

Abstract
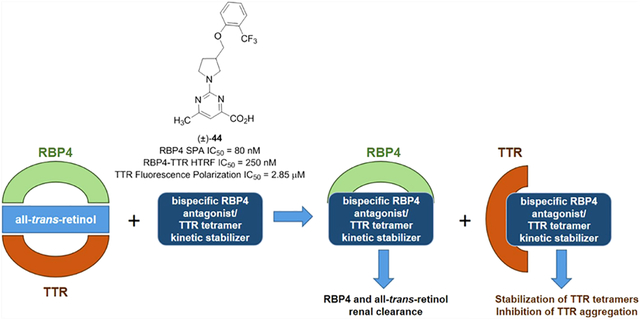
Accumulation of cytotoxic lipofuscin bisretinoids may contribute to atrophic age-related macular degeneration (AMD) pathogenesis. Retinal bisretinoid synthesis depends on the influx of serum all-trans-retinol (1) delivered via a tertiary retinol binding protein 4 (RBP4)–transthyretin (TTR)–retinol complex. We previously identified selective RBP4 antagonists that dissociate circulating RBP4–TTR–retinol complexes, reduce serum RBP4 levels, and inhibit bisretinoid synthesis in models of enhanced retinal lipofuscinogenesis. However, the release of TTR by selective RBP4 antagonists may be associated with TTR tetramer destabilization and, potentially, TTR amyloid formation. We describe herein the identification of bispecific RBP4 antagonist–TTR tetramer kinetic stabilizers. Standout analogue (±)-44 possesses suitable potency for both targets, significantly lowers mouse plasma RBP4 levels, and prevents TTR aggregation in a gel-based assay. This new class of bispecific compounds may be especially important as a therapy for dry AMD patients who have another common age-related comorbidity, senile systemic amyloidosis, a nongenetic disease associated with wild-type TTR misfolding.
Graphical Abstract
INTRODUCTION
All-trans-retinol (vitamin A, 1) (Figure 1) is an essential vitamin that serves as a precursor for the biosynthesis of retinoic acid (2),1 11-cis-retinal (3),2 and many other key retinoids involved in multiple cellular processes and numerous critical biological functions throughout the body. Extracellular delivery of fatsoluble 1 from the liver to vitamin A-dependent tissues is accomplished via a protein transport complex involving the lipocalin protein retinol binding protein 4 (RBP4) and transthyretin (TTR, thyroxine binding prealbumin).3 The majority of circulating RBP4 is synthesized in the liver (~60%), where it requires the binding of 1 (holo-RBP4) prior to secretion. The tertiary complex between holo-RBP4 and TTR is required as the small size of RBP4 (21 kDa) makes it susceptible to rapid glomerular filtration.4 Formation of the complex depends on binding of 1 to RBP4 as apo-RBP4 associates with TTR poorly.
Figure 1.
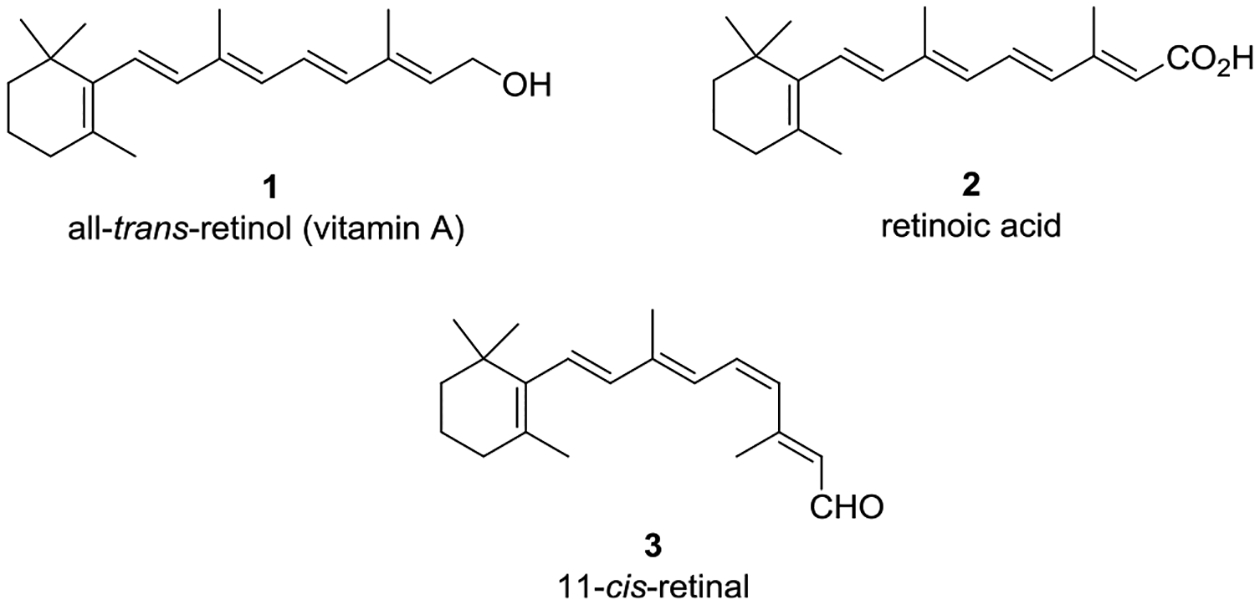
All-trans-retinol (1), retinoic acid (2) (a vital retinoid involved in morphogenesis), and 11-cis-retinal (3) (a key retinoid required for phototransduction).
TTR is a 55 kDa homotetrameric protein largely synthesized in the liver for systemic circulation and by the choroid plexus for secretion into the cerebral spinal fluid (CSF).5 In plasma, TTR mainly functions as a transporter of holo-RBP4 while also serving as a secondary transporter for the thyroid hormone thyroxine (T4, 4) (Figure 2). Thyroxine binding globulin (TBG) is the major T4 transport carrier in plasma with approximately 10–15% bound to TTR.
Figure 2.
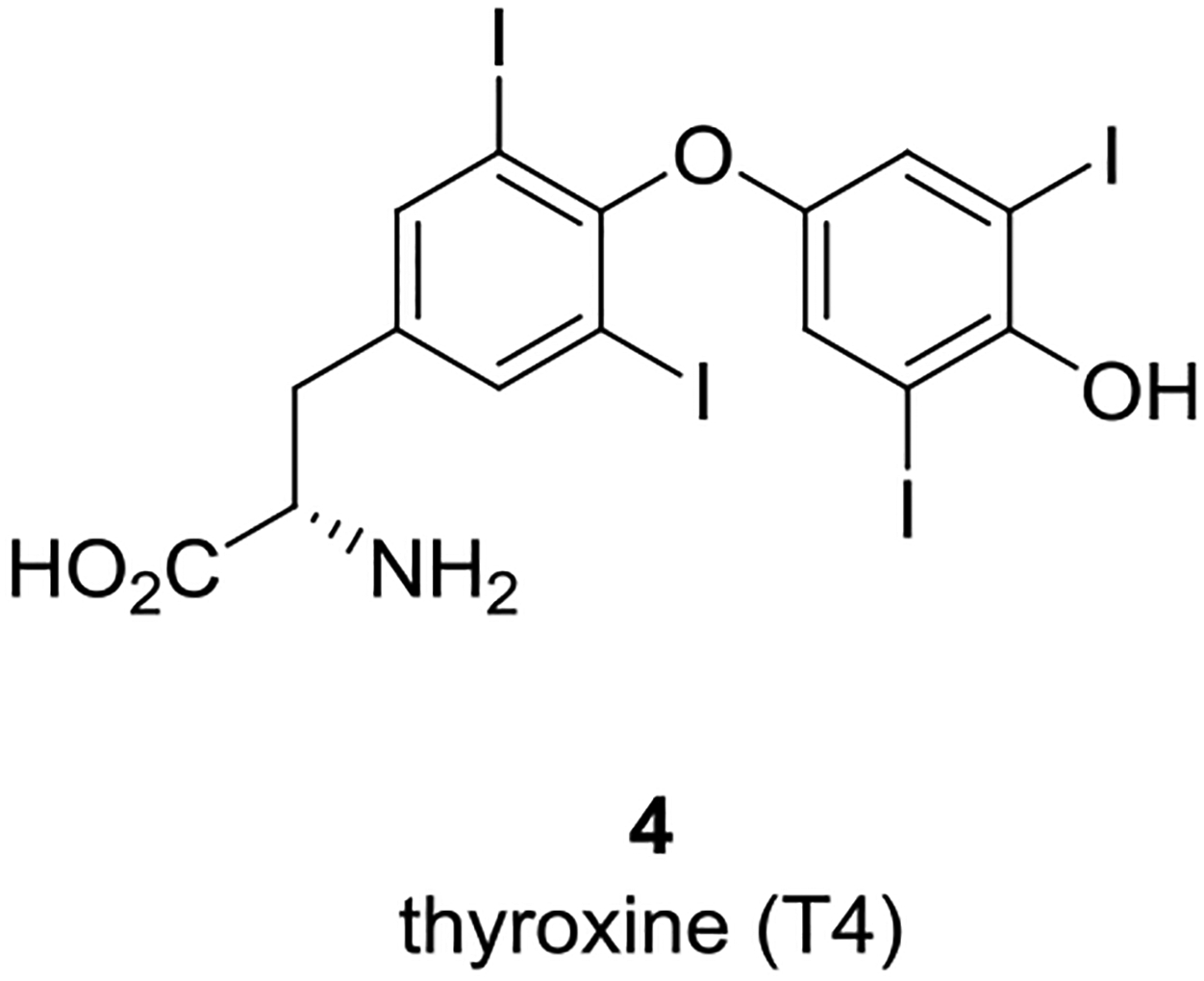
Thyroid hormone thyroxine (T4) (4).
The quaternary structure of TTR is composed of a dimer of dimer arrangement that features two identical C2 symmetric binding sites for T4 at the central channel of the tetramer formed, where the dimers interface (Figure 3A).5 Each dimer subunit is constructed from two 127-residue β-sheet-rich polypeptide monomers that associate via their edge β-strands. The dimer–dimer interface at the T4 binding sites is weak, and the breakage of it constitutes the first step in the TTR tetramer dissociation process.6
Figure 3.
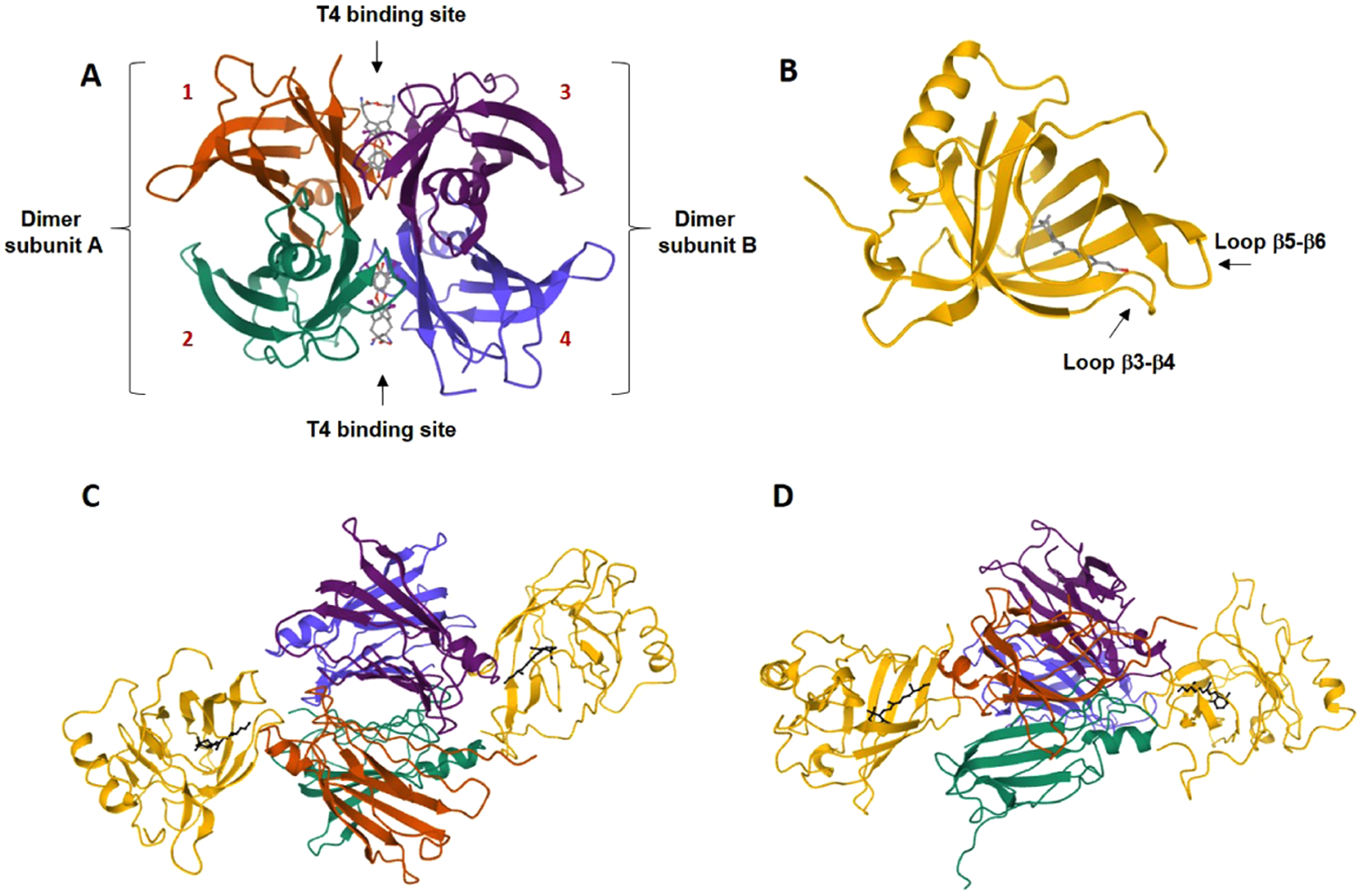
Three-dimensional structures of TTR, holo-RBP4, and the holo-RBP4–TTR tertiary complex. (A) Ribbon diagram of the quaternary homotetrameric structure of TTR with 4 bound (Protein Data Bank (PDB) 2ROX).5c The dimers are shown as dimer subunit A and dimer subunit B, and each monomer subunit of the tetramer (labeled 1–4) is shown with its secondary structural elements and colored differently. The monomer components of dimer A are shown in orange (monomer 1) and cyan (monomer 2). The monomer components of dimer B are shown in violet (monomer 3) and blue (monomer 4). The dimers are associated back-to-back creating a large channel through the center of the tetramer that presents two C2 symmetrical T4 binding sites. The binding of 4 in both T4 binding pockets of TTR is shown in a ball-and-stick format (gray). (B) Ribbon diagram of holo-RBP4 (PDB 1RBP).7 RBP4 is shown as yellow, and 1 is depicted in a ball-and-stick format (gray) bound within the RBP4 hydrophobic β-barrel binding cavity. (C) The holo-RBP4–TTR tertiary complex (PDB 1QAB)10 colored by a chain and viewed from the front. The TTR tetramer is located at the center of the complex with two holo-RBP4 molecules docked at a twofold axis of symmetry that is orthogonal to the T4 binding sites. Compound 1 is depicted in a ball-and-stick format (black). (D) Holo-RBP4–TTR tertiary complex (PDB 1QAB)10 colored by a chain and viewed from the side. Compound 1 is depicted in a ball-and-stick format (black).
Complexation between RBP4 and TTR requires that 1 be initially bound to RBP4. The structure of RBP4 contains a cylindrical binding cavity for 1, and analysis of Protein Data Bank (PDB) X-ray crystallographic data for holo-RBP4 (PDB 1RBP)7 shows the isoprene tail of 1 occupying the tunnel with its pendant trimethyl cyclohexene ring projecting within the inner hydrophobic β-ionone cavity. The hydroxyl group of 1 resides near the binding cavity opening and is exposed to solvent. Binding of 1 to RBP4 induces conformational changes to exterior loops8 that provide favorable protein–protein interaction surfaces for facile TTR engagement. These conformational changes permit a twofold axis of symmetry docking to TTR (Figure 3B). TTR features two equivalent binding sites for holo-RBP4, providing a complex stoichiometry of one TTR tetramer to two holo-RBP4 molecules (Figure 3C).5 However, a 1:1 molar complex is typically observed due to limiting concentrations of plasma RBP4. Finally, a H-bond between the hydroxyl group of 1 and TTR further stabilizes the protein transport complex9 and allows it to fully conceal the hydrophobic vitamin as it is transported in serum.
It was hypothesized that reducing systemic levels of circulating RBP4 and 1 via selective antagonists could impede the biosynthesis of retinal bisretinoids and prevent geographic lesion formation and growth associated with late-stage dry (atrophic) age-related macular degeneration (AMD) and Stargardt disease.11,12 All-trans-retinol-competitive antagonists of RBP4 prohibit holo-RBP4–TTR complexation, thereby inducing reductions with circulating RBP4 and 1 levels via rapid renal clearance. A diminished influx of 1 to the RPE results in a reduction of cytotoxic bisretinoid accumulation in the retina, which is believed to underlie parts of the pathophysiology of dry AMD and Stargardt disease.11a,13–16 The approach is supported by proof-of-concept data obtained for fenretinide (5) (Figure 4), which was studied preclinically with Abca4−/− knock-out mice and in a phase II proof-of-concept study with dry AMD patients.17 Thorough analysis of the human data with 5 indicated that a reduction in serum RBP4 levels below the 1 μM threshold was required to significantly hinder the expansion of demarcated atrophic lesions. The nonretinoid RBP4 antagonist A11208 (6) was reported to disrupt holo-RBP4–TTR complexation in vitro and reduce rodent RBP4 plasma levels by >70% and reduce retinal bisretinoid accumulation in Abca4−/− mice.13 Our selective and orally bioavailable non-retinoid RBP4 antagonists 718 and BPN-1413619 (8) displayed favorable pharmacokinetic (PK) profiles and induced dose-dependent circulating RBP4 reductions in rodents. Compound 8 also robustly lowered serum RBP4 levels and exhibited excellent pharmacokinetic–pharmacodynamic (PK–PD) correlations in nonhuman primates upon oral administration.20 Finally, 8 inhibited the production of fluorophore bisretinoid N-retinylidene-N-retinylethanolamine (A2E) while restoring homeostatic complement system protein expression in the Abca4−/− mouse retina without altering visual cycle kinetics.14
Figure 4.
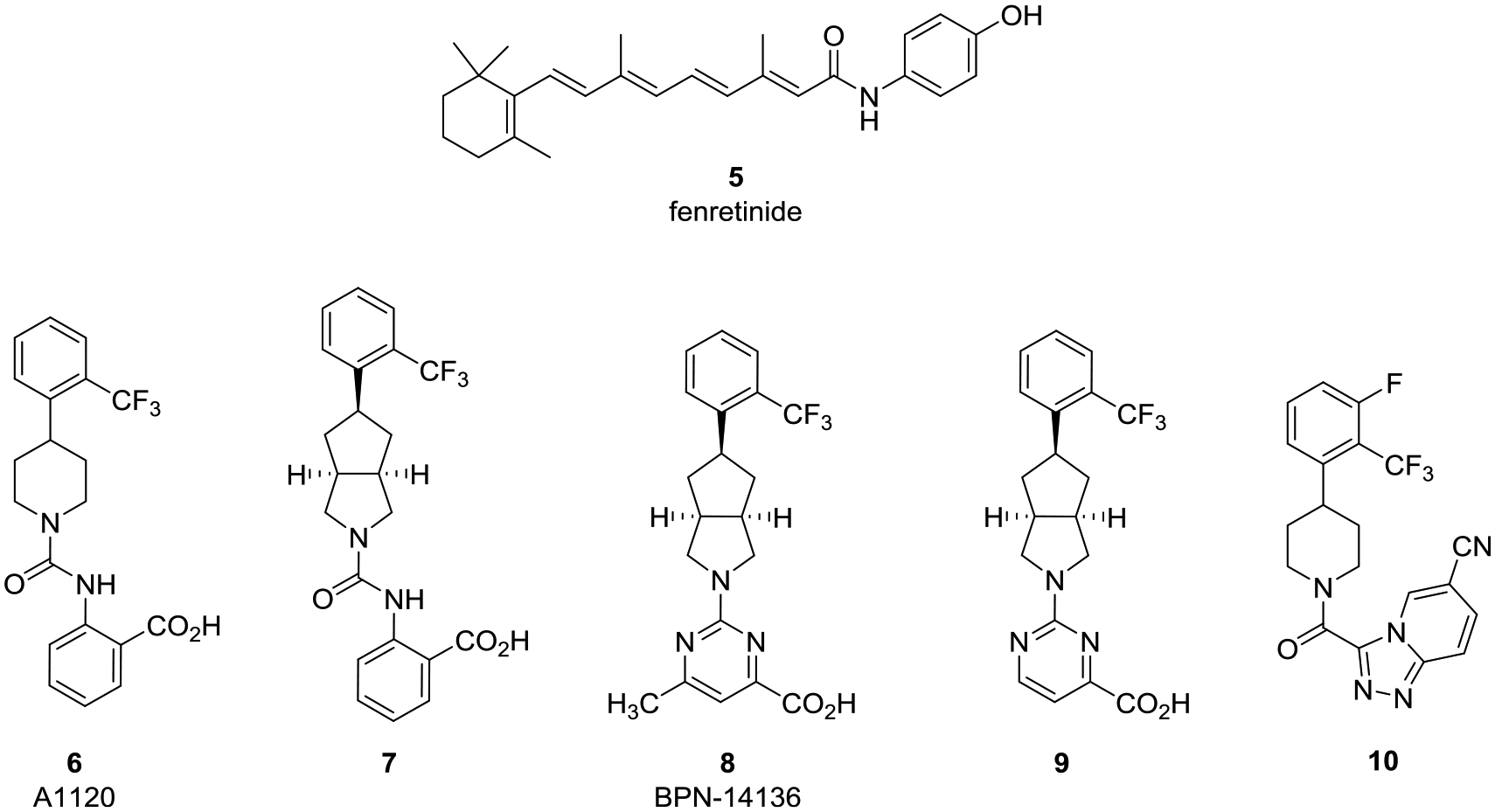
Examples of previously reported RBP4 antagonists that include fenretinide (5), A1120 (6), and previously identified RBP4 antagonists 7, BPN-14136 (8), 9, and 10.
In addition to transporting 1 to targeted tissues, RBP4 has also been identified as an adipokine potentially involved in metabolic disorders including type 2 diabetes,21 obesity,22 insulin resistance,23 cardiovascular disease,24 and hepatic steatosis.25 Thus, the pharmacological reduction of circulating RBP4 serum levels may also hold promise for the treatment of a myriad of metabolic diseases. Indeed, we have recently reported that our antagonist 10 significantly lowered serum RBP4 levels in rodents (>80%), reduced the concentration of circulating RBP4 produced in the adipose tissue, and ameliorated hepatic steatosis in transgenic adi-hRBP4 mice. These data provide evidence that RBP4 antagonists may hold therapeutic promise for treating nonalcoholic fatty liver disease (NAFLD).26
The formation of amyloid aggregates derived from either mutant or wild type underlies TTR amyloidosis (ATTR) diseases such as senile systemic amyloidosis (SSA), peripheral polyneuropathy (ATTR-PN), and cardiomyopathy (ATTR-CM).6,27 As was noted above, the breakage of the dimer–dimer interface in TTR tetramers constitutes the first step in the TTR tetramer dissociation process that leads to TTR misfolding. Approximately 50% of serum TTR is associated with RBP4, and it is suggested that tertiary holo-RBP4–TTR complexation serves to stabilize this fraction of serum TTR tetramers and prevent them from dissociation and misfolding.28,29 Based on the in vitro observation that RBP4–TTR interaction is capable of conferring an additional stabilization to tetrameric TTR,28,29 it seems plausible that the release of TTR tetramers from RBP4–TTR–retinol complexes induced by selective RBP4 antagonists may lead to tetramer destabilization and its enhanced dissociation to dimer subunits. The resulting dimers may then further dissociate into monomers that can misfold, aggregate, oligomerize, and eventually form insoluble TTR amyloid fibrils.28,29 While selective RBP4 antagonists can be a safe and effective therapy for the majority of dry AMD patients, this class of compounds may potentially be counterindicated for a fraction of AMD patients who may be prone to developing ATTR. In addition to individuals with rare genetic forms of ATTR caused by pro-amyloidogenic TTR mutations, the use of selective RBP4 antagonists may not be optimal in patients with SSA, a late-onset nongenetic disease associated with misfolding and aggregation of wild-type TTR. SSA affects approximately 25% of patients over the age of 80,30 and based on the high-population frequency of this disease and dry AMD, significant comorbidity between the two conditions is expected. In addition, the use of selective RBP4 antagonists may not be optimal in older African-American patients with dry AMD who have an increased chance of carrying a relatively high-frequency pro-amyloidogenic V122I mutation in the TTR gene.31 It is undesirable for an effective chronic treatment for one of the two conditions to be counterindicated for the use in patients with another one, and developing an optimal therapy for dry AMD that can be safely used in patients with ATTR comorbidities is an important objective.
