

Abstract
In this review, transition metal-catalyzed methodologies and applications that exploit C–C bond cleavage of vinylcyclopropanes (VCPs) are summarized with a focus on cycloaddition and related addition reactions. Transition metals, including palladium, nickel, iron, ruthenium, rhodium, cobalt, and iridium, can catalyze the cleavage of C–C bonds in activated or non-activated VCPs. Additionally, these bond-breaking reactions can occur as intra- or intermolecular processes. The properties of activated and non-activated VCPs are discussed in the introduction. Various transition metal-catalyzed cycloaddition and addition reactions involving the cleavage of C–C bonds in activated VCPs are then discussed in the next chapter. The transition metal-catalyzed cycloadditions involving the cleavage of C–C in non-activated VCPs are summarized in the following chapter. Finally, challenges and potential opportunities are outlined in the last chapter.
Graphical Abstract
1. INTRODUCTION
Cycloaddition reactions, such as the classic Diels–Alder reaction, are some of the most frequently used methods in organic synthesis, providing atom-economical,1–2 efficient, and practical access to complex molecules from simple starting materials.3–6 Most cycloadditions can be mediated by Lewis acids, heat, or light, but transition-metal catalysts are also used to promote formal cycloadditions through coordination to unsaturated moieties such as alkynes, alkenes, allenes, or dienes. C–C bond cleavage is a common process involved in transition-metal-catalyzed cycloadditions.7–10 Due to the high energy required to cleave C–C bonds, several strategies have been developed to facilitate transition-metal-mediated selective C–C bond cleavage.11 One common strategy involves using strained small-rings (three- and four-membered), where the strain-release energy associated with the cleavage event provides a major driving force, which makes them ideal candidates for transition-metal-catalyzed C–C bond cleavage methodologies.12–15
Vinylcyclopropanes (VCPs) have been widely used in transition-metal-catalyzed cycloaddition reactions because they possess an olefin ligand that directs the transition-metal for the selective C–C bond cleavage.16–19 Moreover, the high strain energy of VCPs (ca. 28 kcal/mol, Scheme 1, a) makes the ring opening of the cyclopropane energetically favorable20 and generates active π-allyl-metal complexes. As a result, the transition-metal-catalyzed activation of VCPs, in combination with a broad variety of unsaturated acceptors, has led to the development of novel cycloaddition patterns in organic synthesis.
Scheme 1.
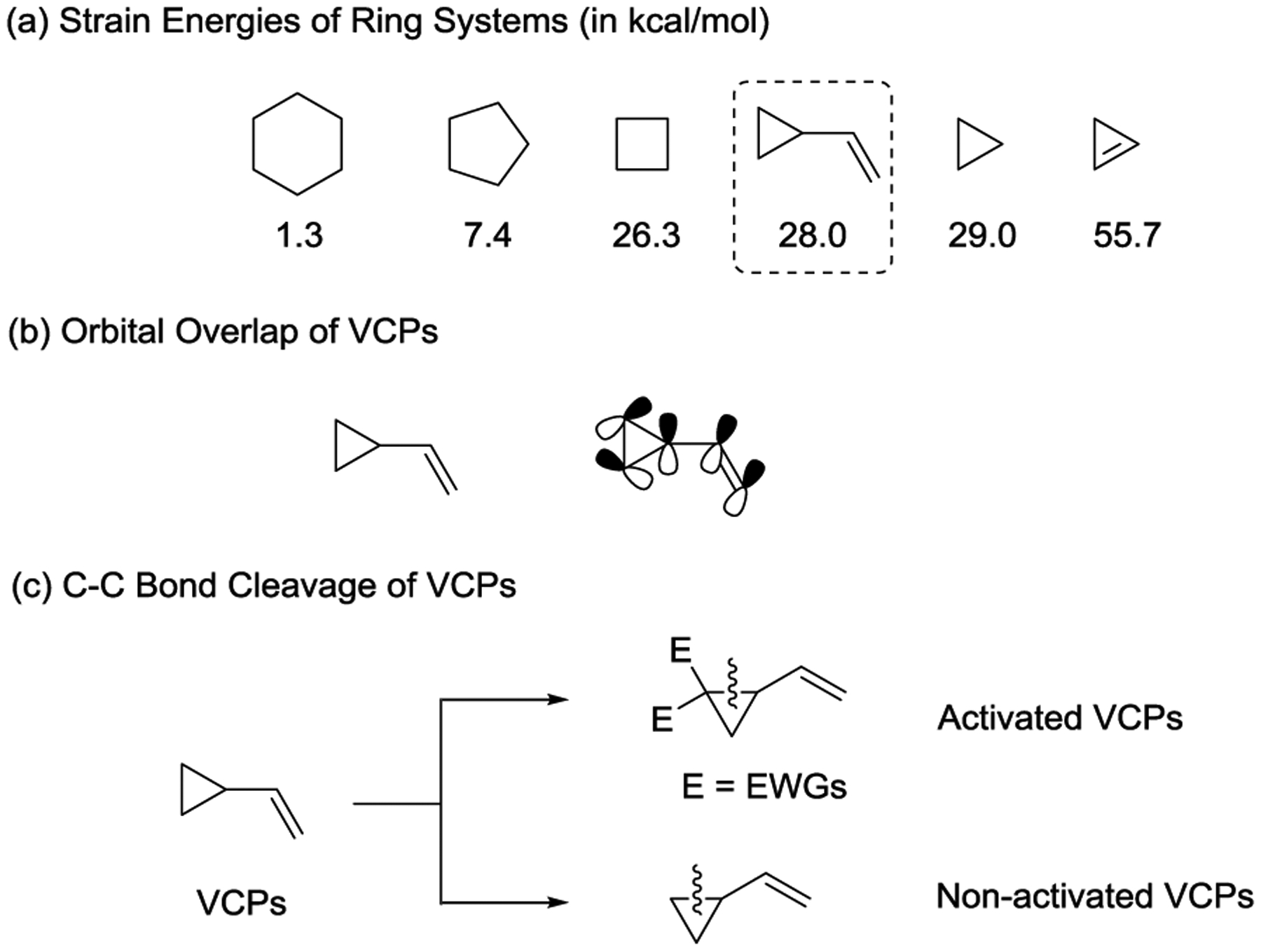
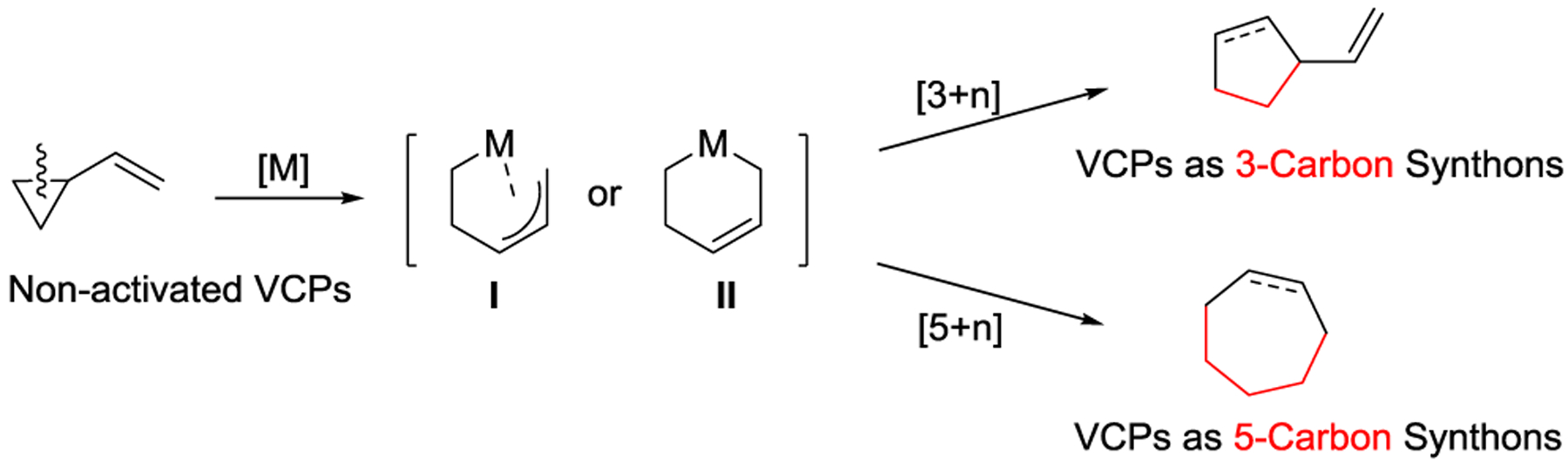
The bonding in VCPs is such that an s-trans-gauche conformational equilibrium exists to allow for maximum orbital overlap of the p-orbitals of the cyclopropane ring with the π- or π*-orbitals of the ethylene unit (Scheme 1, b). In general, VCPs are divided into two categories with respect to the substitution pattern on the cyclopropane - activated VCPs feature strong electron-withdrawing groups (EWGs) while non-activated VCPs lack these EWGs (Scheme 1, c). Depending on whether the vinyl substituent participates in the cycloaddition, VCPs can serve as three- or five-carbon synthons. As three-carbon synthons, VCPs mainly participate in [3 + 2] and [3 + 2 +1] cycloadditions that yield five- and six-membered rings.21 As five-carbon synthons, VCPs participate in a wide range of cycloadditions, such as [5 + 1], [5 + 2], [5 + 2 + 1], and [5 + 1 + 2 + 1]. These result in the formation of mono- or fused six-, seven-, and even eight-membered ring systems. By using VCPs as either three- or five-carbon synthons, both intramolecular and intermolecular cycloaddition reactions have been realized to efficiently produce diversified skeletons.
During the past three decades, significant progress has been made using transition-metal catalysis to cleave VCP C-C bonds for the development of cycloaddition reactions. For example, a recently published book provides in-depth discussion of processes involving C–C bond cleavage.11 Earlier developments of transition-metal-catalyzed C–C bond cleavage of cyclopropanes (until 2007)12 and cycloadditions of VCPs (until 2013) have also been reviewed.16 The purpose of this review is to present a comprehensive picture of the development of cycloaddition and related addition reactions involving VCPs from 1970 to the present day, even if areas of this field have been previously covered.16–19 The review is organized according to the transition metal used in the C-C bond cleavage of activated and non-activated VCPs, in both intramolecular and intermolecular cycloaddition and related addition reactions. Two sections that highlight the power of these transformations as the key scaffold-building step for the preparation of bioactive natural products and pharmaceutical agents are also presented in the hope that they will inspire continued investigation into this fascinating research topic.
2. C-C BOND CLEAVAGE OF ACTIVATED VCPs
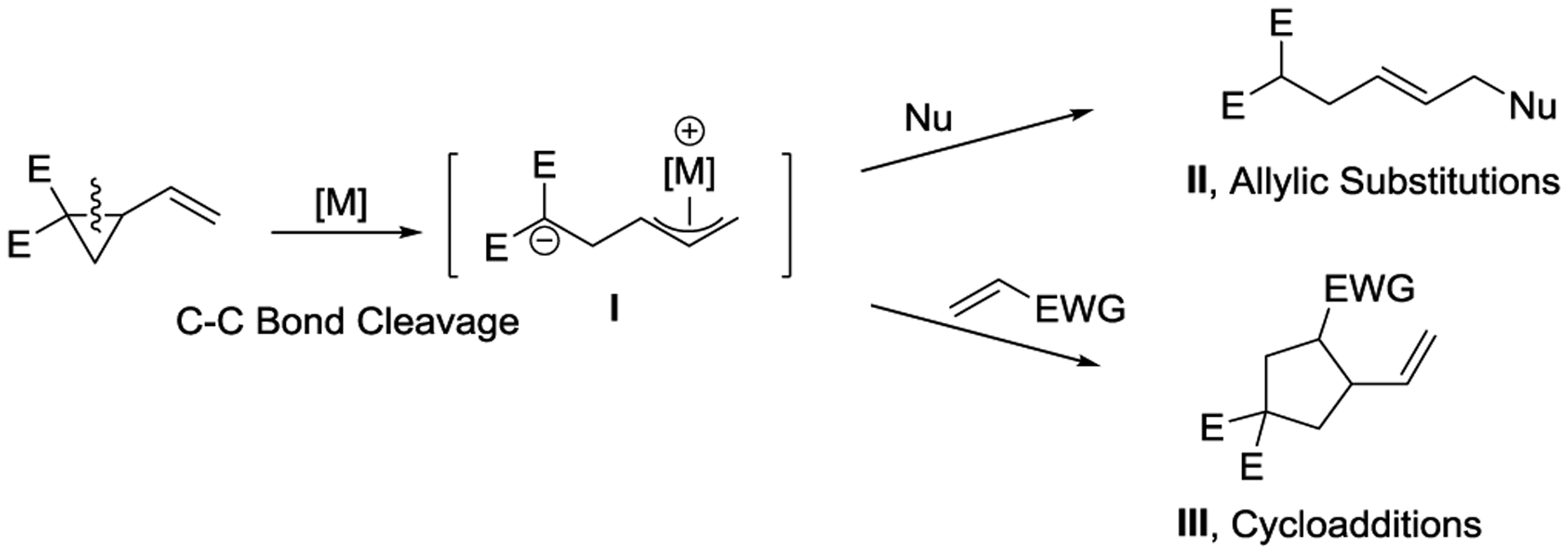
VCPs can be activated by EWGs directly attached to the cyclopropane ring. The presence of EWGs on VCPs facilitate ring opening reactions, and these VCPs are also known as Tsuji-type VCPs, due to Tsuji’s pioneering work in this area,22,23 or donor– acceptor VCPs.21 Donor–acceptor VCPs have a vinyl group that acts as an electron-donating group (donor) and typically feature a geminal diester as the EWG (acceptor). In the presence of transition-metal catalysts, these VCPs are readily activated to form zwitterionic π-allylmetal species I. These species are capable of allylic substitution processes or addition reactions as well as cycloaddition reactions with various π-systems, yielding allylic products II and cycloadducts III, respectively. (Scheme 2).
Scheme 2.
2.1. Metal-Catalyzed Intramolecular Processes
2.1.1. Palladium Catalysis
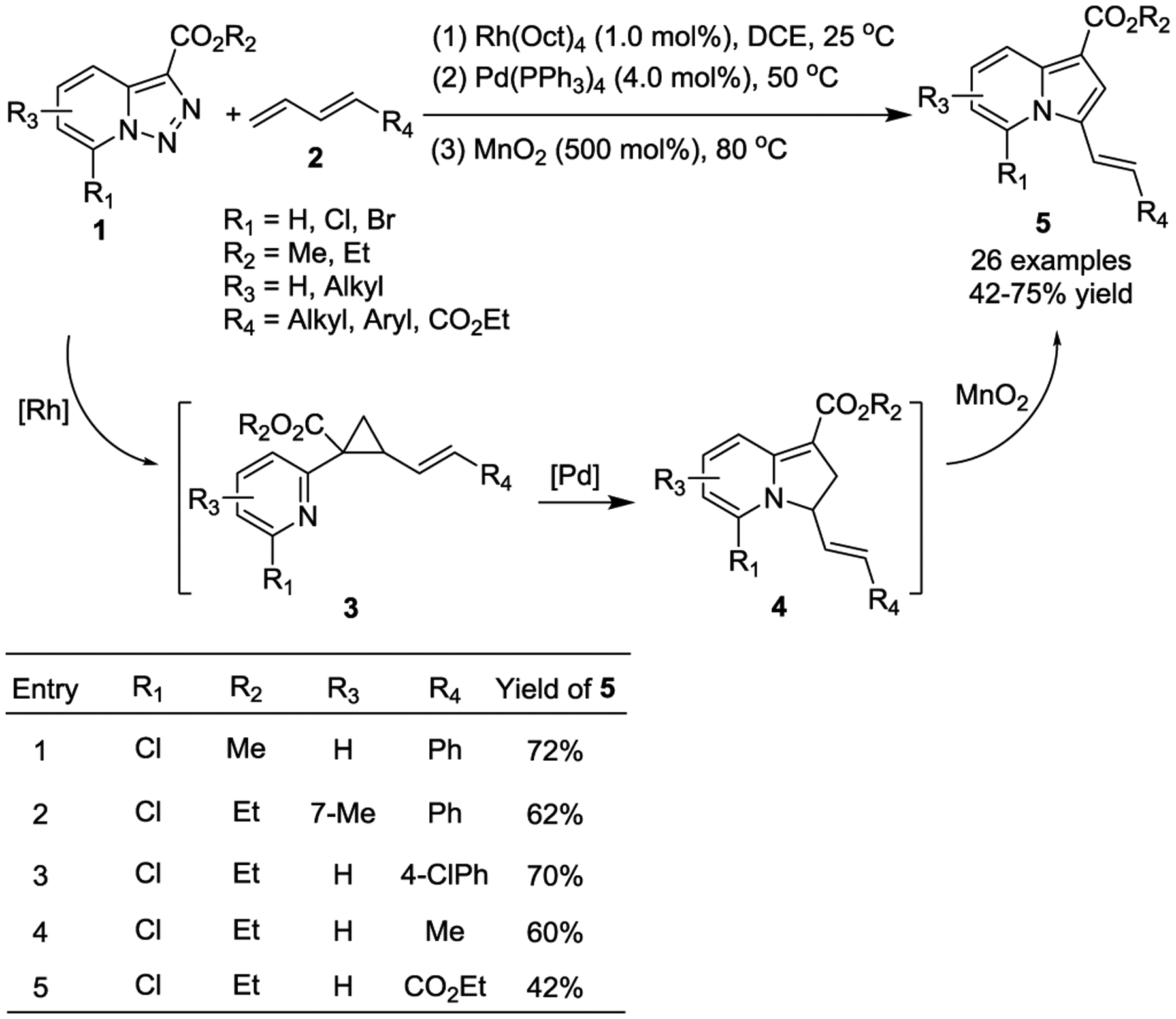
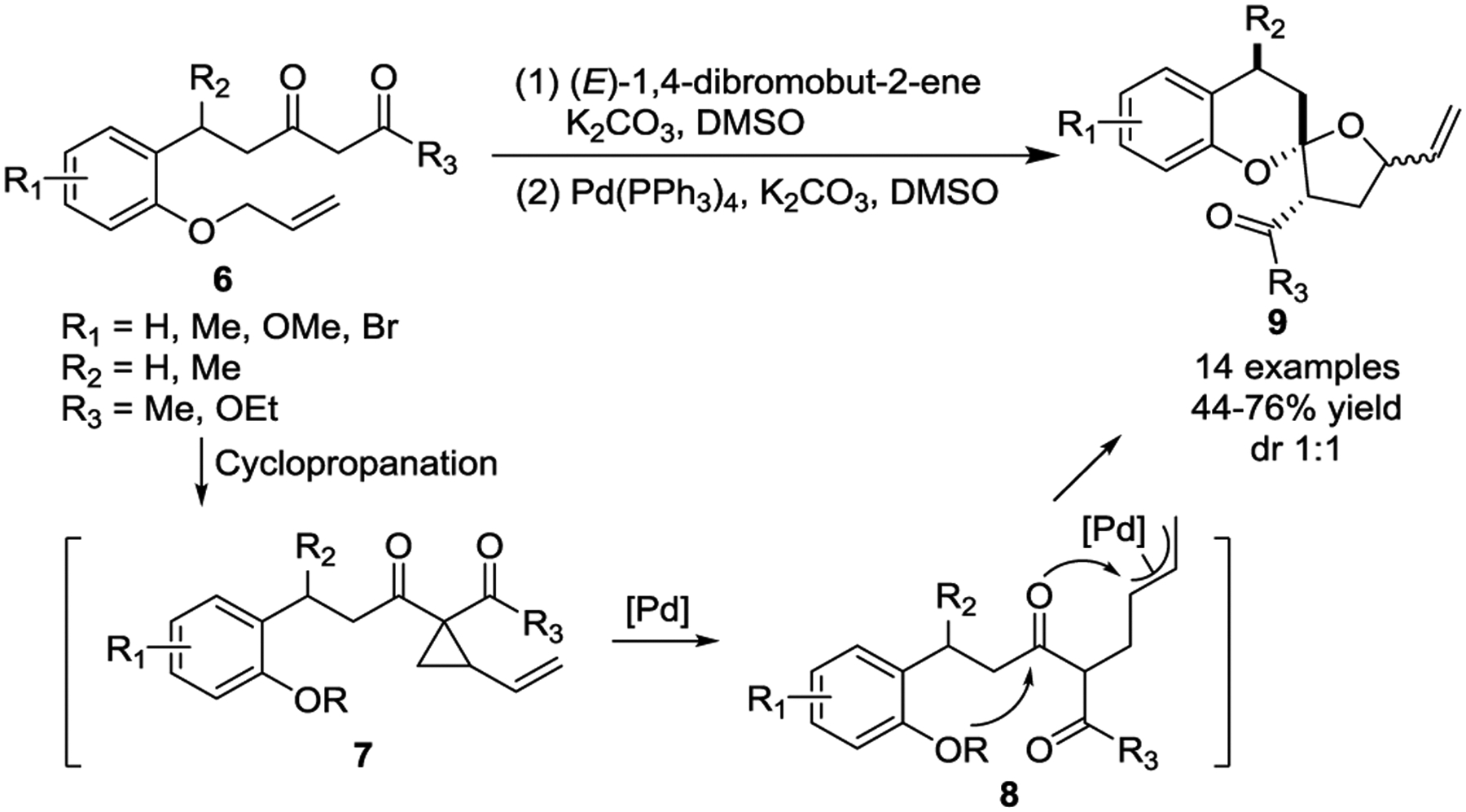
Thus far, only a few literature examples demonstrate activated VCPs undergoing transition-metal-catalyzed intramolecular cycloadditions, which may be attributed to difficulty in substrate preparation. In 2017, the Lee group reported an efficient, one-pot synthetic method that used pyridotriazoles 1 and 1,3-dienes 2 for the preparation of functionalized indolizine derivatives 5 (Scheme 3).24 This tandem three step process involves a Rh(II)-catalyzed cyclopropanation of 1,3-dienes, a Pd(0)-catalyzed ring expansion of in-situ generated VCPs 3 through addition of the nitrogen nucleophile to the Pd-π-allyl, and subsequent oxidation by MnO2 to afford indolizine compounds 5 in good yields. In 2019 the Hawkins group reported an efficient synthesis of benzannulated 6,5-spiroketals 9 from vinyl 1,1-diacylcyclopropanes 7 (Scheme 4).25 The in-situ generated VCPs 7 undergo Pd(0)-catalyzed C-C bond cleavage to form π-allyl intermediates 8, followed by ring closure to afford spiroketal complexes 9 in good yields. However, the products were isolated as a 1:1 mixture of diastereomers with the relative stereochemistry set except at the vinyl group, but both isomers could be readily separated by column chromatography. The utility of this method was further demonstrated by its application in the construction of the Berkelic acid core.
Scheme 3.
Scheme 4.
2.2. Metal-Catalyzed Intermolecular Processes
2.2.1. Palladium Catalysis
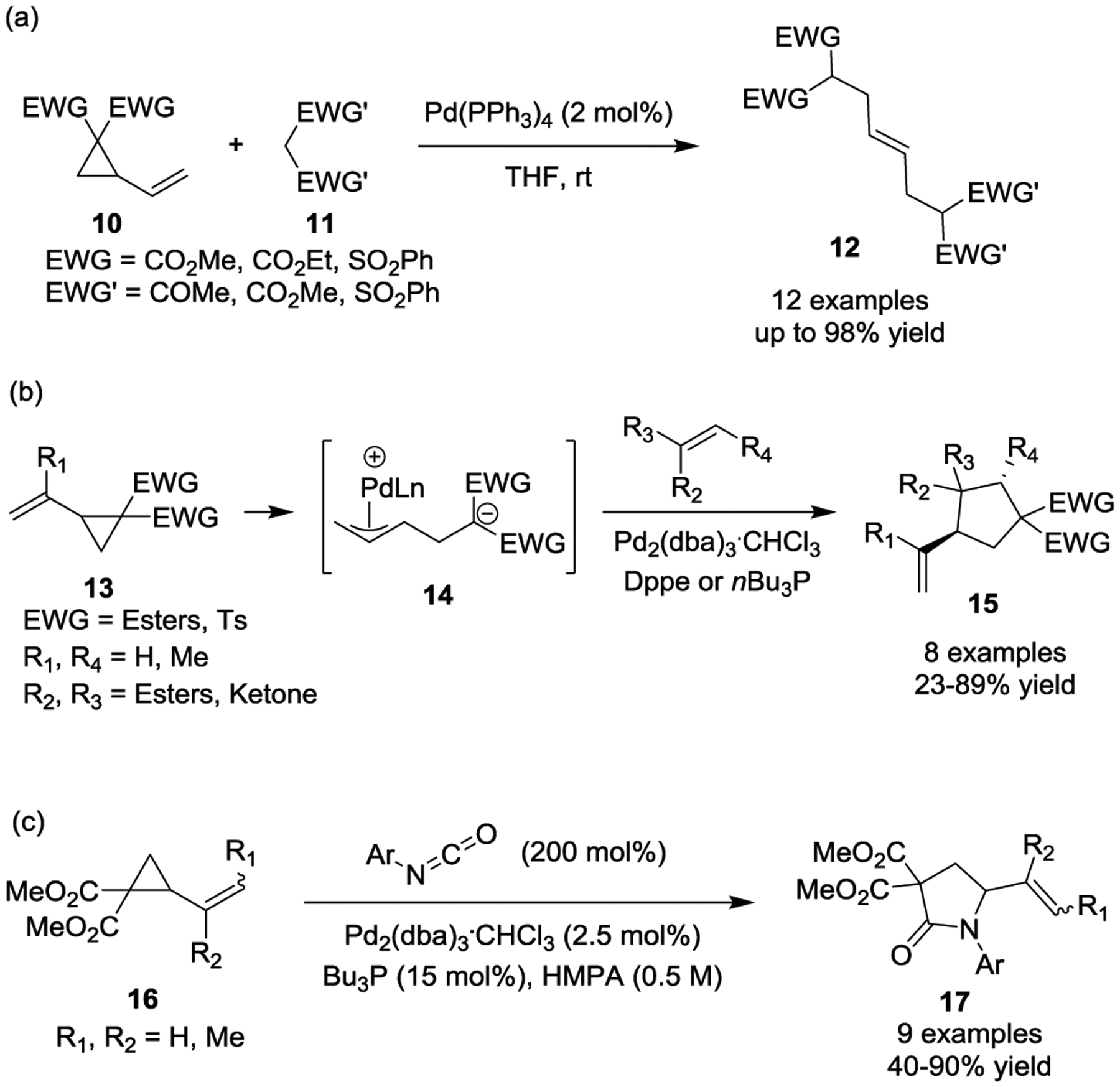
More than 30 years ago, Burgess first demonstrated the nucleophilic trapping of zwitterionic π-allylpalladium species generated from the ring-opening of activated VCPs 10 (Scheme 5, a).22 Various nucleophiles including malonates, 1,3-diketones, and bis(phenylsufonyl)methane were used for this process. At almost the same time, the Tsuji group independently reported its seminal study of a Pd(0)-catalyzed [3 + 2] cycloaddition of activated VCPs 13 with α,β-unsaturated esters or ketones to provide vinylcyclopentanes 15 in good yields under mild conditions (Scheme 5, b).23 In the presence of Pd(0) and a bis(diphenylphosphino)ethane (dppe) or tributylphosphine ligand, C-C bond cleavage of cyclopropanes by oxidative addition generated acyclic zwitterionic π-allylpalladium species 14. This activation mode has been found to be generally effective for other nucleophiles or formal cycloaddition processes involving polarized π-systems, such as isocyanate electrophiles, to afford the corresponding γ-lactams 17 in good yields (Scheme 5, c).26
Scheme 5.
In 2005, the Szabó group reported that Pd(0)-pincer-complex 19 catalyzed the synthesis of functionalized allylboronate derivatives 20 from activated VCPs 18 with tetrahydroxydiboron (Scheme 6, a).27 The reaction provided an efficient route to diversified allyl boronic acids, which were subsequently converted to the more stable potassium trifluoro(allyl)borates 21 in the presence of aqueous KHF2 in high yields under mild conditions. Notably, no desired products were observed by using Pd2(dba)3 or Pd(PPh3)4 catalysts for this transformation.
Scheme 6.
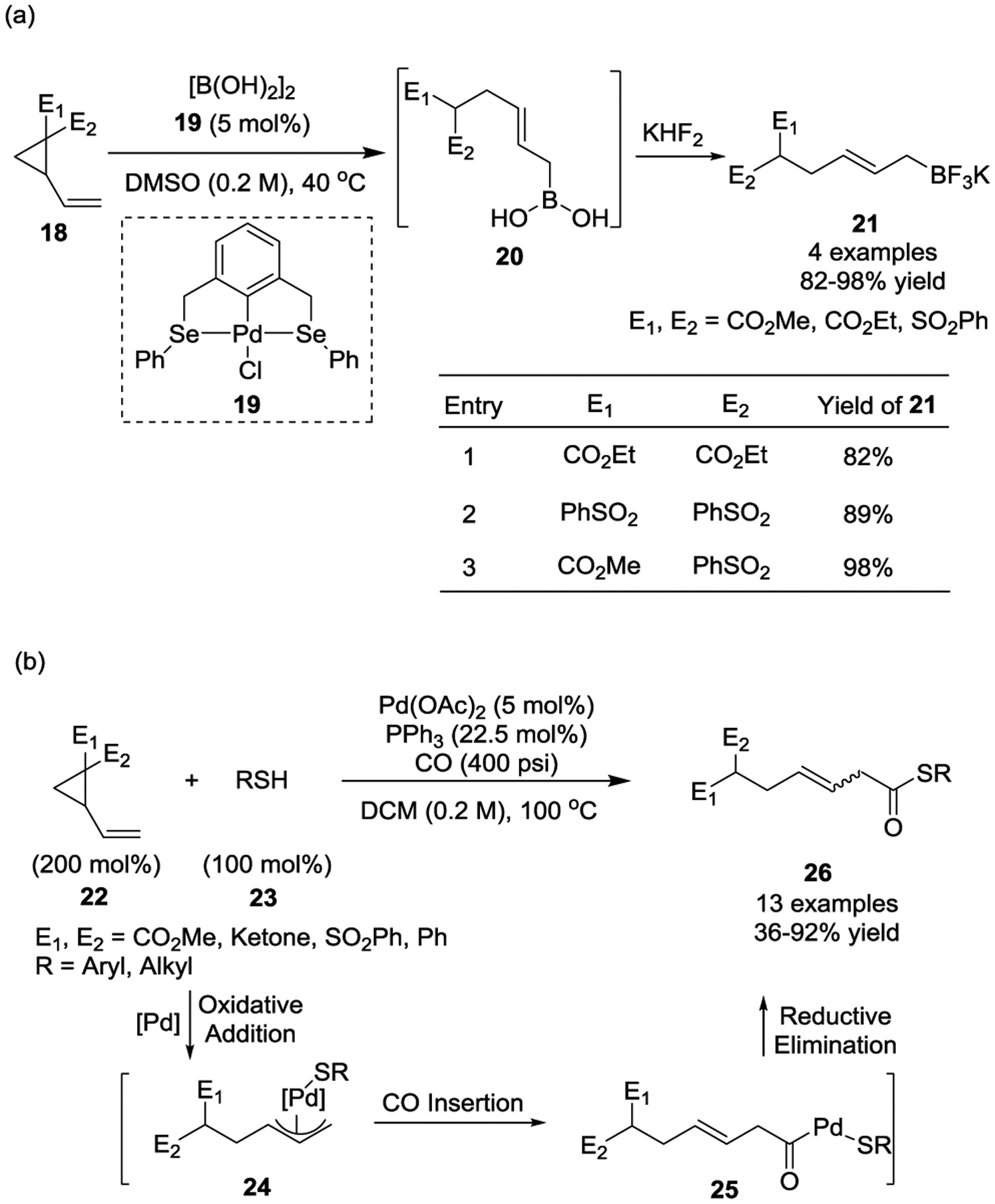
Other nucleophiles have also been reported to be effective for this type of transformation. For instance, in 2008 Alper and Xiao described a palladium-catalyzed thiocarbonylative ring-opening of activated VCPs with thiols and carbon monoxide (Scheme 6, b).28 This reaction provided a general method for the synthesis of β, γ-unsaturated thioesters 26 in moderate to excellent yields. Mechanistically, the oxidative addition of activated VCPs 22 in the presence of a Pd(0) catalyst gives η3-allylpalladium complex 24, followed by CO insertion to generate acylpalladium intermediate 25. This intermediate then undergoes subsequent reductive elimination to afford thioesters 26 and regenerates the Pd(0) catalyst. Further isomerization of the β,γ-unsaturated thioesters provided the α, β-unsaturated isomers as the major products in most cases.
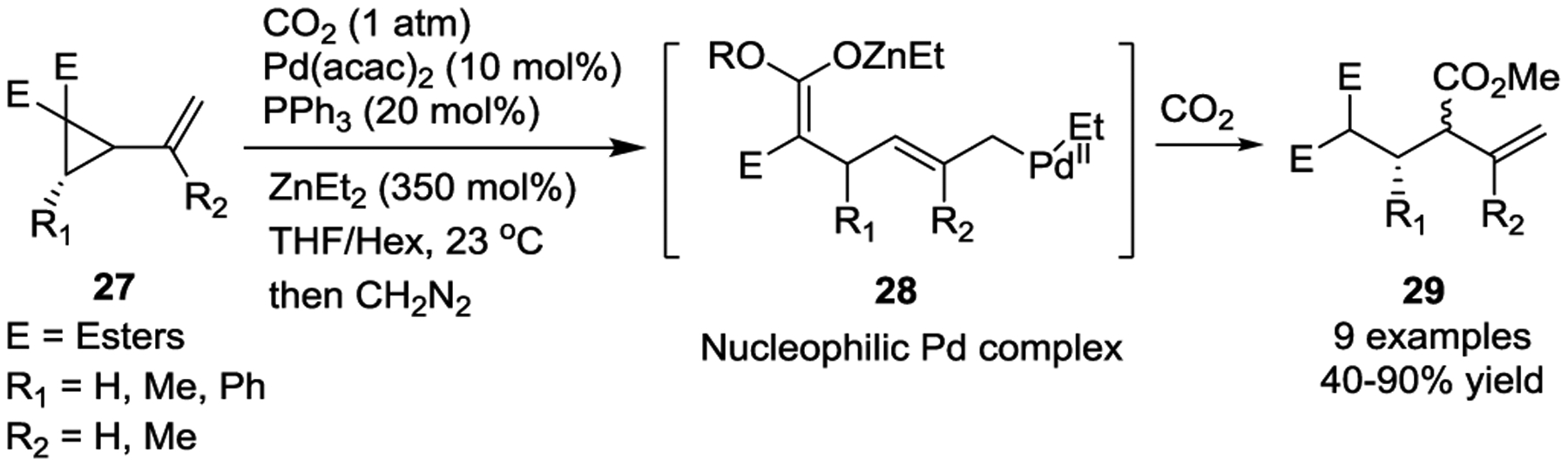
In 2016, Mita and colleagues reported the Pd(II)-catalyzed carboxylation of activated VCPs with ZnEt2 to afford the corresponding β,γ-unsaturated carboxylic acids in high yields under a CO2 atmosphere (1 atm), as shown in Scheme 7.29 The active intermediate in this reaction is thought to be a nucleophilic η1-allylethylpalladium species 28, which was produced from π-allylpalladium and ZnEt2.
Scheme 7.
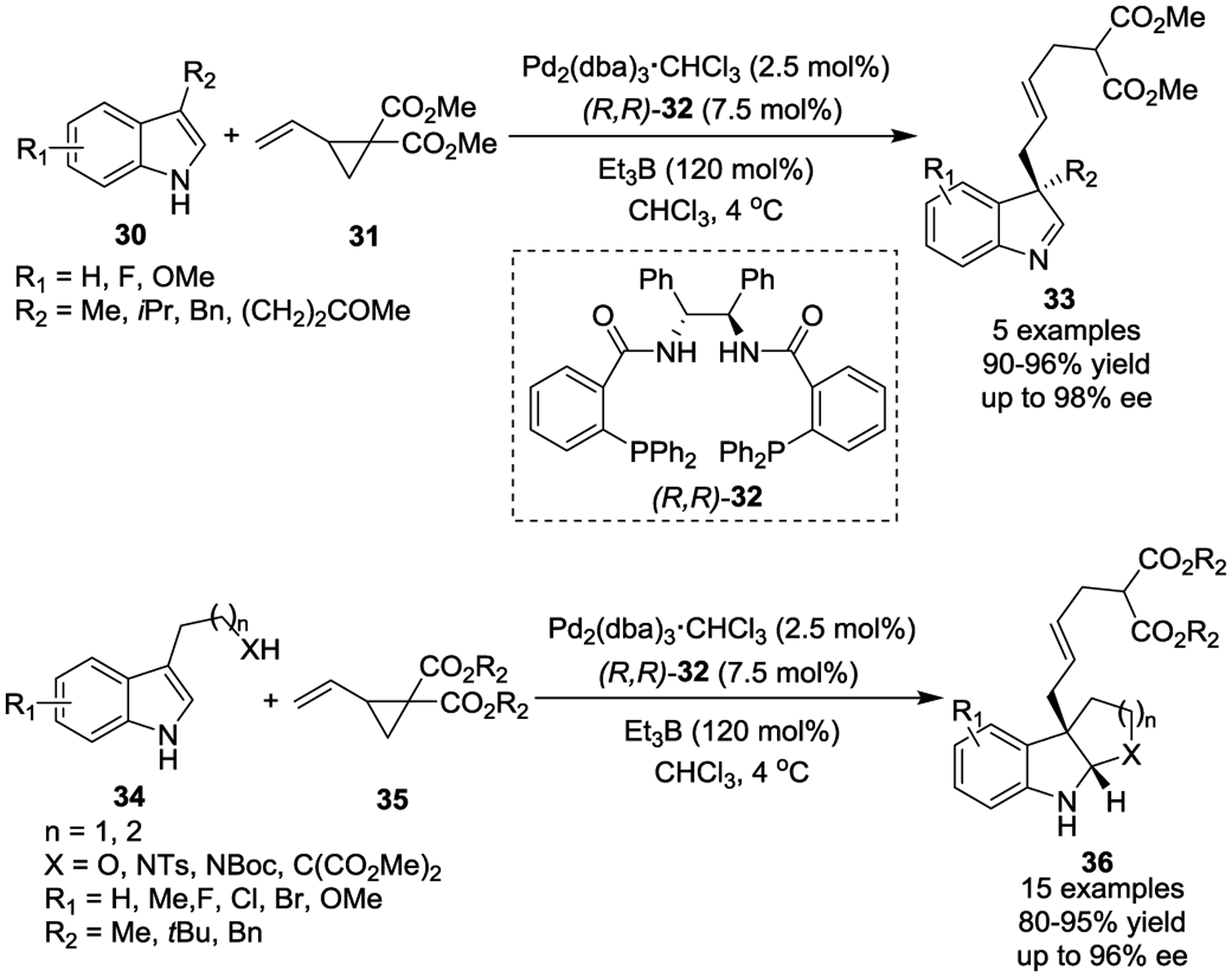
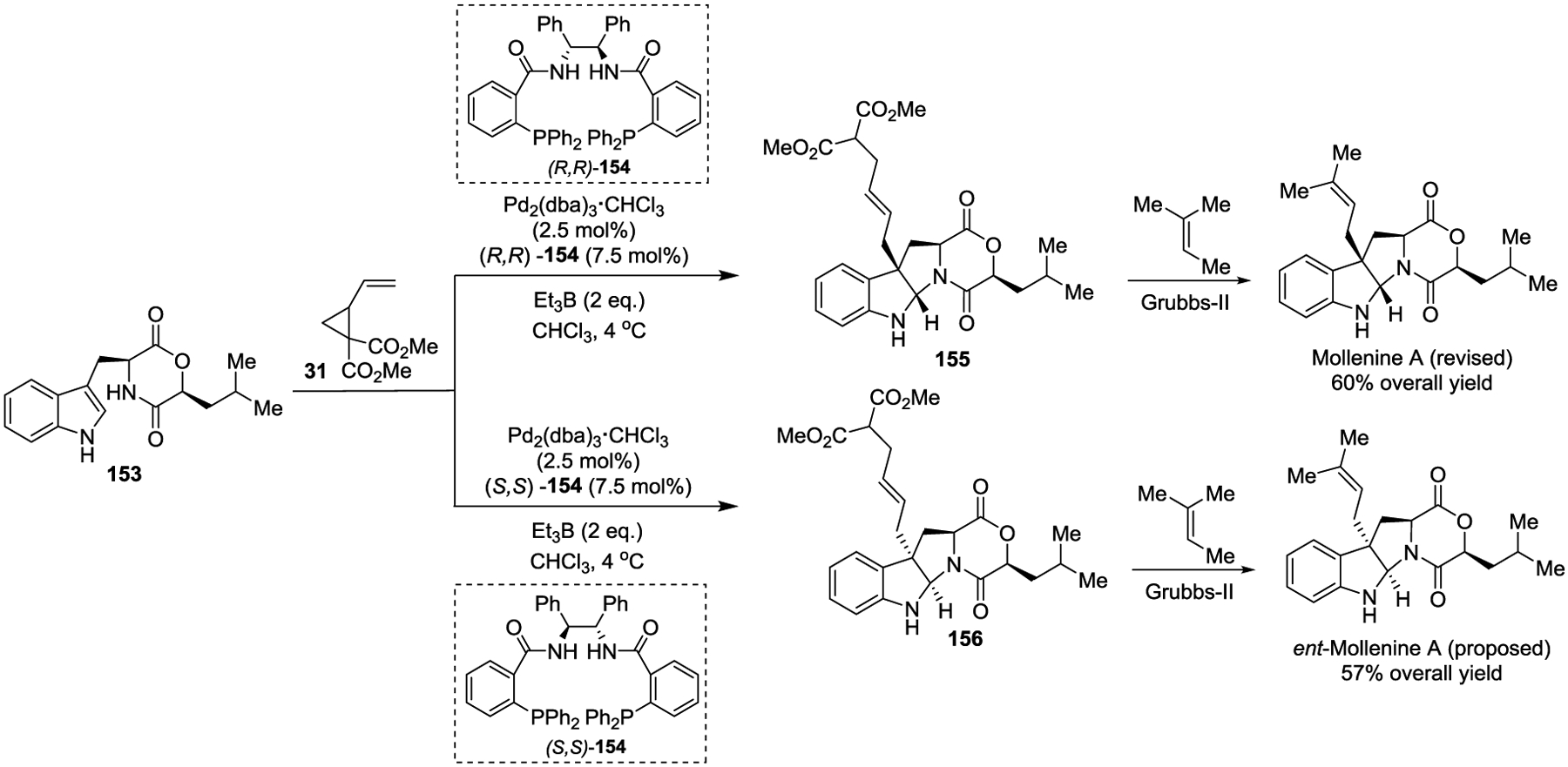
In 2018, the Trost group reported a completely atom-economical preparation of 3-substituted 1H-indoles and tryptophan derivatives via a Pd-catalyzed asymmetric allylic alkylation (Pd-AAA)30 with the stilbene-derived Trost ligand. In this reaction, activated VCPs serve as the electrophiles while 3-alkylated indoles function as the nucleophiles (Scheme 8).31 This transformation provides a variety of indolenine 33 and indoline 36 products in excellent yields with high chemo-, regio-, and enantioselectivities. Et3B is crucial for this reaction and linear selectivity. The diverse functional groups found in the products serve as useful handles that can be conveniently converted into complicated polycyclic skeletons. The significance of this method is further demonstrated by the efficient asymmetric total synthesis of Mollenine A.
Scheme 8.
In 2008, Johnson and coworkers reported that aldehydes were also suitable partners for activated VCPs in Pd(0)-catalyzed [3 + 2] cycloadditions (Scheme 9, a).32 A series of electron-deficient aryl aldehydes 37 were used in this transformation to afford 2,5-cis disubstituted tetrahydrofuran derivatives 38. These products were obtained in high yields and excellent diastereoselectivities, and with Pd catalyst loading as low as 0.5 mol%. Electron-rich aldehydes, such as p-anisaldehyde, could not be tolerated for this reaction. The complete degradation of enantio-enrichment from the enantioenriched VCPs (er = 99 : 1) to the products (er = 52.5 : 47.5) was observed under the standard reaction conditions, which is consistent with the generation of an achiral intermediate in the mechanism.
Scheme 9.
