

Abstract
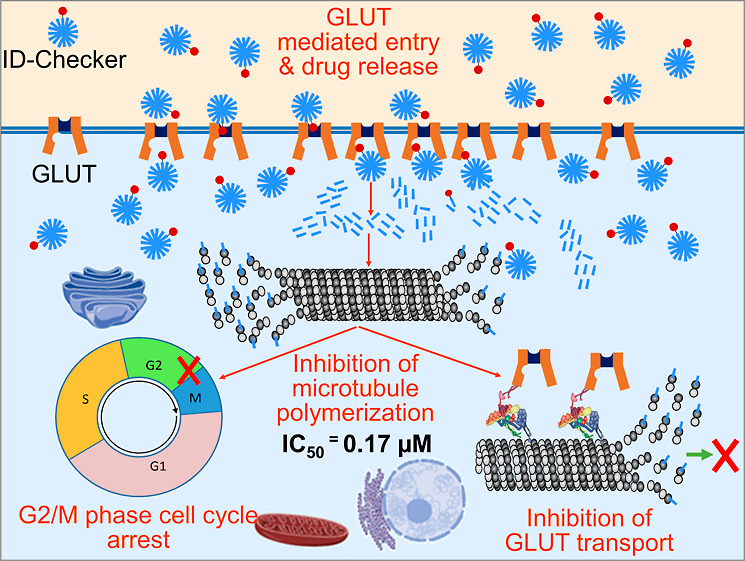
Cancer cells deploy several glucose transport protein (GLUT) channels on the cell membranes to increase glucose uptake. Cancer cells die within 24 h in the absence of glucose. Thus, preventing the deployment of GLUT channels can deprive them of glucose, resulting in apoptosis within 24 h. Herein, we developed the ID-Checker with a glucose tag that ensures its highly specific macroscale delivery of anticancer agents to the cancer cells through the GLUT channels. ID-Checker presented here showed IC50 values of 0.17–0.27 and 3.34 μM in cancer and normal cell lines, respectively. ID-Checker showed a selectivity index of 12.5–20.2, which is about 10–20 times higher than that of known anticancer agents such as colchicine. ID-Checker inhibits the microtubule formation, which results in the prevention of the deployment of GLUT channels in 6 h and kills the cancer cells within 24 h without any damage to normal cells.
1. Introduction
Anticancer drug discovery has always been a hot topic.1 Despite decades of efforts, an anticancer drug with high cancer cell selectivity and almost no side effects has been a distant goal.2 The drugs currently in practice for cancer therapy are known to be highly toxic and require several months of treatment without the desired outcomes in many cases.3,4 Furthermore, the recent research on using stem cells to treat incurable diseases has shown promising results, but with the drawback that such patients eventually develop cancer.5,6 Due to the unavailability of nontoxic chemotherapy, such efforts are in vain. Therefore, developing a highly selective anticancer drug that can kill cancer cells within 24 h without damaging normal cells is highly awaited.
There has been tremendous research on cancer cell metabolism and the development of anticancer drugs.7−11 Despite the outstanding accomplishments in achieving knowledge on cancer cell metabolism, a breakthrough in the cancer-cell selective and nontoxic anticancer drug is long-awaited. The inefficient energy production through anaerobic glycolysis requires cancer cells to enhance glucose uptake. Cancer cells achieve this by elevating the expression and relocation of glucose transport proteins (GLUTs) such as GLUT1 and GLUT4 to the plasma membrane compared to normal cells.12−17 The exemplary work in crystallization of the GLUT1 channel in 2014 and the identification of the mechanism behind glucose recognition and transport in the following year18 has significantly contributed to the field of anticancer drug discovery by exploiting the GLUT channels.19−22 The conformational change in the GLUT channel induces the alternate exposure of the glucose-binding site(s) to either side of the plasma membrane and allows glucose transport into the cells.23,24 Therefore, drug–glucose conjugates are developed to use GLUTs to boost membrane permeation and site-selective delivery of anticancer drugs.25−27 Even though the drug–glucose conjugates show higher anticancer activity than the drug itself, they are ineffective in terminating the tumor. The primary reason behind this anomaly is that the drug–glucose conjugates cross the GLUTs at a single molecular level. They do not achieve enough intracellular levels to shut down the targeted cancer cell mechanism. Therefore, a novel method for macroscale delivery of anticancer agents to the cancer cells through GLUT is essential to terminate tumors.28−30
The GLUT channels are deployed into the plasma membrane through a multistep process involving microtubule cytoskeletons. The polymerization and depolymerization process of tubulin (intracellular concentration of ∼5 μM) into microtubules is crucial for several intracellular processes. One of these processes is the GLUT channel transport to and from the plasma membrane. Therefore, the microtubule-targeting agents (MTAs) are gaining high interest in recent years because the stabilization or destabilization of microtubules results in the collapse of cellular mechanisms, including GLUT relocation to and from the plasma membrane, resulting in eventual cancer cell apoptosis. Cancer cells die within 24 h in glucose-depleted media, which has attracted our attention to develop a novel MTA that destabilizes the microtubule network and induces apoptosis in cancer cells.
Nocodazole (NOZ), fenbendazole (FBZ), mebendazole (MBZ), and albendazole (Al) like anthelmintic drugs containing the benzimidazole scaffold are recently reported to exhibit anticancer activity by inhibiting the formation of microtubules.31−35 The dividing cells are thus arrested in the G2/M phase, leading to the phagocytic removal of cancer cells. However, these drugs lack cancer cell selectivity and are bound to demonstrate high toxicity to normal cells as they are designed to cross the cell membrane. There are no reports on glucose conjugates of these drugs. The glucose conjugates of the drug molecules are known to have higher cancer cell selectivity than the nude ones.36−39
Herein, we report on the design and development of the ID-Checker for highly selective macroscale delivery of an anticancer agent to cancer cells. The ID-Checker is obtained by constituting aggregates of the albendazole amine glucose conjugate (Al–G) and albendazole ammonium ion (Al–NH3+) in a 1:100 ratio. The glucose tag on the surface of the ID-Checker ensures their highly specific entry to the cancer cells through the GLUT channels. The ID-Checker provides enough Al–NH3+ in the cancer cells to completely inhibit the polymerization of tubulins into microtubules, resulting in cancer cell apoptosis. The obtained results indicate that the ID-Checker technology presented here can prevent the deployment of GLUT channels in 6 h and kill the cancer cell in 24 h.
2. Results and Discussion
2.1. Development of ID-Checker for a Highly Specific Anticancer Therapy
The conjugation of glucose to the drug molecules imparts the cancer cell selectivity as compared to the drug itself. Therefore, we synthesized Al–G [a glucose conjugate of albendazole–amine (Al–NH2)] to develop an MTA–glucose conjugate for highly selective cancer cell targeting. Figure 1A depicts the reaction pathway for Al–G synthesis (Supporting Information, Figures S1–S9). The hydrolytic removal of the carbamate group of Al at 70 °C using an aqueous solution of potassium hydroxide in methanol generates Al–NH2 with over 72% yield. The glycosylation of Al–NH2 was carried out by a slight variation in the reported method for glycosylation of the aromatic amines. In brief, a hydrochloric acid solution (pH = 3.45) containing Al–NH2 (1.2 mM) and β-d-glucose (0.5 mM) in a Teflon-coated rubber-stoppered glass vial was kept at 40 °C for 4 h. During this time, aliquots of the reaction mixture were subjected to thin-layer chromatography (TLC) using 10% methanol in dichloromethane to check the progress of the reaction. The reaction was quenched by diluting the vial contents with acetate buffer (0.5 M, pH = 5.8). The solvent was evaporated under reduced pressure at 40 °C. The crude residue was then dissolved in an ample amount of methanol and purified by column chromatography using 10% methanol in dichloromethane as an eluent. The aliquots of the eluent were concentrated under vacuum to obtain Al–G with a yield of over 62%. We prepared the Al–NH3+Cl– (yield = 100%), a hydrochloride salt, by bubbling the HCl gas in the acetone solution containing Al–NH2 at 25 °C. All compounds are determined to be >95% pure by high-performance liquid chromatography (HPLC) analysis (Supporting Information, Figures S10–S12).