ATTR pathophysiology begins with the sequential dissociation of TTR tetramers into pro-amyloidogenic monomers with tetramer dissociation into dimer subunits as the rate-limiting step in the process.6 While thyroxine binding was reported to stabilize TTR tetramers,32 the majority of TTR in circulation (up to 90%), including TTR in a complex with holo-RBP4, is not bound to its natural ligand.28 Current therapeutic approaches to treat ATTR-CM include small-molecule kinetic stabilizers of TTR tetramers that bind at the T4 binding sites and increase the energy barrier of tetramer dissociation.33 Two orally bioavailable kinetic stabilizers clinically investigated to date include the Food and Drug Administration (FDA)-approved tafamidis (11) (Figure 5)34 and AG10 (12).35 TTR stabilizer 11 is currently approved to treat familial amyloid polyneuropathy and ATTR-CM patients, and 12 has demonstrated a near-complete stabilization of TTR in ATTR-CM patients with symptomatic, chronic heart failure in phase II clinical trials (a phase III trial is ongoing).36 In addition, the repurposed FDA-approved nonsteroidal anti-inflammatory drug diflunisal (13)37 and catechol-O-methyl transferase inhibitor tolcapone (14)38 are examples of additional small molecules that also exhibit TTR tetramer kinetic stabilization activity and have been investigated for clinical efficacy against ATTR-PN. Finally, the recently reported saturation transfer difference (STD) NMR data by Gimeno and co-workers show that β-amyloid (Aβ) peptides bind at the RBP4 binding pockets of TTR and that stabilization of the TTR tetramer by iododiflunisal (structure not shown) may be facilitating the TTR–Aβ interaction.39 These STD-NMR binding experiments may shed some light on the mechanism by which iododiflunisal produces its ameliorating effects in animal models for Alzheimer’s disease.40
Figure 5.
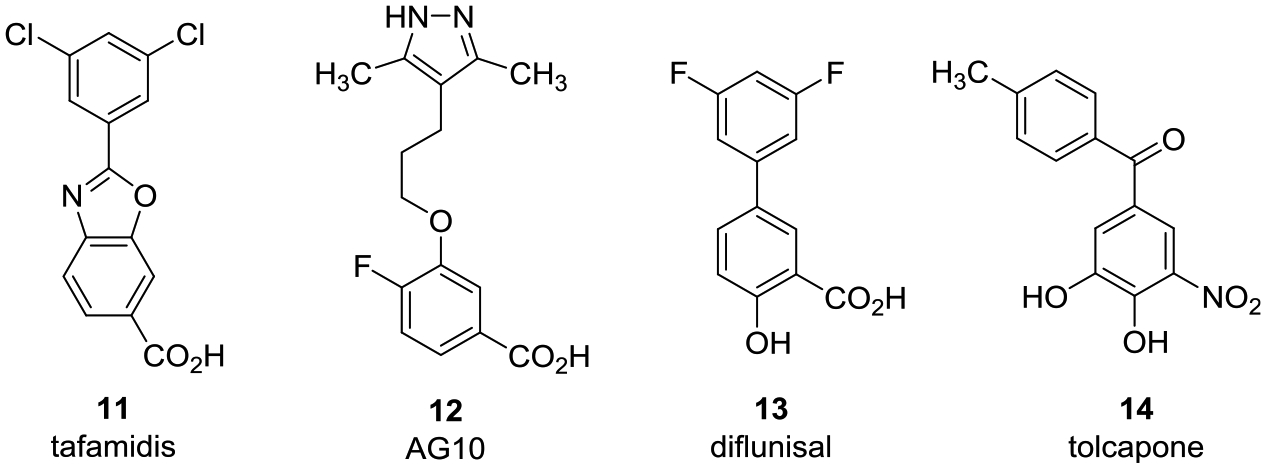
Examples of reported small-molecule TTR tetramer kinetic stabilizers that include tafamidis (11), AG10 (12), diflunisal (13), and tolcapone (14).
Our goal was to develop a treatment for dry AMD and ATTR comorbidities based on bispecific ligands that can serve as both an RBP4 antagonist and TTR tetramer kinetic stabilizer. Such drugs are predicted to provide therapeutic benefits associated with reducing circulating RBP4 levels while simultaneously stabilizing unliganded TTR tetramers released from the holo-RBP4–TTR complex, thus circumventing potential risks of amyloid fibril formation, as schematized in Figure 6. Furthermore, a polypharmacological approach consisting of a single bispecific molecule capable of exhibiting dual activity for both targets may present advantages over the coadministration of a single agent for each target. Such advantages include improving patient compliance, minimizing complex PK, and avoiding potential drug–drug interactions that could arise from multiple drug intakes.41
Figure 6.
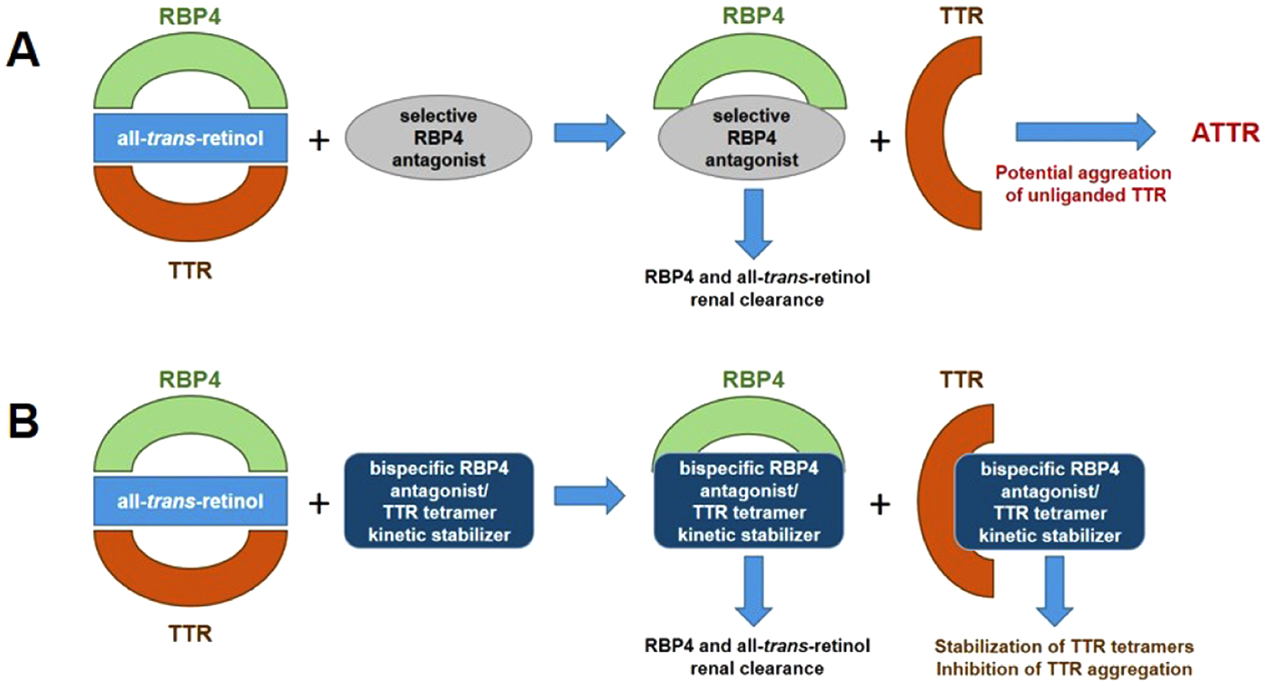
Bispecific RBP4 antagonists and TTR tetramer kinetic stabilizers for treating RBP4 indications with potential ATTR comorbidities. (A) Schematic illustrating the use of selective RBP4 antagonists for disrupting the holo-RBP4–TTR protein–protein interaction and inducing serum reduction of 1 and RBP4. The concomitant release of unliganded TTR may induce its aggregation, potentially contributing to ATTR in predisposed patients. (B) Bispecific ligands with dual RBP4 antagonist and TTR tetramer kinetic stabilization activity may induce reductions in circulating levels of RBP4 and 1 while also preventing potential TTR aggregation and insoluble amyloid fibril formation.
In our work reported herein, we initiated a structure-based drug design effort to identify a novel class of nonretinoid bispecific compounds capable of exhibiting dual RBP4 antagonist and TTR tetramer kinetic stabilization activity. The primary desirable attributes for the newly designed analogues were (1) to contain a nonretinoid framework so as to avoid potential off-target activity at retinoic acid receptors, (2) to effectively compete with 1 and antagonize the all-trans-retinol-dependent RBP4–TTR interaction, (3) to induce robust in vivo serum RBP4 reduction by >70%, and (4) to bind to unliganded TTR tetramers and effectively prevent TTR dissociation and aggregation as judged in a gel-based wild-type TTR aggregation assay. It is for these reasons in our program that compounds were measured for binding potency to non-TTR-associated RBP4 via competition with 1 (scintillation proximity assay (SPA) to generate dose–response IC50 values in the presence of fixed concentrations of [3H]-all-trans-retinol), compound binding potency to unliganded TTR tetramers (fluorescence polarization (FP) assay to generate dose–response IC50 values in the presence of a fixed concentration of a fluorescence probe that binds at TTR tetramer T4 sites), and compound functional antagonist potency for disruption of holo-RBP4–TTR complex formation (homogeneous time-resolved fluorescence assay (HTRF) to generate dose–response IC50 values in the presence of fixed concentrations of all-trans-retinol).
DESIGN OF BISPECIFIC ANALOGUES
We discovered that our previously disclosed series of RBP4 antagonists could serve as suitable benchmark scaffolds for our novel bispecific analogue design efforts. RBP4 antagonists 718 (RBP4 SPA IC50 = 72.7 nM, RBP4–TTR HTRF IC50 = 0.294 μM) and 919 (RBP4 SPA IC50 = 20.8 ± 0.5 nM, RBP4–TTR HTRF IC50 = 79.7 nM) were found to exhibit significant TTR tetramer binding potency (7 TTR FP IC50 = 2.4 μM; 9 TTR FPIC50 = 2.5 μM). These data, in addition to the exemplary PK–PD properties19,20 and in vivo Abca4−/− transgenic mouse model efficacy14 exhibited by standout analogue 8 (RBP4 SPA IC50 = 12.8 ± 0.4 nM, RBP4–TTR HTRF IC50 = 43.6 ± 10.5 nM), led us to focus our initial bispecific drug design efforts starting from their common [3.3.0]-bicyclic core scaffold. Thus, our optimization strategy was to identify novel compounds that presented the following attributes: (1) retaining or improving in vitro activity observed for 7, 8, and 9, (2) exhibiting TTR binding potency that could provide adequate TTR tetramer stabilization and prevent aggregation at therapeutic concentrations required for >70% serum RBP4 reduction, (3) maintaining the largely favorable absorption, distribution, metabolism, and excretion (ADME) profile observed for 7, 8, and 9, and (4) maintaining the favorable PK–PD properties of 7 and 8. A potential drawback for 8 was its ancillary agonist activity at the nuclear peroxisome proliferator-activated receptor-γ (PPARγ) (PPARγ IC50 = 3.6 μM in the agonist-induced corepressor nuclear receptor corepressor (NCoR) release assay). PPARγ plays a critical role in regulating energy homeostasis, metabolism, and inflammation, and we sought to avoid such off-target activity due to potential side effects often associated with PPARγ agonists, which include risk for fluid retention, weight gain, bone loss, and congestive heart failure.42
Our structure-based optimization approach sought to exploit potential similarities presented within the binding pockets of both proteins. X-ray crystallographic data of 4 bound to TTR (PDB 2ROX)5c shows that the T4 binding sites are segregated by an inner and outer binding cavity. The twofold axis symmetry of the T4 binding cavity presents six halogen binding pockets (HBPs) designated as HBP1, HBP1′, HBP2, HBP2′, HBP3, and HBP3′ (Figure 7).5c The HBPs are defined according to the relative positions of the iodine atoms of 4 that occupy them. The inner T4 binding pockets contain HBP3 and HBP3′ and are located between the Ser117, Leu110, Thr119, and Ala108 residues of both subunits. The innermost Ser117 hydroxyl groups may either participate in bridging hydrogen bond interactions with a resident structural water molecule or they may mediate hydrogen bond interactions with bound TTR tetramer kinetic stabilizers, such as 12.35 The HBP2 and HBP2′ pockets are located between the inner and outer binding cavities and are highly lipophilic as they contain Leu17, Ala108, Ala109, and Leu110 residues. Finally, the HBP1 and HBP1′ pockets reside near the outer binding cavity and feature residues Lys15, Glu54, Leu17, Thr106, and Val121 of both subunits. The binding of 4 typically involves a forward binding mode, which projects the bis-iodo phenol “head group” deep within the inner cavity where the iodine atoms occupy HBP2 and HPB3 pockets. The linker phenyl ring resides within the hydrophobic channel bridging the inner and outer cavities, with one of its iodine atoms occupying a HBP1/HBP1′ binding pocket. Finally, the amino acid “bottom group” motif of 4 resides near the periphery of the binding site opening and forms salt-bridge interactions with ionizable residues Lys15/Lys15′ and Glu54/Glu54′.5c
Figure 7.
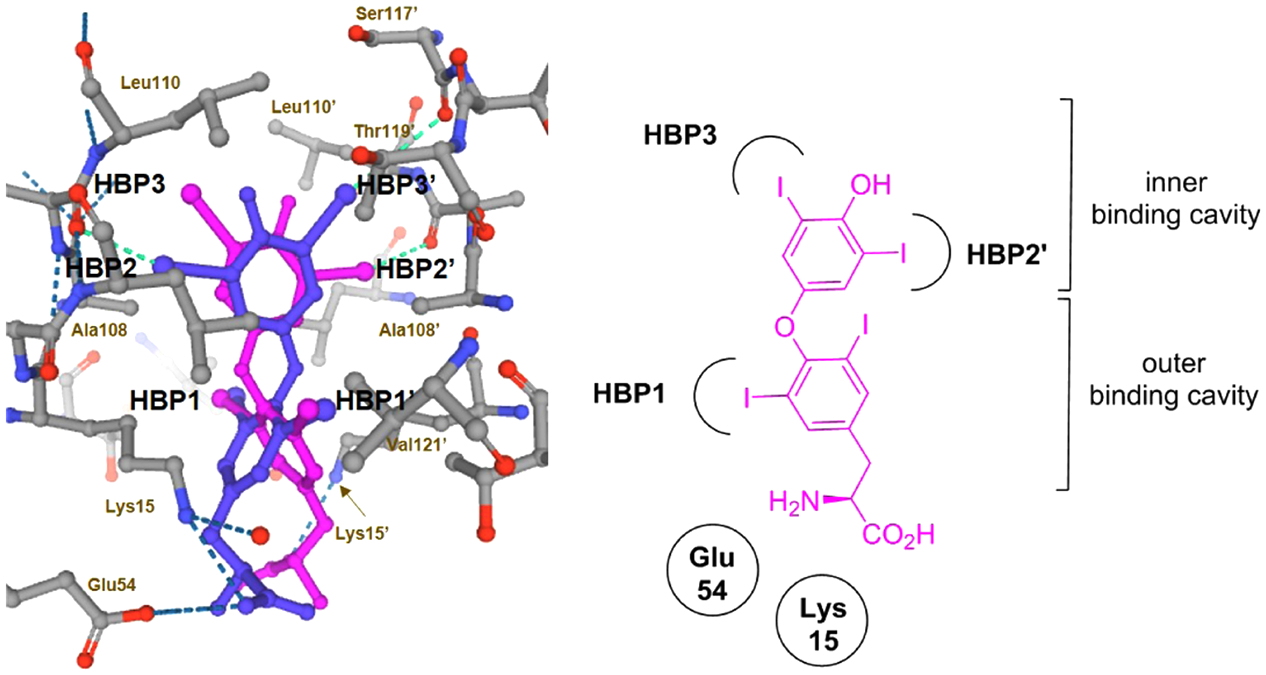
Halogen binding pockets (HBPs) of the T4 binding site with 4 bound (PDB 2ROX).5c Compound 4 is shown in a ball-and-stick format, and both symmetry-related binding modes related by a twofold axis are presented (4 is shown in violet and magenta). The iodine atoms of 4 occupy the HBP1, HBP2, and HBP3 pockets, while the amino acid appendage engages in ionic salt-bridge interactions Lys15 and Glu54 near the opening of the binding site. Contacting amino acids are designated and depicted in a ball-and-stick form. The dashed blue lines represent H-bonds and the dashed cyan lines represent halogen bonds.
A common pharmacophore among small-molecule RBP4 antagonists and TTR tetramer kinetic stabilizers is a “bottom group” carboxylic acid appendage connected through a core linker scaffold to an aromatic “head group” moiety. The X-ray crystallographic data PDB 3FMZ8 provides insight as to how 6 and related analogues 7 and 8 may bind to RBP4 (Figure 8A). The binding pose of 6 involves the piperidine core linker residing within the central cavity, while the lipophilic phenyl ring bearing a trifluoromethyl group extends into the inner lipophilic cavity in an orthogonal manner. The ionizable anthranilic acid forms electrostatic binding interactions with Arg121 and a pair of H-bonds with neighboring residues Tyr90 and Gln98. The structural data for 4 bound to the T4 binding site (PDB 2ROX)5c reveals how both molecular targets present binding pockets of comparable geometries and van der Waals surface areas of hydrophobicity and polarity. Ligand 4 binds in the aforementioned forward binding mode with its lipophilic phenolic head group projecting into the inner binding cavity and oriented orthogonally (Figure 8C). The linker phenyl ring of 4 resides within the hydrophobic channel bridging the inner and outer cavities, and similar to RBP4 antagonist 6, the ionizable amino acid appendage resides near the opening of its respective binding site where it also engages in salt-bridge interactions.
Figure 8.
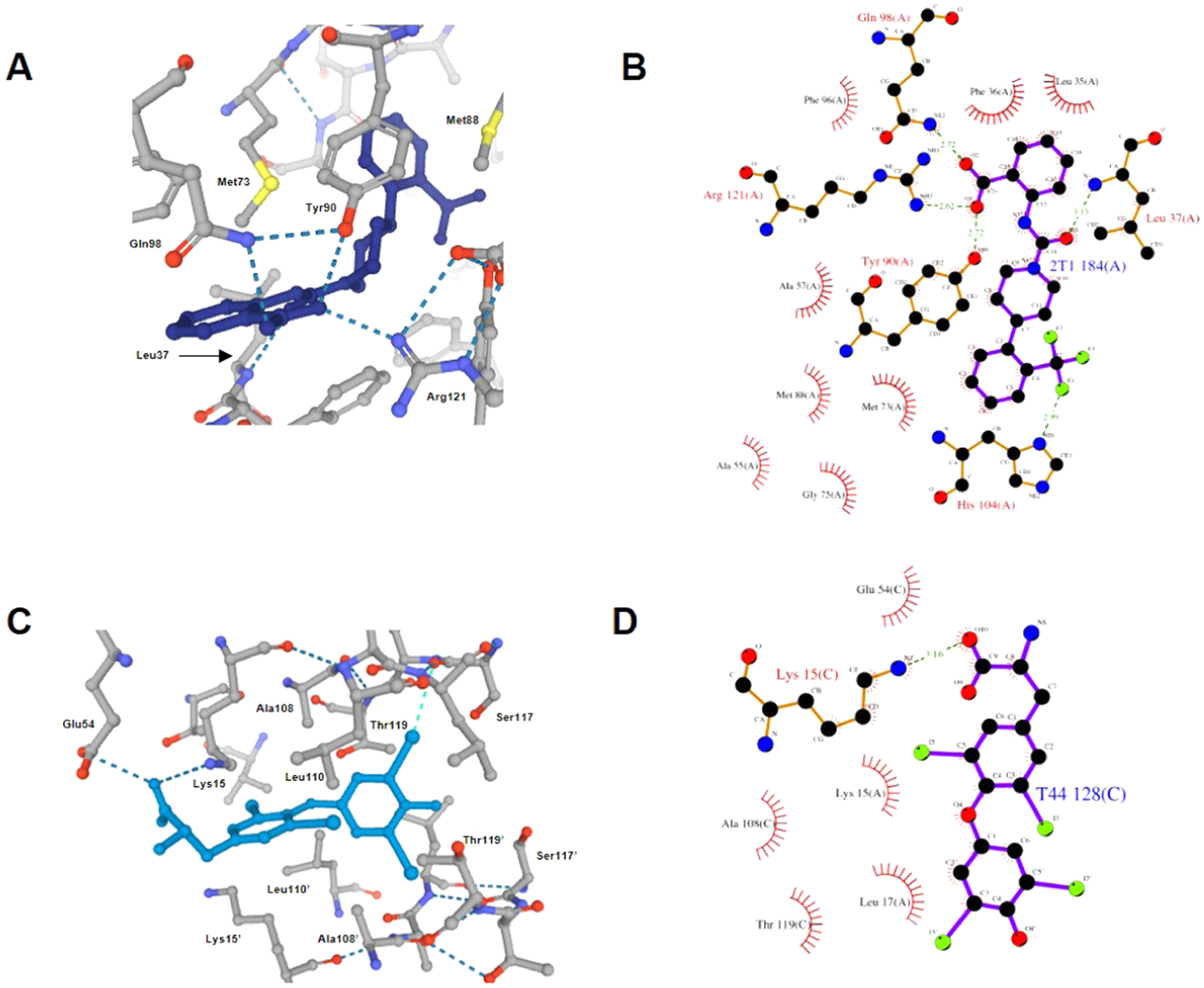
X-ray crystallographic data showing similar binding poses for 6 bound to RBP4 (PDB 3FMZ)8 and 4 bound to the T4 binding site (PDB 2ROX).5c (A) RBP4 antagonist 6 is shown in a ball-and-stick format (dark blue) with polar binding interactions (e.g., H-bonds and salt bridges) shown as the blue dotted lines. (B) Ligand interaction diagram of 6 with RBP4 indicating key H-bonds and salt-bridge interactions. (C) TTR ligand 4 is shown in a ball-and-stick format (light blue) with the phenolic aryl appendage projecting into the inner cavity in an orthogonal manner. The iodine atoms of the head group occupy the HBP2 and HBP3 pockets, while the iodine atoms of the central core phenyl ring occupy the HBP1 pockets. The amino acid appendage engages in ionic salt-bridge interactions Lys15 and Glu54 near the opening of the binding site. Polar binding interactions (e.g., H-bonds and salt bridges) are shown as the dark blue dotted lines, and halogen bonds are shown as the light-blue dotted lines. (D) Ligand interaction diagram of 4 with TTR indicating the key salt-bridge interaction.
We planned to develop novel bispecific ligands that retain the key pharmacophoric elements described for ligands of both targets. Importantly, critical salt-bridge interactions with the basic Arg121 and Lys15 residues of RBP4 and TTR, respectively, are mediated by an aromatic carboxylic acid for many known RBP4 antagonists and TTR ligands. Hence, this motif was not altered in our novel bispecific analogue design. From an examination of PDB 3FMZ8 and 2ROX,5c we speculated that varying the length and conformational flexibility in the [3.3.0]-bicyclic core linker of 8 might maintain RBP4 antagonist potency while potentially enhancing TTR activity. Figure 9 highlights how we executed our sequential structure–activity relationship (SAR) plan by examining three critical regions of our novel scaffolds.
