

Summary
Precisely measuring the number and somatic volume of neurons in the central nervous system at single-cell resolution is technically challenging. Here, we combine multiple techniques to address this challenge in optically cleared mouse spinal cords. We describe in vivo neuron labeling approaches, tissue-clearing technology, light sheet fluorescence microscopy, and machine learning-guided imaging analysis. This combination provides a precise determination of the cell number and somatic volume of any neuron population in the spinal cords.
Subject areas: Cell Biology, Microscopy, Neuroscience, Molecular/Chemical Probes
Graphical abstract
Highlights
-
•
Retrograde labeling of a specific group of motor neurons in the mouse spinal cord
-
•
CUBIC tissue clearing of spinal cord samples for high-resolution volumetric imaging
-
•
Light sheet fluorescence microscopy-based imaging and machine learning-based analysis
-
•
Precise determination of the number and somatic volume of neurons in the spinal cords
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Precisely measuring the number and somatic volume of neurons in the central nervous system at single-cell resolution is technically challenging. Here, we combine multiple techniques to address this challenge in optically cleared mouse spinal cords. We describe in vivo neuron labeling approaches, tissue-clearing technology, light sheet fluorescence microscopy, and machine learning-guided imaging analysis. This combination provides a precise determination of the cell number and somatic volume of any neuron population in the spinal cords.
Before you begin
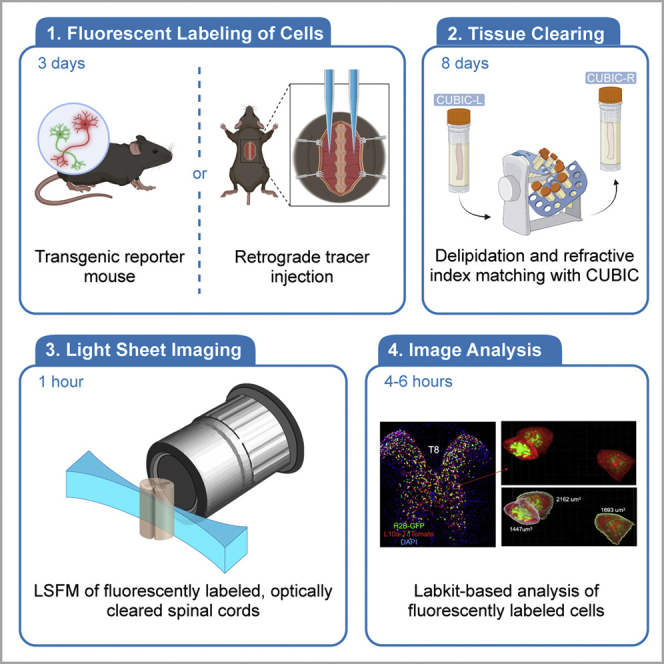
Standard histology techniques are widely used to estimate cell number and size; however, the compression of complicated three-dimensional cell bodies into a two-dimensional image introduces imprecision in cell number and cell size (one large cell could be counted multiple times for very thin sections, cells could be missed for thicker sections, and there is no reason to expect the cell bodies to be sectioned in a way that the 2D area is directly related to its total volume). Even when 3D imaging is performed on live thick tissue sections, transitions between sections can be difficult to handle computationally. Light sheet florescence microscopy on cleared mouse spinal cords allows the direct measurement of 3D structure without physically sectioning the entire spinal cord. This protocol (Figure 1) below describes the specific steps for labeling, imaging, and quantifying the number and somatic volume of neurons in intact mouse spinal cord segments (3–6mm). Use of the Labkit pixel classifier with Imaris visualization tools allows segmentation of cells in 3D even when fluorescent labeling is non-uniform as is sometimes the case with neuronal tracers.
Figure 1.

Schematic overview of animal preparation, tissue clearing, imaging, and analysis workflow, including hands-on- and waiting- time
I: neuron labeling; II & III: tissue and organ clearing; IV: 3D images taken by LSMF; V & VI: neuronal number and somatic volume measurement.
Institutional permissions
This study was conducted at the University of Texas Medical Branch (UTMB) and was reviewed and approved by the Institutional Animal Care and Use Committee of University of Texas Medical Branch.
Software installation
Timing: 10–30 min
-
1.Download and install Imaris 9.9.0 or greater (Bitplane, Oxford Instruments, requires license).
-
a.Follow the instructions on https://imaris.oxinst.com/support/installation-instructions to install Imaris.
-
a.
-
2.Download and install FIJI (Image J version 1.8.0_172) that includes the Labkit plugin).
-
a.Follow the instructions on https://inagej.net/software/fiji/downloads.
-
a.
-
3.Setup Labkit (Image J plugin) for Imaris.
-
a.Follow the instructions on https://imaris.oxinst.com/learning/view/article/configuring-imaris-9-9-to-work-with-labkit to set up Labkit.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Ketamine HCl | Covetrus | REF: 11695-6840-1 |
| AnaSed (Xylazine) | Akorn | REF: 59399-110-20 |
| 96% PFA | Thermo Scientific | REF: AC416780030 |
| D-PBS, 1X, without calcium or magnesium | Corning | REF: MT21031CV |
| Sodium azide | Sigma | REF: 2002-25G |
| Agarose Low EEO | Sigma | REF: A9539-10G |
| CUBIC-L | TCI America | T3740-100ML |
| CUBIC-R+(M) | TCI America | T3741-100ML |
| ddH2O | N/A | N/A |
| Sucrose | Ward’s Science | REF: 470302-804 |
| Tissue-Plus OCT Compound | Fisher | REF: 23-730-571 |
| Cholera Toxin Subunit B, Alexa Fluor 647 Conjugate | Invitrogen | REF: C34778 |
| Sterile PBS | Thermo Scientific | REF: J61196-AP |
| 0.9% Saline | Hospira | REF: RL-7302 |
| Nair | Fisher | REF: NC0132811 |
| Artificial Tears | Akorn | REF: 59399-162-35 |
| Triple Antibiotic Ointment | Med-Vet International | REF: CUR001231H |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J, female, 10 weeks old | The Jackson Laboratory | 000664 |
| Mouse: Ai14, female, 10 weeks old | The Jackson Laboratory | 007914 |
| Software and algorithms | ||
| Zen Black | ZEISS | Version: 3.1 |
| Imaris | Oxford Instruments | Version: 9.9 |
| Windows 10 Professional for Workstations (Image analysis workstation) | Microsoft | Version: 21H2 (Build 19044.1586) |
| FIJI/ImageJ (1.8.0_172) |
Schindelin et al. (2012) https://doi.org/10.1038/nmeth.2019 |
https://imagej.net/software/fiji/ |
| Labkit | https://doi.org/10.3389/fcomp.2022.777728 | https://imagej.net/plugins/labkit/index |
| CLIJ | https://doi.org/10.1038/s41592-019-0650-1 | https://clij.github.io/clij-docs/ |
| CLIJ2 | https://doi.org/10.1038/s41592-019-0650-1 | https://clij.github.io/ |
| Other | ||
| Zeiss Z7 Light Sheet Fluorescence Microscope | ZEISS | REF: Z7 |
| Illumination lenses (5X, 0.1 NA, n=1.33–1.58) | ZEISS | REF: 400900-9030-000 |
| Detection lens (5X, 0.16 NA, n=1.33–1.58) | ZEISS | REF: 420330-8220-000 |
| Solid state laser 561 nm (50 mW) |
ZEISS | REF: 400600-9210-000 |
| Solid state laser 638 nm (75 mW) |
ZEISS | REF: 400600-9230-000 |
| Laser blocking Notch filter 405/488/561/638 nm |
ZEISS | REF: 404900-9101-000 |
| Emission filters | ZEISS | REF: 404900-9322-000 |
| PCO.Edge 5.5 sCMOS cameras | PCO | PN: 70108070129 |
| Dell Precision Workstation 7920 | Dell | SKU: 210-AMRM |
| Micropipette Beveler | Sutter Instruments | REF: BV-10 |
| Flaming/Brown Micropipette Puller | Sutter Instruments | REF: P-97 |
| Stereoboom Microscope | Laxco | N/A |
| Ophthalmic Surgical Microscope | ZEISS | REF: OPMI S5 |
| Small Animal Stereotaxic Instrument | KOPF | REF: Model 940 |
CRITICAL: PFA is toxic. Perform all steps using PFA in a fume hood. Avoid inhalation and contact with skin or eyes.
