

Summary
Protein-protein interaction (PPI) prediction is essential to understand the functions of proteins in various biological processes and their roles in the development, progression, and treatment of different diseases. To perform economical large-scale PPI analysis, several artificial intelligence-based approaches have been proposed. However, these approaches have limited predictive performance due to the use of in-effective statistical representation learning methods and predictors that lack the ability to extract comprehensive discriminative features. The paper in hand generates statistical representation of protein sequences by applying transfer learning in an unsupervised manner using FastText embedding generation approach. Furthermore, it presents “ADH-PPI” classifier which reaps the benefits of three different neural layers, long short-term memory, convolutional, and self-attention layers. Over two different species benchmark datasets, proposed ADH-PPI predictor outperforms existing approaches by an overall accuracy of 4%, and matthews correlation coefficient of 6%. In addition, it achieves an overall accuracy increment of 7% on four independent test sets. Availability: ADH-PPI web server is publicly available at https://sds_genetic_analysis.opendfki.de/PPI/
Subject areas: Association analysis, Bioinformatics, Computational bioinformatics
Graphical abstract
Highlights
-
•
Protein sequences representation generation through unsupervised transfer learning
-
•
A unique paradigm for sequence fixed length generation
-
•
Development of a robust, precise, and interpretable classifier
-
•
Development of a public web server to predict protein-protein interactions on the go
Association analysis; Bioinformatics; Computational bioinformatics
Introduction
Proteins are large and complex biomolecules that perform a multitude of crucial functions within living organisms mostly by interacting with other proteins (Berggård et al., 2007). Protein-protein interaction (PPI) analysis is important to understand diverse biological processes including cell proliferation (Nooren and Thornton, 2003), signal transduction (Pawson and Nash, 2000), DNA transcription, replication (Zhang et al., 2012; Vickers, 2017), hormone regulation (Zhao, 2015), cycle control (Kulminskaya and Oberer, 2020), and neuro-transmission (Südhof, 1995). It also helps to identify disease-related signaling pathways and symbolize unfamiliar targets for therapeutic intervention (You et al., 2010). In-depth exploration of PPIs is critical for a thorough understanding of protein functionalities, genetic mechanisms (Wang et al., 2007; Alberts, 1998), discovery of new drug targets (Andrei et al., 2017), and development of effective preventive or therapeutic strategies to combat diseases (Petta et al., 2016).
A number of experimental approaches including tandem affinity purification (Gavin et al., 2002), mass spectrometric protein complex identification (Ho et al., 2002), protein chips (Zhu, 2003), and yeast two-hybrid (Y2H) (Ito et al., 2000; Krogan et al., 2006) have been utilized to infer PPIs. However, these experimental methods are expensive and time-consuming (Schoenrock et al., 2014). Furthermore, because of high specificity between proteins, these experimental approaches produce significant false positive results which marks the need of additional methodologies to cross-check the obtained results. Due to slow sequence analysis process, these approaches have been typically applied to identify intra-species PPIs, whereas inter-species interactome remained comparatively understudied (Schoenrock et al., 2014). Advancements in high-throughput approaches and the influx of PPI data related to different species have given rise to many databases including the Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi (Salwinski et al., 2004), the Molecular Interaction Database (MINT): RRID:SCR_003546; http://integrativebiology.org (Licata et al., 2012), and the Human Protein References Database (HPRD): RRID:SCR_007027; http://www.hprd.org (Peri et al., 2004). The public availability of such humongous annotated data has opened new horizons for the development of computational approaches for economical, fast, and more accurate analysis of PPIs.
In order to predict PPIs, to date, a plethora of computational approaches have been developed (Joshi et al., 2004; Qi et al., 2005) which can be broadly segregated into three classes: 1) Structure based, 2) Network based, and 2) Sequence based. Structure-based approaches estimate the likelihood of PPIs by leveraging primary and higher level spatial structures like secondary, tertiary, or quarternary structures (Northey et al., 2018). Those proteins are more likely to interact in which compatibility levels of interacting regions are high or in which spatial structures more oftenly appear on protein-protein binding-motif regions (Northey et al., 2018; Espadaler et al., 2005; Singh et al., 2010). Following this principle, (Hue et al., 2010) performed the pioneer work to predict PPIs in which they fed structural information of protein pairs to support vector machine (SVM) classifier. (Zhang et al., 2012) performed similar work by using protein structural information and Bayesian classifier for PPI interaction prediction. (Hosur et al., 2012) utilized protein structural information to compute the interaction confidence score for each protein pair using a boosting classifier. Structural information-based PPI predictors neglect the mutual influence of local structures (Fu et al., 2019; Asim et al., 2020a). Such approaches are more vulnerable to overlook important information for accurate PPI prediction which might be present in primary sequences and likely to get lost while extracting structural information (Fu et al., 2019; Asim et al., 2020a).
Network-based PPI prediction approaches utilize the link information present in existing PPI networks. PPI networks are hierarchical illustrations of interacting proteins and exist in form of ontologies where each node represents a particular protein and interaction of two different proteins is represented by an association link. Proteins resided in upper hierarchy act as parents and their attached interacting partners of lower hierarchy act as child’s. Network-based PPI prediction approaches extract the names of proteins from existing ontologies to find their biological characteristics in other resources and heterogeneous relations between proteins in order to predict interactions between new proteins on the basis of prior learning. Initial network-based approaches considered that those proteins are more likely to interact which share more common interacting partners in PPI network (Kovács et al., 2019). However, these approaches have become obsolete after the discovery of (Kovács et al., 2019) that two proteins are more likely to interact if at least one of them is very similar to other’s interacting partners. But (Kovács et al., 2019) approach has limited practical significance as it lacks to determine the interactions between the long distant proteins. To address this problem, (Wang et al., 2020b) predicted PPIs without defining the length of different network paths in advance; however, their approach heavily relies on the quality of PPI network. Most recent paradigm of network-based approaches considers that proteins of same functional module are more likely to interact as compared to the proteins of different functional module (Hu et al., 2021b). Using the already known information of the functional modules, (Hu et al., 2021a) integrated biological information of proteins, particularly Gene Ontology into PPI network to predict PPIs. Likewise, Ioan et el. (Ieremie et al., 2022) proposed attention-based deep learning model which used graph-based embeddings to learn deep semantic relations of Gene Ontology to distinguish interactive and non-interactive protein sequence pairs. A closer look at different network-based PPI prediction approaches reveals that these approaches completely rely on pre-computed PPI networks and biological information, both of which need periodic updates to cater huge proteins-related data produced by high-throughput technologies. Furthermore, such resources are characterized by high false-positive as well as false negative rates which eventually hamper the performance of PPI predictors. Therefore, raw sequence-based PPI prediction approaches are widely considered more appropriate to perform large-scale PPI analysis.
To date, several raw sequence-based machine and deep learning-based approaches have also been proposed (Yu et al., 2021; Jiang et al., 2020; Wang et al., 2020a) for PPI prediction. For example, most recently, (Yu et al., 2021) proposed a machine learning-based PPI predictor GcForest-PPI. It utilized residues composition information and physicochemical characteristics to generate statistical representation of protein sequences. It used elastic net (Zou and Hastie, 2005) to extract discriminative set of features that were passed to an ensemble classifier based on three different models namely XGBoost, Random Forest, and Extra-Tree. GcForest-PPI achieved the accuracy of 95.44% and 89.26% on benchmark Saccharomyces cerevisiae (S.cervisiae) and Helicobacter pylori (H.pylori) datasets. (Kong et al., 2020) presented another machine learning approach namely FCTP-WSRC. They utilized amino acid physicochemical properties, composition, and transition information to generate statistical representations of protein sequences. They utilized principal component analysis to reduce redundant features and generate better feature space. Using reduced statistical representations and WSRC (Kong et al., 2020) classifier, they managed to achieve the accuracy of 86.73% and 78.70% on 2 benchmark S.cervisiae and H.pylori datasets. (Jia et al., 2015) also proposed a machine learning-based PPI predictor namely “iPPI-Esml”. They combined residue composition information, physicochemical characteristics, and protein chain-specific wavelet transform information to generate statistical representations of protein sequences which were passed to a deep forest classifier. The iPPI-Esml approach achieved the accuracy of 95% on benchmark S.cervisiae and 90% on H.pylori datasets.
Apart from machine learning-based PPI predictors, (Yao et al., 2019) proposed a deep learning-based predictor namely DeepFE-PPI. They utilized Word2vec-based embedding generation approach (Mikolov et al., 2013) to generate statistical representations of protein sequences which were passed to a multilayer perceptron model for PPI prediction. DeepFE-PPI achieved the accuracy of 95% on benchmark S.cerevisiae dataset. (Du et al., 2017) presented DeepPPI which utilized residues physicochemical properties to generate statistical representations of protein sequences. They utilized a multilayer perceptron model which extracted the high-level discriminative features from statistical vectors to make accurate PPI prediction. DeepPPI achieved the accuracy of 94% and 86% on 2 benchmark S.cervisiae and H.pylori datasets.
Critical analysis of machine and deep learning-based PPI predictors (i.e GcForest-PPI (Yu et al., 2021) WSRC (Kong et al., 2020), DeepFE-PPI (Yao et al., 2019)) reveals that residue composition or physicochemical properties-based protein sequence encoding methods overlook the relationships that exist between different amino acid segments as a function of context of long protein sequences (Jia et al., 2015; Yu et al., 2021). Furthermore, selecting an optimal set of physicochemical properties from a huge available collection requires extensive empirical evaluation (Jia et al., 2015; Yu et al., 2021). Besides, concatenation of statistical representations generated through different types of encoding methods also gives birth to redundant features. To remove redundant features, existing PPI predictors (Kong et al., 2020; Yu et al., 2021) utilize dimensionality reduction or feature selection approaches to generate an effective feature space. However, dimensionality reduction approaches generally prove in-efficient for large and weakly nonlinear data (Sorzano et al., 2014; Chao et al., 2019). Also, determining the number of principal components for the generation of compressed representation varies across different datasets, indicating that optimal principal components are found through comprehensive empirical evaluation. Similarly, major disadvantage of using elastic net as a feature selection approach (Yu et al., 2021) is the high computational cost as one needs to cross validate the relative weights of L1 and L2 penalty. Elastic-net leverages a combination of L1 and L2 penalty in order to shrink coefficient of un-important features to near zero, which is a computationally expensive and a time-consuming process (Sanchez-Pinto et al., 2018).
Furthermore, Word2vec (Mikolov et al., 2013)-based PPI prediction approaches (Yao et al., 2019) also lack to generate effective statistical representation of protein sequences. Because, Word2vec (Mikolov et al., 2013) treats k-mers as atomic entities to generate their distinct vectors in which it neglects the distribution of amino acids within each k-mer. FastText (Bojanowski et al., 2017) is an extension of Word2vec (Mikolov et al., 2013) where vector of each k-mer is computed by considering the distribution of k-mers and distribution of amino acids inside the k-mers. Also, our previous work (Asim et al., 2020b) found that among three different neural embedding generation approaches namely Word2Vec, FastText, and Glove, FastText approach most effectively captures semantic information of k-mers.