Figure 9.
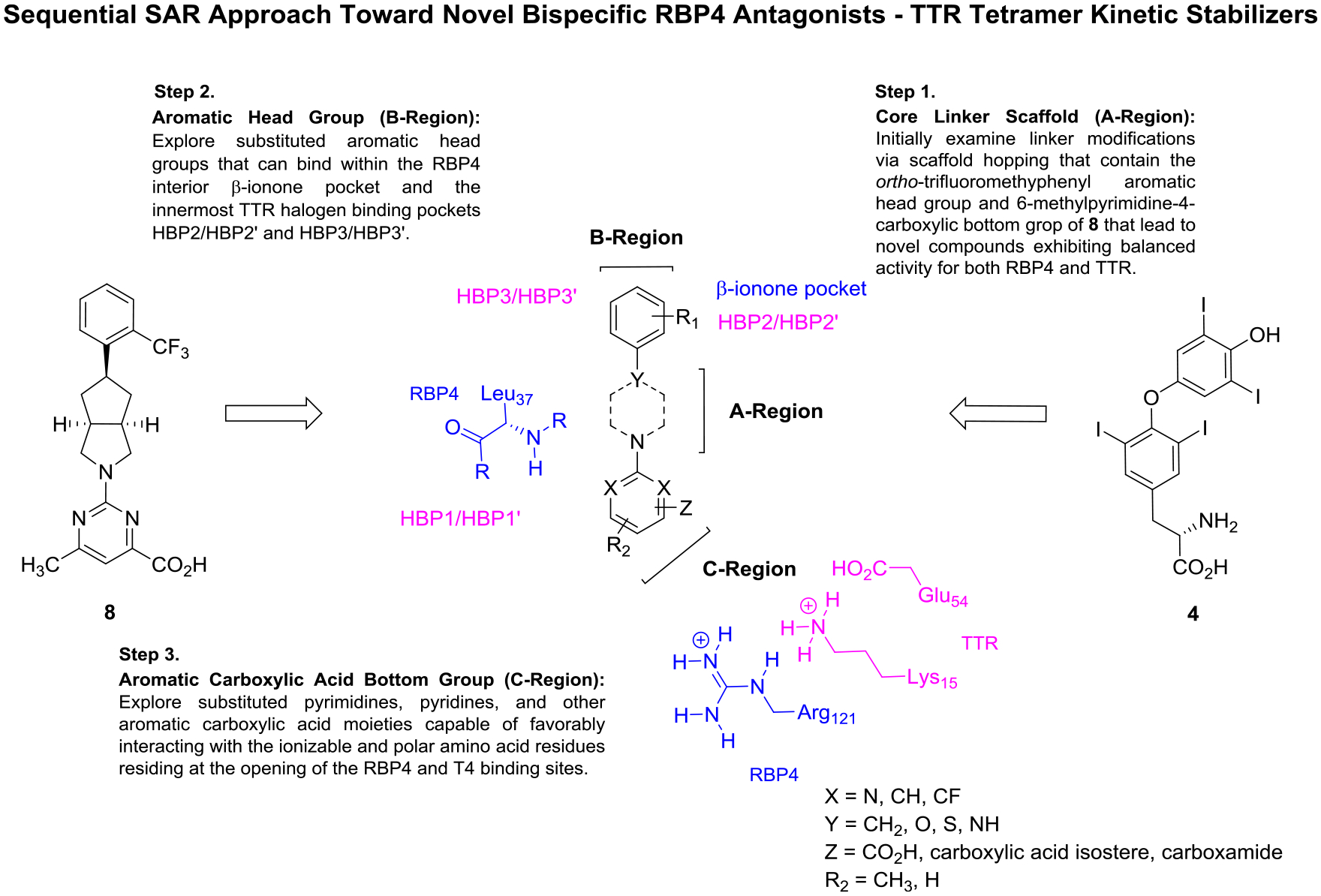
Medicinal chemistry strategy to identify bispecific RBP4 antagonist–TTR tetramer kinetic stabilizers. Novel analogues were rationally designed from benchmark RBP4 antagonist 8 and endogenous TTR ligand 4. The initial SAR campaign involved scaffold hopping of the core linker (A-region) while maintaining the aromatic head group and aromatic carboxylic acid bottom group appendages of 8. A subsequent SAR campaign of the RBP4 β-ionone and T4 inner cavities involved exploration of the aromatic head group (B-region) with a newly identified A-region core linker. Finally, with an optimal A-region and B-region scaffolds identified, aromatic carboxylic acid analogues (C-region) were then investigated. These analogues were designed to potentially engage in critical RBP4 H-bonding interactions with Arg121, Gln98 (not shown), and backbone Leu37 (all RBP4 interactions shown in blue) while also engaging in key TTR binding interactions with Lys15, Glu54, and within the HBP1/HBP1′ (all TTR interactions shown in magenta).
We initially executed a scaffold hopping campaign that explored how altering the shape and flexibility of the linker core of 8 (A-region) would affect potency at both targets. We hypothesized that subtle changes to this region of the molecule may better position an appropriately substituted aromatic head group to fully occupy both the RBP4 β-ionone pocket and inner T4 binding cavity. We synthesized initial illustrative examples of 8 containing alternative linker scaffolds that retained the ortho-trifluoromethylphenyl head group and the 6-methylpyrimidine-4-carboxylic acid bottom group moieties. We chose to maintain the ortho-trifluoromethylphenyl head group as it is an established head group motif for optimal RBP4 binding affinity and it has served as a suitable head group for previously disclosed oxime43 and N-phenyl phenoxazine44 TTR amyloid fibril inhibitors (structures not shown). We postulated that the 6-methylpyrimidine-4-carboxylic acid bottom group provides functionality for the aforementioned key ionic interactions while also providing additional structural components that could also engage in favorable binding interactions. For example, we hypothesized that the pyrimidine nitrogen para to the carboxylic acid may be in a similar position as the urea carbonyl of 6 (PDB 3FMZ), which hydrogen-bonds with backbone Leu37.8 Furthermore, the nitrogen ortho to the carboxylic acid group could also be within suitable proximity to H-bond with Glu98/Tyr80 of RBP4 (PDB 3FMZ)8 and/or Glu54 of TTR (PDB 2ROX).5c Analysis of PDB 2ROX suggests that the pyrimidine ring 6-position, which previously provided informative RBP4 SAR,19 could also potentially provide a handle to further probe the HBP1/HBP1′ pocket within the T4 binding site.5c
The various scaffold hopping core linkers explored presented similar distances between the aromatic head group and carboxylic acid bottom group of 4 and bicyclic analogues 6–8. Once a novel A-region core linker with suitable activity at both targets was identified, it was then used to explore the SAR of alternatively substituted aromatic head groups (B-region). Finally, we subsequently studied carboxylic acid bottom group (C-region) SAR with an identified optimal A- and B-region scaffold. This C-region SAR sample set included analogues featuring picolinic and nicotinic acids, positional variation of the carboxylic acid, carboxylic acid isosteres, and carboxamides that were prepared and analyzed for potency effects at both targets.
CHEMISTRY
The preparation of alkoxypiperidine analogue 21 shown in Scheme 1 begins with a sodium borohydride (NaBH4) reduction of tert-butyl 4-oxopiperidine-1-carboxylate (15) to give alcohol 16. Sulfonylation of 16 followed by bimolecular nucleophilic (SN2) displacement of the corresponding sulfonate ester 17 with 2-(trifluoromethyl)phenol led to the formation of phenoxyether 18. Treatment of 18 with trifluoroacetic acid (TFA) provided Boc-deprotected amine 19, which, when reacted with methyl 2-chloro-6-methylpyrimidine-4-carboxy-late, afforded pyrimidine methyl ester 20. Hydrolysis of 20 with lithium hydroxide (LiOH) followed by subsequent acidification of the lithium carboxylate salt with 2 N aqueous (aq) hydrochloric acid (HCl) provided the desired carboxylic acid 21.
Scheme 1.
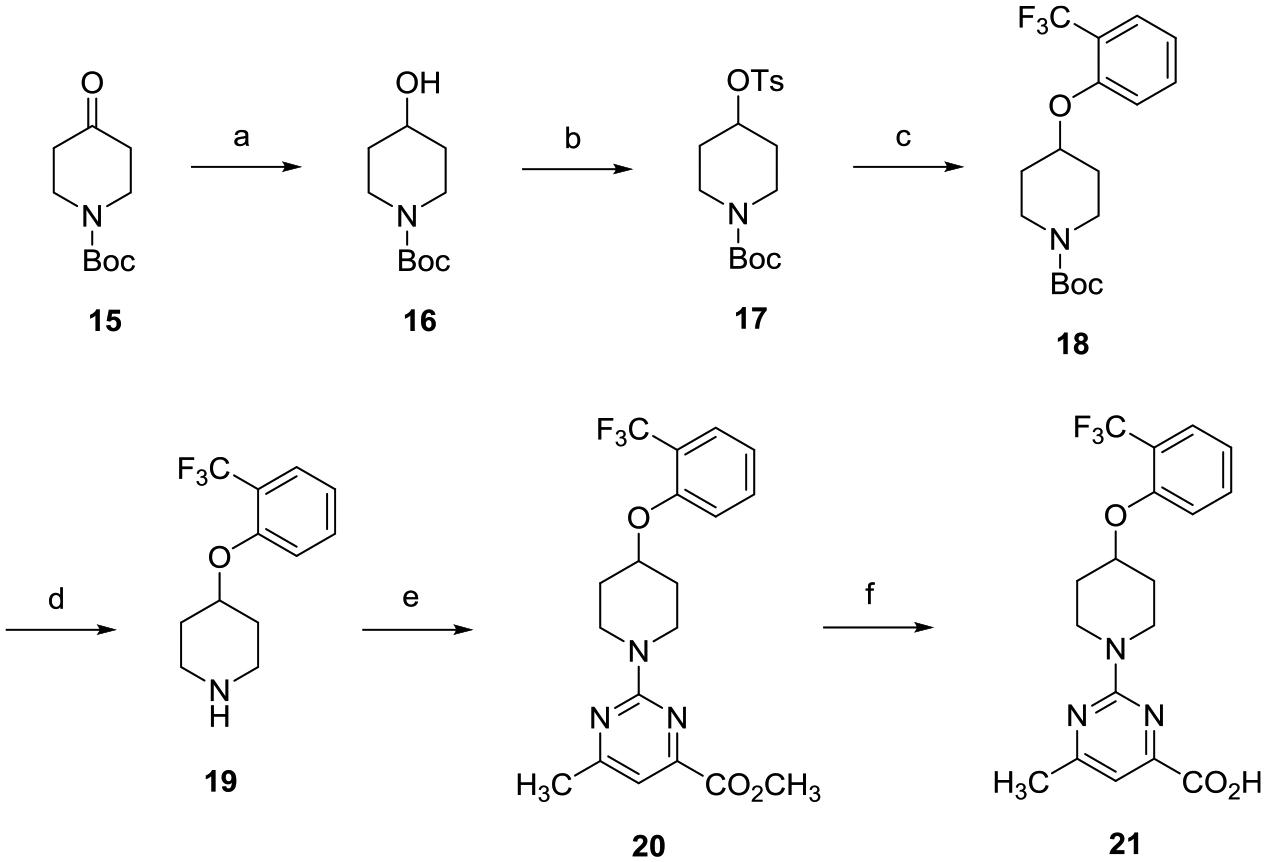
aReagents and conditions: (a) NaBH4, CH3OH, 0 °C to room temperature (rt), 8 h; (b) TsCl, 4-dimethylaminopyridine (DMAP), Et3N, CH2Cl2, 0 °C to rt, 16 h; (c) 2-(trifluoromethyl)phenol, Cs2CO3, dimethylformamide (DMF), 80 °C, 16 h; (d) TFA, CH2Cl2, 0 °C to rt, 8 h; (e) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, tetrahydrofuran (THF), reflux, 16 h; and (f) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Scheme 2 highlights the preparation of racemic alkoxypyrrolidine analogue (±)-27. tert-Butyl 3-(tosyloxy)pyrrolidine-1-carboxylate ((±)-23) was generated via tosylation of commercially available racemic tert-butyl 3-hydroxypyrrolidine-1-carboxylate ((±)-22). Treatment of sulfonate ester (±)-23 with 2-(trifluoromethyl)phenol provided ether (±)-24, which was deprotected with TFA affording amine (±)-25. Conversion of (±)-25 to the desired carboxylic acid (±)-27 followed the same two-step process outlined in Scheme 1.
Scheme 2.
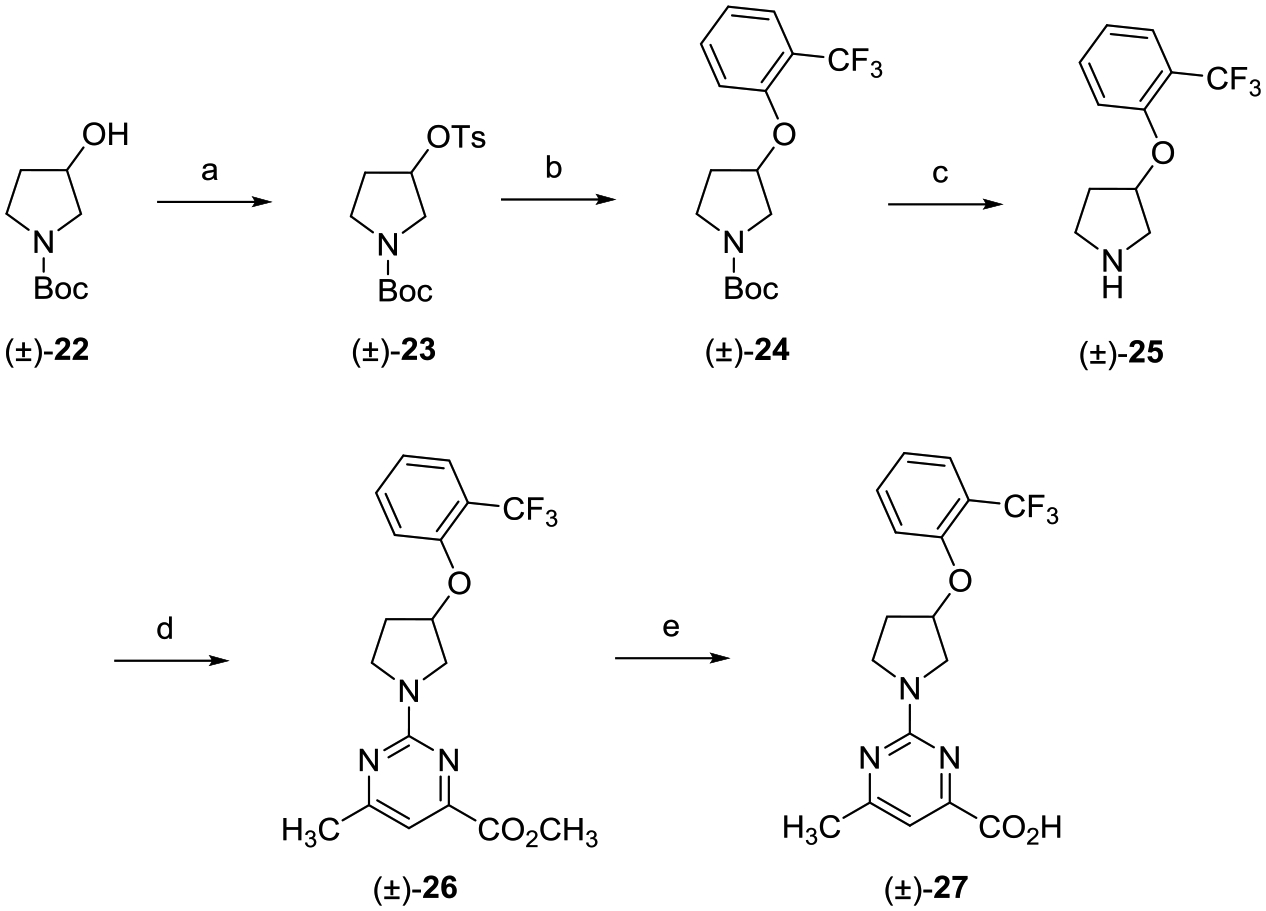
aReagents and conditions: (a) TsCl, DMAP, Et3N, CH2Cl2, 0 °C to rt, 16 h; (b) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (c) TFA, CH2Cl2, 0 °C to rt, 8 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (e) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
O-Alkylation of tosylated azetidine 28 with 2-(trifluoromethyl)phenol afforded phenyl ether 29 (Scheme 3). TFA-mediated Boc-deprotection of 29 followed by treatment with methyl 2-chloro-6-methylpyrimidine-4-carboxylate yielded pyrimidine 31, which was hydrolyzed with LiOH to provide acid 32.
Scheme 3.
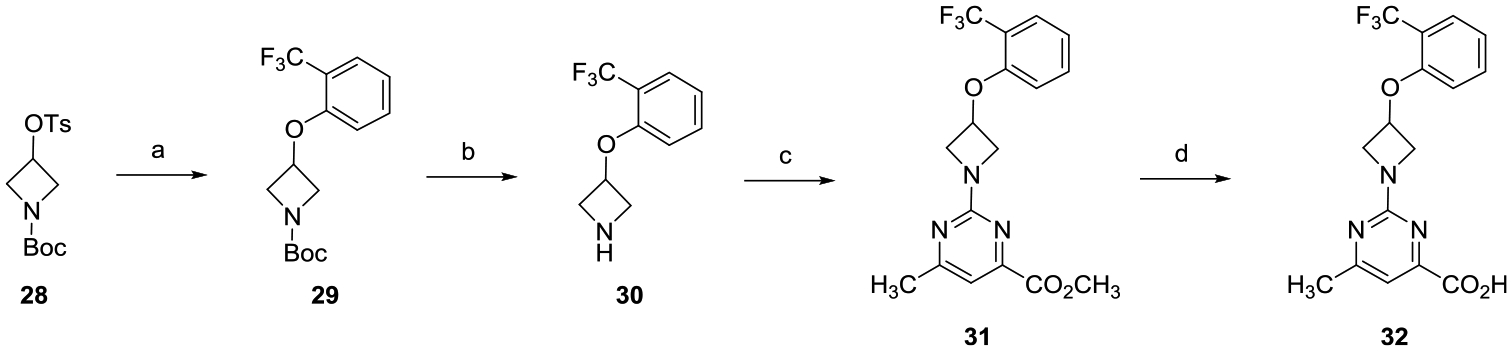
aReagents and conditions: (a) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (b) TFA, CH2Cl2, 0 °C to rt, 8 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (d) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Construction of (hydroxymethyl)azetidine analogue 38 followed the route depicted in Scheme 4. Tosylation of alcohol 33 gave sulfonate ester 34, which was subsequently treated with 2-(trifluoromethyl)phenol to provide phenyl ether 35. TFA-mediated deprotection of 35 followed by reaction with methyl 2-chloro-6-methylpyrimidine-4-carboxylate provided analogue 37. LiOH promoted hydrolysis of methyl ester 37, and subsequent acidification with 2 N aqueous HCl gave the desired carboxylic acid 38.
Scheme 4.
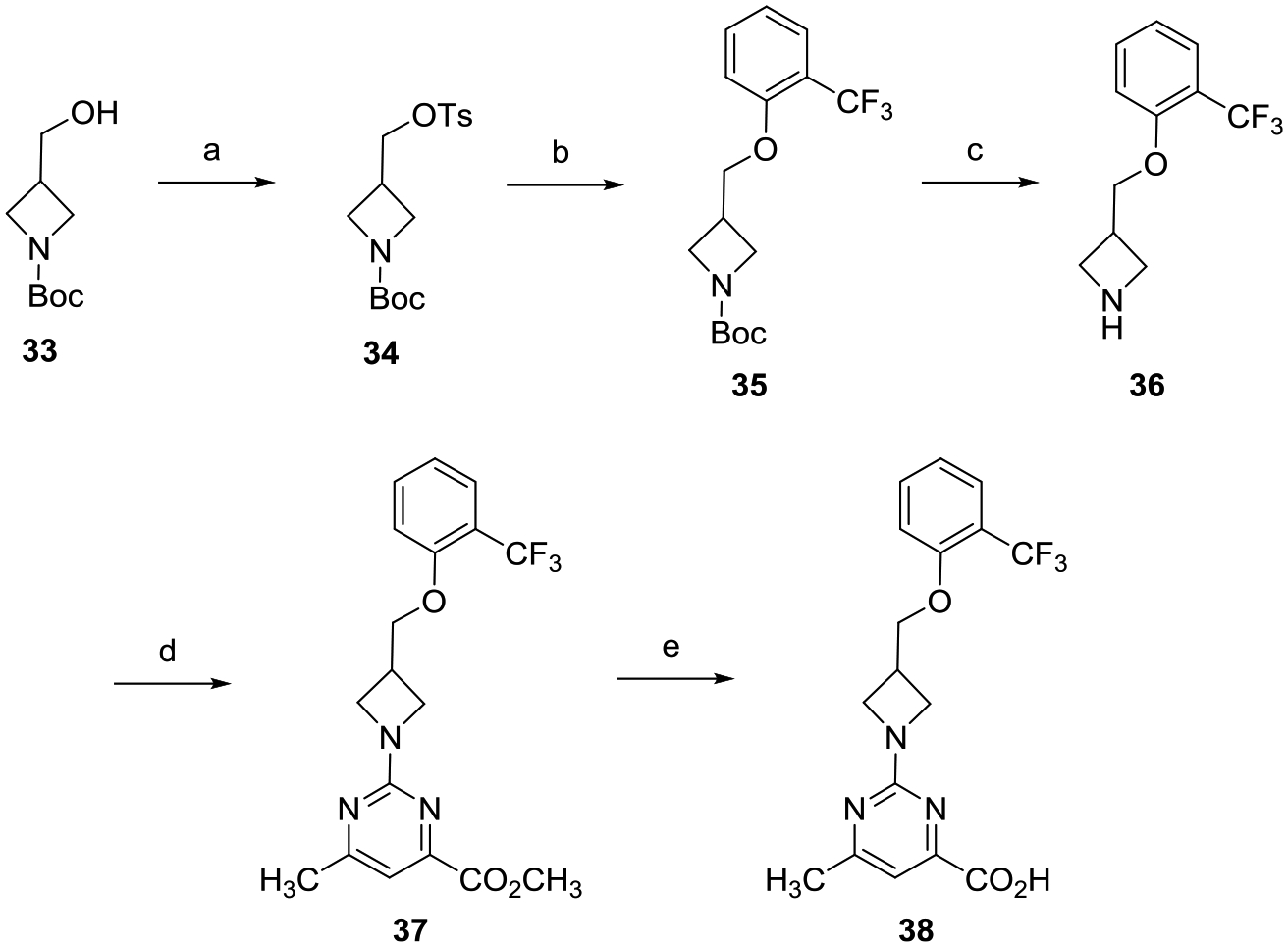
aReagents and conditions: (a) TsCl, DMAP, Et3N, CH2Cl2, 0 °C to rt, 16 h; (b) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (c) TFA, CH2Cl2, 0 °C to rt, 8 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (e) (i) LiOH, CH3OH, THF,H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Racemic 3-(hydroxymethyl)pyrrolidine (±)-44 was achieved via the route shown in Scheme 5. Sulfonate ester formation of racemic alcohol (±)-39 followed by SN2 displacement with 2-(trifluoromethyl)phenol in the presence of cesium carbonate (Cs2CO3) gave O-alkylated product (±)-41. Treatment of (±)-41 with TFA followed by reaction with methyl 2-chloro-5-methylpyrimidine-4-carboxylate yielded (±)-43, which was saponification with LiOH to provide the desired racemic carboxylic acid (±)-44.
Scheme 5.
