

Abstract
Although cumulative genetic and epigenetic changes in cancer cells are correlated with tumor malignancy, accumulating evidence supports that tumor cell-extrinsic mechanisms play an essential role in driving tumor progression. The tissue architecture surrounding tumor cells evolves during disease progression and becomes a significant barrier to cancer treatments. The functional traits of the tumor microenvironment (TME), either tumor suppressive or supportive, are defined by the distribution of various stromal cells and their sequential and reciprocal cellular interactions. Recent studies have uncovered a significant heterogeneity in stromal cells and identified specific subpopulations correlated with clinical outcomes, providing novel insights into the complex TME system that drives tumor progression and therapy resistance. Moreover, a small population of tumor cells with tumor-initiating and drug-resistant capabilities, cancer stem cells (CSCs), is maintained by the specialized TME, the so-called CSC niche. The crosstalk between CSCs and niche cells is an attractive avenue for identifying the vulnerability of difficult-to-treat cancers. Here, we review the recent advance in understanding TME biology and its impact on CSCs. We then focus on a newly identified niche signaling loop by which CSCs promote malignant progression and drug resistance of squamous cell carcinoma. The CSC niche is a promising research field that needs more attention and could facilitate the development of durable cancer treatment.
Keywords: cancer stem cells (CSCs), tumor microenvironment (TME), CSC niche; cellular crosstalk, TGF-β, IL-33, macrophages, cancer progression, drug resistance
Introduction
Most tumors are of a clonal origin but display significant intratumor heterogeneity at clinical diagnosis. For many decades, phenotypic heterogeneity among tumor cells has been considered a major driver of tumor progression and treatment failure, limiting patient survival [1]. Understanding the mechanisms that cause tumor heterogeneity can profoundly impact the development of effective treatments. Chromosomal instability and somatic mutations promote the genetic diversity and malignant phenotypes of tumor cells [2,3]. Together with tumor cell-intrinsic changes, surrounding stromal cells, including fibroblasts, endothelial cells, and immune cells, functionally diversify through interactions with neighboring tumor cells [4,5]. Accumulating evidence indicates that interactions with the tumor microenvironment (TME) have significant effects on malignant properties of tumor cells.
Most adult organs harbor stem cells that maintain tissue functions by replenishing the lost differentiated cells during homeostatic tissue turnover and injuries [6]. Tissue-resident stem cells are preferential targets for oncogenic transformation because they are long-lived and thus accumulate many mutations that will lead to cancer [7]. Intrinsic properties of stem cells, such as self-renewal potential, seem to be a necessary prerequisite for cancer development. Moreover, because stem cells reside in specialized microenvironments or niches, extrinsic factors enriched in the niche are probably utilized by tumor-initiating cells to establish early tumors [8].
The concept of cancer stem cells (CSCs) was proposed to explain intratumor heterogeneity many decades ago [9], and researchers have identified CSCs in various types of human cancer [10–12]. CSCs are a small subset of long-lived tumor cells with the ability to self-renew, differentiate into defined progenies, and initiate and sustain tumor growth. Because CSCs are highly resistant to conventional therapies and can be a source of tumor recurrence and metastasis, CSCs are considered a rational target for cancer treatment. However, the development of effective CSC-targeted therapies is not as successful as was initially hoped due to the lack of targetable vulnerabilities. Importantly, it has become evident that numerous extrinsic factors, signaling pathways, and non-genetic mechanisms can transiently induce drug-resistant CSC-like phenotypes in tumor cells [13]. Therefore, the cellular crosstalk between CSCs and neighboring stromal cells that regulates malignant properties of CSCs holds great potential for the development of innovative therapeutic strategies.
The CSC niche is considered a distinct TME that supports the self-renewal and survival of CSCs, which is composed of a specific set of functionally distinct stromal cells, extracellular matrix (ECM) proteins, and signaling factors located near CSCs [14]. Although reciprocal interactions between CSCs and the niche cells seem critical for maintaining their C relationship [15], how the niche microenvironment emerges during malignant tumor progression is still poorly understood.
Cellular crosstalk that drives tumor progression and drug resistance
Because cancers are often refractory to targeted drugs designed to inhibit oncogenic drivers, therapeutic strategies to disrupt non-genetic mechanisms of tumor progression are urgently needed. Here, the stromal cell types comprising the TME are summarized and recent studies that identified previously unappreciated cell populations and signaling pathways regulating CSCs, tumor progression, and drug resistance are highlighted (Figure 1).
Figure 1.
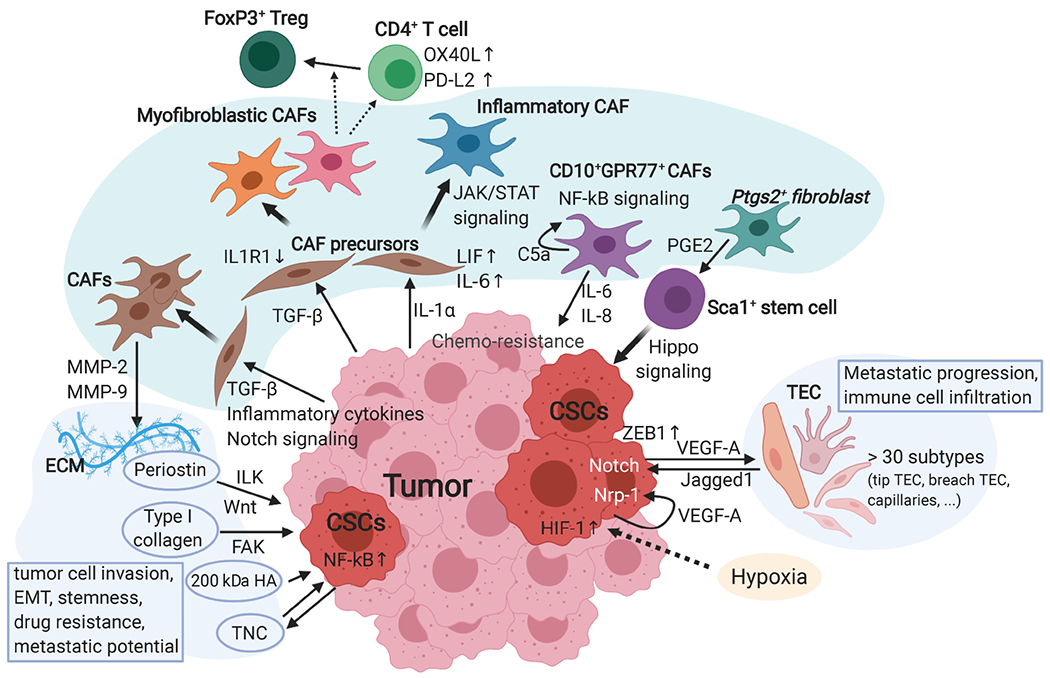
The crosstalk between tumor cells and non-immune stromal cells and the TME conditions. Cellular interactions between tumor cells and the surrounding microenvironment influence tumor initiation, malignant progression, therapy resistance, and metastasis. This figure focuses on the heterogeneity of non-immune stromal cells and biochemical interactions. Fibroblasts have several key subtypes that interact with tumor cells, CSCs, and other stromal cells differentially. By secreting TGF-β and IL-1α, the tumor regulates CAF precursors’ differentiation into myofibroblastic CAFs and inflammatory CAFs, respectively. In addition to promoting tumor cell invasion, myofibroblastic CAFs contribute to immunosuppression by elevating OX40L and PD-L2 expression in CD4+ T cells and promoting differentiation of FoxP3+ regulatory T cells (Treg). In contrast, inflammatory CAFs induced by LIF-JAK/STAT signaling recruit myeloid cells and Treg. By secreting IL-6/8, CD10+CPR77+ CAFs generated by a self-reinforced C5a and NF-kB loop promote stemness and chemoresistance of CSCs. Ptgs2+ fibroblasts secrete metabolites, such as PGE2, induce the expansion of Sca1+ quiescent stem cells and tumor initiation through the Hippo-YAP pathway. Endothelial cells have more than 30 different states based on expression profiling. The tip and breach subtypes are predictive of poor prognosis and sensitive to VEGF-A blockade. Endothelial cells and CSCs form a feedback VEGF-A-Jagged1 loop through Notch signaling. The ECM produced by fibroblasts, tumor cells, and other stromal cells, provides the framework of the TME. CAFs and tumor cells secrete MMPs, such as MMP-2/9, to remodel the ECM. Some specific ECM proteins, including periostin, type I collagen, laminin-332, the 200 kDa hyaluronic acid (HA), and tenascin C (TNC), maintain and promote CSC phenotypes through activating various signaling pathways, such as Wnt, integrin/focal adhesion kinase (FAK), and NF-kB pathways.