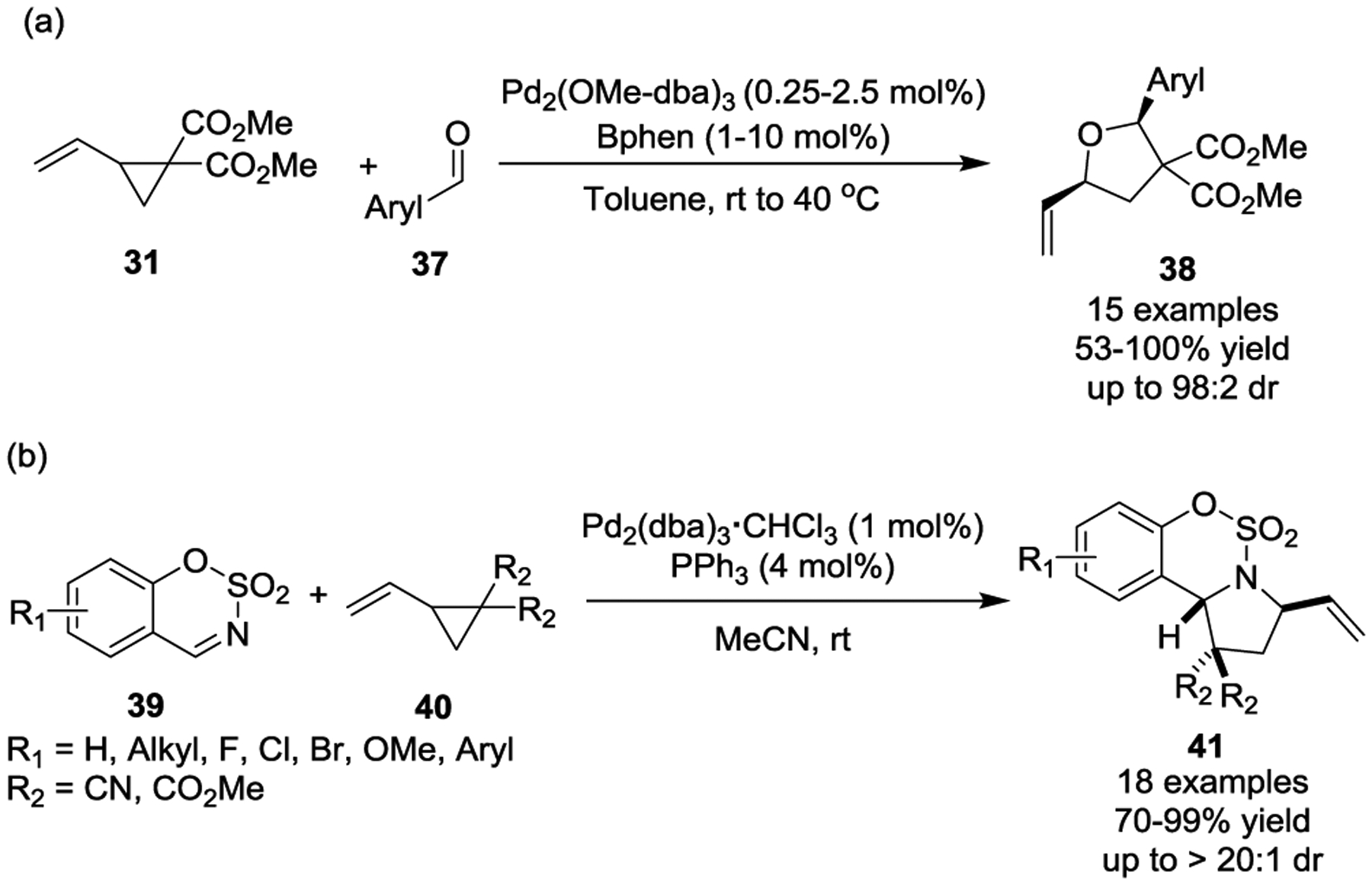
Later studies by the Guo group showed that sulfamate-derived cyclic imines 39 could be reactive partners in Pd(0)-catalyzed diastereoselective [3 + 2] cycloaddition reactions, providing polycyclic systems 41 in good to excellent yields at room temperature (Scheme 9, b).33 The cycloaddition components in this transformation were further extended to include 1-azadienes.34
In 2011, Trost and coworkers disclosed a Pd(0)-catalyzed diastereo- and enantioselective [3 + 2] cycloaddition of activated VCPs and alkylidene azlactones 43 for the synthesis of diversified cyclopentanes 44. This reaction occurred via a dynamic kinetic process and used (S, S)-(−)-1,2-Diaminocyclohexane-N,N’-bis(2’-diphenylphosphinobenzoyl) [(S, S)-DACH-Phenyl Trost Ligand] as the chiral ligand (Scheme 10, a).35 In most cases, excellent diastereo- and enantioselectivity was obtained for alkenyl and aryl substituted azlactones, such as 43. The observed stereoselectivity is rationalized by the “wall and flap” model.36 The scope of electron-deficient olefins for the Pd-catalyzed asymmetric [3 + 2] cycloaddition with activated VCPs was further extended from azlactones 43 to Meldrum’s acid alkylidenes 46 (Scheme 10, b).37 Using these alkylidenes, functionalized cyclopentanes 47 were obtained in good yields with high levels of diastereo- and enantioselectivity when R = Aryl group. Interestingly, the alkynyl-substituted Meldrum’s acid alkylidenes were well tolerated for this reaction in contrast to the alkyl-substituted ones, which were found to be poor substrates.
Scheme 10.
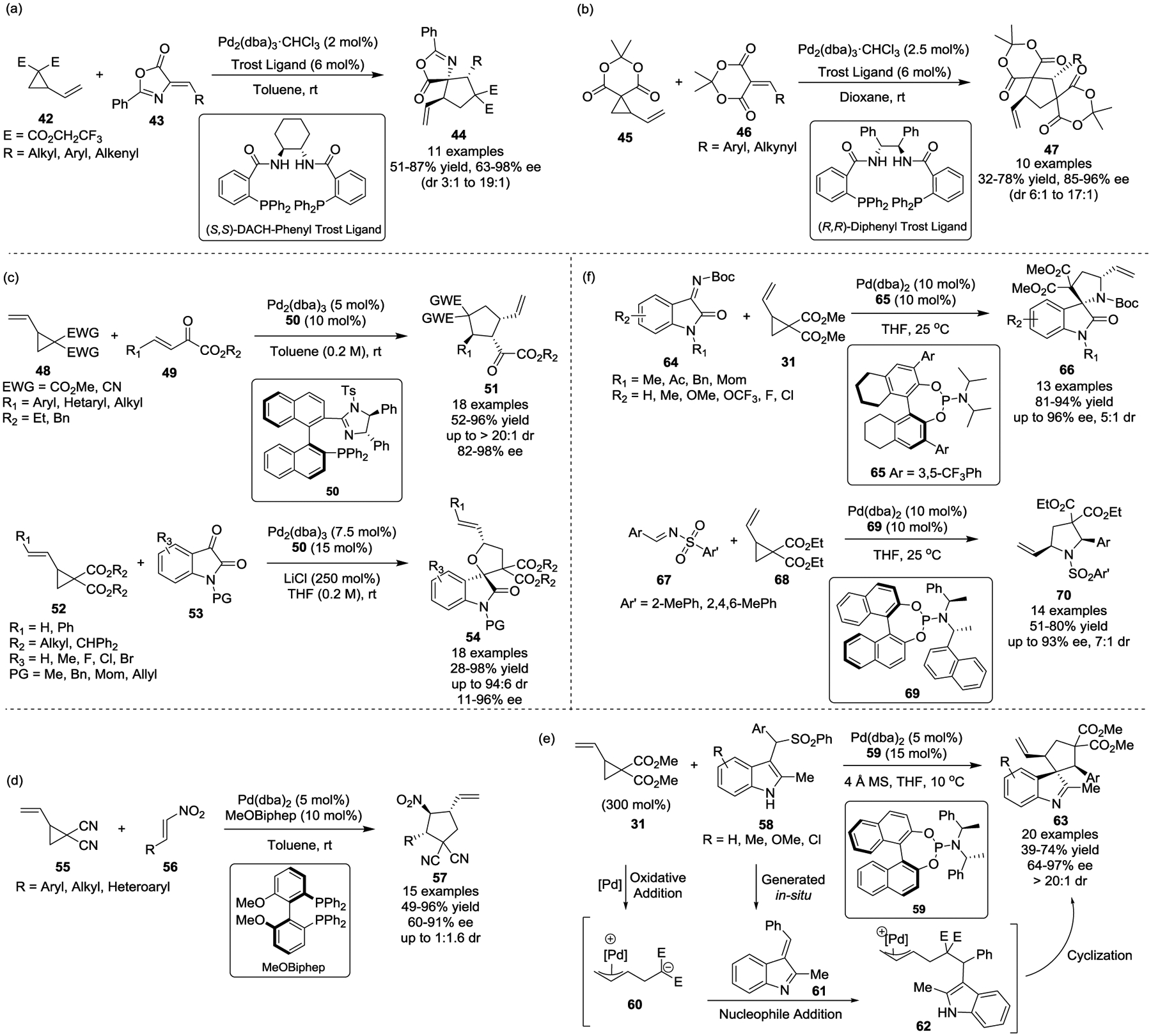
Since then, a wide range of π-systems have been used for diastereoselective and enantioselective [3 + 2] cycloadditions with activated VCPs in the presence of chiral ligands. For instance, Shi and Xu reported that Pd(0)- and chiral imidazoline–phosphine ligands catalyzed an asymmetric formal [3 + 2] cycloaddition of activated VCPs with β,γ-unsaturated α-keto esters 49, affording the corresponding highly functionalized cyclopentanes 51 in good yields along with high diastereo- and enantioselectivities under mild conditions (Scheme 10, c).38 A range of aryl, heteroaryl and alkyl α-keto esters are compatible for this transformation. Later, the substrate scope of this reaction was further extended to isatins 53.39 A variety of diversified oxindole-fused spirotetrahydrofuran frameworks 54 were also obtained in good yields with high diastereo- and enantioselectivities.
In 2014, Liu, He, and Kang developed the Pd-catalyzed asymmetric cycloaddition of dicarbonitrile VCP 55 and nitroolefins 56 for the construction of highly functionalized nitrocyclopentanes 57 (Scheme 10, d).40 Both alkyl and aryl substituted nitro-olefins worked well to give optically enriched nitrocyclopentanes 57 with three consecutive chiral centers in good yields and up to 92% ee. The diastereomeric ratios remained modest; however, the two diastereomers could be readily separated by column chromatography in all cases, allowing for the preparation of both diastereomers in optically enriched form. Later, they applied the same strategy for the enantioselective cycloaddition of activated VCPs and α, β-unsaturated imines 61, which were generated in-situ from aryl sulfonyl indoles 58 (Scheme 10, e).41 Various spiroindolenines 63, which contain an all-carbon quaternary center and two tertiary stereocenters, were obtained in good yields with excellent diastereo- and enantioselectivity.
Mechanistically, in the presence of Pd(0), oxidative addition of activated VCPs occurs to form zwitterionic π-allylpalladium complex 60. A subsequent conjugate addition to the in-situ formed unsaturated imine 61 generates intermediate 62, which undergoes a Pd(0)-catalyzed intramolecular C3-allylation of the indole to provide spiro-indole derivatives 63.
Just recently in 2019, Liu and He described a Pd(0)-catalyzed asymmetric formal [3 + 2] cycloaddition of activated VCPs and isatin-derived ketimines 64 or aldimines 67 with chiral phosphoramidite ligands (Scheme 10, f).42 A wide range of ketimines or aldimines proceeded smoothly to afford the corresponding highly functionalized and optically enriched spiro-oxindole derivatives 66 or pyrrolidines 70 in high yields with good diastereo- and enantioselectivity at room temperature.
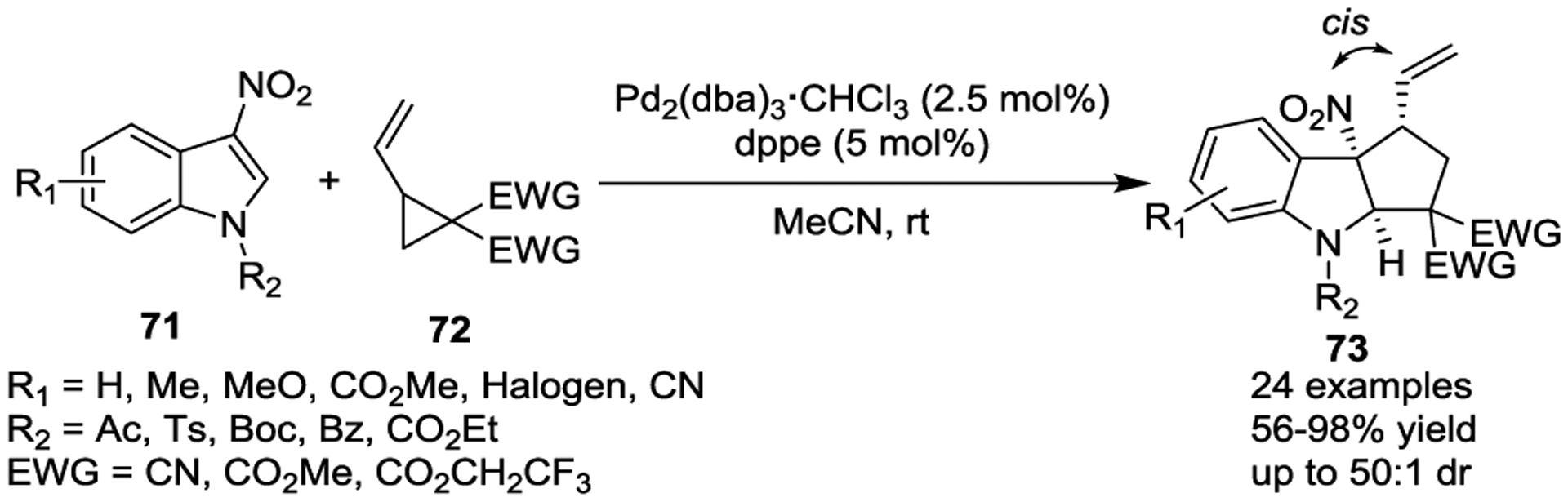
3-Nitroindoles, a particular class of electron-poor indole derivatives, are interesting two-carbon synthons that have been used in a variety of dearomative [3 + 2] cycloaddition processes.43–45 In 2017, Vitale and colleagues reported the diastereoselective synthesis of functionalized nitroindolines 73 through a Pd(0)-catalyzed dearomative [3 + 2] cycloaddition of 3-nitroindoles 71 with VCPs 72 (Scheme 11).43 This study provided a straightforward and highly atom-economical method to construct a wide range of 2,3-fused cyclopentannulated indoline scaffolds 73 in good yields and excellent cis diastereoselectivities.
Scheme 11.
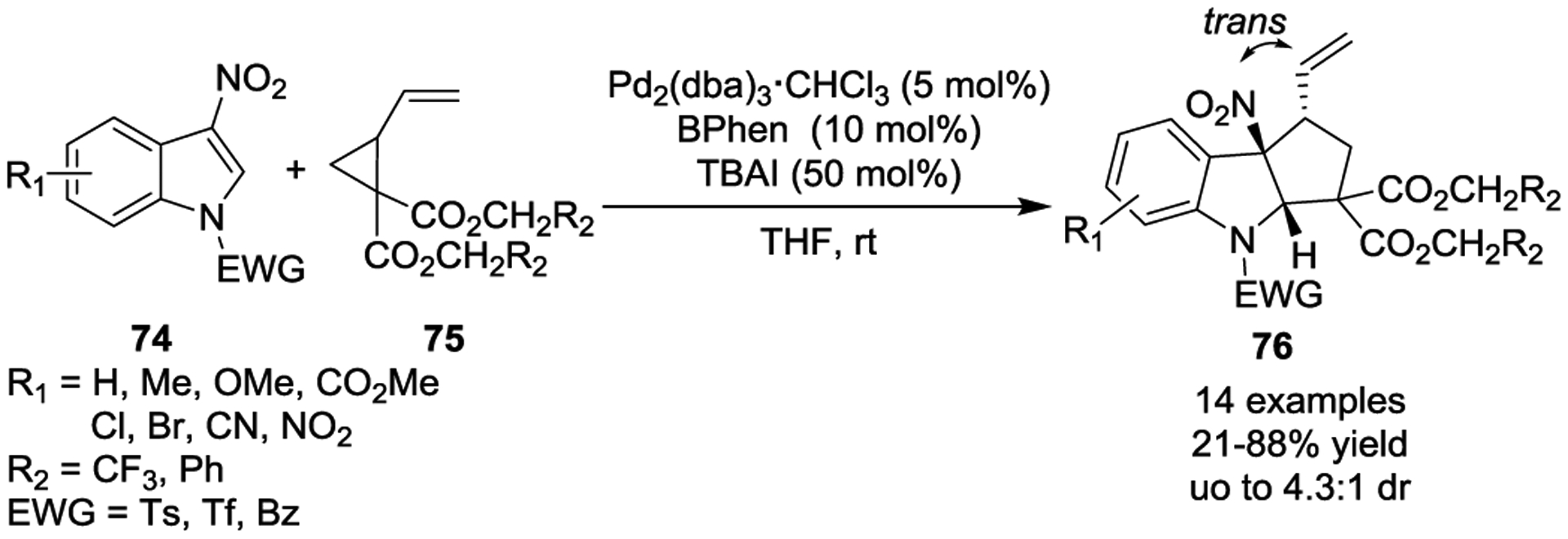
Additionally, the Hyland group developed an asymmetric version of this transformation that used Pd(0) with a chiral BPhen ligand (Scheme 12).44 The reaction provided highly functionalized cyclopenta[b]indolines 76, which contained three contiguous chiral centers, in good yields with satisfactory regio-, chemo-, and enantioselectivity under mild conditions. Interestingly, a trans-selectivity was observed for vinyl and nitro-groups in final product 76, which is complementary to the previous work done by Vitale. The diastereoselectivity of the reaction, which was proposed to be a result of a rapid π-σ-π interconversion between the π-allylic intermediates allowing for Curtin–Hammett control, was governed by the addition of a halide. Later, a related asymmetric study published by the Wang group reported that dearomative [3 + 2] cycloadditions could occur in the presence of a chiral box/Pd(0) complex.45 Besides 3-nitroindoles, in 2018 Vitale and coworkers reported that 2-nitrobenzofurans 77 were also suitable substrates for efficient dearomative [3 + 2] cycloadditions with VCPs (Scheme 13) in the presence of Pd(0) with bisphosphine ligand 1,2- bis(diphenylphosphino)ethane (dppe).46
Scheme 12.
Scheme 13.
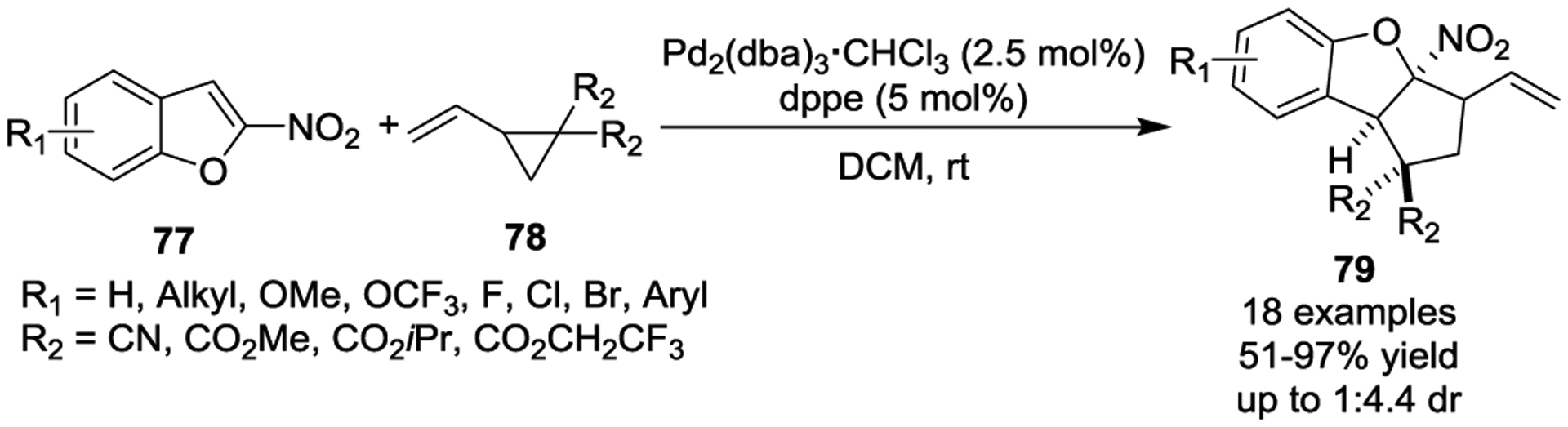
Recently in 2019, Xiao and Lu reported a novel Pd(0)-catalyzed [3 + 2] cycloaddition of VCPs and ketenes 81, which were generated from a Wolff rearrangement of α-diazo ketones 80 via a synergistic strategy that combined visible light photoactivation and Pd catalysis (Scheme 14).47 This sequential transformation worked well to provide a series of highly functionalized tetrahydrofurans 83 that displayed predominantly O-allylic alkylation (58–99% yields) in the presence of a hybrid P,S-ligand at room temperature. Additionally, the same group performed an asymmetric photoinduced Wolff rearrangement/Pd-catalyzed [3 + 2] cycloaddition that afforded the chiral trahydrofuran 87 in 91% yield and 75:25 er with chiral ligand 86.
Scheme 14.
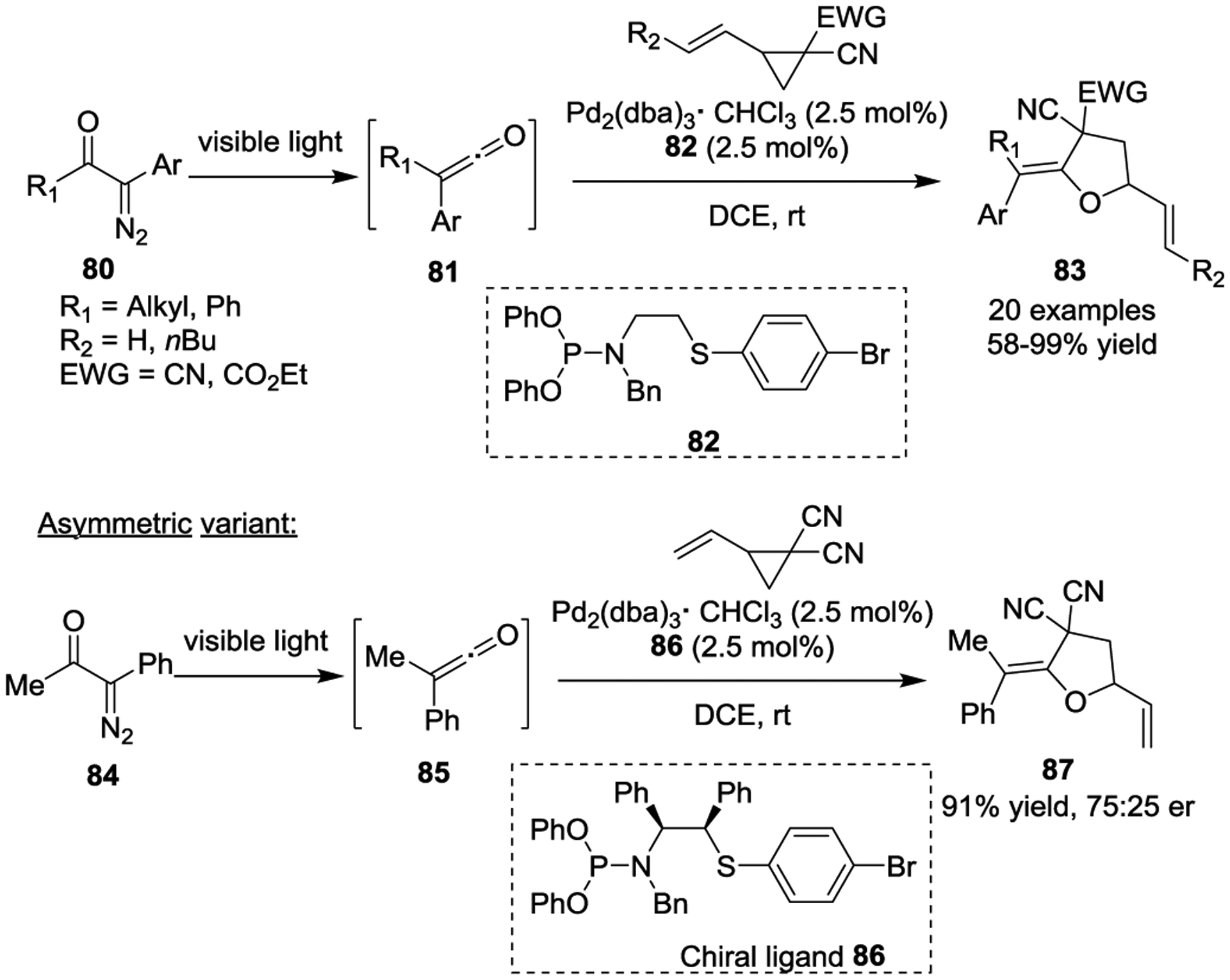
In 2019, the You group disclosed a Pd(0)-catalyzed asymmetric dearomative [4 + 3] cycloaddition of anthranils 88 with activated VCPs 89 in the presence of a chiral Ph-Phox ligand (Scheme 15).48 The resulting bridged cyclic products 90 were obtained in moderate yields with excellent stereo- and enantioselectivities by using a catalytic amount of Et3B as a key activator. Preliminary NMR studies confirmed the formation of a borane–anthranil complex, which supports the thought that borane is a crucial component needed for reactivity.
Scheme 15.
2.2.2. Nickel Catalysis
In 2006, the Johnson group reported Ni(0)-catalyzed rearrangements of activated VCPs 91 to access dihydrofuranproducts 92 at room temperature (Scheme 16).49 Notably, treatment of enantioenriched VCP 93 with Ni(cod)2 and Ph3P afforded dihydrofuran 94 as a single diastereomer in 94% yield with the complete preservation of enantioselectivity (88% ee). In terms of the substitution pattern, the methyl and vinyl substituents of the five-membered ring were displayed in a trans relationship to one another.
Scheme 16.
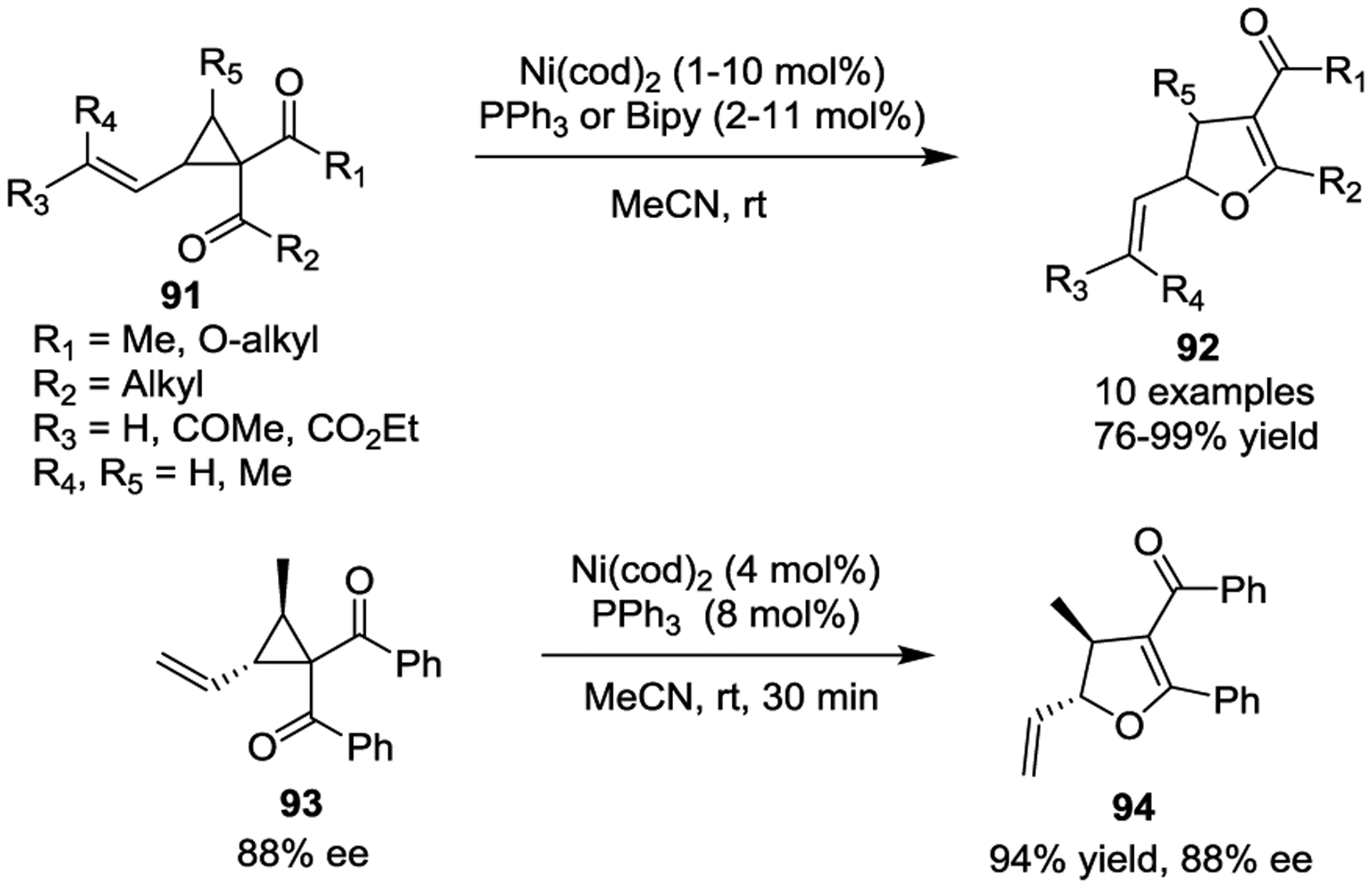
In 2008, Yorimitsu and Oshima reported a Ni(0)-catalyzed synthesis of allylic boronates 99 by a borylative ring opening reaction of activated VCPs 95 with bis(pinacolato)-diboron (Scheme 17).50 A variety of allylic boronates were obtained in good yields and a high degree of E-stereoselectivity. The tandem reaction involves an oxidative addition of VCPs to form π-allyl(oxa-π- allyl)nickel species 96, transmetalation, and subsequent reductive elimination to yield boron enolates 98, which were transferred to allylic boronates 99 via protonolysis. The allylic boronate product 100 reacted smoothly with benzaldehyde to afford the corresponding homoallyl alcohol 101 in 91% yield with high anti selectivity. The authors also outlined a one-pot procedure involving sequential borylative ring-opening/allylation reactions.
Scheme 17.
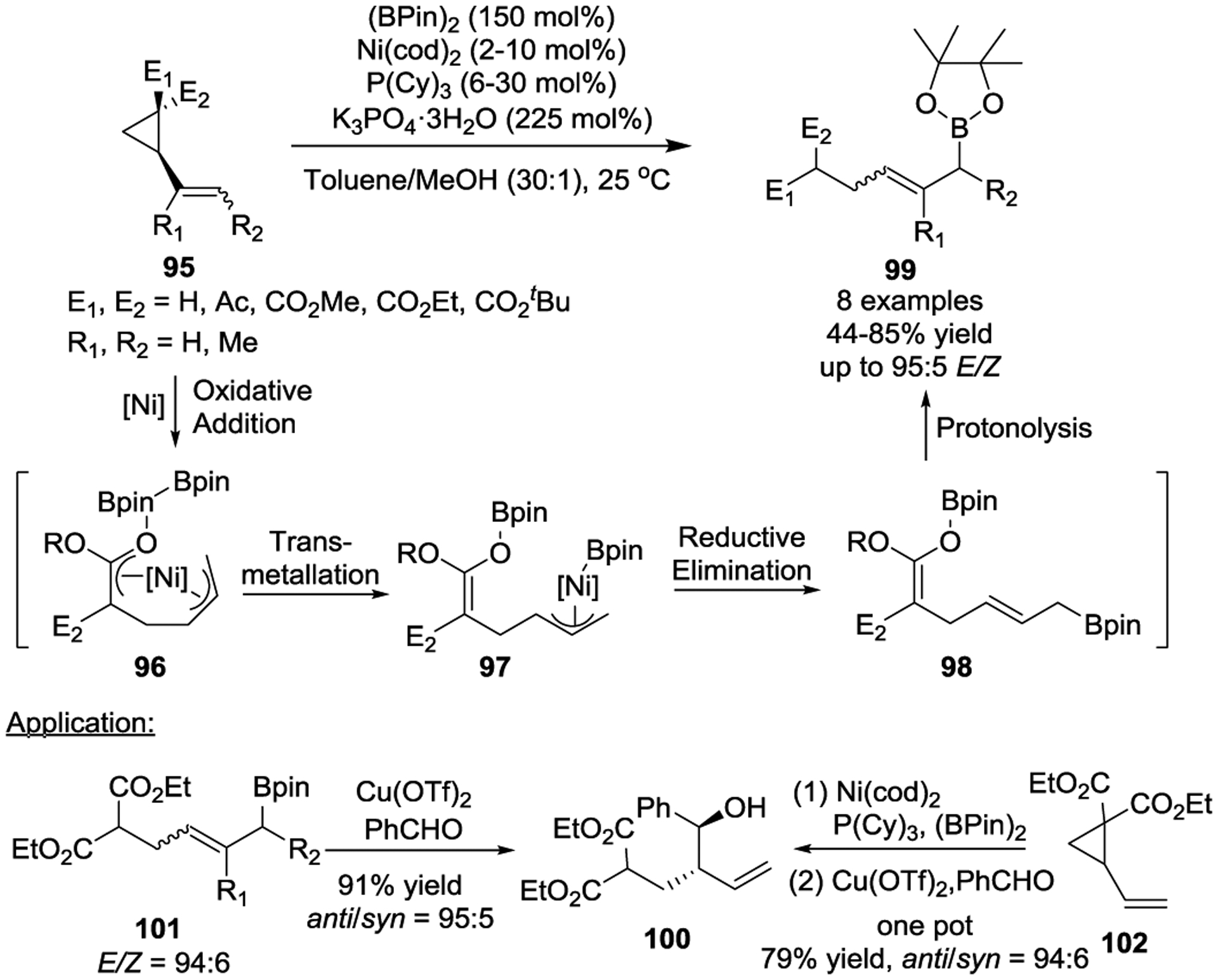
Later in 2011, Kimura and colleagues realized a Ni(II)-catalyzed three-component reductive coupling reaction of VCP, alkynes, and dimethylzinc that provided skipped dienes in high yields with excellent E-stereoselectivity at room temperature (Scheme 18).51 Internal alkynes that participated in the coupling reaction giving rise to dimethyl (α-heptadienyl)malonates 106, while terminal alkynes such as phenylacetylene and trimethylsilylacetylene afforded the dienynes 109a and 109b, which involved the dimerization of the terminal alkynes 107. Allylnickel species 104 and 108 were proposed as intermediates for this transformation, which is distinct from the metal π-allyl intermediate 14 outlined in Scheme 5, b. In the case of the terminal alkyne, instead of insertion of the internal alkyne to the allylic terminus of the allylnickel complexes 104, the formed zinc acetylides preferentially engage with allylnickel species 108 via transmetalation to give rise to diene-ynes 109 as the major products.
Scheme 18.
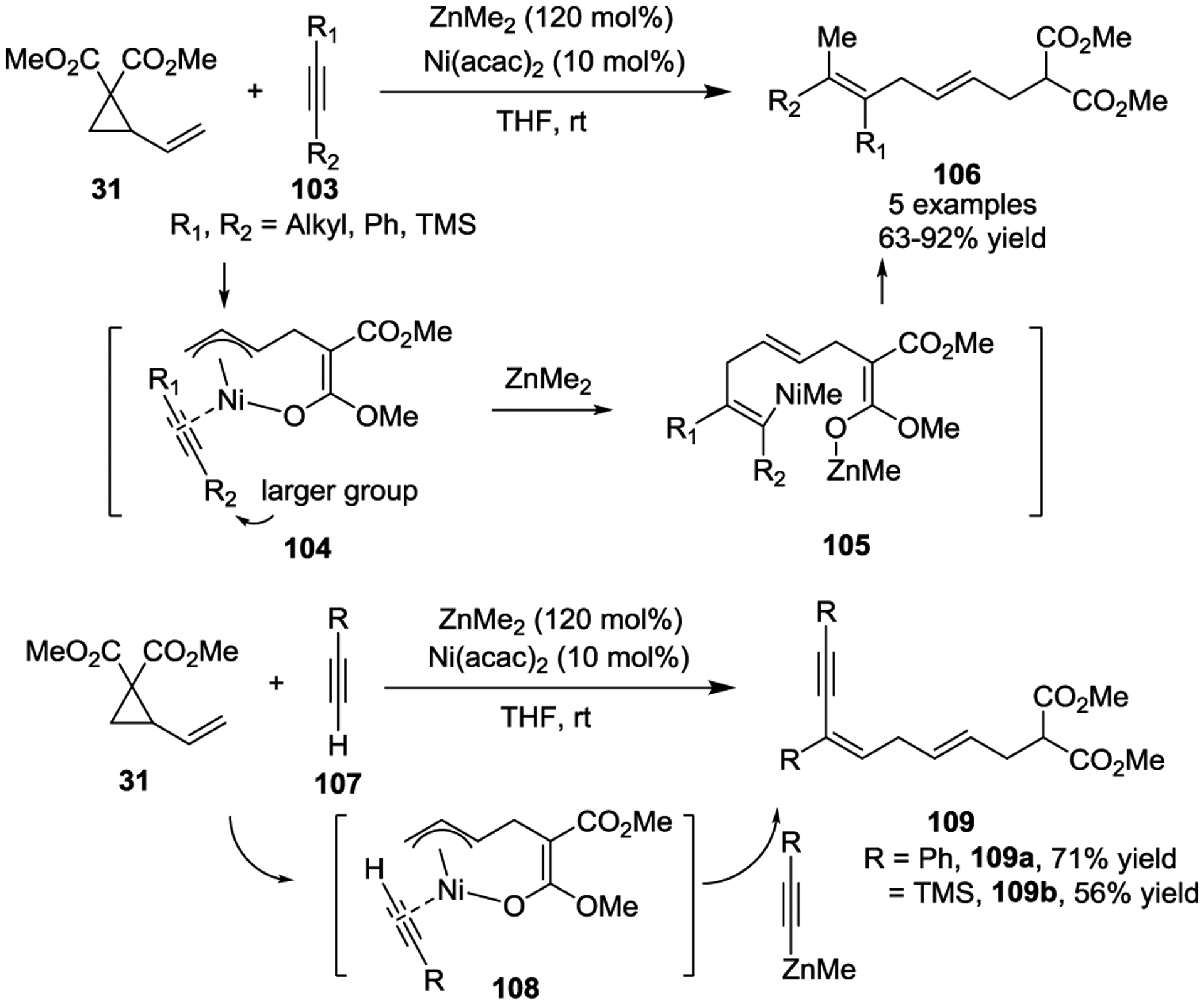
In 2013, the Ni(0)-catalyzed intermolecular [3 + 2] cycloaddition of VCPs with imines for the preparation of pyrrolidine derivatives was developed by Matsubara and coworkers (Scheme 19).52 A variety of imines 111 were used in this transformation to generate functionalized pyrrolidines 113 in high yields, with good regio- and diastereoselectivities under mild reaction conditions. Preliminary asymmetric studies of the reaction afforded the single diastereomer 115 in 83% yield with 56% ee by using (R,R)-iPr-Duphos as the chiral phosphine ligand. The authors suggested that this process involved a nickelacyclic complex 112 as a key intermediate, which is similar to Kimura’s earlier study.51
Scheme 19.
2.2.3. Iron Catalysis
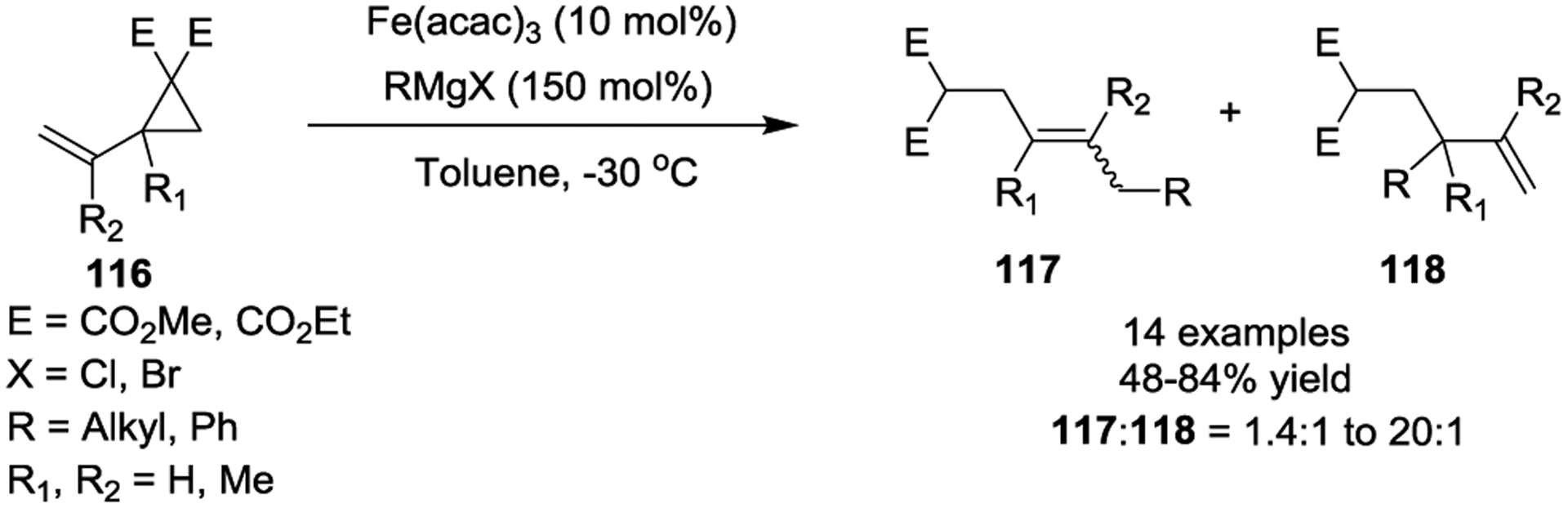
Fe-catalyzed reactions have received considerable attention in organic synthesis due to the associated economic and environmental benefits.53 In 2009, the Fürstner group reported a highly regioselective Fe(acac)3-catalyzed conjugate addition of Grignard reagents to activated VCPs 116. They obtained the resulting 1,7-addition products 117 in good yields as predominately the E-isomers (Scheme 20). This instance was the first example of the catalytic addition of hard organometallic nucleophiles to VCPs by using an inexpensive and environmentally benign precatalyst.54
Scheme 20.
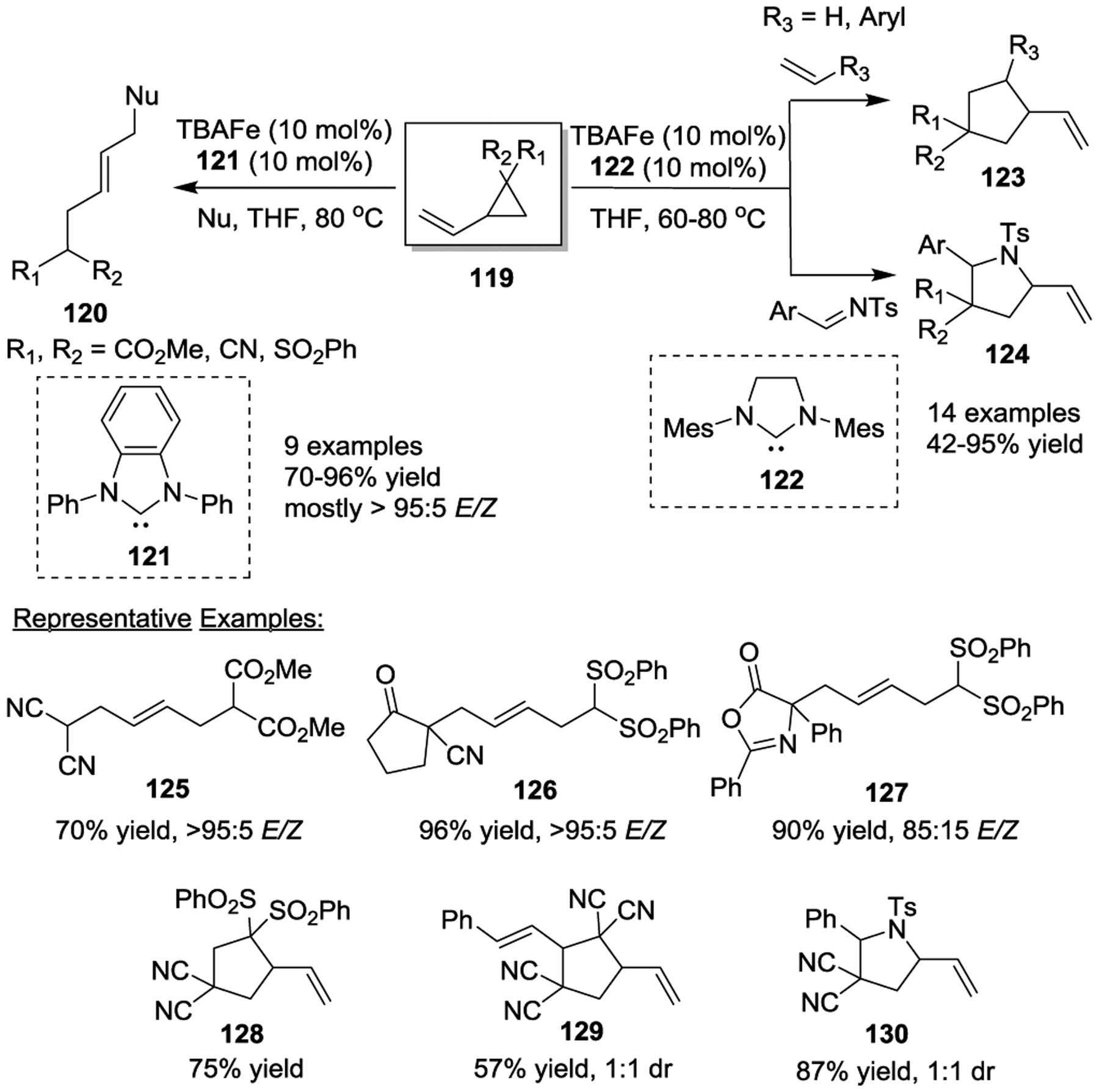
In 2012, Plietker and colleagues further explored the Fe-catalyzed allylic substitution within the context of a formal [3 + 2] cycloaddition of VCPs with electron-deficient olefins and imines. This reaction affords either functionalized acyclic products 120 or densely substituted cyclopentanes 123 and pyrrolidines 124 in high yields and with good regioselectivities (Scheme 21).55 The low-valency iron complex Bu4N[Fe-(CO)3(NO)] (TBAFe) was the superior catalyst, while the versatile reactivities were governed by different NHC-ligands. For example, a series of pronucleophiles such as esters, dinitriles, cyanocyclopentanones, or phenylazlactones were successfully employed in this transformation with NHC-ligand 121. In general, E-configured double bonds were observed with high selectivity, favoring the linear substitution products 120. In addition, functional groups, such as esters, nitriles, ketones, sulfones and amides, are well tolerated in the [3 + 2] cycloaddition process in the presence of NHC-ligand 122.