Figure 1.

Preparation of ID-Checker. (A) Scheme for the synthesis of Al–NH2, Al–NH3+Cl–, and Al–G and (B) their use in the preparation of the Al–NH3+ aggregate, Al–G aggregate, and ID-Checker. (C,E,G) HPLC spectra for Al–NH2, Al–NH3+Cl–, and Al–G samples prepared in methanol. (D,F,H) HPLC spectra for Al–NH3+ aggregate, Al–G aggregate, and ID-Checker samples prepared in 0.9% saline solution. The aqueous solution containing 40% methanol was used as an eluent.
As shown in Figure 1B, the Al–NH3+ aggregate, Al–G aggregate, and ID-Checker were prepared separately. The 1.0 M Al–NH3+ ethanolic stock solution was diluted to 200 mM using 0.9% saline solution to prepare the Al–NH3+ aggregate. The obtained solution was sonicated for 30 min and then allowed to stand for 2 h. Then, the solvent (about 1.5 times the amount of ethanol in the solution) was evaporated under reduced pressure to eliminate the ethanol. Then, the volume was matched with the distilled water to maintain the salinity (0.9%) of the final solution containing 200 mM Al–NH3+, which was left overnight at 25 °C to produce a solution containing the Al–NH3+ aggregate. Similarly, the 200 mM Al–G aggregate solution was prepared from the 1.0 M Al–G ethanolic stock solution by following the process mentioned for the Al–NH3+ aggregate. For ID-Checker preparation, the ethanolic stock solutions of Al–NH3+ and Al–G were mixed in a 100:1 ratio and diluted to 200 mM solution using 0.9% saline. The obtained mixture was sonicated for 30 min, and then, the solvent was evaporated to eliminate the ethanol from the solution, as mentioned earlier. The volume of the solution was matched with double-distilled water. The obtained solution was left overnight at 25 °C to produce a solution containing 200 mM ID-Checker.
The formation of the Al–NH3+ aggregate, Al–G aggregate, and ID-Checker was confirmed by HPLC, as shown in Figure 1C–H. As shown in Figure 1C,E,G, the methanolic solutions of Al–NH3+, Al–G, and 100:1 Al–NH3+, Al–G mixture, when eluted with 40% aqueous methanol, show two distinct peaks corresponding to the respective monomers (26.863, 20.768, and 26.865 min) and aggregates (27.957, 22.565, and 27.967 min). However, the Al–NH3+ aggregate (27.969 min), Al–G aggregate (22.684 min), and ID-Checker (27.977 min) samples showed a single peak for respective particles when eluted with 40% aqueous methanol, as shown in Figure 1D,F,H. The HPLC spectra indicate the formation of the particles of the Al–NH3+ aggregate and Al–G aggregate from their respective monomers. Furthermore, the formation of ID-Checker containing Al–NH3+ and Al–G with a 100:1 ratio is evident from its HPLC spectra as the peak for the Al–G aggregate (22.513 min) is less than 0.1%, and the peak corresponding to the ID-Checker (27.977 min) is over 99.9%.
The Al–NH3+ aggregate, Al–G aggregate, and ID-Checker were characterized by dynamic light scattering (DLS) using the Malvern Zetasizer NanoZS90, at 25 °C and a fixed angle of 90°. As shown in Table 1 (Supporting Information Figure S13), the sizes of the Al–NH3+ aggregate and ID-Checker were around 2.0 nm. The size of the Al–G aggregate was found to be around 2.5 nm. These results indicate that the Al–NH3+ aggregate, Al–G aggregate, and ID-Checker are nearly spherical in nature.
Table 1. Characterization of Aggregates (n = 3).
| aggregate | Al–NH3+/Al–G ratio | size (nm)a |
|---|---|---|
| Al–NH3+ aggregate | 100:0 | 2.03 ± 0.89 |
| Al–G aggregate | 0:100 | 2.50 ± 1.05 |
| ID-Checker | 100:1 | 2.02 ± 0.71 |
Mean diameter measured by dynamic light scattering (DLS).
2.2. In Vitro Antiproliferation Activity of ID-Checker Compared to Al, Al–NH3+, and Al–G
The compounds Al, Al–NH3+ aggregate, Al–G aggregate, and ID-Checker were studied for in vitro anticancer activities using five cell lines, including pancreatic cancer (ASPC-1), lung cancer (A549), breast cancer (MCF-7), colon cancer (HCT 116), and lung normal (MRC-5, IMR-90, CCD-18Co) cell lines. The IC50 (μM) values for 24 h incubation obtained through MTT assay are presented in Table 2. Colchicine (CLC) and NOZ were used as reference standards. The IC50 values for Al were in the range of 52.7–72.4 μM in cancer cell lines and 96.1 μM in a normal cell line (MRC-5). Al showed 7–10 times lower activity than the CLC and NOZ. Moreover, Al did not show significant selectivity for cancer cells (A549) over normal cells (MRC-5), indicated by a selectivity index (SI) of 1.32. The Al–NH3+ aggregate had IC50 values in the range of 64.6–86.7 μM in cancer cell lines and 178.9 μM in a normal cell line (MRC-5), which is about 2–2.8 times cancer cell selectivity. Interestingly, the Al–G aggregate demonstrated the IC50 values in the range of 131.0–182.5 μM in cancer cell lines and 895.1 μM in a normal cell line. The Al–G aggregate showed five to seven times cancer cell selectivity over normal cells (MRC-5).
Table 2. In Vitro Antiproliferation Activity of Al, Al–NH3+ Aggregate, Al–G Aggregate, and ID-Checker in Various Cell Lines After 24 h Incubation.
| IC50 ± SD (μM)a |
|||||||
|---|---|---|---|---|---|---|---|
| compound | ASPC-1 | A549 | MCF-7 | HCT 116 | MRC-5 | IMR-90 | CCD-18Co |
| Al | 56.3 (±4.81) | 72.4 (±6.15) | 59.0 (±9.01) | 52.7 (±1.95) | 96.1 (±2.33) | 284.7 (±21.2) | 119.0 (±15.5) |
| Al–NH3+ | 86.7 (±8.84) | 70.9 (±7.62) | 64.6 (±11.3) | 73.2 (±4.01) | 178.9 (±7.32) | >1000 | 337.9 (±12.3) |
| Al–G | 182.5 (±12.5) | 138.6 (±17.7) | 131.0 (±14.4) | 156.3 (±16.0) | 895.1 (±13.7) | >1000 | 701.1 (±11.2) |
| ID-Checker | 0.27 (±0.02) | 0.21 (±0.03) | 0.17 (±0.002) | 0.18 (±0.01) | 3.34 (±0.10) | 15.9 (±2.25) | 11.6 (±2.45) |
| CLC | 10.4 (±0.12) | 9.63 (±0.25) | 8.34 (±0.62) | 7.81 (±0.34) | 14.0 (±1.15) | * | * |
| NOZ | 10.5 (±0.76) | 10.8 (±0.53) | 7.41 (±0.33) | 6.82 (±0.75) | 12.0 (±1.18) | * | * |
Data are the average of three independent assays and presented as mean (±SD); *, not determined; Al, albendazole; CLC, colchicine; and NOZ, nocodazole.
ID-Checker showed IC50 values of 0.17–0.27 and 3.34 μM in cancer cell lines and normal cell line (MRC-5), respectively, with an SI of 12.5–20.2. The SIs for CLC and NOZ were in the range of 1.3–1.7 and 1.1–1.8, respectively. As shown in Table 2, it is crucial to note that the ID-Checker efficiently inhibits glucose uptake in cancer cells with highly selective macroscale delivery of anticancer agents to cancer cells without damage to normal cells.