Alternatives: Low EEO agarose from other manufacturers can be used to make CUBIC-R+(M) gel. For example, Nacalai Tesque - Cat no. 01163-76, was used in the CUBIC tissue clearing protocol by (Matsumoto et al., 2019).
Materials and equipment
Anesthetic: Ketamine and Xylazine
| Reagent | Final concentration | Amount | Storage temperature |
|---|---|---|---|
| Ketamine | 15% v/v | 1 mL | Room temperature (20°C) |
| Xylazine | 5% v/v | 0.5 mL | Room temperature (20°C) |
| Saline | N/A | 8.5 mL | Room temperature (20°C) |
| Total | N/A | 10 mL | Room temperature (20°C) |
Storage: Non-diluted Ketamine is stable for 1 year. Non-diluted Xylazine is stable for 2 years. Once combined and diluted, the shelf life is 6 months from the mix date.
Analgesic: Buprenorphine (administered at 0.1 mg/kg)
| Reagent | Final concentration | Amount | Storage temperature |
|---|---|---|---|
| Buprenorphine (0.3 mg/mL) | 0.03mg/mL | 1 mL | Room temperature (20°C) |
| Saline | N/A | 9 mL | Room temperature (20°C) |
| Total | N/A | 10 mL | Room temperature (20°C) |
Storage: Diluted buprenorphine has a shelf life of 2 months from the mix date.
Spinal neuron labeling: Cholera Toxin Subunit B (CTB)
| Reagent | Final concentration | Amount | Storage temperature |
|---|---|---|---|
| CTB-Alexa647 | 20 mg/mL | 500 μg | −20°C |
| PBS | N/A | 100 μL | Room temperature (20°C) |
| Total | N/A | 100 μL | −20°C |
Storage: Store at -20°C for 3 months max.
Step-by-step method details
Part 1: Cholera toxin subunit B (CTB) muscle injection surgery
Part 1 of this protocol describes how to use dye-conjugated CTB to retrogradely label a specific group of motor neurons in the spinal cord. We have chosen a longer-wavelength conjugate which reduces scattering and improves image quality in deeper areas of cleared tissues. We describe how to prepare the CTB solution for injection and describe the surgical procedure to retrogradely label motor neurons from the external oblique (trunk) muscles (Goetz et al., 2015; Tani et al., 1994) (Figure 2).
-
1.Prepare anesthetic solution.
-
a.Mix 1 mL of ketamine (100 mg/mL), 0.1 mL of xylazine (100 mg/mL) and bring to 10 mL with normal 0.9% saline together to create 10 mL of anesthetic solution.
-
b.The final solution should contain 10 mg/mL of ketamine and 1 mg/mL of xylazine.
-
c.When solution is prepared the day of surgery, store at room temperature (20°C) until ready to use.
-
a.
-
2.Prepare a 0.5% (wt/vol) CTB tracer solution for injection.
-
a.To prepare 0.5% (wt/vol) concentration, dissolve 500 μg of solid CTB tracer in 100 μL of 1x PBS, pH 7.2.
-
b.Gently mix the solution until CTB tracer is fully dissolved.
-
c.Quickly spin the solution in a centrifuge for 2 s (1000 × g) to help mix the solution and to move any residue to the bottom.
-
d.Keep the tracer solution on ice until ready to use.
-
e.Aliquot the CTB tracer solution into 2.5 μL aliquots and store at -20°C for 3 months max.
-
a.
Note: Allow the CTB tracer solution to warm to room temperature (20°C) before injecting.
-
3.Prepare surgical equipment and the operating area.
-
a.Sterilize operating equipment according to institutional guidelines.
-
b.Pull several glass capillaries (1.0 mm Outer Diameter) into needles with a micropipette puller (Program 14). For muscle injections, the tip of the pulled needle should be around 7 mm long. Trim any excess length.
-
c.Smoothen needle tips using a micropipette beveler.
-
a.
-
4.Prepare the mouse for surgery.
-
a.Intraperitoneally inject the anesthetic solution into the mouse using a 26G 3/8 needle (0.1 mL/10 g body weight).
-
b.Once the mouse is deeply anesthetized, shave the animal’s back until the operating area (lower neck to hip) is devoid of hair. Remove any hair stubs using Nair.
-
c.Disinfect the region of exposed skin by scrubbing several times with 10% povidone iodine followed by 70% ethanol.CRITICAL: All subsequent steps must be conducted under aseptic conditions appropriate to the species and must follow any guidelines and procedures of the relevant institutional animal care committees.
-
d.Apply triple antibiotic ointment to the mouse’s eyes.
-
e.Properly secure the animal in the stereotaxic device.
-
i.Place a blunt ear bar into the mouse’s right ear canal and tighten in place.
-
ii.Once the right ear bar is secured, hold the animal in place and repeat the process with the left ear. If both ear bars are correctly inserted, the animal will not be able to move its head side to side.
-
iii.Secure the mouse’s upper incisors in the mouth holder.
-
iv.Adjust the apparatus until the mouse head position is level.CRITICAL: The animal’s head should be as secure and level as possible to ensure accurate injections.
-
i.
-
f.Disinfect the surgical site using povidone iodine and ethanol again. Drape sterile towels over the operating area.
-
a.
-
5.Expose the external oblique muscle.
-
a.Perform a sagittal incision along the midline of the back where the external oblique is located.
-
b.Retract the skin away from the midline to expose the muscle layer.Note: There might be a thin, transparent layer of connective tissue over the muscle layer. The presence of this layer will not affect CTB tracer injection.
-
c.Secure the retracted skin with surgical hooks and identify the external oblique muscles.
-
a.
-
6.Load CTB tracer solution into the tracer injection apparatus.
-
a.Connect a glass needle to the Hamilton syringe to create the injection apparatus.
-
b.Slowly load a minimum of 5 μL of tracer solution into the micropipette.CRITICAL: Do not introduce air bubbles into the tracer solution during the loading process.
-
c.Ensure there are no air bubbles in the injection apparatus before proceeding.
-
d.Attach the injection apparatus to the syringe pump.
-
a.
-
7.Inject CTB tracer into the external oblique muscle (Figure 2A), bilaterally.
-
a.Before starting the injections, determine the amount of tracer solution to inject and set the automated syringe pump accordingly. For mice, we set the pump to inject 2 μL of tracer. Each muscle will be injected 4 times on each side (8 CTB injections total).
-
b.Activate the pump to test for needle patency and satisfactory flow.
