Fig 3c, d). We found further that the p62 aggregates were enriched for LC3 and Ubiquitin (Fig 3f–g, j–k), but not for Rab7 or the aggresome marker, Vimentin (Fig 3h–i, l–m) (Johnston et al., 1998, Moriya et al., 2015). Thus, in the absence of associated RILP, ubiquitinated substrates positive for p62 tend to accumulate in the cytoplasm and fail to reach the late endosomal compartment, further highlighting the role of RILP in the regulation of early stages in the autophagy pathway. Together, these results show that RILP is essential for autophagic turnover of p62/Sequestosome-1, and this function requires the specific interaction of RILP with autophagosomes through its LIR motifs.
Figure 3. RILP Depletion Prevents Autophagic Clearance of p62/Sequestosome-1.

As a test for a physiological RILP role in autophagic turnover, we evaluated the distribution of Myc-tagged p62/sequestosome-1 (SQSTM1) with or without RILP RNAi in C6 cells. (A) p62 exhibited diffuse cytoplasmic distribution (top) in control cells. Upon RILP RNAi, p62 showed extensive aggregation (bottom), quantified in (B) (n>90). (C, D) GFP alone, wt RILP or LIR3-mutant RILP were transfected into C6 cells and endogenous p62 puncta were counted after 24 hours of expression (n=105 per condition). (C) Using immunostaining, comparable numbers of p62 puncta were detected in GFP and wt RILP expressing cells, but substantially higher numbers were observed in LIR3-mutant RILP expressing cells, quantified in (D). (E) Scatter plot shows relative increase in p62 aggregates upon RILP RNAi in C6 cells. (F-M) Further characterization of p62 aggregates shows that they are positive for endogenous LC3 (F) and ubiquitin (G), but mostly negative for Rab7 (H) and aggresome marker vimentin (I). Quantification of relative localization of p62 aggregates with LC3 (J), Ubiquitin (K), Rab7 (L) and Vimentin (M). P-value: ***=0.001.
Induction of Autophagy by mTOR Inhibition Upregulates RILP Expression and Alters its Subcellular Behavior
In view of the substantial range of roles we identified for RILP in autophagy, it was of interest to determine how RILP’s behavior in cells was affected by mTOR function. For this purpose, we used a small molecule-mediated inhibition of mTOR kinase to induce autophagy. mTOR is the major nutrient level and stress sensor in cells, and is the key negative regulator of mammalian autophagy. Inhibition of mTOR by Rapamycin or the more potent inhibitor Torin1, is known to induce autophagy in non-neuronal cells, as monitored by formation of LC3-II positive autophagosomes and inhibition of S6K phosphorylation (Thoreen and Sabatini, 2009). Although mTOR kinase plays critical roles in processes underlying neurodevelopment and degeneration, the mechanisms by which it regulates neuronal autophagy remain unclear (Maday and Holzbaur, 2016).
To evaluate the effects of mTOR on RILP function, we treated C6 glioma cells and rat cortical neurons with Torin1. Inhibition of mTOR kinase activity and induction of autophagy were confirmed by a decrease in the level of S6K phosphorylation and an increase in the ratio of converted (lipidated) to full length LC3, respectively (Sup Fig 3a). We found that short-term (500nm, 6 hr) Torin1 treatment in C6 Glioma cells increased RILP mRNA levels by 2.36 +/− 0.45 -fold (Sup Fig 3b). There was also an upregulation in RILP protein levels as early as 2 hrs post-Torin1 treatment (Sup Fig 3c). To determine whether mTOR-mediated upregulation was unique to RILP, we immunoblotted for other well-known motor protein adaptors, including the dynein adaptors HOOK1, HOOK3 and BicD2, as well as a kinesin adaptor FYCO1, the latter shown to bind LC3 and to recruit Kinesin-1 to APs (Pankiv et al., 2010, Raiborg et al., 2015) (Sup Fig 3d–g). However, we detected no alteration in the total protein levels of these adaptors, which suggested that RILP expression is specifically responsive to changes in mTOR activity in cells.
Next, we tested the effect of mTOR inhibition on endogenous RILP behavior using immunocytochemistry in rat NRK-F cells. In these cells, endogenous RILP and LC3 exhibited a mostly diffuse cytoplasmic staining, with occasional puncta observable, consistent with low levels of basal autophagy (Sup Fig 4 c(i)). Short-term Torin1 treatment combined with Bafilomycin A1, which neutralizes lysosomal pH and prevents AP-LE fusion (Mauvezin and Neufeld, 2015), resulted in robust stimulation of autophagy, as observed by an increase in LC3-II/I ratio and a dramatic redistribution of RILP to discrete perinuclear puncta, a predominant subset of which were also positive for LC3 (Sup Fig (a-b), c(iv)). Importantly, this phenomenon was not observed upon Bafilomycin A1 treatment alone (Sup. Fig. 4 c (iii)), suggesting that RILP localization to AP membranes is specifically induced by mTOR inhibition, rather than reflecting a defect in fusion of RILP-positive APs with the lysosomal compartment (Sup Fig 4 c(ii)).
We further tested whether RILP shows similar behavior in neurons as well. In cortical neurons treated with Torin1, we observed a more modest increase in RILP mRNA levels (Sup Fig 4d). We reasoned that this difference might reflect a higher basal level of mTOR activity in neurons due to a significantly higher concentration of insulin in the B-27 supplement added to neuronal culture medium compared to non-neuronal cell culture medium, which, itself stimulates mTOR (Vander Haar et al., 2007, Brewer and Cotman, 1989, Chen et al., 2008, Brewer et al., 1993). Indeed, short-term Insulin withdrawal revealed a similar ~2.1- fold Torin1-mediated increase in neuronal RILP mRNA levels, comparable to the effect we observed in glioma cells (Sup Fig 4d). We also found that cytoplasmic RILP behavior in these neurons was similar to that seen in NRK-F cells. In axons of untreated neurons, RILP exhibited a mostly diffuse cytoplasmic distribution, however mTOR inhibition resulted in a clear increase in RILP-positive vesicles, in some of which a clear lumen could be discerned (Sup Fig 4e).
Together, these results show that mTOR-dependent induction of autophagy increases RILP expression and promotes its recruitment to newly forming autophagosomes.
RILP is Recruited to Isolation Membranes through ATG5
In view of its unexpected effect on regulating autophagosome number, we hypothesized that RILP might be involved in the early stages of autophagy, i.e. in some aspect of biogenesis. To test this hypothesis, we used live imaging to probe for a RILP association with early, pre-closure autophagosomal intermediates in rat cortical neurons, using double-labeling for the nucleation site and isolation membrane markers DFCP1 and ATG5, respectively (Fig 4, Sup Fig 5a–e, j–k). RILP was present at each of these structures. Close inspection of the distribution pattern for RILP vs DFCP1 staining revealed RILP-positive membranes to be juxtaposed to and extending from the DFCP1 puncta in DIV7–8 neurons (Sup Fig 5 j–k). In contrast, wt RILP and ATG5 were coincident along the isolation membranes (Fig 4b). RILP, thus, appears to be present at both nascent and more mature stages of isolation membrane formation in neurons. In contrast, GFP alone showed a diffuse cytoplasmic distribution (Fig 4a, sup fig 5j), and had no detectable effect on DFCP1 or ATG5 distribution.
Figure 4. RILP is Recruited to Isolation Membranes in Neurons.