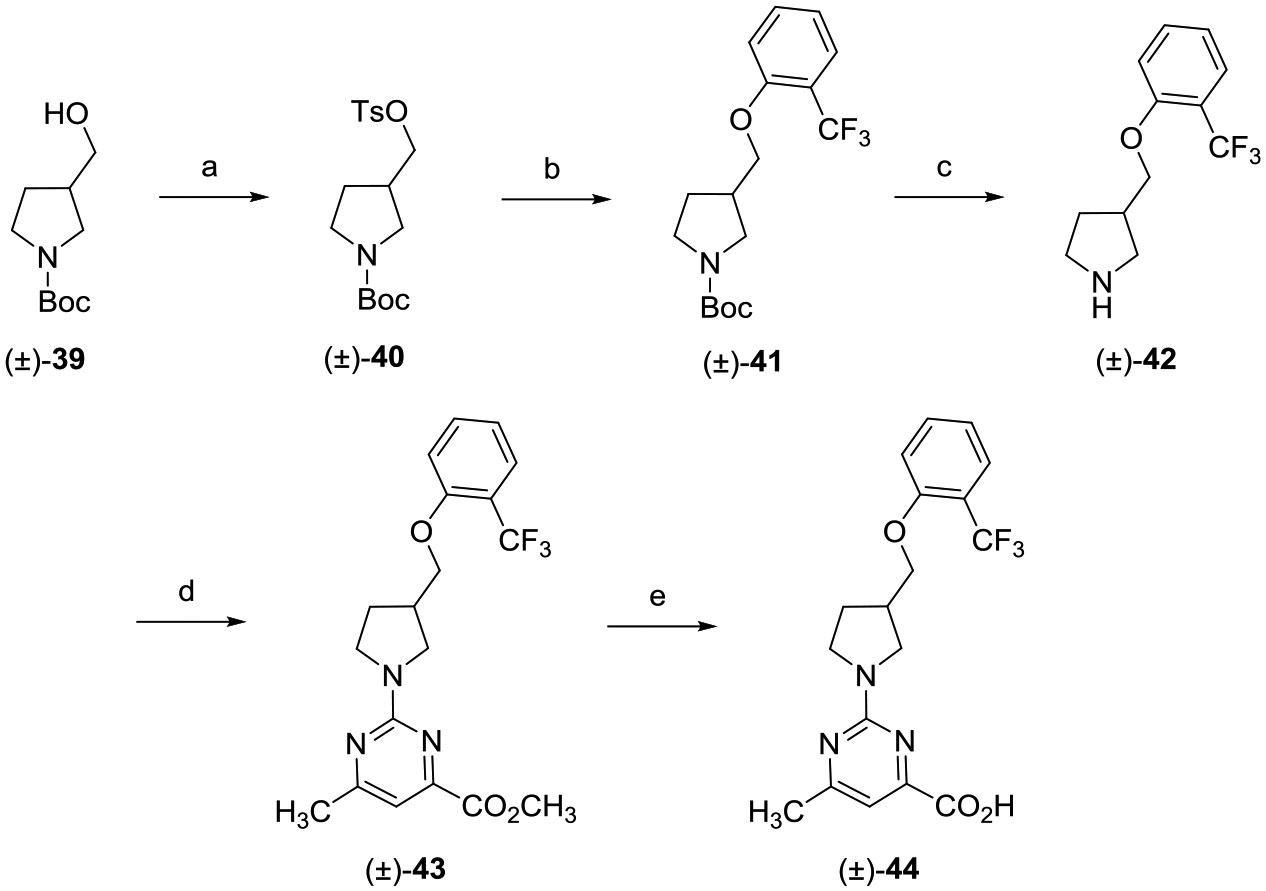
aReagents and conditions: (a) TsCl, DMAP, Et3N, CH2Cl2, 0 °C to rt, 16 h; (b) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (c) TFA, CH2Cl2, 0 °C to rt, 8 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (e) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Preparation of enantiomers (R)-50 and (S)-56 is highlighted in Scheme 6. The synthesis of the enantiopure analogues was achieved using the same route outlined in Scheme 5 but starting with either tert-butyl (R)-3-(hydroxymethyl)pyrrolidine-1-carboxylate ((R)-45) or tert-butyl (S)-3-(hydroxymethyl)-pyrrolidine-1-carboxylate ((S)-51).
Scheme 6.
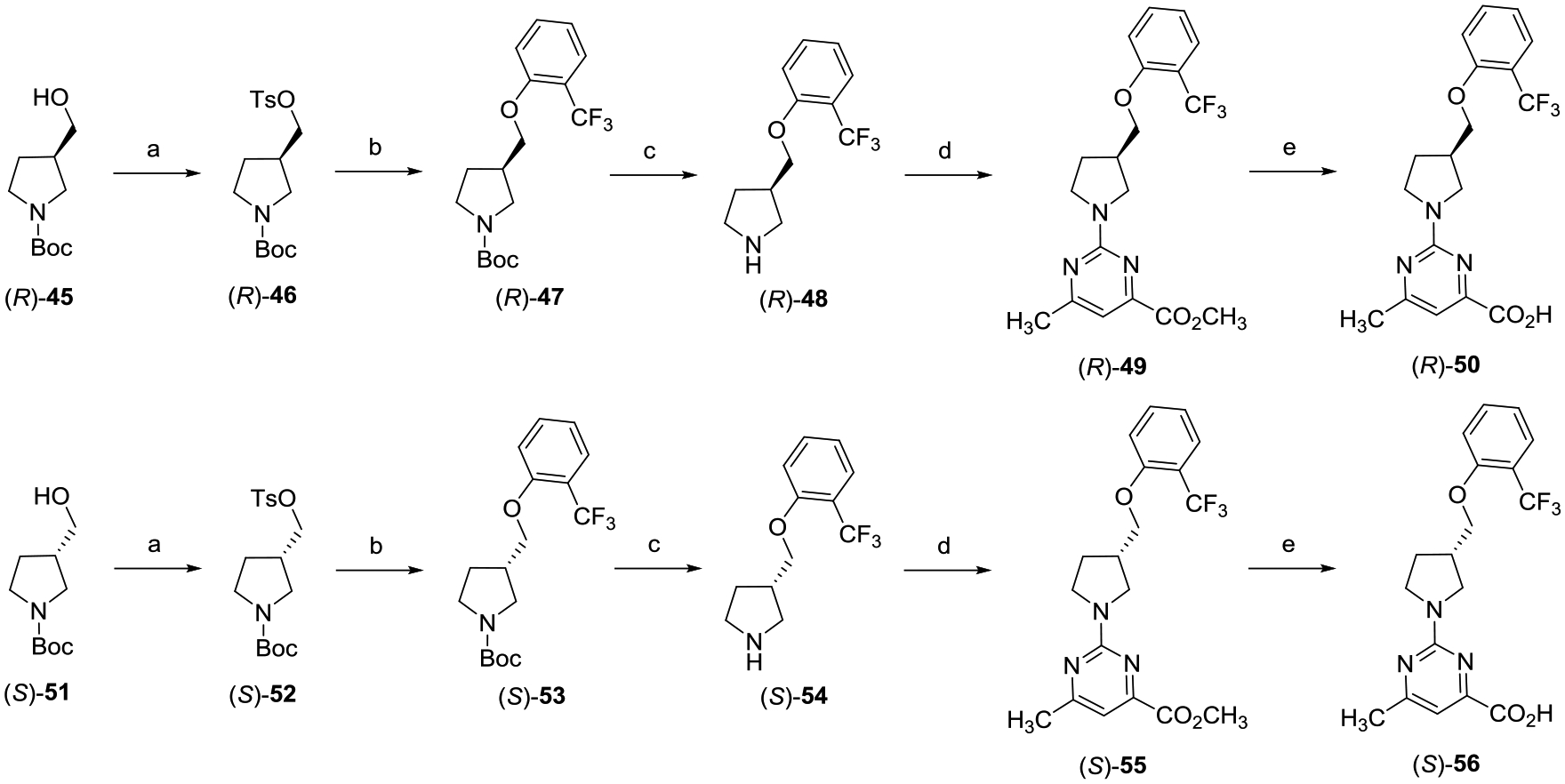
aReagents and conditions: (a) TsCl, DMAP, Et3N, CH2Cl2, 0 °C to rt, 16 h; (b) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (c) TFA, CH2Cl2, 0 °C to rt, 8 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (e) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
The preparation of the racemic piperidine carboxylic acid analogue (±)-62 begins with the SN2 displacement of tosylate (±)-58 with 2-(trifluoromethyl)phenol followed by treatment with TFA, which gave secondary amine (±)-60 (Scheme 7). Reaction of amine (±)-60 with methyl 2-chloro-5-methylpyr imidine-4-carboxylate yielded (±)-61, which upon saponification provided the desired carboxylic acid (±)-62.
Scheme 7.
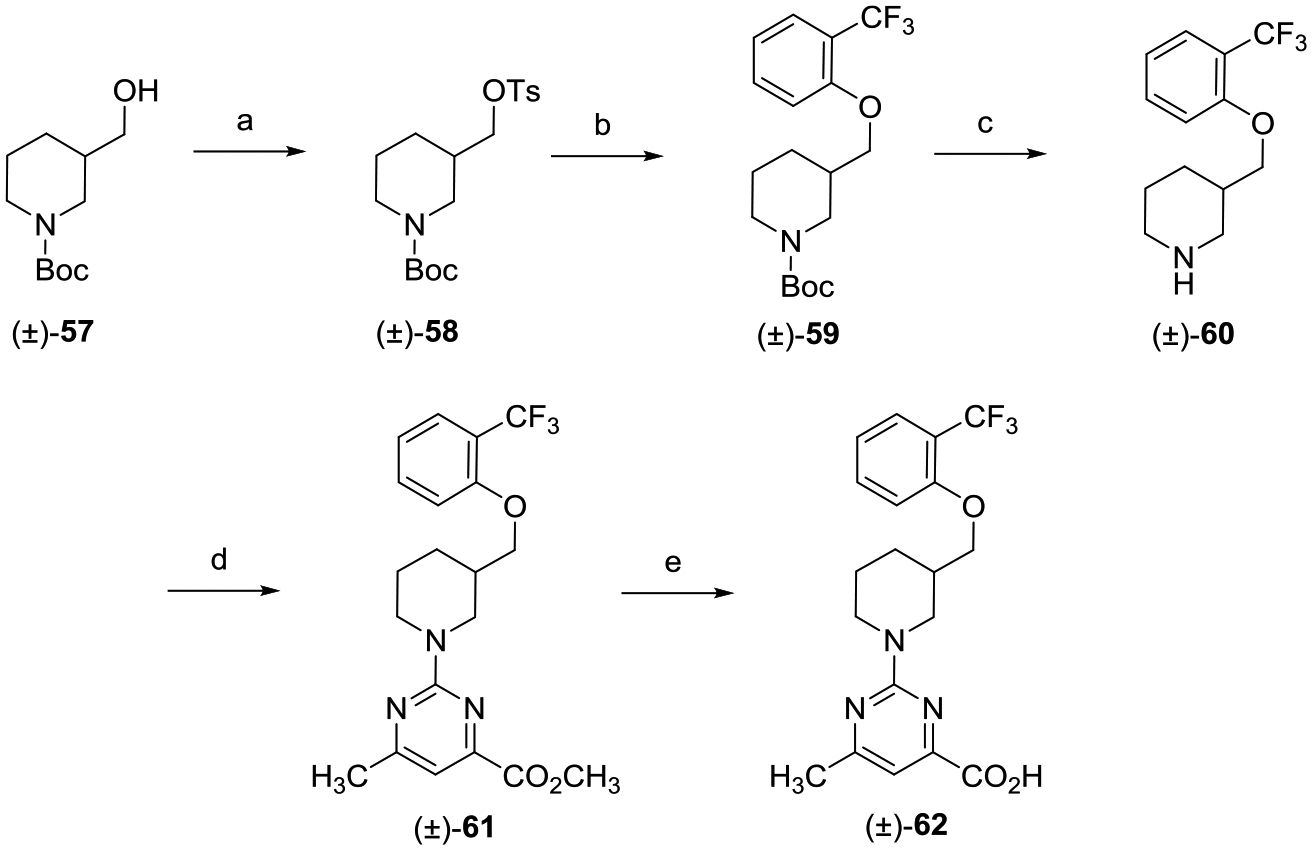
aReagents and conditions: (a) TsCl, DMAP, Et3N, CH2Cl2, 0 °C to rt, 16 h; (b) 2-(trifluoromethyl)phenol, Cs2CO3, DMF, 80 °C, 16 h; (c) TFA, CH2Cl2, 0 °C to rt, 8 h; (d) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (e) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Racemic 2,7-diazaspiro[4.4]nonane analogue (±)-67 was designed to explore the SAR effects of introducing a ring-constrained core (Scheme 8). Aniline (±)-64 was generated via a palladium-catalyzed amination between (±)-63 and 1-bromo-2-(trifluoromethyl)benzene using Tris(dibenzylideneacetone)-dipalladium(0) (Pd2(dba)3) and 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos). Boc-deprotection of (±)-64 with TFA gave amine (±)-65, which was subsequently reacted with methyl 2-chloro-5-methylpyrimidine-4-carboxylate to give pyrimidine (±)-66. Saponification of (±)-66 with LiOH followed by acidification of the lithium carboxylate salt with 2 N HCl provided the desired spirocycle (±)-67.
Scheme 8.
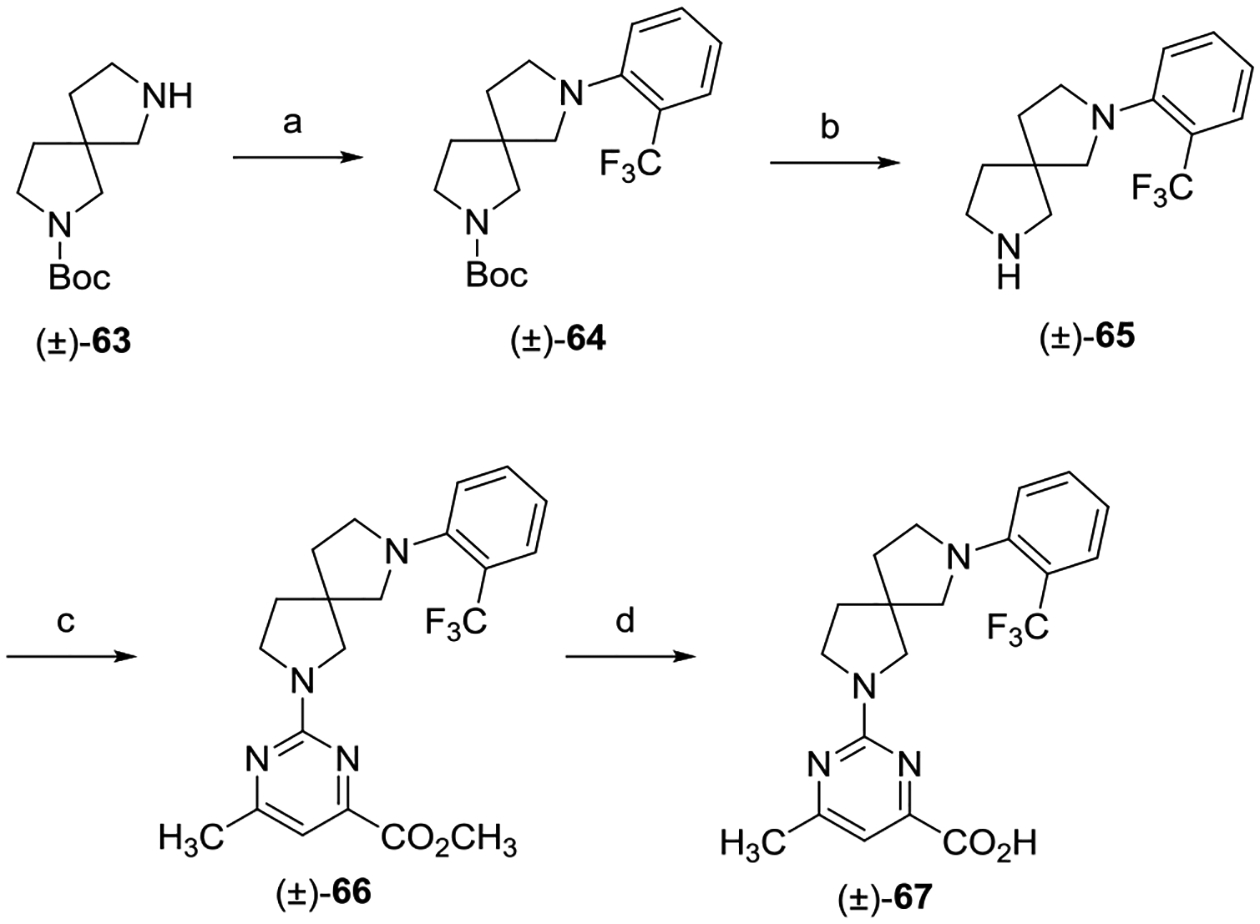
aReagents and conditions: (a) 1-bromo-2-(trifluoromethyl)benzene, XPhos, Pd2(dba)3, Cs2CO3, 1,4-dioxane, 110 °C, 16 h; (b) TFA,CH2Cl2, 0 °C to rt, 8 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (d) (i) LiOH, CH3OH, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
The racemic thioether analogue (±)-71 construction is depicted in Scheme 9. The route used for the synthesis of (±)-71 follows the aforementioned route described for (±)-44 except that the SN2 displacement of sulfonate ester (±)-39 was conducted with 2-(trifluoromethyl)benzenethiol.
Scheme 9.
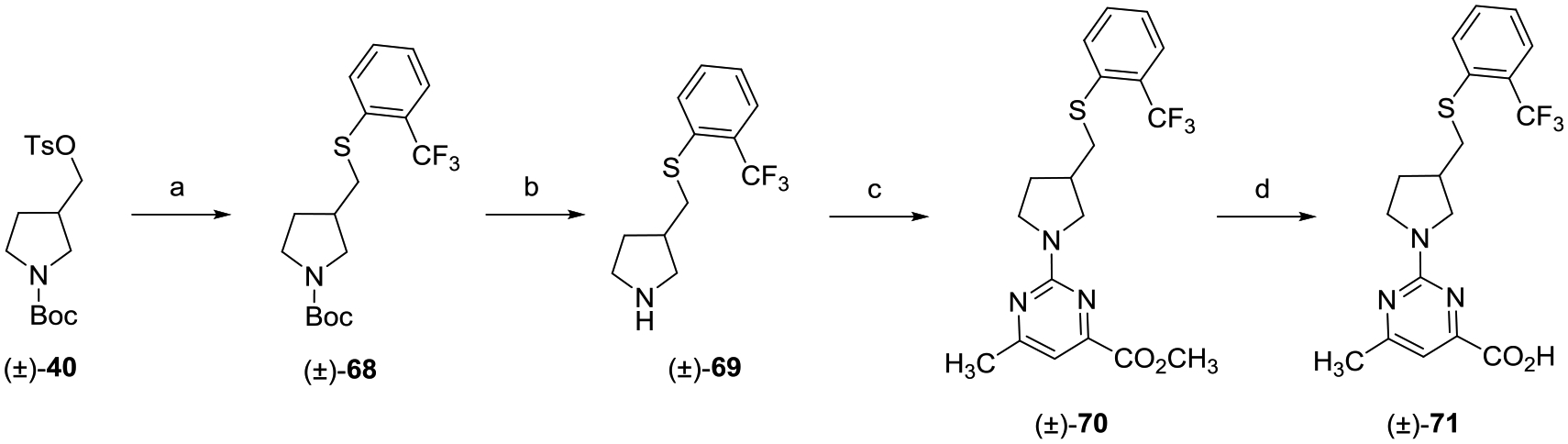
aReagents and conditions: (a) 2-(trifluoromethyl)benzenethiol, Cs2CO3, DMF, 80 °C, 16 h; (b) TFA, CH2Cl2, 0 °C to rt, 8 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (d) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
Synthesis of amine linker (±)-76 was achieved via the synthetic route provided in Scheme 10. Reductive amination of tert-butyl 3-formylpyrrolidine-1-carboxylate ((±)-72) with 2-(trifluoromethyl)aniline with sodium triacetoxyborohydride (NaBH(OAc)3) and acetic acid (HOAc) yielded compound (±)-73. Boc-deprotection of (±)-73 with TFA gave bis-amine (±)-74, which was converted to pyrimidine methyl ester (±)-75 via treatment with methyl 2-chloro-5-methylpyrimidine-4-carboxylate. Ester (±)-75 was hydrolyzed with LiOH, which yielded the desired (±)-76 upon acidification.
Scheme 10.
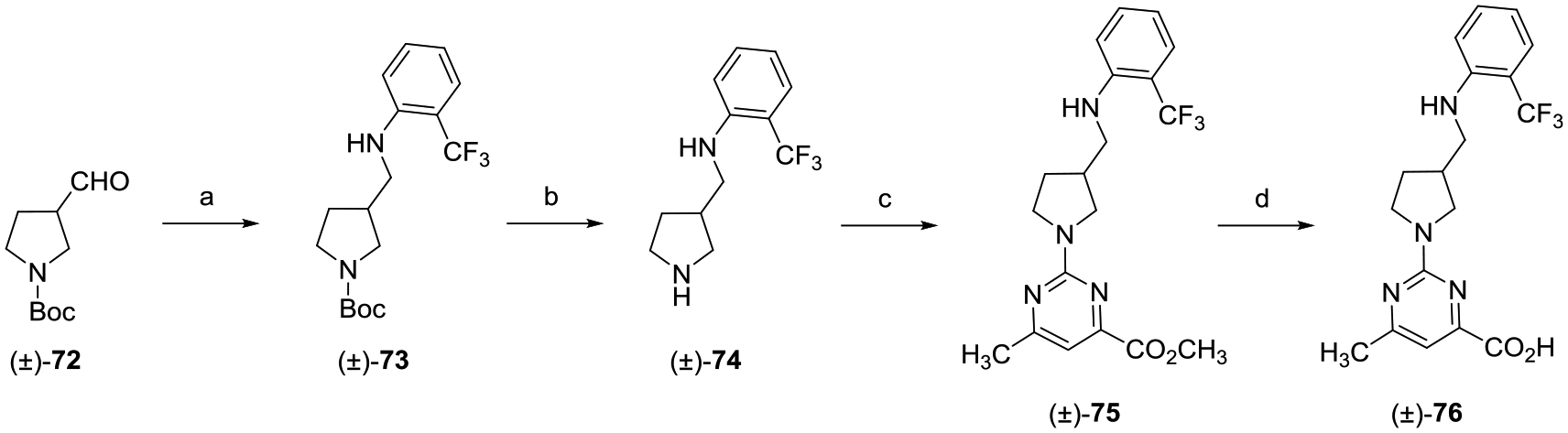
aReagents and conditions: (a) 2-(trifluoromethyl)aniline, NaBH(OAc)3, HOAc, CH2Cl2, rt, 16 h; (b) TFA, CH2Cl2, 0 °C to rt, 12 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, DMF, 80 °C, 16 h; and (d) (i) LiOH, CH3OH, THF, H2O, rt, 12 h; (ii) 2 N aqueous HCl.
The synthesis of racemic ether (±)-80 was accomplished via the route depicted in Scheme 11. Intermediate (±)-77 was produced via a Williamson ether synthesis between 1-(bromomethyl)-2-(trifluoromethyl)benzene and the sodium salt of racemic alcohol (±)-22. Treatment of ether (±)-77 with TFA provided Boc-deprotected (±)-78, which was subsequently reacted with methyl 2-chloro-5-methylpyrimi-dine-4-carboxylate to give (±)-79. Saponification of (±)-79 followed by acidification with 2 N aqueous HCl provided the desired carboxylic acid (±)-80.
Scheme 11.