We use FastText approach to generate comprehensive contextual information aware statistical vectors for k-mers present in protein sequences. Furthermore, we generate fixed length protein sequences using six traditional and four robust fixed length generation approaches. We propose a robust attention-based deep hybrid model namely ADH-PPI which makes best use of different neural network layers and optimization strategies for accurate PPI prediction. ADH-PPI makes use of long short-term memory, convolutional, and attention layers to find most discriminative features along with their short- and long-range dependencies important to effectively distinguish interactive protein sequence pairs from non-interactive protein sequence pairs. To avoid under-fitting and over-fitting, training of the ADH-PPI is optimized using different kinds of dropout, normalization, and learning rate decay strategies.
A comprehensive empirical evaluation indicates that proposed ADH-PPI approach outperforms several machine and deep learning-based PPI predictors across 6 different species benchmark datasets with a decent margin. To better describe the decisions of proposed ADH-PPI approach, we map the weights of statistical feature space to potential k-mer distributions which contribute the most for accurate PPI prediction through the reverse engineering strategy.
Results
Key idea
We generate an effective statistical representation of protein sequences which is based on comprehensive local and global contextual information of sequence residues captured by applying transfer learning in an unsupervised manner using FastText-based embedding generation approach. To generate fixed length proteins sequences without losing residues distributions important for protein-protein interaction prediction, we use six traditional and four robust fixed length generation methods. Using optimized fixed length protein sequences, we develop a robust classifier which makes best use of heterogeneous neural layers such as long short-term memory layer, convolutional layer, and attention layer to capture most informative hidden features and their long-range dependencies essential to effectively distinguish interactive proteins from non-interactive proteins.
Summary of results
We comprehensively describe the performance produced by 6 traditional pre-processing strategies used to generate fixed length sequences. Furthermore, we compare the performance of 4 distinct settings based on subsequences to showcase which region of protein sequences contains most crucial information about PPI prediction. We perform a performance comparison of proposed predictor using traditional pre-processing strategies and proposed subsequence generation settings. We quantify the performance impact of CNN layer in proposed predictor. Finally, we compare the performance of proposed ADH-PPI approach with existing PPI predictors using two core datasets and four independent test sets.
A comprehensive performance analysis of traditional sequence pre-processing strategies
Figure 1 illustrates the performance values produced by proposed ADH-PPI predictor under the hood of 6 traditional copy padding and sequence truncation approaches used to generate fixed length sequences across two benchmark core datasets.
Figure 1.

Comparison of the performance of proposed ADH-PPI approach across 2 different S.cerevisiae and H.pyloir datasets using 6 traditional sequence fixed length generation approaches
Performance analysis of 6 commonly used pre-processing strategies over Saccharomyces cerevisiae (S.cerevisiae) dataset indicates that mapping protein sequence to maximum possible length and applying copy padding at the end of protein sequences marks best performance of 92% in terms of accuracy and F1-score. Mapping protein sequences to average length and applying padding or sequence truncation trick at the end of protein sequences achieve second best performance. Among all 3 settings which applies copy padding or sequence truncation at the end of protein sequences, mapping protein sequences to minimum possible length attains lowest performance across both evaluation measures. On the other hand, from 3 settings where copy padding or sequence truncation trick is applied at the start of protein sequences, once again mapping protein sequences to maximum possible length achieves overall third best and slightly better performance than other 2 settings based on average and minimum length.
Contrarily, over Helicobacter pylori (H.pylori) dataset, mapping protein sequences to average sequence length and applying copy padding or sequence truncation at the ending region of protein sequence marks best performance followed by the performance produced by maximum sequence length setting where copy padding is applied at the starting region of protein sequences across both evaluation metrics. Setting based on maximum length where copy padding is applied at the end of protein sequence and setting based on minimum length where sequence truncation is applied at the end of sequence length achieve almost similar performance of around 87% in terms of accuracy and F1-score. Whereas, average sequence length-based setting where copy padding or sequence truncation is applied at starting region of protein sequences marks slightly better performance than minimum sequence length-based setting.
Among all 6 traditional copy padding or sequence truncation approaches, sequence fixed length generation approaches which apply copy padding or sequence truncation at the ending regions of protein sequences using average or maximum sequence length mark better performance across both core datasets.
Performance analysis of proposed subsequence-based pre-processing approaches
To showcase the impact of 4 different subsequence-based fixed length generation strategies on the performance of proposed classifier, Figure 2 illustrates which protein regions contain most informative distribution of residues for PPI predictions across core datasets of 2 distinct species.
Figure 2.

Proposed classifier performance analysis under the hood of 4 different subsequences-based strategies used to generate fixed length sequences
Here PA represents protein A and PB refers to protein B, whereas S indicates the starting amino acids of protein sequence and E represents the ending amino acids of protein sequence.
A critical analysis indicates that over S.cerevisiae dataset, performance of 2 settings where amino acids taken from the start of protein A are combined with the amino acids taken from the end of protein B, and amino acids of starting region of protein A are combined with amino acids of starting region of protein B marks similar performance trends across different thresholds of residues. While former setting achieves the performance of 95.5%, latter setting attains the performance of 94% until 20 residues. With the increase of amino acids, performance of both settings slightly fluctuates before finishing at 95% and 93.5%, respectively, at 70 residues across both evaluation metrics. Former setting achieves the peak performance using 40 amino acids whereas latter setting marks the best performance with 10 amino acids. Performance of setting-5 which explores the start-end regions of protein A and protein B almost gradually declines until 30 amino acids, increases up to 94% with 40 amino acids before flattening off across rest of amino acids thresholds. Likewise, performance of setting-3 which selects amino acids from the ending regions of protein A and protein B also progressively decreases from the peak of 93.5% until 30 amino acids before leveling off until 50 amino acids and finishing at 93% at 70 amino acids in terms of accuracy and F1-score. Among all 4 settings, setting-4 which selects amino acids from starting region of protein A and ending region of protein B marks best performance followed by setting-5 which explores the start-end region of both proteins. Whereas, setting-3 marks the lowest performance among all settings based on protein discriminative subsequences.
Over H.pylori dataset (Figure 2), performance of setting-4 remains around 86% until 30 residues before jumping to the peak of 91% with 40 amino acids which declines afterward and finished at 87% with 70 amino acids. Here, performance of setting-2 slightly fluctuates until 40 amino acids before declining and leveling off at 87%. Performance of setting-2 almost gradually decreases from 86% to 84% until 30 amino acids, jumps to the peak of 88.5% until 50 amino acids before decreasing and ending around 86%. Setting-5 performance shows upward trend at most amino acids thresholds and finishes around 86% across both evaluation metrics. Like S.cerevisiae dataset, once again, setting-4 which explores the starting region of protein A and ending region of protein B marks best performance in terms of accuracy and F1-score. However, for H.pylori dataset, peak performances of all 4 setting are comparatively lower than the figures achieved over S.cerevisiae dataset across both evaluation metrics.
Overall, among all protein subsequence-based settings, setting-4 which selects amino acids from starting region of protein A and ending region of protein B marks best performance across both core PPI datasets in terms of accuracy and F1-score.
Performance comparison of proposed subsequence approaches with traditional sequence fixed length generation approaches
In order to compare the performance of traditional copy padding or sequence truncation-based settings with 4 other settings which explores the performance potential of distinct regions of protein sequences by selecting different number of residues, Figure 3 indicates area under receiver operating characteristics (AU-ROC) produced by 5 different settings over S.cerevisiae and H.pylori datasets. As is indicated by the Figure 3, over S.cerevisiae dataset, in setting-1, applying traditional copy padding or sequence truncation approaches at the ending region of protein sequence slightly achieve better degree of separability as compared to those approaches which pad or truncate starting region of protein sequence. Former approaches attain the peak of 95% and latter approaches acquire the peak of 94%. Among all 6 approaches, mapping protein sequences to average length and applying copy padding or sequence truncation at the end of protein sequences mark best performance followed by another ending region-based setting which maps protein sequences to minimum length.
Figure 3.

Impact of 5 different settings on the performance of proposed ADH-PPI approach across 2 different S.cerevisiae and H.pyloir datasets for the task of PPI prediction in terms of area under receiver operating characteristics
Setting 1 is based on traditional copy padding, sequence truncation, and hybrid approaches. Setting 2, 3, 4, and 5 are based on subsequences criteria where x number of amino acids from staring and ending regions of Protein A and Protein B are taken. The value of x varies from 10 to 70 amino acids with the difference of 10 amino acids. Setting 2 takes x number of amino acids from starting region of Protein A and ending region of Protein B. Setting 3 takes x number of amino acids from ending region of Protein A and Protein B. Setting 4 takes x number of amino acids from starting region of Protein A and Protein B. Setting 5 takes x number of amino acids from starting and ending region of Protein A and starting and ending region of Protein B.
Furthermore, in setting-2 based on partial protein sequences, with the influx of residues, ADH-PPI degree of separability gets improved up to the peak of 98% until 30. Afterward, ADH-PPI performance fluctuates across different residue thresholds before finishing at 97%. However, all setting-2 residue variants achieve better performance than traditional copy padding or sequence truncation approaches (setting-1), indicating the prime performance potential of protein subsequences.
In setting-3 which explores the performance potential of merely ending region of protein pairs, varying the residues from 10 to 70, performance of ADH-PPI remains almost constant at 96% which is still better than the performance attained by most commonly used sequence fixed length generation approaches (setting-1). Likewise, in setting-4 which selects different residues from starting region of protein A and ending region of protein B, ADH-PPI achieves the degree of separability of 98% across 7 different residue thresholds, showing best AU-ROC among all 5 settings. Whereas setting-5 based on start-end region of protein pairs attains the performance of 97% across all 7 residue thresholds, indicating degree of separability comparable to setting-2.
On the other hand, over H.pylori dataset, applying copy padding or sequence truncation approaches at the starting region of protein pairs attain slightly superior degree of separability as compared to approaches based on ending region of protein sequences. However, unlike S.cerevisiae dataset, here, both kind of approaches mark better performance by mapping the protein sequences to minimum length. In setting-2, performance of ADH-PPI declines from 93% to 91% until 30 residues however jumps to 95% until 50 residues before finishing at 93%. Overall, it outperforms traditional copy padding or sequence truncation approaches by 2% in terms of AU-ROC. In setting-3 which merely selects residues from ending region of protein pairs, performance of ADH-PPI fluctuates by the figure of 1%. ADH-PPI attains the degree of separability of 93% with 40 residues, indicating overall better performance than setting-1 but slightly lower performance than setting-2. Like S.cerevisiae dataset, here once again, setting-4 based on starting region of protein A and ending region of protein B achieves best degree of separability among all 5 settings. With the influx of residues, ADH-PPI performance jumps to 96% until 40 residues before slightly fluctuating and ending at 93%. Whereas, performance of setting-5 based on start-end region of protein pairs increases upto 92% until 30 residues and gets flatten afterward across rest of the residue thresholds.