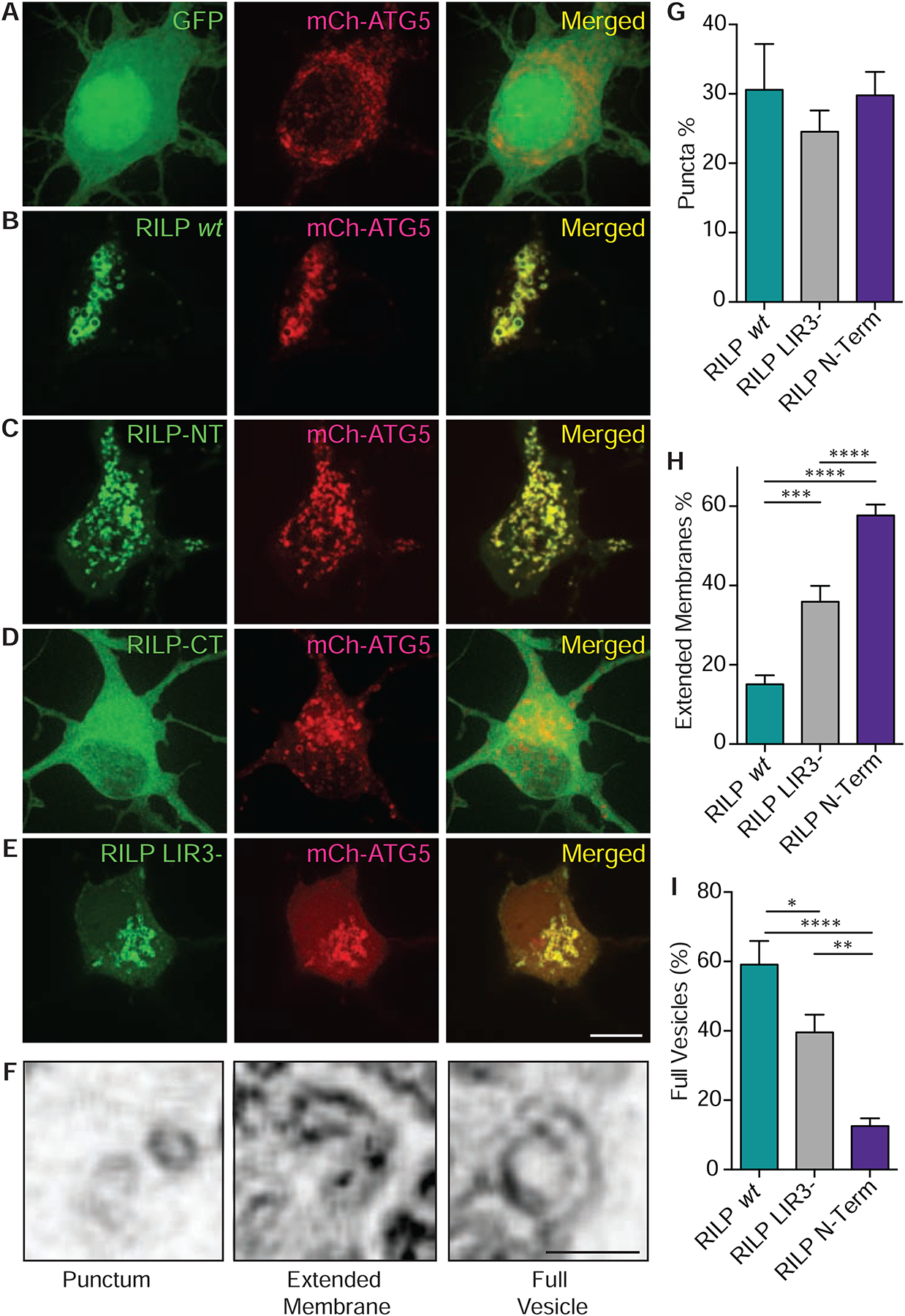
(A-E) GFP-tagged RILP, RILP mutants, and sub-fragments were co-expressed with ATG5, a marker for isolation membranes, in DIV7–8 cortical neurons. (A) GFP control vector shows diffuse cytoplasmic distribution unaffected by mCh-ATG5 expression. (B) GFP-RILP, (C) N-terminal RILP (1–185aa) and (E) LIR3- mutant RILP show specific localization to isolation membranes. (D) C-terminal RILP (210–401aa) shows no detectable localization to isolation membranes (n>20) (Scale bar: 5 μm) (F) Three distinct classes of ATG5 and RILP dual-positive structures were observed using live spinning disk confocal microscopy at 200X magnification: solid puncta, extended membranes and full vesicles (Scale bar: 1 μm). (G) Comparable number of ATG5 puncta observed in wt, LIR3- mutant and N-terminal RILP fragment expressing neurons. (H) Striking increase in extended ATG5 membranes in LIR3- mutant and N-terminal RILP expressing neurons. (I) Striking decrease in fully-formed ATG5 structures in LIR3-mutant and N-terminal RILP expressing neurons. P-values: *=0.05, **=0.01, ***=0.001, ****=0.0001.
To address the molecular basis for RILP recruitment to isolation membranes, we examined the ability of our N- and C-terminal RILP fragments as well as the LIR3-mutant RILP to localize to these structures in cortical neurons expressing mCh-ATG5. A high percentage of colocalization was observed for both the N-terminal and LIR3-mutant RILP constructs, but almost none for the RILP C-terminal fragment alone (Fig 4c–e, Sup Fig 5c–e). In the case of N-terminal RILP expression, a striking increase in crescent-shaped ATG5-positive isolation membrane profiles was observed (Fig 4c, Sup Fig 5c). This suggests an arrest in maturation of these structures. Similar structures were also observed upon expression of the LIR3-mutant RILP (Fig 4e, Sup Fig 5e). In both cases, there was a corresponding decrease in the mature isolation membranes (Fig. 4i). Wild type full-length RILP, in contrast, exhibited a high degree of localization to more mature, vesicular isolation membrane profiles (Fig. 4 f–i). These data suggest that the RILP N-terminal domain is sufficient for its recruitment to isolation membranes, whereas the C-terminal domain is required for extension and successful closure of these structures. The localization of the LIR3-mutant RILP to isolation membranes is presumably via the RILP N-terminal ATG5-binding site. These results also argue that LC3 is not the primary anchor for wt RILP on isolation membranes. This conclusion is supported by the failure of the RILP C-terminal fragment alone, which includes LC3- and Rab7-binding sites, to localize to isolation membranes (Fig 4d). Together, these results suggest that RILP binds to a distinct anchor protein on isolation membranes through an N-terminal site, independent of LC3 and Rab7. We further found that RILP C-terminus showed distinct colocalization with both, mCh-Rab7 positive LEs and RFP-LC3 positive APs in glioma cells, which is consistent with an active role for the C-terminal Rab7- and LC3- binding sites in RILP (Sup Fig 5 f, g). Conversely, the RILP N-terminal fragment, which contains dynein and ATG5-binding sites, showed a diffuse cytoplasmic distribution and no detectable recruitment to the LE or AP membranes (Sup Fig 5h, i).
To investigate the association of RILP with ATG5-positive isolation membranes further, we tested for a potential RILP-ATG5 interaction biochemically. We found clear RILP co-immunoprecipitation with endogenous ATG5 in glioma cell extracts (Fig 5a). Furthermore, recombinant bacterially-expressed full length or N-terminal RILP pulled down purified recombinant ATG5 supporting a direct RILP-ATG5 interaction through the RILP N-terminal domain (Fig 5b). His-ATG5 also efficiently pulled down GST-tagged full length and N-terminal RILP (Sup Fig 6a, b), as well as the LIR3-mutant RILP (Sup Fig 6c, d). These results further indicated that the RILP LIR motifs are dispensable for the interaction with ATG5. Conversely, ATG5 failed to pull down the RILP C-terminal domain alone, which contains the Rab7- and LC3-binding sites (Sup Fig 6c, d). Together, our findings show that LC3 and Rab7 do not serve as anchors for RILP recruitment to isolation membranes. Instead, we find the well-established isolation membrane component, ATG5, which is essential for autophagosome biogenesis, to function as a critical RILP interactor, serving to recruit RILP to growing isolation membranes.