-
c.Identify and mark (Figure 2B) the first injection site on one side using a marker. slowly insert the needle into the muscle and inject 2 μL of tracer (Figure 2C). This injection serves as a marker for the other 3 injections (Figure 2D).CRITICAL: Remove the needle slowly from the muscle to prevent tracer solution backflow due to negative pressure.
-
d.Using a marker, label the desired contralateral injection site.
-
e.Inject 2 μL of tracer into the labeled site.
-
f.Perform 3 additional injections of tracer solution at the initial injection site. All injections should fall along the same line.
-
g.Repeat the process for the contralateral muscle (Figure 2E).
-
a.
-
8.
Close surgical incisions using staples and apply antibiotics along the incision line.
-
9.
Wait 3 days before perfusing for tissue clearing.
Alternatives: Here retrograde CTB was used to label motor neurons. Please note that viral-based anterograde and retrograde intraspinal tracers (Inquimbert et al., 2013; Liu et al., 2020; Nectow and Nestler, 2020) can also be used for neuronal labeling.
Figure 2.

Key steps of CTB tracer injection surgery
(A) The back of the mouse is exposed. Identify rib 10-12.
(B and C) (B) The injection region is identified (yellow outline), (C) the injection sites are marked.
(D) CTB tracer solution is injected into the muscle.
(E) CTB tracer solution is injected into both sides in a mouse. Scale bars: 2mm.
Part 2: Perfusion and tissue clearing
The following section describes a rapid and high-performing CUBIC tissue clearing method (Matsumoto et al., 2019) that prepares spinal cord samples for high-resolution volumetric imaging. After whole body perfusion, spinal cords are post-fixed in 4% PFA. This is followed by a two-step clearing procedure involving delipidation in CUBIC-L and refractive index matching in CUBIC-R+(M). After clearing, the sample is mounted in an agarose gel suitable for imaging using a light sheet fluorescence microscope (Figures 3 and 4).
-
10.Prepare D-PBS and 4% PFA for whole body perfusion.
-
a.To make 2 liters (L) of 4% PFA, add 160 g of PFA to 2 L of ultrapure H2O in a 4 L Erlenmeyer flask.
-
b.Add a stir bar to the flask and wrap the top of the flask with aluminum foil.
-
c.Place the flask on a hot plate at 60°C and stir until the PFA is dissolved.
-
d.Allow the mixture to cool to room temperature (20°C) before proceeding.
-
a.
Note: Step 10 should be done in a fume hood.
-
11.Anesthetize the mouse prior to perfusion.
-
a.Prepare 10 mL of anesthetic solution by mixing 1 mL of ketamine (100 mg/mL), 0.1 mL of xylazine (100 mg/mL), and 8.5 mL of normal saline.
-
b.Intraperitoneally inject the anesthetic solution into the mouse using a 26G 3/8 needle (0.1 mL/10g body weight).
-
c.Wait for the mouse to become anesthetized before proceeding with perfusion.
-
a.
-
12.Perform whole body perfusion of mouse.CRITICAL: All animal experiments must follow relevant governmental and institutional guidelines.CRITICAL: Cold D-PBS and 4% PFA is critical for successful perfusion. Keep solutions in an ice bath during the procedure.
-
a.Pre-chill D-PBS and 4% PFA solutions before beginning.
-
b.Fill the perfusion system tubing with cold D-PBS.
-
c.Secure the mouse and open the chest cavity to expose the heart.
-
d.Insert the perfusion needle into the left ventricle.CRITICAL: Do not apply excessive pressure or torque to the heart. Avoid advancing the needle excessively. Deep needle placement can puncture the ventricular septum and result in poor fixation.
-
e.Begin perfusion with cold D-PBS according to the following:
-
i.18 mL/min for 30 s, total 9 mL.
-
ii.10 mL/min for 60 s, total 10 mL.
-
i.
-
f.Continue perfusion with cold PFA according to the following:
-
i.10 mL/min for 2 m, total 20 mL.Note: Perfusion is successful when the liver becomes pale and the animal becomes stiff.
-
i.
-
g.Immediately dissect the spinal cord and place it in 10 mL of cold 4% PFA in preparation for fixation (The spinal cord structure as shown in Figure 3). For this step, use 15 mL conical tubes. Leave the dura intact which will be easier to dissect without damaging the spinal cord after post-fixation.
-
a.
-
13.Tissue post-fixation.
-
a.Remove spinal cords in 4% PFA from the ice bath and gently shake at 150 RPM at 4°C for 12–24 h.CRITICAL: Keep fixation duration and temperature as specified to avoid overfixation, which lowers clearing efficiency.CRITICAL: Minimize cord exposure to light to prevent degradation of the fluorescence of the CTB tracer (and fluorescent reporters if planning to use endogenous fluorescence).
-
b.Remove the spinal cord from the PFA. Carefully dissect the spinal cord dura with scissors and forceps without damaging the spinal cord (Figure 3).Note: We chose to dissect the spinal cord after the fixation step as the hardened dura is easier to remove.
-
a.
-
14.PBS Wash.
-
a.Rinse the sample with 1x PBS to remove debris.
-
b.Isolate the segment of interest. If interested in the cervical or lumbar regions region, dissect the spinal region with the cervical or lumbar enlargement. If interested in the thoracic region, isolate the spinal segment between the cervical and lumbar enlargements (Figure 3C). We routinely clear spinal segments up to 7 mm long (3–4 mm is the most common length) (Figure 4A).
-
c.Transfer the spinal cord segments into a 2 mL cryotube vial.
-
d.Wash the samples to remove the PFA according to the following schedule:
-
i.1st wash: Add 2 mL of PBS with 0.01% (wt/vol) sodium azide by shaking at 150 RPM on an orbital shaker at room temperature (20°C) for 2 h.
-
ii.2nd wash: Replace with 2 mL of fresh PBS and continue washing for an additional 2 h.
-
iii.3rd wash: Replace with 2 mL of fresh PBS and continue washing for a final 2 h.
-
i.
-
a.
Pause Point: The fixed spinal cords can be stored at −80°C. First, place the spinal cords in a 15 mL conical tube with 10 mL of 30% (wt/vol) sucrose and gently shake at 4°C. Once the spinal cords sink to the bottom (can take up to 24 h), embed them OCT compound and store at −80°C for up to 6 months. To start delipidation, thaw the samples at room temperature (20°C), and repeat the PBS wash to remove OCT (step 14c).
-
15.Delipidation using CUBIC-L.Note: For this step it is necessary to prepare a half-diluted CUBIC-L solution. This is made by mixing equal volumes of CUBIC-L with ultrapure H2O. Following delipidation, the sample is washed to remove excess CUBIC-L. CUBIC-L can be stored at room temperature (20°C). Make fresh half-diluted CUBIC-L each time.Note: CUBIC treated samples become soft and fragile. To avoid damaging samples, minimize sample handling and perform the following steps in the same vial.
-
a.Prepare half-diluted CUBIC-L: Prepare 2 mL of half-diluted CUBIC-L for each 4 mm spinal cord segment.
-
i.Add a small stir bar to a 50 mL jar. Add 10 mL ultrapure H2O and 10 mL CUBIC-L and stir for 5 min.
-
i.
-
b.Half-diluted CUBIC-L: Immerse the sample in 2 mL of half-diluted CUBIC-L and shake at 150 RPM at 37°C for 12–24 h.
-
c.1st CUBIC-L: Replace with 2 mL of fresh 1x CUBIC-L and shake at 150 RPM at 37°C for 24 h.
-
d.2nd CUBIC-L: Replace with 2 mL of fresh 1x CUBIC-L and shake at 150 RPM at 37°C for an additional 24 h.