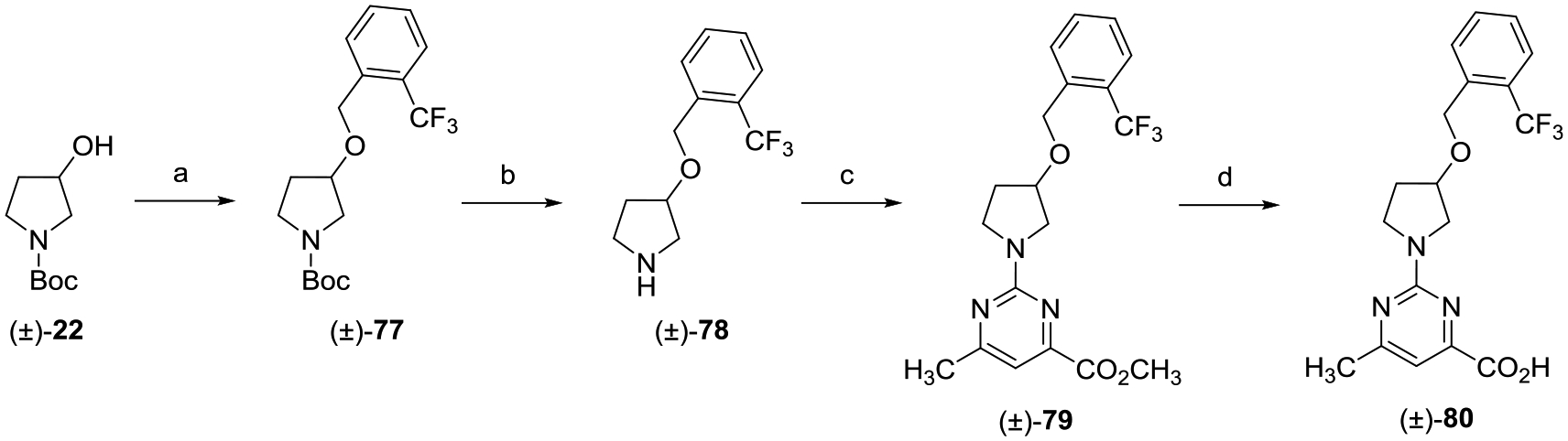
aReagents and conditions: (a) 1-(bromomethyl)-2-(trifluoromethyl)benzene, NaH, DMF, 0 °C to rt, 16 h; (b) TFA, CH2Cl2, 0 °C to rt, 8 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (d) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
A series of analogues derived from (±)-44 that present varied aryl head groups ((±)-83–(±)-93) were prepared in the manner depicted in Scheme 12. O-Alkylation with tosylate (±)-40 using an appropriately substituted phenol provided aryl ether intermediates (±)-81a–k. Boc-deprotection provided secondary amine intermediates (±)-82a–k, which, when treated with 2-methyl 2-chloro-6-methylpyrimidine-4-carboxylate followed by LiOH promoted saponification, gave the desired acids (±)-83–(±)-93.
Scheme 12.
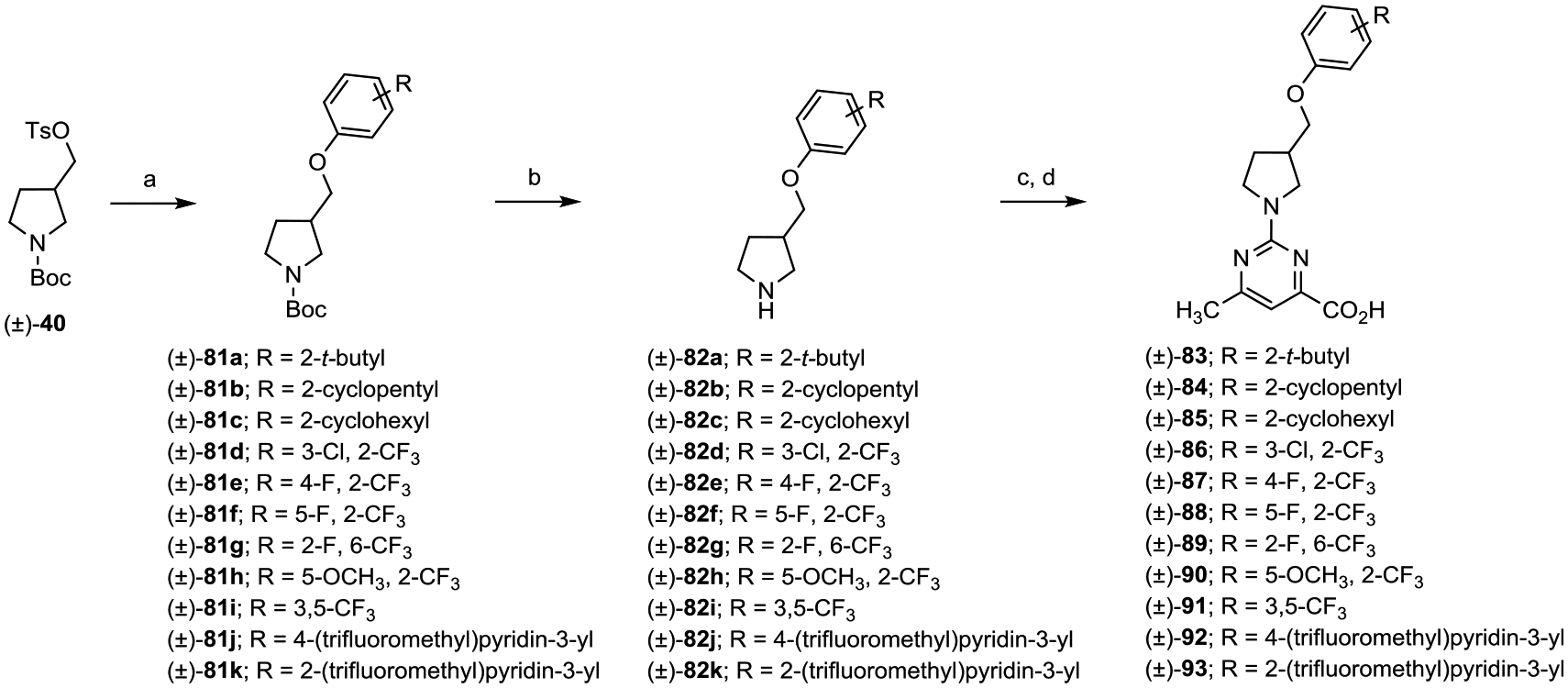
aReagents and conditions: (a) substituted phenol, Cs2CO3, DMF, 80 °C, 16 h; (b) TFA, CH2Cl2, 0 °C to rt, 8 h; (c) methyl 2-chloro-6-methylpyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; and (d) (i) LiOH, CH3OH, THF, H2O, rt, 16 h; (ii) 2 N aqueous HCl.
The production of the set of pyrimidine-substituted analogues (±)-94–(±)-98 is shown in Scheme 13. Pyrimidine analogue (±)-94 was prepared in the same manner as (±)-44 starting from methyl 2-chloropyrimidine-4-carboxylate. For analogues (±)-95–(±)-98, the corresponding substituted chloro picolinic or nicotinic methyl esters and methyl 3-bromo-4-fluorobenzoate underwent palladium-catalyzed amination with amine (±)-42, Pd2(dba)3, and either 4,5-bis(diphenylphosphino)-9,9-dime-thylxanthene (XantPhos) or XPhos in the presence of Cs2CO3. The resulting esters were hydrolyzed with LiOH, and the lithium carboxylate salts were acidified with 2 N aqueous HCl.
Scheme 13.
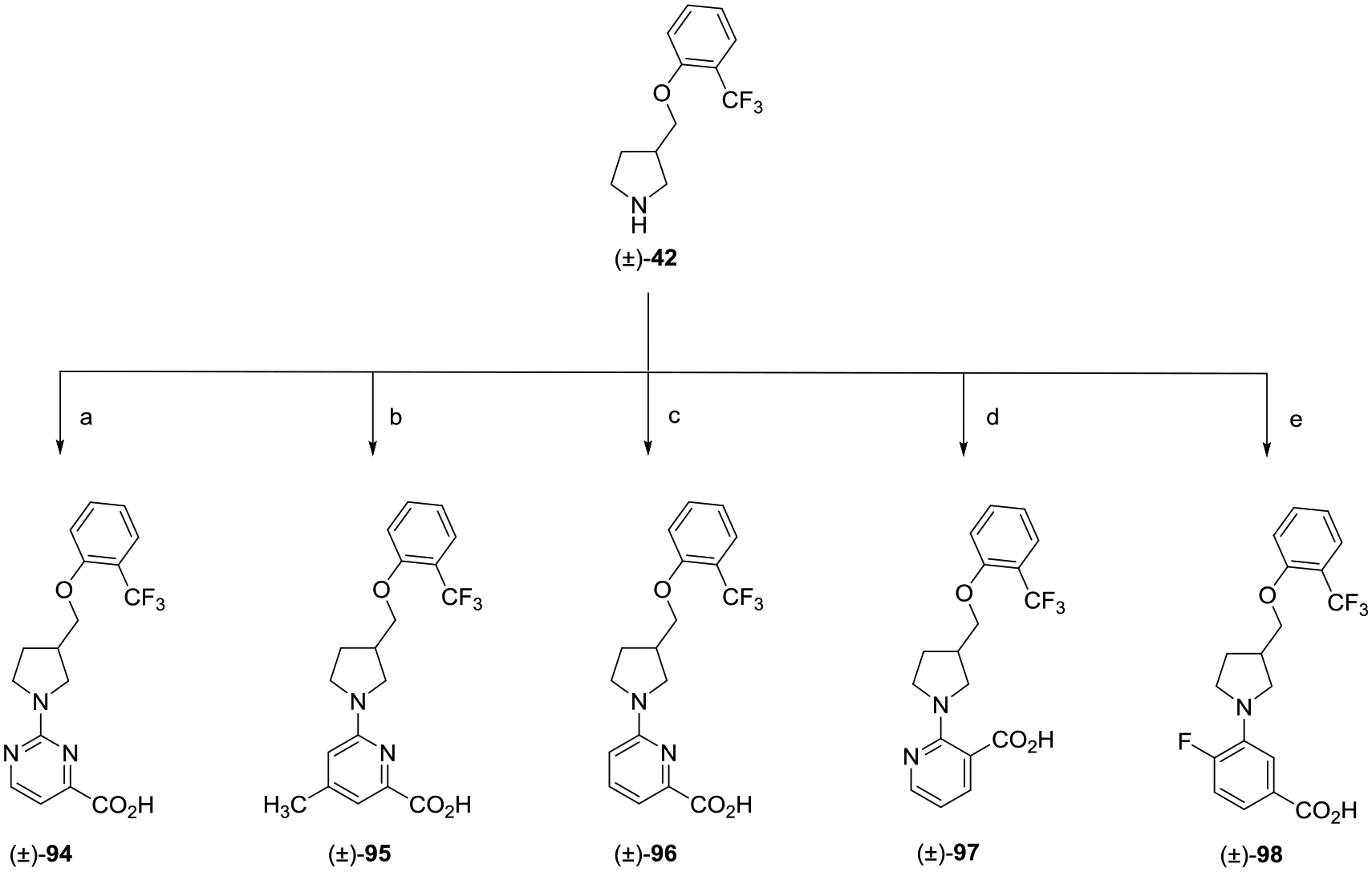
aReagents and conditions: (a) (i) methyl 2-chloropyrimidine-4-carboxylate, i-Pr2NEt, THF, reflux, 16 h; (ii) LiOH, CH3OH, H2O, rt, 16 h; (ii) 2 N aqueous HCl; (b) (i) methyl 6-chloro-4-methylpicolinate, XantPhos, Pd2(dba)3, Cs2CO3, 1,4-dioxane, 80 °C, 16 h; (ii) LiOH, CH3OH, H2O, rt, 16 h; (iii) 2 N aqueous HCl; (c) (i) methyl 6-chloropicolinate, XantPhos, Pd2(dba)3, Cs2CO3, 1,4-dioxane, 80 °C, 16 h; (ii) LiOH, H2O, CH3OH, THF, rt, 16 h; (iii) 2 N aqueous HCl; (d) (i) methyl 2-chloronicotinate, XantPhos, Pd2(dba)3, Cs2CO3, 1,4-dioxane, 80 °C, 16 h; (ii) LiOH, H2O,CH3OH, THF, rt, 16 h; (iii) 2 N aqueous HCl; and (e) (i) methyl 3-bromo-4-fluorobenzoate, XPhos, Pd2(dba)3, Cs2CO3, 1,4-dioxane, 110 °C, 16 h; (ii) LiOH, H2O, CH3OH, THF, rt, 16 h; (iii) 2 N aqueous HCl.
Preparation of analogues designed to explore the SAR of carboxylic acid isosteres and carboxamide analogues of (±)-44 is shown in Schemes 14 and 15. Generation of acyl sulfonamide (±)-99 was achieved via a peptide coupling involving (±)-44, methane sulfonamide, and N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU). Similarly, carboxamide (±)-100 was synthesized from acid (±)-44 and ammonium chloride using HBTU. NMethyl amide analogue (±)-101 was prepared from (±)-44 and methyl amine hydrochloride using T3P. Cyclopropyl amide (±)-102 was synthesized in a similar manner as carboxamide (±)-100 but with cyclopropylamine.
Scheme 14.
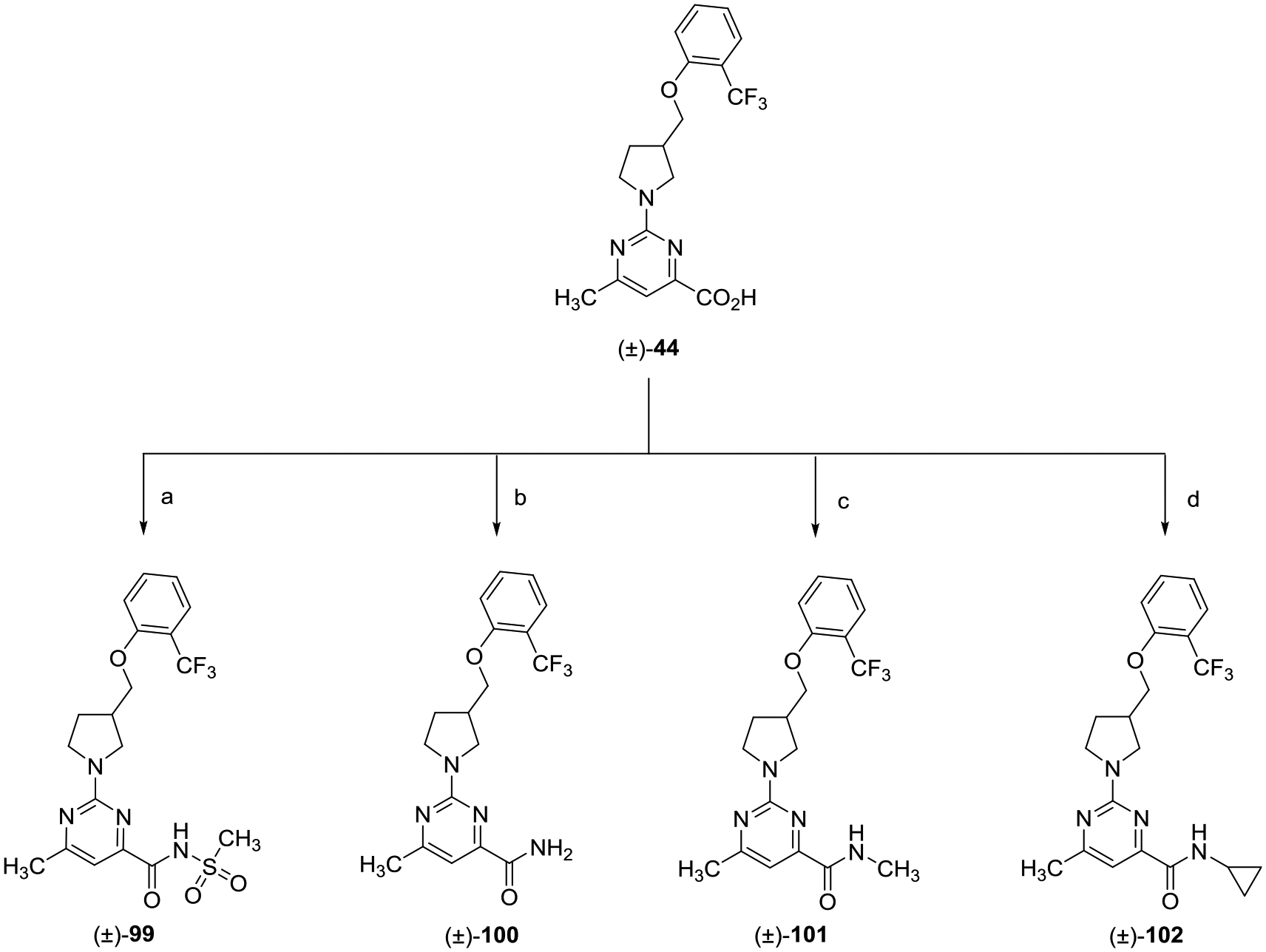
aReagents and conditions: (a) methane sulfonamide, HBTU, i-Pr2NEt, DMF, rt, 18 h; (b) NH4Cl, HBTU, i-Pr2NEt, DMF, rt, 18 h; (c) NHCH3· HCl, T3P, i-Pr2NEt, DMF, rt, 18 h; and (d) cyclopropylamine, HBTU, i-Pr2NEt, DMF, rt, 18 h.
Scheme 15.
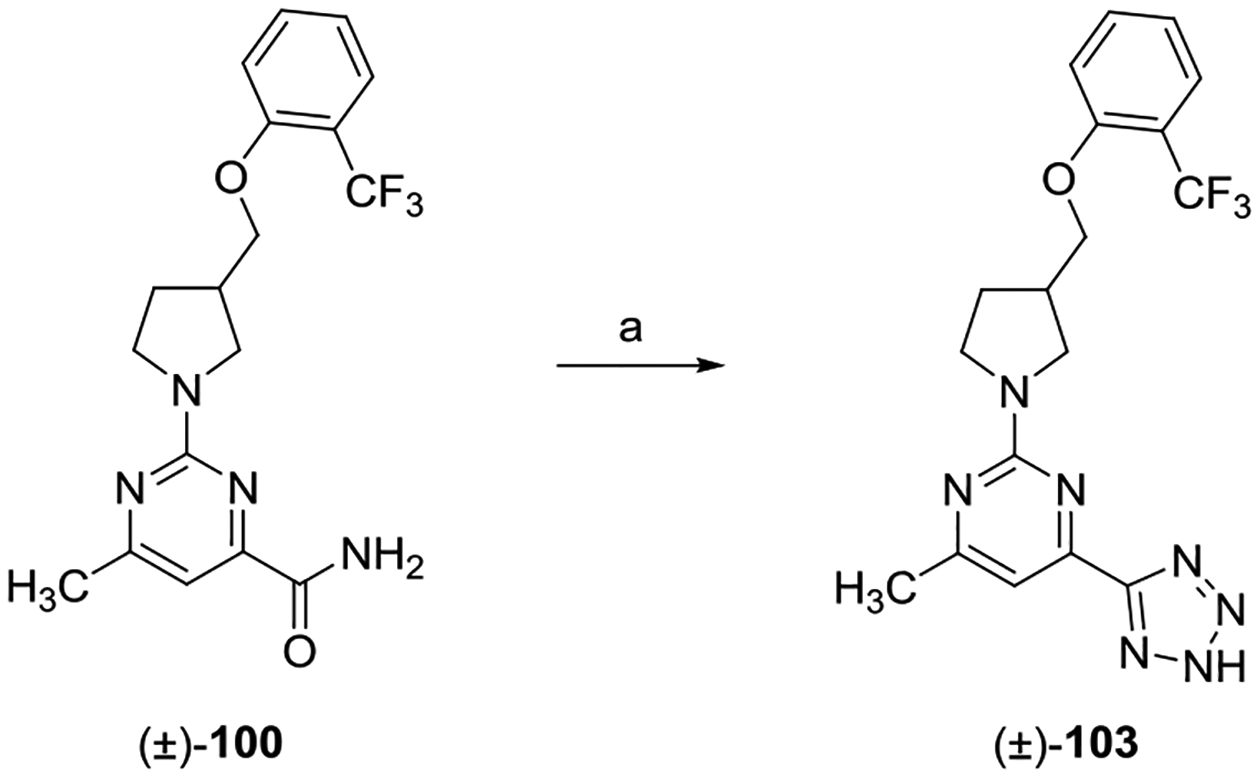
aReagents and conditions: (a) NaN3, tetrachlorosilane, CH3CN, 80 °C, 18 h.
The preparation of tetrazole analogue (±)-103 was achieved via treatment of carboxamide (±)-100 with tetrachlorosilane and sodium azide (NaN3) in anhydrous CH3CN heated at 80 °C.
RESULTS AND DISCUSSION
Structure–Activity Relationships.
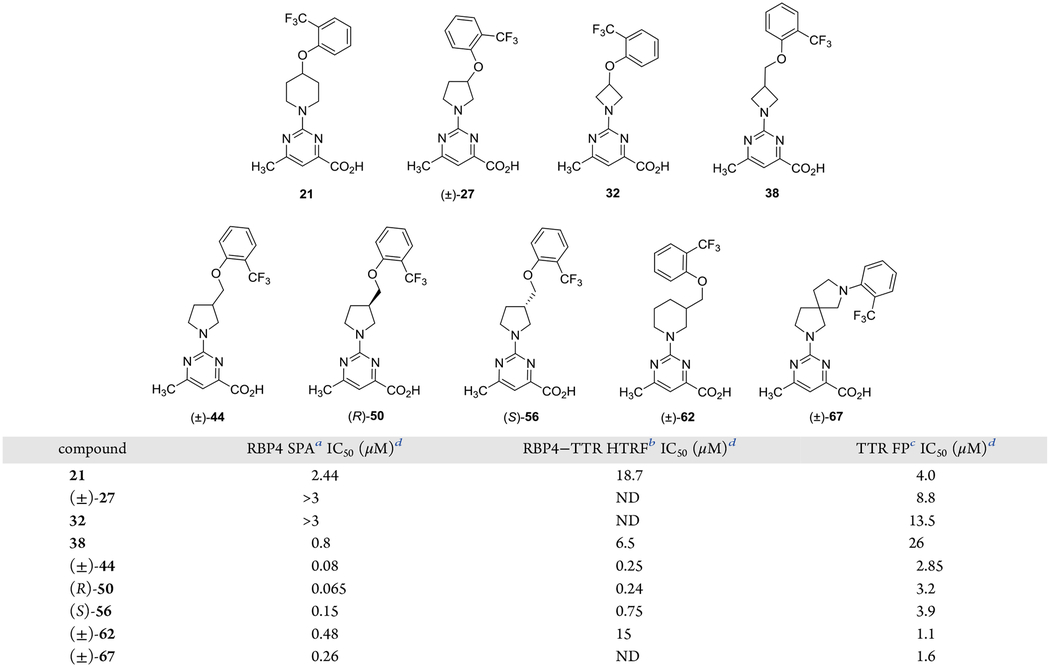
A series of scaffold hopping A-region analogues bearing various isosteric replacements for the linker of 8 were initially prepared. In vitro RBP4 binding potency and functional activity, as well as TTR tetramer binding potency data for the scaffold hopping congeners of 8, are given in Table 1. Of this sample set, racemic (±)-44 emerged as an initial analogue of interest. Although less potent at RBP4 than benchmark 8 ((±)-44 RBP4 SPA IC50 = 80.0 nM; RBP4–TTR HTRF IC50 = 0.25 μM), novel (±)-44 did present an attractive balance of dual activity for both targets ((±)-44 TTR FP IC50 = 2.85 μM). A subtle enantiopreference was observed for the R-enantiomer of (±)-44 ((R)-50 RBP4 SPA IC50 = 65.0 nM; (S)-56 RBP4 SPA IC50 = 150.0 nM); however, there was no delineation between the enantiomers with regard to RBP4–TTR HTRF or TTR FP activity. The remaining analogues of this sample set demonstrated diminished potency for RBP4, TTR, or both targets. In light of these findings, the pyrrolidin-3-ylmethanol core scaffold of (±)-44 was selected for subsequent SAR exploration.
Table 1.
In Vitro RBP4 and TTR Data for A-Region Scaffold Hopping Core Analogues
|
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of [3H]-all-trans-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 μM concentration of all-trans-retinol.
IC50 values for the FP assay obtained in the presence of a fixed, 25 μM concentration of fluorescein isothiocyanate (FITC)-coupled TTR FP probe.
IC50 data is represented as the mean ± standard deviation (SD) or compounds tested more than twice the IC50 or as the mean of two independent experiments if only run twice. ND = not determined.