Scheme 21.
2.2.4. Iridium Catalysis
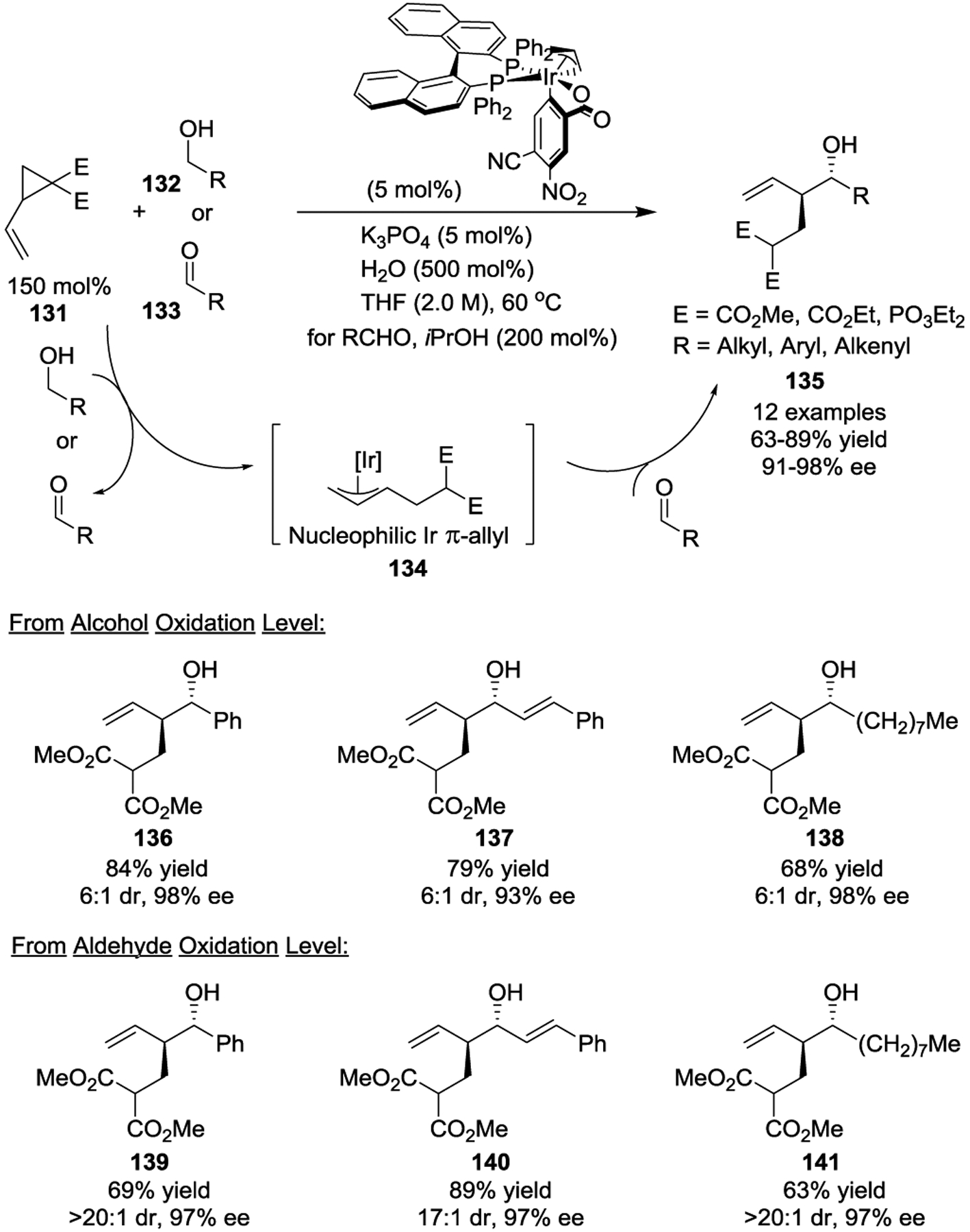
In 2011, Johnson, Krische, and coworkers reported polarity inversion of activated VCPs in the presence of a cyclometalated Ir catalyst (Scheme 22).56 In these cases, the generation of nucleophilic Ir π-allyls 134, which are distinct from the usual generation of electrophilic π-allyl intermediates of activated VCPs, demonstrated the unique reactivity of the π-allyliridium catalyst.57 The coupling of in-situ generation carbonyl electrophiles with Ir π-allyls provided homoallylic alcohols 135 in good yields with high diastereo- and enantioselectivities. Identical products were obtained upon isopropanol-mediated transfer hydrogenation in the presence of aldehydes 133. The authors further demonstrated the potential utility of this transformation by readily converting the reaction products to enantioenriched cis-4,5-disubstituted δ-lactones.
Scheme 22.
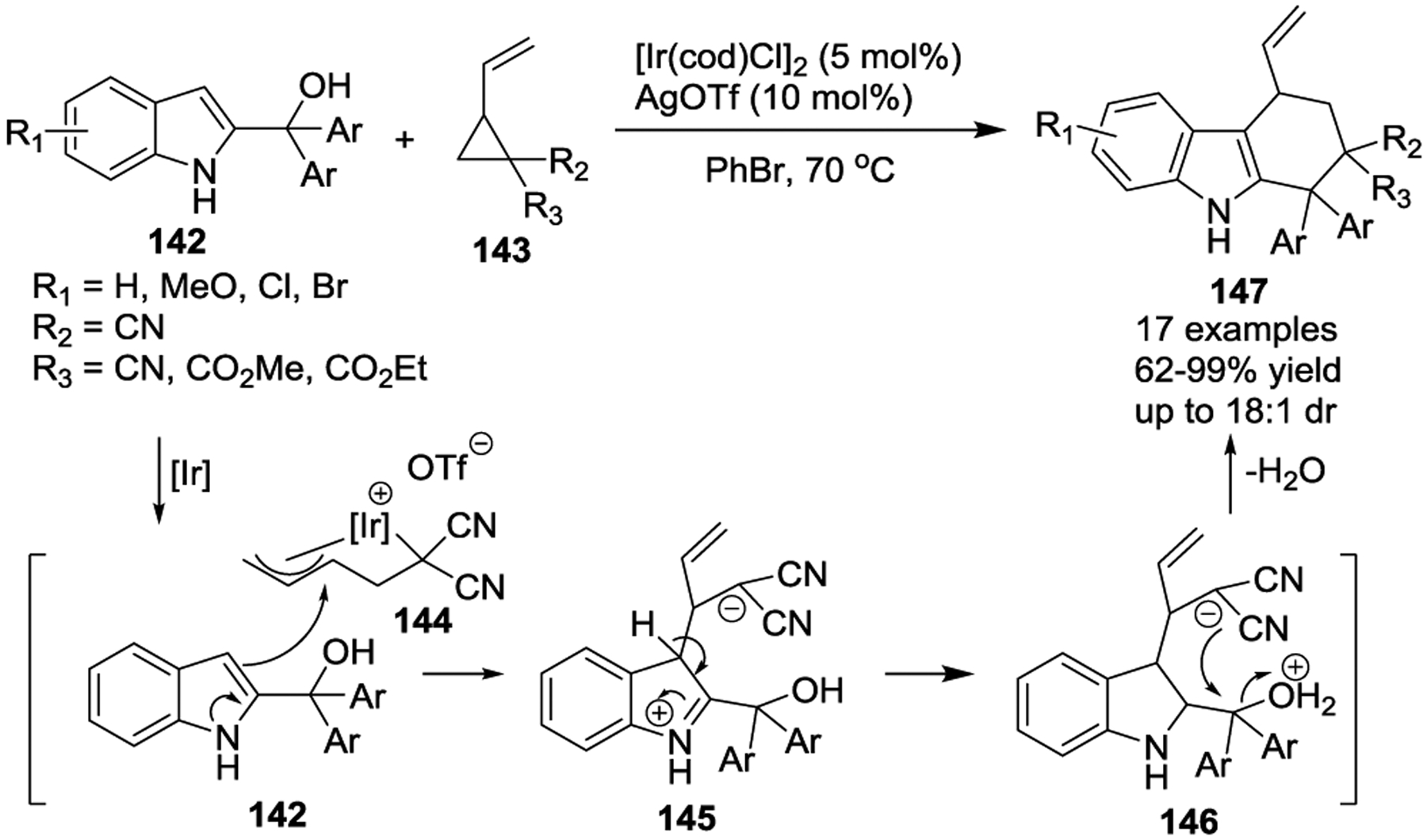
In 2018, the Shi group described the first Ir(I)-catalyzed [3 + 3] cyclization of 2-indolylmethanols and VCPs with AgOTf additive (Scheme 23).58 This reaction generated tetrahydrocarbazole frameworks 147 bearing two vicinal quaternary stereocenters in high yields with excellent regioselectivity. In the presence of a cationic iridium catalyst, VCPs 143 could transform into (π-allyl)-Ir intermediates 144 via oxidative addition, followed by the nucleophilic addition of 2-indolylmethanols 142 to generate intermediates 145. These intermediates could rapidly undergo proton transfer. Finally, 146 undergoes an intramolecular nucleophilic substitution via dehydration to provide final products 147. Notably, the nucleophilicity of the C3-position of indoles 142 induced the observed reversed regioselectivity compared to other 2-indolylmethanol-involved transformations.59
Scheme 23.
2.2.5. Rhodium Catalysis
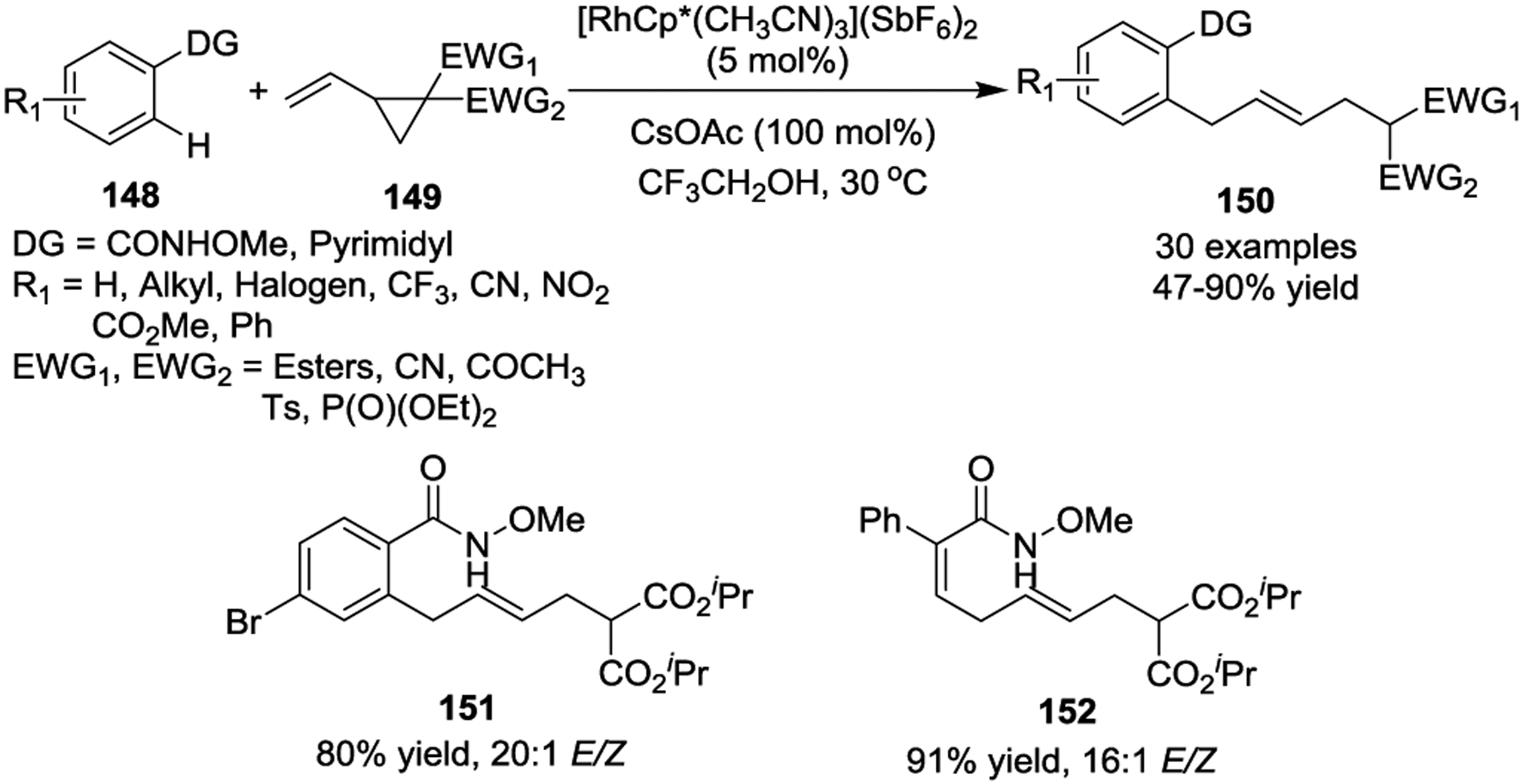
In 2015, Wang and coworkers reported the synthesis of allylated arenes and skipped dienes 150 through a Rh(III)-catalyzed sequential C–H activation and C–C activation reaction with activated VCPs 149 as the coupling partners (Scheme 24).60 N-methoxybenzamides and pyrimidyl groups were chosen as suitable directing groups. The reaction boasted an excellent substrate scope and good stereoselectivity. Computational studies suggested that a formal β-carbon elimination was involved in the reaction mechanism, which represents a novel ring opening method for activated VCPs. This reaction was also applicable to an alkenyl substrate, affording skipped diene 152, which contained valuable handles for further transformations. Later, a similar transformation was also realized by the Glorius group.61
Scheme 24.
2.3. Applications
Although transition-metal-catalyzed [3 + 2] cycloadditions of activated VCPs with π-systems produce diversified scaffolds, only one report demonstrated their application in the total synthesis of biologically active natural products. In 2018, the Trost group validated the synthetic utility of their newly established Pd-catalyzed [3 + 2] cycloaddition method to quickly access and verify the absolute stereochemistry of Mollenine A (Scheme 25).31 The cycloaddition between indole derivative 153 and VCP 31 gave the tetracyclic pyrroloindolines 155 and 156 in high yields and >10:1 dr, respectively, by using chiral ligand [(R, R)]-154 and [(S, S)]-154. The three-pot synthesis provided Mollenine A in an impressive 60% overall yield, demonstrating the utility of this method to build diketopiperazine- or diketomorpholine-containing natural products.
Scheme 25.
It is well-known that Lewis acids can catalyze cycloadditions of donor-acceptor cyclopropanes21 with aldehydes or imines. These cycloadditions are highly reliable transformations as they have been used as one of the key steps in total syntheses of various natural products, such as (+)-Nakadomarin A,62 (−)-Allosecurinine,63 FR901483,64 (+)-Isatisine A.65 However, these cycloadditions do not involve VCPs so they are not covered in this review.
3. C-C BOND CLEAVAGE OF NON-ACTIVATED VCPs
Due to the absence of strong EWGs, non-activated VCPs generally do not form 1,3-dipoles in the presence of metal catalysts. Instead, these VCPs typically undergo oxidative addition to generate metallacycle I or II, which may initiate a subsequent cycloaddition process (Scheme 26).66 These different reactivities, such as the ability to use these non-activated VCPs as 5-carbon synthons, provide alternate pathways to synthesize carbocycles or heterocycles not accessible from activated VCPs.
Scheme 26.
3.1. Metal-Catalyzed Intramolecular Processes
3.1.1. Rhodium Catalysis
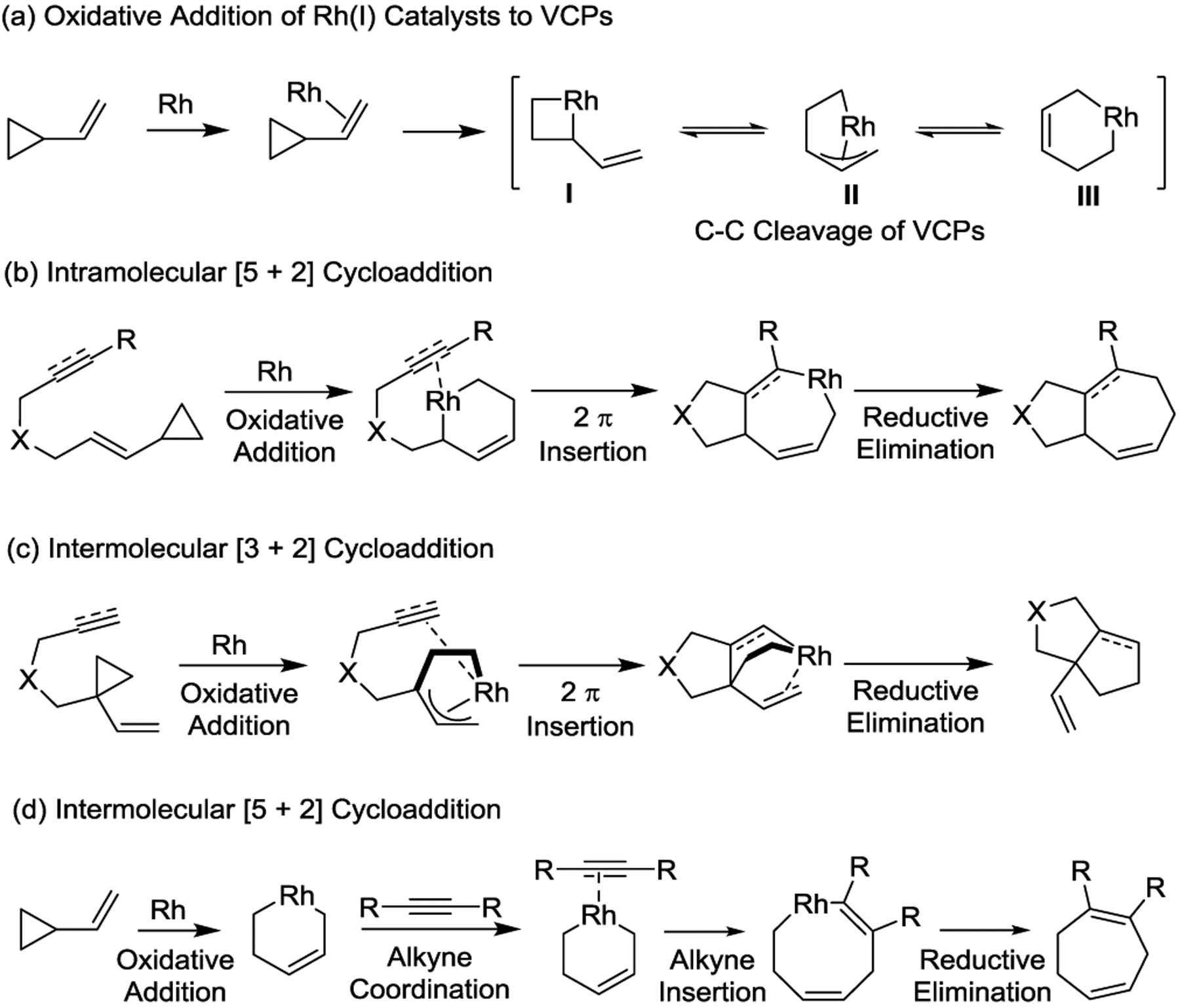
In general, both the Rh(I)-catalyzed intra- and intermolecular cycloadditions follow a similar mechanism, as shown in Scheme 27.66–70 These processes are generally initiated by the π-coordination of Rh(I) to the VCPs, which triggers C–C bond cleavage of the cyclopropanes to provide π-allyl rhodacycles I, II, or III (Scheme 27, a). Subsequent migratory insertion affords metallacyclooctene intermediates, which can undergo reductive elimination to yield a wide range of cycloadducts. Furthermore, in the resulting cycloadditions, either all five-carbon units of the VCP or the three-carbon unit of the cyclopropane can be transferred to the ring products via intra- or intermolecular transformations. This affords the corresponding seven-membered or five-membered ring systems, respectively (Scheme 27, b, c, d). Notably, the position of vinyl moiety in the VCPs could dramatically alter the cycloaddition pathways. For example, the geometry of the 1-ene-VCP substrate forces the VCP moiety to act as a three-carbon unit to favor a [3 + 2] formal cycloaddition since the tethered unit must approach the VCP on the opposite face of the vinyl group, which makes the competitive [5 + 2] cycloaddition impossible (Scheme 27, c).71–72 Rate limiting steps may vary depending on the 2π-unit used in the cycloaddition processes.
Scheme 27.
In 1995, the Wender group reported its seminal work with Rh(I)-catalyzed intramolecular [5 + 2] cycloadditions of VCPs with alkynes. This provided a conceptually new method for the synthesis of a variety of fused 5,7-bicyclic cycloheptadienes 158 (Scheme 28).73 [Rh(PPh3)3Cl] (Wilkinson’s catalyst) and silver triflate were used to initiate this reaction, and the authors observed that substitution pattern on the alkyne and alkene moieties had little effect on the efficiency of the cycloaddition. This transformation represented the first example of non-activated VCPs serving as five-carbon synthons for [5 + 2] cycloaddition reactions, which inspired further developments in transition-metal catalysis for VCP cycloadditions.74–75 Shortly after this study, the Wender group quickly expanded the substrate scope to include tethered alkenes, such as 159, under the same reaction condition (Scheme 29).76–77
Scheme 28.
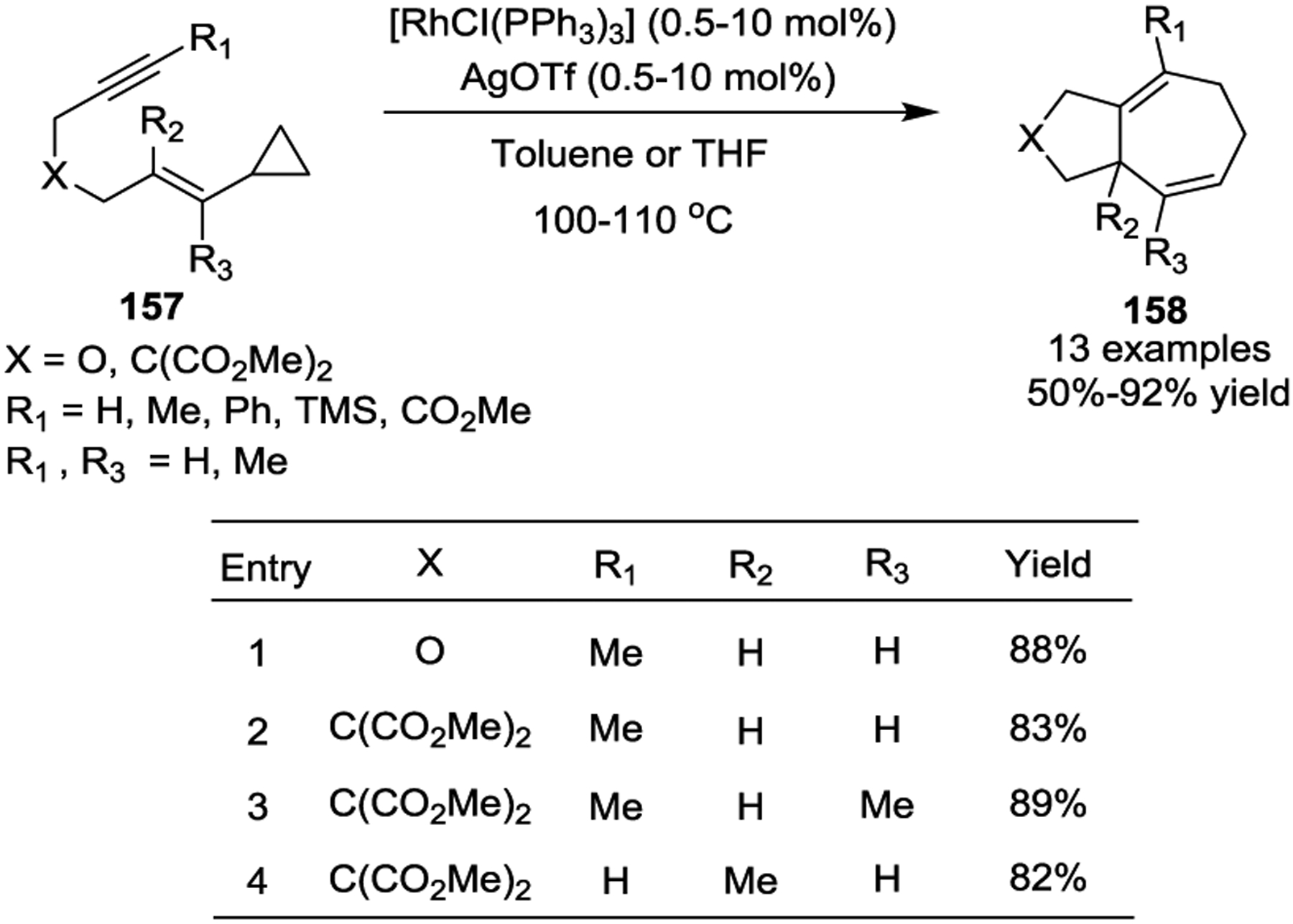
Scheme 29.
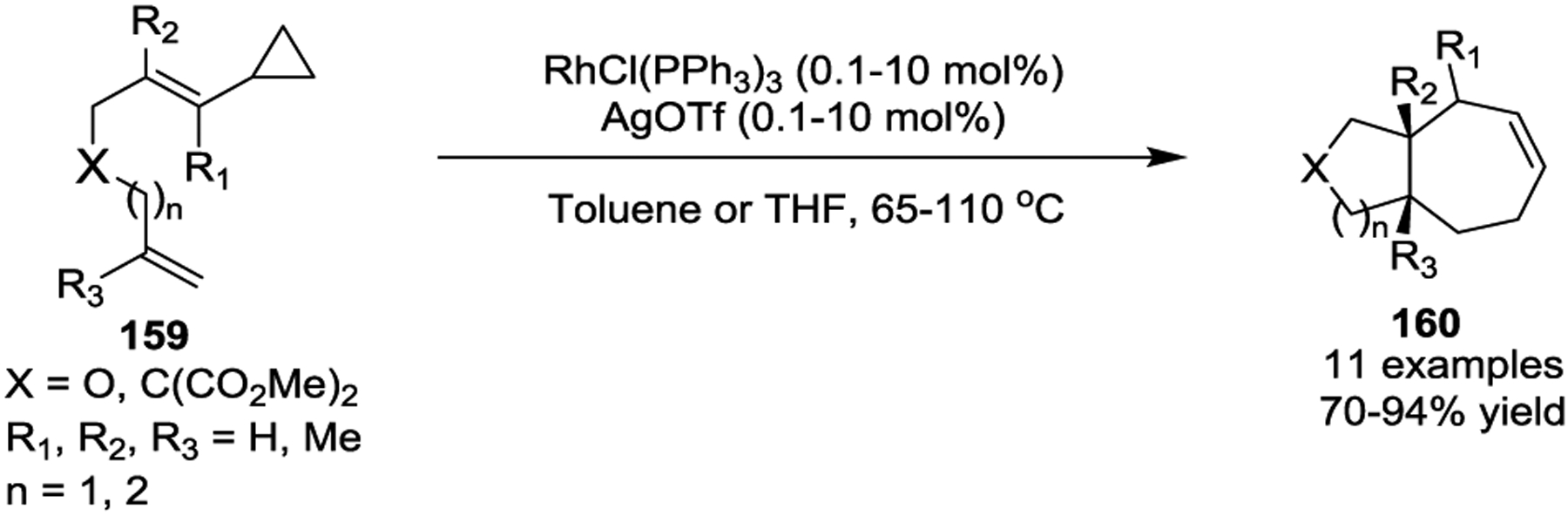
Since then, Wender and coworkers have extensively studied intramolecular Rh(I)-catalyzed [5 + 2] cycloadditions of VCPs with various π-systems, such as alkynes, alkenes, and allenes. For instance, they investigated the regio- and stereoselectivity of the reaction by using cis and trans VCPs 161 (Scheme 30, a).78 In all cases, both cis and trans substrates 161 afforded cycloadducts 162 or 163 as a single regio- and diastereoisomer in high yields. In addition to 1,2-disubstituted cyclopropanes, 1,1-disubstituted VCPs 164 were also well tolerated under these conditions (Scheme 30, b). Notably, alkoxy substitution at the 1-position of VCPs has been shown to significantly accelerate their rearrangements in both thermal and metal-mediated reactions. Later, the authors demonstrated that excellent control over the regioselectivity of the [5 + 2] cycloaddition can be achieved through the selection of appropriate cyclopropyl substituents and/or catalyst modifications.79
Scheme 30.
The previous [5 + 2] cycloaddition studies were completed using Wilkinson’s catalyst [(PPh3)3RhCl] with silver activation. A new and selective catalyst, [Rh(CO)2Cl]2, then proved to be more effective than Wilkinson’s catalyst and generally allowed for the reaction to proceed in better yields under mild conditions (Scheme 30, c).80 The author’s attributed the improved reactivity of the [Rh(CO)2Cl]2 catalyst to the lower ligand count and reduced steric hindrance compared to Wilkinson’s catalyst, both of which facilitate coordination and subsequent cleavage of the cyclopropane ring.
The use of allenes as 2π-units in Rh(I)-catalyzed [5 + 2] cycloadditions with VCPs was also explored (Scheme 30, d).81 This reaction worked with mono-, di-, and trisubstituted allenes and provided the bicyclic products 169 in good to excellent yields, even when quaternary centers were formed (R3 = Me in 169). In all case, the cycloaddition preferentially occurred at the internal alkene of the allenyl system. Chirality in the cyclopropane substrates could be completely conserved throughout the course of the cycloaddition process.
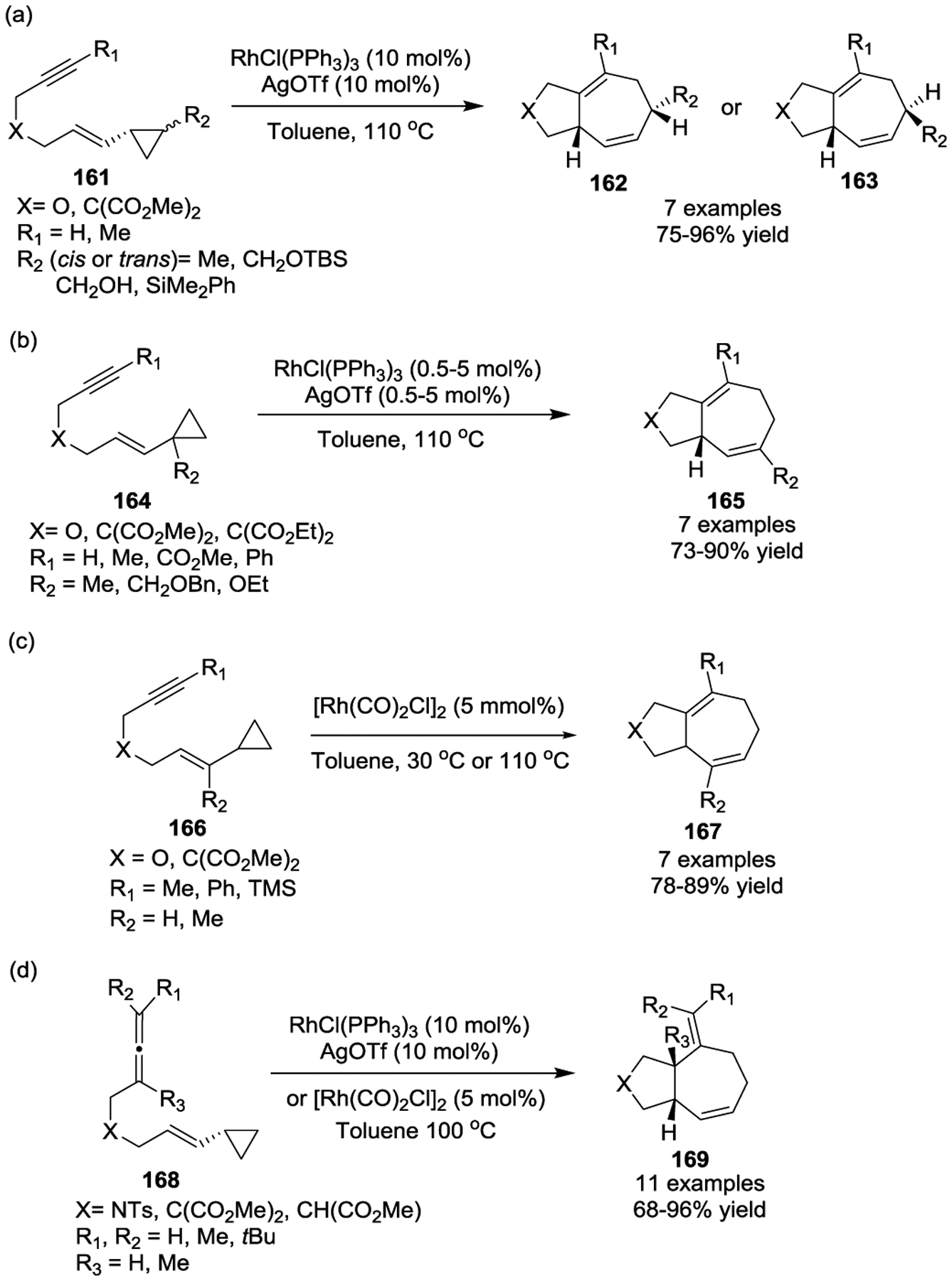
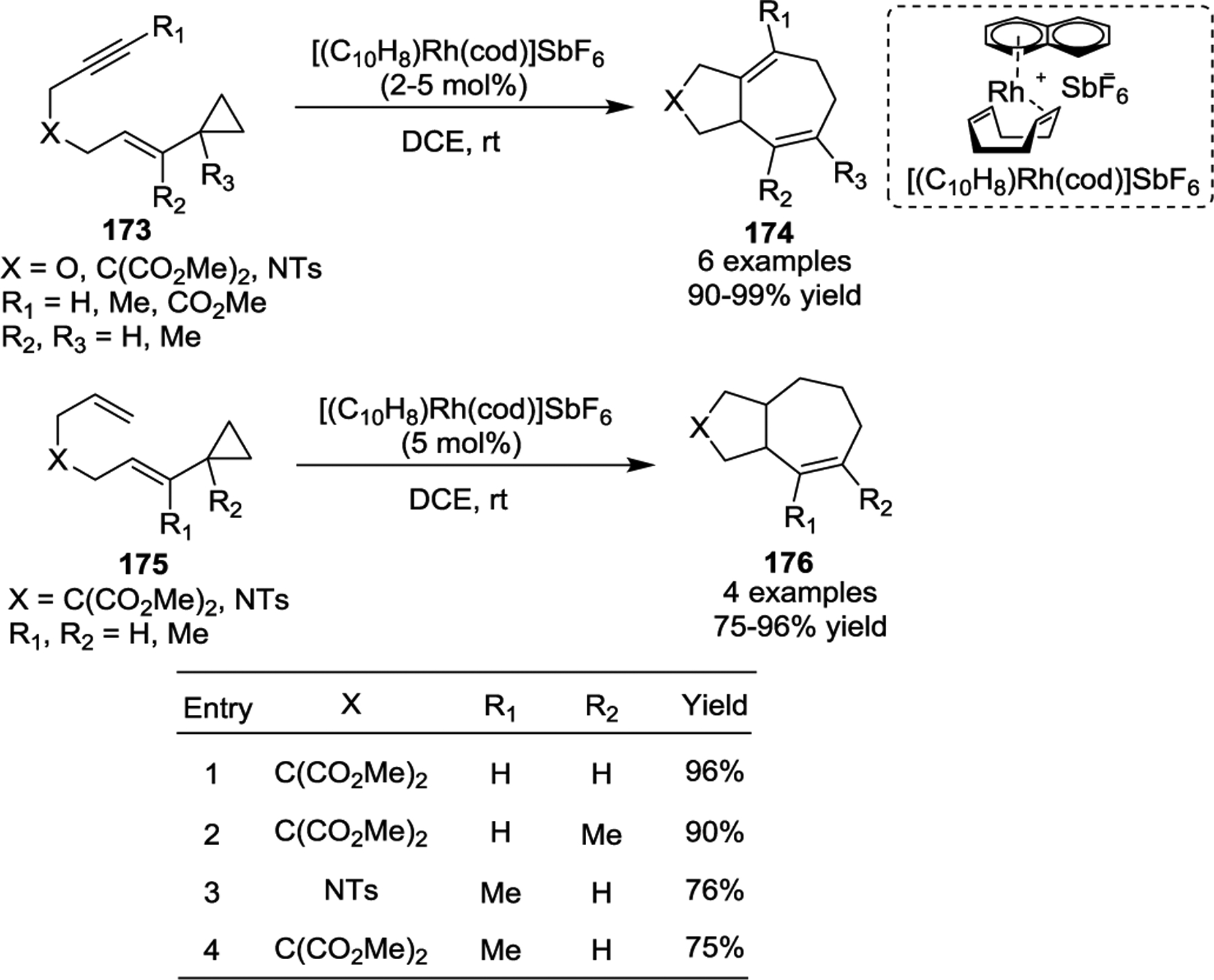
The success of rhodium catalysis in [5 + 2] cycloadditions of VCPs inspired other chemists, and soon other groups discovered a wide variety of rhodium reagents capable of catalyzing this transformation under even milder conditions. Based on bisphosphine ligands, the rhodium catalyst [Rh(dppb)Cl]2 (dppb: 1,4-Bis(diphenylphosphino)butane) was demonstrated to be an efficient catalyst [5 + 2] cycloadditions with tethered substrates 170, resulting in the production of 172 in good yields at room temperature (Scheme 31).82 Later, the Chung group demonstrated a Rh-NHC catalyst was exceptionally effective for the same cycloaddition.83 The Wender group then reported a naphthalene complex of rhodium ([(C10H8)Rh(cod)]SbF6), which provided cycloadducts in minutes with excellent yields at room temperature in all cases studied (Scheme 32).84 Notably, alkene VCPs 175, which underwent cycloadditions with Wilkinson’s catalyst under previously reported harsh conditions,76 were readily converted to cycloadducts 176 with the new catalyst at room temperature.
Scheme 31.
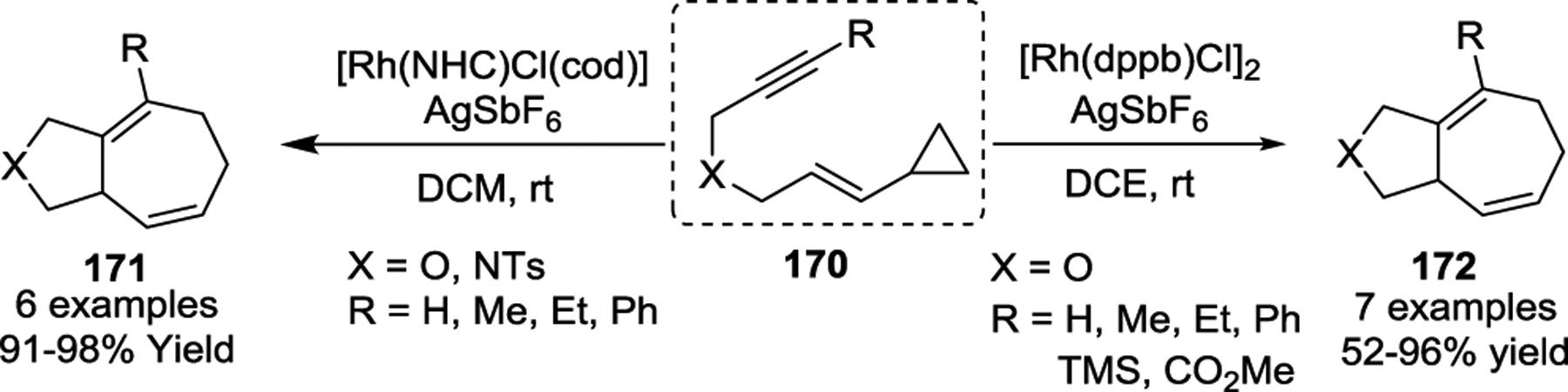
Scheme 32.
In 2005, the Wender group developed the first catalyst system for the asymmetric [5 + 2] cycloaddition of VCPs with tethered alkenes (Scheme 33).85 Good yields and high enantioselectivities were obtained with the chiral rhodium catalyst [Rh(R-BINAP)]SbF6. However, replacing the alkene acceptor group with an alkyne resulted in reduced enantioselectivities ranging from 22 to 56%. Later, Hayashi and coworkers realized the asymmetric intramolecular [5 + 2] cycloaddition of VCPs with alkynes by using of a rhodium complex coordinated to a chiral phosphoramidite ligand (Scheme 34).86
Scheme 33.

Scheme 34.
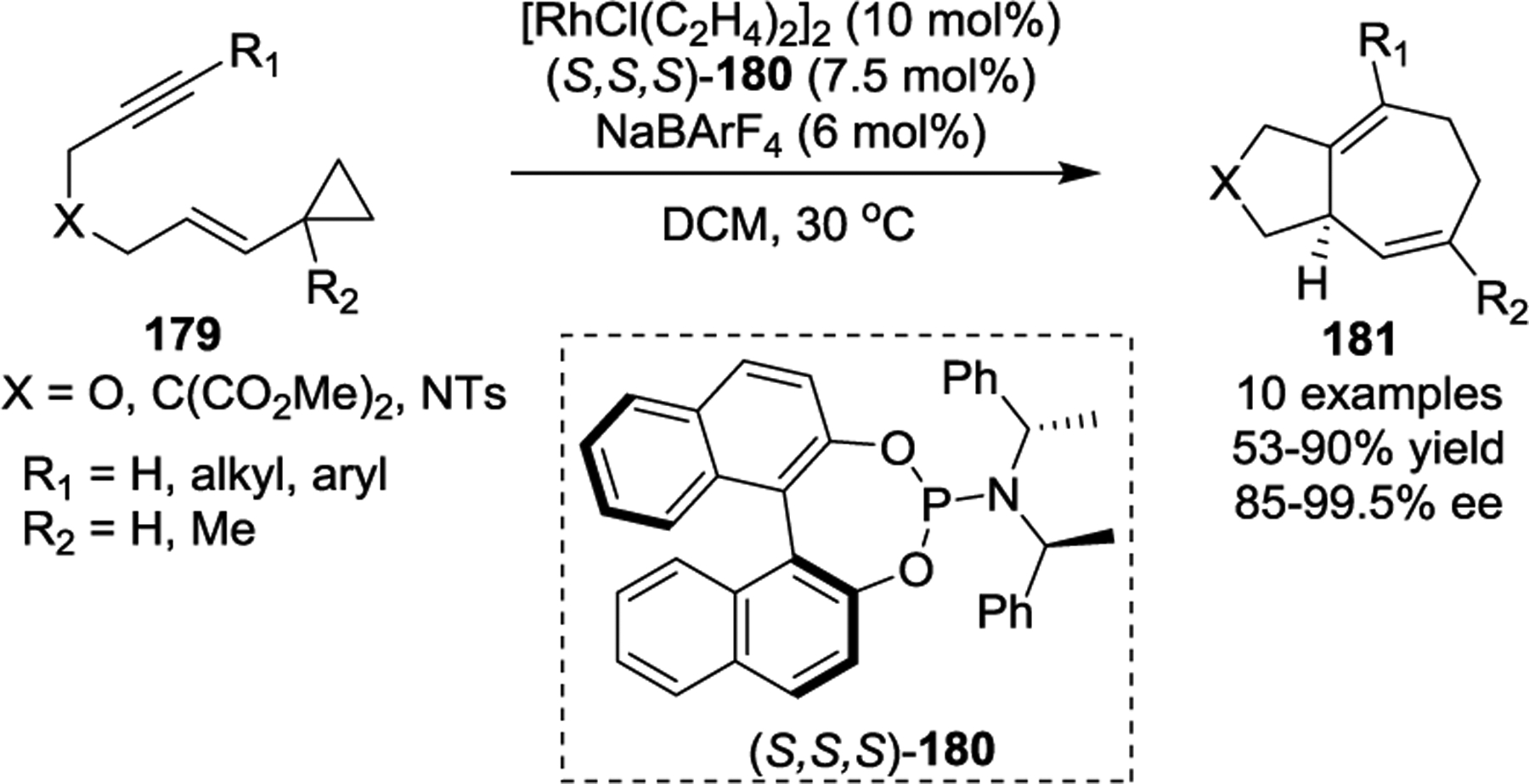
In 2016, the Anderson group designed a highly reactive phosphoramidite-complexed rhodium catalyst for the stereoselective and enantioselective cycloisomerization of ynamide-VCPs 182 to [5.3.0]-azabicycles 185 via a combined theoretical and experimental approach (Scheme 35).87 The designed p-fluorobenzyl ligand 183 promoted a dramatic increase in the reaction rate, as well as enhanced diastereo- and enantioselectivity in the cyclization step. Interestingly, computational studies supported a mechanism that involves β-carbon elimination of the cyclopropane C–C bond after the formation of rhodium intermediate 184, which is distinct from Wender’s Rh-catalyzed intramolecular VCP [5 + 2] cycloaddition.66 Notably, high levels of catalyst-controlled diastereoselective cycloisomerizations of 182 were also observed in this study.