We believe that the high SI of ID-Checker for cancer cells is because of the tagged glucose on its surface. As shown in Scheme 1, the glucose of the ID-Checker acts as an identity (ID) key for the GLUT channel. The GLUT channels on the cell membrane recognize the glucose of ID-Checker in its glucose-binding site and allow the entry of ID-Checker into the cell. The dynamics of opening and closing of the GLUT through glucose recognition enable the transport of ID-Checker across the cell membrane. There are many more GLUT proteins on the cancer cell membrane than normal cells, deployed originally to uptake glucose in large amounts. The uptake of ID-Checker exclusively through the GLUT channel endows higher selectivity for cancer cells than normal cells. The Al–G aggregate contains only Al–G molecules. Thus, this closely packed glucose in the Al–G aggregate prohibits it from recognition by GLUT channels. The Al–NH3+ aggregate does not enter the cell through GLUT channels. The Al–NH3+ aggregate and Al–G aggregate enter the cell through the cell membrane, probably by endocytosis, and show high toxicity toward the normal cells. However, due to the high selectivity of GLUT channels, the ID-Checker selectively kills cancer cells within 24 h without significant harm to normal cells.
Scheme 1. Scheme Depicting the (A) ID-Checker Entering the Cell through the GLUT Channel by ID (Glucose) Recognition, (B) Al–G Aggregate, and (C) Al–NH3+ Aggregate That Cannot Enter through the GLUT Channel or Directly through the Cell Membrane.

2.3. In Vivo Antitumor Activity of ID-Checker
We evaluated ID-Checker’s in vivo antitumor activity using the xenograft model in BALB/c mice. The xenograft model was established by subcutaneous injection of 4T1 cells into the left mammary adipose tissues of the BALB/c mice. Results of the 11 day animal study are presented in Figure 2A–E, and the results of the 3 day study are shown in Figure 2F.
Figure 2.

ID-checker treatment causes tumor regression in BALB/c mice. Representative H&E-stained tumor sections from (A) outside and (B) inside the tumor tissues. The white area indicates necrotic tumor cells. All images were acquired at 20× magnification. (C) Changes in the tumor volume. (D) Average mass (g) of the excised tumor tissue at the endpoint. Photographs of excised tumors from each animal in the respective groups after (E) 11 day treatment (G1, vehicle control; G2, ID-Checker) low-dose group (0.711 mg/kg for day 1–3 and 14.22 mg/kg for day 4–11); G3, ID-Checker high-dose group (1.422 mg/kg for day 1–3 and 28.44 mg/kg for day 4–11); G4, 2 mg/kg doxorubicin (once in 3 days), and (F) 3 day treatment (G1, vehicle control; G3, 28.44 mg/kg ID-Checker). (G) MCF-7 cells were treated with vehicle (0.9% saline), 0.50, 0.25, 0.50, and 1.0 μM ID-Checker for 6, 12, 18, and 24 h. Cells were incubated with the anti-α-tubulin antibody followed by anti-mouse IgG/FITC antibody (green fluorescence) to stain the microtubules, and nuclei were stained by subsequent incubation of cells with DAPI (blue fluorescence). Bottom, DAPI; middle, microtubule network; top, merged images observed by confocal microscopy.
When the tumor volume had grown about 50 mm3, mice were arbitrarily allocated to four groups. Group 1 (G1) was assigned as a control group and was subjected to intraperitoneal (IP) administration of the vehicle (0.9% saline). Mice in group 2 (G2) received the ID-Checker by IP administration at a dose of 0.711 mg/kg for days 1–3 and 14.22 mg/kg for days 4–11. Mice in group 3 (G3) were treated with IP injection of ID-Checker at a dose of 1.422 mg/kg for days 1–3 and 28.44 mg/kg for days 4–11, whereas mice in group 4 (G4 DOX) were given IP injection of 2 mg/kg doxorubicin once in 3 days. Animals in all groups were sacrificed, and solid tumors were excised on day 12 (Figure 2E). The excised tumor tissues were dissected and stained with hematoxylin–eosin (H&E, Figure 2A,B) after measuring the respective volume (Figure 2C) and mass (Figure 2D). There was no significant difference in the weight of animals in each group measured at the beginning and the endpoint (Supporting Information, Table S1).
As depicted in Figure 2A,B, the outside and inside H&E-stained tumor sections demonstrate nonsignificant necrosis in G1. However, H&E-stained tumor sections from G2 and G3 showed a higher degree of necrosis than the doxorubicin-treated G4 DOX group. Interestingly, cancer cells inside the tumor undergo several folds higher degree of necrosis upon treatment with the 28.44 mg/kg ID-Checker compared to doxorubicin-treated mice. We believe that one of the reasons behind necrotic tumor cells is their inability to uptake glucose. As mentioned earlier, ID-Checker enters cells through the GLUT channels and inhibits the tubulin polymerization into microtubules. The disruption of the microtubule network causes a collapse in GLUT channel translocation to the cell membranes responsible for the necrosis in glucose-deprived tumor cells.
As shown in Figure 2C, the lower dose (G2 = 0.711 and G3 = 1.422 mg/kg) treatment for 3 days with ID-Checker did not show a significant change in the tumor volume compared to mice in G1 and G4 DOX. Thus, we increased the dose by 20-fold from the fourth day of treatment. The tumor volume showed about 30–40% decrease during just 3 days (day 4–day 6) in mice treated with higher doses of ID-Checker (G2 = 14.22 and G3 = 28.44 mg/kg) compared to mice in G1. At the endpoint, the average tumor volume for G3 showed rescission by 37.2% compared to the G1. Mice in G3 showed 2× and 1.3× lower reduction in tumor volumes compared to G2 (16.4%) and G4 DOX (29.4%) group mice. As shown in Figure 2D, the average mass of tumor tissue for mice in G3 (24.6%) showed a significant reduction compared to the mice in G2 (9.2%) and G4 DOX (14.5%). However, it is noticeable that the decrease in tumor volumes was not expressed well in reducing tumor masses. One of the reasons behind this discrepancy is the severe necrosis inside the tumor, resulting in lower chances for dead cell clearance.
The promising results of the initial in vivo investigation of the anticancer activity of ID-Checker prompted us to evaluate its effectiveness in a short study. Hence, we conducted a new 3 day study by administering 28.44 mg/kg ID-Checker to mice designated as G3 and vehicle to the control group (G1). The pictures of excised tumor tissues in both groups on day 4 are presented in Figure 2F. The treatment with ID-Checker resulted in a 61% tumor mass decrease in G3 (0.033 g) compared to untreated group G1 (0.085 g). The tumor volume in treated group G3 (41.9 mm3) was about 50% lower compared to that in G1 (84.5 mm3). These results indicate that the ID-Checker shows excellent results in killing the cancer cells in just 3 days. Even at a 14 times higher dose than doxorubicin, ID-Checker kills cancer cells without observable toxicity to mice.
2.4. Investigation of the Mode of Action for Anticancer Activity of ID-Checker
2.4.1. ID-Checker Inhibits Tubulin Polymerization
We investigated the mode of action for anticancer activity of ID-Checker through an in vitro tubulin polymerization assay (Supporting Information, Figure S14). Paclitaxel (PCL) and NOZ are known microtubule-stabilizing and microtubule-destabilizing agents, respectively. Hence, these compounds were used as positive controls. The control experiments were carried out without adding any compounds to the vehicle (DMSO). As expected, the PCL (20 μM) enhanced the tubulin polymerization, and NOZ (20 μM) inhibited it compared to the vehicle control. The incubation of tubulin with 20 μM each of Al, Al–NH3+ aggregate, and Al–G aggregate and 0.20 μM ID-Checker demonstrated the inhibition of tubulin polymerization similar to Al and NOZ. These results indicate that the ID-Checker binds to the tubulin and inhibits the tubulin polymerization, making it a microtubule destabilizing agent.