-
e.3rd CUBIC-L: Replace with 2 mL of fresh 1x CUBIC-L and shake at 150 RPM at 37°C for a final 24 h.
-
f.Wash the samples to remove residual CUBIC-L.
-
i.1st wash: Replace CUBIC-L with 2 mL of PBS with 0.01% (wt/vol) sodium azide and shake at 150 RPM at room temperature (20°C) for 2 h.
-
ii.2nd wash: Replace with 2 mL of fresh PBS and continue washing for an additional 2 h.Pause Point: CUBIC-L treated spinal cords can be stored for up to 1 wk at 4°C in PBS-0.01% (wt/vol) sodium azide. After storage, you can start the RI matching step without additional washes.
-
i.
-
a.
-
16.RI matching using CUBIC-R+(M).Note: For this step it is necessary to prepare a half-diluted CUBIC-R+(M) solution. This is made by mixing equal volumes of CUBIC-R+(M) with ultrapure H2O.CRITICAL: Fluorescent-protein labeled tissue may experience diminished signal intensity in CUBIC-R+(M), therefore the RI-matching steps should be performed in the dark for light-sensitive fluorophores. CUBIC-R+(M) can be stored at room temperature (20°C). Make fresh half-diluted CUBIC-R+(M) each time.
-
a.Prepare half-diluted CUBIC-R+(M): Prepare 2 mL of half-diluted CUBIC-R+(M) for each 4 mm spinal cord segment.
-
i.Add a small stir bar to a 50 mL jar. Add 10 mL ultrapure H2O and 10 mL CUBIC-R+(M) and stir for 5 min.
-
i.
-
b.Half-diluted CUBIC-R+(M): Immerse the samples in 2 mL of half-diluted CUBIC-R+(M) and shake at 150 RPM at room temperature (20°C) for 12–24 h.
-
c.1st CUBIC-R+(M): Replace with 2 mL of CUBIC-R+(M) and shake at 150 RPM at room temperature (20°C) for 24 h.
-
d.2nd CUBIC-R+(M): Replace with 2 mL of CUBIC-R+(M) and shake at 150 RPM at room temperature (20°C) for a final 24 h (Figure 4C).Pause Point: Samples can be left in CUBIC-R+(M) for up to 1 month at room temperature (20°C) in a dark environment. The stability of your fluorophores in CUBIC-R+(M) should be determined empirically. In our hands, TdTomato and CTB-647 are stable for over a month.
-
a.
-
17.Gel embedding sample for imaging.Note: A two-layer gel inside a plastic 1 mL syringe was used to embed the sample. Allowing the first layer to solidify provides a solid base to position the sample during embedding. Creating a gel using CUBIC-R+(M) minimizes the difference in the refractive indices of the embedded sample and the CUBIC-R+(M) solution in the microscope chamber.
-
a.Cut off the end of a 1 mL syringe to create an even cylinder (Figures 4D and 4E).
-
b.Smoothen out the edges of the open end of the syringe.Note: Sharp edges can deform during handling and puncture the embedded sample when lowering the gel to image (Flood et al., 2013).Note: Make sure to keep a syringe length appropriate to maintain compatibility with the microscope sample chamber. Removing too much from the distal end of the syringe will prevent the embedded sample from being lowered far enough into the sample chamber when it is installed on the overhead stage. We made our cuts at the 0.1 mL mark.
-
c.Thoroughly dissolve 2% (wt/wt) agarose powder in CUBIC-R+(M) at room temperature (20°C) by vigorous stirring.
-
i.To make 5 mL of gel, we added 0.11 g of agarose to 5.4 mL of CUBIC-R+(M) solution in a lidded glass jar.
-
i.
-
d.Loosen the lid and heat the mixture using a microwave until it just starts to boil. Immediately remove the jar from the microwave and cover it.CRITICAL: Keep heating times as short as possible. Boiling the solution for too long will cause excess water evaporation which can change the gel concentration and cause the gel to crystallize. troubleshooting: Problem 2.
-
e.Vigorously stir the mixture until the agarose dissolves and the gel becomes transparent. Hot agarose is volatile however, so minimize the introduction of air to the gel while stirring to avoid excess bubbling and splashes.Note: The gel will maintain the yellow hue of the CUBIC-R+(M) solution when fully transparent.
-
f.Make a gel base in a 1 mL syringe (Figure 4F):
-
i.Let the gel cool, then pipette 400 μL of agarose-CUBIC-R+(M) solution into the 1 mL syringe to make a 20 mm (0.3 mL) long base.
-
ii.Gently pipette out any bubbles that may be introduced.
-
i.
-
g.Cover the syringe with parafilm and leave at room temperature (20°C) for 1 h or until the gel base solidifies.Note: The amount of gel used to make the base depends on the length of the syringe and the length of the sample. The shorter the sample, the longer the gel base required. Do not let the gel cool too rapidly to avoid CUBIC-R+(M) crystallization.
-
h.Make a gel top layer in the 1 mL syringe (Figure 4G):
-
i.Transfer 100–200 μL of agarose-CUBIC-R+(M) solution into the 1 mL syringe to make a 5–10 mm long top layer. This part of the gel will keep the sample coupled to the syringe when the sample is pushed into the imaging chamber.
-
ii.Gently pipette out any bubbles that may be introduced.
-
iii.Without covering the syringe, allow the gel to cool slightly and become more viscous.
-
i.
-
i.Embedding the sample in the top layer:
-
i.Remove the sample from its vial using a soft brush (Figure 4H).
-
ii.Using the brush, gently guide the sample towards the center of the syringe and slide it into the gel.
-
iii.Manipulate the sample with the brush until it is in the desired orientation.
-
i.
-
j.Cover the syringe with parafilm and leave in the dark at room temperature (20°C) for 1 h or until the gel top layer solidifies (Figure 4I).CRITICAL: The gel embedded sample must be kept in the dark to prevent fluorophore bleaching.Note: We recommended making a new batch of gel for each run. Reheating CUBIC-R+(M) can cause the gel to crystallize.
-
a.
Figure 3.

Isolating spinal segment of interest
(A) The dissected spinal cord left in the PFA for overnight, and then the fixed spinal cord was held and the dura was gently removed using a pair of tweezers.
(B) The segment of interest is identified and measured.
(C) Two incisions are made to isolate the segment of interest. Scale bars: 5mm.
Figure 4.

Tissue clearing overview
(A–C) Images showing the stages of tissue clearing, scale bars: 3 mm. The initially opaque spinal cord (A) becomes transparent after delipidation with CUBIC-L (B). The sample will reach maximum transparency after RI-matching in CUBIC-R+(M) (C).
(D–G) (D) A 1 mL syringe is used as a mold for the sample by first (E) removing the tip. 2% CUBIC-R+(M) agarose is pipetted into the syringe (F) and a cleared sample is embedded in the agarose (G).
(H) After gel polymerization, the embedded sample can be lowered for imaging.
(I) A syringe prepared for gel embedding.
Part 3: 3D light sheet fluorescence imaging
In this step, cleared spinal cords are imaged using the Zeiss Z7 LSFM (McCreedy et al., 2021). Volumetric tiles were used to image the entire specimen. For example, a 1 mm wide, 2 mm long spinal cord segment was separated into 2–3 tiles in the x direction (width, left-right axis) and 5–6 tiles in the y direction (length, rostral-caudal axis) with 10% overlap. Tile size should be optimized so tile registration is accurate (by avoiding aberrations when FOV is too large), to ensure features of interest are imaged with sufficient resolution for image analysis, and to minimize imaging time and data size (Figure 5).