Scheme 35.
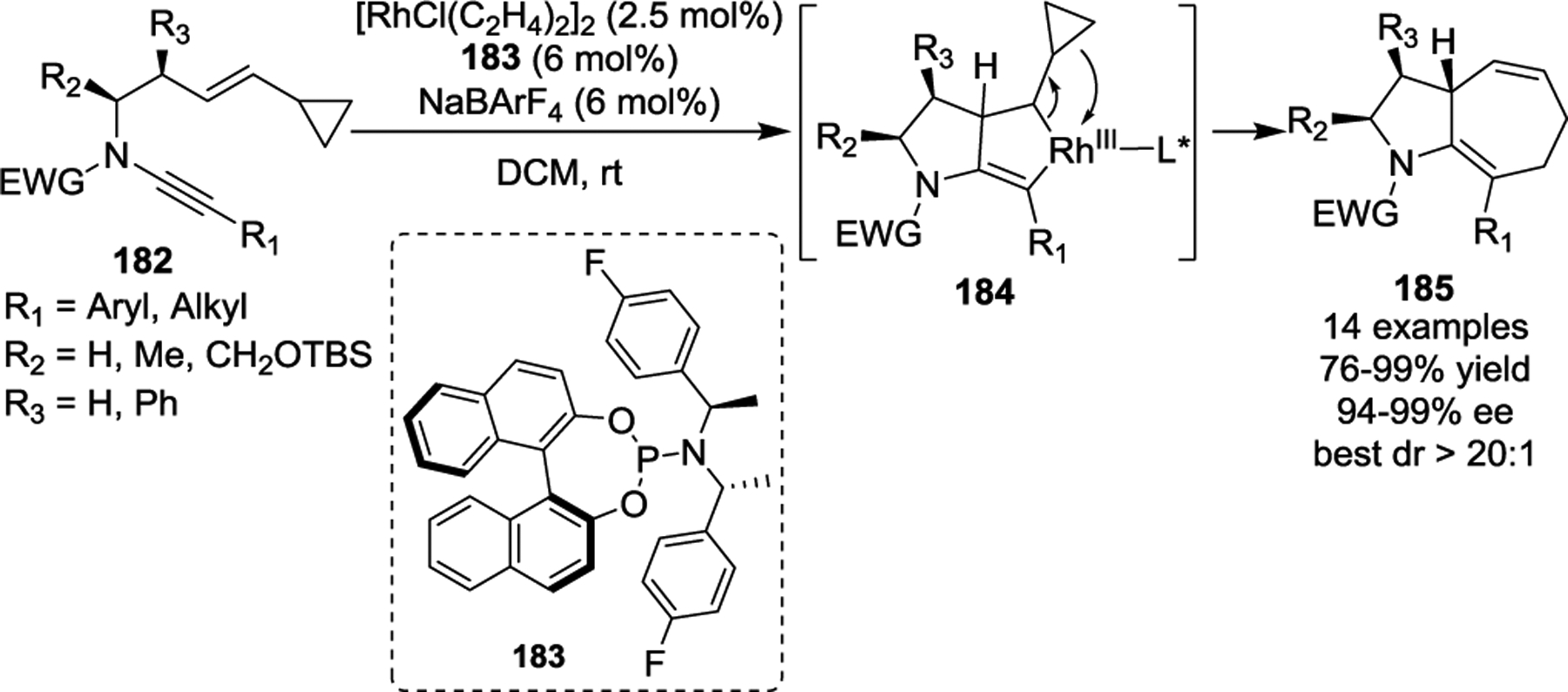
Recent developments pertaining to the intramolecular [5 + 2] cycloaddition have demonstrated that allenylcyclopropanes exhibit reactivity similar to VCPs (Scheme 36).81, 88–89 In 2009, the Mukai group described the straightforward preparation of the bicyclo[5.4.0]undecatrienes and bicyclo[5.5.0]dodecatrienes 187. These products were prepared via cycloaddition of alkyne-allenylcyclopropane derivatives 186, catalyzed by either a [Rh(CO)2Cl]2 or [Rh(CO)(dppp)Cl]2 catalyst (Scheme 36, a).88 Both terminal and internal alkynes were well tolerated in this reaction, affording the fused 6/7 or 7/7 bicyclic products 187 in good yields. Interestingly, an 86% yield was obtained for one substrate that contained a methyl group at the R1 position under the standard conditions, which suggested that a phenylsulfonyl substituent on the allenyl moiety was not essential (Scheme 36, a, entry 4). Later, the authors investigated the cycloaddition of substrates 188, which contained allenylcyclopropanes with alkenes instead of alkynes. During this work they discovered a novel and highly stereoselective cascade transformation that led to the corresponding bicyclic compounds 192 (Scheme 36, b).89 The formation of these bicyclic compounds begins with the oxidative addition of Rh(I) to allenylcyclopropanes 188 to generate the rhodacyclohexenes 189. Next, β-H elimination with ring opening of the rhodacyclohexene rings occurs to provide the tetraene intermediates 190, followed by C(sp2)-Rh(III) bond insertion into the double bond to form intermediates 191. Then, the C(sp3)-Rh(III) bond inserts into the terminal double bond, and a subsequent reductive elimination affords the products 192.
Scheme 36.
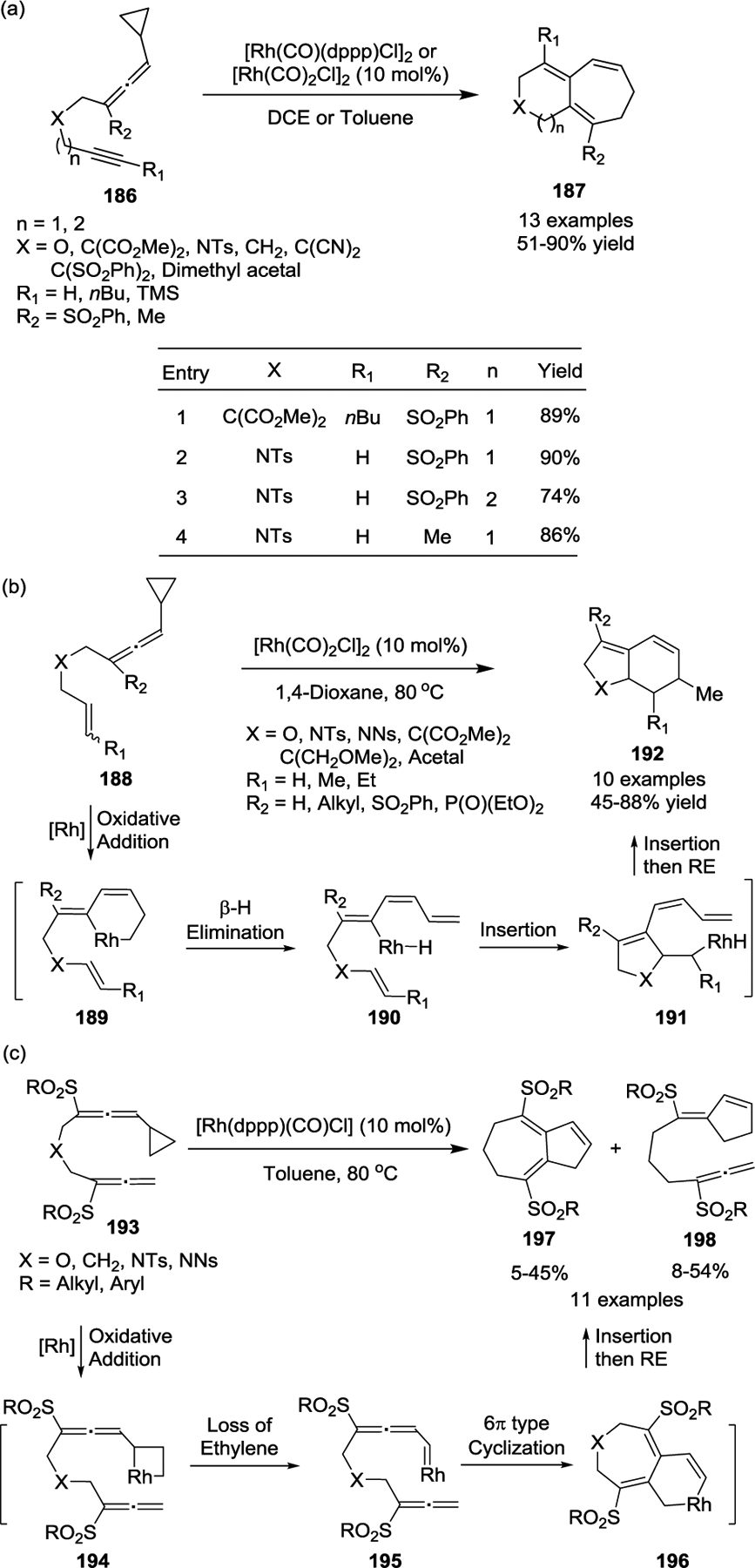
In 2015, the Mukai group executed a unique Rh(I)-catalyzed intramolecular [5 + 2 + 2]-type cycloisomerization of allene–allenylcyclopropanes 193 (Scheme 36, c).90 In contrast to the [5 + 2] formal cycloadditions of VCPs, a fused 7,5-ring system 197, in which the heteroatom substituent is contained within the 7-member ring, was synthesized in less than 50% yield. Based on deuteration experiments, the authors proposed a reaction mechanism that starts with C-C bond cleavage of cyclopropanes 193 to generate intermediates 194. Next, ethylene is liberated to afford rhodium carbenoid species 195. This species can then undergo a 6π-electrocyclic type ring closure to afford rhodabicyclo[5.4.0] intermediates 196, followed by reductive elimination to finally yield the tetrahydroazulene derivatives 197. This novel transformation was significantly affected by the properties of the substituents on both of the two allenyl moieties, and sulfonyl or phosphonate groups were required on the internal position of at least one of the allenes.
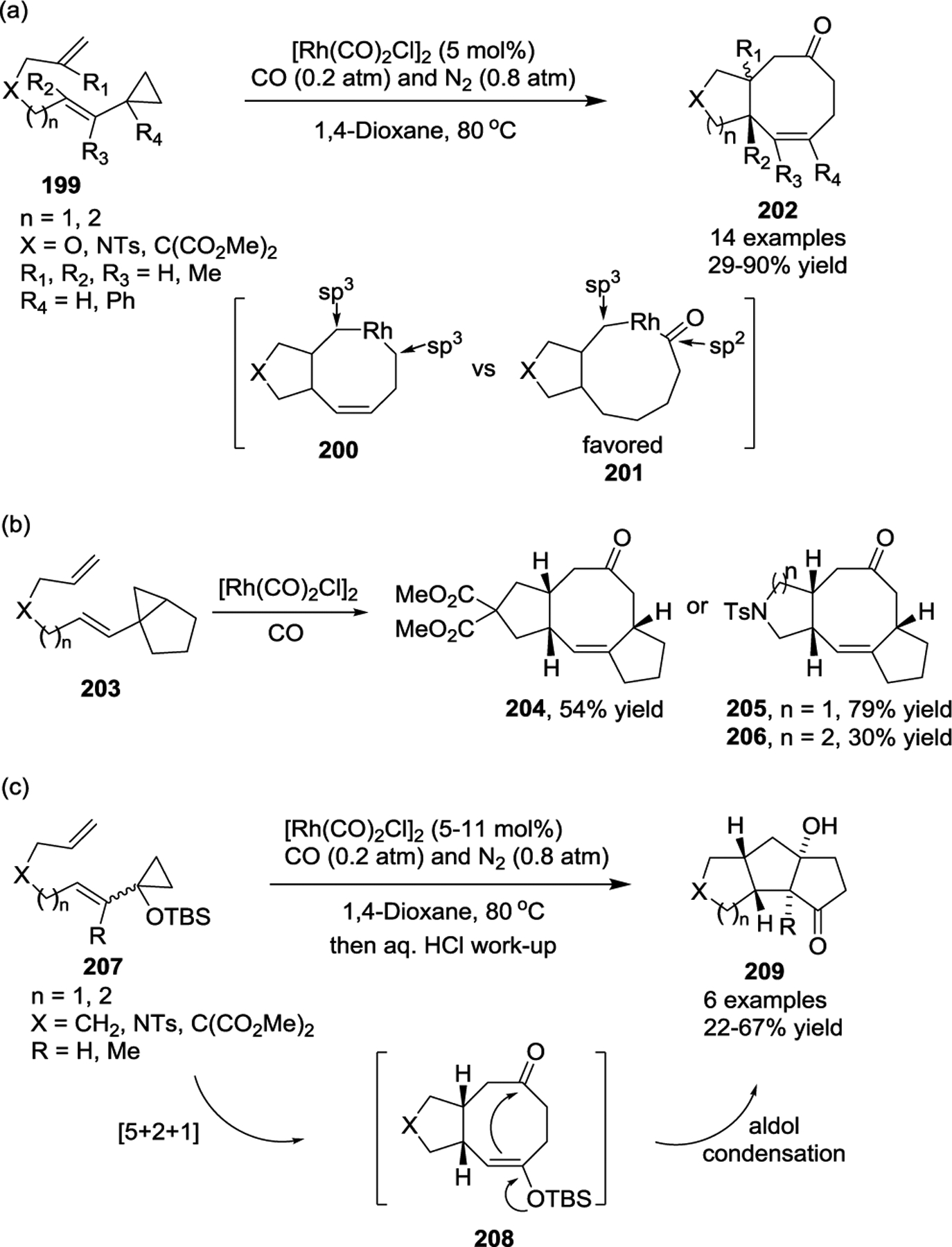
Based on DFT calculations, the Yu group designed a Rh(I)-catalyzed intramolecular [5 + 2 + 1] cycloaddition of tethered ene-VCPs with CO (Scheme 37, a).91 This study provided a flexible and efficient method for constructing functionalized 5/8- and 6/8-fused bicyclic cyclooctenones 202 in good yields, even those containing quaternary centers. The calculations supported the possibility that the addition of CO to the [5 + 2] formal cycloaddition may occur rapidly, converting the eight-membered rhodacycle 200 into a nine-membered rhodacycle 201, which then undergoes an easy C(sp2)–C(sp3) reductive elimination process to afford the [5 + 2 + 1] products.92 Interestingly, tricyclic [5-8-5] skeletons, such as 204–206, could be prepared from the [5 + 2 + 1] cycloaddition by introducing a five-membered carbocycle ring fused with the cyclopropane rings of ene-VCPs 203 under the same conditions (Scheme 37, b).93 Inspired by Wender’s [5 + 2 + 1] cycloaddition,94 in 2008 the Yu group also reported a tandem reaction involving a Rh(I)-catalyzed [5 + 2 + 1] cycloaddition and subsequent aldol condensation (Scheme 37, c).95 Siloxy activated ene-VCPs 207 underwent a [5 + 2 + 1] cycloaddition/aldol process readily to produce a single diasteromer tricyclic cycloadducts 209 in acceptable yields. The present strategy enabled a straightforward approach to access the linear triquinane skeletons, as demonstrated by the concise total syntheses of (±)-Hirsutene and (±)-1-Desoxyhypnophilin.
Scheme 37.
Until now, various types of non-activated VCPs have been designed for use in Rh(I)-catalyzed [m + n] cycloadditions, and dramatically different reaction patterns have been observed (Scheme 38, a). The Yu group was the first to report a Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition of non-activated VCPs 210 to produce bicyclic five-membered ring systems 211, where the VCPs served as the three-carbon synthons (Scheme 38, b).71 Interestingly, the [3 + 2] cycloaddition is limited to trans-ene-VCPs, while the [5 + 2] cycloaddition that uses cis-ene-VCPs could afford 5,7-bicyclic compounds instead. Complete chirality transfer was observed in the present [3 + 2] cycloaddition, providing an efficient method to synthesize optically pure fused compounds. This study demonstrates that VCPs without electron-withdrawing activation groups can act as three-carbon synthons in transition-metal-catalyzed cycloadditions. Further investigation showed that trans-2-allene-VCPs 212 also underwent intramolecular [3 + 2] cycloadditions, yielding bicyclo[3.3.0]octane derivatives 213 in the presence of a [Rh(CO)(PMe3)2]Cl catalyst (Scheme 38, c).96
Scheme 38.
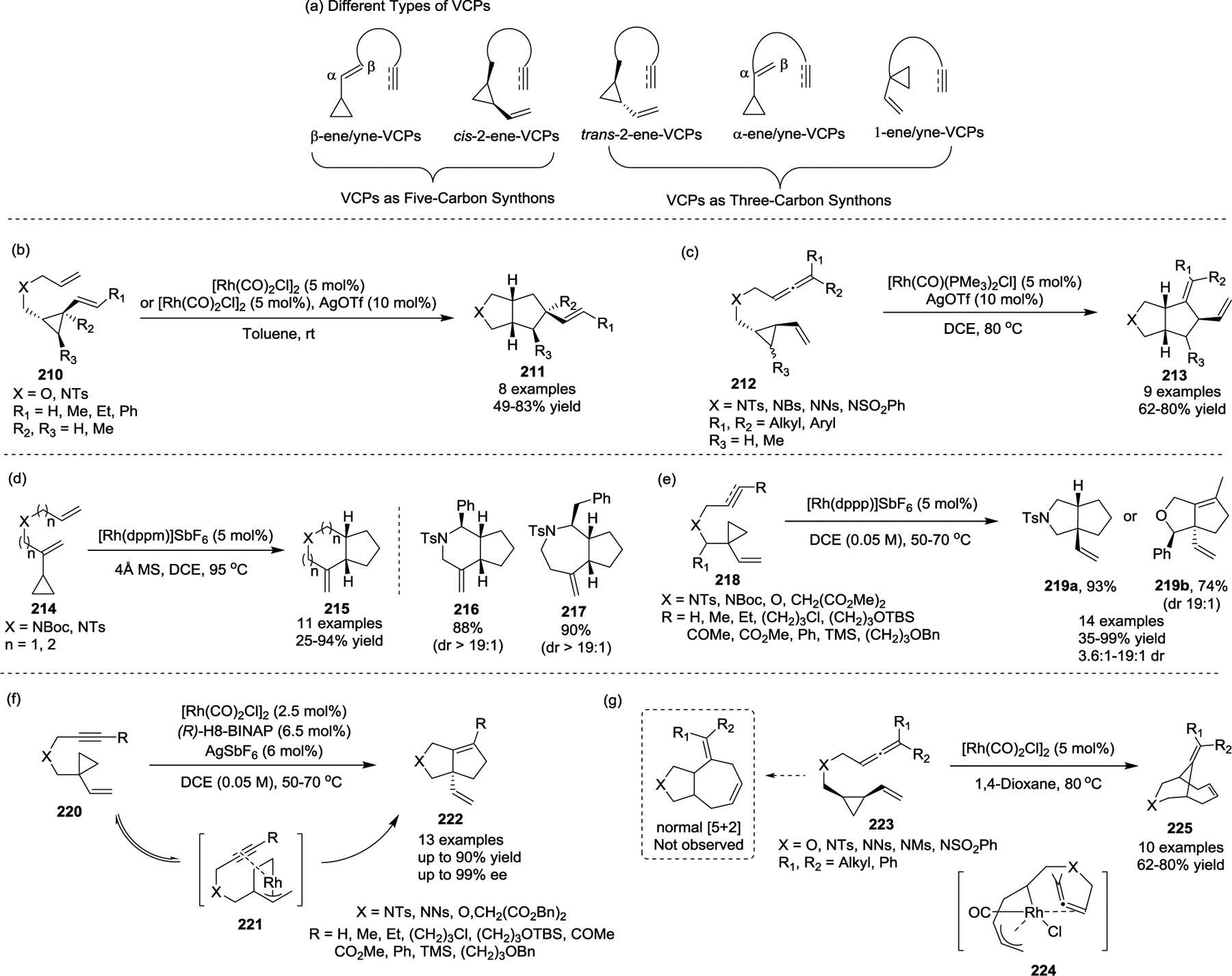
In general, the strategic difference between the intramolecular [5 + 2] and [3 + 2] cycloadditions of non-activated VCPs is the placement of the tethered 2π-units with respect to the cyclopropane ring.92 For example, if the alkene is placed in a β-ene-VCP, or a cis-2-ene-VCP configuration, as reported by Wender and other groups, a [5 + 2] cycloaddition can take place. However, if a tethered alkene is placed in a trans-2-ene-VCP, α-ene-VCP, or a 1-ene-VCP position, the [3 + 2] formal cycloaddition can occur.
In 2010, Yu and colleagues disclosed another new type of α-ene-VCP that extended the range of processes where α-ene-VCPs can be used as the three-carbon components in cycloadditions. [Rh(dppm)]SbF6 catalyzed the intramolecular [3 + 2] cycloaddition of α-ene-VCPs with alkenes, resulting in the efficient construction of 5/6- and 5/7-bicyclic compounds 215, while the anticipated [5 + 2] products were not observed (Scheme 38, d).97 Substrates with substitutions on the alkene could not be tolerated in this transformation. By switching the attachment position of the tethered π-systems in VCPs, related transformations involving alkenes, alkynes, and allenes tethered VCPs 218 were also reported, which provided an efficient and versatile method for the synthesis of cyclopentane- and cyclopentene-embedded bicyclic structures 219 (Scheme 38, e).98
The mechanism of this transformation was studied by DFT calculations.72 The computational results revealed that the relative orientation of the tethers in the 1-ene/yne-VCPs plays a key role in controlling the stereochemistry of the [3 + 2] cycloadducts. Alkene/alkyne insertion is the rate- and stereoselectivity determining step of these multistep [3 + 2] cycloadditions.
In 2012, the Yu group developed the first asymmetric Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition of 1-yne-VCPs to produce bicyclo[3.3.0] compounds 222 in high yields and enantiomeric excess (Scheme 38, f).99 This asymmetric reaction was realized by the combination of a cationic Rh(I) system with (R)-H8-BINAP, which generated the fused ring systems 222 bearing an all-carbon chiral quaternary stereocenter at the bridgehead. Notably, alkyne substituted VCPs are crucial for this transformation, and attempts for the asymmetric cycloaddition of 1-ene-VCPs failed. Furthermore, computational analysis of the alkyne-insertion transition states supported the reversible formation of rhodacycle 221 in advance of the rate- and stereochemistry-determining alkyne insertion step where the R group in the alkyne moiety of the substrates affects the enantioselectivity.
In 2017, a novel Rh(I)-catalyzed intramolecular bridged [5 + 2] cycloaddition of cis-allene-VCPs 223 was developed to synthesize the challenging bicyclo[4.3.1]decane skeletons 225 (Scheme 38, g).100 Preliminary DFT calculations revealed that the inverse allene insertion was favored over the expected normal [5+2] cycloaddition in order to reduce the repulsion between the allene and VCP moiety in the transition state 224, which resulted in the formation of unexpected bridged [5 + 2] cycloadducts.
The Yu group also investigated the multicomponent cycloaddition of 1-ene/yne-VCPs with CO. In 2010, they reported a novel Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition for the synthesis of six-membered carbocycles 228 (Scheme 39).101 Both tethered alkene and alkyne moieties were compatible in this study, and this work enabled a new approach to generate bicyclic cyclohexanones and cyclohexenones. In a later publication, the Yu group also incorporated two CO units to execute a novel Rh(I)-catalyzed formal [5 + 1]/[2 + 2 + 1] cycloaddition of 1-yne-VCPs (Scheme 40).102 Various 1-yne-VCPs 229 were examined under the optimized conditions, which allowed the development of an efficient way to construct functionalized tricyclic 5/5/6 skeletons 232 with good yields in a single step. Significantly, when R1 = Me, this transformation allowed the synthesis of angular tricyclic 5/5/6 skeletons with two adjacent bridgehead quaternary all-carbon stereocenters.
Scheme 39.
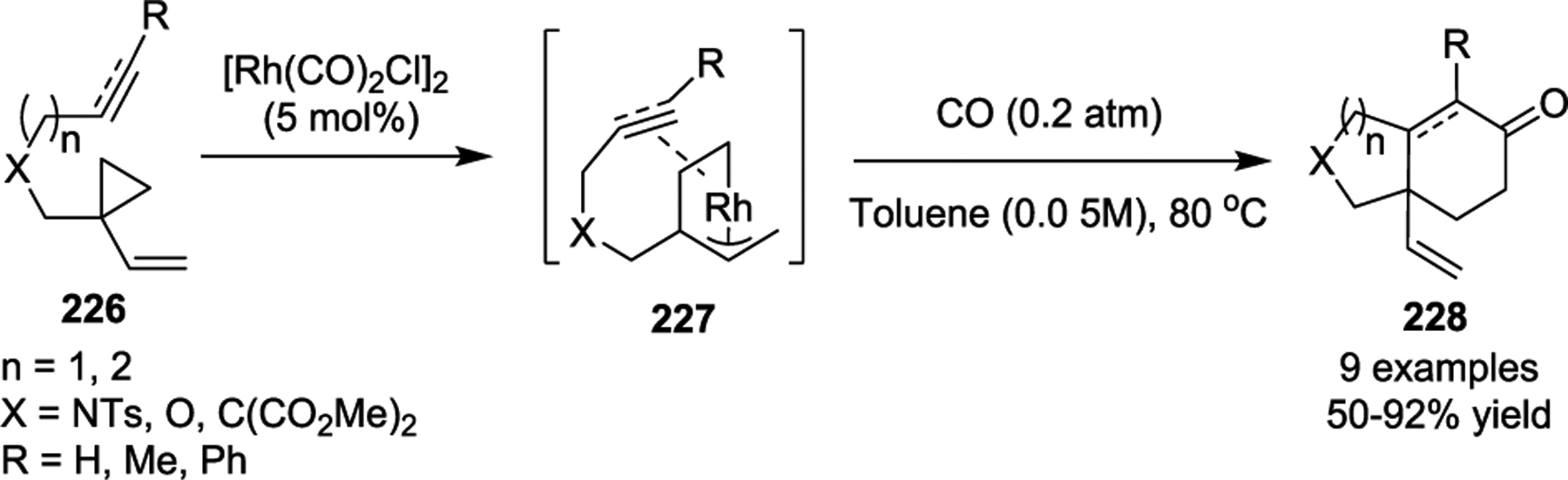
Scheme 40.
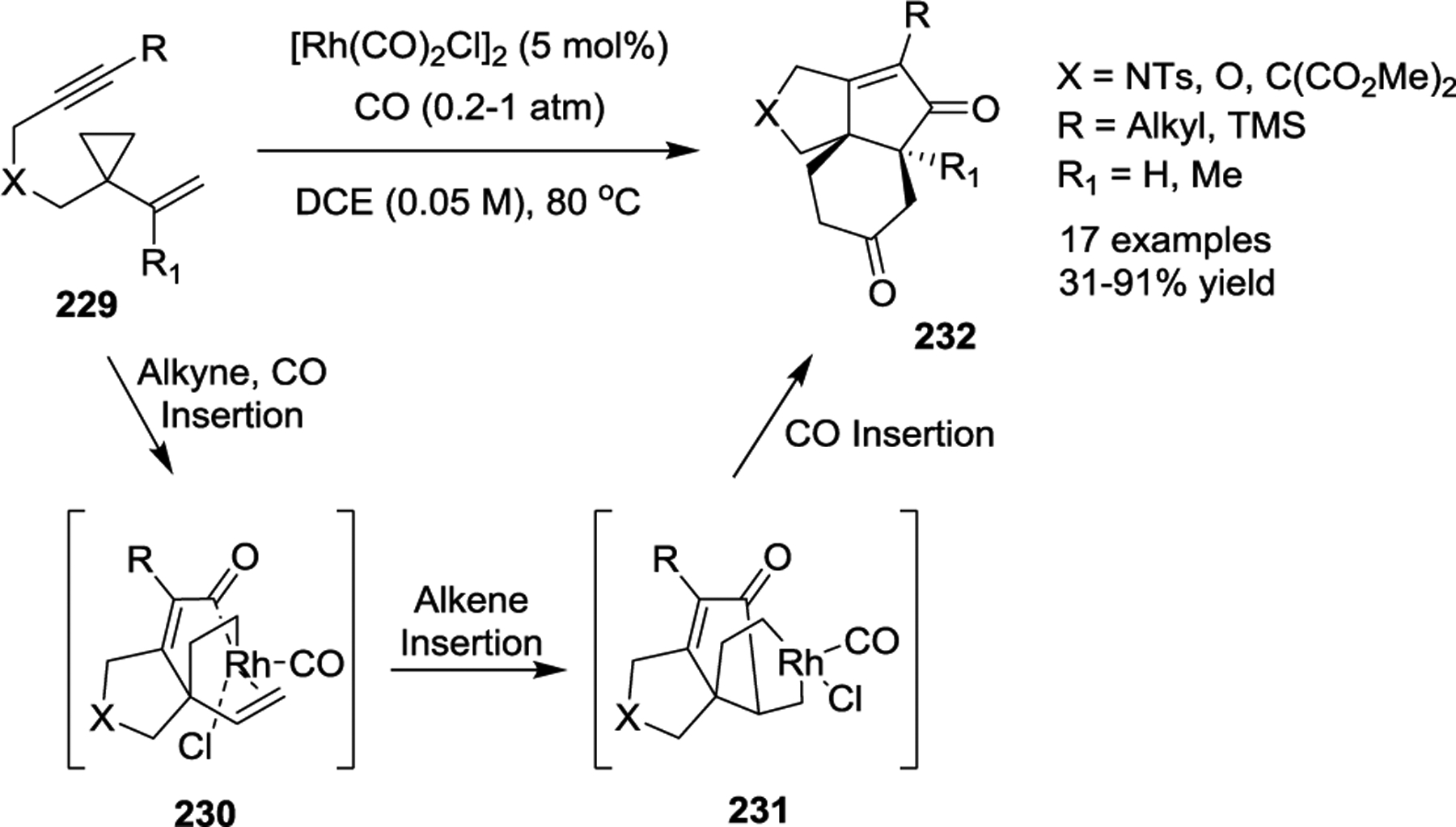
Preliminary DFT calculations indicated that the alkyne substituent R of the 1-yne-VCPs 229 makes the formal [5 + 1]/[2 + 2 + 1] pathway gradually preferred over the [(3 + 2) + 1] pathway. Thus, rather than C–C reductive elimination of intermediates 230 to afford the [(3 + 2) + 1] byproducts, alkene migratory insertion occurs to provide new rhodacycles 231. Next, these rhodacycles readily undergo carbonylation and then C–C reductive elimination to generate the products 232.
Similar to VCPs, allenylcyclopropanes are also active intermediates in Rh(I)-catalyzed cycloadditions. In 2011, the Tang group reported an interesting example that involved the generation of allenylcyclopropanes in-situ (Scheme 41).103–104 This transformation was initiated by the Rh (I)-catalyzed 1,3-acyloxy migration (Saucy–Marbet rearrangement)105 of propargyl esters 233, which lead to the formation of allenylcyclopropanes 233. Subsequently, ring expansion of the allenyl esters 233 occurs to form alkylidene metallacyclohexene intermediates 234. Next, CO insertion and reductive elimination generate functionalized cyclohexenones 236. Good regioselectivity was generally observed in both cis- and trans-substituted cyclopropanes, and higher regioselectivity (16:1 to >20:1) was obtained for all trisubstituted cyclopropanes. Interestingly, chirality in the cyclopropane starting material could be completely transferred to the product. The acyloxy group not only eased the access to allene intermediates but also provided a handle for further functionalization of the products.
Scheme 41.
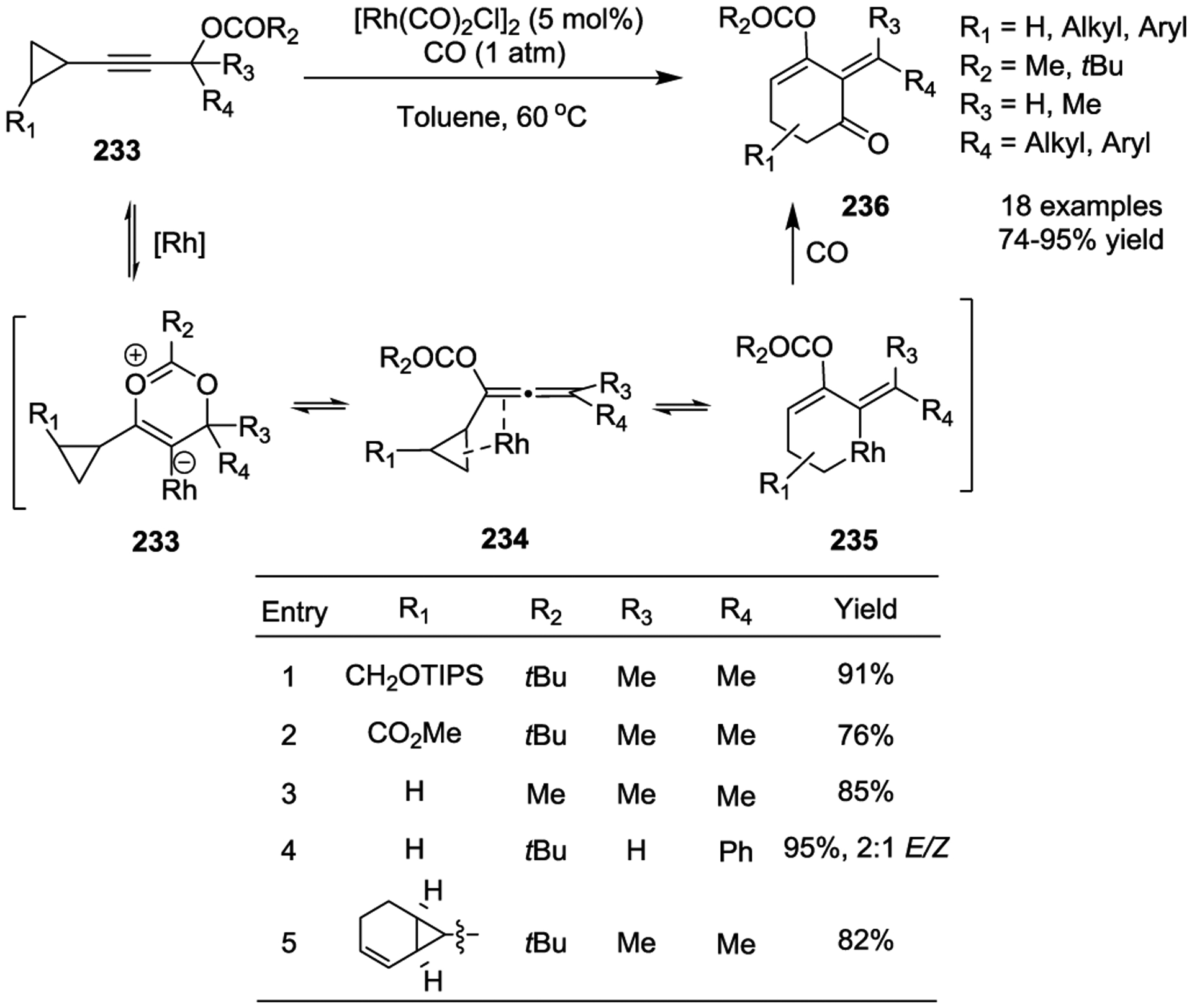
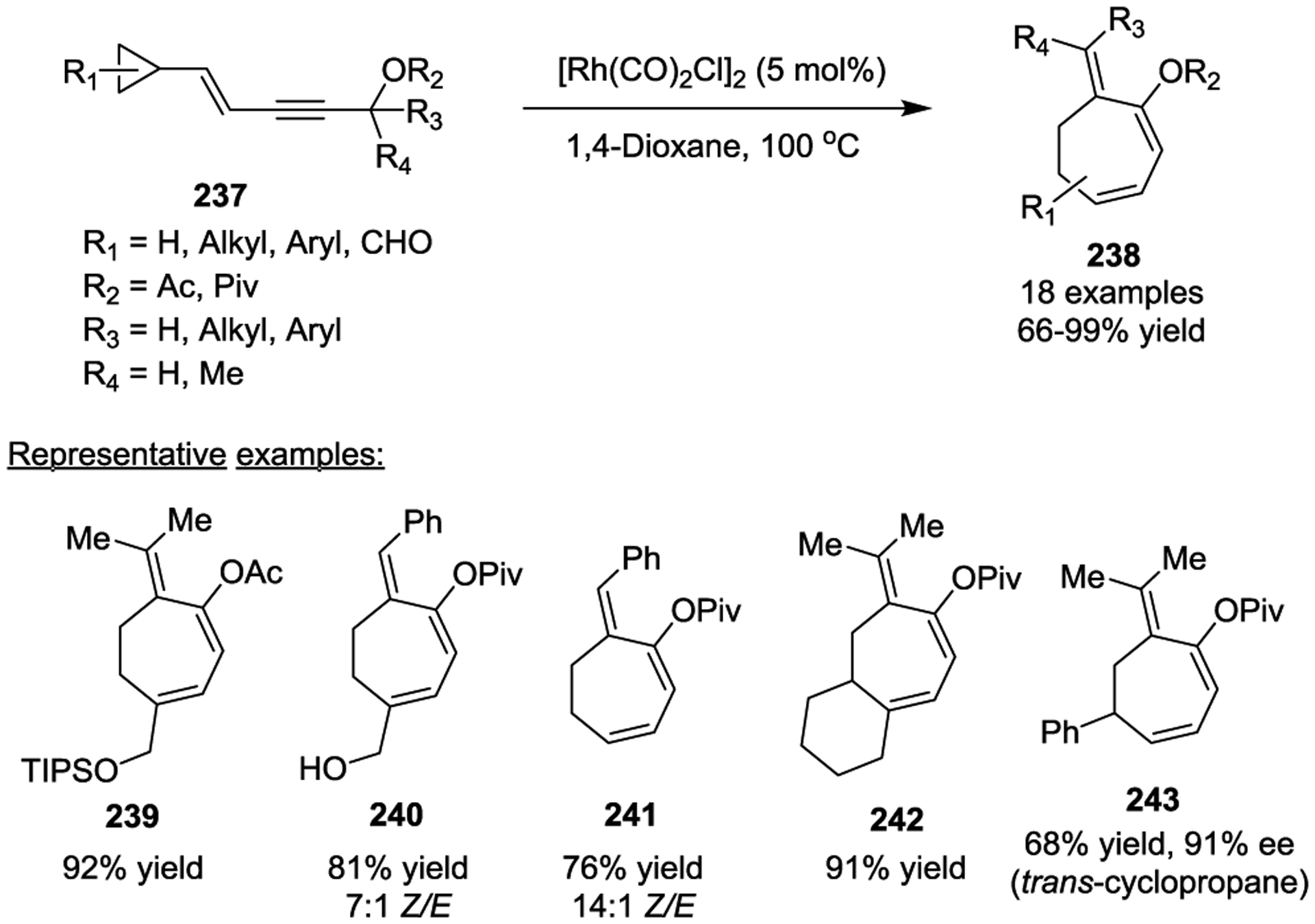
In 2011, Tang and colleagues then reported a Rh(I)-catalyzed ring expansion of VCPs to seven-membered rings 238 based on a similar strategy (Scheme 42).106 In this case, good Z/E selectivity was obtained when R is an aryl group, such as 240. Only one regioisomer was observed for trans-substituted cyclopropanes. 5/7- and 6/7-fused bicyclic complexes were prepared in 85% and 91% yields, respectively. Notably, chirality in the starting cyclopropane (92% ee) could be completely transferred to the optically pure product 243 (91% ee). Preliminary mechanistic studies suggested that the allene group is critical for the ring expansion reaction since a dienyl cyclopropane did not yield any desired product under the standard conditions.
Scheme 42.
3.1.2. Ruthenium Catalysis
In 2000, the Trost group reported a cationic ruthenium catalyst-promoted intramolecular [5 + 2] cycloaddition between non-activated VCPs and alkynes 244 (Scheme 43, a).107 The cycloaddition worked at room temperature to provide bicyclic ring systems in good yields and excellent regioselectivity. The authors also further investigated the regioselectivity of the cyclopropyl ring opening (Scheme 43, b).108 They determined that when using trans cyclopropyl substrates 250, the cleavage of the more substituted C-C bond on the cyclopropyl ring was favored to afford product 252, while opposite regioselectivity was observed for those substrates with bulky silyl groups. When using cis cyclopropyl substrates 253, in most cases, a single regioisomer was generated, which suggested the dominance of steric effects for these starting materials. Alternatively, this regioselectivity was opposite to that observed by the Wender group when using a [Rh(CO)2Cl]2 catalyst,78 which indicates that the Ru(II)-catalyzed [5 + 2] process likely proceeds via a distinct mechanism. When the authors attempted to expand the substrate scope beyond alkynes to other π-systems, they isolated a novel ruthenium complex 257 from the attempted intramolecular formal cycloaddition of an alkene-tethered VCP 256 in the presence of their [RuCp(CH3CN)3]PF6 catalyst (Scheme 43, c). This helped shed light on the difference in reactivity between ruthenium and rhodium catalysis.109 Further experimental and theoretical studies by Trost and Houk revealed that oxidative cyclization in advance of a β-carbon elimination generates metallacyclopentanes 246. Subsequent β-carbon elimination results in the cyclopropane C–C bond cleavage and forms metallacyclooctenes 247.110 The advantage of the Ru(II)-catalyzed intramolecular [5 + 2] cycloaddition was further demonstrated by the efficient construction of 5/7/6-, 5/5/7-, and 6/5/7-fused tricyclic compounds 259 and 261, which all contained seven-membered rings and were obtained in high yields and diastereoselectivities (Scheme 43, d).111–112
Scheme 43.