2.4.2. ID-Checker Disrupts the Microtubule Cytoskeleton and Blocks GLUT Channel Transport in Cancer Cells
We investigated the effect of ID-Checker on the tubulin polymerization into the microtubule network, its distribution, and arrangement in MCF-7, A549, and MRC-5 cells by in situ immunofluorescence assay. As shown in Figure 2G, treatment of MCF-7 with ID-Checker at various concentrations (0, 0.05, 0.25, 0.50, and 1.0 μM) for various time intervals (6, 12, 18, and 24 h) revealed its ability to inhibit the tubulin polymerization in tumor cells. Interestingly, ID-Checker disrupts the microtubule cytoskeleton in 6 h at 1.0 μM, and a similar result is observed for 0.05 μM when cells were treated for 24 h. The results for immunofluorescence staining of microtubules in A549 cells also confirm ID-Checker’s ability to disrupt the microtubule cytoskeleton (Supporting Information, Figure S15). In both MCF-7 and A549 cell lines, ID-Checker inhibited tubulin polymerization upon treatment for 6–12 h at lower concentrations and killed most cells upon treatment with 0.5 and 1.0 μM in 18–24 h.
The disruption of the microtubule network hampers the transport of GLUT channels to and from the cell membrane, resulting in cancer cell apoptosis as they cannot uptake glucose. Thus, we investigated the effect of ID-Checker on the GLUT1 and GLUT4 translocation process by in situ immunofluorescence assay in MCF-7 and A549 cells. As shown in Figure 3A–D, the number of GLUT channels on the cancer cells decreases with increasing ID-Checker concentrations from 0.05 to 0.50 μM, and all cells die at a concentration of 1.0 μM upon 24 h treatment. Furthermore, the time-dependent study including 6, 12, 18, and 24 h treatment of A549 cells with various concentrations of ID-Checker revealed that GLUT channel transport is blocked in just 6 h by 1.0 μM ID-Checker (Supporting Information, Figure S16). These results indicate that the disruption of the microtubule network by ID-Checker collapses the GLUT channel transport, leading to the shortage in intracellular glucose and eventual cell death in 24 h.
Figure 3.

ID-Checker disrupts the transport of GLUT channels in cancer cells. Immunofluorescence staining of GLUT channels in (A,B) MCF-7, (C,D) A549 cells, and (F) MRC-5 cells. Cells were treated with vehicle (0.9% saline) and 0.05, 0.25, 0.50, and 1.0 μM ID-Checker for 24 h followed by successive incubation with rabbit anti-GLUT1 or anti-GLUT4 antibodies and the donkey anti-rabbit IgG/ALEXA488 antibody (green fluorescence) to stain the GLUT channels. The nuclei were stained by subsequent incubation of cells with DAPI (blue fluorescence). (E) MRC-5 cells were incubated with the anti-α-tubulin antibody followed by the anti-mouse IgG/FITC antibody (green fluorescence) to stain the microtubules, and the nuclei were stained by subsequent incubation of cells with DAPI (blue fluorescence). Bottom, DAPI; middle, GLUT channels; top, merged images observed by confocal microscopy.
As shown in Table 2, the IC50 (3.34 ± 0.11) value of ID-Checker in a normal cell line (MRC-5) was 16 times higher than that of A549 cancer cell lines, indicating that it would not show any toxicity even at 1.0 μM concentration upon 24 h treatment. An SI of 1.2 for NOZ and CLC indicated that these drugs enter the cell through the cell membrane. An SI of 16 for ID-Checker indicated that ID-Checker enters the cell through the GLUT channel and not the cell membrane. Thus, we investigated the effect of ID-Checker (0, 0.5, 0.25, 0.50, and 1.0 μM) on the microtubule cytoskeleton and GLUT1 channel in normal cells (MRC-5) in a 24 h study. ID-Checker did not affect the microtubule cytoskeleton (Figure 3E) or translocation of GLUT1 channels (Figure 3F) in normal cells even at the highest concentration of 1.0 μM. These results indicate that the ID-Checker is not absorbed well by normal cells, thus showing almost no toxicity. As mentioned earlier, we believe that one of the reasons behind the selectivity of ID-Checker toward cancer cells is its ability to enter cells through the GLUT channels. Cancer cells present a lot more GLUT channels than normal cells. Therefore, the selectivity of ID-Checker for cancer cells is apparent. Reported glucose-conjugated drugs enter cells through GLUT channels as individual molecules. Thus, these drugs do not reach enough intracellular concentration to kill cancer cells. However, ID-Checker delivers a large amount of Al–NH3+ at once that effectively disrupts the microtubule cytoskeleton in 6–12 h, resulting in cell cycle arrest and ensuing apoptosis.
ID-Checker technology presented here can completely inhibit glucose uptake in cancer cells with highly selective macroscale delivery of anticancer agents to cancer cells. Therefore, ID-Checker prevents the deployment of GLUT channels in 6 h and kills the cancer cells within 24 h without any damage to normal cells.
2.4.3. ID-Checker Arrests the Cell Cycle in the G2/M Phase
Almost all MTAs induce cell-cycle arrest at the G2/M phase, thereby influencing cell division and cell proliferation.40,41ID-Checker exhibited a potent antiproliferation activity against a panel of breast and lung cancer cell lines and in vivo antitumor activity. Thus, we investigated its effect on the cell cycle distribution in A549 and MCF-7 cells by flow cytometry after staining the cells with propidium iodide.
A549 and MCF-7 cells were treated with 0, 0.5, 0.25, 0.50, and 1.0 μM Al, Al–NH3+, Al–G, and ID-Checker for 24 h. The cell cycle phase distributions (G1/G0, S, and G2/M) were compared among the cells from each group. A549 (Figure 4A) and MCF-7 (Supporting Information, Figure S17) cell lines treated with vehicle displayed the normal cell cycle progression. The treatment of cells with Al, Al–NH3+, and Al–G resulted in the slight accumulation of cells in the G2/M phase. However, these changes were insignificant compared to cells treated with the vehicle. In contrast, cells treated with ID-Checker resulted in a pronounced accumulation of cells in the G2/M phase and associated decrease in cells in the G1 and S phases.
Figure 4.

ID-Checker arrests the cell cycle in the G2/M phase. (A) Effects on the cell cycle distribution of A549 cells upon treatment with Al, Al–NH3+, Al–G, and ID-Checker at various concentrations (0, 0.05, 0.25, 0.50, and 1.0 μM) for 24 h followed by staining with PI to determine the proportion of DNA by flow cytometry, (B,C) Bar graph shows the percentage of A549 and MCF-7 cells in each phase of the cell cycle.
As shown in Figure 4A, 5 μM ID-Checker arrested over 80% of cells in the G2/M phase, and almost no living cell was found at 1.0 μM in the A549 cell line. In a study with the MCF-7 cell line, 0.25 μM ID-Checker arrested over 60% of cells in the G2/M phase, and no living cell was found when treated with 1.0 μM ID-Checker (Supporting Information, Figure S17). As shown in Figure 4B,C, the treatment of A549 cells and MCF-7 with ID-Checker for 24 h at various concentrations resulted in cell cycle arrest at the G2/M phase with higher percentages than Al, Al–NH3+ aggregate, and Al–G aggregate. Overall, these results establish that ID-Checker efficiently induced the G2/M phase arrest of both A549 and MCF-7 cells.
2.4.4. Molecular Docking Reveals a Higher Affinity for Al–NH3+ in CBS and NBS Compared to Al–G
The molecular docking results for the binding of Al, Al–NH3+, and Al–G in the CLC binding site (CBS, PDB: 4O2B) and NOZ binding site (NBS, PDB: 5CA1) are presented in Supporting Information, Table S2. Overall, Al, Al–NH3+, and Al–G showed higher docking scores for NBS (−6.61––7.31 kcal mol–1) compared to CBS (−6.20––6.64 kcal mol–1). The CBS is on the interface between α- and β-dimeric complexes with CLC binding mostly in the β-tubulin cavity with the minor interactions with the α-tubulin. Analysis of docking results indicates that the Al, Al–NH3+, and Al–G best fit in the cavity of the β-tubulin, and none of the atoms of these molecules perturbed up to the α-tubulin (Supporting Information, Figure S18).