-
18.Initialize the microscope.
-
a.Power on the microscope and acquisition system.
-
b.Start Zen Black 3.1 software with the appropriate hardware database (.mdb).
-
c.Check the “Maintain” tab to see that the correct detection and illumination lenses are selected (5X, 0.16 NA variable refractive index detection objective and 5X, 0.1 NA variable refractive index illumination objective).CRITICAL: To avoid damaging the sample and imaging chamber, ensure that there is no sample in the imaging chamber when starting the software for the first time, as the stage calibrates itself during initialization. If the software needs to be restarted while system is still on, the stage position is retained, and the stage will not move doing software startup.
-
d.Open the chamber door and install the 5X, 0.16 NA variable refractive index imaging lens into the microscope with the refractive index set to n=1.52, the refractive index of CUBIC-R+(M).
-
e.Install the 5X, 0.1 NA variable refractive index illumination lenses (left and right) with the same refractive index as the imaging lens (n=1.52).
-
f.Install the imaging chamber on the dovetail track and secure with the set screw.
-
g.Fill a 50 mL Luer Lok syringe with fresh CUBIC-R+(M) solution. Use tubing to connect the syringe to the microscope imaging chamber.
-
h.Fill the imaging chamber to cover the imaging coverglass windows with CUBIC-R+(M) and close the chamber door.Note: To prevent spills, do not overfill the chamber.
-
a.
-
19.Locate the tissue sample and orient it in front of the microscope’s imaging lens.
-
a.Set the microscope stage to “Load” position.
-
b.Open the microscope’s top door,insert the 1 mL syringe into the appropriate sample holder, and couple it to the microscope stage using the magnetic clips, with the white lines on the sample holder and stage aligned. Close the top door.
-
c.Navigate to the “Locate” tab and start “Overview”. A webcam in the door of the microscope will give you an image of inside the chamber.
-
d.Using the specimen navigator to control the stage (Figure 5C), lower the syringe until just a small tip is visible in the overview panel. If you run out of travel on the stage, the syringe may be too short to reach the imaging medium. Leave some travel on the y-axis rather than lowering the syringe completely.
-
e.Open the top door, and gently push the syringe’s plunger down until the region of the gel containing the sample is centered in the overview panel. Close the top door.
-
f.Press the “Overview” button once more to shut down the webcam.Note: When manipulating the syringe, make sure to keep the agarose column and plunger within the syringe. It might be helpful to mark start and end positions on the plunger before mounting it in the microscope.
-
g.Press “Locate Sample” to illuminate the sample with near-infrared LEDs in the door of the Z7 and capture transmitted light images with the imaging objective.
-
h.Zoom out to increase field of view.
-
i.Move the sample near the illustrated focus of the imaging objective (marked with a blue sheet in the specimen navigator) and adjust the light intensity and display settings to provide good contrast until you see a focused image of the spinal cord.
-
j.Using the specimen navigator, orient the sample until the desired orientation for fluorescence imaging is reached. We typically orient the ventral spinal cord toward the imaging objective to minimize the amount of tissue between the objective and the planes of interest as well as flatten the imaging volume to minimize the number of slices and acquisition time. If you encounter bubbles and other defects in the agarose, or especially scattering or absorbing structures that might obscure this view of labeled neurons, try rotating to a different angle.
-
k.Turn off “Locate Sample”.
-
a.
-
20.Initialize the lasers and set up the acquisition parameters.
-
a.Turn on the lasers with the best excitation lines (Figure 5A).
-
i.To visualize ChAT-Ai14 samples (tdTomato labeled motor neurons), the 561 nm laser is used.
-
ii.To visualize CTB-647 samples (retrogradely labeled motor neurons), the 631 nm laser is used.
-
i.
-
b.Enumerate the correct “Light Path” (Figure 5B). For single-channel imaging you will have one “Track” to configure. For dual-channel imaging, you can choose to have one or more tracks to help discriminate fluorescent emissions if need be.
-
i.Ensure the laser blocking filter is selected “LBF405/488/561/638”.
-
ii.Select the appropriate emission filter cube, such as the “LP 640/BP 575-615/LP 660”.
-
iii.In the light path, check the camera (channel 1 or channel 2) that you will be imaging with.
-
i.
-
c.Set acquisition mode parameters as follows (Figure 5D):
-
i.Bit depth to 16 bit.
-
ii.Light sheet to “Dual side when experiment”. When this mode is selected, the live acquisition will use single side illumination from the direction of your choice but will use dual side illumination during data acquisition.
-
iii.Direction to either “Left” or “Right”.
-
iv.Select “online dual side fusion” and pick the “mean” fusion method.
-
v.Ensure “Pivot Scan” is selected.
-
vi.Zoom will be at 1X by default, which is fine for light sheet alignment.
-
vii.Frame size to 1920 × 1920.
-
i.
-
d.Set Channels parameters as follows (Figure 5E):
-
i.Select the desired colormap for each channel.
-
ii.Ensure the correct excitation laser(s) is/are checked on for the correct tracks(s).
-
iii.Set the laser power to a low value initially, between 5–10% laser power (lasers in the Z7 range from 50–75 mW output power).
-
iv.Note the camera exposure time, which you can adjust as needed after light sheet alignment. Faster exposure time makes alignment easier, but not if it requires increasing the laser power and by extension, photobleaching.
-
i.
-
a.
Note: We used default light sheet thickness, but this parameter can be adjusted if desired.
-
21.Align the light sheets. Light sheet offset values are associated with individual tracks so you can optimize light sheets of different wavelengths to the same focus with different settings for each track (to help reduce chromatic aberration). We find it is not necessary to do this when acquiring images at cellular resolution with fluorophores on the red side of the visible spectrum, where chromatic aberration is less severe.CRITICAL: To avoid bleaching the sample, laser power should be low when positioning the sample and aligning the light sheets. If the image is difficult to see, the brightness and contrast can be increased under the display panel. troubleshooting: Problem 3.
-
a.Under channels, select the appropriate track corresponding to the laser used. Each track will need to be aligned to the same focal plane in z. We do this sequentially one track, then the next, with a final check with a test data set.
-
i.For tdTomato labeled samples, we selected the track for the 561 nm laser and set the display color to red.
-
ii.For CTB-647 samples, we selected the track for the 638 nm laser and set the display color to gray.
-
i.
-
b.Turn on “Continuous” mode and adjust the laser intensity and camera exposure time until the sample is clearly visible with low laser power but the exposure is not too long.
-
c.Navigate to a superficial position within the sample, using single-side illumination (left or right).
-
d.Bring the illumination laser from one side into focus by clicking in the box where the light-sheet offset is located and using the up and down arrow keys to move the light sheet through the sample, adjusting the sample position if it runs out of range. When the image of the sample begins to come into focus, continue moving the light sheet through the focus and to where the image begins to defocus. Once you begin to defocus again, switch directions and bring the light sheet back to the middle of the focus.
-
e.Once one side is in focus, choose a group of cells to serve as reference markers for bringing the other side into the same focal plane. Digitally zooming in can be helpful at this step.
-
f.Switch to the other side illumination and adjust the light sheet offset, aiming to match the image to the reference image from step e.
-
g.Once you have the images matched as best as possible (due to minor refraction even in cleared tissues, it may not be possible to get a perfect match). Move to other locations within the sample and switch between left/right illumination to check the quality of co-alignment of the image planes.