Fibroblasts
Fibroblasts comprise a large part of the tumor stroma and provide its structural framework by synthesizing ECM proteins. Fibroblasts also play a central role in tumor development by releasing multiple signaling molecules, metabolites, and exosomes. Tumor cells promote the conversion of normal fibroblasts to cancer-associated fibroblasts (CAFs) via various tumor-derived factors, including TGF-β, inflammatory cytokines, and Notch signaling [16]. In turn, CAFs produce matrix metalloproteinases, such as MMP-2 and MMP-9, and remodel the ECM to promote tumor cell invasion and epithelial–mesenchymal transition, stemness, drug resistance, and metastatic potential [17]. Although CAFs are a promising therapeutic target, depletion of CAFs in pancreatic ductal adenocarcinoma (PDAC) resulted in more aggressive and poorly differentiated tumor formation, with reduced stromal content and survival [18,19]. These results suggest significant functional heterogeneity among CAFs, representing a substantial barrier for their therapeutic application. Recent studies have revealed the molecular basis of the heterogeneity and plasticity of CAFs.
Using PDAC mouse models, Tuveson and colleagues [20] identified two distinct CAF subtypes, i.e. myofibroblastic and inflammatory CAFs. Tumor-derived IL-1α induces the production of proinflammatory factors, including leukemia inhibitory factor (LIF) and IL-6, in quiescent CAF precursors. Autocrine LIF activates the JAK/STAT signaling pathway in the precursors, promoting differentiation into inflammatory CAFs. In contrast, tumor-derived TGF-β inhibits IL-1 signaling by downregulating the IL-1 receptor (IL1R1), restricting inflammatory CAF differentiation but promoting differentiation toward myofibroblastic CAFs. These data suggest that the spatially limited, tumor-derived factors produce heterogenous CAF phenotypes [20]. By transcriptomic analysis of human PDAC patient-derived CAFs, Neuzillet et al [21] identified four fibroblast subtypes, including ones associated with poor prognosis. They also found that culture medium from cancer cell lines could induce non-tumoral pancreatic stellate cells into CAF-like phenotypes, demonstrating that CAF phenotypes are regulated by tumor-derived factors.
Fibroblast heterogeneity is also described in the TME of breast cancers. Costa et al [22] identified four CAF subsets with differential properties and activation status. A subset of myofibroblastic CAFs induces OX40L and PD-L2 expression on CD4+ T cells and differentiation of FoxP3+ regulatory T cells (Tregs), indicating that the heterogeneity among CAFs reflects the formation of immunosuppressive TMEs. Moreover, Su et al [23] used two cell surface markers, CD10 and GPR77, to identify a subset of CAFs that supports CSCs in breast and lung cancers and induces chemo-resistance by secreting IL-6 and IL-8. CSC-supporting phenotypes of CD10+GPR77+ CAFs are maintained by a complement C5a-induced NF-κB signaling mediated by GPR77.
Using single-cell RNA-seq (scRNA-seq) analysis, Roulis et al [24] recently identified a fibroblast subset in the intestinal stem cell niche that highly expresses Ptgs2, the gene encoding cyclooxygenase-2 (COX-2), an enzyme that processes arachidonic acid into inflammatory lipid mediators, such as prostaglandin E2 (PGE2). The fibroblast-derived PGE2 induces the expansion of quiescent stem cells, facilitating early tumor formation. Mechanistically, PGE2 binds to its receptor EP4 (Ptger4) expressed on stem cells and induces a strong regenerative/tumorigenic program through the Hippo-YAP signaling pathway [24]. YAP is also involved in multiple CAF functions, inducing matrix stiffening, cancer cell invasion, and angiogenesis. Matrix stiffening further enhances YAP transcriptional activity by activating Src family kinases, establishing a feed-forward loop that helps to maintain the CAF phenotype. Notably, Rho kinase inhibition can disrupt this feed-forward loop and destabilize the CAF phenotypes [25].
Extracellular matrix
The mixture of ECM proteins secreted by fibroblasts, tumor cells, and other stromal cells defines the framework of the TME. In particular, CSCs interact with specific ECM with distinct physical and biochemical properties [26]. The ECM’s mechanical properties, such as stiffness and topological alignment, and hypoxia could induce CSC phenotypes. Pang et al [27] showed that stiff substrate and oxygen tension regulate integrin expression and integrin-linked kinase activity to induce CSC properties and marker expression, including CD44 and Nanog.
In addition to supporting CSCs mechanically, the CSC-associated ECM promotes the self-renewal and survival of CSCs. Periostin, encoded by the gene POSTN, is an ECM protein that binds to integrins to support adhesion and migration of tumor cells. Periostin is known to sustain CSCs in metastatic sites through physically recruiting Wnt ligands [28]. Type I collagen was found to increase tumor-initiating potential, self-renewal, and the frequency of PDAC CSCs through the activation of focal adhesion kinase [29]. Govaere et al [30] showed that laminin-332, also known as laminin-5, plays a critical role in promoting CSC quiescence and chemoresistance [30]. In head and neck cancer, a specific hyaluronic acid with a size of 200 kDa promotes a CSC expression signature [31]. Moreover, in glioblastoma, tumor cells with elevated NF-κB activity express a higher level of tenascin C, which promotes self-renewal of glioblastoma CSCs. Furthermore, tenascin C also induces glioma cells to differentiate into tumor-derived endothelial cells [32]. These studies highlight specific interactions between ECM proteins and CSCs, which could be manipulated as therapeutic strategies.