Figure 5. ATG5 Directly Binds RILP and Prevents Dynein Recruitment to Isolation Membranes.

(A) Full-length GFP-RILP immunoprecipitated endogenous ATG5:12 from C6 glioma cell lysates, visualized by an anti-ATG5 antibody. (B) Full- length and N-terminal GST-RILP pulled down recombinant His- ATG5 in vitro. Live cell time-lapse imaging was used to monitor GFP-RILP behavior at mCh-ATG5-labeled isolation membranes in DIV7–8 cortical neurons. (C) Single frames for individual axons are shown at top for wt GFP-RILP and mCh-ATG5. Shown below are kymographs which indicate the positions of individual isolation membranes over time. Scale bars: x = 5μm; y= 30sec. RILP-positive isolation membranes were mostly immotile. (D) Immunostaining for dynein LIC1 subunit showed that ATG5-positive RILP structures were devoid of dynein (Top, (i)), while ATG5-negative RILP structures were enriched for dynein (bottom, (ii)) in soma as well as (E) neurites. (F) Percent relative localization of RILP with ATG5 and/or dynein. (G) Majority of RILP-positive isolation membranes are devoid of dynein. Scale bars: 5 μm. p-values: *=0.05, ***= 0.001.
ATG5-mediated RILP Recruitment to Isolation Membranes is Independent of Dynein
In striking contrast to the highly motile RILP-positive autophagosomes, live imaging of RILP-labeled isolation membranes revealed them to be mostly immotile (Fig 5c; Video S6). As noted, ATG5 interacts with the N-terminal domain of RILP (Fig 5b), which also has a well-established role in dynein binding (Scherer et al., 2014). To compare the relative localization of dynein vs ATG5 on isolation membranes, we directly tested for the presence of dynein on isolation membranes using an antibody to the dynein LIC1 (Light Intermediate Chain 1) subunit. In DIV8 neurons co-expressing GFP-RILP and mCh-ATG5, we found that dynein was absent from RILP-positive isolation membranes. In contrast, dynein was enriched at ATG5-negative fully-formed RILP-containing autophagosomes (Fig 5d–e, quant. f-g). Thus, when recruited to isolation membranes via ATG5, RILP fails to bind dynein. An in vitro competitive binding assay between RILP and ATG5 vs dynein LIC1 showed that RILP binding to ATG5 was strikingly decreased in the presence of LIC1 (Sup Fig 6e). In contrast, LC3 binding to RILP was unaffected by ATG5. These data suggest that, although the dynein LIC1 and ATG5 may compete for binding to RILP, LC3 and ATG5 interact with RILP independently (Sup Fig 6f). We did note a small population of RILP–positive structures in cortical neurons that were enriched for both, ATG5 and dynein, which might represent a transition from ATG5- to dynein-bound structures. In view of the transition of APs from immotile to motile structures, we speculate that this is likely controlled by dynein displacement of ATG5 on RILP.
Discussion
Autophagy plays fundamental roles in yeast and mammalian homeostasis. Neuronal autophagy is of particular interest for its involvement in nervous system development and neurodegeneration among other roles (Cecconi and Levine, 2008, Sarkar et al., 2009, Tang et al., 2014). However, the molecular mechanisms mediating and regulating neuronal autophagy have remained poorly understood. Although the core yeast autophagy machinery is conserved in mammals, additional mechanisms might have evolved to regulate autophagosome behavior in the morphologically more complex, highly compartmentalized and long-lived mammalian cells, especially neurons. Our study now identifies a neuronal autophagy mechanism involving the dynein adaptor protein RILP. Surprisingly, we find that RILP performs multiple distinct functions during autophagosome biogenesis and subsequent retrograde transport, and is essential for autophagic clearance. Interestingly, we also find these RILP functions to be responsive to induction of autophagy by mTOR inhibition.
RILP Interaction with Autophagosomes through LC3
We have found that the dynein adaptor protein RILP associates with two core autophagosomal membrane proteins, ATG5 and LC3, through distinct N- and C-terminal binding sites, respectively. RILP showed extensive colocalization and co-transport with LC3-positive autophagosomes in cortical neurons. This behavior was severely inhibited by RILP depletion, supporting the physiological significance of the RILP-LC3 interaction. We identified three LC3-binding (LIR) motifs within the RILP C-terminal domain. Mutational analysis of these motifs showed that all three are functional and necessary for LC3- binding, RILP recruitment to neuronal APs, and their retrograde transport. In contrast, the previously identified RILP Rab7-binding site was dispensable for RILP recruitment to APs, though essential for its recruitment to LEs. RILP’s dual interaction with LC3 and Rab7 suggests that this protein may associate with and transport both early (LC3-positive) and late (LC3-Rab7 dual-positive) autophagosomal structures in neurons (Figure 6c–e).
Figure 6. A Model for RILP Coordination of Autophagosome Biogenesis with Retrograde Transport.

Stages in autophagy progression are shown, along with stage-specific protein markers, RILP, and dynein associated with the microtubule at the bottom. Nucleation, expansion of isolation membrane and maturation of the autophagosome are depicted. RILP shown as an elongated coiled coil-containing dimer, is first detected in juxtaposition with nucleation sites (a), but concentrated at nascent and more fully formed isolation membranes, associated with ATG5 (b). RILP then appears at mature autophagosomes, linked through LC3 (c, d). RILP remains associated with AP-LE membranes through LC3 and Rab7 (e), potentially until lysosomal fusion. This pathway is activated by mTOR regulation of RILP expression, and functions in autophagic clearance. (i-iii) Effects of RILP perturbations on autophagy pathway: (i) Full-length wt RILP binds ATG5 on isolation membranes through N-terminus and may recruit or stabilize LC3 at these structures through the C-terminal LIR motifs. This allows for normal autophagosome formation and eventual retrograde transport to lysosomes. (ii) RILP N-terminal domain is recruited to ATG5 on isolation membranes, but fails to bind LC3. This results in an arrest in the maturation of isolation membranes into fully-formed autophagosomes. (iii) RILP LIR-mutant expression or RNAi also result in an arrest in the maturation of isolation membranes and an aggregation of p62/Sequestosome-1 in cells.
RILP Control of Autophagosome Biogenesis
Surprisingly, the RILP LIR mutations resulted in a striking decrease not only in AP transport, but also in autophagosome number, an effect not observed for the Rab7-binding mutation. RILP knockdown also showed a similar phenotype, supporting an unexpected and independent RILP role in AP formation. Our data suggest that RILP may affect AP biogenesis at the stage of isolation membrane maturation and/or closure. Our colocalization analysis revealed that RILP associates with early, pre-closure autophagosomal intermediates in neurons, including DFCP1-positive nucleation sites as well as ATG5-positive isolation membranes (Fig 6 a, b). RILP was observed to be slightly off-set from DFCP1 at the nucleation sites, but more clearly co-localizing with the nascent isolation membranes extending from these sites. RILP and ATG5, on the contrary, exhibited clear coincidence in distribution along the isolation membrane structure.
We found further, as a basis for this behavior, physical interaction between RILP and ATG5 in vitro as well as in cell lysates. Unlike LC3 and Rab7, however, ATG5 bound to the RILP N-terminal domain consistent with distinct functional roles for the ATG5 vs LC3 and Rab7 interactions. Although the RILP N-terminal domain was sufficient for recruitment to isolation membranes, it was not sufficient for their maturation into fully-formed autophagosomes. Expression of the RILP N-terminal domain alone, which lacked the three LIR motifs, caused a notable change in isolation membrane morphology, resulting in an apparent extension of the isolation membranes without closure. A similar defect was observed when the full length LIR3-mutant RILP was expressed in neurons. Thus, we speculate that the RILP-LC3 interaction may play an additional critical role in the proper maturation of isolation membranes into autophagosomes. Our data are consistent with a model according to which RILP would first become anchored to the isolation membrane through ATG5. The RILP C-terminal LIR motifs might then serve to accumulate LC3 molecules for their lipidation and incorporation into the growing isolation membranes by components of the ATG5–12:16 complex.