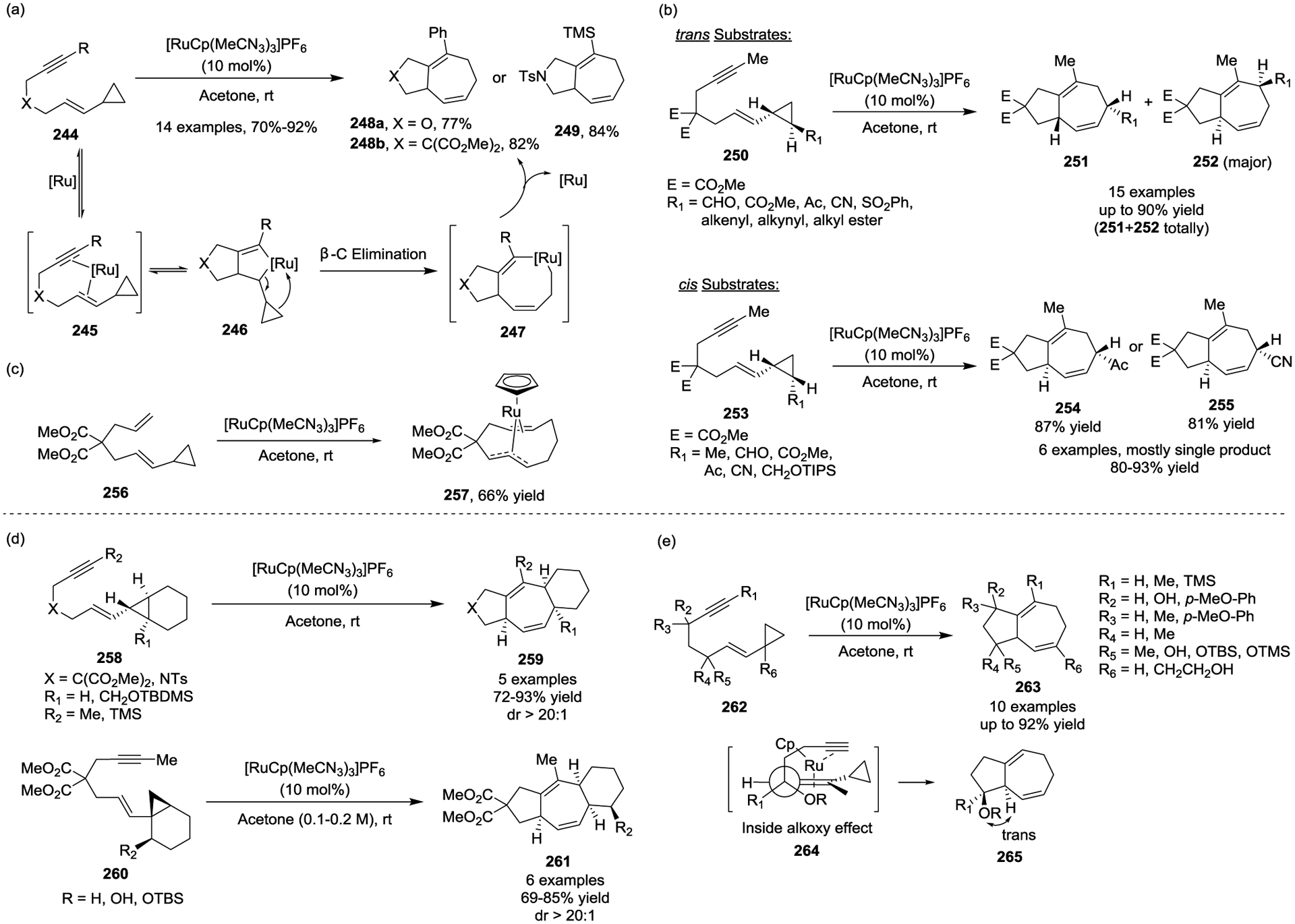
The diastereoselectivity of the Ru(II)-catalyzed [5 + 2] cycloaddition was also examined by the Trost group (Scheme 43, e).113 Their study demonstrated that the existing stereogenic centers in the tether can influence the configuration of the newly installed chiral center at the bridgehead carbon atom, which affords high diastereoselectivity in all cases. Particularly high levels of diastereoselectivity resulted for the substrates bearing bulky substituents. In addition, the angular hydrogen atom preferentially shows a trans relationship to the hydroxyl group in the favored diastereomeric cycloadduct 265. This is in agreement with the “inside alkoxy” model 264 (Stork/Houk-Jäger model), which describes how the antibonding orbital of the alkoxy group destabilizes the transition state, while the electron-donating orbitals of the C–H or C–R bond are stabilizing, and thus lead to the observed trans relationship.114
3.1.3. Nickel Catalysis
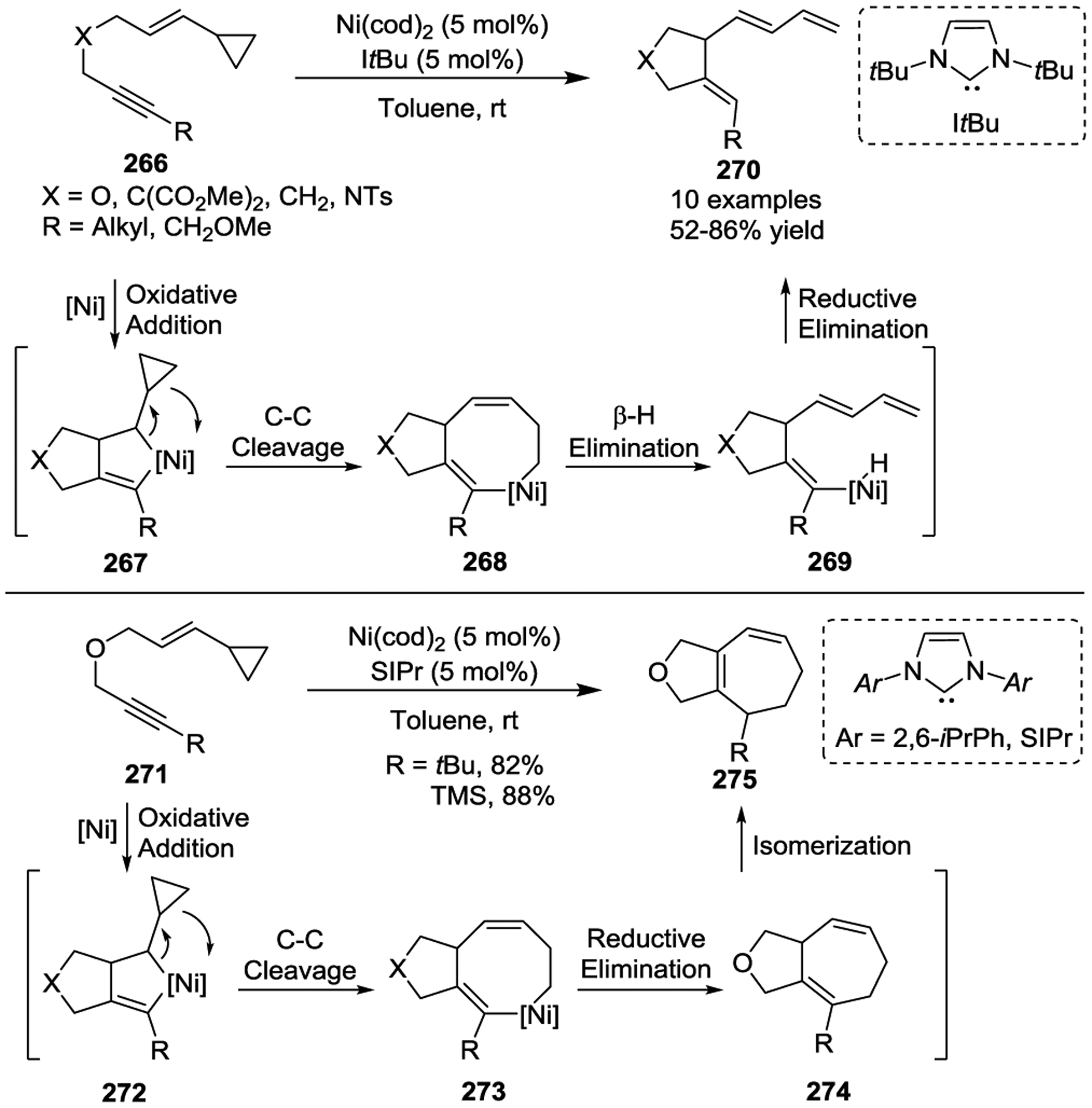
In 2005, the Louie group demonstrated that Ni(0) could catalyze either isomerization of VCPs 266 to afford cyclopentenes 270 or a [5 + 2] cycloaddition to produce bicyclic 5/7 cycloadducts 275 in excellent yields under mild conditions, depending on the selection of N-heterocyclic carbene (NHC) ligands (Scheme 44).115 This study showed that the cycloisomerization process was controlled with a less sterically demanding NHC (ItBu: 1,3-di-tert-butylimidazolin-2-ylidene), while the [5 + 2] cycloaddition was promoted by a slightly bulkier NHC [SIPr: 1,3-Bis-(2,6-diisopropylphenyl) imidazolin-2-ylidene]. A preliminary mechanism was proposed to analyze the selectivity, and the Houk group later performed computational calculations to determine a detailed mechanism.116 The energetically favored pathway involves oxidative alkyne–alkene cyclization to form metallacyclopentene intermediates 267 or 272, in contrast to the cyclopropane cleavage pathway with Rh(I) catalysts.66 C-C bond cleavage of cyclopropanes in 267 or 272 affords an eight-membered metallacycles 268 or 273, followed by either β-H elimination and reductive elimination to provide dienes 270 or direct reductive elimination to yield seven-membered ring systems 275. The competition between β-H elimination and reductive elimination was rationalized by the slightly different steric environments created by the NHC ligands.
Scheme 44.
3.1.4. Iron Catalysis
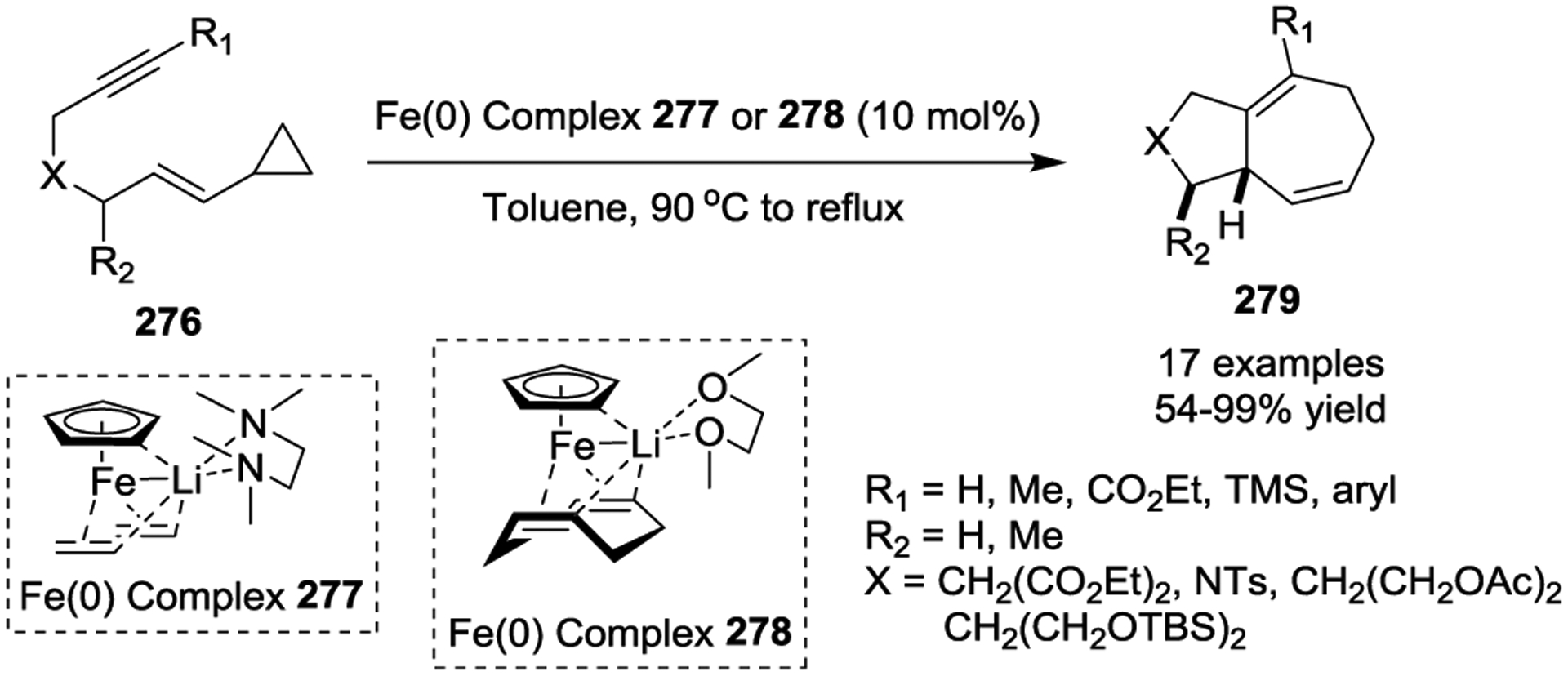
Besides the noble metal catalysts, Fe-based catalysts have also been used for intramolecular [5 + 2] cycloadditions of non-activated VCPs (Scheme 45).117 Inexpensive ferrate complexes 277 and 278 were shown to be excellent catalysts for this reaction featuring VCPs 276. This reaction was compatible with a variety of substitution patterns on the tethered alkyne, and it has so far proved to be analogous to its Rh(I) counterparts which indicates that these iron complexes can be powerful alternatives to the commonly used noble metal catalysts in the realm of cycloaddition chemistry.
Scheme 45.
3.2. Metal-Catalyzed Intermolecular Processes
3.2.1. Iron Catalysis
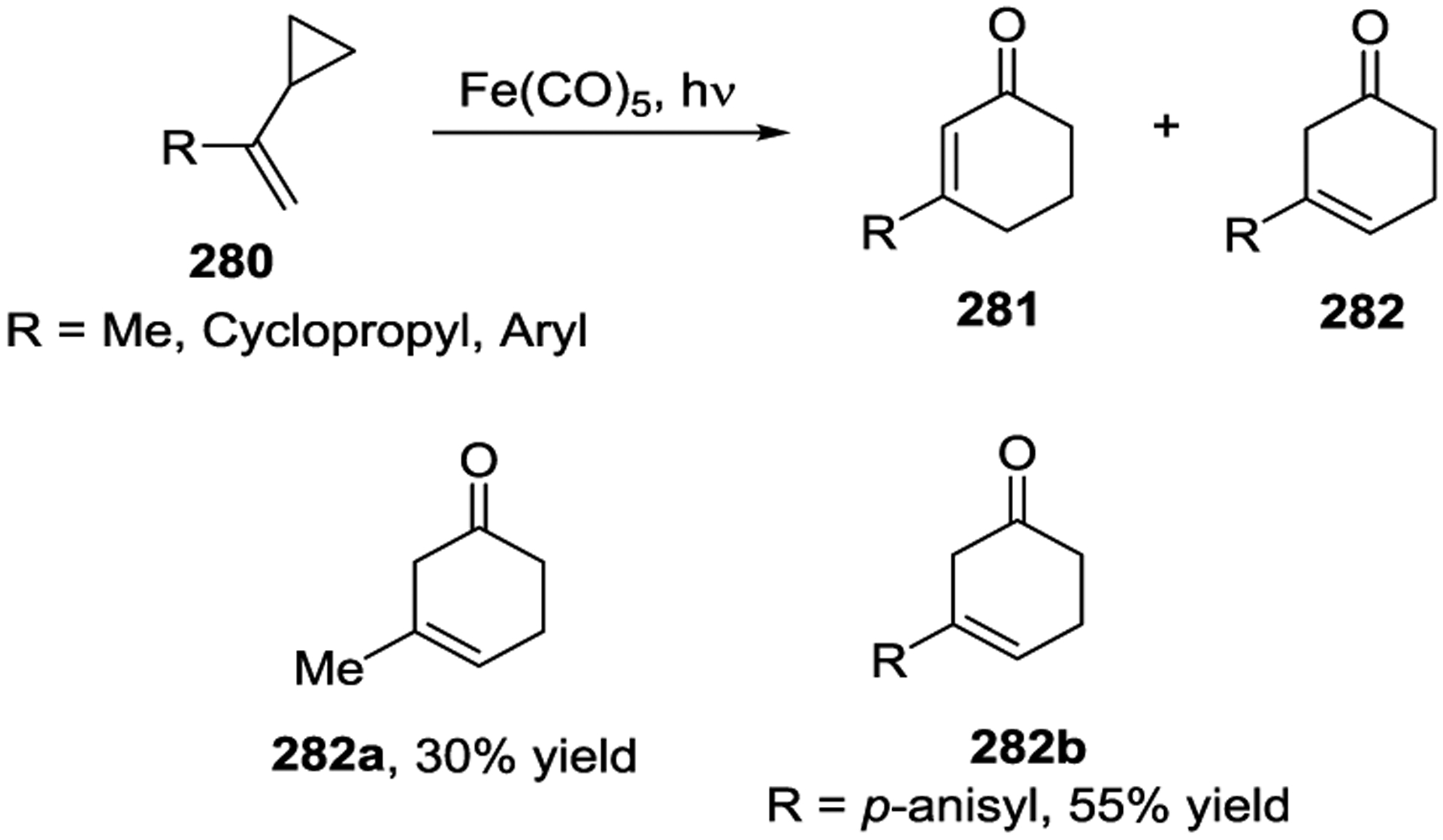
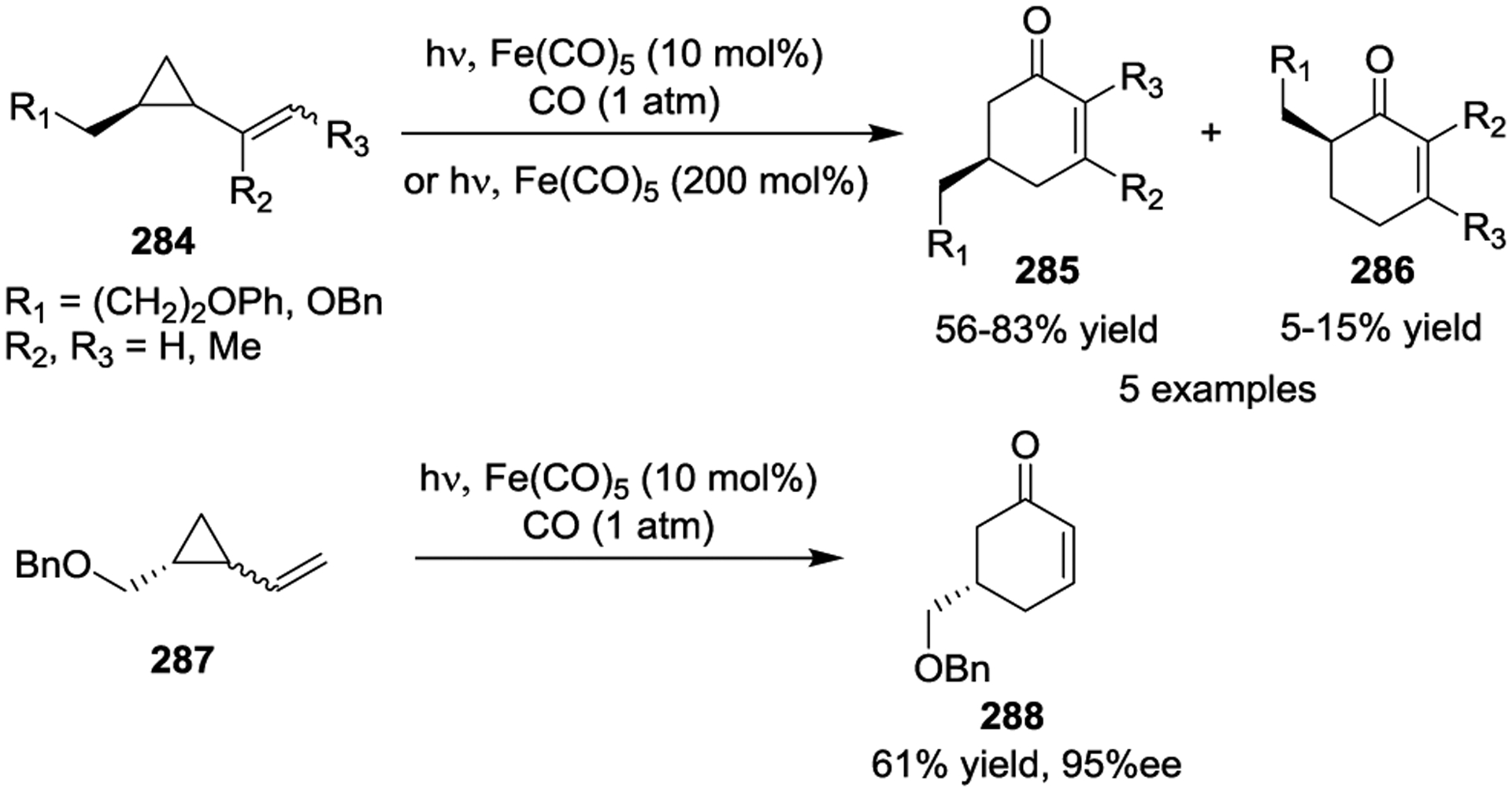
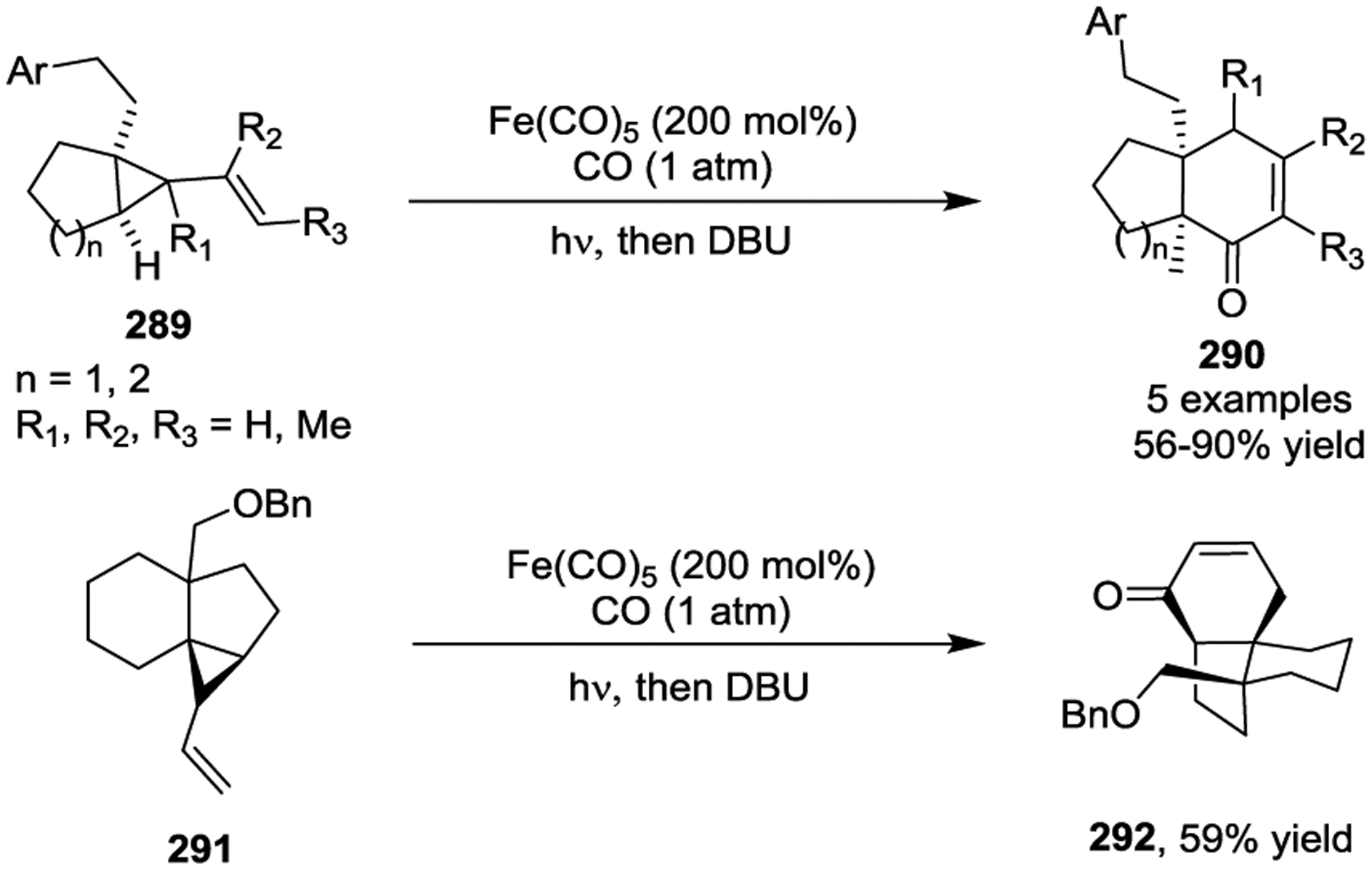
Iron has been known to interact with non-activated VCPs for several decades.118–119 The photoinitiated 1,5-carbonylation of VCPs 280 in the presence of Fe(CO)5, which resulted in cyclohexenones 281 and 282, was first reported by Sarel in 1969 (Scheme 46).120 Stochiometric amounts of Fe complex were necessary for the reaction under UV light. In 2000, the Taber group reported a general method for the synthesis of optically pure 5-alkyl cyclohexenones 285 from enantiomerically pure VCPs such as 284 in the presence of Fe(CO)5 and ultraviolet irradiation (Scheme 47).121 Similar yields were obtained either with 2 equivalents of Fe(CO)5 or 10 mol% Fe(CO)5 under a CO atmosphere. High regioselectivity was observed in this reaction, which provided a general method for the preparation of chiral substituted cyclohexenones, such as 288.122 This transformation was further expanded to the preparation of multicyclic ring systems through an Fe(CO)5 mediated [5 + 1] cycloaddition of bicyclic VCPs 289 with CO (Scheme 48).123 The reaction provided carbobicyclic 5/6-systems 290 in good yields with excellent regioselectivities. Interestingly, carbonylation of 291 even delivered the more complex tricyclic compound 292 in 59% yield. Later, a three-step synthesis of the tricyclic core of estrone methyl ether, as seen in 295 and 296, was established, which involved a Wittig reaction, an Fe-mediated cyclocarbonylation, and subsequent cyclization processes (Scheme 49).124
Scheme 46.
Scheme 47.
Scheme 48.
Scheme 49.
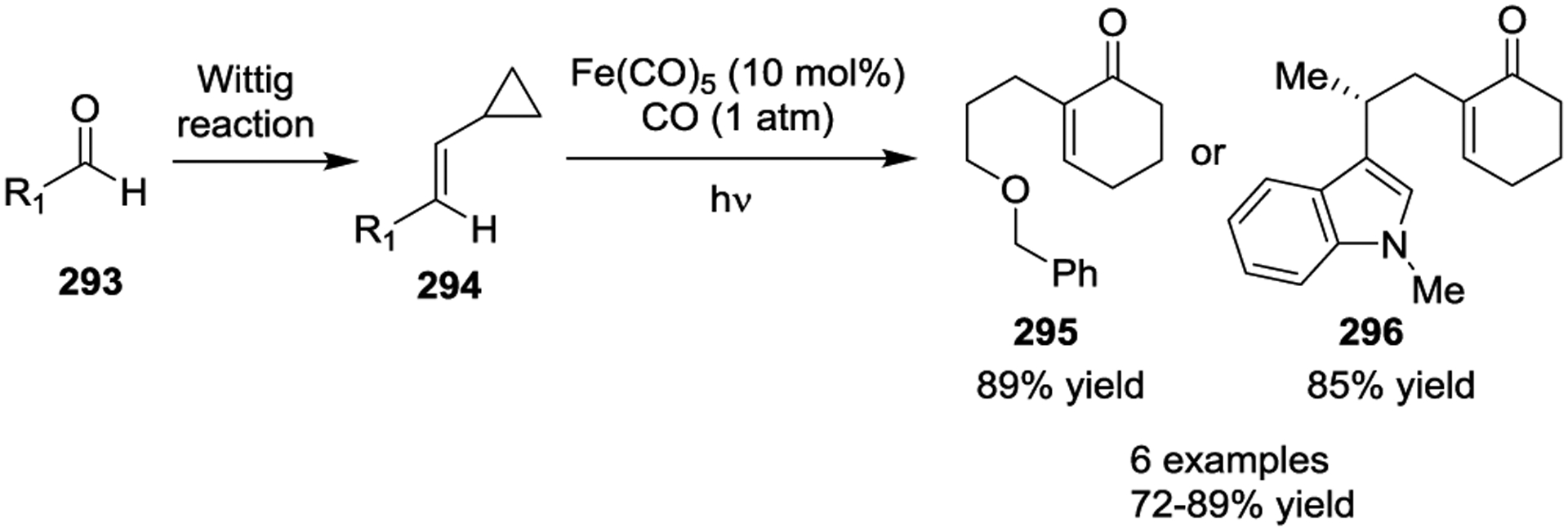
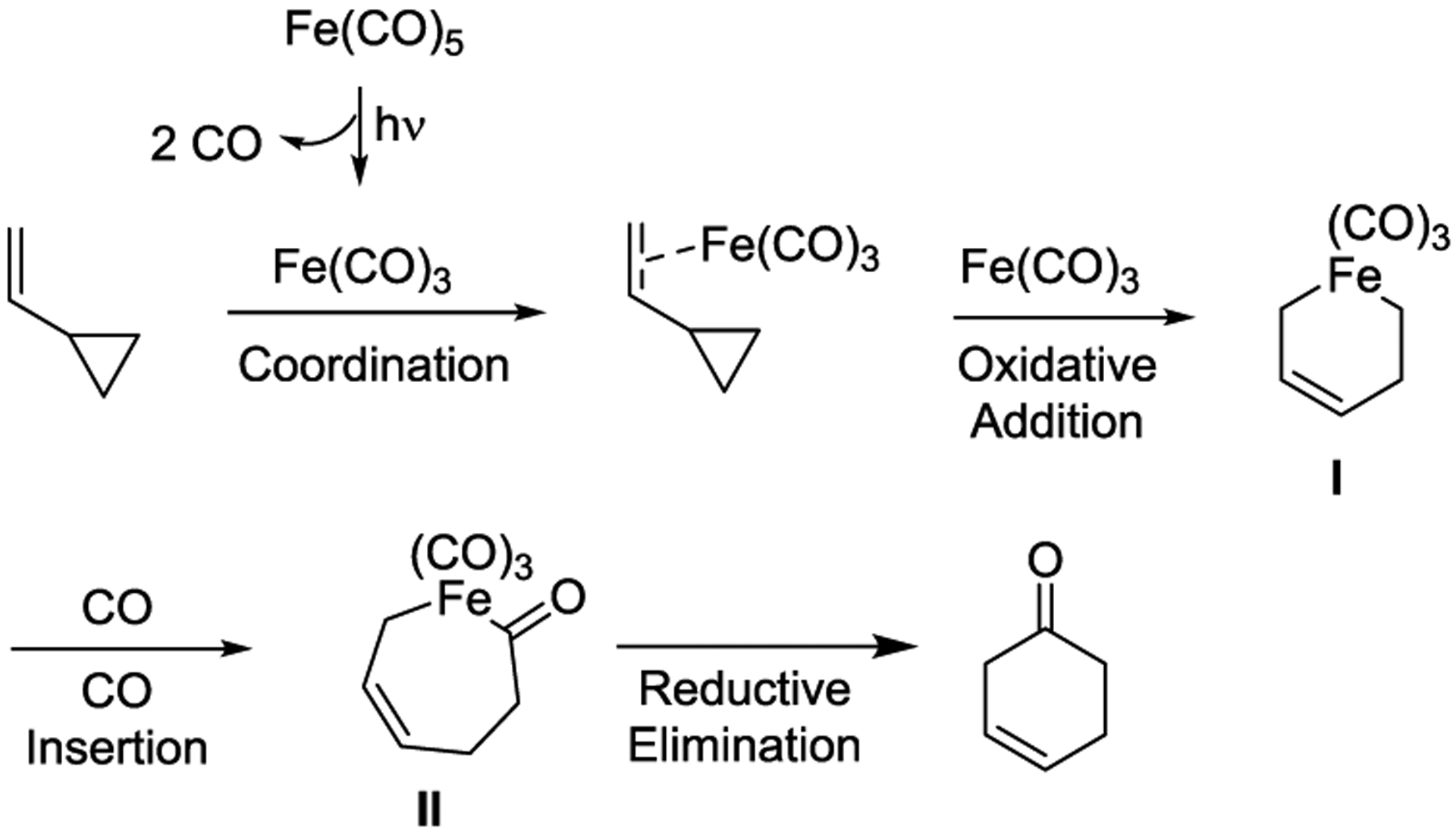
Mechanistic studies indicated that the active Fe(CO)3 catalyst is generated from the photolytic dissociation of Fe(CO)5. After coordinating to the olefin of the VCP, C-C cleavage of the cyclopropyl ring occurs to afford ironmetallocycle I, followed by CO insertion and reductive elimination to form the cyclohexenones II. The regeneration of the active Fe(CO)3 catalyst can occur if the reaction is performed under a CO atmosphere (Scheme 50).125
Scheme 50.
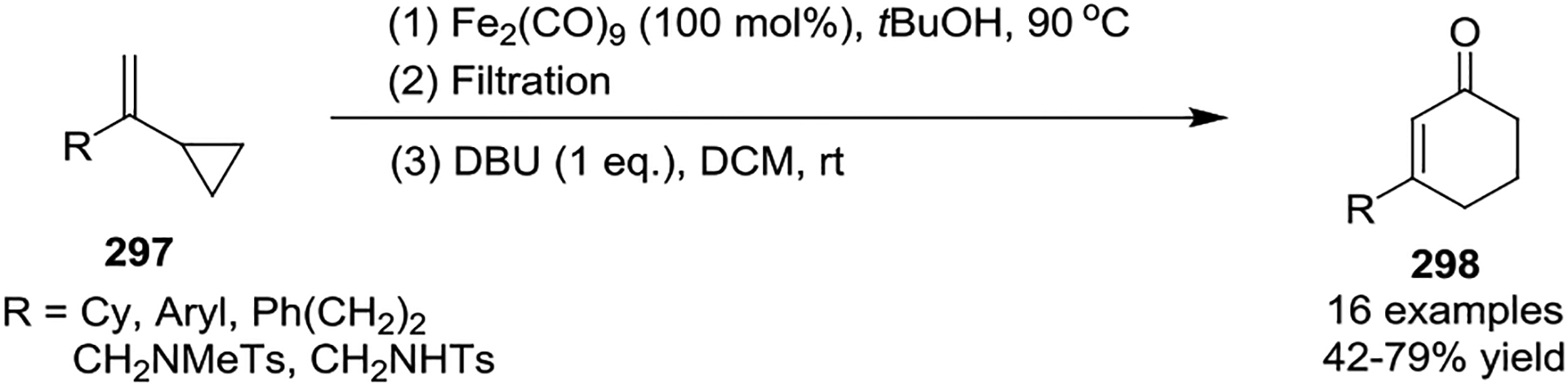
In 2016, the Yu group developed a Fe2(CO)9-mediated [5 + 1] cycloaddition of VCPs 297 to prepare α, β-cyclohexenones 298 (Scheme 51).126 The reaction tolerated aryl and aliphatic groups at the α-position of VCPs as well as bicyclic rings. The advantage of this reaction was demonstrated by the use of less expensive and safer Fe2(CO)9 compared to the volatile Fe(CO)5, which also avoided the photo-irradiation conditions.
Scheme 51.
3.2.2. Cobalt Catalysis
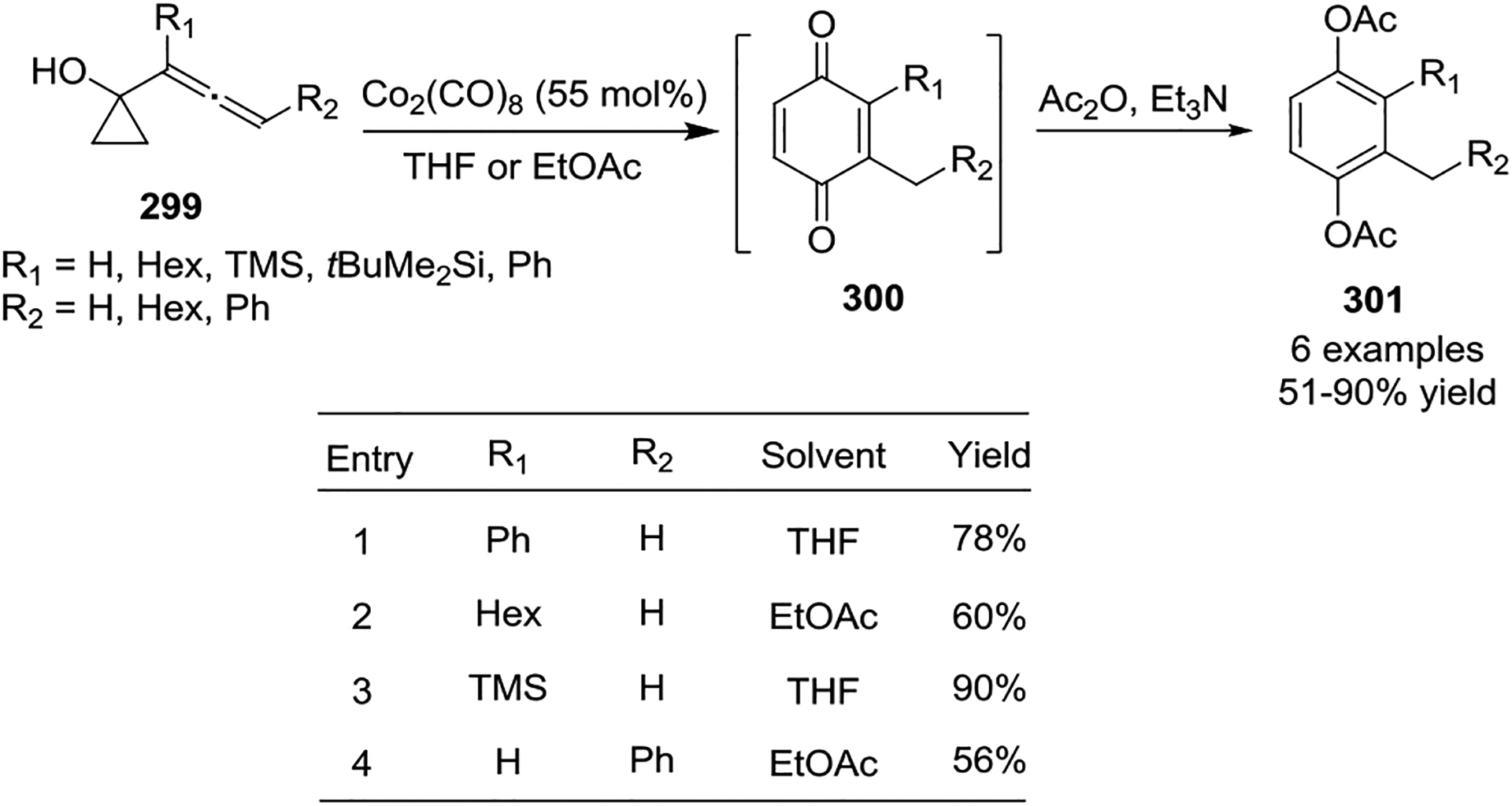
In 1997, Iwasaw and coworkers reported a Co(0)-catalyzed [5 + 1] formal cycloaddition of allenylcyclopropane 299 for the synthesis of hydroquinones 301 under very mild conditions (Scheme 52).127 Stoichiometric amounts of Co2(CO)8 were required for the cobalt-mediated [5 + 1] cycloaddition, even under an atmosphere of CO. Interestingly, the intermediate 2-cyclohexene-1,4-dione derivatives 300 can exist for a relatively long period before tautomerizing into the corresponding hydroquinones 301.
Scheme 52.
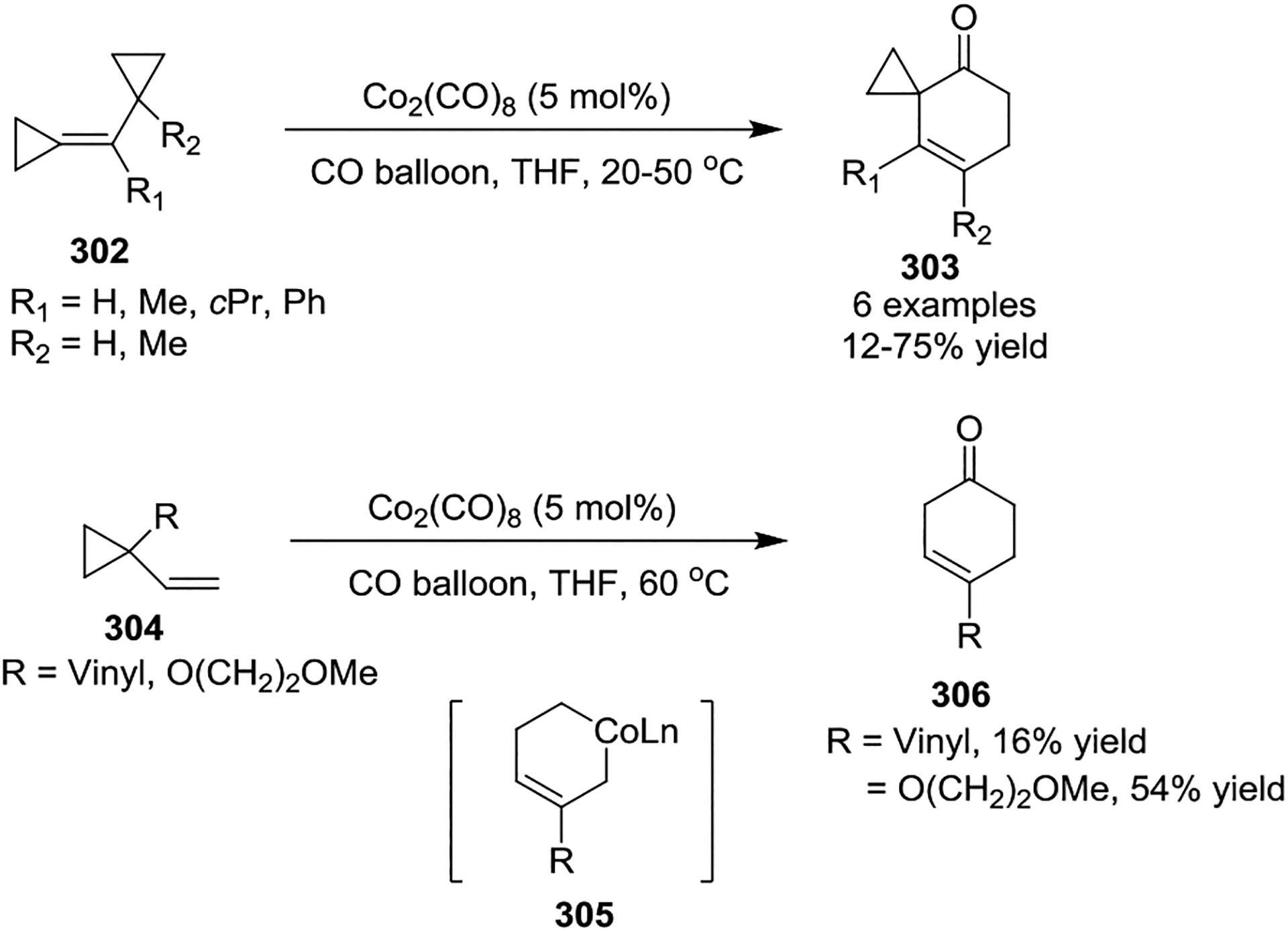
In 2005, Meijere and coworkers realized the [5 + 1] cycloaddition of non-activated VCPs 302 or 304 with catalytic amounts of Co2(CO)8 under an atmosphere of CO (Scheme 53).128 Mechanistically, cobaltcyclohexene intermediate 305 was proposed to participate in this reaction. VCPs react with Co2(CO)8 stoichiometrically to give cyclohex-3-enones in moderate to good yields under mild conditions, while catalytic amounts of the Co2(CO)8 or [Rh(CO)2Cl]2 could also mediate [5 + 1] carbonylation when a CO balloon was present at an elevated temperature (100 °C).
Scheme 53.
3.2.3. Rhodium Catalysis
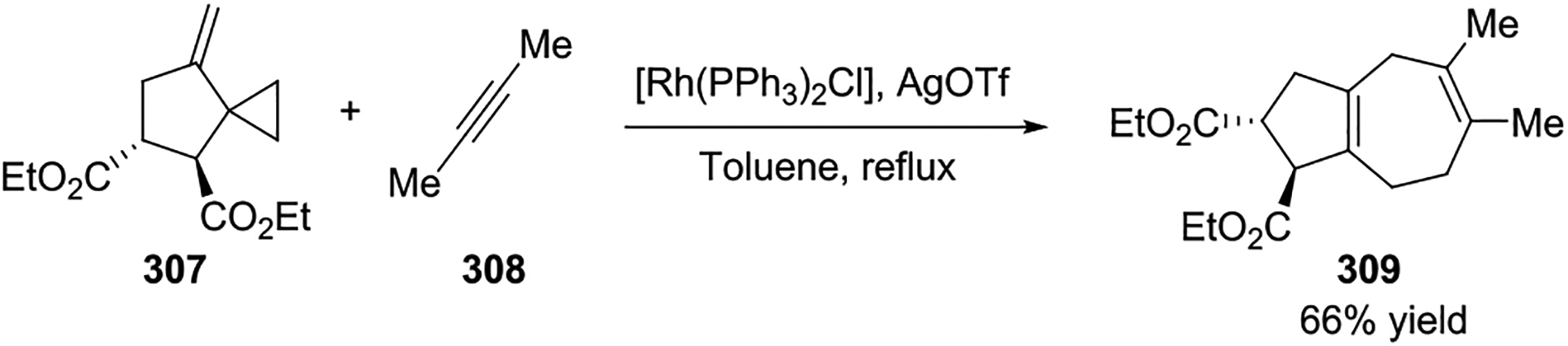
Homologous to the Diels–Alder reaction, intermolecular Rh(I)-catalyzed [5 + 2] cycloadditions of non-activated VCPs have also been realized. In 1998, the Meijere group reported one example of an intermolecular [5 + 2] cycloaddition of ethenylcyclopentene derivative 307 with 2-butyne to give the bicyclo[5.3.0]decadiene derivative 309 by using Wilkinson’s catalyst with silver triflate (Scheme 54).129 Soon after, Wender reported the first general method for this transformation between siloxycyclopropanes 400 and alkynes 401 (Scheme 55, a).130 A broad range of alkynes, even acetylene itself, were amenable to this reaction to provide cycloadducts with various functional groups in good to excellent yields by using a [Rh(CO)2Cl]2 catalyst. The siloxy substituent on the cyclopropane could not only facilitate the cycloaddition process, but it also had the special synthetic advantage of providing access to cycloheptanone derivatives 402 upon hydrolysis of silylenol ether cycloadducts. Later, a new VCP 403 was readily prepared by a concise and cost-effective procedure. This substrate was used in intermolecular [5 + 2] cycloadditions to provide high-yielding cycloadducts in minutes with minimal catalyst loading. This reaction could also be performed on a preparative scale (Scheme 55, b).131 Further studies demonstrated to ability to use simple, non-activated VCPs 406 with a [Rh(CO)2Cl]2 catalyst (Scheme 55, c).132 It is noteworthy that a single regioisomer is observed with all the di- and trisubstituted VCPs, which is consistent with the minimization of steric effects during C-C bond formation in the transition state. In 2001, a tandem [5 + 2]/[4 + 2] cycloaddition was reported to synthesize complex bicyclo[5.4.0]undecane derivatives 411 from readily available starting materials. This reaction created four new bonds and up to four new stereocenters in just one step (Scheme 55, d).133 A variety of enynes 409 and dienophiles 410 were tolerated in this study, and this three-component process could be performed on a preparative scale as well (415, 100 mmol thus far).
Scheme 54.
Scheme 55.