In the absence of docking models for both targets, we can only speculate as to why the pyrrolidin-3-ylmethanol core scaffold of (±)-44 provided the best balance of potency for both target proteins. First, the pyrrolidin-3-ylmethanol core scaffold of (±)-44 shares length and shape complementarity with the core linker of 8, which previous RBP4 docking models revealed was favorably aligned within the all-trans-retinol binding site.19 Previous docking models depict the carboxylic acid of 8 engaged in key H-bond and electrostatic interactions near the binding site opening as the core linker extends through the β-barrel channel projecting the aryl head group deep into the inner cavity.19 We hypothesize that (±)-44 similarly projects the aryl head group ring deep into the inner hydrophobic cavity; however, the additional rotational freedom of its pyrrolidin-3-ylmethanol core may impart entropic penalties that lead to the observed slight reduction in potency relative to rotationally constricted 8. Thus, it is possible that the core scaffolds of analogues 21–38 are either too long (i.e., 21), too short (i.e., 32), or of less than optimal geometry (i.e., (±)-27 and 38) to achieve a desirable level of potency. The core scaffolds of analogues (±)-62 and (±)-67 do present geometries that are of somewhat better alignment with 8 and (±)-44 in terms of length and geometry, which may explain the observed improved potencies for them relative to 21–38. Of this sample set, (±)-44–(±)-67 also present the best potency at TTR. Although these novel analogues do not present the biphenyl ether motif present in T4, they do share some pharmacophoric similarity with the endogenous TTR ligand. It is anticipated that the pyrimidine acid appendages of (±)-44–(±)-67 engage in electrostatic binding interactions with Lys15 and Glu54. Furthermore, (±)-44–(±)-67 are of similar length to T4 and can confer an orthogonal orientation among the head group and bottom group appendages that T4 adopts upon binding to TTR. We postulate that the B-region aryl head groups of (±)-44–(±)-67 are capable of projecting into the T4 inner binding site where they are within proximity to position their respective ortho-trifluoromethyl groups to potentially occupy resident HBP2 or HBP3 pockets. Future X-ray crystallographic studies will determine if our proposed binding orientations of (±)-44 at both proteins are indeed accurate.
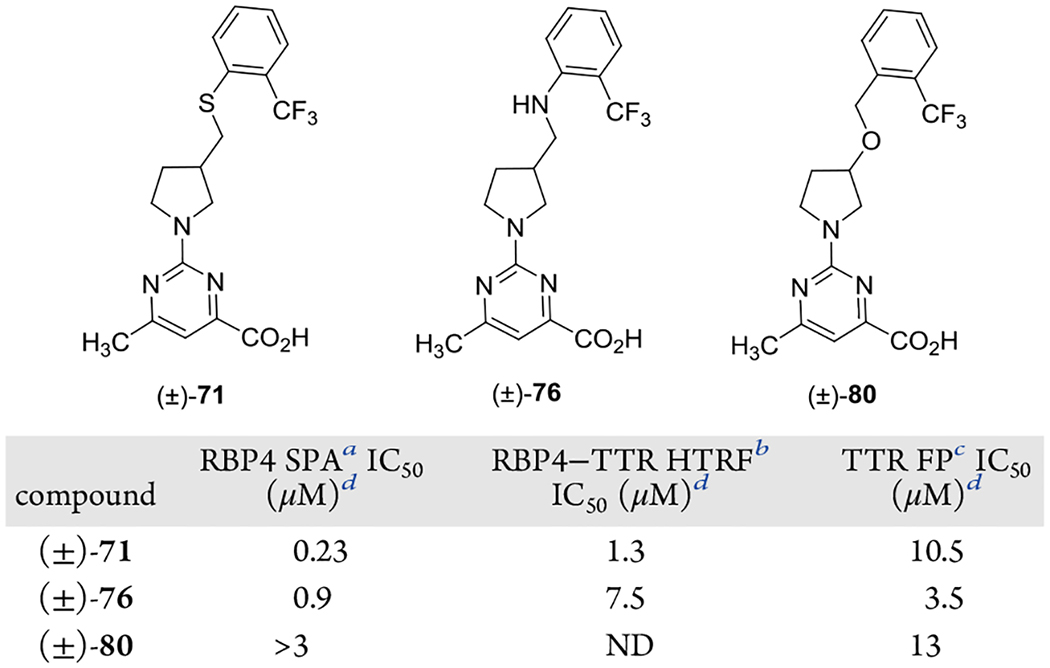
Prior to exploring the aromatic head group B-region SAR, we first explored the effects of either replacing the ether oxygen that connects the aromatic head group with the pyrrolidine core of (±)-44 with other heteroatoms or by repositioning the ether oxygen connectivity from the aromatic ring to the pyrrolidine ring (Table 2). Interestingly, replacing the (±)-44 ether oxygen with sulfur or NH led to significantly diminished potency for either RBP4 or TTR (or both). Furthermore, repositioning the ether oxygen to give (±)-80 led to complete loss of activity at RBP4 with a concomitant precipitous drop in TTR potency. The requirement for an ether linker with the oxygen connectivity presented by (±)-44 may be due to the following factors (or a combination thereof): (1) the oxygen of (±)-44 is better able to optimally modulate electron density of the aromatic head group for potential π–π interactions in the Phe-rich RBP4 β-ionone cavity relative to thioether (±)-71, aniline (±)-76, and alkoxymethyl analogue (±)-80, (2) loss of potency for aniline (±)-76 may be attributed to a more polar, H-bond donating NH group residing in a lipophilic area of both binding pockets, leading to unfavorable interactions and incurred desolvation penalties, (3) the repositioning of the oxygen atom in (±)-80 may also adversely affect hydrogen-bond-accepting (HBA) binding interactions of the aminopyrimidine fragment via the inductive effect, and (4) the ether oxygen of (±)-44 may stereoelectronically stabilize the bioactive conformation of the aromatic head group for binding at RBP4 and TTR. The results obtained from this focused probe led to (±)-44 B-region SAR exploring similarly ether-linked aromatic head groups.
Table 2.
In Vitro RBP4 and TTR Data for (±)-44 Analogues Bearing Alternative Linkers to the 2-Trifluoromethylphenyl Aromatic Head Group
|
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of [3H]-all-trans-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 μM concentration of all-trans-retinol.
IC50 values for the FP assay obtained in the presence of a fixed, 25 μM concentration of fluorescein isothiocyanate (FITC)-coupled TTR FP probe.
IC50 data is represented as the mean ± standard deviation or compounds tested more than twice the IC50 or as the mean of two independent experiments if only run twice. ND = not determined.
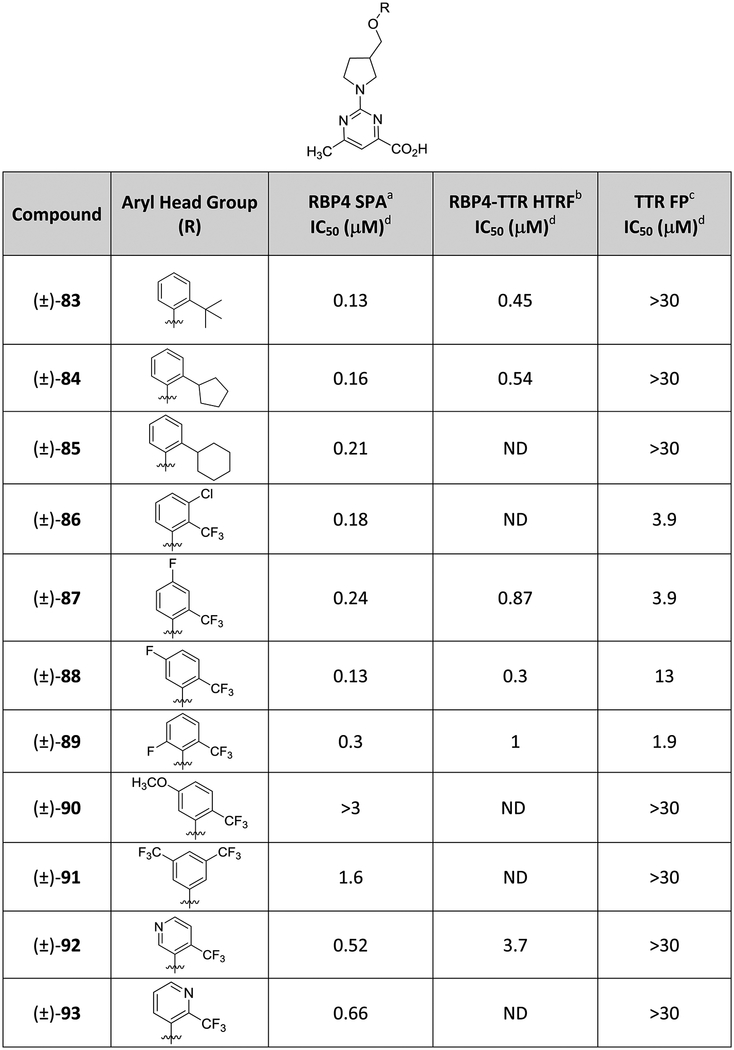
The B-region aromatic head group SAR exploration for analogue (±)-44 was conducted with a set of varyingly substituted phenyl-bearing analogues ((±)-83–(±)-93) (Table 3). The focused sample set was designed to explore how accommodative the inner binding cavities of both binding sites would be to analogues presenting multiple aromatic appendages of varying steric and electronic characteristics. Previously reported SAR by us and others has shown that large and lipophilic groups are required at the ortho position of the aromatic head group for any appreciable activity at RBP4. Specifically, the trifluoromethyl (CF3) group has served as an excellent moiety for generating potent RBP4 antagonists, which is attributed to its size (Hansch parameter ES = −2.4) and lipophilicity (Hansch parameter π = 0.88), which appears to be optimally suited for β-ionone pocket occupancy. Replacement of the ortho-trifluoromethyl group with other groups that are sterically smaller in size or are less lipophilic (i.e., Cl, CH3) can result in a significant loss in potency at RBP4 (data not shown). Similarly, varying the substitution pattern of the CF3 group can also have an undesirable effect on RBP4 potency. It is for these reasons that we largely maintained an ortho-substitution pattern for many of our B-region analogues and that any alternative ortho-substituents explored were typically larger and more lipophilic than CF3. Thus, our initial set of analogues replaced the ortho-trifluoromethyl group with large, lipophilic alkyl and cycloalkyl appendages ((±)-83–(±)-85). These analogues were approximately 2-fold less potent than (±)-44 in the RBP4 SPA binding assay and were inactive at TTR. These results indicate that the RBP4 β-ionone is more vacuous than the inner T4 binding cavity and that the sterically encumbered substituents of analogues (±)-83–(±)-85 may simply have been too large for the inner HBPs to accommodate. The data also suggests that there is a limit to the size of the ortho-substituent and that the CF3 was optimal for further SAR exploration. We next probed various halogenated aromatic head groups bearing a CF3 group ortho to the ether oxygen ((±)-86–(±)-89) with the goal of identifying a substitution pattern that may better interact with the inner T4 HBPs while more fully occupying the RBP4 β-ionone cavity. These halogenated analogues did demonstrate activity for TTR. Interestingly, the positioning of fluorine had a significant impact on TTR potency as analogue (±)-88 was 6-fold less potent than (±)-89 ((±)-88 TTR FP IC50 = 13 μM; (±)-89 TTR FP IC50 = 1.9 μM). This may be attributed to the CF3 group and the fluorine of (±)-89 together occupying HBPs, whereas the positioning of fluorine in (±)-88 is suboptimal and does not permit it to efficiently occupy an inner HBP and/or does not allow the CF3 group to do so. Notably, analogue (±)-89 may be poised for replacement of its fluorine with an iodine atom. Such an iodo analogue may better occupy the inner HBPs of TTR (in a similar manner as T4) as well as position the iodine for a σ-hole halogen binding interaction with the sulfur atom of Met73 residing in close proximity to the RBP4 β-ionone pocket. The 3,5-bis-CF3 analogue (±)-91 mimics the bis-iodine substitution pattern of the aromatic head group of 4. However, the compound neither improved affinity for RBP4 nor was it active at TTR. It is for this reason that the 3,5-bis-iodo congener that would more closely mimic T4 was not explored. Finally, we explored the effects of incorporating H-bond-accepting groups in the B-region ((±)-90, (±)-92, and (±)-93) that could potentially (1) engage in H-bond interactions with the Ser117 and Ser117′ residues buried deep within the inner T4 binding cavity and (2) engage in a potential H-bond interaction with the His104 residue located with the RBP4 β-ionone pocket. Anisole (±)-90 was inactive at both targets, and while pyridyl analogues (±)-92 and (±)-93 did exhibit moderate potency at RBP4 ((±)-92 RBP4 SPA IC50 = 0.52 μM; (±)-93 RBP4 SPA IC50 = 0.66 μM), they too were inactive at TTR. None of the B-region analogues showed improved potency for both targets relative to parent (±)-44; thus, the C-region aromatic carboxylic acid bottom group SAR campaign was conducted using the (±)-3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidine core scaffold intact.
Table 3.
In Vitro RBP4 and TTR Data for (±)-44 B-Region Analogues Bearing Alternative Substituted Aryl Head Groups
|
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of [3H]-all-trans-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 μM concentration of all-trans-retinol.
IC50 values for the FP assay obtained in the presence of a fixed, 25 μM concentration of fluorescein isothiocyanate (FITC)-coupled TTR FP probe.
IC50 data is represented as the mean ± standard deviation or compounds tested more than twice the IC50 or as the mean of two independent experiments if only run twice. ND = not determined.
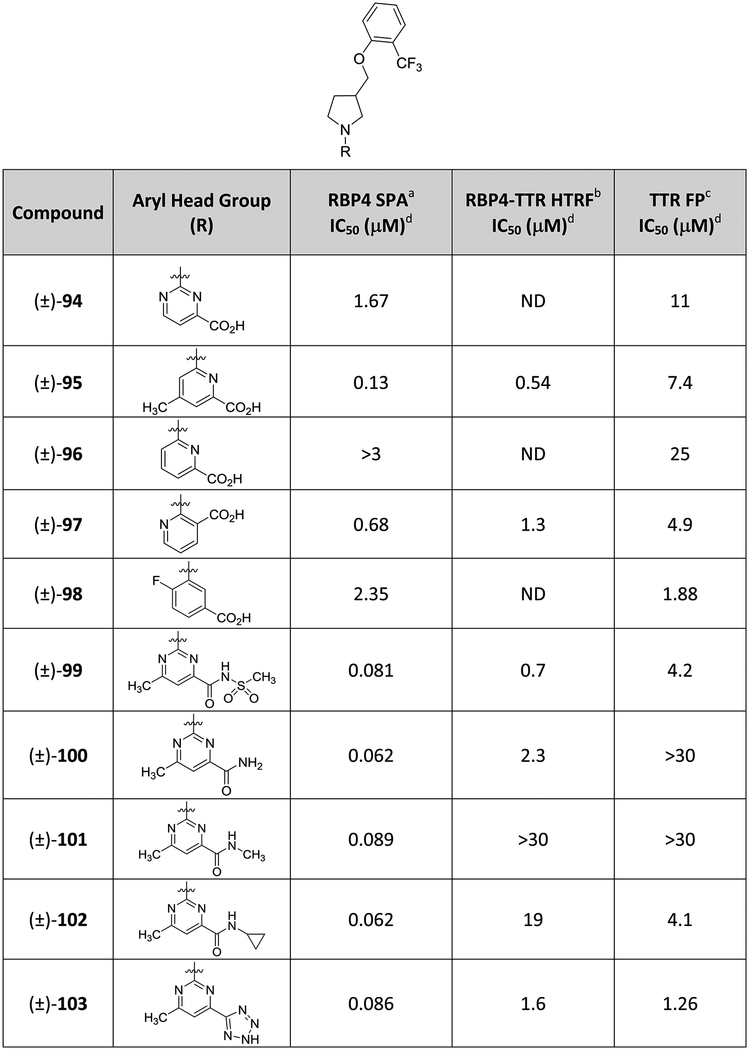
The SAR effects of pyrimidine-4-carboxylic acid, picolinic, nicotinic, and benzoic acid moieties used to replace the pyrimidine ring of (±)-44 are provided in Table 4. Removal of the pyrimidine 6-methyl group had a deleterious effect for both targets as the des-methyl analogue ((±)-94) demonstrated a significant loss in RBP4 potency and a ~5-fold loss in TTR potency relative to (±)-44 (RBP4 SPA IC50 = 1.67 μM; TTR FPIC50 = 11 μM). Selective removal of a pyrimidine nitrogen ((±)-95 and (±)-96) or repositioning the carboxylic acid adjacent to the pyrrolidine core ((±)-97) did not yield analogues with improved binding profiles relative to (±)-44. We also explored the fluorinated benzoic acid appendage featured in TTR ligand 12. X-ray crystallographic data of 12 bound to TTR (PDB 4HIQ) shows the fluorine projecting into HBP1.33 Incorporation of this appendage ((±)-98) did yield a slight improvement in TTR potency relative to (±)-44 (TTR FP IC50 = 1.88 μM); however, the improvement in TTR potency came at the expense of RBP4 (RBP4 SPA IC50 = 2.35 μM). Finally, we also pursued isosteric replacements of the (±)-44 carboxylic acid with an acyl sulfonamide ((±)-99) as well as replacing the acid with carboxamides ((±)-100–(±)-102). Collectively, these analogues provided compounds that were equipotent to (±)-44 for RBP4; however, they were either moderately less potent or inactive at TTR. Interestingly, replacement of the carboxylic acid with a tetrazole isostere ((±)-103) did furnish an analogue that was equipotent to (±)-44 in the RBP4 SPA binding assay (RBP4 SPA IC50 = 86.0nM) and ~2-fold more potent in the TTR FP assay (TTR FP IC50 = 1.26 μM). However, (±)-103 was approximately significantly less potent for RBP4 functional activity than (±)-44 ((±)-103 RBP4–TTR HTRF IC50 = 1.6 μM).
Table 4.
In Vitro RBP4 and TTR Data for (±)-44 Analogues Bearing Alternative C-Region Bottom Group Aromatic Carboxylic Acid Appendages, Carboxylic Acid Isosteres, and Carboxamides
|
IC50 values for the SPA assay obtained in the presence of a fixed, 10 nM concentration of [3H]-all-trans-retinol.
IC50 values for the HTRF assay obtained in the presence of 1 μM concentration of all-trans-retinol.
IC50 values for the FP assay obtained in the presence of a fixed, 25 μM concentration of fluorescein isothiocyanate (FITC)-coupled TTR FP probe.
IC50 data is represented as the mean ± standard deviation or compounds tested more than twice the IC50 or as the mean of two independent experiments if only run twice. ND = not determined.
Analogue (±)-44 emerged as a lead upon completion of our SAR campaign and was further profiled (Table 5). Compound (±)-44 possesses very good kinetic solubility and robust microsomal stability as evidenced by the very low intrinsic clearance (CLint) values. The compound is highly protein-bound and lacks limiting inhibitory activity in a standard cytochrome P450 (CYP) panel and at the human ether-a-go-go (hERG) channel. Importantly, unlike 8, which possesses ancillary PPARγ agonist activity, (±)-44 was found to be devoid of such activity.
Table 5.
ADME Profile Obtained for Advanced Analogue (±)-44
| microsomal CLint (μL/(min mg))b | liver microsomal stability (% remaining at 30 min)c | %PPBe | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| solubility (μM)a | H | R | M | cyno | HLM | RLM | MLM | cyno LM | CYP inhibition (% inhibition at 10, μM) 2C9, 2C19, 2D6, 3A4 | hERGd (IC50) (μM) | PPARγ (IC50) (μM) | H | R | M |
| 187.4 | <0.0231 | 105 | 93 | 95 | 103 | 2C9—0% | >30 | >100 | 99.8 | 96 | 96 | |||
| 2C19—(−)8% | ||||||||||||||
| 2D6—18.1% | ||||||||||||||
| 3A4—(−)2.2% | ||||||||||||||
Kinetic solubility measured in PBR (pH = 7.4).
Microsomal intrinsic clearance (CLint); H = human; R = rat; M = mouse; cyno = cynomolgus monkey.
Liver microsomal metabolic stability, % of parent drug remaining after a 30 min incubation in the presence of the microsomes; HLM = human liver microsomes; RLM = rat liver microsomes; MLM = mouse liver microsomes; cyno LM = cynomolgus monkey liver microsomes.
CiPA hERG QPatch assay; compounds were tested (n = 2) in a five-point concentration–response study.
%PPB = plasma protein binding; H = human, R = rat, M = mouse.
(±)-44 PK Characteristics in CD-1 Male Mice.
(±)-44 was dosed intravenously (IV) (2 mg/kg) and per os (PO) (5 mg/kg) in CD-1 male mice, and the calculated PK parameters are given in Table 6. Compound (±)-44 was found to be relatively stable (CLint = 0.0499 L/(h kg)) with a long half-life of 9.9 h. Significant absorption was achieved (Cmax of 3033 ng/mrelatively rapidly (Tmax = 0.83 h). High levels of exposure were attained (AUCINF was 52439 h ng/mL), and the estimated %F was 52%.
Table 6.
(±)-44 PK Data with CD-1 Male Micea
| route | dose (mg/kg) | C0b (ng/mL) | CLc (L/(h kg)) | t1/2d (h) | Vsse (L/kg) | AUClastf (h ng/mL) | AUCINFg (h ng/mL) | %Fj |
|---|---|---|---|---|---|---|---|---|
| IV | 2 | 9129 (661) | 0.0499 (0.005) | 8.6 (1.15) | 0.518 (0.069) | 39 523 (3665) | 40 336 (4021) | NA |
| route | dose (mg/kg) | Cmaxh (ng/mL) | Tmaxi (h) | t1/2d (h) | Vsse (L/kg) | AUClastf (h ng/mL) | AUCINFg (h ng/mL) | %Fj |
| PO | 5 | 3033 (692) | 0.83 (0.29) | 9.9 (2.0) | NA | 50 400 (5898) | 52 439 (7214) | 52.0 (7.15) |
Data are represented as the mean with standard deviation in parentheses (mean (SD)). Dosing groups consisted of three drug naïve adult male CD-1 mice. IV administration: Test article was administered at the 2 mg/kg dose; test article vehicle = 3% dimethylacetamide (DMA)/45% poly(ethylene glycol) (PEG)300/12% ethanol/40% sterile water; PO administration: test article was administered at the 5 mg/kg dose, vehicle = 2% Tween 80 in 0.9% saline.
Observed initial concentration of compound in blood at time zero.
Total body clearance.
Apparent half-life of the terminal phase of elimination of compound from blood.
Volume of distribution at steady state.
Area under the blood concentration versus time curve from 0 to the last time point that the compound was quantifiable in blood.
Area under the blood concentration versus time curve from 0 to infinity.
Maximum observed concentration of compound in blood.
Time of maximum observed concentration of compound in blood.
Bioavailability; F = (AUCINFpo × Doseiv)/(AUCINFiv × Dosepo).
(±)-44 PK–PD Correlations in Mouse.
Confirmation of target engagement for (±)-44 was assessed by measuring serum RBP4 level dynamics in mice after a single oral dose. A maximum of the 81% reduction in murine serum RBP4 levels was observed 6 h post oral administration of 25 mg/kg of (±)-44 (Figure 10A). Effects on serum TTR levels were not observed (data not shown). The dynamics of the in vivo serum RBP4 reduction correlated with (±)-44 systemic exposures after oral dosing (Figure 10B) and a reduction in serum RBP4 (Figure 10A). The observed maximal reduction (81%) and duration of effect (64% reduction observed at 24 h) correlate well with (±)-44 PK properties such as high Cmax, long exposure, and slow clearance (Table 6). This data supports a conclusion on a generally good correlation between PK characteristics and PD effects between (±)-44 exposures and degree of RBP4 lowering in mice.
Figure 10.