RILP Coordination of Autophagosome Formation with Motility
Using high temporal resolution live imaging, we found that RILP contributes to axonal transport of LC3- positive autophagosomes in the retrograde direction. Although RILP can be detected on isolation membranes, these structures are immotile, as demonstrated here by live imaging analysis. Direct in situ analysis of dynein distribution showed an absence of the motor protein from ATG5-positive isolation membranes, but a clear association with ATG5-negative RILP-containing structures. These observations suggest an unusual mechanism for RILP activation of dynein-mediated transport in concert with completion of isolation membrane closure and departure of the ATG12:5–16 complex.
As RILP binds to both ATG5 and dynein through the N-terminal domain, but rarely colocalizes with both markers, we propose that RILP function at the isolation membrane is dynein-independent. Our data suggest that RILP is initially recruited to the isolation membranes through its N-terminal interaction with ATG5. Dynein is detected predominantly on ATG5 negative membranes, suggesting that dynein may displace ATG5 from a common RILP N- terminal binding site. At this stage, RILP C-terminal LIR motifs may facilitate LC3 sequestration close to growing isolation membrane. Indeed, in vitro binding experiments show that RILP is able to simultaneously bind both ATG5 and LC3 and, unlike dynein, the presence of LC3 does not affect the efficiency of RILP binding to ATG5. Upon isolation membrane closure and departure of the ATG5:12–16 complex (Carlsson and Simonsen, 2015), RILP, through its C-terminal LIR motifs, may remain associated with the newly recruited LC3 on the autophagosomes. At this stage, we observe that RILP recruits dynein and confers motility to the fully-formed APs. Thus, the sequential RILP interactions with ATG5, LC3 and dynein that we identify here would serve to coordinate isolation membrane extension with the start of retrograde autophagosome motility. This would ensure that, wherever within the cell autophagic events occur, the completed AP should immediately begin its journey to the cell body for degradation.
mTOR Regulation of RILP Function and Autophagic Cargo Clearance
Autophagy is negatively regulated by mTOR kinase in yeast and non-neuronal mammalian cells. However, a functional link between mTOR activity and neuronal autophagosome behavior has remained incompletely explored. Perturbations of mTOR function have been implicated in several neurodevelopmental processes as well as neurodegenerative disorders (Abe et al., 2010, Kwon et al., 2006, Nie et al., 2010, Crino, 2013, Tang et al., 2014). Despite this, the underlying cellular and molecular mechanisms, including the contributions of autophagy, remain to be fully understood.
We find that induction of autophagy by mTOR inhibition results in transcriptional and translational upregulation of RILP, but not of several other known dynein or kinesin adaptors. Under these conditions, we observe an increase in RILP mRNA as well as protein levels and a clear increase in RILP-positive vesicles in neuronal and non-neuronal cells. Our data suggest, therefore, that upon mTOR inhibition RILP may be induced in order to participate in autophagosome formation and transport. Previous work has shown that upon mTOR inhibition, LAMP1-positive vesicles in non-neuronal cells show a redistribution from cell periphery towards the nucleus. Our data now suggest that this redistribution might also be under RILP control, though this possibility remains to be explored. Conceivably, RILP expression or function might also be dysregulated in neuropathological disorders with known mTOR dysfunction, which in turn could affect the RILP-mediated autophagy pathway. Evidence for such a role is so far lacking, and further work is likely needed to test this possibility more directly.
Subcellular aggregation of p62 has also been reported in several neurodegenerative disorders, but the molecular mechanisms leading to this pathology have remained incompletely understood. We find that RILP depletion or loss of its LC3-binding capacity potently interfere with autophagic clearance of Sequestosome1/p62, leading to its cytoplasmic aggregation. The p62 aggregates are enriched for ubiquitin as well as LC3, but are mostly negative for Rab7. This suggests that RILP depletion arrests p62 turnover during the early stages of the autophagy pathway, prior to its delivery to the Rab7-positive late endosomal compartment. Mutations of the three RILP LIR motifs also inhibit p62 turnover, which suggests that RILP’s association with autophagosomes, in particular, is required in this function. Furthermore, it has been reported that apart from binding to LC3, the LIR motif in p62 may play a role in establishing a crosstalk between the ubiquitinated substrates and the autophagy machinery to assist AP formation (Zaffagnini et al., 2018a, Danieli and Martens, 2018). Whether RILP can potentially play a role in stabilizing this crosstalk and in assisting cargo capture by the autophagy machinery acting in concert or independently of p62, remains to be tested. Over all, our data lead to the conclusion that RILP is required for autophagic clearance, presenting the interesting possibility that enhancement of RILP function, for example through mTOR inhibition, might help alleviate neurodegenerative pathology.
Together our results strongly support a new model for neuronal autophagy, according to which dynein adaptor RILP coordinates neuronal autophagosome biogenesis and transport to maintain neuronal homeostasis. The ability of RILP to associate with both growing isolation membranes and fully-formed autophagosomes, as well as Rab7-positive post-fusion autophagosomal membranes identifies a highly unusual range of interactions within a common pathway. Conceivably, RILP expression or function might be deregulated in neuropathological disorders, especially those known to have an aberrant mTOR activity. Further insights into RILP behavior in these conditions would help identify roles for RILP-mediated autophagy in neurodevelopmental and degenerative pathways, with potential therapeutic implications.
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard B. Vallee (rv2025@cumc.columbia.edu).
Plasmids generated in this study will be made available on request but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rat Cortical Neuron Culture and Transfection
All experiments involving animals were performed in accordance with protocols approved by the Columbia University Medical Center Institutional Animal Care and Use Committee. E19 embryonic brains from pregnant Sprague-Dawley female rats were used to prepare cortical neuronal cultures as described previously (Yi J., Khobrekar N. et.al. 2016). Briefly, embryonic brains were dissected in ice-cold HBSS (ThermoFisher Scientific #24020117) supplemented with 10mM HEPES (pH 7.4), 10mM MgCl2, 1% P/S and 0.5mM L-glutamine. The hippocampal tissue was discarded and only the cortical tissue was trypsinized for 15mins at 37°C in water bath and resuspended in DMEM with 10% FBS and 1% P/S. The dissociated neurons were spun down to remove debris, counted and plated onto MatTek 50mm glass bottom dishes, pre-coated with Poly-L-Lysine (PLL) (1mg/mL) and Laminin, at a density of 180,000 for live imaging experiments. ~12–15 hours post-plating, the medium was changed to neurobasal (Gibco) supplemented with 0.5mM L-Glutamine, B-27 serum free supplement (Gibco) and 1% P/S. The media were changed every 2–3 days. During Torin1 treatment, the medium was changed to neurobasal with insulin-free B27 supplement (Gibco). For immunofluorescence experiments, neurons were plated on 18×18mm coverslips or 35mm Glass bottom dishes coated with PLL and Laminin.
For live imaging experiments, DIV7–8 cortical neurons were transfected using magnetofectamine kit (OZ biosciences # MTX0750). 0.8μg of total DNA, 5μL of Lipofectamine 2000 and 0.75μL magnetic bead slurry were used to transfect one 50mm glass bottom dish of neurons, half of the medium was changed after 15 minutes and neurons were imaged after 15 hr. Cells with low expression of tagged proteins were selected for motility analysis. 2μg of RILP shRNA (rat RILP shRNA: 5’-GGAGGCTTAACTCTGGGTTCC- 3’) was used to transfect 1×106 cortical neurons with nucleofector kit (Lonza) and an amaxa nucleofector II (program O-003). Successfully transfected neurons were imaged 72 hr after knockdown at DIV3.