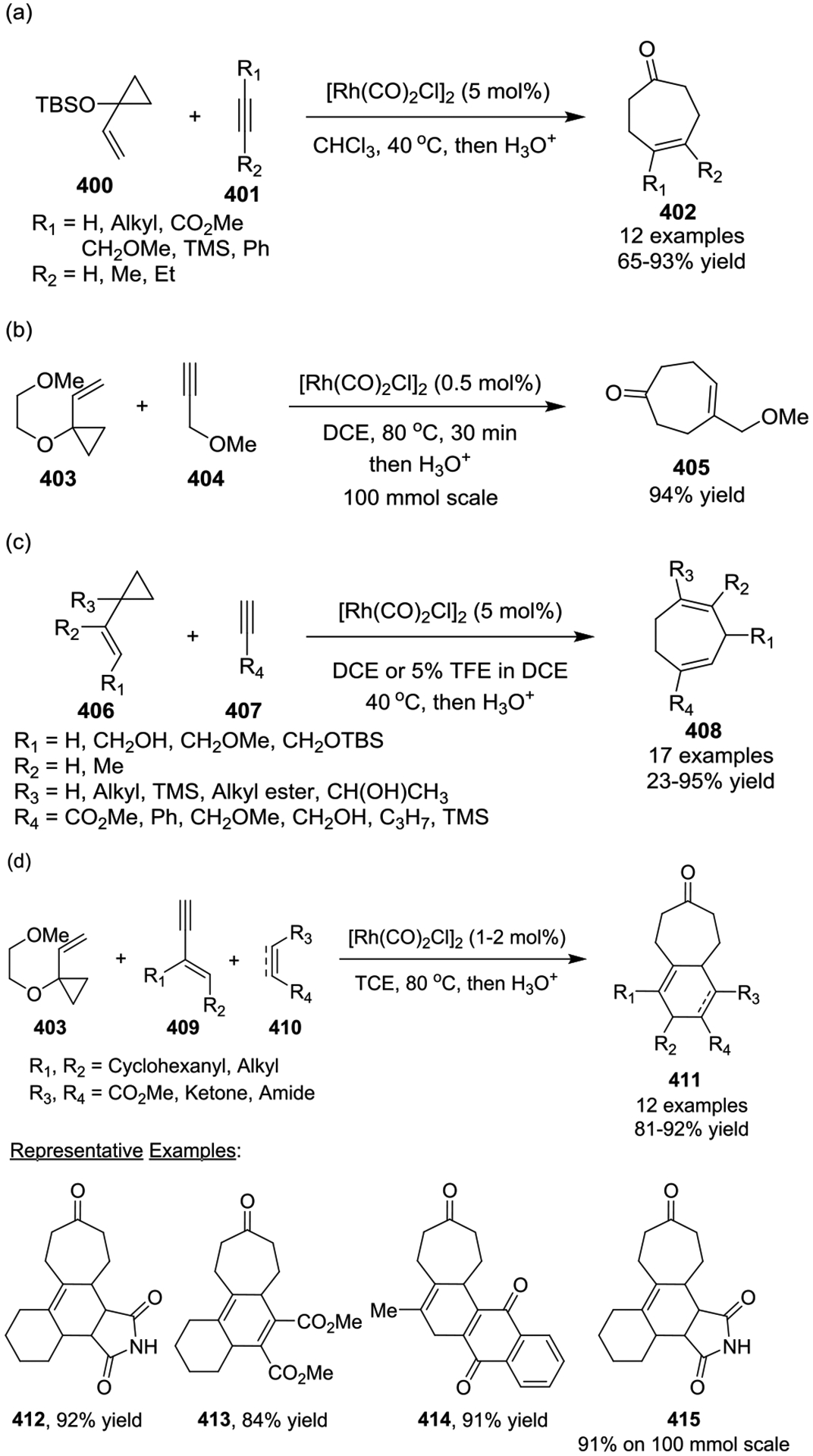
In 2002, the Wender group reported the first Rh(I)-catalyzed [5 + 2 + 1] cycloaddition reactions of VCPs with alkynes under an atmosphere of CO, which successfully utilized VCPs as the five-carbon components in this intermolecular cycloaddition process (Scheme 56, a).94 A variety of bicyclo[3.3.0]octenone cycloadducts 419 could be obtained in good to excellent yields with high or complete regioselectivities, which resulted from the transannular closure of the intermediate eight-membered rings 417. Further investigation demonstrated that alkoxy-substituted VCPs, such as 403, are especially effective in multicomponent [5 + 2 + 1] or [5 + 1 + 2 + 1] cycloadditions with alkynes (Scheme 56, a and b)134 and [5 + 2 + 1] or [5 + 2] cycloadditions with allenes (Scheme 56, c), respectively.135 The mechanism of the intermolecular [5 + 1 + 2 + 1] cycloaddition was recently investigated by the Ylijoki group.136 The study revealed the existence of two mechanistic pathways, [5 + 1 + 2 + 1] and [5 + 1 + 1 + 2], which differed by only 2 kcal/mol. The tandem process involves the generation of cyclononadiendiones 422 via VCPs, alkynes, and 2 equivalent of, followed by tautomerization, disrotatory 6π-electrocyclization to afford 424, and aromatization to yield the final hydroxydihydroindanone products 425, which have been verified as slightly energetically preferred (Scheme 56, b). Additionally, in the case of allenes (Scheme 56, c), the authors found that the allene tethered VCPs must have a pendant moiety, such as an alkyne, alkene, or cyano group. No reaction was observed with a terminally unsubstituted allenes. A methyl substituted allene, such as 428, located opposite of the coordinating group had positive effect on the reactivity and generated the [5 + 2 + 1] carbonylation product 429 in 38% yield as a single E-isomer under a CO atmosphere. A computational study later found that the low reactivity of terminally unsubstituted allenes was associated with a competing allene dimerization that could form a stable rhodium complex, which effectively poisoned the catalyst.137 With the addition of the methyl groups to the allenes, steric repulsion between the terminal substituents significantly increased the barrier of allene dimerization, while the [5 + 2] pathway remained mostly unaffected.
Scheme 56.
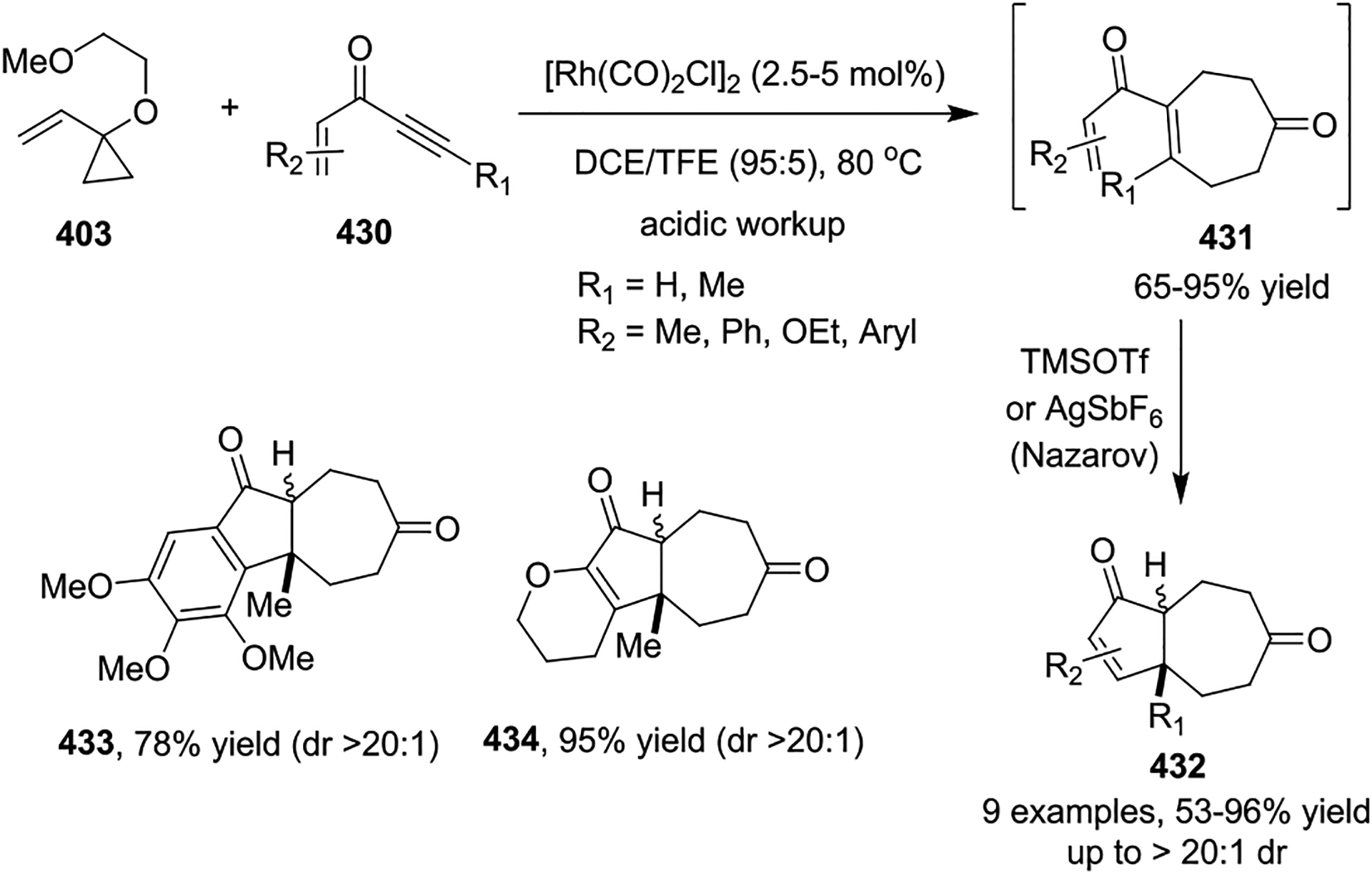
In 2010, the Wender group developed a novel, two-step strategy to synthesize bicyclo[5.3.0]decane derivatives 432 by using a Rh(I)-catalyzed [5 + 2] cycloaddition of VCPs 403 with enynones 430 and subsequent Nazarov cyclization (Scheme 57).138 The [5 + 2] cycloaddition proceeded smoothly with a variety of ene- and aryl-ynones 430 to afford the corresponding enones 432 in 65–95% yields after hydrolytic workup. The following Nazarov cyclization was catalyzed by a Lewis acid, such as TMSOTf or AgSbF6, to provide cis-fused bicyclic systems, such as 433 and 434, in good selectivity. Interestingly, in many cases, this cycloaddition/cyclization process can be carried out in a single operation without negatively affecting the yield or diastereoselectivity. The rationale for the regioselectivity of Rh(I)-catalyzed [5 + 2] cycloadditions between VCPs and alkynes have been analyzed computationally.70
Scheme 57.
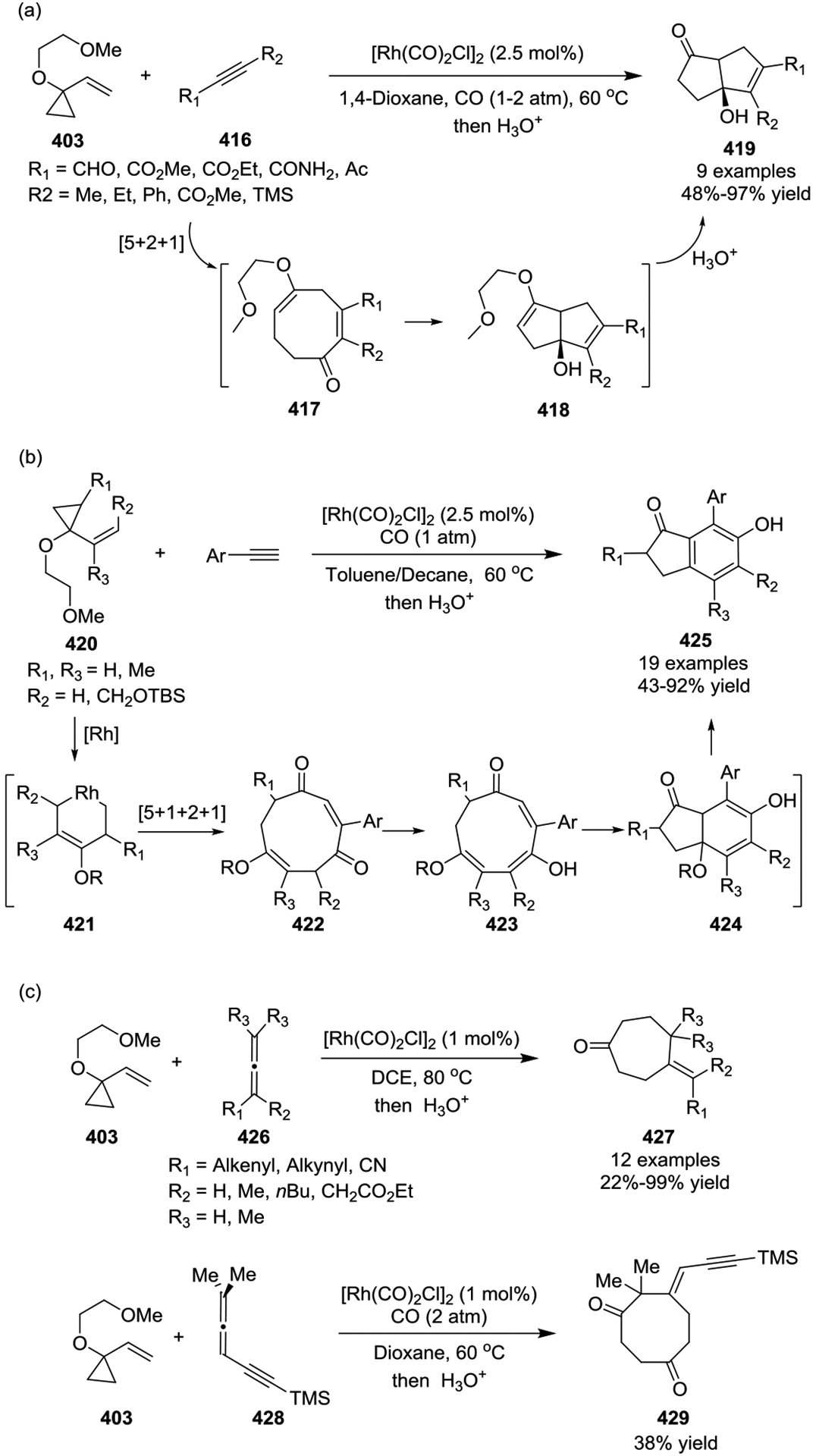
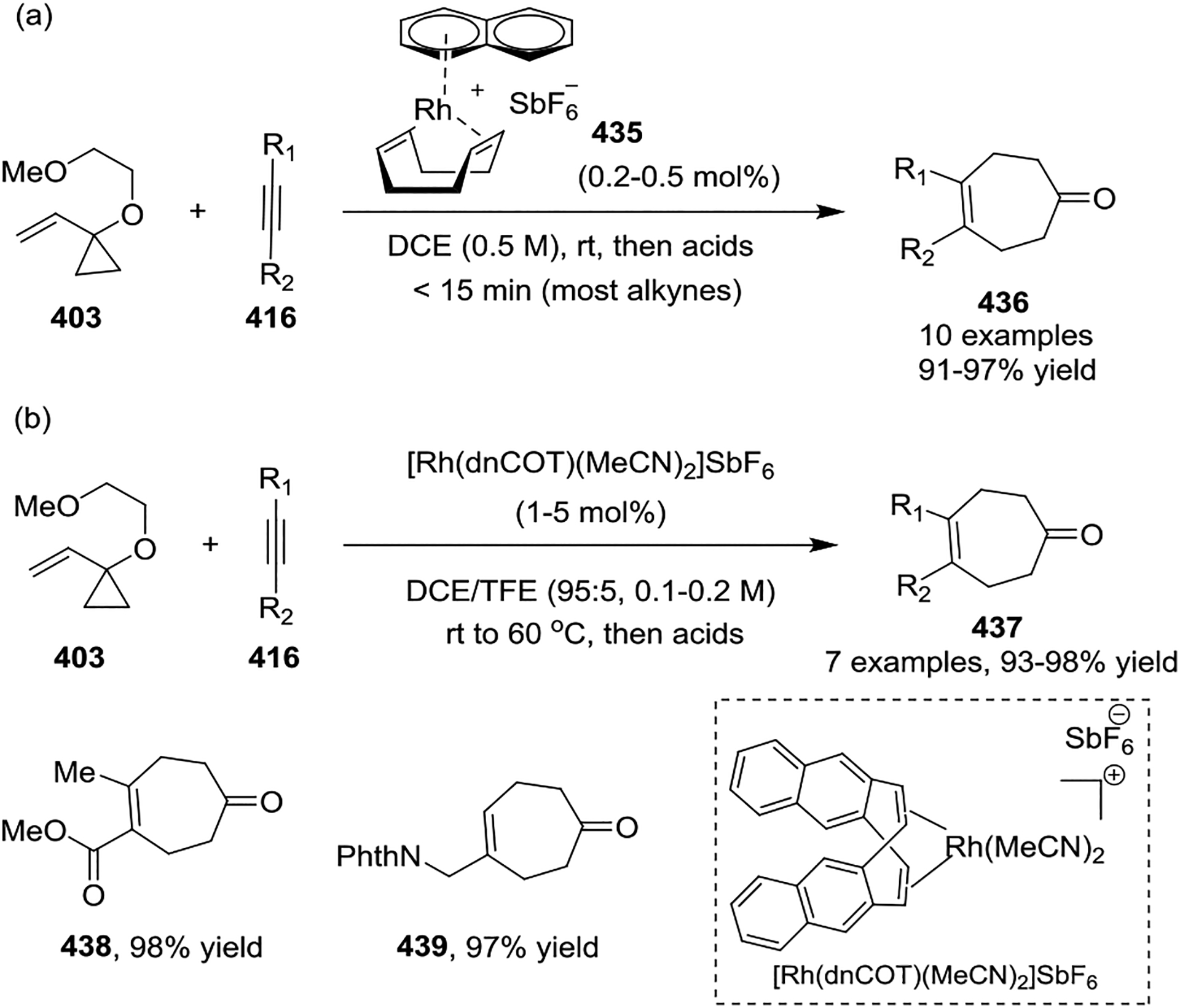
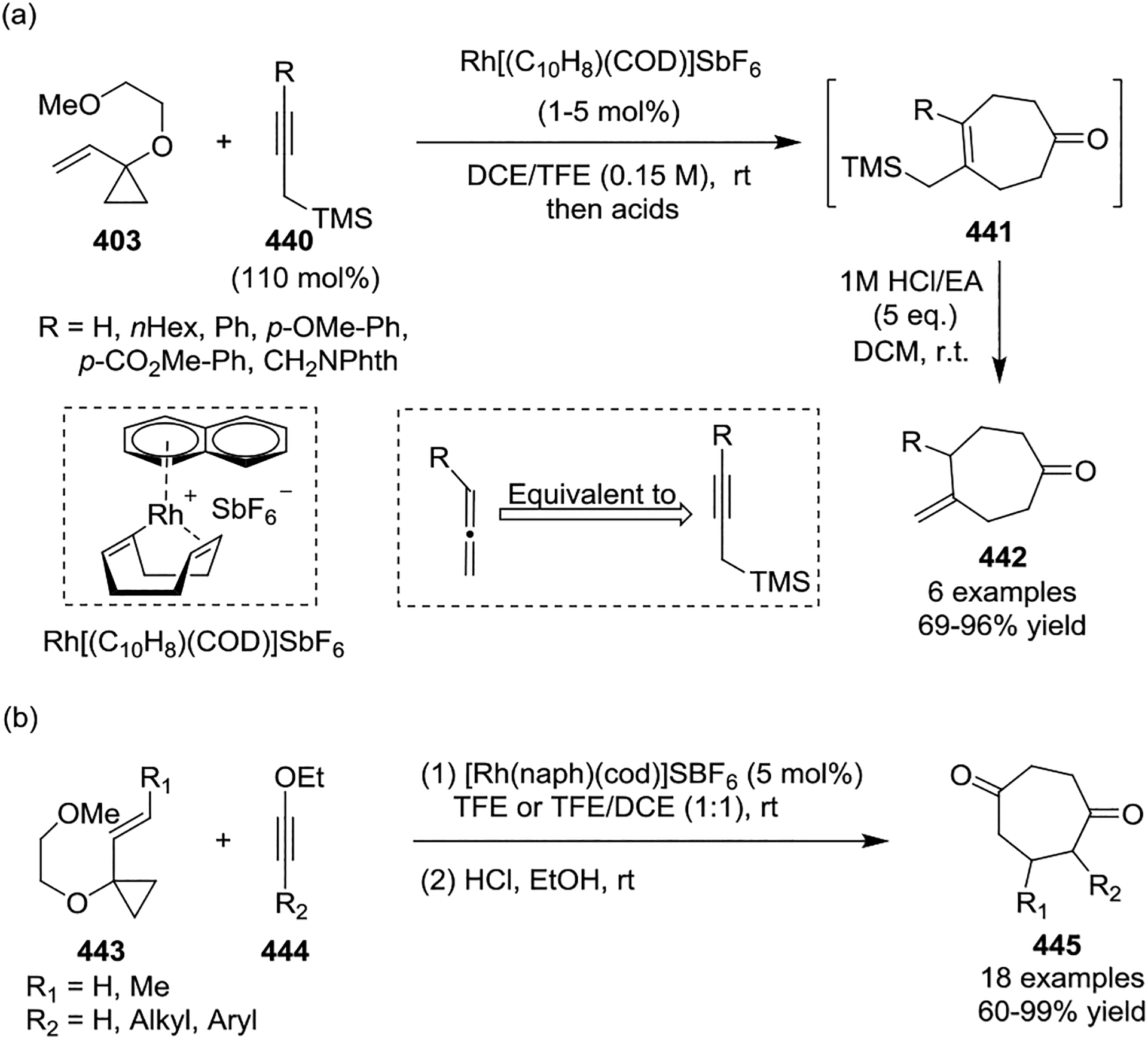
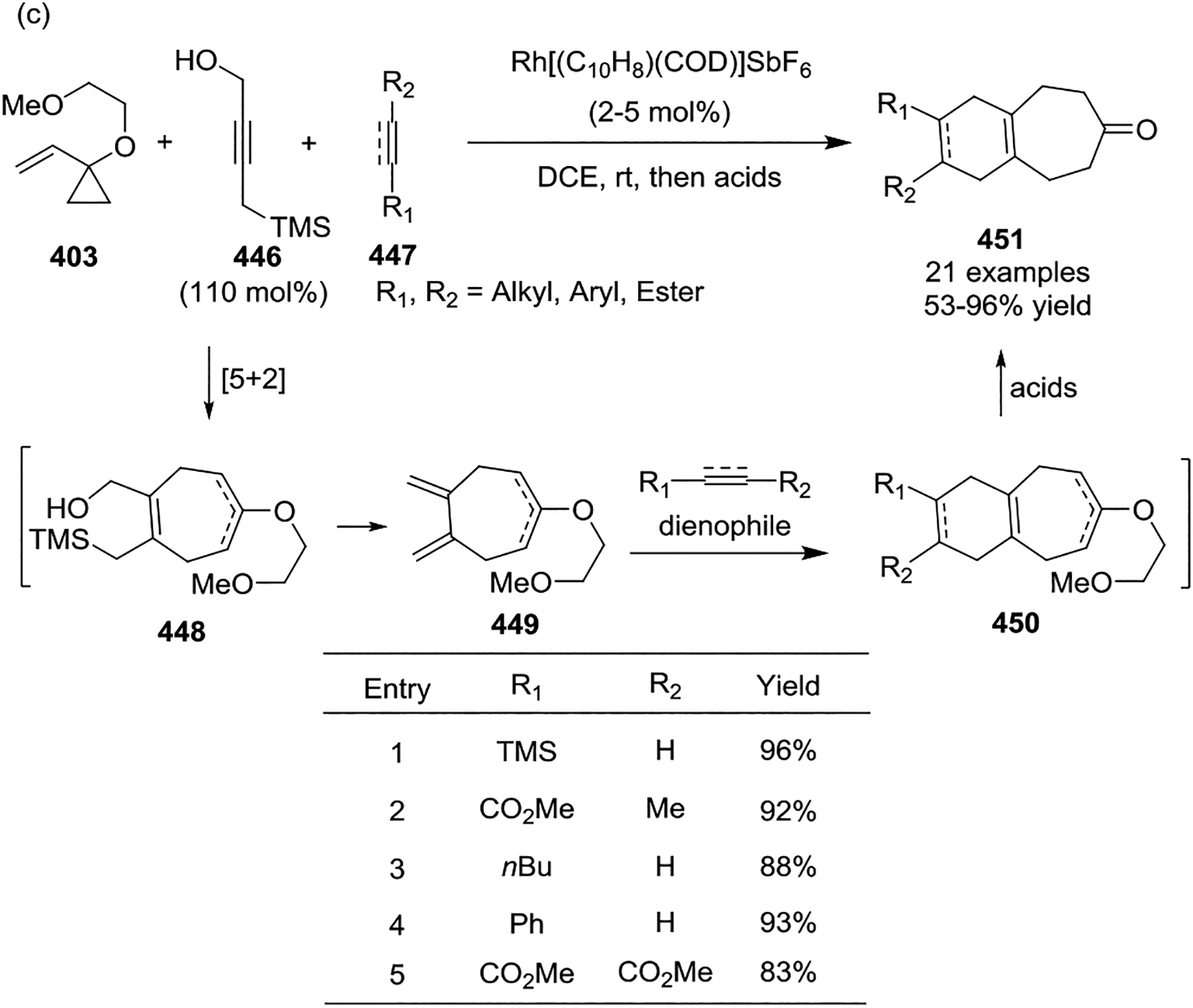
In most cases, [Rh(CO)2Cl]2 served as the sole catalyst for the intermolecular [5 + 2] cycloadditions of non-activated VCPs with limited efficiency. This led to the development of a cationic rhodium(I) complex [{(C10H8)Rh(cod)}SbF6] 435 by the Wender group, which was found to be a highly efficient catalyst for the intermolecular [5 + 2] cycloaddition of 1-alkoxy-VCPs 403 with alkynes 416, providing cycloheptenones 436 at room temperature in all cases (Scheme 58, a).139–140 Subsequent rational design of cationic complex [Rh(dnCOT)(MeCN)2]SbF6 proved to be a highly efficient catalyst for a variety of VCPs-based cycloadditions, such as providing [5 + 2] cycloadducts 437 in high yields with a wide of functional group tolerance. Additionally, this transformation often occurred in minutes at room temperature (Scheme 58, b).141–142 Later on, Wender also disclosed that propargyltrimethylsilanes 440 function as safe, easily handled synthetic equivalents of allenes in cationic rhodium-catalyzed [5 + 2] cycloadditions (Scheme 59, a).143 This reaction proceeded via a two-step or one-pot procedure where a [5 + 2] cycloaddition of propargyltrimethylsilanes and VCPs provided intermediates 441 and was followed by protodesilylation under acidic condition to provide a new, facile, and practical method for accessing formal allene [5 + 2] cycloadducts 442. In 2017, the Wender group showed that ynol ether 444 can act as a ketene equivalent, providing a highly efficient, direct route to enediones 445 at room temperature (Scheme 59, b).144 High regioselectivity resulted from the use of methyl substituted VCPs 443. Later, Wender and coworkers extended the multicomponent process involving a novel 4-(trimethylsilyl)but-2-yn-1-ol 446 as a 1,2,3-butatriene equivalent in a [5 + 2] and [4 + 2] cycloaddition sequence (Scheme 59, c).145 Reagent 446 functioned as a two-carbon alkyne component in the rhodium-catalyzed [5 + 2] cycloaddition with an alkoxy-substituted VCP 403 to produce an intermediate 448. Compound 448 then undergoes a rapid 1,4-Peterson elimination of the TMS and OH groups to afford the diene intermediate 449, which is readily intercepted in either a rhodium-catalyzed or thermal [4 + 2] cycloaddition. After hydrolysis, this generates bicyclic or tricyclic ring systems 451 in good yields with high chemoselectivity. Both activated and non-activated alkyne or alkene dienophiles 447 can be used in this cascade cycloaddition, which allows flexible access to highly diversified polycycles.
Scheme 58.
Scheme 59.
In 2012, the Yu group reported a cationic Rh(I)-catalyzed carbonylative [5 + 1] cycloaddition of VCPs 452 and CO (0.2 atm), affording either β,γ-cyclohexenones 453 as the major products or α,β-cyclohexenones 454 exclusively, depending on the reaction conditions (Scheme 60, a).146 In this study, most β,γ-cyclohexenones could isomerize to the α,β- cyclohexenones quantitatively with DBU at room temperature. In 2016, Yu and colleagues also outlined a similar carbonylative [5 + 1] strategy that used allenylcyclopropanes 456 to generate of 2-methylidene-3,4-cyclohexenones 457 (Scheme 60, b).147
Scheme 60.
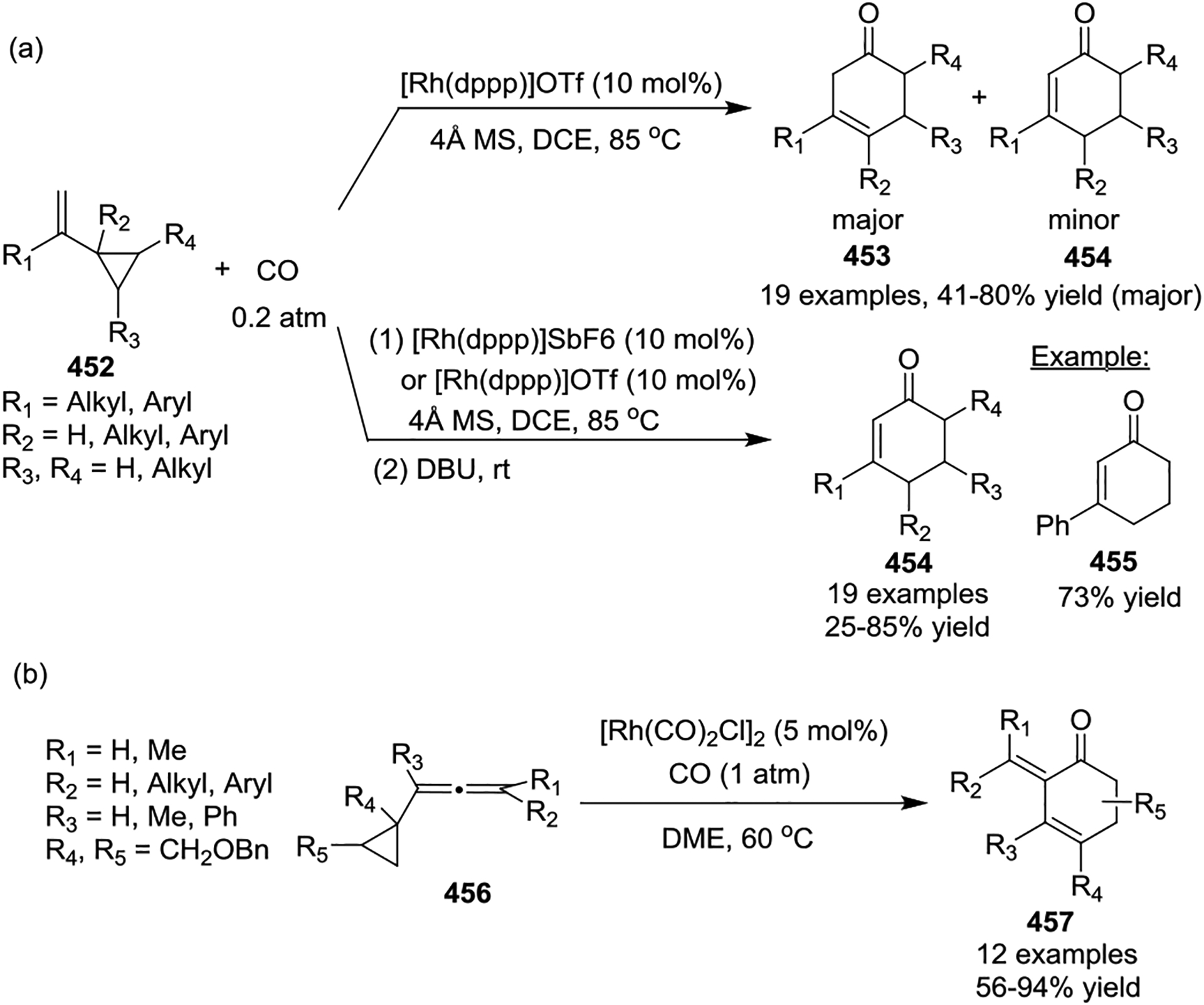
In 2011, the Yu group also developed a new seven-carbon building block from buta-1,3-dienylcyclopropanes (BDCPs) for the synthesis of a Rh(I)-catalyzed [7 + 1] cycloaddition products 462 and 463 under the catalysis of [Rh(CO)2Cl]2 and CO (Scheme 61).148 In the presence of a Rh(I) complex, BDCPs coordinate to Rh, followed by the C-C bond cleavage of cyclopropanes to give intermediates 459 or 460. Reductive elimination from 460 gives a seven-membered carbocyclic product, which is expected to be difficult since this step involves the formation of a C(sp3)-C(sp3) bond.66 Therefore, the migratory insertion of a CO unit is preferred leading to complex 461, and the subsequent reductive elimination and formation of the energetically favored C(sp2)–C(sp3) bond yields eight-membered rings 462 and 463. This reaction provides an efficient method to access the synthetically challenging eight-membered carbocyclic skeletons, which complements the known transition-metal-catalyzed cycloadditions.
Scheme 61.
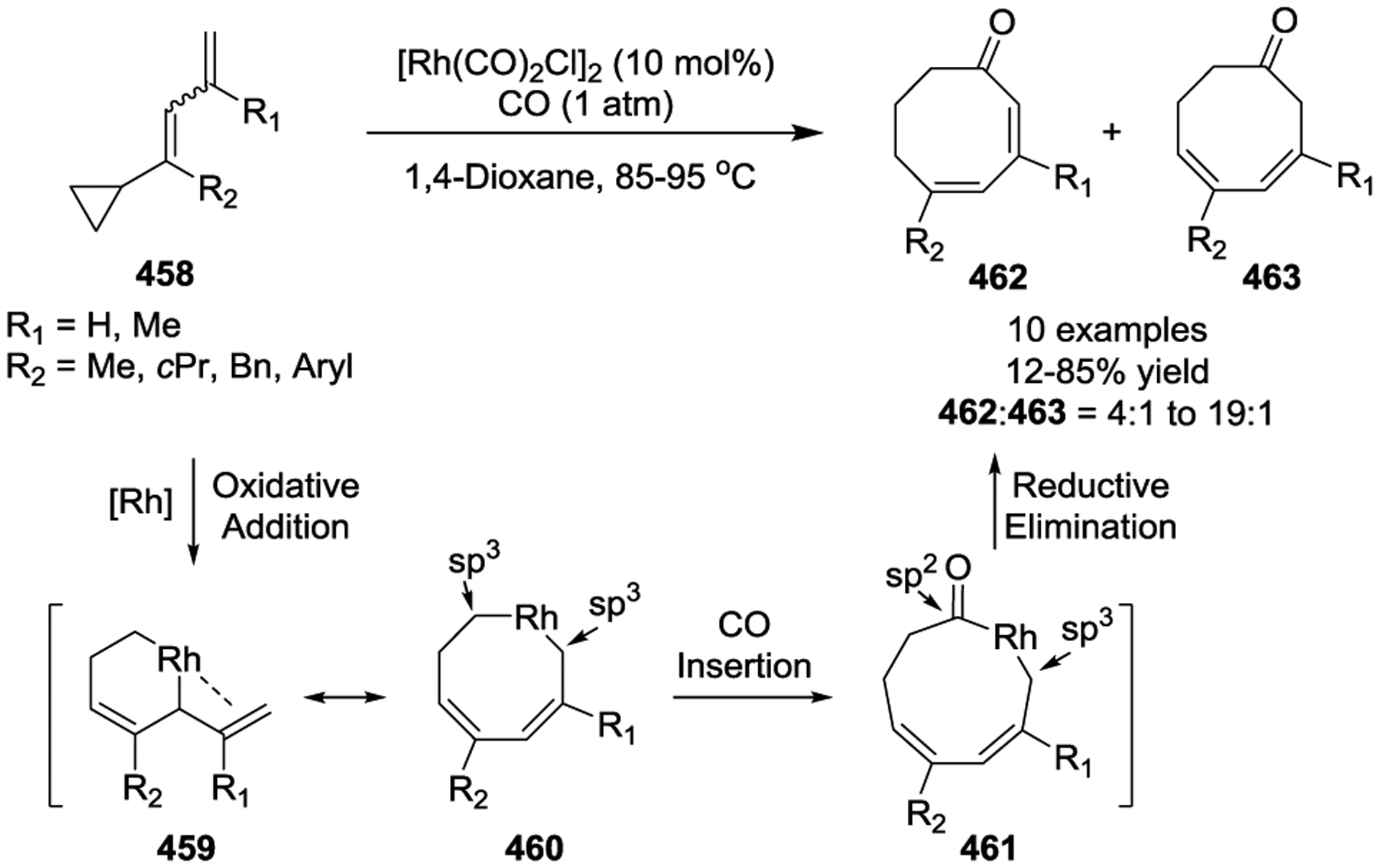
In 2015, a new Rh(I)-catalyzed [7 + 1] cycloaddition between intermediates 465 and CO was developed to produce benzocyclooctenones 468 (Scheme 62).149 In this reaction, a thermal electrocyclic ring opening of the four-membered rings in cyclopropyl-benzocyclobutenes 464 (CP-BCBs) generates the seven-carbon synthons 465. A Rh-catalyzed oxidative addition then converts 465 into eight-membered metallacycles 466. CO migratory insertion and subsequent reductive elimination gives the benzocyclooctenones 468 in good yields. Two C-C bond cleavages were involved in this tandem process: one is through the thermal opening of the four-membered ring in benzocyclobutenes, and the other through a Rh(I)-catalyzed cyclopropane opening. Particularly impressive was the fact that substrates 464, which contained five- and six-membered ring fused cyclopropanes, were suitable substrates to give benzo/[7 + 1] cycloadducts in good yields, respectively. Unfortunately, this method did not tolerate di-substitutions on the cyclobutene ring due to unfavorable steric interactions.
Scheme 62.
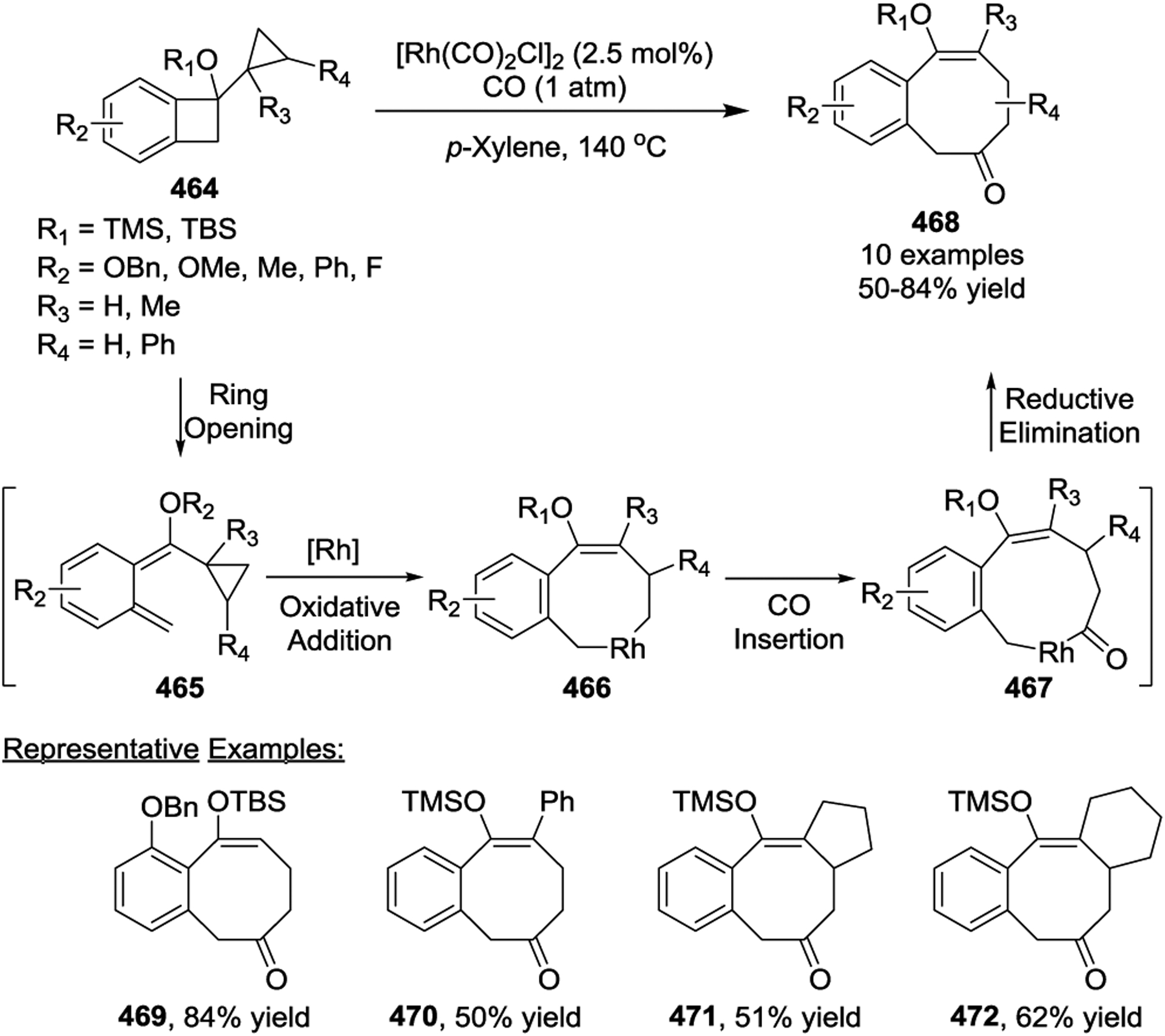
A two-step sequence that includes an enyne cycloisomerization/[5 + 1] cycloaddition to form tetrahydroisoquinolinones 475 was developed in 2017 by the Yu group.150 The Au(I)-catalyzed cycloisomerization of linear enyne-ene 473 was used to generate the six-membered-ring-fused VCP 474. Then a Rh catalyzed [5 + 1] cycloaddition under 0.2 atm of CO occurred to afford the desired tetrahydroisoquinolinone framework 475 in acceptable yields with a quaternary center at the R3 position (Scheme 63). This two-step reaction could also be carried out in one-pot without isolating the requisite bicyclic-VCP 474, which resulted in the formal [3 + 2 + 1] cycloaddition.
Scheme 63.
3.2.4. Iridium Catalysis
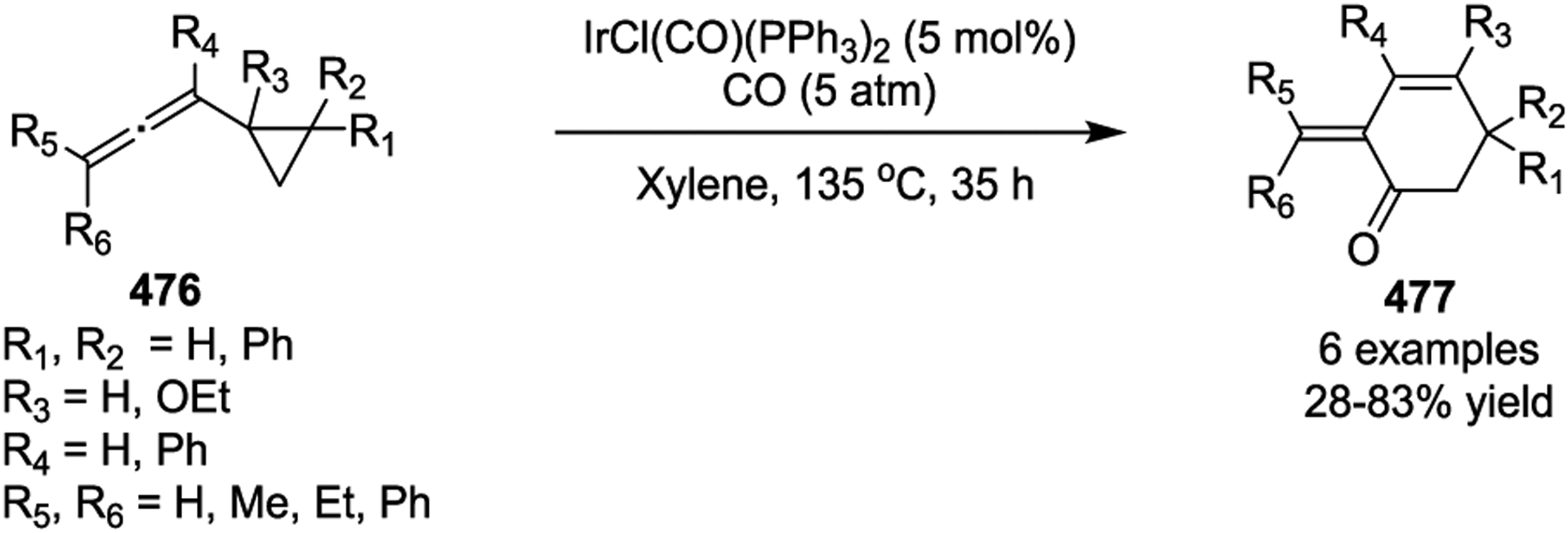
In 1998, Murakami and Ito reported an IrCl(CO)(PPh3)2-catalyzed [5 + 1] cycloaddition of allenylcyclopropanes 476 to incorporate a five-carbon unit into the resulting six-membered rings 477 under 5 atm of CO (Scheme 64).151 When the allene was switched with alkene adjacent to the cyclopropyl group, neither formation of a [5 + 1] cycloadduct nor isomerization to cyclopentene was observed under the standard conditions. This result demonstrated the importance of the substituted allene functionality for the initial C-C bond cleavage of cyclopropane with an iridium catalyst. In 2014, the Strand group reported the first iridium-catalyzed inter- and intramolecular [5 + 2] cycloadditions of non-activated VCPs (Scheme 65).152 [Ir(cod)Cl]2 was found to be the optimal catalyst for the intermolecular [5 + 2] cycloaddition (Scheme 65, a), while [Ir(dbcot)Cl]2 worked best for the intramolecular variant (Scheme 65, b). This transformation afforded various seven-membered cycloadducts 482, 483 or 485 with a range of functional groups. DFT calculations suggested that the mechanism of the Ir(I)-catalyzed intermolecular formal cycloaddition proceeds through similar transition states as rhodium catalysis, which involves oxidative addition of Ir(I) to VCPs, migratory insertion of alkynes to form metallocycles 481, and subsequent reductive elimination. The Ir-mediated process was shown to have lower energetic barriers, enabling substantially faster catalysis under milder reaction conditions compared to its rhodium counterpart.