In a nutshell, prime objective of developing artificial intelligence-based predictors is to make best use of raw protein sequences, and extract distinct distribution of amino acids in the sequences in order to discriminate interactive protein sequences from non-interactive protein sequences. However, protein sequences are highly variable in length and deep learning models require fixed length input sequences. Commonly used sequence fixed length generation approaches are copy padding and sequence truncation. In copy padding approach, all sequences are mapped to maximum sequence length by padding certain letter to shorter sequences, whereas in sequence truncation approach, all sequences are mapped to minimum sequence length by eliminating extra amino acids from longer sequences. Distribution of amino acids varies in different sub regions of sequences and the performance of deep learning algorithms primarily relies on the extraction of discriminative distribution of amino acids. Copy padding approach creates unnecessary bias through the repetition of same padding letter which make sequences quite similar to each other; similarly, sequence truncation approach is vulnerable to loose most informative distribution of amino acids. Subsequences-based fixed length generation is more effective as it does not insert any hypothetical letter. Furthermore, it skips constant regions that usually lie in center of the sequences and does not loose informative distribution because it takes both starting and ending regions of the sequences into account. Experimental results reveal that most discriminative distribution of amino acids lies in first 40 amino acids of protein A and last 40 amino acids of protein B, indicating the success of subsequence-based setting for capturing the informative and discriminative essence of protein sequences.
Performance impact of CNN layer
To better illustrate the necessity of convolutional (CNN) layer in proposed ADH-PPI predictor, we have performed experimentation on H.pylori dataset under the hood of two different settings. In first setting, we take long short-term memory (LSTM), CNN, and Attention layers, whereas in second setting, we only take LSTM and attention layers. Table 1 illustrates the predictive performance of both settings in terms of five different evaluation measures namely accuracy, precision, recall, F1-score, and MCC.
Table 1.
Performance analysis of proposed model using LSTM, CNN, and attention layers and only LSTM and attention layers over H.pylori species dataset to quantify the impact of CNN layer
| Evaluation measures | Proposed model with LSTM, CNN, and attention layers | Proposed model with only LSTM and attention layers | Performance difference |
|---|---|---|---|
| Accuracy | 0.926 | 0.919 | Around 1% |
| Precision | 0.928 | 0.921 | Around 1% |
| Recall | 0.961 | 0.945 | Around 2% |
| F1-score | 0.944 | 0.912 | Around 3% |
| MCC | 0.855 | 0.848 | Around 1% |
Among different subsequence-based settings, using 40 residues from starting region of protein A and 40 residues from ending region of protein B, proposed model with LSTM and attention layers achieves the accuracy of 0.919, recall of 0.945, precision of 0.921, F1-score of 0.912, and MCC of 0.848. However, this performance is less than the performance achieved using LSTM, CNN, and attention layers in proposed predictor by the F1-score of 3%, accuracy of 2%, precision, recall, and MCC of 1%. Overall, exclusion of CNN layer slightly drops the predictive performance, and better performance is achieved when LSTM, CNN, and attention layers are used in proposed predictor. This proves the necessity of CNN layer in proposed predictor that essentially captures local dependencies and translational invariance of amino acids present in protein subsequences which complement predictive performance.
Performance assessment of ADH-PPI robustness for different order protein sequence pairs
Empirical evaluation reveals that proposed ADH-PPI achieves the highest performance on 2 core benchmark datasets and 4 independent test sets on account of protein sequence pairs generated by combining the subsequence of protein A with subsequence of protein B. Among different subsequence generation settings, setting-4 (Figure 11) which focuses on the starting region of protein A and ending region of protein B develops most informative residue distribution-based protein sequence pairs. However, it is important to note that we have randomly chosen one protein as protein A and other protein as protein B. Building on the equal possibility of generating conversely ordered protein sequence pairs, here we validate the idea that regardless of protein sequence order, starting region of one protein and ending region of other protein contains the most informative residue distribution for PPI prediction.

A variety of settings based on traditional copy padding or sequence truncation, and proposed bag of most informative amino acids distribution-based tricks used to generate fixed length sequences
Mainly, experimentation is performed with optimal subsequence generation setting across 2 core datasets and 4 independent test sets by treating one protein subsequence as protein A, other protein subsequence as protein B, and exchanging the the order of protein subsequences. Furthermore, we use same parameters (e.g subsequence window size, model parameters (Table 1) values described in previous sections 2.5 for each dataset across both kind of paradigms in order to accurately reveal the robustness of ADH-PPI approach.
Figures 4 and 5 illustrate the performance produced by setting-4 using protein sequence pairs generated by treating one protein as protein A and other protein as protein B as well as reversing the order on 2 core benchmark datasets and 4 independent test sets, respectively. As is indicated by the Figures 4 and 5, ADH-PPI achieves almost same performance across all datasets with protein sequence pairs generated using 2 different protein subsequence orders. This indicates that although changing the combination order of protein-subsequences changes the characteristic of protein-sequence pairs, however, proposed ADH-PPI is robust enough to capture most informative distribution of protein sequences important for PPI prediction.
Figure 4.

Performance assessment of optimal most informative subsequence generation setting using 2 differently ordered protein sequence pairs over S.cerevisiae and H.pyloir core datasets
Here P_A represents the Protein A and P_B refers to Protein B, whereas start and end represent the starting and ending region of respective protein.
Figure 5.

Performance assessment of optimal most informative subsequence generation setting using 2 differently ordered protein sequence pairs over C.elegans, H.sapiens, M.musculus, and E.coli independent test sets after training the model on core S.cerevisiae dataset
Performance comparison of proposed ADH-PPI predictor with existing PPI predictors using two benchmark core datasets
In order to prove the integrity of proposed ADH-PPI predictor, rich performance comparison with existing PPI predictors is performance using two core datasets in terms of 4 different evaluation metrics.
Table 2 compares the performance of proposed ADH-PPI predictor with 12 machine and deep learning-based predictors over S.cerevisiae dataset. As indicated by the Table 2, proposed ADH-PPI predictor outperforms auto co-variance and SVM-based PPI prediction methodology (Guo et al., 2008a) by 7%, 5%, and 7%, and KNN-based methodology (Guo et al., 2008a) by 10%, 13%, and 6% in terms of accuracy, recall, and precision, respectively. It outperforms WSRC classifier (Kong et al., 2020) by 9%, 4%, and 14% in terms of accuracy, recall, and MCC and ippi-esml (Jia et al., 2015) approach by 3%, 5%, 3%, and 4% in terms of accuracy, recall, precision, and MCC, respectively. Multi-scale continuous and discontinuous feature representation and SVM classifier-based approach (You et al., 2014) takes the previous best accuracy of 89%–91%, amino acid substitution matrix-based feature representation and RF classifier-based approach (You et al., 2017) attains the accuracy of 94%. RF classifier achieves the accuracy of 95% using multivariate mutual information of protein feature representation (Ding et al., 2016) and 94% using multiscale local descriptor (MLD)-based feature representation (You et al., 2015a). Proposed ADH-PPI predictor outperforms SVM and random forest-based PPI prediction methodologies by the comparable margin. From existing machine learning-based PPI predictors, GcForest-PPI (Yu et al., 2021) achieves top performance in terms of most evaluation metrics. Proposed ADH-PPI predictor surpasses the performance of GcForest-PPI (Yu et al., 2021) by 1% in terms of accuracy and recall and equalizes the MCC performance value.
Table 2.
Performance comparison of proposed ADH-PPI predictor with 12 existing PPI predictors on benchmark S. cerevisiae dataset, where results of existing PPI predictors are taken from (Yu et al., 2021) paper
| Method | ACC (%) | Recall (%) | Precision (%) | MCC |
|---|---|---|---|---|
| ACC+SVM (Guo et al., 2008a) | 0.8933 ± 2.67 | 0.8993 ± 3.68 | 0.8887 ± 6.16 | N/A |
| Code4+KNN (Guo et al., 2008a) | 0.8615 ± 1.17 | 0.8103 ± 1.74 | 0.9024 ± 1.34 | N/A |
| MCD+SVM (You et al., 2014) | 0.9136 ± 0.36 | 0.9067 ± 0.69 | 0.9194 ± 0.62 | 0.8421 ± 0.0059 |
| MLD+RF (You et al., 2015a) | 0.9472 ± 0.43 | 0.9434 ± 0.49 | 0.9891 ± 0.33 | 0.8599 ± 0.0089 |
| PR-LPQ+RF (You et al., 2015b) | 0.9392 ± 0.36 | 0.9110 ± 0.31 | 0.9645 ± 0.45 | 0.8856 ± 0.0063 |
| MIMI + NMBAC+ RF (Ding et al., 2016) |
0.9501 ± 0.46 | 0.9267 ± 0.50 | 0.9716 ± 0.55 | 0.9010 ± 0.0092 |
| LRA+RF (You et al., 2017) | 0.9414 ± 1.8 | 0.9122 ± 1.6 | 0.9710 ± 2.1 | 0.8896 ± 0.026 |
| DeepPPI (Du et al., 2017) | 0.9443 ± 0.30 | 0.9206 ± 0.36 | 0.9665 ± 0.59 | 0.8897 ± 0.0062 |
| ippi-esml (Jia et al., 2015) | 0.9515 ± 0.25 | 0.9221 ± 0.36 | 0.9797 ± 0.60 | 0.9045 ± 0.0053 |
| WSRC (Kong et al., 2020) | 0.8673 | 0.8993 | NA | 0.7693 |
| DeepFE-PPI (Yao et al., 2019) | 0.9478 | 0.9299 | 0.9645 | 0.8962 |
| GcForest-PPI (Yu et al., 2021) | 0.9544 | 0.9272 | 0.9805 | 0.9102 |
| ADH-PPI | 0.9573 | 0.9394 | 0.9575 | 0.9144 |
Turning toward existing deep learning-based PPI prediction methodologies, DeepPPI (Du et al., 2017) and DeepFE-PPI (Yao et al., 2019) achieve almost similar performance on S.cerevisiae dataset in terms of four different evaluation metrics. Proposed ADH-PPI predictor outperforms deep learning-based PPI predictors by 1% in terms of accuracy, recall, and MCC.
Moreover, performance produced by proposed ADH-PPI predictor and ten existing PPI predictors on H.pylori dataset is shown in Table 3. Analysis of Table 3 reveals that proposed ADH-PPI predictor achieves even more promising figures than existing PPI predictors across all evaluation metrics. Proposed ADH-PPI predictor outshines best performing machine learning-based PPI predictor namely GcForest-PPI (Jia et al., 2015) by 7%, 7%, 4%, and 4% in terms of recall, MCC, precision, and accuracy, respectively. It outperforms another top performing MIMI and Random forest-based PPI predictor (Ding et al., 2016) by Matthews correlation coefficient of 13%, recall of 10%, precision of 5%, and accuracy of 6%. In comparison to deep learning-based PPI predictors, proposed ADH-PPI predictor outperforms DeepPPI (Du et al., 2017) predictor by 7%, 7%, 9%, and 12% in terms of accuracy, recall, precision, and MCC.
Table 3.