Cell Culture
C6 rat glioma cells (ATCC # CCL-107) were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (P/S), at 37°C with 5% C O2. Transfection of C6 cells was performed using a nucleofector kit V (Lonza) with an Amaxa Nucleofector II set to program U-030 or U-009, according to the manufacturer’s instructions. Cells for qRT-PCR analysis were collected 72 hours following transfection and sorted using fluorescence-activated cell sorting (FACS) to isolate GFP/RFP-positive cells.
METHOD DETAILS
Drug Treatments
mTOR inhibitor Torin1 (Tocris Biosciences) was resuspended in DMSO and used at final concentrations of 0.5 μM for short term 1–2 Hr treatments in C6 cells or rat cortical neurons (DIV13) for 6 Hr at 37°C. Bafilomycin A1 (Selleck Chemicals, #S 1413) was resuspended in DMSO and used at final concentration of 100nm for 2–6 Hr in combination with Torin1 in C6 glioma cells.
Immunoprecipitation and Western Blots
For immunoprecipitation experiments, C6 cells were transfected with 5μg plasmid DNA, harvested between 15–17 Hr post-transfection and pelleted at 1200 rpm for 10 minutes. Cell pellets were flash-frozen in liquid nitrogen and then lysed in IP buffer (50mM HEPES, 50mM PIPES, 2mM MgCl2, 1mM EDTA) supplemented with 1% NP-40, 1mM DTT, 1:100 protease inhibitor cocktail (Sigma-Aldrich #P8340) and 1:100 Phosphatase Inhibitors (ThermoFisher Scientific #78420) for 30 minutes on ice. Lysates were spun at 14,000 rpm at 4°C for 15 minutes and the supernatants were used for immunoprecipitations. For one immunoprecipitation, 30–40μL of GFP Trap-MA bead slurry (anti-GFP VHH coupled to magnetic agarose beads, ChromoTek) was used for 1–1.25 mg of total protein lysates. The pre-clearing of the lysate was performed using 40μL protein A Dynabeads (ThermoFisher Scientific) for 1 Hr at 4°C with constant mixing. The GFP-trap beads were then incubated with pre-cleared cell lysates for 2 hr at 4°C, with constant mixing. The beads were washed three times with lysis buffer and boiled at 95°C for 3 minutes in 1X laemmli buffer supplemented with beta-mercaptoethanol.
For Western blotting, cells were lysed on ice for 30min in RIPA Buffer (50mM Tris-HCl; 100mM NaCl; 1mM EGTA; 1% NP-40, pH 7.4), supplemented with 1:100 protease inhibitor cocktail, 1mM DTT and 1% NP-40. The lysates were loaded onto Bis-Tris gradient (4–20%) SDS–polyacrylamide gels (Life Sciences), along with Precision Plus Protein Dual Color Standards (161-0374; Bio-Rad). Proteins were transferred to 0.2μM polyvinylidene difluoride (PVDF) membranes (Immobilon-P #ISEQ00010; Millipore) overnight, and blocked in 5% milk for 2 hours at room temperature. Membranes were probed with primary antibodies diluted in 1% milk at 4°C, overnight. After washing with PBS, membranes were incubated with fluorescently tagged secondary antibodies (Rockland and Life Sciences) and scanned in an Odyssey system (LICOR Biosciences) version 3.0. Protein levels were quantified using ImageJ (NIH, Bethesda, MD).
In Vitro Binding Assays
Vectors for GST-tagged Full length and N-terminal RILP fragment were transformed into BL21-Gold(DE3) pLysS Escherichia coli (Agilent Technologies) and expressed for 4 hours at 18°C after induction with 0.1mM IPTG. Purification was performed following standard procedures involving application of the bacterial lysates to MagneGST Glutathione particles for 3Hr at 4°C on rotation in binding buffer (300mM NaCl, 50mM Tris, 1mM DTT, Lysozyme 1 mg/ml, cOmplete protease inhibitor EDTA-free, Roche at 1 mg/mL, DNase 10μg/mL and 0.1% NP-40, pH 7.4). Bound proteins were eluted in elution buffer (150mM NaCl, 50mM Tris and 1mM DTT, supplemented with 50mM reduced glutathione, pH 7.4). Overnight dialysis was performed in the elution buffer to remove glutathione from the eluates.
In vitro binding was performed using 0.5mM GST-tagged RILP constructs and recombinant human His-tagged ATG5 (Abcam # ab103787) in binding buffer (100mN NaCl, 50mM Tris, 1mM DTT and 1% NP-40) for 3 Hr at 4°C. The proteins were blotted for using anti-GST and anti-ATG5 antibodies.
In vitro competition assay was performed using 1μm GST-tagged RILP, His-tagged ATG5 and Myc-tagged LIC1 in Tris binding buffer (pH 7.4). Magnetic Glutathione beads were used for pulldowns after 3 Hr incubation at 4°C.
Immunofluorescence
Coverslips were washed twice with Tissue culture grade PBS to remove traces of culture medium. Cells were fixed in 4% Paraformaldehyde in PHEM buffer (120mM PIPES, 50mM HEPES, 20mM EGTA, 4mM MgSO4, pH = 7.4) for 12–15min at room temperature (RT), followed by permeabilization with 0.3% Saponin in PHEM buffer for 10 minutes at RT. Cells were blocked in 1% BSA in PBS or PHEM for 1 hr, stained with primary antibodies O/N at 4°C, washed for 30 minutes in PBS and then stained with fluorophore-conjugated secondary antibodies (Jackson laboratories) for 1 hr at room temperature. The coverslips were mounted in Aqua Poly/Mount (PolyScience #18606) and allowed to cure overnight at room temperature before imaging. Images were acquired using an IX83 Andor Revolution XD Spinning Disk Confocal with 60x silicone oil objective (NA 1.30) and a 2x magnifier coupled to an iXon Ultra 888 EMCCD Camera. Final images were compiled using ImageJ (NIH, Bethesda, MD) or Fiji.
Quantitative RT-PCR analysis
Cell lysis, mRNA to cDNA synthesis and quantitative PCR were performed using the Power SYBR Green Cells-to-Ct Kit (Ambion/ThermoFisher). Reactions were performed in a QuantStudio 6 Flex Real- Time PCR System (ThermoFisher). Primers were designed to have TMs around 60 degrees Celsius and to generate amplicons of 70 to 200 base pairs, ideally separated by at least one intron. Three replicates were performed for each condition per experiment and the experiments were carried out at least in triplicates. Target cDNA levels were analyzed by the comparative cycle (Ct) method and values were normalized against β-ACTIN, HPRT and YWHAZ housekeeping gene expression levels.
Live Imaging and Motility Analysis
Live imaging was performed using an IX83 Andor Revolution XD Spinning Disk Confocal System with an environmental chamber at 37°C, a 60x oil objective (NA 1.30) and a 2x magnifier coupled to an iXon Ultra 888 EMCCD Camera. Images were taken at the rate of 1 frame/sec for 3 or 5 minutes. Kymographs were generated with ImageJ (NIH) to quantify the distance travelled, velocity and direction of transport of the individual vesicles. Individual runs were counted if the pause between two processive movements was less than 10 seconds. If the pause was longer, then the runs were counted as separate.