Scheme 64.
Scheme 65.
3.3. Applications
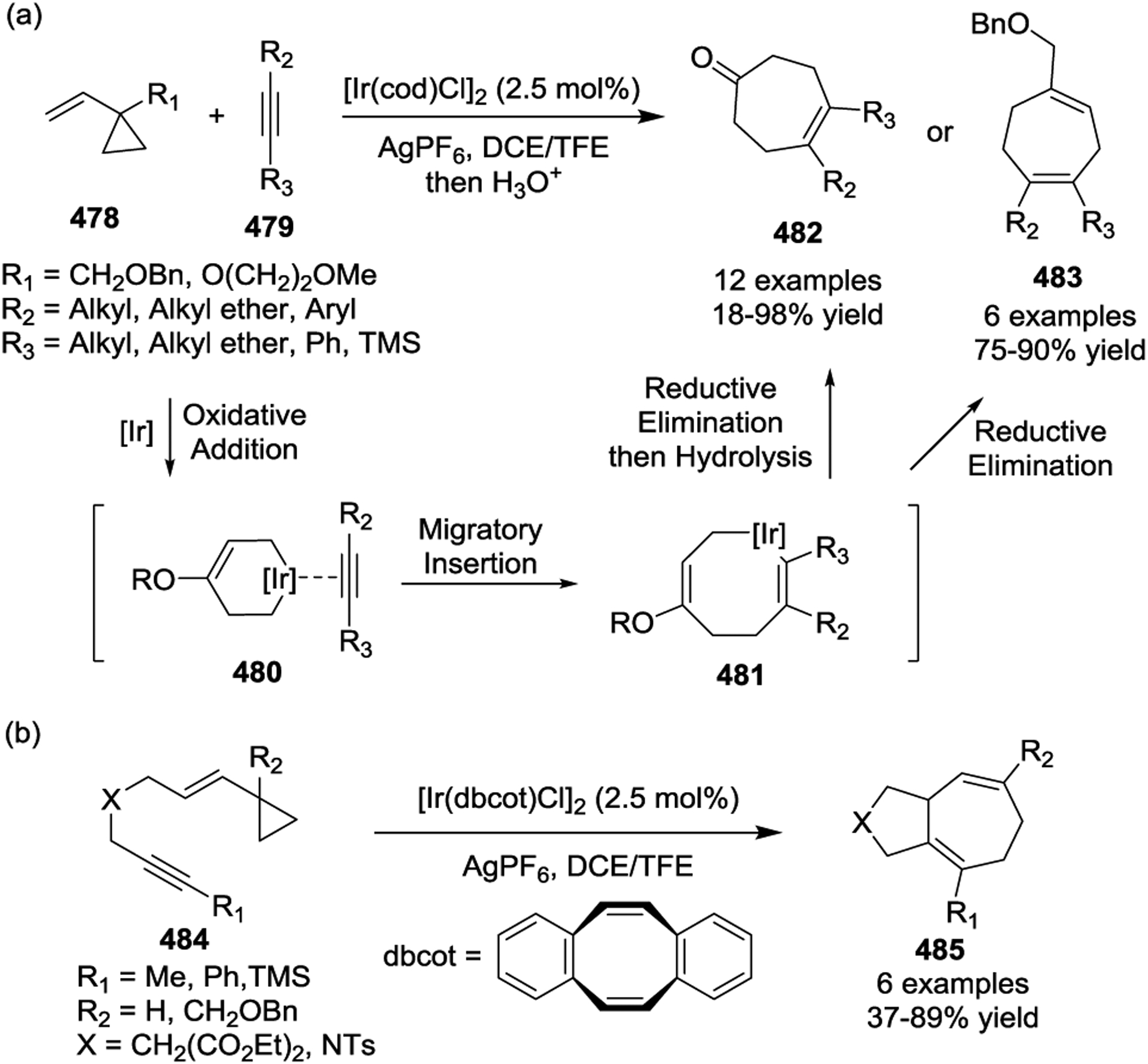
Due to the prevalence of seven-membered rings in a wide range of natural products and biologically important active agents, [5 + 2] cycloadditions have been a key step in the total synthesis of many natural products. In 1999, the Wender group described the asymmetric synthesis of (+)-Dictamnol based on the Rh-catalyzed intramolecular [5+ 2] cycloaddition of allenyl-vinylcyclopropane 486 (Scheme 66).153 Later, a similar strategy was applied to the concise asymmetric total synthesis of (+)-Aphanamol I (Scheme 67).154 The [Rh(CO)2Cl]2-catalyzed [5 + 2] cycloaddition of precursor 488 proceeds exceptionally well, even at 0.5 mol % catalyst loading, to provide the desired bicyclo[5.3.0]decane cycloadduct 489 in 93% yield with complete chemo-, endo-/exo-, and diastereoselectivity. Inspired by Wender’s work with diastereoselective Rh(I)-catalyzed [5 + 2] cycloadditions,78 the Martin group reported the first enantioselective syntheses of the two representative tremulane sesquiterpenes Tremulenolide A and Tremulenediol A (Scheme 68).155 The precursor 490, which was generated from an enantioselective Rh(II)-catalyzed intramolecular cyclopropanation followed by a regioselective allylic alkylation, underwent facile intramolecular [5 + 2] cycloaddition upon heating in the presence of [Rh(CO)2Cl]2 to give the key intermediate 491 as the sole isolated product in an 85% yield.
Scheme 66.
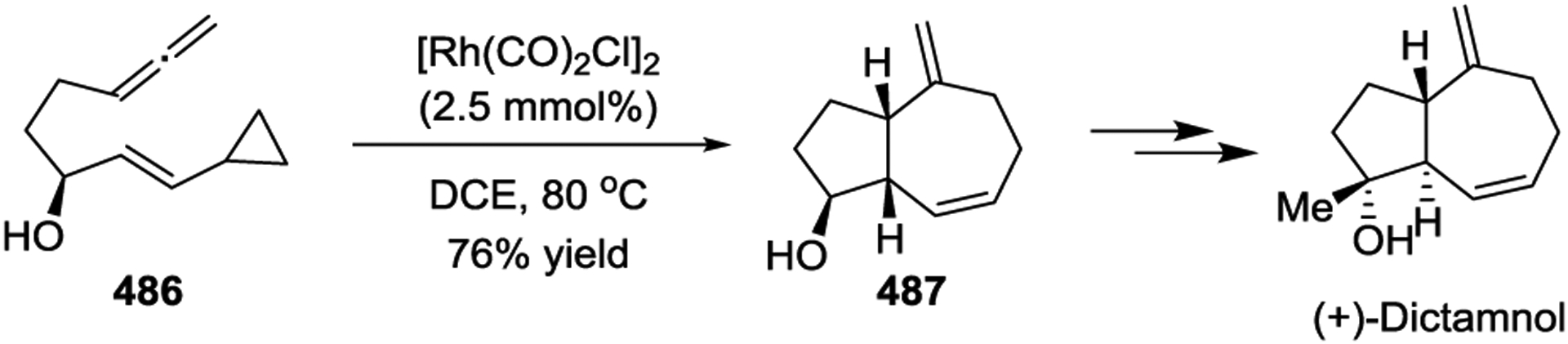
Scheme 67.
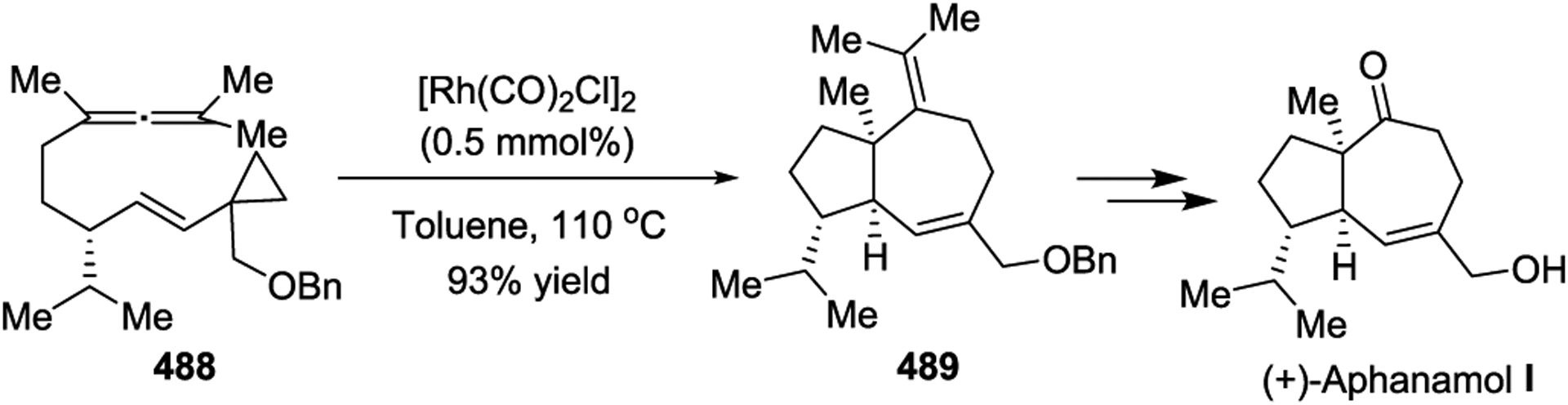
Scheme 68.
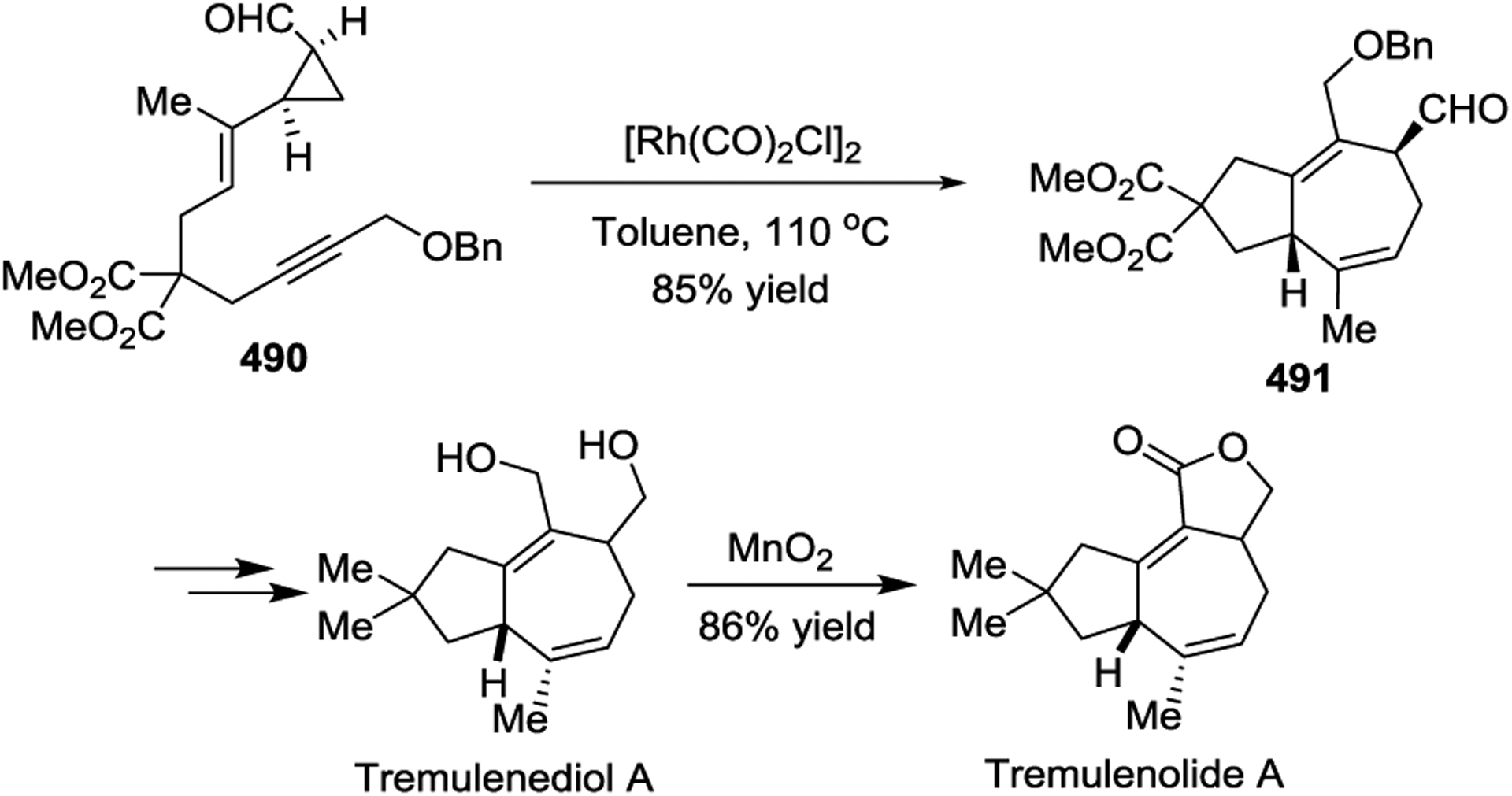
The efficiency of the [5 + 2 + 1] reaction/ aldol reaction strategy was demonstrated by the concise synthesis of four sesquiterpene natural products (Scheme 69, a, b and c), including (±)-Pentalenene,156 (+)-Hirsutene, (±)-1-Desoxyhypnophilin,95 and (±)-Hirsutic acid C,157 which contained linear or branched triquinane skeletons. Furthermore, the Rh(I)-catalyzed [5 + 2 + 1] cycloaddition of VCP 499 to build its 5/8-bicyclic carbon framework 500 was the key step of Yu’s enantioselective total synthesis of (+)-Asteriscanolide (Scheme 69, d).158–159
Scheme 69.
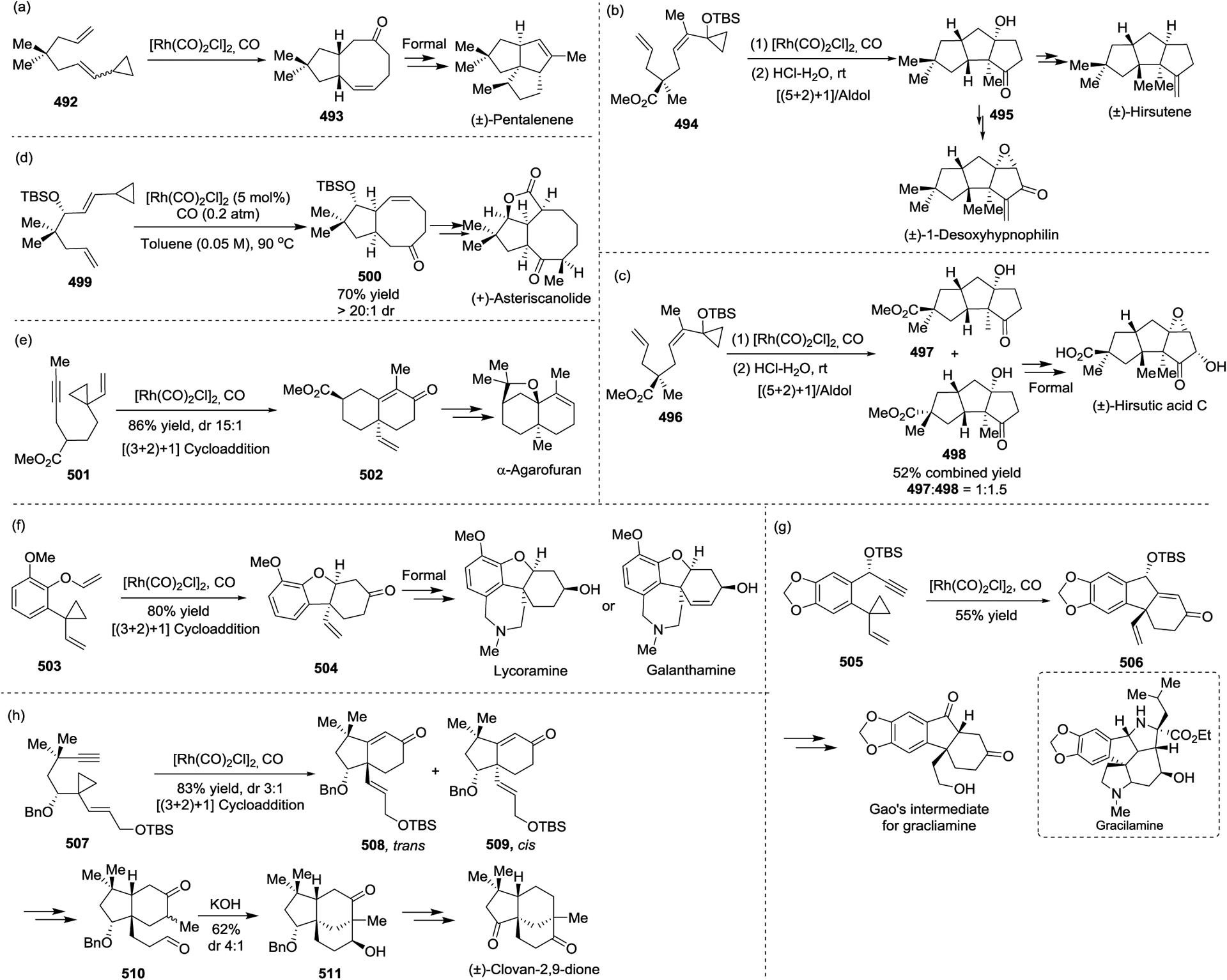
Demonstrating the importance of the [(3 + 2) + 1] reaction of 1-yne-VCP 501 and CO, in 2010 the Yu group realized the total synthesis of a furanoid sesquiterpene natural product α-Agarofuran (Scheme 69, e).101 The key [(3 + 2) + 1] reaction proceeded very well under the optimized conditions, affording bicyclic cyclohexenone 502 in 86% yield with good diastereoselectivity (trans : cis = 15 : 1). A similar strategy has also been used to build the cis-hydrodibenzofuran skeleton 504 and the A–B–C core structure of gracilamine, which was applied to the formal synthesis of (±)-Galanthamine and (±)-Lycoramine (Scheme 69, f)160 and the formal synthesis of (±)-Gracilamine (Scheme 69, g).161 Recently, the first asymmetric total synthesis of (−)-Clovan-2,9-dione was accomplished by using a Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition of 1-yne-VCP and CO, followed by intramolecular aldol reaction as the key steps (Scheme 69, h).162 The [(3 + 2) + 1] cycloaddition of 507 and CO proceeded smoothly under 0.2 atm CO atmosphere to afford the desired products (−)-trans-508 and (−)-cis-509 in an 83% combined yield with a diastereomeric ratio of approximately 3:1, which provided a new way to construct the Clovane skeletons.
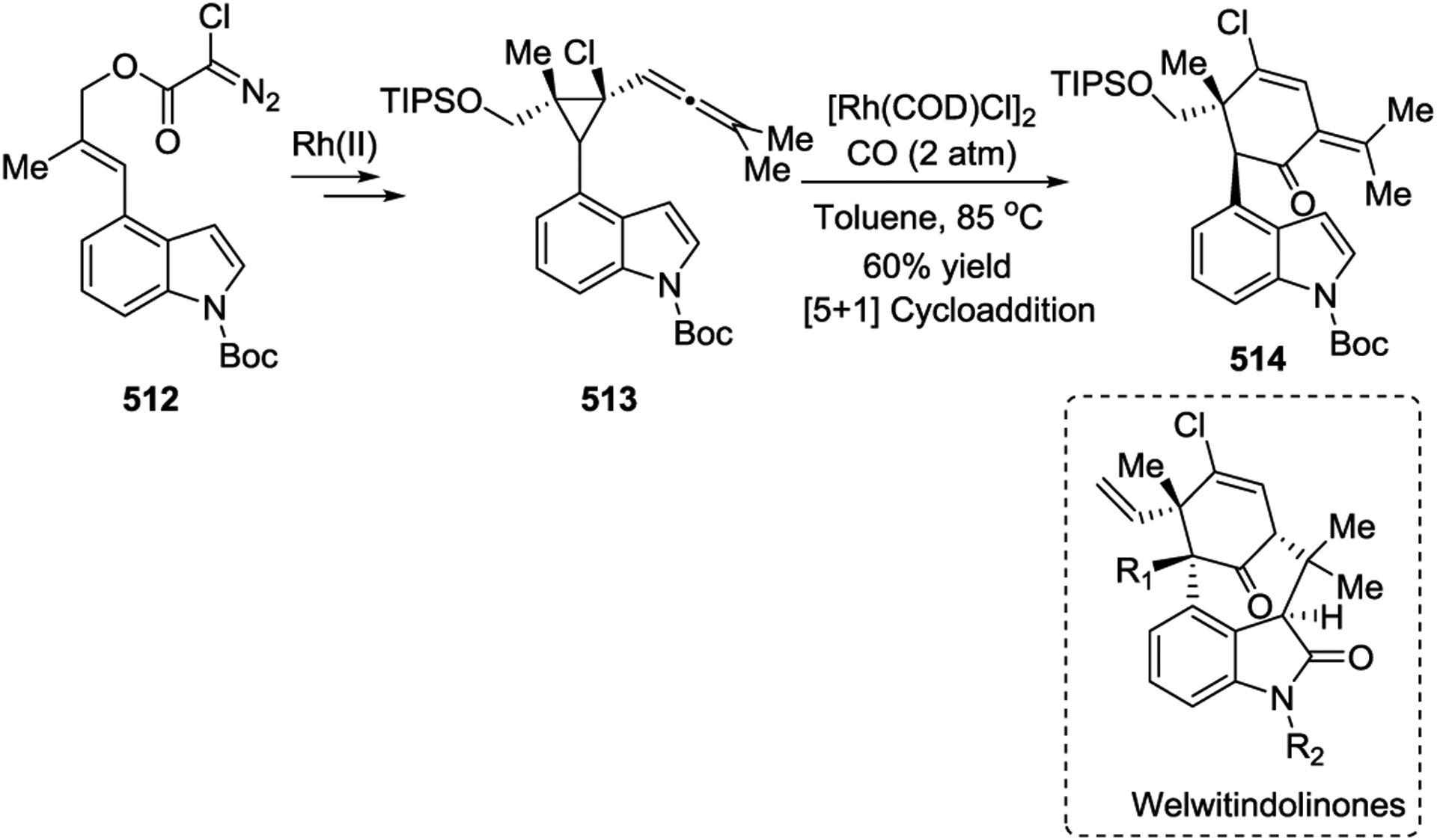
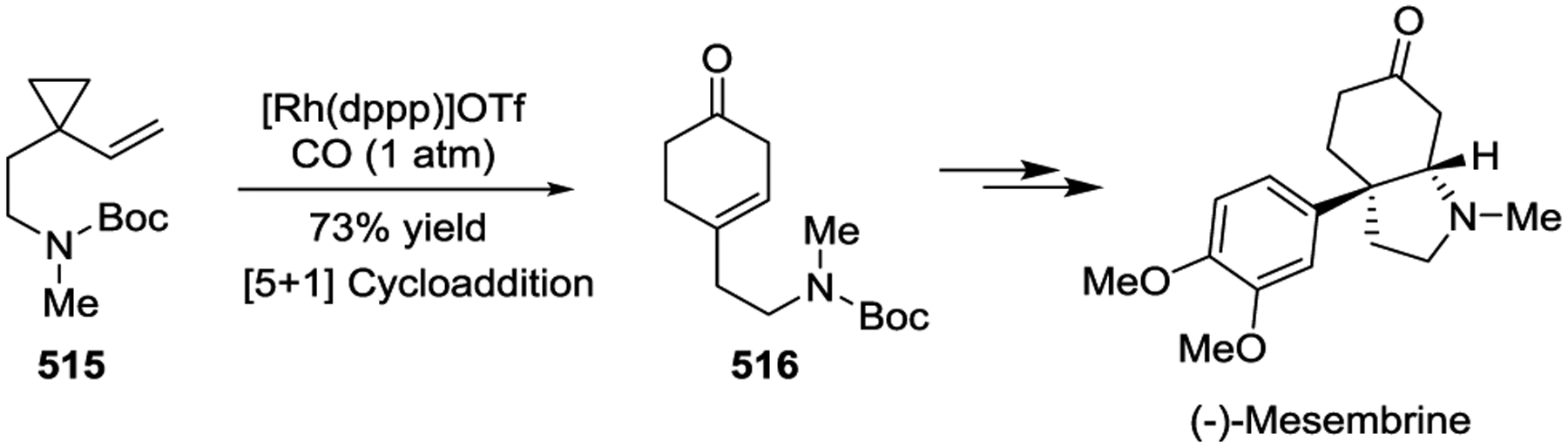
In 2012, the Tang group developed an efficient strategy to synthesize the highly functionalized cyclohexenone core 514 of Welwitindolinones by the Rh(I)-catalyzed [5 + 1] cycloaddition of an allenylcyclopropane 513 with 2 atm of CO in the presence of [Rh(COD)Cl]2 (Scheme 70).163 In 2016, the Yu group reported a concise asymmetric total synthesis of (−)-Mesembrine in four steps from the N-Boc protected VCP 515, with an overall yield of 18% (Scheme 71).164
Scheme 70.
Scheme 71.
In 2007, the Trost group reported the first application of a Ru(II)-catalyzed [5+2] cycloaddition in the total synthesis of (+)-Frondosin A (Scheme 72, a).165 The reaction between an enantioenriched cyclopropyl enyne 517 was employed as the key step to construct the bicyclo[5.3.0] ring system 518 in 80% yield with high regio- and diastereoselectivity. Later, the Ru-catalyzed [5 + 2] intramolecular cycloaddition reaction of an alkyne tethered VCP 519 was applied to the construction of the (−)-Pseudolaric Acid B core 520 (Scheme 72, b).166
Scheme 72.
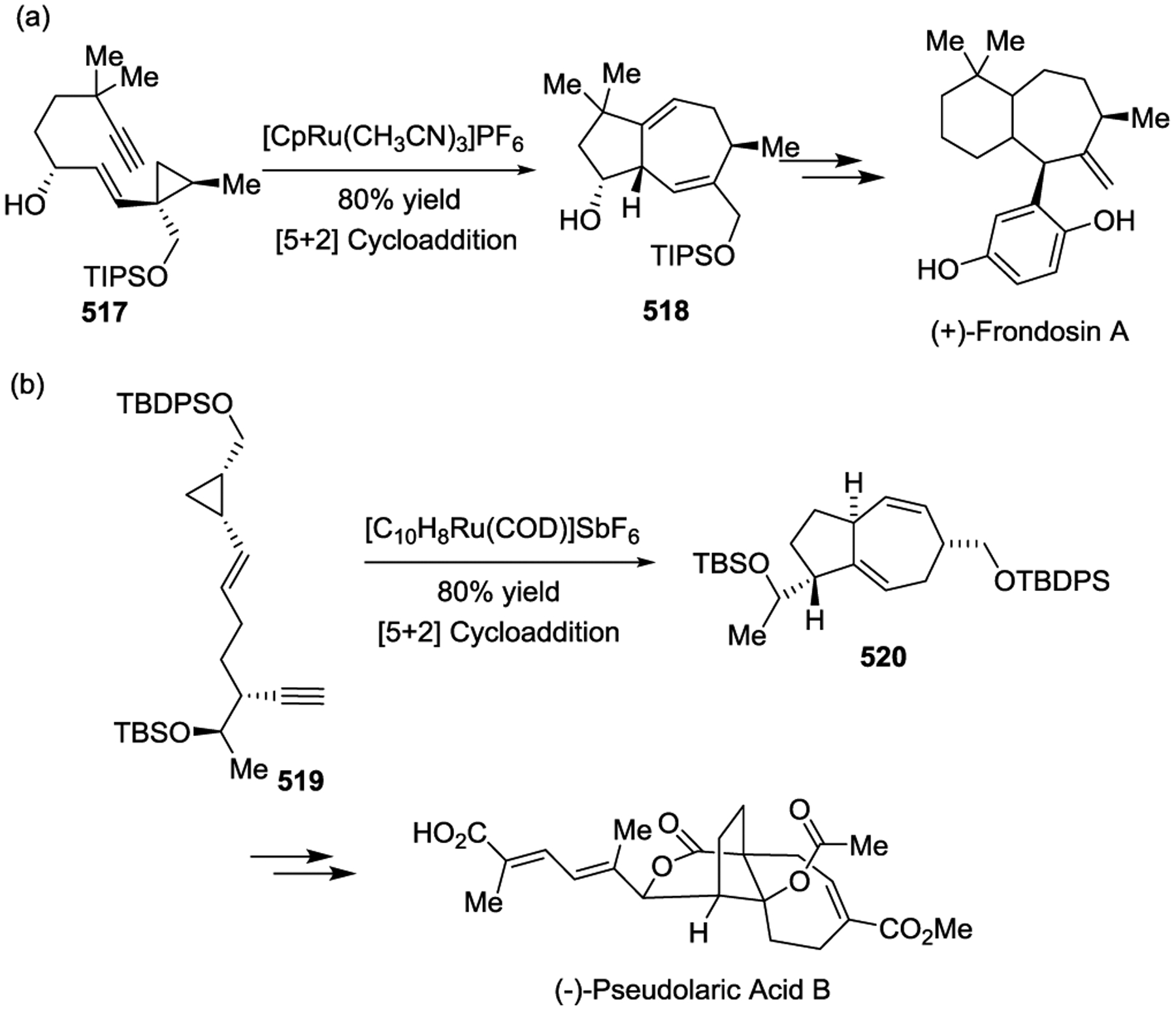
The Taber group synthesized several natural products by using Fe(CO)5 catalyzed [5 + 1] cycloadditions of VCPs with CO under irradiation (Scheme 73). For instance, the alkenyl cyclopropane 521 underwent smooth ring expansion in the presence of Fe(CO)5 under a CO atmosphere, which generated the sesquiterpene (−)-Delobabone with control of both relative and absolute configuration.167 The enantioselective synthesis of (+)-Coronafacic acid was also achieved using Fe-mediated cyclocarbonylation of 522 as the key step.168
Scheme 73.
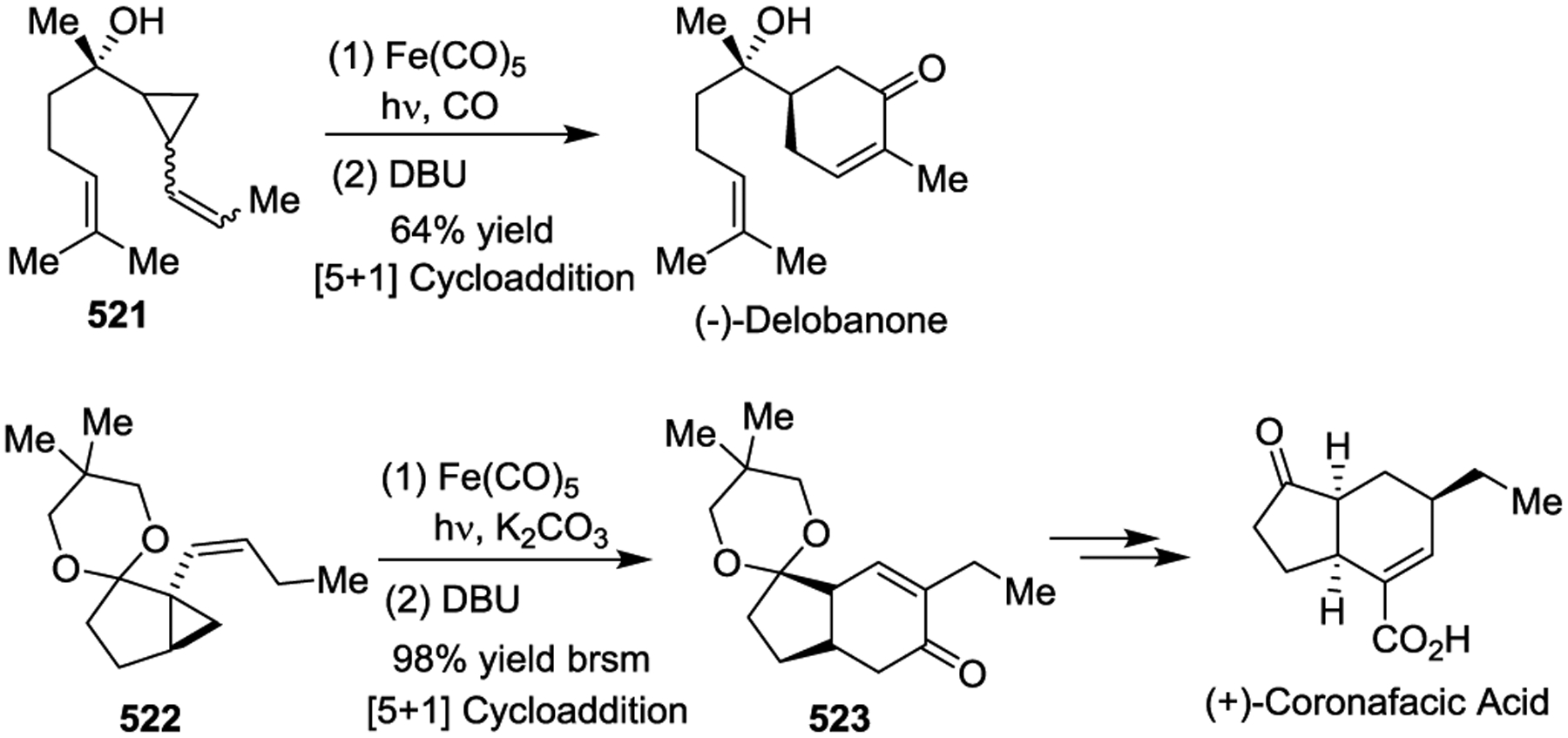
In 2011, the Stoltz group demonstrated the use of a Pd(0)-catalyzed [3 + 2] cycloaddition of a VCP 31 and a, β-nitrostyrene 524 as the key step to rapidly assemble the cyclopentane core 525 of the Melodinus alkaloid (+)-Scandine (Scheme 74).169
Scheme 74.
4. CONCLUSIONS AND OUTLOOK
Transition-metal-catalyzed selective C-C bond cleavage of VCPs has become an increasingly versatile and valuable tool for building functionalized complex molecular structures in organic synthesis. In this review, we highlighted all of the developments and the recent advances in the field of transition-metal-catalyzed cycloadditions and related addition reactions involving activated or non-activated VCPs in both intramolecular and intermolecular processes. The synthetic utility of VCPs in the construction of complex natural products and pharmaceutical agents has demonstrated the potential of metal catalysis to induce reactivity in challenging substrates. Although extensive efforts have been committed to formal [3 + 2] cycloaddition of activated VCPs, the cycloaddition partners are still restricted to two-carbon synthons, such as aldehydes, imines, or electron-deficient olefins. One particularly attractive application is the use of such intermediates to enable the synthesis of medium-sized ring systems. In 2019, the seminal work published by You48 demonstrated the unique properties of activated VCPs for the preparation of diversified medium-sized compounds, especially in an enantioselective manner. Nevertheless, the use of other metals besides palladium in transition-metal-catalyzed asymmetric [3 + n] (n > 2) cycloadditions has remained, until recently, relatively limited, despite the success of asymmetric [3 + 4] cycloadditions.
Pioneered by the Wender group, non-activated VCPs have become ubiquitous partners in a wide range of cycloadditions, as either three-carbon or five-carbon synthons. Concomitantly, enantioselective cycloadditions have seen an exponential increase in use. In particular, Rh catalysts have been the most developed catalysts for non-activated VCP cycloadditions. When new reactions are reported or new catalysts are developed, such as Ru or Ir catalysts, their metrics are frequently compared to those of their rhodium counterparts. While other metals are currently less explored for non-activated VCP cycloadditions, they provide complementary and alternative synthetic routes due to their different mechanistic pathways.
Challenges going forward include moving away from rare and expensive metals, such as Rh, and identifying other metal catalyst systems and control strategies that can exploit a wider range of VCP reactivity patterns. For example, recent progress with Ni, Fe, and Co catalysis shows promise in using abundant first row transition metals, although considerable scope limitations remain with these catalysts. Furthermore, metal catalyzed VCP reactions, in combination with recent progress in C-H activation,170–171 visible light catalysis,172–173 electrochemical methods,174–175 or mechanochemical tools176–177 would open more and more opportunities for the design of new reactions and discovery of new reactivities, especially in the execution of tandem cyclizations to efficiently generate polycyclic scaffolds. Ideally, transformations of VCPs induced by first-row transition-metals should be accessible in good regio-, diastereo-, and enantioselectivity and in an atom-economical manner. It is our hope that this Review will inspire chemists to invent new and creative reaction patterns involving the metal catalyzed C-C bond cleavage of VCPs, which will continue to provide an extensive area for the investigation and applications to complex and bioactive molecule synthesis.
ACKNOWLEDGMENTS
Xiaoxun Li gratefully acknowledge the National Natural Science Foundation of China (21901142), the Natural Science Foundation of Shandong Province (ZR2019QB001), Key Research and Development Program of Shandong Province (2017CXGC1401), the Natural Science Foundation of Jiangsu Province (BK20180227), and the Fundamental Research Funds of Shandong University (21310088963023, 2020QNQT007 and 2020QNQT009) for generous financial support. Stephanie A. Blaszczyk acknowledges the NSF Graduate Research Fellowship Program for its financial support. Weiping Tang thanks NIH (R01 GM088285) for the financial support from 2009 to 2015 for some of the work covered in this review.
Biographies
Jianhua Wang was born in Wuhu, China. He received his B.S. degree in applied chemistry and M.S. degree in medicinal chemistry from Hunan University in 2011. He then started his Ph.D. studies in medicinal chemistry under the supervision of Professor Xiaoxun Li at Shandong University. His current research focuses on asymmetric catalysis.
Stephanie Blaszczyk is a science writer/communicator, the editor-in-chief of Chembites, a former AAAS Mass Media Science and Engineering Fellow for the Milwaukee Journal Sentinel, and a former WISCIENCE Public Service Fellow. Stephanie studied chemistry at Rockford University, where she earned her bachelor’s degree in 2013. She then worked in industry before attending the University of Wisconsin–Madison to pursue graduate studies under the direction of Professor Weiping Tang, where she graduated with her PhD in 2020. Her graduate research focused on synthetic methodology development.
Professor Xiaoxun Li obtained his B.S. and M.S. degrees from Tianjin University. He then moved to the University of Wisconsin-Madison where studied for a Ph.D. degree (2009–2014) under the guidance of Professor Weiping Tang. After that, he undertook a postdoctoral appointment with Professor Barry M. Trost at Stanford University (2015–2017). In December of 2017, he began his independent research at Shandong University. His research interest focuses on asymmetric catalysis, natural product synthesis and medicinal chemistry.