Performance comparison of proposed ADH-PPI predictor with 10 existing predictors on benchmark H. pylori dataset, where results of existing PPI predictors are taken from (Yu et al., 2021) paper
| Method | ACC (%) | Recall (%) | Precision (%) | MCC |
|---|---|---|---|---|
| SVM [6] | 0.8340 | 0.7990 | 0.8570 | N/A |
| WSR (Nanni, 2005) | 0.8370 | 0.7900 | 0.8700 | N/A |
| Ensemble of HKNN (Nanni and Lumini, 2006) |
0.8660 | 0.8670 | 0.8500 | N/A |
| DCT+WSRC (Huang et al., 2016) | 0.8674 | 0.8643 | 0.8701 | 0.7699 |
| MCD+SVM (You et al., 2014) | 0.8491 | 0.8324 | 0.8612 | 0.7440 |
| MIMI+ NMBAC+RF (Ding et al., 2016) |
0.8759 | 0.8681 | 0.8823 | 0.7524 |
| DeepPPI (Du et al., 2017) | 0.8623 | 0.8944 | 0.8432 | 0.7263 |
| ippi-esml (Jia et al., 2015) | 0.9047 ± 0.84 | 0.9115 ± 1.42 | 0.8999 ± 2.06 | 0.8100 ± 0.0163 |
| WSRC (Kong et al., 2020) | 0.7870 | 0.7321 | NA | 0.7693 |
| GcForest-PPI (Yu et al., 2021) | 0.8926 | 0.8971 | 0.8895 | 0.7857 |
| ADH-PPI | 0.9263 | 0.9609 | 0.9284 | 0.8547 |
To summarize, proposed ADH-PPI predictor outperforms both machine and deep learning-based PPI prediction methodologies by decent margin for S.cerevisiae and by significant margin for H.pylori dataset. It is important to mention that (Kong et al., 2020) proposed FCTP-WSRC predictor results are not comparable to proposed ADH-PPI predictor. Generally, dimensionality reduction approaches such as principal components analysis (PCA) is applied on training data to learn the reduced matrix and the transformation is applied on testing data where test data is projected to reduce feature space. However, (Kong et al., 2020) applied PCA on training and testing data separately which introduces biaseness. In our experimentation, to find the valid performance figures of FCTP-WSRC predictor (Kong et al., 2020), we have applied the PCA in correct manner and reported the valid results on 2 core benchmark datasets (Tables 2 and 3) and independent test sets (Figure 6).
Figure 6.

Accuracy comparison of ADH-PPI and recent PPI predictors on four independent test sets
Performance comparison of ADH-PPI with existing PPI predictors using four independent test sets
To further prove the effectiveness of proposed ADH-PPI predictor, comparison between 6 existing PPI predictors and proposed ADH-PPI predictor is performed. Following experimentation criteria of existing predictors, we train the proposed predictor over core S.cerevisiae dataset and perform evaluation over 4 different independent test sets belonging to C.elegans, E.coli, H.sapiens, and M.musculus species (Yu et al., 2021; Huang et al., 2015a). Figure 6 compares the accuracy of proposed ADH-PPI predictor with existing predictors. As shown by the Figure 6, proposed ADH-PPI predictor achieves best performance across all four independent test sets which is higher than the peak performance achieved by deep learning-based PPI predictor DeepPPI (Du et al., 2017) by 4% for C.elegans, 6% for E.coli, 5% for H.sapiens, and 9% for M.musculus species test sets.
Furthermore, proposed ADH-PPI predictor outperforms machine learning-based state-of-the-art PPI predictor namely GCForest-PPI (Yu et al., 2021) by 3%, 2%, 1%, and 1%, achieving more than 98% performance over all four different species independent test sets.
In a nutshell, over two core datasets and four independent test sets, among all existing PPIs predictors, machine learning-based PPI predictors perform better than deep learning-based predictors. Proposed ADH-PPI predictor outshines state-of-the-art PPI predictor across all 6 datasets of different species including humans, Drosophila, Yeast, Bacterium, Caenorhabditis elegans, and Escherichia coli in terms of most evaluation metrics. The paradigm of considering both k-mer distribution as well as amino acid distributions within k-mer best characterizes the protein sequences. Furthermore, the utilization of LSTM, CNN, and attention ensures the extraction of comprehensive discriminative features along with long-range dependencies which are essential to accurately predict PPIs across different species. The best utilization of multiple strategies not only enhances the predictive power of proposed ADH-PPI approach but also makes the decisions of proposed ADH-PPI predictor interpretable. Therefore, we believe ADH-PPI will prove a great computational asset for biological researchers and practitioners which can be used to find protein-protein interactions, protein non-coding RNA interactions, or even interaction between different biomolecules.
Explainability of the proposed ADH-PPI model
With an aim to overcome a very common black box modeling issue of deep neural networks by decoding the importance of individual amino acids and k-mers, we analyze the attention weights associated with different k-mers to illustrate which k-mers contribute the most in making accurate PPI predictions.
To more precisely demonstrate the explainability of the proposed ADH-PPI approach, we arbitrarily take two protein sequence pairs from test sets of benchmark S.cerevisiae and H.pylori datasets. Following the working paradigm of proposed ADH-PPI predictor, we generate 5-mers of both test protein sequence pairs and feed both test protein sequence pairs 5-mers along with pre-trained embeddings to two different classifiers trained on S.cerevisiae and H.pylori training sets. These classifiers decide whether given protein sequence pairs are interactive or non-interactive. Classifiers make decisions based on the attention weights associated with 5-mers. We extract attention weights and feed these attention weighs to decision explainable module which performs reverse engineering to map these attention weights to different 5-mers. To illustrate better, decision explainable module categorizes different 5-mers into five different groups based on different thresholds applied at attention weights ranging from 0 to 1. Each group is represented with a unique shade of red color, the higher the intensity of red color is the higher the attention weight is for particular k-mer, indicating darkest red color 5-mers and their inherent amino acids contribute the most in making accurate PPI predictions and lightest red color 5-mers and their inherent amino acids make least contributions in making accurate PPI predictions. The attention weights range of five different groups of 5-mers is shown on the x axis of the bar graph (Figure 7) whereas y axis of the bar graph shows the count of 5-mers in each group.
Figure 7.

Most informative and least informative amino acid and K-mers patterns identified by attention layer of proposed ADH-PPI predictor in two test protein sequences belonging to benchmark S.cerevisiae and H.pylori Datasets
It is evident in the Figure 7, for S.cerevisiae dataset, only two 5-mers GGKAG and SAAKA fall in first group which has best range of attention weights 0.90–1.0. Two 5-mers fall in second group which has second best range of attention weights 0.70–0.89. Similarly, one 5-mer falls in third group and three k-mers fall in fourth group which have attention weight ranges of 0.50–0.69 and 0.20–0.49, respectively. Among all groups, fifth group has most eight 5-mers, attention weights of which falls in range of 0.1–0.20. Furthermore, it can be seen that starting 5-mers distribution has the top attention weights where amino acids G and A are most frequent, which contribute the most in making accurate PPI predictions on S.cerevisiae dataset.
Unlike S.cerevisiae dataset, on H.pylori dataset, eighteen 5-mers fall in fifth group, fourteen 5-mers in second group, and four 5-mers in third group. Once again, very few 5-mers fall in first and second group. More specifically, three 5-mers LIFYYF, IFYFL, LDFKG, individual amino acids F and L of central regions of subsequence contribute the most in making accurate PPI predictions on H.pylori dataset.
These attention weight distribution patterns at the k-mer level and amino acid level are quite consistent across most sequences. Furthermore, this is quite consistent with our unique hypothesis of predicting PPIs using only most discriminative subsequences. The starting or central k-mers distribution within subsequences gets the higher attention weight and serves as most influential regions, and the surrounding k-mers distribution gets lower attention weights and exists as supportive and auxiliary information regions. In a nutshell, we validate the ADH-PPI suitability to discover useful patterns in protein sequences, their dependencies, and explainable associations for PPI prediction.
An interactive and user-friendly ADH-PPI web server
We have developed a user-friendly web server for ADH-PPI approach which can be accessed at https://sds_genetic_analysis.opendfki.de/PPI/. Proteomics researchers and practitioners can leverage this web server to determine interactions between proteins solely using raw sequences related to human and mouse species. Researchers can also use this web server to validate experimentally identified PPIs using raw sequences, train and optimize the model from scratch for new species, and perform inference on new sequences belonging to existing or new species.
Conclusion
This paper can be considered a huge milestone toward the accurate prediction of PPIs for a variety of species solely using raw sequences. First, unlike previous methods, it captures comprehensive amino acids order, occurrence, and contextual information by generating k-mer of protein sequences, distributed representations of which are computed as the sum of their embeddings and the embeddings of their inherent amino acid sub-mers using FastText (Bojanowski et al., 2017) approach. Second, instead of feeding entire protein sequences to deep learning models, it explores the discriminative aptitude of multifarious regions of protein sequences to obtain highly informative amino acid distribution-based subsequences. Third, it develops an attention-based deep hybrid neural network which makes best use of heterogeneous layers (LSTM, CNN, and Attention) to make accurate and interpretable PPI predictions. A stringent benchmarking performance comparison of ADH-PPI with existing computational predictors proves that ADH-PPI outperforms existing machine and deep learning-based PPI predictors by decent margin. A compelling future line of current would be to assess the performance potential of ADH-PPI approach for interaction prediction tasks related to other biomolecules.
Limitations of the study
The limitation of current work is that it only identifies key k-mers which contributes the most in making accurate PPI prediction. However, to more comprehensively explain the decisions of proposed ADH-PPI approach, identifying which k-mer distributions map to which hidden features as well as which hidden unit gets activated at what point in time and input scenarios, would further fine-grained the explainability and interpretability of proposed ADH-PPI predictor for biomedical researchers and practitioners.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Desposited data | ||
| S.cerevisiae Dataset | (Guo et al., 2008b) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| H.pylori Dataset | (Martin et al., 2005) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| E.coli Test Set | (Zhou et al., 2011) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| C.elegans Test Setdata | (Zhou et al., 2011) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| Homo Sapiens Test Set | (Zhou et al., 2011) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| Mus Musculus Test Set | (Zhou et al., 2011) | Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| Software and algorithms | ||
| Sub-Sequence Generation Paradigm |
This Study | https://sds_genetic_analysis.opendfki.de/PPI/ |
| Protein Sequence Embeddings (FastText) |
Open Source | https://fasttext.cc/ |
| Python | Open Source | https://www.python.org/ |
| Pytorch | Open Source | https://pytorch.org/ |
| Grid Search | (Bergstra et al., 2011) | http://tinyurl.com/54phrd33 |
| CD-HIT | (Fu et al., 2012) | http://weizhong-lab.ucsd.edu/cd-hit/ |
Resource availability
Lead contact
For more information as well as requests for the materials and code shall be directed to and will be fulfilled by lead contact, Muhammad Nabeel Asim (Muhammad_Nabeel.Asim@dfki.de).
Materials availability
In this study, no new materials are generated.
Method details
Data sources benchmark protein-protein interaction prediction datasets
In order to prove the integrity of proposed ADH-PPI approach and to perform a fair comparison with existing PPIs prediction approaches, we evaluate ADH-PPI performance over PPIs datasets of 6 different species including humans, Drosophila, Yeast, Bacterium, Caenorhabditis elegans, and Escherichia coli.