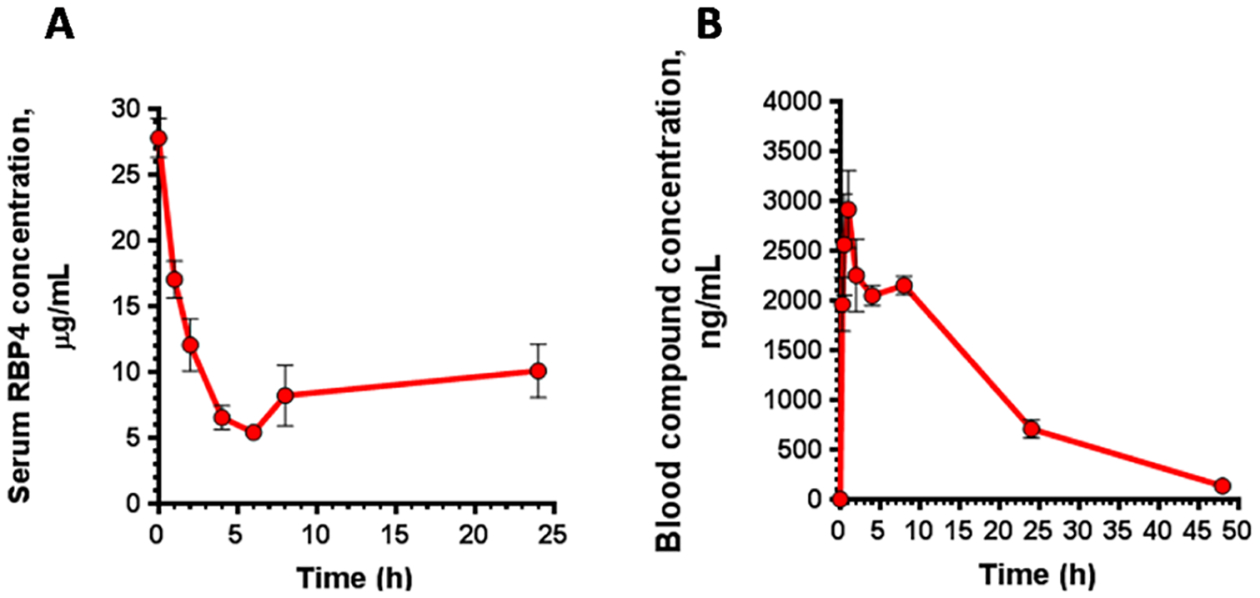
Murine PK–PD correlations of (±)-44. (A) Serum RBP4 levels following a single 25 mg/kg oral administration of (±)-44. (B) Blood compound levels following administration of a single oral 5 mg/kg dose of (±)-44. Data represented as the mean ± SD. Three mice per treatment group were used in the study.
A very good correlation has been previously established between the ability of RBP4 antagonists from different classes to induce serum RBP4 lowering and preclinical efficacy in the Abca4−/− mouse model.11a,13,14 Based on its very good RBP4 lowering activity, it seems reasonable to expect (±)-44 to be efficacious in reducing the formation of cytotoxic lipofuscin retinoids in the retina.
Analogue (±)-44 Attenuates the Formation of High Molecular Forms of TTR.
Inhibition of the acid-induced TTR aggregation in vitro is a well-established approach to characterization of TTR tetramer kinetic stabilizers.45,46 Long-term 72 h incubation of TTR at 37 °C in acidic conditions leads to the tetramer destabilization and dissociation followed by partial monomer misfolding and oligomerization into amyloid fibrils and other high-molecular-weight aggregates.47 To explore the activity of the bispecific analogue (±)-44 as a TTR tetramer kinetic stabilizer, we evaluated the ability of this compound to prevent acid-mediated TTR aggregate formation using modifications of the previously published protocol.48,49 Two compounds were used as a positive control in the aggregation experiments, tafamidis and benzbromarone. Tafamidis (11) is a potent TTR tetramer kinetic stabilizer approved as a therapy for familial amyloid polyneuropathy, while benzbromarone (structure not shown), a uricosuric drug, was found in our prior experiments to be a potent TTR ligand with an IC50 of 293 nM in the FP TTR binding assay, which is on par with the reported potency of 11 in this assay.35a Following 72 h of incubation with dimethyl sulfoxide (DMSO) at pH 4.0, the high molecular forms of TTR were significantly increased, while no such forms were observed after a similar incubation period at neutral pH (Figure 11A). Similar to the activity of two potent TTR ligands, 11 and benzbromarone, (±)-44 significantly reduced the formation of high-molecular-weight TTR species (Figure 11A) indicating that it can act as a TTR tetramer kinetic stabilizer. Higher intensities of the TTR monomer bands in samples treated with 11, benzbromarone, and (±)-44 in comparison to DMSO reflected a corresponding decrease in TTR aggregation conferred by 11, benzbromarone, and (±)-44. Quantitative analysis of band intensity revealed a 3.6-, 5.6-, and 4.7-fold reduction in the formation of high-molecular-weight aggregates induced by tafamidis, benzbromarone, and (±)-44, respectively (Figure 11B). Significant increase in the TTR monomer band intensities associated with the decrease in aggregate formations was evident in samples treated with 11, benzbromarone, and (±)-44 (Figure 11C). Overall, the results of the aggregation experiments established that bispecific analogue (±)-44 can act as a TTR tetramer kinetic stabilizer.
Figure 11.
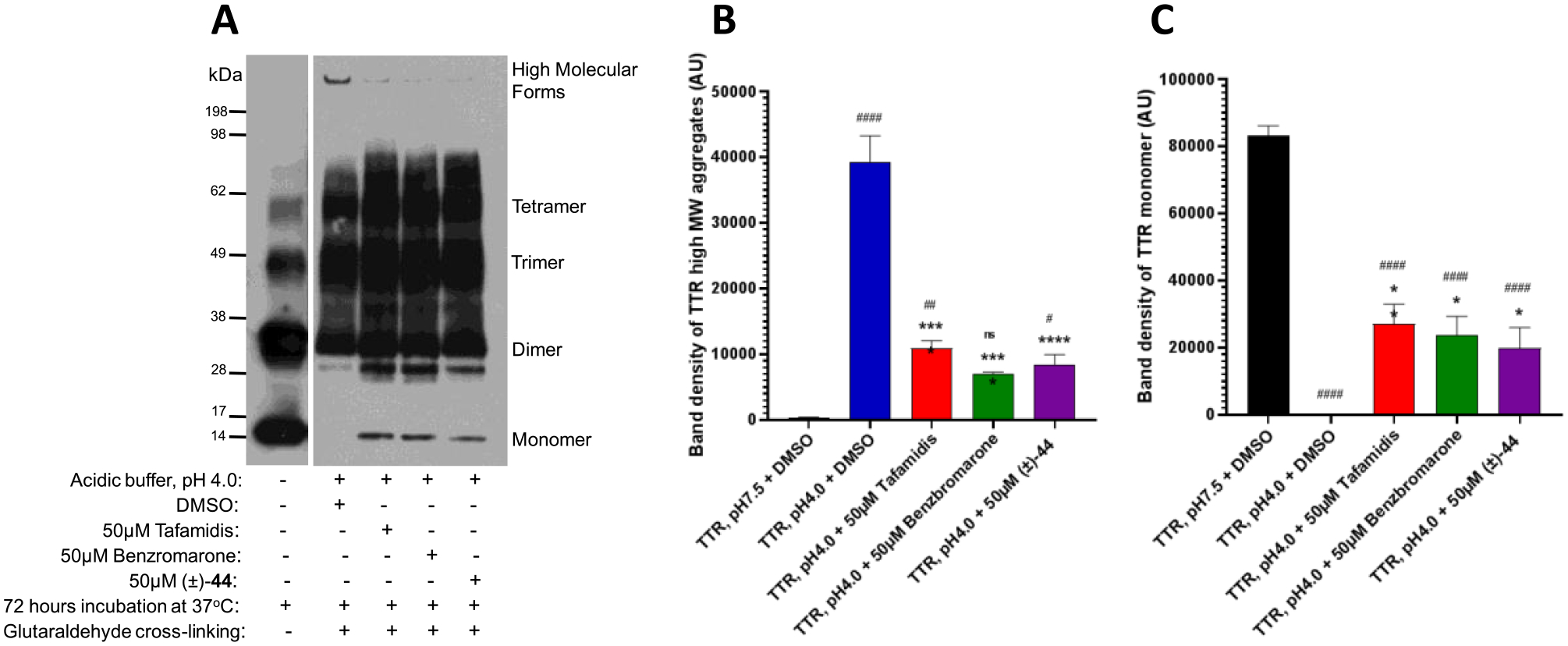
Analogue (±)-44 reduces the formation of high-molecular-weight TTR forms in the acid-induced aggregation assay. (A) TTR protein (5 μg) was aggregated by using acetate buffer (pH 4.0) and incubated for 72 h at 37 °C. After incubation in the presence of 50 μM 11, 50 μM benzbromarone, and 50 μM (±)-44, the samples were cross-linked and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Western blotting with TTR antibodies. The representative blot of at least three independent experiments is presented. Bar graphs represent pixel volumes of TTR high-molecular-weight aggregates (B) and monomers (C). The vertical axes represent the ratio of pixel volume means ± SD of the scanned bands on the immunoblots in arbitrary units. Statistical significance was determined by one-way analysis of variance (ANOVA) with the Holm–Sidak post hoc test; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001 compared to TTR aggregation (pH 4.0) + DMSO group; #, p ≤ 0.05; ##, p ≤ 0.01; ###, p ≤ 0.001; ####, p ≤ 0.0001 compared to TTR without the aggregation (pH 7.5) group.
CONCLUSIONS
AMD is the most common cause of blindness in developed nations that generally afflicts people of 60 years of age or older.50 In the Caucasian population over the age of 80, the frequency of all forms of AMD is 90% in males and 16.4% in females.51 Currently, there are no FDA-approved pharmacotherapies available for the most prevalent dry form of AMD, which affects 90% of AMD patients. SSA is a common late-onset disease associated with the accumulation of wild-type TTR amyloid deposits in extracellular compartments of different tissues and organs. The heart is usually the dominant site of involvement.30a SSA is recognized as a major cause of severe cardiac dysfunction in the elderly, which includes congestive heart failure and cardiac death.52 SSA affects approximately 25% of patients over the age of 80.52 The number of people with AMD is projected to increase to 288 million by 2040.53 The high rate of comorbidity between dry AMD and SSA is expected based on the high-population frequency of both conditions in older individuals. In recent years, we identified and characterized different structural classes of RBP4 antagonists as a potential treatment for macular degeneration and NAFLD.14,18–20,26 We showed that selective RBP4 antagonists can induce a desired drop in serum RBP418,19,26 partially decreasing the concentration of visual cycle retinoids in the retina and significantly inhibiting cytotoxic A2E biosynthesis in Abca4−/− mice, a model that recapitulates the Stargardt disease phenotype.14 However, the long-term use of selective RBP4 antagonists in AMD patients who also have SSA may not be optimal. It is known that holo-RBP4–TTR interaction stabilizes TTR tetramers and prevents the formation of TTR amyloid fibrils. Selective RBP4 antagonists release the unliganded TTR tetramer from the holo-RBP4–TTR complex in circulation, and the release of the unliganded TTR may be associated with destabilization of TTR tetramers and, potentially, with the TTR amyloid fibril formation in a fraction of susceptible individuals prone to developing TTR amyloidosis, such as SSA patients. We sought to identify novel RBP4 antagonists that also exhibited TTR tetramer-stabilizing capability. We utilized previously reported SAR for 8 and reported X-ray crystallographic data for ligands bound to RBP4 (PDB 3FMZ)8 and TTR (PDB 2ROX)5c to enable our structure-based drug design campaign. Our premise was to identify compounds with suitable RBP4 antagonistic potency that could provide adequate TTR tetramer kinetic stabilization. We initially investigated a series of alternative core linker analogues via a scaffold hopping approach starting from 8, which ultimately lead to the discovery of (±)-44. Compound (±)-44 possesses a favorable balance of in vitro potency for both targets, desirable ADME properties, and excellent PK characteristics in mouse (Table 6). In addition, the compound lowered serum RBP4 levels upon oral dosing in mice (>80%) (Figure 10) and significantly decreased TTR high-molecular-weight aggregates in an in vitro TTR aggregate assay (Figure 11). These data suggest that (±)-44 shows potential as an oral treatment for RBP4-related indications while preventing possible TTR amyloid fibril formation and ATTR comorbidities such as SSA or hereditary TTR amyloidosis. One general safety concern for the novel class of bispecific compounds relates to their effect on traffic and delivery of thyroxine and vitamin A. Binding of compounds like (±)-44 to TTR is not expected to significantly affect thyroxine metabolism and function given that less than 1% of circulating TTR carries thyroxine, while another serum protein, TBG, functions as the primary T4 carrier in the blood. TTR knockout mice are phenotypically normal with no signs of growth or developmental abnormalities.54 Similarly, serum RBP4 lowering by bispecific compounds is unlikely to promote systemic vitamin A deficiency symptoms given the ability of alternative RBP4/TTR-independent pathways to provide retinoids to tissues, including the retina.55–61 Rbp4−/− mice present no systemic abnormalities55–57 or retinal degeneration.56 It is hopeful that the class of bispecific compounds exemplified by (±)-44 will be safe and efficacious as an orally administered pharmacotherapeutic for the dry AMD–SSA comorbidity.
EXPERIMENTAL SECTION
In Vitro Binding of Compounds to RBP4.
Compound binding to RBP4 was assessed in the radiometric scintillation proximity (SPA) assay that was previously described.18,19,26 The assay measured competitive displacement of radiolabeled [3H]-all-trans-retinol from native RBP4 purified from human urine (Fitzgerald, 30R-AR022L). The protein was biotinylated using the EZ-link Sulfo-NHS-LC-biotinylation kit from ThermoFisher (cat #21335) as recommended by the manufacturer. Binding assays were implemented in a final volume of 100 μL in SPA buffer (1× PBR, pH 7.4, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% BRA, 0.5% 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS)). The assay reaction included a radioligand, 10 nM [3H]-all-trans-retinol (48.7 Ci/mmol; PerkinElmer, Waltham, MA), along with the 0.3 mg/well streptavidin-PVT beads (PerkinElmer, RPNQ0006) and 50 nM biotinylated human RBP4. Unlabeled retinol (Sigma-Aldrich, cat #95144) at 20 μM was added to control wells to assess a nonspecific binding. Radioactivity counts were measured using a CHAMELEON plate reader (Hidex Oy, Turku, Finland) after 16 h of incubation at rt with mild shaking.
Assessment of Antagonistic Activity in the HTRF RBP4–TTR Interaction Assay.
The ability of analogues to act as antagonists of all-trans-retinol-dependent RBP4–TTR interaction was measured in the homogeneous time-resolved fluorescence (HTRF) assay as described previously.18,19,26 Untagged TTR (Calbiochem, cat #529577) and maltose-binding protein-tagged RBP4 expressed in Escherichia coli were used in this assay. HTRF cryptate labeling kit from CisBio (Cisbio, cat #62EUSPEA, Bedford, MA) was used to label TTR with Eu3+ cryptate. The assay was performed in a final assay volume of 16 μL in the buffer that contained 10 mM Tris–HCl pH 7.5, 1 mM dithiothreitol (DTT), 0.05% NP-40, 0.05% Prionex, 6% glycerol, and 400 mM KF. Other components of the reaction mix included 60 nM MBP–RBP4, 5 nM TTR–Eu, 26.7 nM of anti-MBP antibody conjugated with d2 (Cisbio, cat #61MBPDAA), and 1 μM all-trans retinol (Sigma-Aldrich, cat #95144). All of the reactions were performed under dim red light in the dark. The plates were read in the SpectraMax M5e multimode plate reader (Molecular Devices, Sunnyvale, CA) after overnight incubation at 4 °C. Fluorescence was excited at 337 nm; the emission was measured at 668 and 620 nm with a 75 μs counting delay. The HTRF signal was expressed as the ratio of fluorescence intensity: Flu668/Flu620 × 10 000.
Fluorescence Polarization TTR Tetramer Binding Assay.
Compound binding to TTR was assessed in the fluorescence polarization assay. The assay measured competitive displacement of the fluorescent probe, FITC–diclofenac, from TTR isolated from human plasma (Clabiochem-Millipore, cat. #52957). FITC–diclofenac was synthesized at LeadGen Lab, LLC following the published procedure.59 Each well contained 200 nM TTR and 100 nM FITC–diclofenac in the FP buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.01% CHAPS, 0.01%Prionex) along with test compounds. Nonspecific binding was determined in the presence of 500 μM unlabeled diclofenac (Sigma-Aldrich). Reactions with test compounds were incubated overnight at 4 °C, and FP was measured on a Spectramax M5e plate reader (Molecular Devices).
TTR Aggregation Assay.
The ability of test compounds to prevent TTR aggregation was evaluated under the acidic conditions that favor TTR aggregation and fibril formation. A 2 μL solution of 167 μM human TTR (ACRO Biosystems #H5223) was incubated with 7 μL of 50 mM sodium acetate pH 4.0 (Sigma-Aldrich #S7545) and 100 mM KCl (Sigma-Aldrich #S5405) in the presence or absence of 1 μL TTR inhibitor for 72 h at 37 °C. At the end of the incubation, 3.5 μL of 500 mM sodium phosphate (Sigma-Aldrich #S5136) buffer pH = 8.0 was added to each sample for neutralization and 0.6 μL of 5% CHAPS (Sigma-Aldrich #C5070) as a detergent to prevent reassociation of protein. The cross-linking was performed by adding 1.5 μL of 5% glutaraldehyde solution (Sigma-Aldrich #G6257). After 4 min, the reaction was stopped by the addition of 2.5 μL freshly made 5% NaBH4. The samples were subjected to TTR Western blotting with prealbumin antibodies (1:500; Dako #A0002). Band intensity for TTR monomer and TTR aggregates was quantified from scanned images of the blots.
General Chemistry.
All reactions were performed under a dry atmosphere of nitrogen unless otherwise specified. The indicated reaction temperatures refer to the reaction bath, while room temperature (rt) is noted as 25 °C. Commercial-grade reagents and anhydrous solvents were used as received from vendors, and no attempts were made to purify or dry these components further. Removal of solvents under reduced pressure was accomplished with a Buchi rotary evaporator at approximately 28 mmHg pressure using a Teflon-linked KNF vacuum pump. Thin-layer chromatography (TLC) was performed using 1 in. × 3 in. AnalTech no. 02521 silica gel plates with a fluorescent indicator. Visualization of TLC plates was made by observation with either short-wave UV light (254 nm lamp), 10% phosphomolybdic acid in ethanol or in iodine vapors. Preparative thin-layer chromatography was performed using Analtech, 20 × 20 cm2, 1000 μm preparative TLC plates. Flash column chromatography was carried out using a Teledyne Isco CombiFlash Companion Unit and a Biotage Selekt System with a Teledyne Isco RediSep Rf and Biotage Sfär silica gel columns. If needed, products were purified by reverse phase chromatography, using a Teledyne Isco CombiFlash Companion Unit and a Biotage Selekt System with a RediSep Gold C18 reverse phase column. Proton NMR spectra were obtained on a 400 MHz Varian nuclear magnetic resonance spectrometer. Chemical shifts (δ) are reported in parts per million (ppm), and coupling constant (J) values are given in hertz (Hz), with the following spectral pattern designations: s, singlet; d, doublet; t, triplet, q, quartet; quint, quintet; m, multiplet; dd, doublet of doublets; dt, doublet of triplets; dq; doublet of quartets; br, broad signal. Tetramethylsilane was used as an internal reference. Peak listing, multiplicity designations, and coupling constant calculations were conducted using Mnova v.14 software (Mestrelab Research). Carbon NMR spectra were obtained on a 500 MHz Bruker AV III nuclear magnetic resonance spectrometer, and tetramethylsilane was used as an internal reference. Fluorine NMR spectra were obtained on a 400 MHz Bruker AV III nuclear magnetic resonance spectrometer. Any melting points provided are uncorrected and were obtained using a Stanford Research Systems OptiMelt melting point apparatus (MPA100) with an automated melting point system. Mass spectroscopic analyses were performed using electrospray ionization (ESI) on a Waters AQUITY UPLC MS triple quadrupole mass spectrometer. High-pressure liquid chromatography (HPLC) purity analysis was performed using a Waters Breeze2 HPLC system with binary solvent systems A and B using a gradient elusion [A, H2O with 0.1% formic acid; B, CH3CN with 0.1% formic acid] and flow rate = 0.5 mL/min, with UV detection at 254 nm (system equipped with a photodiode array (PDA) detector). An ACQUITY UPLC BEH C18 column, 130 Å, 1.7 μm, 2.1 mm × 50 mm was used. High-resolution mass spectrometry (HRMS) analysis was performed using an Agilent 6530 Accurate-Mass Q-TOF. All final compounds tested for in vitro and in vivo biological testing were purified to ≥95% purity, and these purity levels were measured by both 1H NMR and HPLC.
6-Methyl-2-(4-(2-(trifluoromethyl)phenoxy)piperidin-1-yl)-pyrimidine-4-carboxylic Acid (21).
Step A: To a 0 °C cooled solution of tert-butyl 4-oxopiperidine-1-carboxylate (15, 5.0 g, 25.1 mmol) in CH3OH (50 mL) was added NaBH4 (1.14 g, 30.1 mmol). The mixture was stirred for 8 h while gradually warming to rt. The mixture was concentrated under reduced pressure, and the resulting residue was diluted with H2O (100 mL) and extracted with CH2Cl2 (2 × 100 mL).The combined organic extracts were dried over Na2SO4, filtered, and evaporated under reduced pressure to give tert-butyl 4-hydroxypiperidine-1-carboxylate (16) as a white solid (4.5 g, 89%): 1H NMR (400 MHz, CDCl3): δ 3.91−3.75 (m, 3H), 3.05−2.96 (m, 2H), 1.90−1.79 (m, 2H), 1.49−1.40 (m, 11H); ES MS: m/z 224 [M + Na]+.
Step B: To a 0 °C cooled solution of tert-butyl 4-hydroxypiperidine-1-carboxylate (16, 4.5 g, 22.3 mmol) in CH2Cl2 (50 mL) were added Et3N (4.7 mL, 33.5 mmol) and DMAP (0.127 g, 1.10 mmol) followed by the addition of TsCl (5.10 g, 26.8 mmol). The resulting solution was stirred for 16 h while gradually warming to rt under an atmosphere of N2. The mixture was diluted with saturated aqueous NaOH (50 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give tert-butyl 4-(tosyloxy)piperidine-1-carboxylate (17) as a colorless liquid (6.6 g, 84%): 1H NMR (400 MHz, CDCl3) δ 7.82 (m, 2H), 7.49 (m, 2H), 4.69 (br, 1H), 3.49 (m, 2H), 3.15 (m, 2H), 2.43 (br, 3H), 1.70 (m, 2H), 1.51 (m, 2H), 1.38 (s, 9H); ESI MS m/z 356 [M + H]+.
Step C: To a solution of tert-butyl 4-(tosyloxy)piperidine-1-carboxylate (17, 0.250 g, 0.703 mmol) in anhydrous DMF (4 mL) were added Cs2CO3 (0.450 g, 1.38 mmol) and 2-(trifluoromethyl)-phenol (95.0 mg, 0.586 mmol), and the resulting solution was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then diluted with H2O (20 mL). The aqueous mixture was extracted with EtOAc (3 × 25 mL), and the combined organic extracts were washed with H2O (3 × 25 mL) and brine (25 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexanes) to give tert-butyl 4-(2-(trifluoromethyl)phenoxy)-piperidine-1-carboxylate (18) as a white solid (0.118 g, 56%): 1H NMR (400 MHz, acetone-d6) δ 7.65−7.55 (m, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.08 (t, J = 7.6 Hz, 1H), 4.92−4.82 (m, 1H), 3.67−3.57 (m, 2H), 3.50−3.40 (m, 2H), 2.0−1.90 (m, 2H), 1.80−1.70 (m, 2H), 1.45 (s, 9H); ESI MS m/z 346 [M + H]+.
Step D: To a 0 °C cooled solution of tert-butyl 4-(2-(trifluoromethyl)phenoxy)piperidine-1-carboxylate (18, 0.118 g, 0.341 mmol) in CH2Cl2 (10 mL) was added TFA (0.33 mL, 4.31 mmol), and the resulting solution was stirred for 16 h while gradually warming to rt. The mixture was neutralized by carefully pouring into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 4-(2-(trifluoromethyl)phenoxy)piperidine (19) as a white solid (80.0 mg, 95%): 1H NMR (400 MHz, acetone-d6) δ 7.70−7.60 (m, 2H), 7.37 (d, J = 6.0 Hz, 1H), 7.14 (t, J = 7.6 Hz, 1H), 5.10−5.03 (m, 1H), 3.50−3.40 (m, 4H), 2.47−2.37 (m, 2H), 2.21−2.11 (m, 2H); ESI MS m/z 246 [M + H]+.