Endothelial cells
The blood vessels supply oxygen and nutrients necessary for tumor cell growth and survival. However, the vascular structure is highly disorganized compared with that in normal tissue. Depending on structural properties and density, it can promote intratumor heterogeneity in the adjacent microenvironment. Hypoxia is an important feature of the TME, which is also a well-known characteristic of the stem cell niche [33]. Tumor hypoxia promotes cancer cell invasion and stimulates CSC phenotypes [15]. In turn, elevated hypoxia-inducible factor 1 (HIF-1) activity in tumor cells stimulates the production of proangiogenic factors, such as vascular endothelial growth factor A (VEGF-A) [34,35].
In a mouse model of skin tumor, tumor-derived VEGF-A is involved in establishing a perivascular niche, in which a VEGF-A–Nrp1 autocrine loop promotes stemness and tumor progression [36]. Tumor endothelial cells (TECs) also promote CSC phenotypes through Notch signaling and nitric oxide signaling pathways [37,38]. Recently, Jiang et al [39] identified a positive feedback loop between breast CSCs and TECs that is mediated by VEGF-A and Notch. They found that TECs express Jagged1, which binds to the Notch receptor on CSCs and upregulates ZEB1 expression. ZEB1 induces VEGF-A secretion from CSCs, which ultimately induces Jagged1 expression in TECs [39]. Additionally, VEGF-A is known to induce the expression of programmed cell death protein 1 (PD-1) and other immune checkpoint molecules involved in CD8+ T cell exhaustion, establishing an immunosuppressive microenvironment [40], which is a preferred feature of the CSC niche.
Cellular heterogeneity among TECs was also reported recently. Using scRNA-seq analysis, Goveia et al [41] identified many different phenotypes in lung TECs, including distinct subsets related to migratory tip, basement membrane breaching, immune cell recruitment, and semiprofessional antigen presentation. They found that tip/breach TEC signatures were correlated with patient survival and were most sensitive to treatment with a VEGF blockade [41]. Such functional heterogeneity in TECs and newly identified angiogenic regulators provide an opportunity to develop novel anti-angiogenic strategies.
The tumor vasculature also serves as an entry site of circulating immune cells into the tumor tissue. Tumor-associated lymphatic vessels are positively correlated with metastatic progression, while they also contribute to anti-tumor immunity by increasing immune cell infiltration into tumors [42]. Notably, intratumor density and the location of T cells and macrophages can be predictive of patient survival and metastasis [43,44]. The mechanisms that regulate immune cell composition and phenotypes are active multidisciplinary research areas to develop durable cancer treatments, including immunotherapy.
T cells
T cells play vital roles in anti-tumor immunity as well as immunosuppression. Naïve CD8+ T cells differentiate into cytotoxic T cells, whereas CD4+ T cells differentiate into an array of different types of helper type 1 cell (Th1), Th2, Th17, or Tregs, depending on microenvironmental cues [45]. Although CD8+ T cell abundance in the tumor correlates with anti-tumor immune responses, most tumors are immunologically tolerant. PD-1, an immune checkpoint receptor, is upregulated in activated T cells to induce immune tolerance [46]. In general, tumor cells overexpress PD-L1, the ligand for PD-1, facilitating escape from the anti-tumor T cell responses [47]. In addition to PD-L1, T cell-inflamed tumors often exhibit high expressions of other immunosuppressive mediators, such as indoleamine-2,3-dioxygenase (IDO) and FoxP3+ Tregs [48].
Although immunotherapies relieving these suppressive mechanisms have improved overall survival, responses to immune checkpoint inhibitors are significantly variable between patients. To elucidate the underlying mechanisms, Oh et al [49] profiled T cells isolated from human bladder tumors and non-malignant tissue using scRNA-seq analysis. Interestingly, they did not find the distinct states in CD8+ T cells between tumors and non-malignant tissues. They found that CD4+ T cell populations showed tumor-specific states, including different subpopulations in Tregs and cytotoxic CD4+ T cells, killing tumor cells in an MHC class II (MHC-II)-dependent manner. Moreover, the cytotoxic CD4+ T cell gene expression signature was predictive for anti-PD-L1 therapy efficacy for metastatic cancers [49].
In contrast, Merad and colleagues [50] recently described the immune landscape of early lung cancers. They found a significant alteration in T cell and NK cell compartments even in stage I lung adenocarcinomas. However, anti-tumor T cell immunity was negatively impacted by tumor-infiltrating myeloid cells already in early-stage cancers [50], suggesting that the emergence of CSC niches may be closely associated with immune cell recruitment and differentiation.
Mariathasan et al [51] and Tauriello et al [52] showed that stromal TGF-β signaling could be a determinant factor of T cell exclusion. They found that T cell exclusion away from the tumors correlated with the presence of peritumoral fibroblasts that express a TGF-β response signature. Pharmacological inhibition of TGF-β signal reversed such T cell exclusion phenotypes and facilitated cytotoxic CD8+ T cell infiltration into tumors. Moreover, therapeutic co-administration of anti-TGF-β and anti-PD-L1 antibodies provoked T cell-mediated anti-tumor immunity and tumor regression.
Fuchs and colleagues [54] recently reported that a CSC population activated a unique immune-resistant mechanism against cytotoxic T cells in skin cancer. They focused on a previously characterized TGF-β-responding CSC population, described in detail below [53], and found that CSCs selectively expressed CD80, a binding partner for CD28 and CTLA4. By binding to CTLA4, CD80+ CSCs directly dampened cytotoxic T cell activity. Moreover, when anti-TGF-β and anti-CTLA4 antibodies were administered, tumor relapse after cytotoxic T cell transfer was diminished [54], suggesting that CSCs are protected from the anti-tumor immune system through various cell-intrinsic and-extrinsic mechanisms.
Myeloid-derived suppressor cells (MDSCs) and other myeloid cells
Tumors recruit immature myeloid cells from the bone marrow and spleen and create a complex landscape of myeloid cells, including monocytes, dendritic cells, macrophages, and granulocytes (neutrophils, eosinophils, basophils, etc.) [55]. Tumor-derived factors also generate pathologically activated immature myeloid cells with antigen-specific and non-specific immunosuppressive roles, known as MDSCs, which can be divided into two populations, monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) (Figure 2). M-MDSCs are more immunosuppressive and exist in various differentiation phases toward tumor-associated macrophages (TAMs). In contrast, PMN-MDSCs are less immunosuppressive than M-MDSCs and functionally related to tumor-associated neutrophils [56].
Figure 2.