From Yeast specie, performance of ADH-PPI is evaluated on a well known public benchmark dataset namely Saccharomyces cerevisiae (S.cerevisiae), which is extensively utilized by several researchers for PPI prediction (Yu et al., 2021; Jiang et al., 2020; Wang et al., 2020a). PPIs of Saccharomyces cerevisiae (S.cerevisiae) were first extracted by (Guo et al., 2008b) from Database of Interacting Proteins (DIP): RRID:SCR_003167; https://dip.doe-mbi.ucla.edu/dip/Main.cgi (Xenarios et al., 2002). Authors eliminated those protein pairs where any one of the protein was comprised of less than 50 residues and obtained a dataset of 5,943 protein pairs with positive interactions. To eliminate redundancy, researchers utilized a renowned program CD-HIT (Fu et al., 2012). From 11,188 PPIs, a total of 5,594 PPIs were retained considering that they had less than 40% pairwise sequence similarity with each other. An equal number of negative PPIs were generated using 3 different approaches. In first approach, non-interacting protein pairs were generated by random pairing of proteins which were not present in the positive dataset. In second approach, negative dataset was generated by combining proteins having similar sub-cellular localization patterns extracted from Swiss-Prot database (Bairoch and Apweiler, 1996). In third approach, negative dataset was generated using data augmentation approach. Another widely used PPI prediction dataset (Yu et al., 2021; Jiang et al., 2020; Wang et al., 2020a) Helicobacter pylori belongs to Bacterium specie, which was compiled by (Martin et al., 2005). It contained 2,916 protein pairs out of which 1,458 protein pairs were positive and 1,458 protein pairs were negative. From a collection of protein pairs which were not explicitly declared as interactive, a bunch of protein pairs were selected as non-interacting proteins. Statistics of both core datasets S.cerevisiae and H.pylori are described in Figure.

Statistics of 2 core protein-protein interaction prediction datasets
In order to perform a fair performance comparison with existing PPI predictors and to further prove the versatility of proposed methodology ADH-PPI, we also evaluate ADH-PPI over 4 independent test sets developed by (Zhou et al., 2011). These datasets have been extensively used in literature (Yu et al., 2021; Huang et al., 2015b; Hashemifar et al., 2018). As the procedure used to develop 4 different independent test sets has been described in existing studies (Zhou et al., 2011; Yu et al., 2021; Huang et al., 2015a; Hashemifar et al., 2018), here we only shed light on the statistics of 4 independent test sets. E.coli consists of 6,954 protein pairs with positive interactions, C.elegans contains 4,103, Homo sapiens (H. sapiens) consists of 1,412, and Mus musculus (M. musculus) is composed of 313 protein pairs with positive interactions.
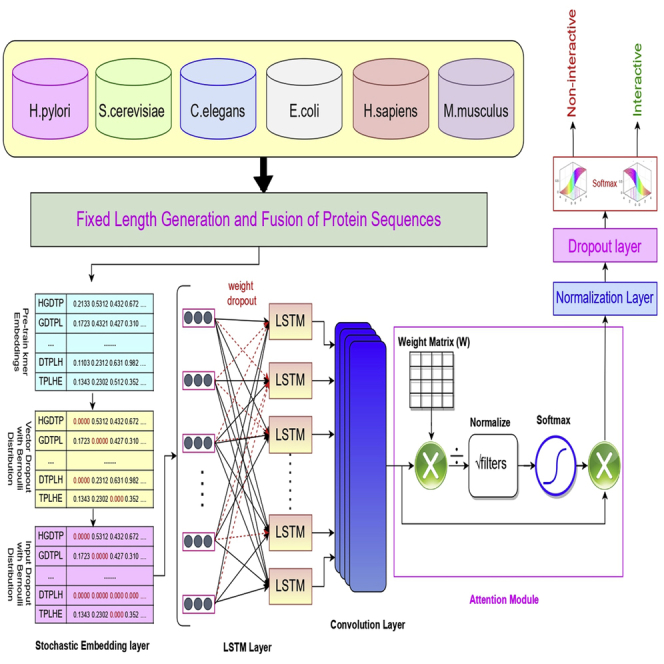
Model architecture
ADH-PPI: An Attention based Deep Hybrid Model for Protein Protein Interaction Prediction
The working of the proposed ADH-PPI predictor can be categorized into three different modules. First module generates effective statistical representations of k-mers present in protein sequences by applying transfer learning in an unsupervised manner. Second module generates fixed length protein sequences using traditional and robust sequence fixed length generation methods. Using fixed length protein sequences and k-mer embeddings, third module trains a robust attention based deep hybrid neural network for PPI prediction. A brief description of each module is provided in the following sub-sections.
K-mer embedding generation using unsupervised transfer learning
To generate k-mer embeddings, first step is to divide the protein sequences into k-mers. Overlapping k-mers are generated by rotating a fixed-size window over a protein sequence where the stride size is always less than the size of window. On the other hand, non-overlapping k-mers are generated by rotating a window with a stride size equal to window size. Figure illustrates the process of generating overlapping and non-overlapping k-mers of a hypothetical protein sequence.

Overlapping and non-overlapping K-mers generation for protein sequences
Protein sequences are made up of 20 distinct amino acids. Hence, in both overlapping or non-overlapping k-mer generation, the unique vocabulary size is equal to . The value of k determines the size of vocabulary which impacts model complexity, memory cost, run time cost, as well as up to what extent amino acid contextual information is taken into account, hence the choice of k is very crucial. Following the work of (Le et al., 2019) and (Asim et al., 2020a), we generate different overlapping and non-overlapping k-mers by varying the window size from 2-to-7 and stride size from 1-to-7.
For different sizes overlapping and non-overlapping k-mers, we generate k-mers embeddings of different dimensions using FastText embedding generation model, working of which is graphically illustrated in Figure.

Workflow of unsupervised transfer learning applied using 6 datasets of distinct species to learn distributed representation of higher order sequence residues
With an aim to capture comprehensive information of amino acids distributions, we take two benchmark S.cerevisiae, H.pylori datasets and four independent test sets in order to most effectively train the FastText model over large dataset of 26,886 protein sequences. For all 26,886 protein sequences, we generate overlapping and non-overlapping k-mers by varying the window size from 2-to-7 and stride size from 1-to-7. This produces number of different k-mers=6 maximum possible different stride size =6 equal to 36 different versions of protein sequences corpus based on different overlapping k-mers and 6 versions of protein sequences corpus based on different non-overlapping k-mers. For each version of protein sequences corpus, we train the FastText model to generate k-mer embeddings of different dimensions d ranging from 100, 120, 240, to 300. For example, by considering non-overlapping 3-mers, 26,886 protein sequences are divided in 3-mers which generates a vocabulary of unique 3-mers and FastText generates d-dimensional statistical vectors for each 3-mer. FastText embedding generation model is an extension of Skipgram model (Mikolov et al., 2013). Given a training k-mer sequence , objective function of Skipgram model can be defined as follows:
| (Equation 1) |
Where represents the collection of surrounding k-mers of current k-mer , given current k-mer , denotes the probability of observing its surrounding k-mer .
| (Equation 2) |
Here represents the scoring function. Skipgram model considers the scoring function as scalar product , where and represent the vectors of two k-mers and respectively. However, SkipGram can only generate a distinct vector for each k-mer without exploiting their sub-kmer information. To overcome this problem, FastText represents a k-mer as a bag of sub-kmers. For instance, k-mer “HGDTP” will be represented by sub-kmers such as #HGD, HGDT, GDTP, DTP# and k-mer itself HGDTP. Unlike Skigpgram model, FastText defines the scoring function as the where denotes the sub-kmers collection of , represents the vector of sub-kmer, and represents the vector of k-mer . In this manner, FastText learns the embeddings of sub-kmers. Using sub-kmers embeddings, a k-mer embedding is learned as the sum of distributed representations of its sub-kmers. Major advantage of FastText embedding generation model is that it takes k-mer distributions as well as distributions of amino acids within k-mers into account to generate effective distributed representation of k-mers. Another advantage is that it shares the distributed representation of sub-kmers across all the k-mers which is extremely useful to generate optimal embeddings for less frequent k-mers. FastText embedding generation model is trained with an objective to maximize the probability of target k-mer over all k-mers present in the vocabulary using a softmax layer. Embedding matrix along with output layer parameters are learned by back propagating the error using stochastic gradient descent and negative sampling approach. Using FastText, we generate effective d-dimensional vectors for k-mers where the value of d is varied from 100, 120, 240, to 300.
Fixed length generation of Protein sequences
Exploratory analysis of 2 core PPI datasets (Figure) indicates that minimum sequence length for both S.cerevisiae and H.pylori datasets is 10 residues, average protein sequence length for S.cerevisiae dataset is around 1100 residues and for H.pylori dataset, average sequence length is around 734 residues. It is evident that protein sequences have high length variability. Considering machine learning approaches require fixed length protein sequences, existing PPI prediction approaches transform variable length protein sequences into fixed length sequences using traditional copy padding or sequence truncation approaches (Yu et al., 2021; Jiang et al., 2020; Wang et al., 2020a). We perform experimentation with 6 different variations of copy padding and sequence truncation approaches in order to quantify their efficacy for PPI prediction. Furthermore, we present a unique way to generate fixed length protein sequences by finding and retaining only most informative amino acids distributions. This section briefly summarizes five different settings to generate fixed length sequences.
In setting, performance of 6 traditional copy padding and sequence truncation is evaluated. In copy padding approach, first maximum length of sequence is computed by comparing corpus sequences. Then, all the sequences having length less than maximum length are extended to make them equal to maximum length by adding certain constant. Sequence truncation is another way to make fixed length sequences where minimum sequence length is computed by comparing corpus sequences. Residues from all those sequences whose length is larger than minimum length are truncated to make them equal to minimum sequence length. Another trend is to utilise both copy padding and truncation approaches where average length is computed by comparing corpus sequences. Certain constant is added in those sequences which are shorter than the average length, whereas, sequences that are larger than the average length are truncated.
In copy padding trick, it is an important question whether the start of the sequences is an ideal location for the addition of constant or the end of the sequences. Likewise, in the sequence truncation approach, it is questionable whether extra residues need to be truncated from start of the sequences or end of the sequences. For copy padding trick, we first add constant at the start of sequences, and in another variation, we add constant at the end of the sequence to find out which approach is more appropriate. Similarly, for the sequence truncation approach, we truncate sequences from the start of corpus sequences and from the end of sequences in other variation. In hybrid sequence fixed length generation paradigm based on average length, we also extend or truncate corpus sequences from the start of sequences or end of the sequences. A graphical representation of all 6 strategies is presented in Figure under the the hood of setting-1.
Considering the vulnerability of traditional copy padding approach to create unnecessary bias through the addition of too many constants and sequence truncation to loose important k-mer distribution while handling flexible protein sequences. Here we propose a unique idea to optimize fixed length sequence generation process where fixed length sequences are generated using only the few residues from different regions of protein sequences which contains the most informative distribution of amino acids for the task of PPI prediction. More specifically, in Figure, under the hood of setting, ADH-PPI selects X residues solely from the starting region of one protein A and Y residues solely from starting region of Protein B. In setting, performance of X residues taken only from the ending region of protein A and Y residues taken merely from the ending region of protein B is evaluated. Whereas, in setting , X residues of protein A taken from the starting region of protein sequence are combined with Y residue of protein B taken from the ending region of protein sequence to assess the discriminative aptitude of start-end region. In last setting, performance is assessed by combining X residues taken from start-end regions of protein A with Y residue taken from start-end region of protein B. To identify up to what number of residues can capture the discriminative essence of protein sequences, in all 4 proposed sub-sequence based fixed length generation settings, we select as minimum number of residues as possible (e.g 10, depending on the minimum sequence length of the benchmark dataset) and iteratively increments this number with a step size of 10 residues up to 50% of average sequence length of benchmark core PPI prediction datasets. In all 4 settings, X and Y range from 10-to-70 residues taken with the difference of 10 residues. By fusing protein A sequences with protein B sequences, the fixed length protein sequence generated through traditional and robust pre-processing strategies are passed to an attention based deep hybrid neural network for PPI prediction.