QUANTIFICATION AND STATISTICAL ANALYSES
For all experiments involving rat neurons, triplicates were obtained from three different pregnant rats. sample sizes are specified in figure legends. All statistical analyses were performed in Prism 6 (GraphPad Software, La Jolla, CA, USA). For comparing two populations, two-tailed unpaired t-test with Welch’s correction was used. For multiple comparisons, ANOVA with Tukey’s multiple comparison test was used. p<0.05, p<0.01, p<0.001 values are labeled with *, ** and ***, respectively. Error bars represent mean + SEM unless stated otherwise.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
Supplemental Video S1 (Related to Figure 1)
GFP-RILP Colocalizes and Co-migrates with RFP-LC3-positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing GFP-RILP wt and RFP-LC3. Within the axon, RILP colocalizes and co-migrates with LC3-positive autophagosomes predominantly in the retrograde direction. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S2 (Related to Figure 1)
LIR3- mutant GFP-RILP Fails to Localize with RFP-LC3- positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing LIR3-mutant GFP-RILP and RFP-LC3. Unlike wt RILP, LIR3- mutant RILP fails to colocalize and co-migrate with LC3-positive autophagosomes. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S3 (Related to Figure 1)
Rab7-mutant GFP-RILP Colocalizes and Co-migrates with RFP-LC3- positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing Rab7-mutant GFP-RILP and RFP-LC3. Unlike LIR3- mutant RILP, Rab7-binding mutant RILP efficiently colocalizes and co-migrates with LC3-positive autophagosomes. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S4 (Related to Figure 1)
LIR3- mutant, but not the Rab7-mutant GFP-RILP Colocalizes and Co-migrates with mCh-Rab7-positive Late Endosomes
Live spinning disk confocal microscopy shows DIV8 rat cortical neurons transiently expressing Rab7-mutant or LIR3- mutant GFP-RILP and mCh-Rab7. Rab7-binding mutant RILP shows a striking decrease in late endosomal localization. On the contrary, LIR3- mutant RILP efficiently associates with Rab7-positive late endosomes. Movies acquired at 1 frame/second and played back at 20× real-time.
Supplemental Video S5 (Related to Figure 2)
RILP Knockdown Results in Decreased Retrograde AP and LE Transport
Live spinning disk confocal microscopy shows control and RILP knockdown DIV3 rat cortical neurons, co-transfected with either RFP-LC3 or mCh-Rab7. In control axons, RFP-LC3-positive Autophagosomes and mCh-Rab7-positive Late Endosomes show processive retrograde transport. Upon RILP depletion, many APs and LEs are stalled within the axon. Movies acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S6 (Related to Figure 5)
GFP-RILP and mCh-ATG5 Colocalize but Remain Stationary in Axons
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing GFP-RILP wt and mCh-ATG5. Within the axon, GFP-RILP wt shows enrichment on mCh-ATG5-positive isolation membranes but these structures do not exhibit directional movement. Movie acquired at 1 frame/second and played back at 20x real-time.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal LC3B | Novus Biologicals | Cat# NB100-2220, RRID:AB_10003146 |
| Rabbit Anti-p70 S6 Kinase Monoclonal Antibody, Unconjugated, Clone 49D7 | Cell Signaling Technology | Cat# 2708, RRID:AB_390722 |
| Rabbit Phospho-p70 S6 Kinase (Thr389) Antibody | Cell Signaling Technology | Cat# 9205, RRID:AB_330944 |
| Mouse Anti-GFP (Green Fluorescent Protein) Monoclonal Antibody, Unconjugated | Abcam | Cat# ab1218, RRID: AB_298911 |
| Rabbit Anti-mCherry antibody | Abcam | Cat# ab167453, RRID: AB_2571870 |
| Chicken anti-Dynein LIC1 antibody | N/A | |
| Rabbit Autophagy APG5L Antibody (N-term) | Abgent | Cat# AP1812a, RRID: AB_2258842 |
| Mouse GST (1E5) antibody | Santa Cruz Biotechnology | Cat# sc-53909, RRID: AB_783586 |
| Rabbit RILP Antibody | Abcam | Cat# ab128616, RRID:AB_11145272 |
| Rabbit Rab7 Antibody | Cell Signaling Technology | Cat# 9367, RRID: AB_1904103 |
| Mouse p62/SQSTM1 Antibody | Novus | Cat# NBP1–48320SS, RRID: AB_10011072 |
| Rabbit Ubiquitin Antibody | Abcam | Cat# ab7780, RRID: AB_306069 |
| Rabbit HOOK1 Antibody | Sigma-Aldrich | Cat# HPA018820, RRID: AB_1850910 |
| Rabbit HOOK3 Antibody | Proteintech | Cat# 15457–1-AP, RRID: AB_2120828 |
| Rabbit FYCO1 Antibody | Sigma-Aldrich | Cat# HPA035526, RRID:AB_10672867 |
| Rabbit BicD2 Antibody | Abcam | Cat# ab117818, RRID:AB_10938165 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Torin1 (mTOR Kinase Inhibitor) | Tocris Biosciences | Cat# 4247 |
| Bafilomycin A1 | Selleck Chemicals | Cat# S1413 |
| protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Phosphatase Inhibitors | ThermoFisher | Cat# #78420 |
| anti-GFP VHH coupled to magnetic agarose beads | ChromoTek | Gtma-10 |
| Recombinant human His-tagged ATG5 | Abcam | Cat# ab103787 |
| Recombinant GST-RILP wt | Scherer et.al. 2014 | N/A |
| Recombinant GST-RILP N-Terminus | Scherer et.al. 2014 | N/A |
| Recombinant GST-RILP C-Terminus | Scherer et.al. 2014 | N/A |
| Recombinant GST-RILP full length LIR Triple mutant | This study | N/A |
| Recombinant Myc-Light Intermediate Chain 1 | Scherer et.al. 2014 | N/A |
| Recombinant Human LC3B/MAP1LC3B | Abcam | Cat# AB103506 |
| Experimental Models: Cell Lines | ||
| C6 Rat Glioma cells | ATCC | CCL-107 |
| HeLaM Cells | Gift of | N/A |
| NRK-F Cells | ATCC | CRL-6509 |
| Oligonucleotides | ||
| Rat RILP shRNA: 5’-GGAGGCTTAACTCTGGGTTCC- 3’ |
IDT | N/A |
| Rat β-ACTIN Forward: 5’-CATGTACGTTGCTATCCAGGC- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat β-ACTIN Reverse: 5’-CTCCTTAATGTCACGCACGAT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat YWHAZ Forward: 5’-GCTACTTGGCTGAGGTTGCT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat YWHAZ Reverse: 5’-TGCTGTGACTGGTCCACAAT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat HPRT Forward: 5’-AGGCCAGACTTTGTTGGATT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat HPRT Reverse: 5’-GCTTTTCCACTTTCGCTGAT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat RILP forward: 5’-GATGCGGCAACCCCAGAT- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Rat RILP Reverse: 5’-GGCTTTGAGCTCATTCCGCTC- 3’ |
IDT | https://pga.mgh.harvard.edu/primerbank/ |
| Recombinant DNA | ||
| GFP-RILP wt | Scherer et.al. 2014 | N/A |
| GFP-RILP N-Terminus | Scherer et.al. 2014 | N/A |
| GFP-RILP C-Terminus | Scherer et.al. 2014 | N/A |
| GFP-RILP LIR1, LIR2, LIR3 Mutants | This paper | N/A |
| GFP-RILP Rab7 Mutant | This paper | N/A |
| RFP-LC3 | Gift from Dr. Gil Di Paolo lab | N/A |
| mCherry-Rab7A | Gift from Gia Voeltz (Rowland et al., 2014) |
Addgene plasmid # 61804; http://n2t.net/addgene:61804; RRID: Addgene_61804 |
| pmCherry-ATG5 | Gift from Roberta Gottlieb (Hamacher-Brady et al., 2007) |
Addgene plasmid # 13095; http://n2t.net/addgene:13095; RRID: Addgene_13095 |
| pmCherry-DFCP1 | This paper | N/A |
| pMXs-puro GFP-p62 | Gift from Noboru Mizushima (Itakura and Mizushima, 2010) |
Addgene plasmid # 38277; http://n2t.net/addgene:38277; RRID: Addgene_38277 |
| pMXs-puro GFP-DFCP1 | Gift from Noboru Mizushima (Itakura and Mizushima, 2010) |
Addgene plasmid # 38269; http://n2t.net/addgene:38269; RRID: Addgene_38269 |
| Software and Algorithms | ||
| ImageJ | ImageJ | |
| FIJI | https://fiji.sc/ | |
| PRISM 6 | GraphPad Software, La Jolla, CA, USA | N/A |
| Photoshop | Adobe | N/A |
| Illustrator | Adobe | N/A |
Highlights.