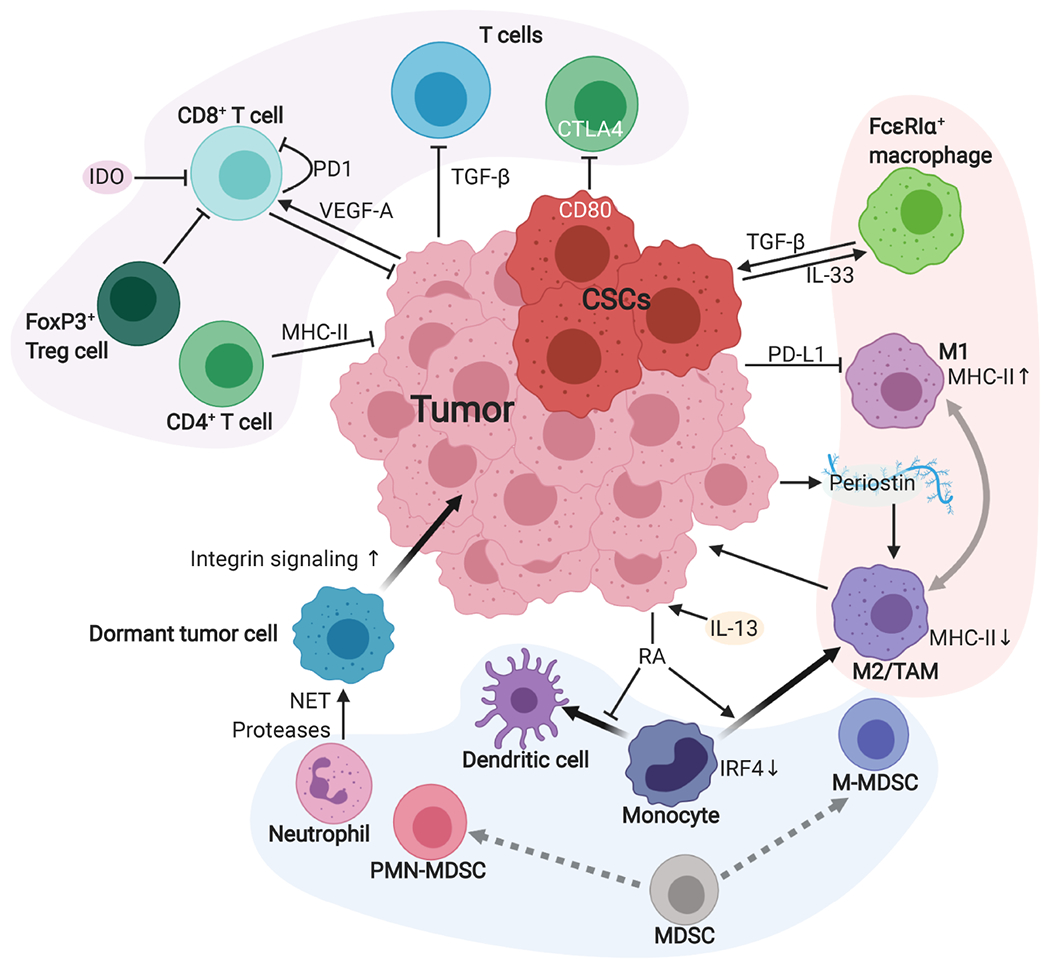
The crosstalk between tumor cells and immune cells in the TME. The TME is composed of heterogeneous immune cells. Here, we summarize some of the major cell types. Various mechanisms suppress the cytotoxic activity of CD8+ T cells. T cell-intrinsic expression of immune checkpoint molecules, such as PD-1 and CTLA4, suppress anti-tumor immunity. Tumor-derived VEGF-A also induces PD-1 expression in CD8+ T cells. CD4+ T cells can also kill tumor cells in an MHC-II-dependent manner. Although TGF-β suppresses T cell activity in general, TGF-β can also upregulate CD80 expression in CSCs and CD80 binds to CTLA4 to suppress anti-tumor responses. Bone marrow-derived myeloid cells are abundantly recruited into the tumor. The MDSCs could differentiate into M-MDSCs and PMN-MDSCs subtypes, which are more similar to TAMs and neutrophils, respectively. Tissue inflammation causes neutrophils to release NET proteases, which cleave laminin in the basement membrane and activate integrin signaling in dormant tumor cells, promoting metastatic tumor formation. Tumor-derived retinoic acid (RA) suppresses monocyte differentiation into dendritic cells, resulting in an increase in TAMs. Macrophages have two interchangeable states, classically activated phenotype (M1) and alternatively activated phenotype (M2). PD-L1 expressed in tumor cells also suppresses macrophage differentiation toward M1 by inhibiting PD-1. M2 macrophages induce angiogenesis and promote metastasis. Periostin secreted from tumor cells also supports M2 macrophage differentiation. CSCs establish an IL-33–TGF-β signaling loop with newly identified FcεRIα+ macrophages to maintain their stemness.
Neutrophils constitute a significant part of the TME and have essential roles linking inflammation and cancer development. Neutrophil density is predictive of metastatic progression and poor overall survival in many cancer types [57]. Recently, Egeblad and colleagues [58] reported that tobacco smoke-induced chronic lung inflammation awakens disseminated, dormant cancer cells to aggressively growing metastases. Mechanistically, sustained inflammation induces the formation of neutrophil extracellular traps (NETs), extracellular fibers composed of nuclear chromatin and granule proteins released from neutrophils. Interestingly, NET DNA acts as a proteolysis scaffold. NET-associated proteases, such as neutrophil elastase and MMP-9, cleave laminin, the most abundant glycoprotein of the basement membrane. Then, proteolytically cleaved laminin activates integrin α3β1 signaling, which induces proliferation of dormant cancer cells [58].
Additionally, Ombrato et al [59] recently characterized a metastatic niche induced by disseminated breast cancer cells in the lung using an adjacent cell-labeling method in vivo. They found that disseminated cancer cells induced de-differentiation of adjacent lung epithelial cells into cancer-associated parenchymal cells. These non-malignant epithelial cells supported tumor cell growth, particularly when neutrophils coexisted, highlighting the contribution of diverse cell types to early metastatic niche formation [59].
Macrophages
Macrophages play a central role in the innate immune system by detecting and ingesting unhealthy cells through phagocytosis. Macrophages promote inflammation and mediate adaptive immunity by presenting antigens bound to the surface MHC-II molecules to naïve CD4+ T cells [60]. Moreover, macrophages play a crucial role in resolving inflammation to facilitate tissue repair and remodeling [61]. Notably, such macrophage plasticity between classically activated ‘tissue-destructive’ phenotypes (often associated with high MHC-II expression) and alternatively activated ‘tissue-regenerative’ phenotypes is also induced by various stimuli and cytokines in the TME. In the tumor, alternatively activated TAMs promote angiogenesis, immunosuppression, tumor cell invasion, and metastasis [62,63].