An attention based deep hybrid neural network (ADH-PPI)
In the marathon of developing robust and precise deep learning based end-to-end frameworks for diverse Genomics and Proteomics sequence analysis tasks, we are witnessing the explosion of deep learning approaches, core architectures of which are mainly formed by deep feed forward neural networks (Li et al., 2019), deep belief networks (Zou et al., 2019), convolutional neural networks (Li et al., 2019), autoencoders (Meyer, 2021), and long short-term memory networks (Meyer, 2021). Predominantly, efforts are being made under the hood of two different paradigms to develop more efficient deep learning models for diverse sequence analysis tasks (Zou et al., 2019; Li et al., 2019; Meyer, 2021). The main focus of one paradigm is to develop deep neural networks based on series of neural layers (i.e convolutional layer, recurrent layer) to effectively capture the non-linearity of genomic and proteomic sequences (Zou et al., 2019; Li et al., 2019; Meyer, 2021; Nusrat and Jang, 2018; Lydia and Francis, 2019). Whereas, other paradigm pays more attention to develop shallow or ensemble neural networks which utilize neural layers (i.e convolutional layer, recurrent layer) in different parallel channels and combine the features extracted by different channels to perform target prediction. The paper in hand develops an attention based deep hybrid model (ADH-PPI) for PPI prediction following the structure of first paradigm. Workflow of proposed ADH-PPI approach is illustrated in Figure, a brief description of different components of ADH-PPI approach is given in the following subsections.

Workflow of proposed attention-based deep hybrid methodology ADH-PPI for protein-protein interaction prediction
Stochastic embedding layer
Stochastic embedding layer takes k-mers of protein sequences and unsupervisedly learned k-mer embeddings (generation of which is explained in section ADH-PPI: An Attention based Deep Hybrid Model for Protein Protein Interaction Prediction) to generate an embedding weight matrix , where the number of rows are equal to unique k-mers and number of columns are equal to k-mers embedding size. To fine-tune embedding matrix in a more generic way, we apply two different kinds of dropouts on the embedding matrix, where there is a probability to fully replace k-mer embedding vectors with zero and probability to replace individual continuous values with zero in remaining k-mer embedding vectors. First kind of dropout drops few k-mer embedding vectors whereas second kind of dropout drops few continuous values of remaining k-mer embedding vectors (Gal and Ghahramani, 2016; Merity et al., 2017). This regularization avoids model over-fitting by ensuring that model does not over-specialize certain k-mers to extract most informative features for various classes. The k-mer embedding vector and dimension dropout probabilities are varied from 0.002-to-0.008 where we find that of 0.004 and of 0.005 performs better.
The optimized embedding matrix containing 120-dimensional embedding vectors for unique k-mers is passed to a Long Short Term Memory layer.
Optimized long short term memory layer
Long short-term memory (LSTM) layer is a special kind of recurrent layer which avoids gradient explosion and gradient disappearance issues faced by the neural network during the modeling of long sequences (Huang et al., 2015a). Furthermore, LSTM is really effective for the extraction of long dependencies of features which is very critical for accurate PPI prediction (Huang et al., 2015b). Unlike a traditional recurrent neural network, LSTM makes use of multiple gates described in following equations to control the flow of comprehensive k-mers information provided by optimized embedding layer.
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
| (Equation 6) |
| (Equation 7) |
| (Equation 8) |
The input, forget, and output gates get activated or deactivated mainly on the basis of their weigh matrices and biases, and work on the basis of their activation functions (sigmoid, tanh) to determine which information need to be retained or discarded from memory cell states (, ). In order to preserve k-mers information for a longer period of time, hidden state h of each cell is saved at every time step t.
To most effectively regularize recurrent layer, unlike existing approaches which arbitrarily drop the hidden states during the update to the memory state , we use DropConnect (Wan et al., 2013) which applies dropout with the probability of 0.4 on the recurrent [, , ] and non-recurrent weight matrices [, , ] of the LSTM layer before the forward and the backward pass to enhance LSTM aptitude to extract informative features along with their long range dependencies. Using LSTM with 120 hidden units, ADH-PPI model extracts the short and long range dependencies of features which are important to distinguish interactive protein sequence pairs from non-interactive protein sequence pairs. The 120-dimensional feature vectors produced by the LSTM layer are passed to the convolutional layer.
Convolutional layer
The convolutional layer has been widely applied in Natural Language Processing and Bioinformatics tasks (Alzubaidi et al., 2021) due to two unique charateristics, local perception as well as parameter sharing (Goodfellow et al., 2016). Like simulating the cells with local receptive fields within human brain, the convolutional layer performs an operation known as convolution which uses local connection and shared weights to extract hidden informative features and reduce the overall complexity of neural network (Goodfellow et al., 2016). Convolution operation applied at a particular layer produces a feature map that can be mathematically expressed as:
| (Equation 9) |
Where represents the weight matrix of convolutional kernel of the layer, symbol denotes the convolutional operation, represents the off-set vector, and f(x) denotes the activation function. We use ReLu as an activation function to sparse the final output of convolutional layer which leads to speed up the training process and maintain the steady convergence rate to prevent vanishing gradient issue. CNN layer use 50 kernels of size 3 to produce 50-dimensional feature vectors which are passed to an attention layer.
Attention layer
Attention layer is widely used to adjust the weights of feature vectors in such a manner that most crucial features are emphasized and less important features are penalized (Goodfellow et al., 2016). Attention function can be considered a mapping from a Query vector (Q), and Key-Value vectors (K-V) to an output vector. Here Q, K, and V are linear projection of given protein sequence statistical representation and output is the new protein sequence statistical representation of same dimensions incorporating comprehensive mutual association of k-mers present in protein sequences. The entire process involves three steps: acquiring Query, Key, and Value linear projections, estimating the weight through placing Query and Key into a certain compatibility function, and obtaining the output by estimating the weighted sum of Value using the pre-computed weight. There are many types of compatibility functions which produces many flavors of attention mechanism. Considering the long length of protein sequences (thousands of k-mers), we use the least space and time efficient version of the compatibility function namely Scaled Dot-Product Attention (SDPA). The SDPA computes the dot product of Query and Key which is divided by where denotes the Key dimension, and finally applies the softmax over it to obtain the weight.
| (Equation 10) |
In Equation 10, Weight represents a square matrix having number of rows/columns equivalent to length of protein sequences calculated in terms of number of k-mers. Each ith row jth column value denotes the interaction intensiveness among ith k-mer and jth k-mer. After computing weight, every row of output that represents the statistical vector of a k-mer, can be estimated as the weighted sum of all k-mers. This is primarily implemented through a single-matrix multiplication which can be mathematically expressed as follows:
| (Equation 11) |
Given, 50-dimensional statistical vectors of protein sequences, attention layer updates the values of statistical features on the basis of their usefulness for PPI prediction.
Normalization layer
Neural network faces the issue of internal co-variance shift which de-stabilizes the neural network due to change in input distribution to hidden layers of neural network when model weights are updated after the execution of every batch (Ioffe and Szegedy, 2015). Internal co-variance shift makes the optimal weights learned by the network during previous iterations obsolete (Ioffe and Szegedy, 2015), disturbs the convergence and generalizability of the model (Ioffe and Szegedy, 2015).
Normalization addresses this issue by standardizing the input before feeding to a hidden layer for every batch. It ensures that input-to-output mapping of a neural network does not over-specialize one particular region of protein sequences, resulting in faster training, convergence and improved generalizability (Ioffe and Szegedy, 2015).
Equation 12 describes the overall paradigm of normalization which normalizes each sequence by tuning 2 parameters γ and β.
| (Equation 12) |
Equation 13 illustrates the way mean of a given batch is computed where represents the current sequence from m sequences present in a given batch b.
| (Equation 13) |
Equation 14 describes the way variance of every batch b is computed where each sequence is subtracted from the mean of entire batch () before aggregating and computing average using m number of sequences present in given batch b.
| (Equation 14) |
Equation 15 subtracts each sequence from mean of the batch and takes fraction by standard deviation to normalize the values between 0 and 1, which is represented with .
| (Equation 15) |
In order to enable the network to adapt mean and variance of distribution, 2 parameters γ and β are learned and updated along with biases and weights during training. Final, normalized, scaled, and shifted version of hidden distribution can be represented using Equation 16.
| (Equation 16) |
Standard dropout layer
Dropout is a de-facto standard to regularize neural networks, which generally improves the quality of the hidden features by alleviating the likelihood of hidden units co-adaptations problem. More specifically, for every hidden unit, dropout avoids co-adaptation by iteratively tweaking the presence and absence of other hidden units to ensure that a hidden unit can not rely on other hidden units to fix its mistakes.
In proposed ADH-PPI methodology, each hidden unit has the probability p to be dropped where the value of p falls in range of of 0.01-to-0.4. Mathematically (Equation 17), likelihood of omitting a hidden unit is done according to the Bernoulli distribution with probability p. Through an element wise product of hidden unit vector with a mask where each element is randomly sampled from Bernoulli distribution, hidden units are dropped during training. Whereas, for testing (Equation 18), instead of dropping the hidden unit, probability for a hidden unit not to be dropped % is estimated.
| (Equation 17) |
| (Equation 18) |
Softmax layer
Using dense 50-dimensional representation of protein sequences, softmax layer discriminates interactive protein pairs from non-interactive protein pairs. Categorical cross-entropy also known as softmax loss is used as a loss function which is a simple softmax activation plus a cross entropy loss. Working of softmax activation and categorical cross entropy are described in Equation 19 and Equation 20 respectively.
| (Equation 19) |
| (Equation 20) |
In these equations, t represents one-hot encoded ground truth, represents probability score for each class in C and refers to softmax activation applied before the computation of cross-entropy loss.
Model training optimization
Proposed ADH-PPI model is implemented using Pytorch (Paszke et al., 2019). To perform a fair performance comparison of proposed ADH-PPI approach with existing PPI predictors on two benchmark core S.cerevisiae and H.pylori datasets, 10-fold cross validation is performed. ADH-PPI is trained on complete core S.cerevisiae dataset to evaluate its performance on four independent test sets belonging to E.coli, C.elegans, H.sapiens, and M. musculus species. To optimize the training of ADH-PPI model, learning rate is the most crucial hyperparameter which controls how much model weights need to be updated in response to error estimated after the execution of every batch. Choosing a learning rate is really challenging as very small value may lead to longer training, a very large value may destabilize the training process or learn a sub-optimal collection of weights quickly, and constant learning rate leads to saturation where validation loss of the model stops improving. To find an optimal learning rate, we use a learning rate or weight decay strategy which iteratively changes the learning rate if validation loss does not improve for specific number of epochs. In this manner, model converges to optimal weights which largely reduce the generalization error. The ADH-PPI approach is trained using the batch size of 64 and ADAMW tweaks initial learning rate defined as 0.01-to-0.07 using decay rate defined as 0.00001-to-0.01 if categorical cross entropy loss on 1% validation sequences stops improving. To facilitate the reproduceability of experimental results, best values of different hyperparameters found through Grid search (Shekar and Dagnew, 2019; Liashchynskyi and Liashchynskyi, 2019) with respect to two core benchmark datasets and 4 independent test sets are provided in Table.