Rab-Interacting Lysosomal Protein RILP binds ATG5, LC3, Rab7 and cytoplasmic dynein
RILP controls autophagosome (AP) biogenesis, retrograde transport and p62 turnover
RILP expression and function are controlled by mTOR kinase
ATG5 – Dynein competition for RILP coordinates AP biogenesis with transport
Acknowledgements
This work was supported by NIH grants (R01GM102347 and P01GM105536-01A1-Reg) to R.V. We thank Drs Ai Yamamoto and Clarissa Waites (Columbia University, New York) for extensive helpful discussions, Dr. Waites for the critical reading of the manuscript and Dr. Alexandre Baffet (Institute Curie, France) for helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interest.
REFERENCES:
- ABE N, BORSON SH, GAMBELLO MJ, WANG F & CAVALLI V 2010. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem, 285, 28034–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALEMU EA, LAMARK T, TORGERSEN KM, BIRGISDOTTIR AB, BOWITZ LARSEN K, JAIN A, OLSVIK H, OVERVATN A, KIRKIN V & JOHANSEN T 2012. ATG8 Family Proteins Act as Scaffolds for Assembly of the ULK Complex: Sequence Requirements for LC3-Interacting Region (LIR) Motifs. Journal of Biological Chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALLY S, LARSON AG, BARLAN K, RICE SE & GELFAND VI 2009. Opposite-polarity motors activate one another to trigger cargo transport in live cells. J Cell Biol, 187, 1071–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AXE EL, WALKER SA, MANIFAVA M, CHANDRA P, RODERICK HL, HABERMANN A, GRIFFITHS G & KTISTAKIS NT 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol, 182, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORDI M, BERG MJ, MOHAN PS, PETERHOFF CM, ALLDRED MJ, CHE S, GINSBERG SD & NIXON RA 2016. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy, 12, 2467–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BREWER GJ & COTMAN CW 1989. Survival and growth of hippocampal neurons in defined medium at low density: advantages of a sandwich culture technique or low oxygen. Brain Res, 494, 65–74. [DOI] [PubMed] [Google Scholar]
- BREWER GJ, TORRICELLI JR, EVEGE EK & PRICE PJ 1993. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res, 35, 567–76. [DOI] [PubMed] [Google Scholar]
- BRIGHT NA, DAVIS LJ & LUZIO JP 2016. Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr Biol, 26, 2233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANTALUPO G, ALIFANO P, ROBERTI V, BRUNI CB & BUCCI C 2001. Rab-interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. Embo j, 20, 683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARLSSON SR & SIMONSEN A 2015. Membrane dynamics in autophagosome biogenesis. J Cell Sci, 128, 193–205. [DOI] [PubMed] [Google Scholar]
- CECCONI F & LEVINE B 2008. The Role of Autophagy in Mammalian Development: Cell Makeover Rather than Cell Death. Dev Cell, 15, 344–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Y, STEVENS B, CHANG J, MILBRANDT J, BARRES BA & HELL JW 2008. NS21: redefined and modified supplement B27 for neuronal cultures. J Neurosci Methods, 171, 239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG XT, ZHOU B, LIN MY, CAI Q & SHENG ZH 2015. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J Cell Biol, 209, 377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRINO PB 2013. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol, 125, 317–32. [DOI] [PubMed] [Google Scholar]
- DANIELI A & MARTENS S 2018. p62-mediated phase separation at the intersection of the ubiquitin proteasome system and autophagy. Journal of Cell Science, 131, jcs214304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DERUBEIS AR, YOUNG MF, JIA L, ROBEY PG & FISHER LW 2000. Double FYVE-containing protein 1 (DFCP1): isolation, cloning and characterization of a novel FYVE finger protein from a human bone marrow cDNA library. Gene, 255, 195–203. [DOI] [PubMed] [Google Scholar]
- DOOLEY HC, RAZI M, POLSON HE, GIRARDIN SE, WILSON MI & TOOZE SA 2014. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell, 55, 238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FU MM, NIRSCHL JJ & HOLZBAUR ELF 2014. LC3 binding to the scaffolding protein JIP1 regulates processive dynein-driven transport of autophagosomes. Dev Cell, 29, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO J, LANGEMEYER L, KUMMEL D, REGGIORI F & UNGERMANN C 2018. Molecular mechanism to target the endosomal Mon1-Ccz1 GEF complex to the pre-autophagosomal structure. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOWRISHANKAR S, YUAN P, WU Y, SCHRAG M, PARADISE S, GRUTZENDLER J, DE CAMILLI P & FERGUSON SM 2015. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci U S A, 112, E3699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRISON RE, BRUMELL JH, KHANDANI A, BUCCI C, SCOTT CC, JIANG X, FINLAY BB & GRINSTEIN S 2004. Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol Biol Cell, 15, 3146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JIA R, GUARDIA CM, PU J, CHEN Y & BONIFACINO JS 2017. BORC coordinates encounter and fusion of lysosomes with autophagosomes. Autophagy, 13, 1648–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSTON JA, WARD CL & KOPITO RR 1998. Aggresomes: a cellular response to misfolded proteins. J Cell Biol, 143, 1883–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORDENS I, FERNANDEZ-BORJA M, MARSMAN M, DUSSELJEE S, JANSSEN L, CALAFAT J, JANSSEN H, WUBBOLTS R & NEEFJES J 2001. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol, 11, 1680–5. [DOI] [PubMed] [Google Scholar]
- KALVARI I, TSOMPANIS S, MULAKKAL NC, OSGOOD R, JOHANSEN T, NEZIS IP & PROMPONAS VJ 2014. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy, 10, 913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAUSHIK S & CUERVO AM 2018. The coming of age of chaperone-mediated autophagy. Nature Reviews Molecular Cell Biology, 19, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM J, KUNDU M, VIOLLET B & GUAN K-L 2011. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology, 13, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWON CH, LUIKART BW, POWELL CM, ZHOU J, MATHENY SA, ZHANG W, LI Y, BAKER SJ & PARADA LF 2006. Pten regulates neuronal arborization and social interaction in mice. Neuron, 50, 377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAMARK T, KIRKIN V, DIKIC I & JOHANSEN T 2009. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle, 8, 1986–90. [DOI] [PubMed] [Google Scholar]
- MADAY S & HOLZBAUR ELF 2016. Compartment-Specific Regulation of Autophagy in Primary Neurons. J Neurosci, 36, 5933–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAUVEZIN C & NEUFELD TP 2015. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy, 11, 1437–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIZUSHIMA N 2018. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol, 20, 521–527. [DOI] [PubMed] [Google Scholar]
- MIZUSHIMA N, YOSHIMORI T & OHSUMI Y 2011. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol, 27, 107–32. [DOI] [PubMed] [Google Scholar]