-
h.Collect a small sample z-stack using the “dual-side illumination” and “online dual-side fusion” and examine the quality of the resulting image volume. Try the different fusion options to see what gives the best result (for the Ai14 we got the best result from the “mean”).
-
i.If structures are doubled or too elongated along the z-axis, go back and adjust the sheets to bring them closer to the same plane, collect sample z-stack again until result is optimized.
-
j.Switch tracks and enter the offsets found for the track used for alignment.
-
k.Collect a dual-track dataset to check that all illumination sides, color channels, and tracks are imaging the same plane. If the tracks are misaligned, perform alignment as described in b-i above for the second track, aiming for the same focal plane. Collect another test dataset and repeat until both tracks image the same plane.
-
a.
-
22.Set up the vertical z-stacks.
-
a.Under the acquisition tab, check the “Z-stack” box to enable vertical tiling. Under the Z-stack panel, select “First/Last”.
-
b.Move the overview to the top left corner of the sample and set the zoom to 0.36 X. Zooming out at this step allows you to more quickly find the extremes of the larger tissue in the z-dimension.
-
c.Move the sample away from the detection objective until the end of the sample is reached. Set this position as “last”.
-
d.Move the sample through the focus of the detection objective in the opposite direction until the end is reached. Set this position as “first”. Acquisition will be set to begin at the deeper portion of the tissue and end at the superficial surface relative to the detection objective.
-
e.Shift the field of view and check if the previously determined first and last points need to be adjusted to ensure there is no clipping.
-
f.Zoom to 1.6X, or whichever zoom planned for individual tiles.
-
g.Use the optimal slice thickness by clicking the “Optimal” button to ensure proper sampling in the z-dimension based on the resolution limits of the system.
-
a.
-
23.Set up tiling using the bounding box method.
-
a.Under the acquisition tab, check the “Tiling” box to enable horizontal and vertical tiling.
-
b.Select “Bounding grid” method to enumerate the tiles.
-
c.Set “Overlap” to 10%.
-
d.Move the sample to the top left corner and press “add” to record this position as the first point of the bounding box.
-
e.Move the camera to the bottom right corner and “add” it as the second point of the bounding box.
-
a.
Note: During this step, it is useful to scroll horizontally and vertically starting at the top left or bottom right edge of the sample to ensure all of the sample is enclosed before setting one as the point. If you find a protrusion, adding more points will increase the bounding box size.
-
24.
Click on “Start Experiment” to create a file name and begin image acquisition. Depending on number of tiles and channels, data may be 50–200 GB.
Figure 5.

Overview of the Zeiss Zen Black user interface used to control the microscope
Several acquisition settings can be adjusted before imaging begins. The required acquisition lasers are initiated (A) and their light paths are configured (B). The specimen navigator is used to adjust the position of the camera relative to the sample (C). Under acquisition mode, Image size and bit depth can be adjusted along with settings to control how the light sheets are used to acquire the image (D). Channel settings, including the intensity of each acquisition laser and exposure time can be manually set (E).
Part 4: Analyzing fluorescently labeled cells
This step introduces a simple method to quantify the number and volume of individual cells. For this protocol, we simultaneously quantified the cell number and cell volume of fluorescently labeled neurons using the Imaris surfaces tool (Takeoka and Arber, 2019) powered by a machine learning algorithm. The Labkit AI (Arzt et al., 2022) was trained to recognize different cells and exclude background when determining cell boundaries (Figures 6, 7, 8, and 9 & Method video S1).
-
25.Convert the raw image file from .czi to multi-resolution, chunked .ims format.
-
a.Add the raw image files into Imaris File Converter.
-
b.Press “Start All” to convert the files.
-
c.Once converted, unstitched Imaris images will be split into multiple .ims image pyramid files, each representing a single acquisition tile. To prevent file mix-ups, ensure that each raw image’s output folder is unique. Start the file conversion process.
-
a.
-
26.Stitch the individual converted tiles into a single image.
-
a.Add the .ims tiles into Imaris Stitcher.
-
b.Check the positioning of the tiles on the screen. For this step, the goal is to make sure each tile is properly overlapped with each other. If there are any issues with the overlap, manually drag the tiles until you are satisfied with their placement.
-
c.Align and stitch the images with “Align All Images”.
-
d.Check the aligned image for alignment problems and realign if necessary. Save the stitched image when you are satisfied with the alignment.
-
a.
Note: We use Imaris stitcher because it is interactive and flexible with respect to tiling position, and the stitching algorithm is suited for complicated 3D shapes and larger data sets.
-
27.Determine an estimated cell diameter for the population of labeled cells (Figure 6).
-
a.Switch Imaris to “Slice” view and measure the cell diameter of 30 randomly selected cells according to the following steps:
-
i.Adjust the slice viewed until the thickest region of the selected cell is in view.
-
ii.Start the measurement at the midpoint of one edge of the cell, and end at the opposite side, making sure the line crosses the center of the cell. The measured cell diameter will automatically appear on the screen.
-
iii.Repeat this for 30 randomly selected cells and average all their diameters to obtain the estimated average value.
-
i.
-
a.
-
28.Create a new surface object in Imaris and isolate the cells of interest (Figure 7).
-
a.Create a surface object in Imaris.
-
b.To ensure the fastest and most direct quantification, use the “Segment only a Region of Interest” feature in “3D View” to isolate the volume containing the cells of interest.Note: It is not necessary to manually create a surface that encloses the region of interest for volumetric analysis unless you wish to speed up image processing. For cellular quantification, it is good practice to only enclose objects that will be counted.
-
c.For Region of Interest settings, adjust the yellow overlay to include the cells of interests.
-
d.Add additional regions of interest as necessary.
-
a.
-
29.Select the appropriate source (fluorescence) channel and thresholding method for quantification.
-
a.Select the appropriate channel for each fluorophore.
-
b.We left “Surfaces Detail” at the default value. The default value is always the value where Imaris will average the intensities of two nearby voxels and use their average to perform calculations.
-
a.
Figure 6.

Measuring cell diameter using Imaris
Diameters of labeled cells were measured in slice mode (A) by drawing a line starting from the midpoint of one edge of the cell and ending at the opposite end, making sure to cross the center point, as shown here in a CTB labeled motor neuron (B). Similarly, diameters of Ai14 (tdTomato) labeled cells can be determined (C).
Figure 7.

Surface creation setup
Surface creation begins with enabling segmenting a region of interest (A) and manually dragging the boundaries until the region of interest is completely enclosed (B). We kept surface detail for smoothing at the default value for pixel doubling and used the Labkit pixel classifier algorithm for determining soma boundaries (C).
Figure 8.

Marking foreground and background pixels in Labkit
Pixels that make up the cell soma are marked as foreground using the red foreground label with a brush size of 1 (A). Background pixels and those that make up the axons are marked using the blue background label with a brush size of 1(B). After labeling, the machine learning algorithm can be trained to detect the cell soma (C).
Figure 9.

Examples of finished quantification
Cell number for the ventral horns of a T7-T10 mouse spinal cord segment was quantified using the Labkit pixel classifier algorithm.
(A and B) (A) Ventral view and (B) side view.
(C) There were 393 cells identified on the left side (cyan) and 386 cells on the right side (green).
(D) Schematic showing oblique trunk muscle motor neurons labeled with CTB.
(E) 3D view of individual neurons in the spinal cord from T5 to L1.