As indicated by the docking scores, Al and Al–NH3+ demonstrated similar binding strengths in CBS (−6.64 and −6.44 kcal mol–1) and NBS (−7.31 and −7.14 kcal mol–1). Al showed several hydrophobic interactions and four hydrogen-bonding interactions in the NBS. The hydrogen-bonding interactions included ring N–H with VAL236 (2.16 Å), ring N with two amino acids including TYR200 (2.29 Å), GLU198 (2.08 Å), and carbamate carbonyl with ASN165 (2.62 Å), whereas Al–NH3+ displayed several hydrophobic interactions and two hydrogen bonds, including ring N–H with VAL236 (1.97 Å) and −NH2 hydrogen with GLU198 (2.51 Å) in the NBS. In contrast, instead of the benzimidazole nitrogens Al–G, hydroxyl groups in glucose showed hydrogen bonds with TYR200, VAL236, and CYS239. It is important to note that the extremely hydrophilic nature of the glucose residue in Al–G would have a very high desolvation penalty. Thus, it is unlikely that the glucose residue will participate in binding interactions with binding regions in NBS. These results are also reflected in the IC50 values of Al–G that are about two times higher than for Al and Al–NH3+. We believe that ID-Checker shows highly selective anticancer activity because it can deliver a large amount of Al–NH3+ with every ID-Checker particle that enters the cell. Complete inhibition of glucose uptake by cancer cells with highly selective macroscale delivery of anticancer agents to cancer cells can kill them within 24 h without damage to normal cells.
3. Conclusions
Here, we presented a method to kill the cancer cells in 24 h by destroying the framework required for glucose uptake in cancer cells. The results presented here agree with Otto Warburg’s discovery on the complete dependency of cancer cells on glucose. The glucose tag on the ID-Checker ensures its highly specific macroscale delivery of anticancer agents to the cancer cells through the GLUT channels. Anticancer agents inhibit the microtubule formation and thus the deployment of GLUT channels on membranes to block cancer-cell glucose uptake completely. We achieved this remarkable feat with ID-Checker technology presented here for the highly selective macroscale delivery of anticancer agents to cancer cells. Therefore, ID-Checker technology is a powerful tool for highly selective anticancer therapy through highly selective macroscale delivery of anticancer agents to cancer cells.
4. Experimental Section
4.1. Materials and Methods
All chemicals and reagents were procured from Sigma-Aldrich (South Korea), Thermo Fisher Scientific (South Korea), TCI Chemicals (South Korea) and used as received unless otherwise stated. All deuterated solvents were purchased from Cambridge Isotope Laboratories. The successful formation of the product was confirmed by monitoring the reaction using TLC on Merck Silica Gel 60 F254 plates. The developed plates were visualized under UV light (254 nm). All solvents were of HPLC grade from SK Chemicals, Korea. Ultrapure water (18 MΩ/cm) was obtained from a Milli-Q purification system (Millipore, USA). Cell viability was assessed by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin, and 3-(4,5-dimethyl thiazol2yl)-2,5-diphenyl tetrazolium bromide were purchased from Thermo Fisher Scientific. The tubulin polymerization assay kit (006P) was procured from Cytoskeleton, Inc. (Denver, USA). Cell culture plates and glass coverslips were purchased from SPL Life Sciences. Pancreatic cancer (ASPC-1), lung cancer (A549), breast cancer (MCF-7), colon cancer (HCT 116), and lung normal (MRC-5) cell lines were obtained from the Korea Cell Line Bank.
The synthesized compounds were characterized by 1H and 13C NMR on a Jeol FT-NMR spectrometer (400 MHz; JEOL, Japan). Spectra were visualized and analyzed using MestReNova (version 10.0). The chemical shifts (δ) are reported in parts per million (ppm), and the coupling constants (J) are quoted in Hz. The mass spectra were recorded on a JMS-700 MStation mass spectrometer (JEOL, Japan) or Agilent Technologies 7820A GC/5977E MSD (Agilent Technologies, USA). The Shimadzu UV-24500 (Shimadzu, Tokyo, Japan) spectrometer and Agilent Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, Santa Clara, CA, USA) were used to record UV–visible spectra and fluorescence emission spectra, respectively. A JMS-700 MStation mass spectrometer (JEOL, Tokyo, Japan), a microplate reader ABS Plus (Molecular Devices, San Jose, CA, USA), and a confocal laser scanning microscope (Carl Zeiss LSM710, Osnabrück, Germany) were also used in the present study. The aggregate sizes were recorded by the DLS technique using the Malvern Zetasizer NanoZS90 (Malvern Instruments Ltd, Malvern, U.K.), at 25 °C and a fixed angle of 90°. No statistical methods were used to predetermine sample size in animal experiments. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
4.1.1. Synthesis of Al–NH2
To a suspension of albendazole (886 mg, 3.34 mmol) in methanol (25 mL) and water (6.5 mL) was added potassium hydroxide (375 mg, 6.68 mmol). The resultant mixture was heated at reflux, giving a yellow solution. After heating for 72 h, TLC analysis showed that the starting material was present. A further portion of potassium hydroxide (375 mg, 6.68 mmol) was added, and the mixture was stirred for a further 24 h. The mixture was allowed to cool to room temperature before removing methanol in vacuo. The remaining aqueous was then diluted with CH2Cl2 (3 × 20 mL) before combined organic layers were washed with brine (50 mL), dried over sodium sulfate, and concentrated in vacuo to afford Al–NH2 (502 mg, 72.5%) as a light gray solid. 1H NMR (400 MHz, CD3OD): δ 7.25 (s, 1H1), 7.10 (d, J = 8.13 Hz, 2H2), 7.05 (d, J = 8.12 Hz, 2H3), 2.80 (t, J = 7.16 Hz, 2H4), 1.58 (sextet, J = 7.32 Hz, 2H5), 0.99 (t, J = 7.36 Hz, 3H6). 13C NMR (100 MHz, CD3OD): δ 157.20, 139.76, 138.52, 127.62, 125.77, 116.64, 112.95, 39.21, 23.67, 13.53. HRMS(EI) m/z: [M]+ calcd for C10H13N3S, 207.0830; found, 207.0829. HPLC purity 99.77%.
4.1.2. Synthesis of Al–NH2·HCl (Al–NH3+Cl)
To a suspension of Al–NH2 (5 g, 24 mmol) in acetone (30 mL) was bubbled excess HCl gas generated using sodium chloride (4.2 g, 72 mmol) and sulfuric acid (3.9 mL, 72 mmol). The resultant mixture was stirred for 30 min at rt. The mixture was filtered to obtain Al–NH2·HCl (4.7 g, 80%) as a light ivory solid. 1H NMR (400 MHz, DMSO-d6): δ 12.58 (s, 2H1), 8.58 (s, 2H2), 7.34–7.29 (m, 1H3, 1H4), 7.20 (dd, J = 8.31, 1.70 Hz, 1H5), 2.90 (t, J = 7.14 Hz, 2H6), 1.55 (sextet, J = 7.26 Hz, 2H7), 0.95 (t, J = 7.33 Hz, 3H8). 13C NMR (100 MHz, DMSO-d6): δ 150.69, 130.45, 130.05, 128.43, 124.78, 112.35, 111.90, 35.76, 21.96, 13.10. HRMS(ESI) m/z: [M + H]+ calcd for C10H14N3S+, 208.0903; found, 208.0930. HPLC purity 99.95%.