- MORIYA S, KOMATSU S, YAMASAKI K, KAWAI Y, KOKUBA H, HIROTA A, CHE XF, INAZU M, GOTOH A, HIRAMOTO M & MIYAZAWA K 2015. Targeting the integrated networks of aggresome formation, proteasome, and autophagy potentiates ER stress-mediated cell death in multiple myeloma cells. Int J Oncol, 46, 474–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NARENDRA D, TANAKA A, SUEN DF & YOULE RJ 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol, 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIE D, DI NARDO A, HAN JM, BAHARANYI H, KRAMVIS I, HUYNH T, DABORA S, CODELUPPI S, PANDOLFI PP, PASQUALE EB & SAHIN M 2010. Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci, 13, 163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PANKIV S, ALEMU EA, BRECH A, BRUUN JA, LAMARK T, OVERVATN A, BJORKOY G & JOHANSEN T 2010. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol, 188, 253–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAIBORG C, WENZEL EM, PEDERSEN NM, OLSVIK H, SCHINK KO, SCHULTZ SW, VIETRI M, NISI V, BUCCI C, BRECH A, JOHANSEN T & STENMARK H 2015. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature, 520, 234–8. [DOI] [PubMed] [Google Scholar]
- RICHTER B, SLITER DA, HERHAUS L, STOLZ A, WANG C, BELI P, ZAFFAGNINI G, WILD P, MARTENS S, WAGNER SA, YOULE RJ & DIKIC I 2016. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A, 113, 4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SARKAR S, RAVIKUMAR B & RUBINSZTEIN DC 2009. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol, 453, 83–110. [DOI] [PubMed] [Google Scholar]
- SAXTON RA & SABATINI DM 2017. mTOR Signaling in Growth, Metabolism, and Disease. Cell, 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHERER J, YI J & VALLEE RB 2014. PKA-dependent dynein switching from lysosomes to adenovirus: a novel form of host-virus competition. J Cell Biol, 205, 163–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SETO S, MATSUMOTO S, TSUJIMURA K & KOIDE Y 2010. Differential recruitment of CD63 and Rab7-interacting-lysosomal-protein to phagosomes containing Mycobacterium tuberculosis in macrophages. Microbiol Immunol, 54, 170–4. [DOI] [PubMed] [Google Scholar]
- STARR T, NG TW, WEHRLY TD, KNODLER LA & CELLI J 2008. Brucella intracellular replication requires trafficking through the late endosomal/lysosomal compartment. Traffic, 9, 678–94. [DOI] [PubMed] [Google Scholar]
- TAKEI N & NAWA H 2014. mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci, 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN SC, SCHERER J & VALLEE RB 2011. Recruitment of Dynein to late endosomes and Lysosomes through light intermediate chains. Mol Biol Cell, 22, 467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TANG G, GUDSNUK K, KUO SH, COTRINA ML, ROSOKLIJA G, SOSUNOV A, SONDERS MS, KANTER E, CASTAGNA C, YAMAMOTO A, YUE Z, ARANCIO O, PETERSON BS, CHAMPAGNE F, DWORK AJ, GOLDMAN J & SULZER D 2014. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron, 83, 1131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOREEN CC & SABATINI DM 2009. Rapamycin inhibits mTORC1, but not completely. Autophagy, 5, 725–6. [DOI] [PubMed] [Google Scholar]
- UEMURA T, YAMAMOTO M, KAMETAKA A, SOU Y-S, YABASHI A, YAMADA A, ANNOH H, KAMETAKA S, KOMATSU M & WAGURI S 2014. A Cluster of Thin Tubular Structures Mediates Transformation of the Endoplasmic Reticulum to Autophagic Isolation Membrane. Molecular and Cellular Biology, 34, 1695–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANDER HAAR E, LEE SI, BANDHAKAVI S, GRIFFIN TJ & KIM DH 2007. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol, 9, 316–23. [DOI] [PubMed] [Google Scholar]
- WOZNIAK AL, LONG A, JONES-JAMTGAARD KN & WEINMAN SA 2016. Hepatitis C virus promotes virion secretion through cleavage of the Rab7 adaptor protein RILP. Proc Natl Acad Sci U S A, 113, 12484–12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU M, WANG T, LOH E, HONG W & SONG H 2005. Structural basis for recruitment of RILP by small GTPase Rab7. Embo j, 24, 1491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAP CC, DIGILIO L, MCMAHON LP, GARCIA ADR & WINCKLER B 2018. Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YI JY, ORI-MCKENNEY KM, MCKENNEY RJ, VERSHININ M, GROSS SP & VALLEE RB 2011. High-resolution imaging reveals indirect coordination of opposite motors and a role for LIS1 in high-load axonal transport. J Cell Biol, 195, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAFFAGNINI G, SAVOVA A, DANIELI A, ROMANOV J, TREMEL S, EBNER M, PETERBAUER T, SZTACHO M, TRAPANNONE R, TARAFDER AK, SACHSE C & MARTENS S 2018a. p62 filaments capture and present ubiquitinated cargos for autophagy. Embo j, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAFFAGNINI G, SAVOVA A, DANIELI A, ROMANOV J, TREMEL S, EBNER M, PETERBAUER T, SZTACHO M, TRAPANNONE R, TARAFDER AK, SACHSE C & MARTENS S 2018b. Phasing out the bad-How SQSTM1/p62 sequesters ubiquitinated proteins for degradation by autophagy. Autophagy. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video S1 (Related to Figure 1)
GFP-RILP Colocalizes and Co-migrates with RFP-LC3-positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing GFP-RILP wt and RFP-LC3. Within the axon, RILP colocalizes and co-migrates with LC3-positive autophagosomes predominantly in the retrograde direction. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S2 (Related to Figure 1)
LIR3- mutant GFP-RILP Fails to Localize with RFP-LC3- positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing LIR3-mutant GFP-RILP and RFP-LC3. Unlike wt RILP, LIR3- mutant RILP fails to colocalize and co-migrate with LC3-positive autophagosomes. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S3 (Related to Figure 1)
Rab7-mutant GFP-RILP Colocalizes and Co-migrates with RFP-LC3- positive Autophagosomes
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing Rab7-mutant GFP-RILP and RFP-LC3. Unlike LIR3- mutant RILP, Rab7-binding mutant RILP efficiently colocalizes and co-migrates with LC3-positive autophagosomes. Movie acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S4 (Related to Figure 1)
LIR3- mutant, but not the Rab7-mutant GFP-RILP Colocalizes and Co-migrates with mCh-Rab7-positive Late Endosomes
Live spinning disk confocal microscopy shows DIV8 rat cortical neurons transiently expressing Rab7-mutant or LIR3- mutant GFP-RILP and mCh-Rab7. Rab7-binding mutant RILP shows a striking decrease in late endosomal localization. On the contrary, LIR3- mutant RILP efficiently associates with Rab7-positive late endosomes. Movies acquired at 1 frame/second and played back at 20× real-time.
Supplemental Video S5 (Related to Figure 2)
RILP Knockdown Results in Decreased Retrograde AP and LE Transport
Live spinning disk confocal microscopy shows control and RILP knockdown DIV3 rat cortical neurons, co-transfected with either RFP-LC3 or mCh-Rab7. In control axons, RFP-LC3-positive Autophagosomes and mCh-Rab7-positive Late Endosomes show processive retrograde transport. Upon RILP depletion, many APs and LEs are stalled within the axon. Movies acquired at 1 frame/second and played back at 20x real-time.
Supplemental Video S6 (Related to Figure 5)
GFP-RILP and mCh-ATG5 Colocalize but Remain Stationary in Axons
Live spinning disk confocal microscopy shows a DIV8 rat cortical neuron transiently expressing GFP-RILP wt and mCh-ATG5. Within the axon, GFP-RILP wt shows enrichment on mCh-ATG5-positive isolation membranes but these structures do not exhibit directional movement. Movie acquired at 1 frame/second and played back at 20x real-time.
Data Availability Statement
This study did not generate any unique datasets or code.