Professor Weiping Tang received his B.S. degree from Peking University, M.S. degree from New York University, and Ph.D. degree from Stanford University. He was a HHMI postdoctoral fellow at Harvard University. He is currently a professor in the School of Pharmacy and Department of Chemistry at the University of Wisconsin-Madison. His group is interested in developing new synthetic methods, natural product synthesis, medicinal chemistry and chemical biology.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Trost BM The Atom Economy-A Search for Synthetic Efficiency. Science 1991, 254, 1471–1477. [DOI] [PubMed] [Google Scholar]
- (2).Trost BM Atom Economy-A Challenge for Organic Synthesis: Homogeneous Catalysis Leads the Way. Angew. Chem., Int. Ed 1995, 34, 259–281. [Google Scholar]
- (3).Lautens M; Klute W; Tam W Transition Metal-Mediated Cycloaddition Reactions. Chem. Rev 1996, 96, 49–92. [DOI] [PubMed] [Google Scholar]
- (4).Winkler JD Tandem Diels–Alder Cycloadditions in Organic Synthesis. Chem. Rev 1996, 96, 167–176. [DOI] [PubMed] [Google Scholar]
- (5).Methods and Applications of Cycloaddition Reactions in Organic Syntheses. Nishiwaki N Eds. Wiley–VCH, Weinheim, Germany, 2014. [Google Scholar]
- (6).Metal-Catalyzed Cyclization Reactions 1. Ma S; Gao S Eds. Thieme, 2016. [Google Scholar]
- (7).Jun C-H Transition Metal-Catalyzed Carbon-Carbon Bond Activation. Chem. Soc. Rev 2004, 33, 610–618. [DOI] [PubMed] [Google Scholar]
- (8).Murakami M; Matsuda T Metal-Catalysed Cleavage of Carbon- Carbon Bonds. Chem. Commun 2011, 47, 1100–1105. [DOI] [PubMed] [Google Scholar]
- (9).Ruhland K Transition-Metal-Mediated Cleavage and Activation of C–C Single Bonds. Eur. J. Org. Chem 2012, 14, 2683–2706. [Google Scholar]
- (10).Souillart L; Cramer N Catalytic C–C Bond Activations via Oxidative Addition to Transition Metals. Chem. Rev 2015, 115, 9410–9464. [DOI] [PubMed] [Google Scholar]
- (11).Cleavage of Carbon–Carbon Single Bonds by Transition Metals; Murakami M; Chatani N Eds. Wiley–VCH, Weinheim, Germany, 2016. [Google Scholar]
- (12).Rubin M; Rubina M; Gevorgyan V Transition Metal Chemistry of Cyclopropenes and Cyclopropanes. Chem. Rev 2007, 107, 3117–3179. [DOI] [PubMed] [Google Scholar]
- (13).Seiser T; Cramer N Enantioselective Metal-Catalyzed Activation of Strained Rings. Org. Biomol. Chem 2009, 7, 2835–2840. [DOI] [PubMed] [Google Scholar]
- (14).Aïssa C Transition-Metal-Catalyzed Rearrangements of Small Cycloalkanes: Regioselectivity Trends in β-Carbon Elimination Reactions. Synthesis 2011, 21, 3389–3407. [Google Scholar]
- (15).Fumagalli G; Stanton S; Bower JF Recent Methodologies That Exploit C–C Single-Bond Cleavage of Strained Ring Systems by Transition Metal Complexes. Chem. Rev 2017, 117, 9404–9432. [DOI] [PubMed] [Google Scholar]
- (16).Jiao L; Yu Z-X Vinylcyclopropane Derivatives in Transition-Metal-Catalyzed Cycloadditions for the Synthesis of Carbocyclic Compounds. J. Org. Chem 2013, 78, 6842–6848. And reviews cited therein. [DOI] [PubMed] [Google Scholar]
- (17).Ganesh V; Chandrasekaran S Recent Advances in the Synthesis and Reactivity of Vinylcyclopropanes. Synthesis 2016, 48, 4347–4380. And reviews cited therein. [Google Scholar]
- (18).Meazza M; Guo H; Rios R Synthetic Applications of Vinyl cyclopropane Opening. Org. Biomol. Chem 2017, 15, 2479–2490. And reviews cited therein. [DOI] [PubMed] [Google Scholar]
- (19).Brownsey DK; Gorobets E; Derksen DJ Beyond Geminal Diesters: Increasing the Scope of Metal-mediated Vinylcyclopropane Annulations While Decreasing Pre-activation. Org. Biomol. Chem 2018, 16, 3506–3523. And reviews cited therein. [DOI] [PubMed] [Google Scholar]
- (20).Khoury PR; Goddard JD; Tam W Ring Strain Energies: Substituted Rings, Norbornanes, Norbornenes and Norbornadienes. Tetrahedron 2004, 60, 8103–8112. [Google Scholar]
- (21).Schneider TF; Kaschel J; Werz DB A New Golden Age for Donor–Acceptor Cyclopropanes. Angew. Chem., Int. Ed 2014, 53, 5504–5523. [DOI] [PubMed] [Google Scholar]
- (22).Burgess K Conjugate Nucleophilic Ring Opening of Activated Vinylcyclopropanes Facilitated by Homogenous Palladium Catalysis. Tetrahedron Lett. 1985, 26, 3049–3052. [Google Scholar]
- (23).Shimizu I; Ohashi Y; Tsuji J Palladium-Catalyzed [3 + 2] Cycloaddition Reaction of Vinylcyclopropanes with α, β-Unsaturated Esters or Ketones. Tetrahedron Lett. 1985, 3825–3828. [Google Scholar]
- (24).Kim H; Kim S; Kim J; Son J-Y; Baek Y; Um K; Lee PH One-Pot Synthesis of Indolizines via Sequential Rhodium-Catalyzed [2 + 1]-Cyclopropanation, Palladium-Catalyzed Ring Expansion, and Oxidation Reactions from Pyridotriazoles and 1,3-Dienes. Org. Lett 2017, 19, 5677–5680. [DOI] [PubMed] [Google Scholar]
- (25).Gai S; Lucas NT; Hawkins BC Benzannulated 6,5-Spiroketals from Donor–Acceptor Cyclopropanes. Org. Lett 2019, 21, 2872–2875. [DOI] [PubMed] [Google Scholar]
- (26).Yamamoto K; Ishida T; Tsuji J Palladium(0)-catalyzed Cycloaddition of Activated Vinylcyclopropanes with Aryl Isocyanates. Chem. Lett 1987, 16, 1157–1158. [Google Scholar]
- (27).Sebelius S; Olsson VJ; Szabó KJ Palladium Pincer Complex Catalyzed Substitution of Vinyl Cyclopropanes, Vinyl Aziridines, and Allyl Acetates with Tetrahydroxydiboron. An Efficient Route to Functionalized Allylboronic Acids and Potassium Trifluoro-(allyl)borates. J. Am. Chem. Soc 2005, 127, 10478–10479. [DOI] [PubMed] [Google Scholar]
- (28).Li C-F; Xiao W-J; Alper H Palladium-Catalyzed Ring-Opening Thiocarbonylation of Vinylcyclopropanes with Thiols and Carbon Monoxide. J. Org. Chem 2009, 74, 888–890. [DOI] [PubMed] [Google Scholar]
- (29).Mita T; Tanaka H; Higuchi Y; Sato Y Palladium-Catalyzed Carboxylation of Activated Vinylcyclopropanes with CO2. Org. Lett 2016, 18, 2754–2757. [DOI] [PubMed] [Google Scholar]
- (30).Trost BM; Crawley ML Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev 2003, 103, 2921–2943. [DOI] [PubMed] [Google Scholar]
- (31).Trost BM; Bai W-J; Hohn C; Bai Y; Cregg JJ Palladium-Catalyzed Asymmetric Allylic Alkylation of 3-Substituted 1H-Indoles and Tryptophan Derivatives with Vinylcyclopropanes. J. Am. Chem. Soc 2018, 140, 6710–6717. [DOI] [PubMed] [Google Scholar]
- (32).Parsons AT; Campbell MJ; Johnson JS Diastereoselective Synthesis of Tetrahydrofurans via Palladium(0)-Catalyzed [3 + 2] Cycloaddition of Vinylcyclopropanes and Aldehydes. Org. Lett 2008, 10, 2541–2544. [DOI] [PubMed] [Google Scholar]
- (33).Wang Q; Wang C; Shi W; Xiao Y; Guo H Pd-Catalyzed Diastereoselective [3 + 2] Cycloaddition of Vinylcyclopropanes with Sulfamate-derived Cyclic Imines. Org. Biomol. Chem 2018, 16, 4881–4887. [DOI] [PubMed] [Google Scholar]
- (34).Lin Y; Wang Q; Wu Y; Wang C; Jia H; Zhang C; Huang J; Guo H Pd-catalyzed [3 + 2] Cycloaddition of Vinylcyclopropanes with 1-Azadienes: Synthesis of 4-Cyclopentylbenzo [e][1,2,3]oxathiazine 2,2- dioxides. RSC Adv. 2018, 8, 40798–40803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Trost BM; Morris PJ Palladium-Catalyzed Diastereo- and Enantioselective Synthesis of Substituted Cyclopentanes through a Dynamic Kinetic Asymmetric Formal [3 + 2]-Cycloaddition of Vinyl Cyclopropanes and Alkylidene Azlactones. Angew. Chem., Int. Ed 2011, 50, 6167–6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Trost BM; Machacek MR; Aponick A Predicting the Stereochemistry of Diphenylphosphino Benzoic Acid (DPPBA)-Based Palladium-Catalyzed Asymmetric Allylic Alkylation Reactions: A Working Model. Acc. Chem. Res 2006, 39, 747–760. [DOI] [PubMed] [Google Scholar]
- (37).Trost BM; Morris PJ; Sprague SJ Palladium-Catalyzed Diastereo- and Enantioselective Formal [3 + 2]-Cycloadditions of Substituted Vinylcyclopropanes. J. Am. Chem. Soc 2012, 134, 17823–17831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Mei L.-y.; Wei Y; Xu Q; Shi M Palladium-Catalyzed Asymmetric Formal [3 + 2] Cycloaddition of Vinyl Cyclopropanes and β,γ-Unsaturated α-Keto Esters: An Effective Route to Highly Functionalized Cyclopentanes. Organometallics 2012, 31, 7591–7599. [Google Scholar]
- (39).Mei L.-y.; Wei Y; Xu Q; Shi M Diastereo- and Enantioselective Construction of Oxindole-Fused Spirotetrahydrofuran Scaffolds through Palladium-Catalyzed Asymmetric [3 + 2] Cycloaddition of Vinyl Cyclopropanes and Isatins. Organometallics 2013, 32, 3544–3556. [Google Scholar]
- (40).Li W-K; Liu Z-S; He L; Kang T-R; Liu Q-Z Enantioselective Cycloadditions of Vinyl Cyclopropanes and Nitro-olefins for Functionally and Optically Enriched Nitrocyclopentanes. Asian J. Org. Chem 2015, 4, 28–32. [Google Scholar]
- (41).Liu Z-S; Li W-K; Kang T-R; He L; Liu Q-Z Palladium-Catalyzed Asymmetric Cycloadditions of Vinylcyclopropanes and in Situ Formed Unsaturated Imines: Construction of Structurally and Optically Enriched Spiroindolenines. Org. Lett 2015, 17, 150–153. [DOI] [PubMed] [Google Scholar]
- (42).Huang X-B; Li X-J; Li T-T; Chen B; Chu W-D; He L; Liu Q-Z Palladium-Catalyzed Highly Enantioselective Cycloaddition of Vinyl Cyclopropanes with Imines. Org. Lett 2019, 21, 1713–1716. [DOI] [PubMed] [Google Scholar]
- (43).Laugeois M; Ling J; Férard C; Michelet V; Ratovelomanana-Vidal V; Vitale MR Palladium(0)-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles with Vinylcyclopropanes: An Entry to Stereodefined 2,3-Fused Cyclopentannulated Indoline Derivatives. Org. Lett 2017, 19, 2266–2269. [DOI] [PubMed] [Google Scholar]
- (44).Gee YS; Rivinoja DJ; Wales SM; Gardiner MG; Ryan JH; Hyland CJT Pd-Catalyzed Dearomative [3 + 2] Cycloaddition of 3-Nitroindoles with 2-Vinylcyclopropane-1,1-dicarboxylates. J. Org. Chem 2017, 82, 13517–13529. [DOI] [PubMed] [Google Scholar]
- (45).Zhang J-Q; Tong F; Sun B-B; Fan W-T; Chen J-B; Hu D; Wang X-W Pd-Catalyzed Asymmetric Dearomative Cycloaddition for Construction of Optically Active Pyrroloindoline and Cyclopentaindoline Derivatives: Access to 3a-Aminopyrroloindolines. J. Org. Chem 2018, 83, 2882–2891. [DOI] [PubMed] [Google Scholar]
- (46).Ling J; Laugeois M; Michelet V; Ratovelomanana-Vidal V; Vitale MR Palladium(0)-Catalyzed Dearomatization of 2-Nitrobenzofurans through Formal (3+2) Cycloadditions with Vinylcyclopropanes: A Straightforward Access to Cyclopenta[b]benzofurans. Synlett 2018, 29, 928–932. [Google Scholar]
- (47).Liu J; Li M-M; Qu B-L; Lu L-Q; Xiao W-JA Photoinduced Wolff Rearrangement/ Pd-Catalyzed [3+2] Cycloaddition Sequence: an Unexpected Route to Tetrahydrofurans. Chem. Commun 2019, 55, 2031–2034. [DOI] [PubMed] [Google Scholar]
- (48).Cheng Q; Xie J-H; Weng Y-C; You S-L Pd-Catalyzed Dearomatization of Anthranils with Vinylcyclopropanes by [4+3] Cyclization Reaction. Angew. Chem., Int. Ed 2019, 58, 5739–5743. [DOI] [PubMed] [Google Scholar]
- (49).Bowman RK; Johnson JS Nickel-Catalyzed Rearrangement of 1-Acyl-2-vinylcyclopropanes. A Mild Synthesis of Substituted Dihydrofurans. Org. Lett 2006, 8, 573–576. [DOI] [PubMed] [Google Scholar]
- (50).Sumida Y; Yorimitsu H; Oshima K Nickel-Catalyzed Borylative Ring-Opening Reaction of Vinylcyclopropanes with Bis-(pinacolato)diboron Yielding Allylic Boronates. Org. Lett 2008, 10, 4677–4679. [DOI] [PubMed] [Google Scholar]
- (51).Mori T; Nakamura T; Kimura M Stereoselective Coupling Reaction of Dimethylzinc and Alkyne toward Nickelacycles. Org. Lett 2011, 13, 2266–2269. [DOI] [PubMed] [Google Scholar]
- (52).Tombe R; Kurahashi T; Matsubara S Nickel-Catalyzed Cycloaddition of Vinylcyclopropanes to Imines. Org. Lett 2013, 15, 1791–1793. [DOI] [PubMed] [Google Scholar]
- (53).Fürstner A Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes to Make This Base Metal a Multitasking Champion. ACS Cent. Sci 2016, 2, 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sherry BD; Fürstner A Iron-Catalyzed Addition of Grignard Reagents to Activated Vinyl Cyclopropanes. Chem. Commun 2009, 7116–7118. [DOI] [PubMed] [Google Scholar]
- (55).Dieskau AP; Holzwarth MS; Plietker B Fe-Catalyzed Allylic C-C-Bond Activation: Vinylcyclopropanes as Versatile a1,a3,d5-Synthons in Traceless Allylic Substitutions and [3 + 2]-Cycloadditions. J. Am. Chem. Soc 2012, 134, 5048–5051. [DOI] [PubMed] [Google Scholar]
- (56).Moran J; Smith AG; Carris RM; Johnson JS; Krische MJ Polarity Inversion of Donor-Acceptor Cyclopropanes: Disubstituted δ-Lactones via Enantioselective Iridium Catalysis. J. Am. Chem. Soc 2011, 133, 18618–18621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem., Int. Ed 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Zhu Z-Q; Yu L; Sun M; Mei G-J; Shi F Regioselective [3+3] Cyclization of 2-Indolymethanols with Vinylcyclopropanes via Metal Catalysis. Adv. Synth. Catal 2018, 360, 3109–3116. [Google Scholar]
- (59).Zhang H-H; Wang C-S; Li C; Mei G-J; Li Y; Shi F Design and Enantioselective Construction of Axially Chiral Naphthyl-Indole Skeletons. Angew. Chem., Int. Ed 2017, 56, 116–121. [DOI] [PubMed] [Google Scholar]
- (60).Wu J-Q; Qiu Z-P; Zhang S-S; Liu J-G; Lao Y-X; Gu L-Q; Huang Z-S; Li J; Wang H Rhodium(III)-Catalyzed C–H/C–C Activation Sequence: Vinylcyclopropanes as Versatile Synthons in Direct C–H Allylation Reactions. Chem. Commun 2015, 51, 77–80. [DOI] [PubMed] [Google Scholar]
- (61).Lu Q; Klauck FJR; Glorius F Manganese-catalyzed allylation via sequential C–H and C–C/C–Het bond activation. Chem. Sci 2017, 8, 3379–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Young IS; Kerr MA Total Synthesis of (+)-Nakadomarin J. Am. Chem. Soc 2007, 129, 1465–1469. [DOI] [PubMed] [Google Scholar]
- (63).Leduc AB; Kerr MA Total Synthesis of (−)-Allosecurinine. Angew. Chem., Int. Ed 2008, 47, 7945–7948. [DOI] [PubMed] [Google Scholar]
- (64).Carson CA; Kerr MA Total Synthesis of FR901483. Org. Lett 2009, 11, 777–779. [DOI] [PubMed] [Google Scholar]
- (65).Karadeolian A; Kerr MA Total Synthesis of (+)-Isatisine Angew. Chem., Int. Ed 2010, 49, 1133–1135. [DOI] [PubMed] [Google Scholar]
- (66).Yu Z-X; Wender PA; Houk KN On the Mechanism of [Rh(CO)2Cl]2-Catalyzed Intermolecular [5 + 2] Reactions between Vinylcyclopropanes and Alkynes. J. Am. Chem. Soc 2004, 126, 9154–9155. [DOI] [PubMed] [Google Scholar]
- (67).Wang SC; Tantillo DJ Metal Promoted Vinylcyclopropane-Cyclopentene Rearrangements: Reactions Ripe for Mechanism-Based Catalyst Design. J. Organomet. Chem 2006, 691, 4386–4392. [Google Scholar]
- (68).Liu P; Cheong PHY; Yu Z-X; Wender PA; Houk KN Substituent Effects, Reactant Preorganization, and Ligand Exchange Control the Reactivity in RhI-Catalyzed [5+2] Cycloadditions between Vinylcyclopropanes and Alkynes. Angew. Chem., Int. Ed 2008, 47, 3939–3941. [DOI] [PubMed] [Google Scholar]
- (69).Yu Z-X; Cheong PHY; Liu P; Legault CY; Wender PA; Houk KN Origins of Differences in Reactivities of Alkenes, Alkynes, and Allenes in [Rh(CO)2Cl]2-Catalyzed [5 + 2] Cycloaddition Reactions with Vinylcyclopropanes. J. Am. Chem. Soc 2008, 130, 2378–2379. [DOI] [PubMed] [Google Scholar]
- (70).Liu P; Sirois LE; Cheong PH-Y; Yu Z-X; Hartung IV; Rieck H; Wender PA; Houk KN Electronic and Steric Control of Regioselectivities in Rh(I)-Catalyzed [5 + 2] Cycloadditions: Experiment and Theory. J. Am. Chem. Soc 2010, 132, 10127–10135. [DOI] [PubMed] [Google Scholar]
- (71).Jiao L; Ye SY; Yu Z-X Rh(I)-Catalyzed Intramolecular [3 + 2] Cycloaddition of trans-Vinylcyclopropane-enes. J. Am. Chem. Soc 2008, 130, 7178–7179. [DOI] [PubMed] [Google Scholar]
- (72).Jiao L; Lin M; Yu Z-X Density Functional Theory Study of the Mechanisms and Stereochemistry of the Rh(I)-Catalyzed Intramolecular [3+2] Cycloadditions of 1-Ene- and 1-Yne-Vinylcyclopropanes. J. Am. Chem. Soc 2011, 133, 447–461. [DOI] [PubMed] [Google Scholar]
- (73).Wender PA; Takahashi H; Witulski B Transition Metal Catalyzed [5 + 2] Cycloadditions of Vinylcyclopropanes and Alkynes: A Homolog of the Diels-Alder Reaction for the Synthesis of Seven-Membered Rings. J. Am. Chem. Soc 1995, 117, 4720–4721. [Google Scholar]
- (74).Pellissier H Recent Developments in the [5 + 2] Cycloaddition. Adv. Synth. Catal 2011, 353, 189–218. [Google Scholar]
- (75).Ylijoki KEO; Stryker JM [5 + 2] Cycloaddition Reactions in Organic and Natural Product Synthesis. Chem. Rev 2013, 113, 2244–2266. [DOI] [PubMed] [Google Scholar]
- (76).Wender PA; Husfeld CO; Langkopf E; Love JA First Studies of the Transition Metal-Catalyzed [5+2] Cycloadditions of Alkenes and Vinylcyclopropanes: Scope and Stereochemistry. J. Am. Chem. Soc 1998, 120, 1940–1941. [Google Scholar]
- (77).Wender PA; Husfeld CO; Langkopf E; Love JA; Pleuss N The First Metal-Catalyzed Intramolecular [5 + 2] Cycloadditions of Vinylcyclopropanes and Alkenes: Scope, Stereochemistry, and Asymmetric Catalysis. Tetrahedron 1998, 54, 7203–7220. [Google Scholar]
- (78).Wender PA; Dyckman AJ; Husfeld CO; Kadereit D; Love JA; Rieck H Transition Metal-Catalyzed [5+2] Cycloadditions with Substituted Cyclopropanes: First Studies of Regio- and Stereoselectivity. J. Am. Chem. Soc 1999, 121, 10442–10443. [Google Scholar]
- (79).Wender PA; Dyckman AJ Transition Metal-Catalyzed [5 + 2] Cycloadditions of 2-Substituted-1-vinylcyclopropanes: Catalyst Control and Reversal of Regioselectivity. Org. Lett 1999, 1, 2089–2092. [Google Scholar]
- (80).Wender PA; Sperandio D A New and Selective Catalyst for the [5 + 2] Cycloaddition of Vinylcyclopropanes and Alkynes. J. Org. Chem 1998, 63,4164–4165. [Google Scholar]
- (81).Wender PA; Glorius F; Husfeld CO; Langkopf E; Love JA Transition Metal-Catalyzed [5 + 2] Cycloadditions of Allenes and Vinylcyclopropanes: First Studies of Endo–Exo Selectivity, Chemoselectivity, Relative Stereochemistry, and Chirality Transfer. J. Am. Chem. Soc 1999, 121, 5348–5349. [Google Scholar]
- (82).Wang B; Cao P; Zhang XM An efficient Rh-catalyst system for the intramolecular [4+2] and [5+2] cycloaddition reactions. Tetrahedron Lett. 2000, 41, 8041–8044. [Google Scholar]
- (83).Lee SI; Park SY; Park JH; Jung IG; Choi SY; Chung YK Rhodium N-Heterocyclic Carbene-Catalyzed [4 + 2] and [5 + 2] Cycloaddition Reactions. J. Org. Chem 2006, 71, 91–96. [DOI] [PubMed] [Google Scholar]
- (84).Wender PA; Williams TJ [(arene)Rh(cod)]+ Complexes as Catalysts for [5+2] Cycloaddition Reactions. Angew. Chem., Int. Ed 2002, 41, 4550–4553. [DOI] [PubMed] [Google Scholar]
- (85).Wender PA; Haustedt LO; Lim J; Love JA; Williams TJ; Yoon JY Asymmetric Catalysis of the [5 + 2] Cycloaddition Reaction of Vinylcyclopropanes and π-Systems. J. Am. Chem. Soc 2006, 128, 6302–6303. [DOI] [PubMed] [Google Scholar]
- (86).Shintani R; Nakatsu H; Takatsu K; Hayashi T Rhodium-Catalyzed Asymmetric [5+2] Cycloaddition of Alkyne–Vinylcyclopropanes. Chem.–Eur. J 2009, 15, 8692–8694. [DOI] [PubMed] [Google Scholar]
- (87).Straker RN; Peng Q; Mekareeya A; Paton RS; Anderson EA Computational Ligand Design in Enantio- and Diastereoselective Ynamide [5 + 2] Cycloisomerization. Nat. Commun 2016, 7, 10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Inagaki F; Sugikubo K; Miyashita Y; Mukai C Rhodium(I)-Catalyzed Intramolecular [5+2] Cycloaddition Reactions of Alkynes and Allenylcyclopropanes: Construction of Bicyclo- [5.4.0]undecatrienes and Bicyclo[5.5.0]dodecatrienes. Angew. Chem., Int. Ed 2010, 49, 2206–2210. [DOI] [PubMed] [Google Scholar]
- (89).Sugikubo K; Omachi F; Miyanaga Y; Inagaki F; Matsumoto C; Mukai C Rhodium(I)-Catalyzed Cycloisomerization of Alkene-Substituted Allenylcyclopropanes: Stereoselective Formation of Bicyclo-[4.3.0]nonadienes. Angew. Chem., Int. Ed 2013, 52, 11369–11372. [DOI] [PubMed] [Google Scholar]
- (90).Kawamura T; Kawaguchi Y; Sugikubo K; Inagaki F; Mukai C Rhodium(I)-Catalyzed Cycloisomerization of Allene–Allenylcyclopropanes. Eur. J. Org. Chem 2015, 719–722. [Google Scholar]
- (91).Wang YY; Wang JX; Su JC; Huang F; Jiao L; Liang Y; Yang DZ; Zhang SW; Wender PA; Yu Z-X A Computationally Designed Rh(I)-Catalyzed Two-Component [5 + 2 + 1] Cycloaddition of Ene-vinylcyclopropanes and CO for the Synthesis of Cyclooctenones. J. Am. Chem. Soc 2007, 129, 10060–10061. [DOI] [PubMed] [Google Scholar]
- (92).Wang Y; Yu Z-X Rhodium-Catalyzed [5 + 2 + 1] Cycloaddition of Ene–Vinylcyclopropanes and CO: Reaction Design, Development, Application in Natural Product Synthesis, and Inspiration for Developing New Reactions for Synthesis of Eight-Membered Carbocycles. Acc. Chem. Res 2015, 48, 2288–2296. [DOI] [PubMed] [Google Scholar]
- (93).Huang F; Yao ZK; Wang Y; Wang YY; Zhang JL; Yu Z-X RhI -Catalyzed Two-Component [(5 + 2) + 1] Cycloaddition Approach toward [5–8-5] Ring Systems. Chem. –Asian J 2010, 5, 1555–1559. [DOI] [PubMed] [Google Scholar]
- (94).Wender PA; Gamber GG; Hubbard RD; Zhang L Three-Component Cycloadditions: The First Transition Metal-Catalyzed [5 + 2 + 1] Cycloaddition Reactions. J. Am. Chem. Soc 2002, 124, 2876–2877. [DOI] [PubMed] [Google Scholar]
- (95).Jiao L; Yuan CX; Yu Z-X Tandem Rh(I)-Catalyzed [(5 + 2) + 1] Cycloaddition/Aldol Reaction for the Construction of Linear Triquinane Skeleton: Total Syntheses of (±)-Hirsutene and (±)-1-Desoxyhypnophilin. J. Am. Chem. Soc 2008, 130, 4421–4430. [DOI] [PubMed] [Google Scholar]
- (96).Liu C-H; Yu Z-X Rh(I)-Catalyzed Intramolecular [3+2] Cycloaddition of trans-2-Allene-Vinylcyclopropanes. Synlett 2018, 29, 764–768. [Google Scholar]
- (97).Li Q; Jiang G-J; Jiao L; Yu Z-X Reaction of α-Ene-Vinyl cyclopropanes: Type II Intramolecular [5 + 2] Cycloaddition or [3 + 2] Cycloaddition? Org. Lett 2010, 12, 1332–1335. [DOI] [PubMed] [Google Scholar]
- (98).Jiao L; Lin M; Yu Z-X Rh(I)-Catalyzed Intramolecular [3+2] Cycloaddition Reactions of 1-Ene-, 1-Yne-, and 1-Allene-vinyl-cyclopropanes. Chem. Commun 2010, 46, 1059–1061. [DOI] [PubMed] [Google Scholar]
- (99).Lin M; Kang G-Y; Guo Y-A; Yu Z-X Asymmetric Rh(I)-Catalyzed Intramolecular [3 + 2] Cycloaddition of 1-Yne-vinyl-cyclopropanes for Bicyclo[3.3.0] Compounds with a Chiral Quaternary Carbon Stereocenter and Density Functional Theory Study of the Origins of Enantioselectivity. J. Am. Chem. Soc 2012, 134, 398–405. [DOI] [PubMed] [Google Scholar]
- (100).Liu C-H; Yu Z-X Rhodium(I)-Catalyzed Bridged [5+2] Cycloaddition of cis-Allenevinylcyclopropanes to Synthesize the Bicyclo[4.3.1]decane Skeleton. Angew. Chem., Int. Ed 2017, 56, 8667–8671. [DOI] [PubMed] [Google Scholar]
- (101).Jiao L; Lin M; Zhuo L-G; Yu Z-X Rh(I)-Catalyzed [(3 + 2) + 1] Cycloaddition of 1-Yne/Ene-vinylcyclopropanes and CO: Homologous Pauson-Khand Reaction and Total Synthesis of (±)-α-Agarofuran. Org. Lett 2010, 12, 2528–2531. [DOI] [PubMed] [Google Scholar]
- (102).Lin M; Li F; Jiao L; Yu Z-X Rh(I)-Catalyzed Formal [5 + 1]/[2 + 2 + 1] Cycloaddition of 1-Yne-vinylcyclopropanes and Two CO Units: One-Step Construction of Multifunctional Angular Tricyclic 5/5/6 Compounds. J. Am. Chem. Soc 2011, 133, 1690–1693. [DOI] [PubMed] [Google Scholar]
- (103).Shu D; Li X; Zhang M; Robichaux PJ; Tang W Synthesis of Highly Functionalized Cyclohexenone Rings: Rhodium-Catalyzed 1,3-Acyloxy Migration and Subsequent [5 + 1] Cycloaddition. Angew. Chem., Int. Ed 2011, 50, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Shu D; Li X; Zhang M; Robichaux PJ; Guzei IA; Tang W Rhodium-Catalyzed Carbonylation of Cyclopropyl Substituted Propargyl Esters: A Tandem 1,3-Acyloxy Migration [5 + 1] Cycloaddition. J. Org. Chem 2012, 77, 6463–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Shu X-Z; Shu D; Schienebeck CM; Tang W Rhodium-Catalyzed Acyloxy Migration of Propargylic Esters in Cycloadditions, Inspiration from the Recent “Gold Rush”. Chem. Soc. Rev 2012, 41, 7698–7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Li X; Zhang M; Shu D; Robichaux PJ; Huang S; Tang W Rhodium-Catalyzed Ring Expansion of Cyclopropanes to Seven-Membered Rings by 1,5 C-C Bond Migration. Angew. Chem., Int. Ed 2011, 50, 10421–10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Trost BM; Toste FD; Shen H Ruthenium-Catalyzed Intramolecular [5 + 2] Cycloadditions. J. Am. Chem. Soc 2000, 122, 2379–2380. [Google Scholar]
- (108).Trost BM; Shen HC On the Regioselectivity of the Ru-Catalyzed Intramolecular [5 + 2] Cycloaddition. Org. Lett 2000, 2, 2523–2525. [DOI] [PubMed] [Google Scholar]
- (109).Trost BM; Toste FD Synthesis of the First (1–3:6,7-η-Cyclodecadienyl)ruthenium Complex by the Intramolecular Reaction of an Alkene and a Vinylcyclopropane. Angew. Chem., Int. Ed 2001, 40, 1114–1116. [DOI] [PubMed] [Google Scholar]
- (110).Hong X; Trost BM; Houk KN Mechanism and Origins of Selectivity in Ru(II)-Catalyzed Intramolecular [5 + 2] Cycloadditions and Ene Reactions of Vinylcyclopropanes and Alkynes from Density Functional Theory. J. Am. Chem. Soc 2013, 135, 6588–6600. [DOI] [PubMed] [Google Scholar]
- (111).Trost BM; Shen HC; Horne DB; Toste FD; Steinmetz BG; Koradin C Syntheses of Seven-Membered Rings: Ruthenium-Catalyzed Intramolecular [5 + 2] Cycloadditions. Chem. Eur. J 2005, 11, 2577–2590. [DOI] [PubMed] [Google Scholar]
- (112).Trost BM; Shen HC Constructing Tricyclic Compounds Containing a Seven-Membered Ring by Ruthenium-Catalyzed Intramolecular [5+2] Cycloaddition. Angew. Chem., Int. Ed 2001, 40, 2313–2316. [DOI] [PubMed] [Google Scholar]
- (113).Trost BM; Shen HC; Schulz T; Koradin C; Schirok H On the Diastereoselectivity of Ru-Catalyzed [5 + 2] Cycloadditions. Org. Lett 2003, 5, 4149–4151. [DOI] [PubMed] [Google Scholar]
- (114).Haller J; Strassner T; Houk KN Models for Stereoselective Additions to Chiral Allylic Ethers: Osmium Tetroxide Bis-hydroxylations. J. Am. Chem. Soc 1997, 119, 8031–8034. [Google Scholar]
- (115).Zuo G; Louie J Selectivity in Nickel-Catalyzed Rearrangements of Cyclopropylen-ynes. J. Am. Chem. Soc 2005, 127, 5798–5799. [DOI] [PubMed] [Google Scholar]
- (116).Hong X; Liu P; Houk KN Mechanism and Origins of Ligand-Controlled Selectivities in [Ni(NHC)]-Catalyzed Intramolecular (5 + 2) Cycloadditions and Homo-Ene Reactions: A Theoretical Study. J. Am. Chem. Soc 2013, 135, 1456–1462. [DOI] [PubMed] [Google Scholar]
- (117).Fürstner A; Majima K; Martin R; Krause H; Kattnig E; Goddard R; Lehmann CW A Cheap Metal for a “Noble” Task: Preparative and Mechanistic Aspects of Cycloisomerization and Cycloaddition Reactions Catalyzed by Low-Valent Iron Complexes. J. Am. Chem. Soc 2008, 130, 1992–2004. [DOI] [PubMed] [Google Scholar]
- (118).Sarel S Metal-Induced Rearrangements and Insertions into Cyclopropyl Olefins. Acc. Chem. Res 1978, 11, 204–211. [Google Scholar]
- (119).Khusnutdinov RI; Dzhemilev UM Transition Metal Complexes in the Chemistry of Vinylcyclopropanes. J. Organomet. Chem 1994, 471, 1–18. [Google Scholar]
- (120).Victor R; Ben-Shoshan R; Sarel S Photochemically Induced 1,5-Insertion of Carbon Monoxide into Vinylcyclopropane Systems. A Novel Synthesis of Cyclohexenones Mediated by Iron Carbonyl. Tetrahedron Lett. 1970, 4253–4256. [Google Scholar]
- (121).Taber DF; Kanai K; Jiang Q; Bui G Enantiomerically Pure Cyclohexenones by Fe-Mediated Carbonylation of Alkenyl Cyclopropanes. J. Am. Chem. Soc 2000, 122, 6807–6808. [Google Scholar]
- (122).Taber DF; Joshi PV; Kanai K 2,5-Dialkyl Cyclohexenones by Fe(CO)5-Mediated Carbonylation of Alkenyl Cyclopropanes: Functional Group Compatibility. J. Org. Chem 2004, 69, 2268–2271. [DOI] [PubMed] [Google Scholar]
- (123).Taber DF; Guo PF; Guo N Intramolecular [1 + 4 + 1] Cycloaddition: Establishment of the Method. J. Am. Chem. Soc 2010, 132, 11179–11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Taber DF; Sheth RB A Three-Step Route to a Tricyclic Steroid Precursor. J. Org. Chem 2008, 73, 8030–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Schulze MM; Gockel U A Mechanistic Study of Thermal Fe(CO)5 Mediated CO Insertion into Vinylcyclopropanes. J. Organomet. Chem 1996, 525, 155–158. [Google Scholar]
- (126).Liu C-H; Zhuang Z; Bose S; Yu Z-X Fe2(CO)9-Mediated [5 + 1] Cycloaddition of Vinylcyclopropanes and CO for the Synthesis of α,β-Cyclohexenones. Tetrahedron 2016, 72, 2752–2755. [Google Scholar]
- (127).Owada Y; Matsuo T; Iwasawa N Transformation of 1-(1, 2-Propadienyl)cyclopropanols into Substituted Hydroquinones Employing Octacarbonyldicobalt. Tetrahedron 1997, 53, 11069–11086. [Google Scholar]
- (128).Kurahashi T; Meijere A. de. [5+1] Cocyclization of (Cyclopropylmethylene)cyclopropanes and other Vinylcyclopropanes with Carbon Monoxide Catalyzed by Octacarbonyldicobalt. Synlett 2005, 2619–2622. [Google Scholar]
- (129).Binger P; Wedemann P; Kozhushkov SI; Meijere de. Palladium(0)- and Nickel(0)-Catalyzed [3 + 2] Co-Cyclization Reactions of Bicyclopropylidene with Alkenes. Eur. J. Org. Chem 1998, 113–119. [Google Scholar]
- (130).Wender PA; Rieck H; Fuji M The Transition Metal-Catalyzed Intermolecular [5 + 2] Cycloaddition: The Homologous Diels-Alder Reaction. J. Am. Chem. Soc 1998, 120, 10976–10977. [Google Scholar]
- (131).Wender PA; Dyckman AJ; Husfeld CO; Scanio MJC A New and Practical Five-Carbon Component for Metal-Catalyzed [5 + 2] Cycloadditions: Preparative Scale Syntheses of Substituted Cycloheptenones. Org. Lett 2000, 2, 1609–1611. [DOI] [PubMed] [Google Scholar]
- (132).Wender PA; Barzilay CM; Dyckman AJ The First Intermolecular Transition Metal-Catalyzed [5+2] Cycloadditions with Simple, Unactivated, Vinylcyclopropanes. J. Am. Chem. Soc 2001, 123, 179–180. [DOI] [PubMed] [Google Scholar]
- (133).Wender PA; Gamber GG; Scanio MJC Serial [5 + 2]/[4 + 2] Cycloadditions: Facile, Preparative, Multi-Component Syntheses of Polycyclic Compounds from Simple, Readily Available Starting Materials. Angew. Chem 2001, 113, 4013–4015. [DOI] [PubMed] [Google Scholar]
- (134).Wender PA; Gamber GG; Hubbard RD; Pham SM; Zhang L Multicomponent Cycloadditions: The Four-Component [5 + 1 + 2 + 1] Cycloaddition of Vinylcyclopropanes, Alkynes, and CO. J. Am. Chem. Soc 2005, 127, 2836–2837. [DOI] [PubMed] [Google Scholar]
- (135).Wegner HA; Meijere A. de.; Wender PA Transition Metal-Catalyzed Intermolecular [5 + 2] and [5 + 2+1] Cycloadditions of Allenes and Vinylcyclopropanes. J. Am. Chem. Soc 2005, 127, 6530–6531. [DOI] [PubMed] [Google Scholar]
- (136).Mbaezue II; Ylijoki KEO [5 + 1 + 2 + 1] vs [5 + 1 + 1 + 2] Rhodium-Catalyzed Cycloaddition Reactions of Vinylcyclopropanes with Terminal Alkynes and Carbon Monoxide: Density Functional Theory Investigations of Convergent Mechanistic Pathways and Reaction Regioselectivity. Organometallics 2017, 36, 2832–2842. [Google Scholar]
- (137).Hong X; Stevens MC; Liu P; Wender PA; Houk KN Reactivity and Chemoselectivity of Allenes in Rh(I)-Catalyzed Intermolecular (5 + 2) Cycloadditions with Vinylcyclopropanes: Allene-Mediated Rhodacycle Formation Can Poison Rh(I)-Catalyzed Cycloadditions. J. Am. Chem. Soc 2014, 136, 17273–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Wender PA; Stemmler RT; Sirois LE A Metal-Catalyzed Intermolecular [5 + 2] Cycloaddition/Nazarov Cyclization Sequence and Cascade. J. Am. Chem. Soc 2010, 132, 2532–2533. [DOI] [PubMed] [Google Scholar]
- (139).Wender PA; Sirois LE; Stemmler RT; Williams TJ Highly Efficient, Facile, Room Temperature Intermolecular [5 + 2] Cycloadditions Catalyzed by Cationic Rhodium(I): One Step to Cycloheptenes and Their Libraries. Org. Lett 2010, 12, 1604–1607. [DOI] [PubMed] [Google Scholar]
- (140).Mustard TJL; Wender PA; Cheong PH-Y Catalytic Efficiency is a Function of How Rhodium(I) (5 + 2) Catalysts Accommodate a Conserved Substrate Transition State Geometry: Induced Fit Model for Explaining Transition Metal Catalysis. ACS Catal. 2015, 5, 1758–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Wender PA; Lesser AB; Sirois LE Rhodium Dinaphthocyclooctatetraene Complexes: Synthesis, Characterization and Catalytic Activity in [5 + 2] Cycloadditions. Angew. Chem., Int. Ed 2012, 51, 2736–2740. [DOI] [PubMed] [Google Scholar]
- (142).Xu X; Liu P; Lesser A; Sirois LE; Wender PA; Houk KN Ligand Effects on Rates and Regioselectivities of Rh(I)-Catalyzed (5 + 2) Cycloadditions: A Computational Study of Cyclooctadiene and Dinaphthocyclooctatetraene as Ligands. J. Am. Chem. Soc 2012, 134, 11012–11025. [DOI] [PubMed] [Google Scholar]
- (143).Wender PA; Inagaki F; Pfaffenbach M; Stevens MC Propargyltrimethylsilanes as Allene Equivalents in Transition Metal-Catalyzed [5 + 2] Cycloadditions. Org. Lett 2014, 16, 2923–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Wender PA; Ebner C; Fennell BD; Inagaki F; Schroder B Ynol Ethers as Ketene Equivalents in Rhodium-Catalyzed Intermolecular [5 + 2] Cycloaddition Reactions. Org. Lett 2017, 19, 5810–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Wender PA; Fournogerakis DN; Jeffreys MS; Quiroz RV; Inagaki F; Pfaffenbach M Structural Complexity Through Multicomponent Cycloaddition Cascades Enabled by Dual-Purpose, Reactivity Regenerating 1,2,3-Triene Equivalents. Nat. Chem 2014, 6, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Jiang G-J; Fu X-F; Li Q; Yu Z-X Rh(I)-Catalyzed [5 + 1] Cycloaddition of Vinylcyclopropanes and CO for the Synthesis of α,β-and β,γ-Cyclohexanones. Org. Lett 2012, 14, 692–695. [DOI] [PubMed] [Google Scholar]
- (147).Liu C-H; Yu Z-X Rh-Catalyzed [5 + 1] Cycloaddition of Allenylcyclopropanes and CO: Reaction Development and Application to the Formal Synthesis of (−)-Galanthamine. Org. Biomol. Chem 2016, 14, 5945–5950. [DOI] [PubMed] [Google Scholar]
- (148).Yao ZK; Li JJ; Yu Z-X Rh-Catalyzed [7 + 1] Cycloaddition of Buta-1,3-dienylcyclopropanes and CO for the Synthesis of Cyclooctadienones. Org. Lett 2011, 13, 134–137. [DOI] [PubMed] [Google Scholar]
- (149).Fu X-F; Xiang Y; Yu Z-X RhI -Catalyzed Benzo/[7+1] Cycloaddition of Cyclopropyl Benzocyclobutenes and CO by Merging Thermal and MetalCatalyzed C-C Bond Cleavages. Chem. –Eur. J 2015, 21, 4242–4246. [DOI] [PubMed] [Google Scholar]
- (150).Zhuang Z; Li CL; Xiang Y; Wang YH; Yu Z-X An Enyne Cycloisomerization/[5+1] Reaction Sequence to Synthesize Tetrahydroisoquinolinones from Enyne-Enes and CO. Chem. Commun 2017, 53, 2158–2161. [DOI] [PubMed] [Google Scholar]
- (151).Murakami M; Itami K; Ubukata M; Tsuji I; Ito Y Iridium-Catalyzed [5 + 1] Cycloaddition: Allenylcyclopropane as a Five-Carbon Assembling Unit. J. Org. Chem 1998, 63, 4–5. [DOI] [PubMed] [Google Scholar]
- (152).Melcher M-C; Wachenfeldt H. von; Sundin A; Strand D Iridium Catalyzed Carbocyclizations: Efficient (5+2) Cycloadditions of Vinylcyclopropanes and Alkynes. Chem. –Eur. J 2015, 21, 531–535. [DOI] [PubMed] [Google Scholar]
- (153).Wender PA; Fuji M; Husfeld CO; Love JA Rhodium-Catalyzed [5 + 2] Cycloadditions of Allenes and Vinylcyclopropanes: Asymmetric Total Synthesis of (+)-Dictamnol. Org. Lett 1999, 1, 137–139. [Google Scholar]
- (154).Wender PA; Zhang L Asymmetric Total Synthesis of (+)-Aphanamol I Based on the Transition Metal Catalyzed [5 + 2] Cycloaddition of Allenes and Vinylcyclopropanes. Org. Lett 2000, 2, 2323–2326. [DOI] [PubMed] [Google Scholar]
- (155).Ashfeld BL; Martin SF Enantioselective Syntheses of Tremulenediol A and Tremulenolide A. Org. Lett 2005, 7, 4535–4537. [DOI] [PubMed] [Google Scholar]
- (156).Fan X; Zhuo L-G; Tu YQ; Yu Z-X Formal Syntheses of (±)-Asterisca-3(15),6-diene and (±)-Pentalenene using Rh(I)-Catalyzed [(5 + 2) +1] Cycloaddition. Tetrahedron 2009, 65, 4709–4713. [Google Scholar]
- (157).Yuan C; Jiao L; Yu Z-X Formal Synthesis of (±)-Hirsutic Acid C using the Tandem Rh(I)-Catalyzed [(5 + 2) + 1] Cyclo-addition/Aldol Reaction. Tetrahedron Lett. 2010, 51, 5674–5676. [Google Scholar]
- (158).Liang Y; Jiang X; Yu Z-X Enantioselective Total Synthesis of (+)-Asteriscanolide via Rh(i)-Catalyzed [(5 + 2) + 1] Reaction. Chem. Commun 2011, 47, 6659–6661. [DOI] [PubMed] [Google Scholar]
- (159).Liang Y; Jiang X; Fu X-F; Ye S; Wang T; Yuan J; Wang Y; Yu Z-X Total Synthesis of (+)-Asteriscanolide: Further Exploration of the Rhodium(I)-Catalyzed [(5 + 2) + 1] Reaction of Ene-Vinylcyclopropanes and CO. Chem. Asian. J 2012, 7, 593–604. [DOI] [PubMed] [Google Scholar]
- (160).Feng Y; Yu Z-X Formal Synthesis of (±)-Galanthamine and (±)-Lycoramine Using Rh(I)-Catalyzed [(3 + 2) + 1] Cycloaddition of 1-Ene–Vinylcyclopropane and CO. J. Org. Chem 2015, 80, 1952–1956. [DOI] [PubMed] [Google Scholar]
- (161).Bose S; Yang J; Yu Z-X Formal Synthesis of Gracilamine Using Rh(I)-Catalyzed [3 + 2 + 1] Cycloaddition of 1-Yne–Vinylcyclopropanes and CO. J. Org. Chem 2016, 81, 6757–6765. [DOI] [PubMed] [Google Scholar]
- (162).Yang J; Xu W; Cui Q; Fan X; Wang L-N; Yu Z-X Asymmetric Total Synthesis of (−)-Clovan-2,9-dione Using Rh(I)- Catalyzed [3 + 2 + 1] Cycloaddition of 1-Yne-vinylcyclopropane and CO. Org. Lett 2017, 19, 6040–6043. [DOI] [PubMed] [Google Scholar]
- (163).Zhang M; Tang W Synthesis of Functionalized Cyclohexenone Core of Welwitindolinones via Rhodium-Catalyzed [5 + 1] Cycloaddition. Org. Lett 2012, 14, 3756–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Wang L-N; Cui Q; Yu Z-X A Concise Total Synthesis of (−)-Mesembrine. J. Org. Chem 2016, 81,10165–10171. [DOI] [PubMed] [Google Scholar]
- (165).Trost BM; Hu Y; Horne DB Total Synthesis of (+)-Frondosin A. Application of the Ru-Catalyzed [5+2] Cycloaddition. J. Am. Chem. Soc 2007, 129, 11781–11790. [DOI] [PubMed] [Google Scholar]
- (166).Trost BM; Waser J; Meyer A Total Synthesis of (−)-Pseudolaric Acid B. J. Am. Chem. Soc 2008, 130, 16424–16434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Taber DF; Bui G; Chen B Synthesis of (−)-Delobanone. J. Org. Chem 2001, 66, 3423–3426. [DOI] [PubMed] [Google Scholar]
- (168).Taber DF; Sheth RB; Tian WW Synthesis of (+)-Coronafacic Acid. J. Org. Chem 2009, 74, 2433–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Goldberg AFG; Stoltz BM A Palladium-Catalyzed Vinylcyclopropane (3 + 2) Cycloaddition Approach to the Melodinus Alkaloids. Org. Lett 2011, 13, 4474–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Saint-Denis TG; Zhu R-Y; Chen G; Wu Q-F; Yu J-Q Enantioselective C(sp3)–H Bond Activation by Chiral Transition Metal Catalysts. Science 2018, 359, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Gensch T; James MJ; Dalton T; Glorius F Increasing Catalyst Efficiency in C-H Activation Catalysis. Angew. Chem., Int. Ed 2018, 57, 2296–2306. [DOI] [PubMed] [Google Scholar]
- (172).Yi H; Zhang G; Wang H; Huang Z; Wang J; Singh K; Lei A Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev 2017, 117, 9016–9085. [DOI] [PubMed] [Google Scholar]
- (173).Huang X; Meggers E Asymmetric Photocatalysis with Bis-cyclometalated Rhodium Complexes. Acc. Chem. Res 2019, 52, 833–847. [DOI] [PubMed] [Google Scholar]
- (174).Waldvogel SR; Lips S; Selt M; Riehl B; Kampf CJ Electrochemical Arylation Reaction. Chem. Rev 2018, 118, 6706–6765. [DOI] [PubMed] [Google Scholar]
- (175).Robertson JC; Coote ML; Bissember AC Synthetic Applications of Light, Electricity, Mechanical Force and Flow. Nat. Rev. Chem 2019, 3, 290–304. [Google Scholar]
- (176).Kubota K; Pang Y; Miura A; Ito H Redox Reactions of Small Organic Molecules Using Ball Milling and Piezoelectric Materials. Science 2019, 366, 1500–1504. [DOI] [PubMed] [Google Scholar]
- (177).Friščič T; Mottillo C; Titi HM Mechanochemistry for Synthesis. Angew. Chem., Int. Ed 2020, 59, 1018–1029. [DOI] [PubMed] [Google Scholar]