4.1.3. Synthesis of Al–G
The hydrochloric acid solution (pH = 3.45) containing Al–NH2 (1.2 mM) and β-d-glucose (0.5 mM) in a Teflon-coated rubber-stoppered glass vial was kept at 40 °C for 4 h. During this time, aliquots of the reaction mixture were subjected to TLC using 10% methanol in dichloromethane to check the progress of reaction. The reaction was quenched by diluting the contents of the vial with acetate buffer (0.5 M, pH = 5.8). The solvent was evaporated under reduced pressure at 40 °C. The crude residue was then dissolved in ample amount of methanol and purified by column chromatography using 10% methanol in dichloromethane as an eluent. The aliquots of the eluent were concentrated under vacuum to obtain Al–G with the yield over 62%. 1H NMR (400 MHz, CD3OD): δ 7.35 (s, 1H1′), 7.32 (s, 1H1), 7.21–7.16 (m, 1H2), 7.12–7.09 (m, 1H3), 5.39 (d, J = 5.10 Hz,1H4), 4.86 (d, J = 8.88 Hz, 1H4′), 3.89–3.33 (m, 1H5–2H9), 2.82 (t, J = 7.16 Hz, 2H10), 1.58 (sextet, J = 7.32 Hz, 2H11), 0.99 (t, J = 7.33 Hz, 3H12). 13C NMR (100 MHz, CD3OD): δ 156.07, 139.28, 138.03, 128.36, 128.19, 125.99, 125.92, 116.90, 113.47, 85.30, 82.75, 78.92, 74.97, 74.30, 73.64, 71.88, 71.70, 71.34, 62.90, 62.60, 39.05, 23.66, 13.53. HRMS(FAB) m/z: [M + H]+ calcd for C16H24N3O5S+, 370.1431; found, 370.1439. HPLC purity 99.95%.
4.2. HPLC Purity
All compounds are >95% pure by HPLC analysis (Supporting Information, Figures S10–S12).
4.3. Cell Culture
Human cell lines including pancreatic cancer (ASPC-1), lung cancer (A549), breast cancer (MCF-7), colon cancer (HCT 116), and lung normal (MRC-5) were purchased from the Korea Cell Line Bank (Seoul, South Korea). All cell lines were cultured in RPMI 1640 medium (Cat # 10-040-CV, Corning, New York, USA) containing 10% FBS and 1% penicillin/streptomycin in a humidified CO2 incubator at 37 °C (5% CO2/95% air). At about 80–90% confluency, cells were washed with PBS (pH 7.4) and subsequently subcultured by treatment with 0.25% trypsin-2.65 mM EDTA. The medium was replaced every 2 days. The cultured ASPC-1, A549, MCF-7, HCT 116, and MRC-5 cells were used to evaluate the in vitro antiproliferation activity of the test compounds.
4.4. In Vitro Antiproliferation Activity
The cytotoxic activity of ID-Checker, Al, Al–NH3+ aggregate, and Al–G aggregate against a panel of four cancer cell lines including pancreatic cancer (ASPC-1), lung cancer (A549), breast cancer (MCF-7), and colon cancer (HCT 116) cell lines, and three lung normal cell lines (MRC-5, IMR-90, and HEL 299) were evaluated by an MTT assay.42−44 In brief, about 7000 cells/well were seeded in 96-well plates and grown in an incubator for 24 h. 0.9% saline solution was used to prepare 1 mM stock solutions of compounds and then diluted in a cell culture medium and loaded in the wells containing cells at various concentrations. The cells were then incubated for 24 h followed by treatment with MTT for 2 h in the dark. The 96-well plates were transferred to the spectrophotometer (SpectraMAX Plus, Molecular Devices Inc., Sunnyvale, CA, USA), and the absorbance in each well was measured at 490 nm. GraphPad Prism 7 software (San Diego, CA) was used to determine the cell growth inhibition of 50% (IC50) values. All experiments were performed in triplicate, and the results were presented as mean ± SD (standard deviation).
4.5. In Vivo Antitumor Efficacy of ID-Checker
All animal experiments in this study were performed according to the rules of the Animal Experimental Ethics Committee of the Hallym University in accordance with the animal testing regulations (Hallym #2021-51). Five week-old female BALB/c pathogen-free mice were purchased from Dooyeol Biotech Inc. (Seoul, South Korea). After 1 week of quarantine and adaptation period, healthy animals that did not show any weight loss were selected and used for the experiment. Experimental animals were set at 23 ± 3 °C, a relative humidity of 50 ± 10%, ventilation 10–15 times/h, and a light time of 12 h (08:00–20:00). Test animals were allowed to freely ingest solid feed obtained from Cargill Inc. (Minneapolis, USA) and drinking water during the entire study period.
BALB/c mice were injected with 4T1 cells (1 × 105 cells/mice) subcutaneously into the left mammary adipose tissue. After implantation, tumor growth was visualized with the naked eye and measured with a caliper to calculate tumor volume (mm3) = (length × width2)/2, where length and width are the largest and smallest diameters, respectively. When the tumor volume reached about 50 mm3, the mice were classified into four test groups, including control (G1), low-dose group (G2), high-dose group (G3), and doxorubicin-treated group (G4 DOX). Group 1 (G1) was assigned as a control group and was subjected to intraperitoneal (IP) administration of the vehicle (0.9% saline). Mice in group 2 (G2) received the ID-Checker by IP administration at a dose of 0.711 mg/kg for days 1–3 and 14.22 mg/kg for days 4–11. Mice in group 3 (G3) were treated with IP injection of ID-Checker at a dose of 1.422 mg/kg for days 1–3 and 28.44 mg/kg for days 4–11, whereas mice in group 4 (G4 DOX) were given IP injection of 2 mg/kg doxorubicin once in 3 days.
The tumor volume and body weight of each mouse was recorded before the treatment. The experiment was terminated on specified days (study 1, 11 days; study 2, 3 days). Animals were sacrificed at the end of the study, and tumors were dissected. The tumor volumes and weights in the treatment groups G2 and G3 were compared with G1 and G4 DOX groups using one-way ANOVA and multiple comparisons using GraphPad Prism 7 software (San Diego, CA). Statistical significance is presented as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Some of the tumors were then stored at −70 °C for later analysis, and some were fixed in 4% paraformaldehyde (PFA), embedded in paraffin wax, and used for H&E staining. The 5 μm size tissue sections were mounted on the slides and stained with H&E. The H&E-stained slides were digitally scanned using a light microscope (Carl Zeiss, Oberkochen, Germany), and histological changes were observed.
4.6. Tubulin Polymerization Assay
Tubulin polymerization was performed by following the manufacturer’s protocol (BK006P, Cytoskeleton, Inc. Denver, USA). In brief, tubulin proteins (>99% purity) were suspended in G-PEM buffer containing 80 mM PIPES, 2 mM MgCl2, 0.5 mM EDTA, 1.0 mM GTP (pH 6.9), and 5% glycerol in the absence or presence of paclitaxel (PCL), NOZ, Al, Al–NH3+ aggregate, Al–G aggregate, and ID-Checker at concentrations of 20 μM. Then, the 96-well plate containing these mixtures was kept in a spectrophotometer (SpectraMAX Plus, Molecular Devices Inc., Sunnyvale, CA, USA). The absorbance was measured at 380 nm (37 °C) for 90 min at the rate of one measurement per minute.
4.7. Immunofluorescence for Tubulin
The effect of compounds on the microtubule cytoskeleton organization following ID-Checker treatment was visualized by immunofluorescence using the anti-α-tubulin antibody in MCF-7, A549, and MRC-5 cell lines. The respective cells were grown on coverslips for 24 h placed in the six-well plates. The cells were then treated with the ID-Checker at various concentrations (0, 0.05, 0.25, 0.50, and 1.0 μM) for various time intervals, including 6, 12, 18, and 24 h unless otherwise stated. After incubation for a specified time, the cells were washed with 0.1 M PBS (0.5 mL) and fixed by treating them with 1% paraformaldehyde for 20 min at 4 °C. Then, the washed cells were permeabilized with buffer containing 0.025% Triton X-100 for 15 min followed by blocking with 0.1 M PBS buffer containing 1% BSA for 30 min. The cells were then treated with the mouse anti-α-tubulin antibody (Cat # SC-32293, Santa Cruz Biotechnology, Inc., USA) at 37 °C for 2 h. The cells were washed with 0.1 M PBS and incubated with the anti-mouse IgG/FITC antibody (Cat # SC-516140, Santa Cruz Biotechnology, Inc., USA) for 1 h. The nuclei of cells were stained by incubating them in the dark with DAPI (Sigma-Aldrich, Seoul, South Korea) at 25 °C for 10 min. The prepared cells were then visualized under a confocal laser scanning microscope (Carl Zeiss LSM710, Germany).