Optimal values of different hyperparameters of proposed ADH-PPI methodology for 2 core datasets and 4 independent test sets for the task of PPI prediction
| PPI Dataset | Degree of higher order residue (K-mer) |
Stride size | Sequence embedding dimension |
Learning rate | Weight decay | Dropout rate | Subsequence regions |
|---|---|---|---|---|---|---|---|
| S.Cerevisiave | 5 | 5 | FastText-120 | 0.03 | 0.1 | 0.3 | P-A_S-40, P-B_E-40 |
| H.Pyloir | 5 | 1 | FastText-120 | 0.05 | 0.01 | 0.01 | P-A_S-40, P-B_E-40 |
| C.elegans | 5 | 5 | FastText-120 | 0.05 | 1.00 × 10−5 | 0.1 | P-A_S-40, P-B_E-40 |
| H.sapiens | 5 | 5 | FastText-120 | 0.05 | 1.00 × 10−5 | 0.1 | P-A_S-40, P-B_E-40 |
| M.musculus | 5 | 5 | FastText-120 | 0.05 | 1.00 × 10−5 | 0.1 | P-A_S-40, P-B_E-40 |
| E.coli | 5 | 5 | FastText-120 | 0.05 | 1.00 × 10−5 | 0.1 | P-A_S-40, P-B_E-40 |
Model evaluation criterion
Following the evaluation criterion of previous PPI predictors (Yu et al., 2021; Jiang et al., 2020; Wang et al., 2020a), to perform a fair performance comparison of proposed ADH-PPI approach with existing PPI predictors, we use 6 different evaluation measures namely accuracy (ACC), precision, recall, matthews correlation coefficient (MCC), F1-score, and area under receiver operating characteristics (AU-ROC).
| (Equation 21) |
In Equation 21, TP, FP, TN, FN indicate the true positives, false positives, true negatives, and false negatives.
Accuracy (ACC) is the ratio of correctly predicted interactive and non-interactive instances with total predictions. It does not accurately measure the performance of the classifier if the dataset has unbalanced classes. Recall true positive rate, whereas precision computes upto what extent positive predictions are fully correct. F1-score takes the harmonic mean of precision and recall. Evaluation criterion such as (PRE, REC, F1-score) do not take true negatives into account. Whereas, matthews correlation coefficient (MCC) takes all four TP, FP TN, FN into account to compute the performance. High value of MCC shows that classifier is effectively distinguishing all corpus classes even when a class is under or over represented. Area under receiver operating characteristic curve (AU-ROC) measures degree of separability of the model at different thresholds. A high degree of separability indicates that model accurately distinguishes interactive protein sequence pairs from non-interactive protein sequence pairs.
Quantification and statistical analysis
The statistical tests are performed by making use of a python package namely Scipy (version: 1.8.0).
Acknowledgments
We acknowledge that this work was supported by Sartorius Artificial Intelligence Lab.
Author contributions
Conceptualization: M.N.A.; S. A.; Data curation: M. N. A., M. A. I.; Formal analysis: M. N. A., M. A. I.; Investigation: M. N. A., M. A. I.; Methodology: M. N. A., M. A. I.; Software: M. N. A., M. A. I.; Supervision: A. D., S. A., M. I. M.; Validation: M. N. A., M. A. I.; Visualization: M. N. A., M. A. I.; Writing – original draft: M. N. A., M. A. I.; Writing – review & editing: A. D., S. A.
Declaration of interests
The authors declare no competing interests.
Published: October 21, 2022
Data and code availability
-
•
This paper performs PPI prediction over existing benchmark datasets and independent test sets given by (Guo et al., 2008b), (Martin et al., 2005), and (Zhou et al., 2011). All benchmark protein sequences datasets used to predict PPIs are available at https://sds_genetic_analysis.opendfki.de/PPI/Download_PPI/. These datasets are also available on a general-purpose open repository Zenodo https://doi.org/10.5281/zenodo.6973538.
-
•
A Public web server that allows the users to train predictive pipeline from scratch as well as utilize pre-trained predictive pipeline can be publicly accessed at https://sds_genetic_analysis.opendfki.de/PPI/.
-
•
The complete source code of proposed ADH-PPI predictor is available at https://sds_genetic_analysis.opendfki.de/PPI/Download_PPI/.
-
•
Any further information needed to reanalyze the data reported in current study is available from lead contact upon the request.
References
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Alzubaidi L., Zhang J., Humaidi A.J., Al-Dujaili A., Duan Y., Al-Shamma O., Santamaría J., Fadhel M.A., Al-Amidie M., Farhan L. Review of deep learning: concepts, cnn architectures, challenges, applications, future directions. J. Big Data. 2021;8:53–74. doi: 10.1186/s40537-021-00444-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei S.A., Sijbesma E., Hann M., Davis J., O’Mahony G., Perry M.W.D., Karawajczyk A., Eickhoff J., Brunsveld L., Doveston R.G., et al. Stabilization of protein-protein interactions in drug discovery. Expert Opin. Drug Discov. 2017;12:925–940. doi: 10.1080/17460441.2017.1346608. [DOI] [PubMed] [Google Scholar]
- Asim M.N., Ibrahim M.A., Malik M.I., Dengel A., Ahmed S. International Conference on Neural Information Processing. Springer; 2020. Enhancer-dsnet: a supervisedly prepared enriched sequence representation for the identification of enhancers and their strength; pp. 38–48. [Google Scholar]
- Asim M.N., Malik M.I., Dengel A., Ahmed S. 2020 International Joint Conference on Neural Networks (IJCNN) IEEE; 2020. K-mer neural embedding performance analysis using amino acid codons; pp. 1–8. [Google Scholar]
- Bairoch A., Apweiler R. The swiss-prot protein sequence data bank and its new supplement trembl. Nucleic Acids Res. 1996;24:21–25. doi: 10.1093/nar/24.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berggård T., Linse S., James P. Methods for the detection and analysis of protein–protein interactions. Proteomics. 2007;7:2833–2842. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- Bergstra J., Bardenet R., Bengio Y., Kégl B. Algorithms for hyper-parameter optimization. Adv. Neural Inf. Process. Syst. 2011;24 [Google Scholar]
- Bojanowski P., Grave E., Joulin A., Mikolov T. Enriching word vectors with subword information. Trans. Assoc. Comput. Linguist. 2017;5:135–146. [Google Scholar]
- Chao G., Luo Y., Ding W. Recent advances in supervised dimension reduction: a survey. Mach. Learn. Knowl. Extr. 2019;1:341–358. [Google Scholar]
- Ding Y., Tang J., Guo F. Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinf. 2016;17:398–413. doi: 10.1186/s12859-016-1253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X., Sun S., Hu C., Yao Y., Yan Y., Zhang Y. Deepppi: boosting prediction of protein–protein interactions with deep neural networks. J. Chem. Inf. Model. 2017;57:1499–1510. doi: 10.1021/acs.jcim.7b00028. [DOI] [PubMed] [Google Scholar]
- Espadaler J., Romero-Isart O., Jackson R.M., Oliva B. Prediction of protein–protein interactions using distant conservation of sequence patterns and structure relationships. Bioinformatics. 2005;21:3360–3368. doi: 10.1093/bioinformatics/bti522. [DOI] [PubMed] [Google Scholar]
- Fu L., Niu B., Zhu Z., Wu S., Li W. Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Zhu W., Cai L., Liao B., Peng L., Chen Y., Yang J. Improved pre-mirnas identification through mutual information of pre-mirna sequences and structures. Front. Genet. 2019;10:119. doi: 10.3389/fgene.2019.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal Y., Ghahramani Z. Advances in neural information processing systems. 2016. A theoretically grounded application of dropout in recurrent neural networks; pp. 1019–1027. [Google Scholar]
- Gavin A.C., Bösche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.M., Cruciat C.M., et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Goodfellow I., Bengio Y., Courville A. Conference on information and communication systems (ICICS) 2016. Deep learning mit press; pp. 151–156.http://www.deeplearningbook.org [Google Scholar]
- Guo Y., Yu L., Wen Z., Li M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008;36:3025–3030. doi: 10.1093/nar/gkn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Yu L., Wen Z., Li M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008;36:3025–3030. doi: 10.1093/nar/gkn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemifar S., Neyshabur B., Khan A.A., Xu J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics. 2018;34:i802–i810. doi: 10.1093/bioinformatics/bty573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y., Gruhler A., Heilbut A., Bader G.D., Moore L., Adams S.-L., Millar A., Taylor P., Bennett K., Boutilier K., et al. Systematic identification of protein complexes in saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Hosur R., Peng J., Vinayagam A., Stelzl U., Xu J., Perrimon N., Bienkowska J., Berger B. A computational framework for boosting confidence in high-throughput protein-protein interaction datasets. Genome biology. 2012;13:1–14. doi: 10.1186/gb-2012-13-8-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Wang X., Huang Y.A., Hu P., You Z.-H. A novel network-based algorithm for predicting protein-protein interactions using gene ontology. Front. Microbiol. 2021;12:735329. doi: 10.3389/fmicb.2021.735329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Zhang J., Pan X., Yan H., You Z.-H. Hiscf: leveraging higher-order structures for clustering analysis in biological networks. Bioinformatics. 2021;37:542–550. doi: 10.1093/bioinformatics/btaa775. [DOI] [PubMed] [Google Scholar]
- Huang Y.-A., You Z.-H., Chen X., Chan K., Luo X. Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinf. 2016;17:184–211. doi: 10.1186/s12859-016-1035-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.-A., You Z.-H., Gao X., Wong L., Wang L. Using weighted sparse representation model combined with discrete cosine transformation to predict protein-protein interactions from protein sequence. BioMed Res. Int. 2015;2015:902198. doi: 10.1155/2015/902198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z., Xu W., Yu K. Bidirectional lstm-crf models for sequence tagging. arXiv. 2015 doi: 10.48550/arXiv.1508.01991. Preprint at. [DOI] [Google Scholar]
- Hue M., Riffle M., Vert J.-P., Noble W.S. Large-scale prediction of protein-protein interactions from structures. BMC Bioinf. 2010;11:144–149. doi: 10.1186/1471-2105-11-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieremie I., Ewing R.M., Niranjan M. Transformergo: predicting protein–protein interactions by modelling the attention between sets of gene ontology terms. Bioinformatics. 2022;38:2269–2277. doi: 10.1093/bioinformatics/btac104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioffe S., Szegedy C. International conference on machine learning. PMLR; 2015. Batch normalization: accelerating deep network training by reducing internal covariate shift; pp. 448–456. [Google Scholar]
- Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M., Yamamoto K., Kuhara S., Sakaki Y. Toward a protein–protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. USA. 2000;97:1143–1147. doi: 10.1073/pnas.97.3.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Liu Z., Xiao X., Liu B., Chou K.-C. ippi-esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into pseaac. J. Theor. Biol. 2015;377:47–56. doi: 10.1016/j.jtbi.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Jiang T., Wu H., Chen Y., Li H., Qiu J., Lu W., Fu Q. International Conference on Intelligent Computing. Springer; 2020. Prediction of membrane protein interaction based on deep residual learning; pp. 103–108. [Google Scholar]