Devalaraja et al [64] recently uncovered a mechanism by which tumor cells instruct tumor-infiltrated monocytes to induce a tumor-supportive microenvironment. Using murine sarcoma models, they found that a Th2-type cytokine IL-13 facilitated retinoic acid production from tumor cells. Tumor-derived retinoic acid polarized intratumor monocyte differentiation toward TAMs and away from dendritic cells via suppressing transcription factor IRF4. Inhibition of retinoic acid signaling enhanced T cell-dependent anti-tumor responses and synergizes with anti-PD-1 therapy [64].
Macrophages also express PD-1, and its expression level increases with time in mouse models and cancer stages in humans. PD-1 expression on macrophages negatively correlated with phagocytic potency against tumor cells. Moreover, PD-1/PD-L1 blockade promoted macrophage phagocytosis, inhibited tumor growth, and extended survival in mouse models in a macrophage-dependent manner [65].
Macrophages are an established component of the CSC niche [14,15], and cellular interactions between CSCs and TAMs have been actively investigated. Zhou et al [66] reported that CSCs in glioblastoma secrete periostin, an ECM protein often detected in multiple stem cell niches. They found that periostin-rich areas in patient tumor tissues significantly accumulated TAMs. These TAMs were not brain-resident microglia but monocyte-derived macrophages recruited from peripheral blood. Depleting periostin in CSCs reduced TAM density, attenuated alternatively activated phenotypes, and increased survival of mice bearing glioblastoma [66].
Our group recently identified a novel mechanism of the crosstalk between CSCs and TAMs emerging during malignant transformation of cutaneous squamous cell carcinoma (SCC). We found that a signaling loop activated between CSCs and TAMs underlies the CSC niche formation. Below we describe how we have identified TGF-β signaling as a critical factor for malignant properties of CSCs and IL-33 as a crucial mediator to establish an early CSC niche.
TGF-β-responding CSCs and their niche microenvironment
TGF-β-responding tumor cells in squamous cell carcinoma (SCC)
SCC is a prevalent cancer worldwide and has significant risk of metastasis [67]. SCC exhibits high rates of tumor recurrence following conventional therapies. SCC cell populations enriched for CSCs have been identified and characterized. CSCs are typified by elevated integrin expression and other markers, including CD34, CD44, and SOX2. They represent ~1–5% of tumor cells and reside at the tumor–stroma interface, an ideal anatomical location for paracrine interactions with potential niche cells.
TGF-β is an integral component of the crosstalk between stem cells and the niche [68]. TGF-β is secreted as a latent form and sequestered in the ECM (Figure 3A) [69]. After release, TGF-β binds to the TGF-β receptor complex, which phosphorylates intracellular SMAD2 and SMAD3. Phosphorylated SMAD2/3 activates target gene expression with SMAD4 by binding to specific nuclear DNA sequences called SMAD-binding elements (Figure 3A) [71]. TGF-β also induces a series of non-SMAD signaling pathways [72]. TGF-β suppresses tumor formation in premalignant tissues by inducing cytostatic and apoptotic effects. In contrast, TGF-β promotes invasion and metastasis in established tumors [73]. It is noteworthy that TGF-β also exerts tumor supportive effects by affecting various stromal cells [74], including immunosuppression [73]. Therefore, a better understanding of the tumor stage-dependent and cell type-specific role of TGF-β is crucial for the therapeutic application of this potent signaling mediator.
Figure 3.
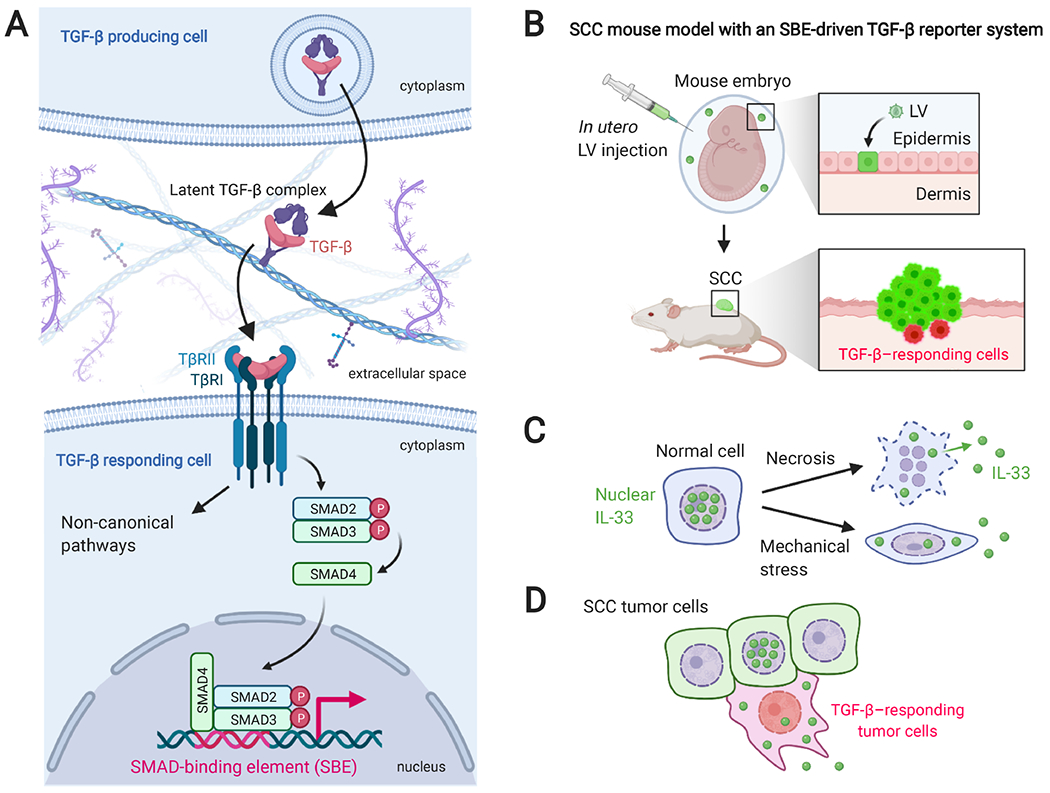
The TGF-β signaling pathway, SCC mouse model, and models of IL-33 extracellular release. (A) A summary of the TGF-β signaling pathway. TGF-β is translated as larger polypeptides and proteolytically cleaved in TGF-β-producing cells. After secretion, TGF-β is sequestered in the ECM as a latent complex. The release of TGF-β ligand is regulated by diverse mechanisms, including protease-mediated cleavage of latent associated protein (LAP), low pH, reactive oxygen species, and conformational changes of LAP by Arg-Gly-Asp (RGD)-binding integrins [69]. TGF-β binds and brings together two transmembrane serine–threonine kinases, TGF-β receptor I (TβRI) and II (TβRII). The receptor complex propagates the signal by phosphorylating intracellular substrates. The canonical downstream effectors are SMAD2 and SMAD3. Phosphorylated SMAD2/3 can complex with SMAD4, translocate to the nucleus, and form an active transcription factor by binding to the specific DNA sequence called SMAD-binding elements (SBEs). Additionally, TGF-β activates SMAD-independent, non-canonical pathways, including PI3K/Akt, MAPK (ERK, JNK, and p38), and Rho/Rho kinase pathways. (B) Schematic illustration of epidermis-specific lentiviral transduction and HRAS-driven SCC mouse model. Lentiviral vectors (LVs) are injected into the amniotic fluid surrounding the TetO-Hras, Rosa-YFP transgenic mouse embryo to transduce the epidermal progenitor cells. Postnatally, doxycycline administration induces HRAS-driven SCC (green), in which TGF-β-responding tumor cells are detected by the reporter protein expression (red). (C) Regulation of the extracellular release of IL-33. After necrotic cell death, full-length IL-33 is released from the nuclei of IL-33-producing cells. Additionally, a cell death-independent, mechanical stress-induced release of IL-33 has been reported [70]. (D) Our group found that NRF2Hi, TGF-β-responding tumor cells in invasive SCC show cytoplasmic translocation of IL-33, which was correlated with the accumulation of FcεRIα+ and the IL-33 receptor ST2+ macrophages in the stroma.