4.8. Immunofluorescence for GLUT1 and GLUT 4 Channels
The effect of compounds on microtubule cytoskeleton organization following ID-Checker treatment was visualized by immunofluorescence using the anti-GLUT1 or anti-GLUT4 antibody in MCF-7, A549, and MRC-5 cell lines. The respective cells were grown on coverslips for 24 h placed in the six-well plates. The cells were then treated with the ID-Checker at various concentrations (0, 0.05, 0.25, 0.50, and 1.0 μM) for various time intervals, including 6, 12, 18, and 24 h, unless otherwise stated. After incubation for a specified time, the cells were washed with 0.1 M PBS (0.5 mL) and fixed by treating them with 1% paraformaldehyde for 20 min at 4 °C. Then, the washed cells were permeabilized with buffer containing 0.025% Triton X-100 for 30 min followed by blocking with 0.1 M PBS buffer containing 1% BSA for 30 min. The cells were then treated with the rabbit anti-GLUT1 or rabbit anti-GLUT4 antibody (Cat # LS-C407646 and LS-C143467, LifeSpan Biosciences, Seattle, USA) at 4 °C for 12 h. The cells were washed with 0.1 M PBS and incubated with the donkey anti-rabbit IgG/ALEXA488 antibody (Cat # A32790, Thermo Fisher Scientific, Waltham, USA) for 90 min. The nuclei of cells were stained by incubating them in the dark with DAPI (Sigma-Aldrich, Seoul, South Korea) at 25 °C for 5 min. The prepared cells were then visualized under a confocal laser scanning microscope (Carl Zeiss LSM710, Germany).
4.9. Cell Cycle Analysis
MCF-7 and A549 cells (2 × 105) were seeded in 100 mm dishes and incubated overnight. The cells were treated with 0.9% saline (vehicle), and various concentrations of ID-Checker (0, 0.05, 0.25, 0.50, and 1.0 μM) for 24 h were detached, collected, and washed with FACS buffer (1% FBS in PBS). The cells were fixed with ice-cold 70% ethanol in PBS for 12 h at 4 °C. The cells were then washed and resuspended in buffer containing RNase (Sigma-Aldrich) and incubated for 30 min at 37 °C. The cells were then incubated with PI for 5 min in the dark and used immediately for cell cycle distribution analysis using FACSCalibur (BD Biosciences, Franklin Lakes, USA).
4.10. Statistical Analysis
Unless otherwise stated, all experiments were performed in triplicates and analyzed using the GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, USA) program. Student’s t-test and one-way analysis variance (ANOVA) were used to compare the difference between the test and control groups. It was judged to be statistically significant only when P < 0.05 or more. Statistical significance is presented as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Acknowledgments
We thank Prof. Dr. Young Jun Hong, M.D. PhD of Department of Laboratory Medicine, Korea Cancer Center Hospital, Seoul, South Korea and Prof. Dr. Kye Young Lee, M.D. PhD of Konkuk University Medical Center, Seoul, South Korea and vice president of Korea Lung Cancer Association for the valuable discussions on the experiments and results during this study. We also thank Dr. Eun Ji Kim and Dr. Jae In Jung of Regional Strategic Industry Innovation Center, Hallym University, South Korea for their valuable support during the animal experiments. This work was partially supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2021R1C1C1008480) to S.B.N.
Glossary
Abbreviations Used
- Al
albendazole
- Al–G
albendazole amine glucose conjugate
- Al–NH2
albendazole–amine
- Al–NH3+
albendazole ammonium ion
- FBZ
fenbendazole
- GLUT
glucose transport proteins
- ID-Checker
an aggregate of Al–G and Al–NH3+ in a 1:100 ratio
- MBZ
mebendazole
- MTAs
microtubule-targeting agents
- NOZ
nocodazole
- CBS
colchicine binding site
- NBS
nocodazole binding site
All relevant data are available from the authors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00646.
NMR spectra, HPLC traces, DLS data, effects on tubulin polymerization and cell cycle distribution, immunofluorescence staining, body weight of experimental animals, docking scores, and docking poses (PDF)
Molecular docking pose of compound Al with CBS (PDB ID: 4O2B); molecular docking pose of compound Al–NH3+ with CBS (PDB ID: 4O2B); molecular docking pose of compound Al–G with CBS (PDB ID: 4O2B); molecular docking pose of compound Al with NBS (PDB ID: 5CA1); molecular docking pose of compound Al–NH3+ with NBS (PDB ID: 5CA1); and molecular docking pose of compound Al–G with NBS (PDB ID: 5CA1) (ZIP)
Molecular formula strings (CSV)
Author Contributions
∥ K.-S.S. and S.B.N. contributed equally to this work. Hence, both should be considered as first authors. Conceptualization, S.B.N., T.K., and T.K.; methodology, K.-S.S., S.B.N., J.K., and T.K.; software, K.-S.S. and T.K.; formal analysis, S.D.W., J.K., K.-S.S., and S.B.N.; investigation, S.D.W., J.K., and K.-S.S.; data curation, K.-S.S., T.K., and S.B.N.; writing—original draft preparation, S.B.N. and K.-S.S.; writing—review and editing, T.K. and S.B.N.; supervision, T.K.; and project administration, K.-S.S. and S.B.N. All authors have read and agreed to the published version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Han T.; Goralski M.; Gaskill N.; Capota E.; Kim J.; Ting T. C.; Xie Y.; Williams N. S.; Nijhawan D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- Dai J.; Su Y.; Zhong S.; Cong L.; Liu B.; Yang J.; Tao Y.; He Z.; Chen C.; Jiang Y. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduction Targeted Ther. 2020, 5, 145. 10.1038/s41392-020-00261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senapati S.; Mahanta A. K.; Kumar S.; Maiti P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduction Targeted Ther. 2018, 3, 7. 10.1038/s41392-017-0004-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E. T. H.; Tong A. T.; Lenihan D. J.; Yusuf S. W.; Swafford J.; Champion C.; Durand J.-B.; Gibbs H.; Zafarmand A. A.; Ewer M. S. Cardiovascular complications of cancer therapy. Circulation 2004, 109, 3122–3131. 10.1161/01.cir.0000133187.74800.b9. [DOI] [PubMed] [Google Scholar]
- Hatzistergos K. E.; Blum A.; Ince T.; Grichnik J. M.; Hare J. M. What is the oncologic risk of stem cell treatment for heart disease?. Circ. Res. 2011, 108, 1300–1303. 10.1161/circresaha.111.246611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Hermann M. Estimation of the cancer risk induced by therapies targeting stem cell replication and treatment recommendations. Sci. Rep. 2018, 8, 11776. 10.1038/s41598-018-29967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Fatangare A.; Svatoš A. Applications of 2-deoxy-2-fluoro-D-glucose (FDG) in Plant Imaging: Past, Present, and Future. Front. Plant Sci. 2016, 7, 483. 10.3389/fpls.2016.00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Sung H.; Ferlay J.; Siegel R. L.; Laversanne M.; Soerjomataram I.; Jemal A.; Bray F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2021, 71, 209–249. 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2020. Ca-Cancer J. Clin. 2020, 70, 7–30. 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- Szablewski L. Expression of glucose transporters in cancers. Biochim. Biophys. Acta Rev. Cancer. 2013, 1835, 164–169. 10.1016/j.bbcan.2012.12.004. [DOI] [PubMed] [Google Scholar]