- Joshi T., Chen Y., Becker J.M., Alexandrov N., Xu D. Genome-scale gene function prediction using multiple sources of high-throughput data in yeast saccharomyces cerevisiae. OMICS A J. Integr. Biol. 2004;8:322–333. doi: 10.1089/omi.2004.8.322. [DOI] [PubMed] [Google Scholar]
- Kong M., Zhang Y., Xu D., Chen W., Dehmer M. Fctp-wsrc: protein–protein interactions prediction via weighted sparse representation based classification. Front. Genet. 2020;11:18. doi: 10.3389/fgene.2020.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács I.A., Luck K., Spirohn K., Wang Y., Pollis C., Schlabach S., Bian W., Kim D.-K., Kishore N., Hao T., et al. Network-based prediction of protein interactions. Nat. Commun. 2019;10:1240–1248. doi: 10.1038/s41467-019-09177-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan N.J., Cagney G., Yu H., Zhong G., Guo X., Ignatchenko A., Li J., Pu S., Datta N., Tikuisis A.P., et al. Global landscape of protein complexes in the yeast saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Kulminskaya N., Oberer M. Protein-protein interactions regulate the activity of adipose triglyceride lipase in intracellular lipolysis. Biochimie. 2020;169:62–68. doi: 10.1016/j.biochi.2019.08.004. [DOI] [PubMed] [Google Scholar]
- Le N.Q.K., Yapp E.K.Y., Ho Q.-T., Nagasundaram N., Ou Y.-Y., Yeh H.-Y. ienhancer-5step: identifying enhancers using hidden information of dna sequences via chou’s 5-step rule and word embedding. Anal. Biochem. 2019;571:53–61. doi: 10.1016/j.ab.2019.02.017. [DOI] [PubMed] [Google Scholar]
- Li Y., Huang C., Ding L., Li Z., Pan Y., Gao X. Deep learning in bioinformatics: introduction, application, and perspective in the big data era. Methods. 2019;166:4–21. doi: 10.1016/j.ymeth.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Liashchynskyi P., Liashchynskyi P. Grid Search, Random Search, Genetic Algorithm: A Big Comparison for Nas. arXiv. 2019 doi: 10.48550/arXiv.1912.06059. Preprint at. [DOI] [Google Scholar]
- Licata L., Briganti L., Peluso D., Perfetto L., Iannuccelli M., Galeota E., Sacco F., Palma A., Nardozza A.P., Santonico E., et al. Mint, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydia A., Francis S. 2019. A Survey of Optimization Techniques for Deep Learning Networks; pp. 2454–9150. [Google Scholar]
- Martin S., Roe D., Faulon J.-L. Predicting protein–protein interactions using signature products. Bioinformatics. 2005;21:218–226. doi: 10.1093/bioinformatics/bth483. [DOI] [PubMed] [Google Scholar]
- Merity S., Keskar N.S., Socher R. Regularizing and optimizing lstm language models. arXiv. 2017 doi: 10.48550/arXiv.1708.02182. Preprint at. [DOI] [Google Scholar]
- Meyer J.G. 2021. ‘Deep Learning Neural Network Tools for Proteomics’, Cell Reports Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolov T., Chen K., Corrado G., Dean J. Efficient estimation of word representations in vector space. arXiv. 2013 doi: 10.48550/arXiv.1301.3781. Preprint at. [DOI] [Google Scholar]
- Nanni L. Fusion of classifiers for predicting protein–protein interactions. Neurocomputing. 2005;68:289–296. [Google Scholar]
- Nanni L., Lumini A. An ensemble of k-local hyperplanes for predicting protein–protein interactions. Bioinformatics. 2006;22:1207–1210. doi: 10.1093/bioinformatics/btl055. [DOI] [PubMed] [Google Scholar]
- Nooren I.M.A., Thornton J.M. Structural characterisation and functional significance of transient protein–protein interactions. J. Mol. Biol. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- Northey T.C., Barešić A., Martin A.C.R. Intpred: a structure-based predictor of protein–protein interaction sites. Bioinformatics. 2018;34:223–229. doi: 10.1093/bioinformatics/btx585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusrat I., Jang S.-B. A comparison of regularization techniques in deep neural networks. Symmetry. 2018;10:648. [Google Scholar]
- Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., Killeen T., Lin Z., Gimelshein N., Antiga L., et al. Pytorch: An imperative style, high-performance deep learning library. arXiv. 2019 Preprint at. arXiv:1912.01703. [Google Scholar]
- Pawson T., Nash P. Protein–protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- Peri S., Navarro J.D., Kristiansen T.Z., Amanchy R., Surendranath V., Muthusamy B., Gandhi T.K.B., Chandrika K.N., Deshpande N., Suresh S., et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004;32:D497–D501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I., Lievens S., Libert C., Tavernier J., De Bosscher K. Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Ther. 2016;24:707–718. doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Klein-Seetharaman J., Bar-Joseph Z. Biocomputing 2005. World Scientific; 2005. Random forest similarity for protein-protein interaction prediction from multiple sources; pp. 531–542. [PubMed] [Google Scholar]
- Salwinski L., Miller C.S., Smith A.J., Pettit F.K., Bowie J.U., Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Pinto L.N., Venable L.R., Fahrenbach J., Churpek M.M. Comparison of variable selection methods for clinical predictive modeling. Int. J. Med. Inform. 2018;116:10–17. doi: 10.1016/j.ijmedinf.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenrock A., Samanfar B., Pitre S., Hooshyar M., Jin K., Phillips C.A., Wang H., Phanse S., Omidi K., Gui Y., et al. Efficient prediction of human protein-protein interactions at a global scale. BMC Bioinf. 2014;15:383. doi: 10.1186/s12859-014-0383-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekar B., Dagnew G. 2019 Second International Conference on Advanced Computational and Communication Paradigms (ICACCP) IEEE; 2019. Grid search-based hyperparameter tuning and classification of microarray cancer data; pp. 1–8. [Google Scholar]
- Singh R., Park D., Xu J., Hosur R., Berger B. Struct2net: a web service to predict protein–protein interactions using a structure-based approach. Nucleic Acids Res. 2010;38:W508–W515. doi: 10.1093/nar/gkq481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano C.O.S., Vargas J., Montano A.P. A survey of dimensionality reduction techniques. arXiv. 2014 Preprint at. arXiv:1403.2877. [Google Scholar]
- Südhof T.C. The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Vickers N.J. Animal communication: when i’m calling you, will you answer too? Curr. Biol. 2017;27:R713–R715. doi: 10.1016/j.cub.2017.05.064. [DOI] [PubMed] [Google Scholar]
- Wan L., Zeiler M., Zhang S., Le Cun Y., Fergus R. International conference on machine learning. PMLR; 2013. Regularization of neural networks using dropconnect; pp. 1058–1066. [Google Scholar]
- Wang L., You Z.-H., Yan X., Zheng K., Li Z.-W. International Conference on Intelligent Computing. Springer; 2020. Gcnsp: a novel prediction method of self-interacting proteins based on graph convolutional networks; pp. 109–120. [Google Scholar]
- Wang R.-S., Wang Y., Wu L.-Y., Zhang X.-S., Chen L. Analysis on multi-domain cooperation for predicting protein-protein interactions. BMC Bioinf. 2007;8:391. doi: 10.1186/1471-2105-8-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Hu P., Hu L. International Conference on Intelligent Computing. Springer; 2020. A novel stochastic block model for network-based prediction of protein-protein interactions; pp. 621–632. [Google Scholar]
- Xenarios I., Salwínski L., Duan X.J., Higney P., Kim S.-M., Eisenberg D. Dip, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y., Du X., Diao Y., Zhu H. An integration of deep learning with feature embedding for protein–protein interaction prediction. PeerJ. 2019;7 doi: 10.7717/peerj.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-H., Chan K.C.C., Hu P. Predicting protein-protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest. PLoS One. 2015;10 doi: 10.1371/journal.pone.0125811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-H., Chan K.C.C., Hu P. Predicting protein-protein interactions from primary protein sequences using a novel multi-scale local feature representation scheme and the random forest. PLoS One. 2015;10 doi: 10.1371/journal.pone.0125811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-H., Lei Y.-K., Gui J., Huang D.-S., Zhou X. Using manifold embedding for assessing and predicting protein interactions from high-throughput experimental data. Bioinformatics. 2010;26:2744–2751. doi: 10.1093/bioinformatics/btq510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z.-H., Li X., Chan K.C. An improved sequence-based prediction protocol for protein-protein interactions using amino acids substitution matrix and rotation forest ensemble classifiers. Neurocomputing. 2017;228:277–282. [Google Scholar]
- You Z.-H., Zhu L., Zheng C.-H., Yu H.-J., Deng S.-P., Ji Z. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinf. 2014;15:1–9. doi: 10.1186/1471-2105-15-S15-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B., Chen C., Wang X., Yu Z., Ma A., Liu B. Prediction of protein–protein interactions based on elastic net and deep forest. Expert Syst. Appl. 2021;176 [Google Scholar]
- Zhang Q.C., Petrey D., Deng L., Qiang L., Shi Y., Thu C.A., Bisikirska B., Lefebvre C., Accili D., Hunter T., et al. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature. 2012;490:556–560. doi: 10.1038/nature11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. Phospholipase d and phosphatidic acid in plant defence response: from protein–protein and lipid–protein interactions to hormone signalling. J. Exp. Bot. 2015;66:1721–1736. doi: 10.1093/jxb/eru540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.Z., Gao Y., Zheng Y.Y. Advances in computer science and education applications. Springer; 2011. Prediction of protein-protein interactions using local description of amino acid sequence; pp. 254–262. [Google Scholar]
- Zhu H., Snyder M. Snyder m. Curr. Opin. Chem. Biol. 2003;7:55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- Zou H., Hastie T. Regularization and variable selection via the elastic net. J. Royal Statistical Soc. B. 2005;67:301–320. [Google Scholar]
- Zou J., Huss M., Abid A., Mohammadi P., Torkamani A., Telenti A. A primer on deep learning in genomics. Nat. Genet. 2019;51:12–18. doi: 10.1038/s41588-018-0295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
This paper performs PPI prediction over existing benchmark datasets and independent test sets given by (Guo et al., 2008b), (Martin et al., 2005), and (Zhou et al., 2011). All benchmark protein sequences datasets used to predict PPIs are available at https://sds_genetic_analysis.opendfki.de/PPI/Download_PPI/. These datasets are also available on a general-purpose open repository Zenodo https://doi.org/10.5281/zenodo.6973538.
-
•
A Public web server that allows the users to train predictive pipeline from scratch as well as utilize pre-trained predictive pipeline can be publicly accessed at https://sds_genetic_analysis.opendfki.de/PPI/.
-
•
The complete source code of proposed ADH-PPI predictor is available at https://sds_genetic_analysis.opendfki.de/PPI/Download_PPI/.
-
•
Any further information needed to reanalyze the data reported in current study is available from lead contact upon the request.