To dissect complex TGF-β roles, we recently generated an HRAS-driven SCC mouse model using epidermal cell-specific lentiviral transduction (Figure 3B) [53]. Our model allows us to examine how TGF-β-responding tumor cells behave during SCC development and drug treatment by an in vivo SMAD-binding element-dependent reporter and the TGF-β signaling-dependent lineage tracing capability (Figure 3B). We have shown that TGF-β-responding tumor cells represent a slow-cycling population that drives invasive SCC progression. Although cisplatin chemotherapy reduced tumor burden significantly in our system, most SCC tumors came back in several weeks. Importantly, lineage tracing of TGF-β-responding cells revealed that recurrent tumors were broadly occupied by their progeny, indicating that TGF-β-responding tumor cells represent drug-resistant CSCs [53]. Transcriptomic analysis revealed that this CSC population has elevation of known CSC markers, including Sox2, Hmga2, Aldh1a3, and Vegfa, and TGF-β target genes, such as Cdkn1a (p21), Cdkn2b (p15), Pthlh, and Gadd45. Notably, the most upregulated biological process was the NRF2-dependent antioxidant response [75], which had a crucial role in chemotherapy resistance [53].
The mechanism by which the CSC niche emerges during malignant tumor progression remains unclear. As abundant TGF-β expression in the stroma is associated with poor prognosis in various types of human cancer [76,77], a ‘TGF-β-rich’ stromal condition might serve as a niche microenvironment for CSCs. We observed that the frequency of TGF-β-responding tumor cells correlates with locally enriched TGF-β in the adjacent stroma. Therefore, understanding how TGF-β-rich microenvironments are established might provide an opportunity for destabilizing drug-resistant CSCs. We recently focused on the cytokine milieu and immune cells surrounding TGF-β-responding CSCs to address the potential mechanism [78].
IL-33 is a CSC-derived cytokine and essential for CSC maintenance
We identified IL-33 as the most highly expressed cytokine in TGF-β-responding CSCs by transcriptomic analysis. The expression of IL-33 was dependent on the presence of TGF-β receptor [78]. IL-33 is a member of the IL-1 family cytokines and has a pivotal role in tumor development [79–81]. The C-terminal region of IL-33 harbors the cytokine domain, whereas the N-terminal contains a DNA-binding domain. In normal tissue, IL-33 is typically detected in the nucleus and released upon necrotic cell death (Figure 3C) [79]. However, it remains unclear how living tumor cells release IL-33 without undergoing cell death.
We found that invasive tumor cells, particularly TGF-β-responding cells, show cytoplasmic IL-33 localization [78], suggesting that CSCs may actively secrete IL-33 in the adjacent stroma. It is reported that IL-33 secretion is induced by mechanical stress (Figure 3C) [70], which is known to activate the antioxidant responses [82]. We showed a correlation between the NRF2 activity and IL-33 cytoplasmic localization in vivo and extracellular release of IL-33 in vitro. Our data suggested that the NRF2-mediated antioxidant response elevated in the TGF-β-responding CSCs facilitates the extracellular release of IL-33 (Figure 3D) [78]. As IL-33 binds to chromatin, we speculate that NRF2 may regulate a CSC-specific chromatin state to facilitate the nuclear export of IL-33. Moreover, as IL-33 lacks a secretory signal peptide, a mechanisms of unconventional protein secretion is probably involved in IL-33 secretion from CSCs.
To determine the role of IL-33, we depleted IL-33 from tumor cells, including TGF-β-responding CSCs. IL-33 depletion resulted in stunted tumor growth with less invasive phenotypes [53,78]. Remarkably, IL-33-depleted tumors have significantly less TGF-β-responding tumor cells. Consistent with diminishing TGF-β signaling, IL-33-depleted tumors were more sensitive to cisplatin chemotherapy [78], suggesting that CSC-derived IL-33 is necessary for drug resistance and is potentially involved in the formation of TGF-β-rich microenvironments.
CSC-derived IL-33 induces FcεRIα+ TAMs to establish a CSC niche during SCC progression
To identify IL-33-responding stromal cells, we searched for immune cells accumulated around TGF-β-responding CSCs. In both human and mouse SCC, we found that macrophages expressing high-affinity IgE receptor FcεRIα existed at high density in proximity to TGF-β-responding CSCs [78]. FcεRIα+ macrophages upregulated a series of alternatively activated macrophage markers, including Arg1, Cd206, and Il10. Importantly, FcεRIα+ macrophages, but not FcεRIαneg counterparts, highly expressed the IL-33 receptor ST2 [83]. Moreover, FcεRIα+ macrophages overlapped with TGF-β-rich stromal areas, which were rarely detected in IL-33-depleted tumors, suggesting that CSCs directly induces FcεRIα+ TAMs to establish the CSC niche [78].
Although macrophage frequency in IL-33-depleted tumors was comparable with that in control tumors, FcεRIα+ TAMs were significantly reduced in IL-33-depleted tumors. Moreover, immature myeloid cells lacking ST2 were similarly recruited to the stroma, indicating that IL-33 is dispensable for macrophage recruitment but required for FcεRIα+ TAM differentiation [78]. We found that IL-33 promotes differentiation of bone marrow-derived cells into FcεRIα+, Arg1Hi alternatively activated macrophages in vitro, in an ST2 and the nuclear factor kB (NF-kB) signaling pathway-dependent manner (Figure 4A). Moreover, IL-33-induced macrophages activated paracrine TGF-β signaling and promoted invasion and epithelial–mesenchymal transition phenotypes in epithelial cells, suggesting that FcεRIα+ TAMs are responsible for TGF-β-rich stroma and SCC progression (Figure 4B) [78].
Figure 4.