- Godoy A.; Ulloa V.; Rodríguez F.; Reinicke K.; Yañez A. J.; García M. d. l. A.; Medina R. A.; Carrasco M.; Barberis S.; Castro T.; Martínez F.; Koch X.; Vera J. C.; Poblete M. T.; Figueroa C. D.; Peruzzo B.; Pérez F.; Nualart F. Differential subcellular distribution of glucose transporters GLUT1–6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell. Physiol. 2006, 207, 614–627. 10.1002/jcp.20606. [DOI] [PubMed] [Google Scholar]
- Cairns R. A.; Harris I. S.; Mak T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Shin E.; Koo J. S. Glucose metabolism and glucose transporters in breast cancer. Front. Cell Dev. Biol. 2021, 9, 2404. 10.3389/fcell.2021.728759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron C.; Tsiani E.; Tsakiridis T. Expression of the glucose transporters GLUT1, GLUT3, GLUT4 and GLUT12 in human cancer cells. BMC Proc. 2012, 6, P4. 10.1186/1753-6561-6-s3-p4. [DOI] [Google Scholar]
- Effert P.; Beniers A. J.; Tamimi Y.; Handt S.; Jakse G. Expression of glucose transporter 1 (Glut-1) in cell lines and clinical specimens from human prostate adenocarcinoma. Anticancer Res. 2004, 24, 3057–3063. [PubMed] [Google Scholar]
- Deng D.; Sun P.; Yan C.; Ke M.; Jiang X.; Xiong L.; Ren W.; Hirata K.; Yamamoto M.; Fan S.; Yan N. Molecular basis of ligand recognition and transport by glucose transporters. Nature 2015, 526, 391–396. 10.1038/nature14655. [DOI] [PubMed] [Google Scholar]
- Mishra R. K.; Wei C.; Hresko R. C.; Bajpai R.; Heitmeier M.; Matulis S. M.; Nooka A. K.; Rosen S. T.; Hruz P. W.; Schiltz G. E.; Shanmugam M. In Silico modeling-based identification of glucose transporter 4 (GLUT4)-selective inhibitors for cancer therapy. J. Biol. Chem. 2015, 290, 14441–14453. 10.1074/jbc.m114.628826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S.; Sheena A.; Poulose N.; Anilkumar G. Molecular dynamics simulation studies of GLUT4: substrate-free and substrate-induced dynamics and atp-mediated glucose transport inhibition. PLoS One 2010, 5, e14217 10.1371/journal.pone.0014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidl S.; Iancu C. V.; Choe J.-y.; Oreb M. Ligand screening systems for human glucose transporters as tools in drug discovery. Front. Chem. 2018, 6, 183. 10.3389/fchem.2018.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan D. A.; Sutphin P. D.; Nguyen P.; Turcotte S.; Lai E. W.; Banh A.; Reynolds G. E.; Chi J. T.; Wu J.; Solow-Cordero D. E.; Bonnet M.; Flanagan J. U.; Bouley D. M.; Graves E. E.; Denny W. A.; Hay M. P.; Giaccia A. J. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70. 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova I.; Kasho V.; Kaback H. R. Lactose permease and the alternating access mechanism. Biochemistry 2011, 50, 9684–9693. 10.1021/bi2014294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu. Rev. Biophys. 2013, 42, 51–72. 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Wozniak M.; Pastuch-Gawolek G.; Makuch S.; Wisniewski J.; Krenacs T.; Hamar P.; Gamian A.; Szeja W.; Szkudlarek D.; Krawczyk M.; Agrawal S. In vitro and in vivo efficacy of a novel glucose-methotrexate conjugate in targeted cancer treatment. Int. J. Mol. Sci. 2021, 22, 1748. 10.3390/ijms22041748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvaresi E. C.; Hergenrother P. J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. 10.1039/c3sc22205e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasarao M.; Galliford C. V.; Low P. S. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat. Rev. Drug Discovery 2015, 14, 203–219. 10.1038/nrd4519. [DOI] [PubMed] [Google Scholar]
- Krueger G. Cancer past, present, and future. Lancet Oncol. 2007, 8, 17. 10.1016/s1470-2045(06)71003-8. [DOI] [Google Scholar]
- Cao Y.; DePinho R. A.; Ernst M.; Vousden K. Cancer research: past, present and future. Nat. Rev. Cancer 2011, 11, 749–754. 10.1038/nrc3138. [DOI] [PubMed] [Google Scholar]
- DeVita V. T. Jr.; Eggermont A. M.; Hellman S.; Kerr D. J. Clinical cancer research: the past, present and the future. Nat. Rev. Clin. Oncol. 2014, 11, 663–669. 10.1038/nrclinonc.2014.153. [DOI] [PubMed] [Google Scholar]
- Dogra N.; Kumar A.; Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci. Rep. 2018, 8, 11926. 10.1038/s41598-018-30158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne C. P.; Jones T.; Bear R. Gemcitabine-(C4-amide)-[anti-HER2/neu] anti-neoplastic cytotoxicity in dual combination with mebendazole against chemotherapeutic-resistant mammary adenocarcinoma. J. Clin. Exp. Oncol. 2013, 2, 1000109. [PMC free article] [PubMed] [Google Scholar]
- Elayapillai S.; Ramraj S.; Benbrook D. M.; Bieniasz M.; Wang L.; Pathuri G.; Isingizwe Z. R.; Kennedy A. L.; Zhao Y. D.; Lightfoot S.; Hunsucker L. A.; Gunderson C. C. Potential and mechanism of mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311. 10.1016/j.ygyno.2020.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Zhao J.; Gao X.; Pei D.; Gao C. Anthelmintic drug albendazole arrests human gastric cancer cells at the mitotic phase and induces apoptosis. Exp. Ther. Med. 2017, 13, 595–603. 10.3892/etm.2016.3992. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nath J.; Paul R.; Ghosh S. K.; Paul J.; Singha B.; Debnath N. Drug repurposing and relabeling for cancer therapy: Emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 2020, 258, 118189. 10.1016/j.lfs.2020.118189. [DOI] [PubMed] [Google Scholar]
- Liu R.; Fu Z.; Zhao M.; Gao X.; Li H.; Mi Q.; Liu P.; Yang J.; Yao Z.; Gao Q. GLUT1-mediated selective tumor targeting with fluorine containing platinum(II) glycoconjugates. Oncotarget 2017, 8, 39476–39496. 10.18632/oncotarget.17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X.; Liu S.; Shi Y.; Huang Z.; Mi Y.; Mi Q.; Yang J.; Gao Q. Mechanistic and biological characteristics of different sugar conjugated 2-methyl malonatoplatinum(II) complexes as new tumor targeting agents. Eur. J. Med. Chem. 2017, 125, 372–384. 10.1016/j.ejmech.2016.09.047. [DOI] [PubMed] [Google Scholar]
- Fu J.; Yang J.; Seeberger P. H.; Yin J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. 10.1016/j.carres.2020.108195. [DOI] [PubMed] [Google Scholar]
- Han J.; Gao X.; Liu R.; Yang J.; Zhang M.; Mi Y.; Shi Y.; Gao Q. Design, synthesis of novel platinum(II) glycoconjugates, and evaluation of their antitumor effects. Chem. Biol. Drug Des. 2016, 87, 867–877. 10.1111/cbdd.12718. [DOI] [PubMed] [Google Scholar]
- Bhalla K. N. Microtubule-targeted anticancer agents and apoptosis. Oncogene 2003, 22, 9075–9086. 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Lee J.-S.; Song I.-h.; Warkad S. D.; Yeom G. S.; Shinde P. B.; Song K.-s.; Nimse S. B. Synthesis and evaluation of 2-aryl-1H-benzo[d]imidazole derivatives as potential microtubule targeting agents. Drug Dev. Res. 2022, 83, 769–782. 10.1002/ddr.21909. [DOI] [PubMed] [Google Scholar]
- Song I.-h.; Torawane P.; Lee J.-S.; Warkad S. D.; Borase A.; Sahoo S. K.; Nimse S. B.; Kuwar A. The detection of Al3+ and Cu2+ ions using isonicotinohydrazide-based chemosensors and their application to live-cell imaging. Mater. Adv. 2021, 2, 6306–6314. 10.1039/d1ma00564b. [DOI] [Google Scholar]
- Lee J.-S.; Warkad S. D.; Shinde P. B.; Kuwar A.; Nimse S. B. A highly selective fluorescent probe for nanomolar detection of ferric ions in the living cells and aqueous media. Arabian J. Chem. 2020, 13, 8697–8707. 10.1016/j.arabjc.2020.09.061. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.