(F) Distribution of CTB+ ventral horn neurons with various somatic volumes from an intact T8-T9 mouse spinal cord segmented. Scale bars: 100 μm (A and B), 200 μm (E) 50 μm (E, cyan box), 15 μm (E, red box).
Cells are identified by rendering matching surfaces over them. These surfaces can be counted for cell number quantification, and their enclosed volumes can be used for volumetric quantification. To determine the boundaries of the cell (segmentation), we used “Labkit for Pixel Classification”.
-
30.Mark the cell as foreground in Labkit using sparse manual labeling (Figure 8).
-
a.Labkit in its current form will open a window in ImageJ and will display the opened image in a slice view.
-
b.Scroll through the image slices and look for cells.
-
c.Labkit comes with two labeling brushes: foreground and background. The foreground brush (in red) is used to label items that you want Imaris to quantify while the background brush (in blue) is used to label anything you want Imaris to consider as background. For this protocol, we want to label the cell soma as foreground and anything that is not the cell soma as the background.
-
d.Scroll through the slices until you find a slice where there are many cells in focus. Using the foreground brush with brush size set to 1, draw a line from one end of the cell to the other, making sure to cross the center point of the cell.Note: Do not outline or shade in the soma of the cell. Do not give the pixel classifier too much information to work with. A simple line from one end of the cell to the other end is enough to start off with. Try to draw the line from edge to edge, but it is not necessary to precisely identify where the edge is.
-
e.Identify other cells on the same slice, and mark at least 2 other cells using the foreground brush using the same technique as the first cell. It would be good at this step to mark cells of different intensity levels as foreground.
-
a.
-
31.Mark the image background in Labkit using sparse manual labels on the same z-stack as the foreground.
-
a.The main three things we marked as background are axons, artifacts, and the image background itself.
-
b.Distinguish axons from cells. Using the background brush with brush size set to 1, trace the axons of at least 3 cells to mark them as background.
-
c.For each cell that has been labeled as “foreground”, draw a short line close to but not touching the soma using the background brush.
-
d.Find three areas of the image that are obviously background. Draw a short line using the background brush in each of the areas.
-
a.
-
32.Repeat sparse labeling steps on two additional slices to help refine training.
-
a.Scroll through the image and find another slice where there are many cells in focus. These cells can be different parts of the cells from the first slice, or completely new cells.
-
b.Using sparse manual labeling, repeat the foreground and background labeling steps. The second slice should be labeled like the first slice.
-
a.
-
33.Continuously train the machine learning algorithm until the desired result is achieved by repeating the labeling and training steps.
-
a.Under segmentation, click on the “Labkit Pixel Classification” button to reveal additional options.
-
b.Click the “Play” button to begin training the algorithm based on the marked foreground and background.
-
c.Once the algorithm finishes training, a red/blue overlay will be superimposed over the original image. The red areas denote foreground, which is what Imaris will base surface creation from. The blue areas denote background, which Imaris will ignore when creating surfaces. This overlay can be turned on or off by clicking on the eye icon for the segmentation panel.
-
d.Scroll through the slices and take note of what the algorithm has denoted as foreground and background. Use either of the tools to correct misidentifications.Note: The most important thing to pay attention to is how well the cell boundaries are defined and if axons have been marked as foreground. When dealing with cell somas of varying intensities, some soma boundaries may be over quantified while others are under quantified. In these cases, it will be necessary to use either the foreground or background brushes to make amendments.
-
e.Train the machine learning algorithm again with the corrections and take note of the result.
-
f.Repeat the correction and training steps until you are satisfied with the results of the algorithm.
-
g.Click “Compute results and send it to Imaris” to send the foreground and background information to Imaris for surface creation.Note: It is possible to save training/classification results and apply them to other data sets. To save the classification results, select Segmentation > Save classifier. To then apply the classification results to other data sets, select Segmentation > Open classifier > navigate to the previously saved segmentation results > then compute the Labkit Pixel Classification (as done in step 9).
-
a.
-
34.Split touching objects using the estimated cell diameter.
-
a.Check “Enable” under “Split touching Objects (Region Growing)”
-
b.Enter the estimated cell diameter in the “Seed Points Diameter” field.
-
c.Filter the seed points using the Quality filter. Adjust the quality threshold until each visible cell is labeled with one spot.
-
a.
Note: It is better to have cells that are split into multiple surfaces over having multiple cells merged into a single surface. It is easier to unify split cells than to split merged cells. See troubleshooting: Problem 5.
-
35.Filter artifacts and incomplete cells by voxel number and volume to finish surface creation.
-
a.On the filter panel, the default filter should be set to “Quality”. The default threshold of this setting should remain unchanged.
-
b.Add a volume filter on top of the “Quality” filter. Set the lower value of this filter to 10,000 μm3 (in our hands the smallest labeled neurons are this size. This may vary based on the clearing technique used). This will exclude all objects smaller than most neurons, leaving all remaining surfaces as potential cells.
-
c.Finish surface creation with “Finish: Execute all creation steps and terminate the wizard”.
Optional: Depending on the estimated cell diameter, some cells might be split into multiple surfaces that need to be unified. Select the single selection tool under the “Navigation” pane on the far right of the user interface. Go to the edit tab in Imaris. Select all parts of the split cell and click on “Unify” to merge them together. -
a.
-
36.Export the volumetric data to an Excel file for analysis.
-
a.Under the “Statistics” main tab, select “Detailed”.
-
b.In the drop-down menu, select “Specific Values”.
-
c.In the second drop down menu that appears, select “Volume”.
-
d.At the bottom, use the “Export Statistics on Tab Display to File” function to export the volumetric data to an Excel file.
-
a.
-
37.Export the numeric data to an Excel file for analysis.
-
a.Under the “Statistics main tab, select “Overall”.
-
b.Under “Variable” the value for “Total Number of Surfaces” gives the number of cells quantified.
-
c.At the bottom, use the “Export Statistics on Tab Display to File” function to export the volumetric data to an Excel file.
-
a.
-
38.Calculate the cell density using the total cell count.
-
a.Switch Imaris to slice view and measure the entire quantified length of the sample. Since we quantified the entire length of the sample, we measured the end-to-end distance.
-
b.Divide the total number of cells with the respective length to get the cell density.
-
a.
Expected outcomes
Successful application of this protocol allows users to label spinal neurons using standard molecular genetic or retrograde tracing approaches and analyze them using easy to use AI-assisted image analysis software. First, crossing various Cre-driver lines (ChAT-Cre, vGlut2-Cre and vGAT-Cre) with fluorescent reporter mice allow users to genetically label specific populations of neurons. For example, we have successfully quantified the number of tdTomato-positive neurons in the ventral horn zone of T7-T10 spinal segments isolated from ChAT::Ai14 (tdTomato) mice. Second, various fluorescent labeling strategies can be used to label specific groups of neurons in the spinal cord. We expect this protocol to be a powerful tool to study cellular responses to external stimuli in the mammalian CNS (Figure 9 & Method video S1).
Limitations
This protocol describes the specific steps needed to reproducibly quantify the number and somatic volume of fluorescently labeled neurons in the spinal cord segments. Here we show a robust analysis and imaging workflow using florescent reporter mice and retrograde dyes to label neurons. However, we have yet to test our analysis pipeline antibody labeled samples, which may require additional optimization and Labkit training sessions. Despite this, with slight modifications, this protocol is suitable for use in broad experiments requiring neuron labeling and analysis.
Troubleshooting
Problem 1
Poor tissue clearing in spinal cords. See step 15: Delipidation using CUBIC-L.