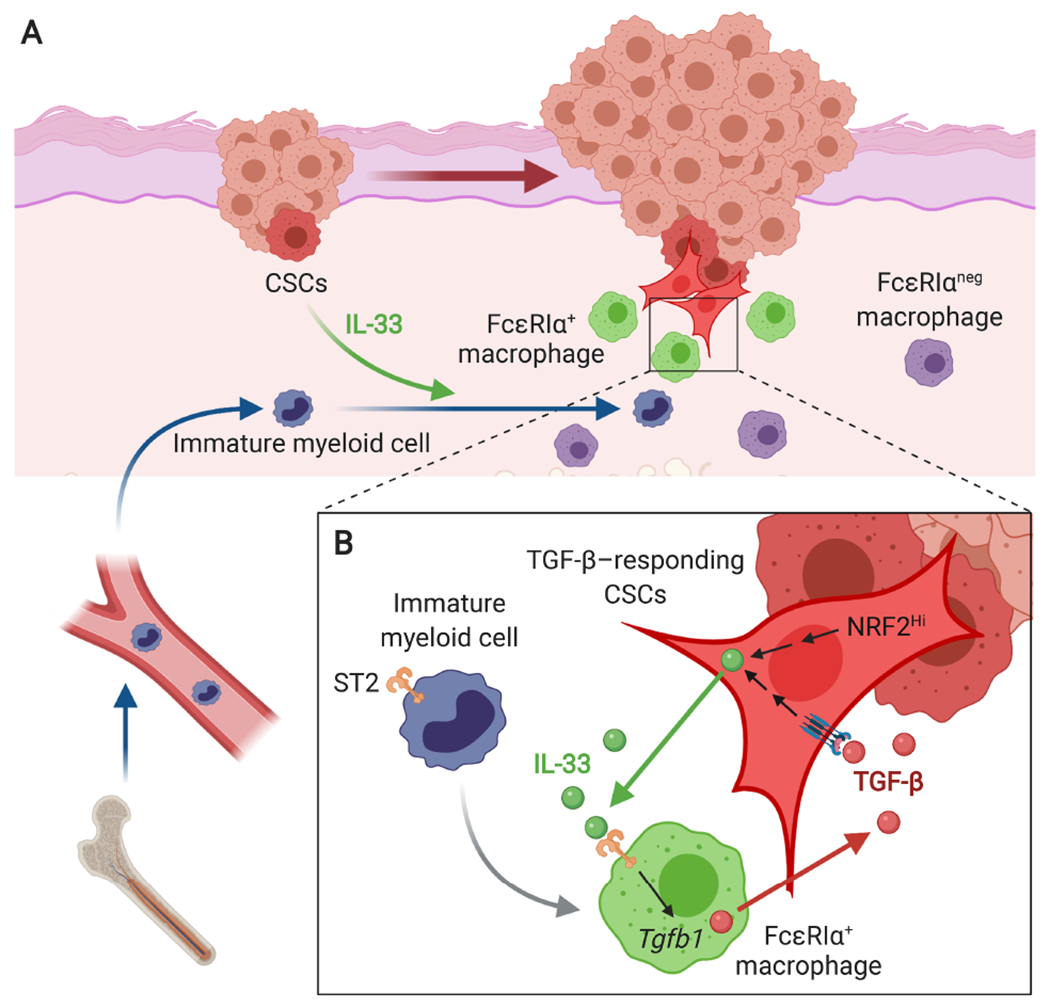
A model of CSC-niche interactions during invasive SCC progression. (A) CSCs, either TGF-β-responding or NRF2-activated, release IL-33 as a short-distant signal, which promotes the differentiation of ST2+ immature myeloid cells into FcεRIα+ alternatively activated macrophages. (B) FcεRIα+ macrophages express high levels of TGF-β and produce ‘TGF-β rich’ niche microenvironments in the proximity to CSCs. The niche sustains paracrine TGF-β signaling necessary to maintain slow-cycling CSCs and promote their invasive and drug-resistant properties. Moreover, TGF-β-responding CSCs upregulate IL-33 expression and activate the extracellular release of IL-33 through the NRF2-mediated antioxidant response. This self-reinforcing IL-33-TGF-β niche signaling loop is a crucial mechanism of malignant progression and drug resistance of SCC.
IL-33 signaling and FcεRIα+ TAMs could be rational targets for destabilizing CSCs. We blocked the IL-33-ST2 signal pathway by ectopic expression of soluble ST2 (sST2), a decoy receptor for IL-33 [83], in vivo. As expected, sST2-expressing tumors had less FcεRIα+ TAMs, grew significantly slowly, and exhibited less malignant phenotypes. The antibody-mediated FcεRIα+ TAM depletion also led to significant reductions in TGF-β reporter activity and tumor growth [78].
Taken together, CSC-derived IL-33 is critical for the enrichment of FcεRIα+ TAMs in the proximity to CSCs and the establishment of the niche for TGF-β-responding CSCs. We propose that CSCs drive a self-reinforcing IL-33–TGF-β signaling loop using CSC-specific antioxidant activities for the maintenance and probably for the expansion of drug-resistant CSCs during tumor progression (Figure 4B).
Conclusions
Recent studies have uncovered specific interactions between CSCs and niche cells from a myriad of crosstalk in the TME. We are starting to see evidence that the perturbation of niche signaling pathways could improve cancer treatment responses. Evolving technologies for single-cell analysis have unveiled previously unappreciated heterogeneity in major stromal cell types, which add additional complexity to our view of tumor heterogeneity. Although our knowledge of the CSC niche is still a collection of fragmented information, these new studies have identified specific stromal subsets that are associated with patient prognosis. Therefore, it will be an active research area to figure out which stromal subpopulations participate in the CSC niche to promote tumor progression and drug resistance. Moreover, because CSC gene expression signatures can predict poor clinical outcomes [84,85], identifying a critical niche component and the signaling pathways responsible for the CSC-specific gene expression program would provide new therapeutic targets to destabilize CSCs and improve cancer treatment efficacy.
Acknowledgements
The figures in this article were created with Biorender.com. This work was supported in part by the following grants and foundations: the NIH R00CA178197 and R01CA245535, the Medical Foundation of Oregon, the Collins Medical Trust, and the start-up support from the Knight Cancer Institute/Oregon Health & Science University to NO.
Footnotes
No conflicts of interest were declared.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 2.Greaves M, Maley CC. Clonal evolution in cancer. Nature 2012; 481: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov 2017; 16: 241–263. [DOI] [PubMed] [Google Scholar]
- 4.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013; 501: 346–354. [DOI] [PubMed] [Google Scholar]
- 5.Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, et al. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun 2020; 11: 5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014; 344: 1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015; 347: 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reagan MR, Rosen CJ. Navigating the bone marrow niche: translational insights and cancer-driven dysfunction. Nat Rev Rheumatol 2016; 12:154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dick JE. Stem cell concepts renew cancer research. Blood 2008; 112: 4793–4807. [DOI] [PubMed] [Google Scholar]
- 10.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367: 645–648. [DOI] [PubMed] [Google Scholar]
- 11.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100:3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 2016; 11: 47–76. [DOI] [PubMed] [Google Scholar]
- 13.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013; 501: 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015; 16: 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prager BC, Xie Q, Bao S, et al. Cancer stem cells: the architects of the tumor ecosystem. Cell Stem Cell 2019; 24: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahai E, Astsaturov I, Cukierman E, et al. A frame work for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 2020; 20: 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannoni E, Bianchini F, Masieri L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res 2010; 70: 6945–6956. [DOI] [PubMed] [Google Scholar]
- 18.Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014; 25: 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014; 25: 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019; 9: 282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuzillet C, Tijeras-Raballand A, Ragulan C, et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J Pathol 2019; 248: 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa A, Kieffer Y, Scholer-Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018; 33: 463–479.e10. [DOI] [PubMed] [Google Scholar]
- 23.Su S, Chen J, Yao H, et al. CD10+GPR77+ cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 2018; 172: 841–856.e16. [DOI] [PubMed] [Google Scholar]
- 24.Roulis M, Kaklamanos A, Schernthanner M, et al. Paracrine orchestration of intestinal tumorigenesis by a mesenchymal niche. Nature 2020; 580: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calvo F, Ege N, Grande-Garcia A, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013; 15: 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nallanthighal S, Heiserman JP, Cheon D-J. The role of the extracellular matrix in cancer stemness. Front Cell Dev Biol 2019; 7: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pang M-F, Siedlik MJ, Han S, et al. Tissue stiffness and hypoxia modulate the integrin-linked kinase ILK to control breast cancer stem-like cells. Cancer Res 2016; 76: 5277–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malanchi I, Santamaria-Martínez A, Susanto E, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012; 481: 85–89. [DOI] [PubMed] [Google Scholar]
- 29.Begum A, Ewachiw T, Jung C, et al. The extracellular matrix and focal adhesion kinase signaling regulate cancer stem cell function in pancreatic ductal adenocarcinoma. PLoS One 2017; 12: e0180181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govaere O, Wouters J, Petz M, et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J Hepatol 2016; 64: 609–617. [DOI] [PubMed] [Google Scholar]
- 31.Shiina M, Bourguignon LY. Selective activation of cancer stem cells by size-specific hyaluronan in head and neck cancer. Int J Cell Biol 2015; 2015: 989070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angel I, Pilo Kerman O, Rousso-Noori L, et al. Tenascin C promotes cancer cell plasticity in mesenchymal glioblastoma. Oncogene 2020; 39: 6990–7004. [DOI] [PubMed] [Google Scholar]
- 33.Mohyeldin A, Garzón-Muvdi T, Quiñones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell 2010; 7: 150–161. [DOI] [PubMed] [Google Scholar]
- 34.Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A 1997; 94: 8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Folkins C, Shaked Y, Man S, et al. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res 2009; 69: 7243–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck B, Driessens G, Goossens S, et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 2011; 478: 399–403. [DOI] [PubMed] [Google Scholar]
- 37.Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell 2010; 6: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu J, Ye X, Fan F, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell 2013; 23: 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, Zhou C, Zhang Z, et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat Commun 2020; 11: 5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voron T, Colussi O, Marcheteau E, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med 2015; 212: 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goveia J, Rohlenova K, Taverna F, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 2020; 37: 21–36.e13. [DOI] [PubMed] [Google Scholar]
- 42.Garnier L, Gkountidi A-O, Hugues S. Tumor-associated lymphatic vessel features and immunomodulatory functions. Front Immunol 2019; 10: 720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006; 313: 1960–1964. [DOI] [PubMed] [Google Scholar]
- 44.Denardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 2011; 1: 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol 2007; 25: 171–192. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura H, Nose M, Hiai H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999; 11: 141–151. [DOI] [PubMed] [Google Scholar]
- 47.Keir ME, Butte MJ, Freeman GJ, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008; 26: 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013; 5: 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh DY, Kwek SS, Raju SS, et al. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 2020; 181: 1612–1625.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lavin Y, Kobayashi S, Leader A, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017; 169: 750–765.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018; 554: 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018; 554: 538–543. [DOI] [PubMed] [Google Scholar]
- 53.Oshimori N, Oristian D, Fuchs E. TGF-beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015; 160: 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miao Y, Yang H, Levorse J, et al. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell 2019; 177: 1172–1186.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12: 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol 2019; 16: 601–620. [DOI] [PubMed] [Google Scholar]
- 58.Albrengues J, Shields MA, Ng D, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018; 361: eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ombrato L, Nolan E, Kurelac I, et al. Metastatic-niche labelling reveals parenchymal cells with stem features. Nature 2019; 572: 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015; 15:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mantovani A, Biswas SK, Galdiero MR, et al. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol 2013; 229: 176–185. [DOI] [PubMed] [Google Scholar]
- 62.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 2012; 21: 309–322. [DOI] [PubMed] [Google Scholar]
- 63.Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014; 41: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Devalaraja S, To TKJ, Folkert IW, et al. Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell 2020; 180: 1098–1114.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gordon SR, Maute RL, Dulken BW, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017; 545: 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou W, Ke SQ, Huang Z, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol 2015; 17: 170–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dotto GP, Rustgi AK. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell 2016; 29: 622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oshimori N, Fuchs E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell 2012; 11: 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116: 217–224. [DOI] [PubMed] [Google Scholar]
- 70.Kakkar R, Hei H, Dobner S, et al. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem 2012; 287: 6941–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hata A, Chen Y-G. TGF-β signaling from receptors to Smads. Cold Spring Harb Perspect Biol 2016; 8: a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Derynck R, Budi EH. Specificity, versatility, and control of TGF-β family signaling. Sci Signal 2019; 12: eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019; 50: 924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer 2013; 13: 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rojo De La Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell 2018; 34: 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012; 22: 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richardsen E, Uglehus RD, Johnsen SH, et al. Immunohistochemical expression of epithelial and stromal immunomodulatory signaling molecules is a prognostic indicator in breast cancer. BMC Res Notes 2012; 5: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taniguchi S, Elhance A, Van Duzer A, et al. Tumor-initiating cells establish an IL-33-TGF-beta niche signaling loop to promote cancer progression. Science 2020; 369: eaay1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liew FY, Girard J-P, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol 2016; 16: 676–689. [DOI] [PubMed] [Google Scholar]
- 80.Wasmer M-H, Krebs P. The role of IL-33-dependent inflammation in the tumor microenvironment. Front Immunol 2016; 7: 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fournié J-J, Poupot M. The pro-tumorigenic IL-33 involved in antitumor immunity: a Yin and Yang cytokine. Front Immunol 2018; 9: 2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aikawa R, Nagai T, Tanaka M, et al. Reactive oxygen species in mechanical stress-induced cardiac hypertrophy. Biochem Biophys Res Commun 2001; 289: 901–907. [DOI] [PubMed] [Google Scholar]
- 83.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov 2008; 7: 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu R, Wang X, Chen GY, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med 2007; 356: 217–226. [DOI] [PubMed] [Google Scholar]
- 85.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 2011; 17: 1086–1093. [DOI] [PubMed] [Google Scholar]