Potential solution
Increase delipidation time (full CUBIC-L). For difficult to clear samples, increasing the delipidation time to a total of 5 days will generally result in successful clearing. This effect is only noticeable upon the completion of the RI matching step. In addition, make sure to keep fixation duration between 12–24 h to avoid the risk of reduced clearing efficiency.
Problem 2
CUBIC-R+(M)-agarose crystallization during preparation of the agarose gel or when creating the gel base/tops. See step 17: Gel embedding sample for imaging.
Potential solution
CUBIC-R+(M) is prone to crystallization due to two different causes: too much water evaporating from the solution or cooling too rapidly. In this protocol, the only step where significant water loss from solution can occur is during the microwave heating step. To solve this issue for smaller volumes of CUBIC+®-agarose solution, microwave in short 5 s intervals and immediately stop when the solution begins to boil. Another way to mitigate this problem is to prepare larger volumes of the CUBIC+®-agarose solution. To prevent the gel base/tops from crystallizing during the cooling process, keep the syringes at room temperature (25°C) and tightly seal with parafilm to prevent water loss.
Problem 3
Only the background signal is visible after light sheet imaging (step 20).
Potential solution
If no cells are visible after imaging:
-
•
If using a fluorescent reporter mouse, confirm the genotype of the experimental animal.
-
•
If using fluorophore conjugated CTB, ensure the CTB solution was prepared correctly. Use a new aliquot of CTB that has not been repeatedly freeze-thawed or vortexed. See: Prepare a 0.5% (wt/vol) CTB tracer solution for injection.
-
•
Poor perfusions are often the culprit for poor labeling. Ensure that 4% PFA is freshly made and that PFA and D-PBS are cold before proceeding performing the perfusion. See: Prepare D-PBS and 4% PFA for whole body perfusion.
Problem 4
CTB tracer labeled samples have large amounts of fluorescent artifacts scattered throughout the sample (steps 20–21).
Potential solution
CTB tracer is known to leak out of labeled cells into surrounding tissues. To mitigate this issue, do not wait too long after injecting CTB before perfusing the mouse, 3 days after CTB injection is recommended. In our hands, CTB-488 was difficult to detect over background autofluorescence. Also, the scattered artifacts from CTB labeling may introduce variation when quantifying cell number. To overcome this, we recommend reducing the concentration/volume of injected CTB or genetically labeling neurons using recombinant viruses or transgenic mice.
Problem 5
Imaris Surfaces merges close cells into one large surface. Using the estimated cell diameter does not successfully split the cells (step 27).
Potential solution
To split merged cells, the simplest way is to enter a smaller value for the estimated cell diameter. If this does not work, it might be necessary to retrain the Labkit pixel classifier and mark the empty spaces between the cells as background. In addition, for precise cellular volume detection, LSMF 20X objective or higher was recommended. We found a combination of the three usually works well for splitting merged cells.
Problem 6
Sample is not aligned with the frame of Imaris, making drawing an accurate region of interest difficult (step 28).
Potential solution
You can align the sample with the frame using the free rotate functionality. We had good results aligning the sample by aligning the sample coronally (axially) with the screen. When the sample is aligned with the screen, it appears as a single coronal section.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bo Chen (Bochen1@utmb.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This study was supported by grants from University of Texas Medical Branch (UTMB) startup funds (B.C.); Mission Connect, TIRR Foundation (B.C.); UT system star award (B.C.); Wings for Life Foundation Research grant (B.C.); Chan Zuckerberg Imaging Scientist Program (H.G.); Clinical and Translational Science Award NRSA (TL1) Training Core (TL1TR001440); and NIH/NCATS (A.S.J.). Light sheet fluorescence microscopy was performed at Texas A&M University’s Microscopy and Imaging Center (RRID: SCR_022128). The graphical abstract and Figure 1 were created with BioRender.com.
Author contributions
H.Y. and B.C. designed research; H.Y., Q.L., A.S.J., T.D., J.L.M., and H.G. performed experiments; H.Y., A.S.J., A.E., and B.C. analyzed data; and H.Y. wrote the paper with the input from all authors.
Declaration of interests
A.E. is an employee of Bitplane.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101759.
Contributor Information
Hao Yu, Email: yu.howard89@yahoo.com.
Bo Chen, Email: bochen1@utmb.edu.
Data and code availability
The Labkit ImageJ plugin requires the following update sites:
clij: https://sites.imagej.net/clij/
CLIJ2: https://sites.imagej.net/clij2/
Imaris-Labkit: https://sites.imagej.net/imarislabkit/
References
- Arzt M., Deschamps J., Schmied C., Pietzsch T., Schmidt D., Tomancak P., Haase R., Jug F. LABKIT: labeling and segmentation toolkit for big image data. Front. Comput. Sci. 2022;4 doi: 10.3389/fcomp.2022.777728. [DOI] [Google Scholar]
- Flood P.M., Kelly R., Gutiérrez-Heredia L., Reynaud E.G. ZEISS Lightsheet Z.1 Sample Preparation. 2013. https://Applications.Zeiss.Com/C125792900358A3F/0/E6504BFCBC3C2D39C1257BD500417CA6/$FILE/EN_41_011_058_LightsheetZ1_Sample-Preparation.Pdf
- Goetz C., Pivetta C., Arber S. Distinct limb and trunk premotor circuits establish laterality in the spinal cord. Neuron. 2015;85:131–144. doi: 10.1016/j.neuron.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Inquimbert P., Moll M., Kohno T., Scholz J. Stereotaxic injection of a viral vector for conditional gene manipulation in the mouse spinal cord. J. Vis. Exp. 2013;1–5 doi: 10.3791/50313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Hegarty S., Winter C., Wang F., He Z. Viral vectors for neuronal cell type-specific visualization and manipulations. Curr. Opin. Neurobiol. 2020;63:67–76. doi: 10.1016/j.conb.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K., Mitani T.T., Horiguchi S.A., Kaneshiro J., Murakami T.C., Mano T., Fujishima H., Konno A., Watanabe T.M., Hirai H., Ueda H.R. Advanced CUBIC tissue clearing for whole-organ cell profiling. Nat. Protoc. 2019;14:3506–3537. doi: 10.1038/s41596-019-0240-9. [DOI] [PubMed] [Google Scholar]
- McCreedy D.A., Jalufka F.L., Platt M.E., Min S.W., Kirchhoff M.A., Pritchard A.L., Reid S.K., Manlapaz R., Mihaly E., Butts J.C., et al. Passive clearing and 3D lightsheet imaging of the intact and injured spinal cord in mice. Front. Cell. Neurosci. 2021;15 doi: 10.3389/fncel.2021.684792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nectow A.R., Nestler E.J. Viral tools for neuroscience. Nat. Rev. Neurosci. 2020;21:669–681. doi: 10.1038/s41583-020-00382-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeoka A., Arber S. Functional local proprioceptive feedback circuits initiate and maintain locomotor recovery after spinal cord injury. Cell Rep. 2019;27:71–85.e3. doi: 10.1016/j.celrep.2019.03.010. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M., Kida M.Y., Akita K. Relationship between the arrangement of motoneuron pools in the ventral horn and ramification pattern of the spinal nerve innervating trunk muscles in the cat (Felis domestica) Exp. Neurol. 1994;128:290–300. doi: 10.1006/exnr.1994.1139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Labkit ImageJ plugin requires the following update sites:
clij: https://sites.imagej.net/clij/
CLIJ2: https://sites.imagej.net/clij2/
Imaris-Labkit: https://sites.imagej.net/imarislabkit/