Step E: A mixture of 4-(2-(trifluoromethyl)phenoxy)piperidine (19, 0.100 g, 0.408 mmol), methyl 2-chloro-6-methylpyrimidine-4-carboxylate (76.1 mg, 0.408 mmol), and i-Pr2NEt (0.21 mL, 1.22 mmol) in THF (10 mL) was heated at reflux for 16 h under an atmosphere of N2. The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0–100% EtOAc in hexanes) to give methyl 6-methyl-2-(4-(2-(trifluoromethyl)-phenoxy)piperidin-1-yl)pyrimidine-4-carboxylate (20) as an off-white solid (0.140 g, 87%): MS (ESI+) m/z 396 [M + H]+.
Step F: A solution of methyl 6-methyl-2-(4-(2-(trifluoromethyl)-phenoxy)piperidin-1-yl)pyrimidine-4-carboxylate (20, 0.100 g, 0.253 mmol) and LiOH (18.1 mg, 0.758 mmol) in CH3OH (5 mL), THF (5 mL), and H2O (5 mL) was stirred at rt for 16 h. The mixture was acidified to pH = 5 with 2 N aqueous HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(4-(2-(trifluoromethyl)phenoxy)piperidin-1-yl)pyrimidine-4-carboxylic acid (21) as an off-white solid (87.8 mg, 91%): 1H NMR (400 MHz, CDCl3) δ 7.56−7.53 (m, 2H), 7.22 (d, J = 6.0 Hz, 2H), 7.02−7.51 (m, 1H), 4.86 (br, 1H), 3.99−3.85 (m, 4H), 2.37 (s, 3H), 2.05−1.95 (m, 2H), 1.81−1.80 (m, 2H); ESI MS m/z 382 [M + H]+; HPLC >99% (AUC), tR = 16.8 min.
(±)-6-Methyl-2-(3-(2-(trifluoromethyl)phenoxy)pyrrolidin-1-yl)-pyrimidine-4-carboxylic Acid ((±)-27).
Step A: To a 0 °C cooled solution of (±)-tert-butyl 3-hydroxypyrrolidine-1-carboxylate ((±)-22, 1.00 g, 5.34 mmol) in CH2Cl2 (20 mL) were added Et3N (1.1 mL, 8.02 mmol) and DMAP (32.0 mg, 0.262 mmol) followed by the addition of TsCl (1.10 g, 5.88 mmol). The resulting solution was stirred for 16 h while gradually warming to rt under an atmosphere of N2. The mixture was diluted with saturated aqueous NaOH (20 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-tert-butyl 3-(tosyloxy)pyrrolidine-1-carboxylate ((±)-23) as a colorless liquid (1.50 g, 82%): 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.9 Hz, 2H), 7.34 (d, J = 7.8 Hz, 2H), 5.05 (br, 1H), 3.43 (m, 4H), 2.43 (br, 3H), 2.06 (m, 2H), 1.45 (s, 9H); ESI MS m/z 342 [M + H]+.
Step B: To a solution of (±)-tert-butyl 3-(tosyloxy)pyrrolidine-1-carboxylate ((±)-23, 0.100 g 0.293 mmol) in DMF (4 mL) were added Cs2CO3 (0.290 g, 0.902 mmol) and 2-(trifluoromethyl)phenol (73.0 mg, 0.450 mmol), and the resulting mixture was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then diluted with H2O (20 mL). The aqueous mixture was extracted with EtOAc (3 × 25 mL), and the combined organic extracts were washed with H2O (3 × 25 mL) and brine (25 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexanes) to give (±)-tert-butyl 3-(2-(trifluoromethyl)phenoxy)-pyrrolidine-1-carboxylate ((±)-24) as a white solid (80.0 mg, 83%): 1H NMR (400 MHz, acetone-d6) δ 7.62 (m, 2H), 7.34 (d, J = 8.0 Hz 1H), 7.15 (t, J = 7.6 Hz, 1H), 5.20 (s, 1H), 3.65−3.40 (m, 4H), 2.19 (m, 2H), 1.44 (s, 9H); ESI MS m/z 332 [M + H]+.
Step C: To a 0 °C cooled solution of (±)-tert-butyl 3-(2-(trifluoromethyl)phenoxy)pyrrolidine-1-carboxylate ((±)-24, 80.0 mg, 0.241 mmol) in CH2Cl2 (5 mL) was added TFA (0.18 mL, 2.41 mmol), and the resulting solution was stirred for 8 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-3-(2-(trifluoromethyl)phenoxy)pyrrolidine ((±)-25) as a white solid (50.0 mg, 90%); ESI MS m/z 232 [M + H]+.
Step D: A mixture of (±)-3-(2-(trifluoromethyl)phenoxy)-pyrrolidine ((±)-25, 0.100 g, 0.432 mmol), methyl 2-chloro-6-methylpyrimidine-4-carboxylate (80.6 mg, 0.432 mmol), and i-Pr2NEt (0.23 mL, 1.29 mmol) in THF (10 mL) was heated at reflux for 16 h under an atmosphere of N2. The reaction was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–100% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(3-(2-(trifluoromethyl)phenoxy)-pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-26) as an off-white solid (0.145 g, 88%): 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.00 (s, 1H), 6.98−6.95 (m, 2H), 5.10 (s, 1H), 4.00−3.86 (m, 3H), 3.89 (s, 3H), 3.76−3.74 (m, 2H), 2.38 (s, 3H), 2.22−2.19 (m, 1H); ESI MS m/z 382 [M + H]+.
Step E: A solution of (±)-methyl 6-methyl-2-(3-(2-(trifluoromethyl)phenoxy)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-26, 48.8 g, 0.128 mmol) and LiOH (9.21 mg, 0.384 mmol) in CH3OH (5 mL), THF (5 mL), and H2O (5 mL) was stirred at rt for 16 h. The mixture was acidified to pH 5 with 2 N aqueous HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-6-methyl-2-(3-(2-(trifluoromethyl)phenoxy)pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-27) as an off-white solid (0.043 g, 91%): 1H NMR (400 MHz, DMSO-d6) δ 7.61−7.54 (m, 2H), 7.34−7.33 (m, 1H), 7.00−6.93 (m, 2H), 5.31 (s, 1H), 3.77−3.86 (m, 3H), 3.50−3.48 (m, 1H), 2.30 (s, 3H), 2.29−2.20 (m, 2H); ESI MS m/z 368 [M + H]+; HPLC >99% (AUC), tR = 14.5 min.
6-Methyl-2-(3-(2-(trifluoromethyl)phenoxy)azetidin-1-yl)-pyrimidine-4-carboxylic Acid (32).
Step A: To a solution of tert-butyl 3-(tosyloxy)azetidine-1-carboxylate (28, 0.500 g 1.52 mmol) in DMF (20 mL) were added Cs2CO3 (990 mg, 3.05 mmol) and 2-(trifluoromethyl)phenol (0.272 g, 1.68 mmol), and the resulting solution was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then diluted with H2O (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (3 × 50 mL) and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexanes) to give tert-butyl 3-(2-(trifluoromethyl)phenoxy)azetidine-1-carboxylate (29) as a colorless liquid (0.400 g, 82%): 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.3 Hz, 1H), 7.61−7.45 (m, 1H), 7.06−7.03 (m, 1H), 6.65 (d, J = 7.9 Hz, 1H), 4.99−4.91 (m, 1H), 4.35−4.30 (m, 2H), 4.10−4.05 (m, 2H), 1.44 (s, 9H); ESI MS m/z 318 [M + H]+.
Step B: To a 0 °C cooled solution of tert-butyl 3-(2-(trifluoromethyl)phenoxy)azetidine-1-carboxylate (29, 0.400 g, 1.20 mmol) in CH2Cl2 (20 mL) was added TFA (0.96 mL, 12.0 mmol), and the resulting solution was stirred for 16 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 3-(2-(trifluoromethyl)phenoxy)azetidine (30) as a white solid (0.240 g, 87%): ESI MS m/z 218 [M + H]+.
Step C: A mixture of 3-(2-(trifluoromethyl)phenoxy)azetidine (30, 0.100 g, 0.460 mmol), methyl 2-chloro-6-methylpyrimidine-4-carboxylate (85.9 mg, 0.460 mmol), and i-Pr2NEt (0.24 mL, 1.38 mmol) in THF (10 mL) was heated at reflux for 16 h under an atmosphere of N2.The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0–100% EtOAc in hexanes) to give methyl 6-methyl-2-(3-(2-(trifluoromethyl)-phenoxy)azetidin-1-yl)pyrimidine-4-carboxylate (31) as an off-white solid (0.152 g, 90%): MS (ESI+) m/z [M + H]+.
Step D: A solution of methyl 6-methyl-2-(3-(2-(trifluoromethyl)-phenoxy)azetidin-1-yl)pyrimidine-4-carboxylate (31, 0.100 g, 0.272 mmol) and LiOH (19.5 mg, 0.816 mmol) in CH3OH (5 mL), THF (5 mL), and H2O (5 mL) was stirred at rt for 16 h. The mixture was acidified to pH 5 with 2 N aqueous HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(3-(2-(trifluoromethyl)phenoxy)azetidin-1-yl)pyrimidine-4-carboxylic acid (32) as an off-white solid (83.6 mg, 87%): 1H NMR (400 MHz, CDCl3) δ 7.59−7.54 (m, 2H), 7.14 (s, 1H), 7.08 (t, J = 7.6 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 5.28 (m, 1H), 4.61−4.60 (m, 2H), 4.15−4.12 (m, 2H), 2.40 (s, 3H); ESI MS m/z 354 [M + H]+; ESI MS m/z 354 [M + H]+; HPLC >99% (AUC), tR = 14.1 min.
6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)azetidin-1-yl)pyrimidine-4-carboxylic Acid (38).
Step A: To a 0 °C cooled solution of tert-butyl 3-(hydroxymethyl)azetidine-1-carboxylate (33, 3.0 g, 16.0 mmol) in CH2Cl2 (50 mL) were added Et3N (4.5 mL, 32.0 mmol) and DMAP (97.0 mg, 0.736 mmol) followed by the addition of TsCl (3.35 g, 17.6 mmol). The resulting solution was stirred for 16 h while gradually warming to rt under an atmosphere of N2. The mixture was diluted with the saturated solution of aqueous NaOH (50 mL) and was extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give tert-butyl 3-((tosyloxy)methyl)azetidine-1-carboxylate (34) as a colorless liquid (5.0 g, 92%): ESI MS m/z 342 [M + H]+.
Step B: To a solution of tert-butyl 3-((tosyloxy)methyl)azetidine-1-carboxylate (34, 5.0 g 14.6 mmol) in DMF (50 mL) were added Cs2CO3 (9.5 g, 29.32 mmol) and 2-(trifluoromethyl)phenol (2.3 g, 14.6 mmol), and the resulting mixture was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then diluted with H2O (100 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (3 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude tert-butyl 3-((2-(trifluoromethyl)-phenoxy)methyl)azetidine-1-carboxylate (35) as a brown liquid (4.5 g, 93%): ESI MS m/z 332 [M + H]+.
Step C: To a 0 °C cooled solution of tert-butyl 3-((2-(trifluoromethyl)phenoxy)methyl)azetidine-1-carboxylate (35, 4.50 g, 13.59 mmol) in CH2Cl2 (50 mL) was added TFA (10.3 mL, 135 mmol), and the resulting solution was stirred for 8 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 3-((2-(trifluoromethyl)phenoxy)methyl)piperidine (36) as a white solid (2.8 g, 90% crude): ESI MS m/z 232 [M + H]+.
Step D: A mixture of 3-((2-(trifluoromethyl)phenoxy)methyl)-azetidine (36, 1.0 g, 4.32 mmol), methyl 2-chloro-6-methylpyrimidine-4-carboxylate (0.807 g, 4.32 mmol), and i-Pr2NEt (2.25 mL, 12.9 mmol) in THF (20 mL) was heated at reflux for 16 h under an atmosphere of N2. The reaction was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0–100% EtOAc in hexanes) to give methyl 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)azetidin-1-yl)pyrimidine-4-carboxylate (37) as an off-white solid (1.64 g, 86%): 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.05 (s, 1H), 7.00−6.95 (m, 2H), 4.33 (t, J = 8.8 Hz, 2H), 4.22−4.21 (m, 2H), 4.08−4.04 (m, 2H), 3.91 (s, 3H), 3.18−3.16 (m, 1H), 2.40 (s, 3H); ESI MS m/z 382 [M + H]+.
Step E: A solution of methyl 6-methyl-2-(3-((2-(trifluoromethyl)-phenoxy)methyl)azetidin-1-yl)pyrimidine-4-carboxylate (37, 1.0 g, 2.62 mmol) and LiOH (0.188 g, 7.86 mmol) in CH3OH (10 mL), THF (10 mL), and H2O (10 mL) was stirred at rt for 16 h. The mixture was acidified to pH = 5 with 2 N aqueous HCl and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give 6-methyl-2-(3-((2-(trifluoromethyl)-phenoxy)methyl)azetidin-1-yl)pyrimidine-4-carboxylic acid (38) as an off-white solid (0.914 g, 95%): 1H NMR (400 MHz, CDCl3) δ7.51 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 1H), 7.00 (s, 1H), 7.00−6.94 (m, 2H), 4.33 (t, J = 8.4 Hz, 2H), 4.24−4.22 (m, 2H), 4.07−4.03 (m, 2H), 3.17−3.15 (m, 1H), 2.42 (s, 3H); ESI MS m/z 368 [M + H]+; HPLC 98.2% (AUC), tR = 13.5 min.
(±)-6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-44).
Step A: To a 0 °C solution of (±)-tert-butyl 3-(hydroxymethyl)pyrrolidine-1-carboxylate ((±)-39, 2.0 g, 9.93 mmol), Et3N (2.8 mL, 83.7 mmol), and DMAP (60.4 mg, 0.494 mmol) in CH2Cl2 (50 mL) was added TsCl (2.27 g, 11.9 mmol). The resulting mixture was stirred for 16 h while gradually warming to rt under an atmosphere of N2. The mixture was diluted with saturated aqueous NaOH solution (50 mL) and then extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexanes) to give (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) as a white solid (3.1g, 88%): 1H NMR (400 MHz, DMSO-d6) δ 7.76 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.4 Hz, 2H), 3.97 (d, J = 6.8 Hz, 2H), 3.28−3.25 (m, 1H), 3.24−3.19 (m, 2H), 3.13−3.10 (m, 1H), 2.85−2.81 (m, 1H), 2.39 (s, 3H), 1.82−1.81 (m, 1H), 1.49−1.47 (m, 1H), 1.33 (s, 9H); ESI MS m/z 356 [M + H]+.
Step B: To a solution of (±)-tert-butyl 3-((tosyloxy)methyl)-pyrrolidine-1-carboxylate ((±)-40, 1.0 g 2.81 mmol) in DMF (10 mL) were added Cs2CO3 (2.74 g, 8.41 mmol) and 2-(trifluoromethyl)-phenol (0.410 g, 2.53 mmol), and the resulting mixture was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then diluted with H2O (30 mL). The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with H2O (3 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexanes) to give (±)-tert-butyl 3-((2-(trifluoromethyl)phenoxy)-methyl)pyrrolidine-1-carboxylate ((±)-41) as a white solid (0.816 g, 84%): 1H NMR (400 MHz, DMSO-d6) δ 7.61−7.56 (m, 2H), 7.22 (d, J = 8.8 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 4.09−4.02 (m, 2H), 3.44−3.35 (m, 1H), 3.36−3.33 (m, 1H), 3.25−3.19 (m, 1H), 3.11−3.01 (m, 1H), 2.62−2.55 (m, 1H), 1.97 (br, 1H), 1.96−1.64 (m, 1H), 1.35 (s, 9H); ESI MS m/z 346 [M + H]+.
Step C: To a 0 °C solution of (±)-tert-butyl 3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidine-1-carboxylate ((±)-41, 0.800 g 2.32 mmol) in CH2Cl2 (10 mL) was added TFA (3.5 mL, 46.3 mmol), and the resulting mixture was stirred for 16 h while gradually warming to rt under an atmosphere of N2. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (50 mL), and the resulting biphasic mixture was separated. The aqueous layer was further extracted with CH2Cl2 (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidine ((±)-42) as a white solid (0.520 g, 90%): 1H NMR (400 MHz, DMSO-d6) δ 9.25 (br, 1H), 7.62−7.57 (m, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.07 (t, J = 7.6 Hz, 1H), 4.16−4.05 (m, 2H), 3.38−3.33 (m, 1H), 3.28−3.16 (m, 2H), 2.98 (t, J = 8.0 Hz, 1H), 2.77−2.69 (m, 1H), 2.11−2.03 (m, 1H), 1.78−1.69 (m, 1H); ESI MS m/z 346 [M + H]+; ESI MS m/z 246 [M + H]+. Step D: To a solution of (±)-tert-butyl 3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidine-1-carboxylate ((±)-42, 0.265 g 1.08 mmol) in THF (5 mL) were added i-Pr2NEt (0.6 mL, 3.24 mmol) and methyl 2-chloro-6-methylpyrimidine-4-carboxylate (0.242 g, 1.29 mmol), and the resulting mixture was stirred at reflux for 16 h. The mixture was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-43) as a white solid (0.362 g, 85%): 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 7.6 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 6.99 (s, 1H), 6.97−6.92 (m, 2H), 4.10−4.06 (m, 1H), 4.00−3.96 (m, 1H), 3.91 (s, 3H), 3.89−3.86 (m, 1H), 3.79−3.76 (m, 1H), 3.66−3.61 (m, 1H), 3.51−3.47 (m, 1H), 2.86−2.82 (m, 1H), 2.39 (s, 3H), 2.24−2.20 (m, 1H), 1.99−1.94 (m, 1H); ESI MS m/z 396 [M + H]+.
Step E: To a solution of (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-43, 0.250 g, 0.632 mmol) in CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (0.151 g, 6.32 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous mixture was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3. The acidified mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-1-(4-(2-(trifluoromethyl)phenyl)piperidine-1-carbonyl)pyrrolidine-2-carboxylic acid ((±)-44) as a white solid (0.160 g, 66%): mp = 105−106 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.59 (m, 2H, H1 and H3), 7.28 (d, J = 8.4 Hz, 1H, H4), 7.09 (t, J = 7.6 Hz, 1H, H2), 6.97 (s, 1H, H5), 4.16−4.11 (dq, J = 16.1, 9.2 Hz, 2H, H6 and H7), 3.77 (dd, J = 11.4, 7.5 Hz, 1H, H8), 3.70 (m, 1H, H9), 3.52 (dt, J = 11.3, 7.6 Hz, 1H, H10), 3.39 (dt, J = 9.2, 7.2 Hz, 1H, H11), 2.76 (quint, J = 7.1 Hz, 1H, H14), 2.35 (s, 3H, 3 × H15), 2.19−2.09 (m, 1H, H12), 1.87 (dq, J = 12.3, 8.0 Hz, 1H, H13);13C NMR (500 MHz, DMSO-d6) δ 169.46, 166.28, 160.16, 156.37, 134.29, 126.74, 124.95, 122.79, 120.33, 116.88, 113.57, 107.71, 69.75, 49.00, 45.82, 27.54, 24.06; 19F NMR (400 MHz, DMSO-d6) δ −59.98 (s, CF3); ESI MS m/z 382 [M + H]+; HRMS (ESI+) C18H18F3N3O3 calcd [M + H]+ = 382.1379, observed [M + H]+ = 382.1380; combustion analysis (%CHN): calcd for C18H18F3N3O3·0.25H2O: %C = 56.03; %H = 4.83; %N = 10.89; found: %C = 56.04; %H = 4.89; %N = 10.86; HPLC 98.7% (AUC), tR = 14.5 min.
(R)-6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((R)-50).
Compound (R)-50 was prepared from tert-butyl (R)-3-(hydroxymethyl)-pyrrolidine-1-carboxylate ((R)-45) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ7.55 (d, J = 7.6 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.12 (s, 1H), 7.01−6.94 (m, 2H), 4.07−4.06 (m, 2H), 3.86−3.77 (m, 2H), 3.62−3.51 (m, 2H), 2.89−2.86 (m, 1H), 2.43 (s, 3H), 2.27−2.23 (m, 1H), 2.14−2.01 (m, 1H); ESI MS m/z 382 [M + H]+; HPLC >99% (AUC), tR = 14.5 min.
(S)-6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((S)-56).
Compound (S)-56 was prepared from tert-butyl (S)-3-(hydroxymethyl)-pyrrolidine-1-carboxylate ((S)-51) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ7.56 (d, J = 8.0 Hz, 1H), 7.46 (t, J = 8.4 Hz, 1H), 7.12 (s, 1H), 7.01−6.94 (m, 2H), 4.07−4.06 (m, 2H), 3.86−3.77 (m, 2H), 3.62−3.51 (m, 2H), 2.90−2.85 (m, 1H), 2.44 (s, 3H), 2.26−2.24 (m, 1H), 2.14−2.01 (m, 1H); ESI MS m/z 382 [M + H]+; HPLC >99% (AUC), tR = 14.5 min.
(±)-6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-piperidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-62).
Step A: To a 0 °C solution of (±)-tert-butyl 3-(hydroxymethyl)piperidine-1-carboxylate ((±)-57, 1.0 g, 4.65 mmol) in CH2Cl2 (20 mL) were added Et3N (0.81 mL, 5.80 mmol) and DMAP (52.0 mg, 0.426 mmol) followed by the addition of TsCl (0.883 g, 4.65 mmol). The resulting solution was stirred for 16 h while gradually warming to rt. The reaction mixture was diluted with saturated aqueous NaOH solution (50 mL) and was extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with H2O (100 mL) and brine (100 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-tert-butyl 3-((tosyloxy)methyl)piperidine-1-carboxylate ((±)-58) as a colorless liquid (1.6 g, 94%): ESI MS m/z 370 [M + H]+.
Step B: To a solution of (±)-tert-butyl 3-((tosyloxy)methyl)-piperidine-1-carboxylate ((±)-58, 0.500 g 1.35 mmol) in DMF (20 mL) were added Cs2CO3 (0.650 g, 2.00 mmol) and 2-(trifluoromethyl)phenol (0.219 g, 1.35 mmol), and the resulting mixture was stirred at 80 °C for 16 h under an atmosphere of N2. The reaction mixture was allowed to cool to rt and then diluted with H2O (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (3 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-tert-butyl 3-((2-(trifluoromethyl)phenoxy)-methyl)piperidine-1-carboxylate ((±)-59) as a brown oil (0.400 g, 82%): ESI MS m/z 360 [M + H]+.
Step C: To a 0 °C solution of (±)-tert-butyl 3-((2-(trifluoromethyl)-phenoxy)methyl)piperidine-1-carboxylate ((±)-59, 0.400 g, 1.11 mmol) in CH2Cl2 (20 mL) was added TFA (0.85 mL, 11.1 mmol), and the resulting solution was stirred for 8 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-3-((2-(trifluoromethyl)phenoxy)methyl)-piperidine ((±)-60) as a white solid (0.250 g, 86%): ESI MS m/z 260 [M + H]+.
Step D: To a solution of (±)-3-((2-(trifluoromethyl)phenoxy)-methyl)piperidine ((±)-60, 0.100 g, 0.385 mmol) in THF (5 mL) were added i-Pr2NEt (0.20 mL, 1.16 mmol) and methyl 2-chloro-6-methylpyrimidine-4-carboxylate (71.8 mg, 0.385 mmol), and the resulting mixture was stirred at reflux for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)piperidin-1-yl)pyrimidine-4-carboxylate ((±)-61) as a white solid (0.137 g, 87%): 1H NMR (400 MHz, DMSO-d6) δ 7.59−7.55 (m, 2H), 7.22 (d, J = 8.4 Hz, 1H), 7.05 (t, J = 7.6 Hz, 1H), 6.94 (s, 1H), 4.79 (d, J = 12.8 Hz, 1H), 4.55 (d, J = 12.8 Hz, 1H), 4.04−3.96 (m, 2H), 3.80 (s, 3H), 2.93−2.83 (m, 3H), 2.29 (s, 3H), 1.85−1.71 (m, 4H); ESI MS m/z 410 [M + H]+.
Step E: To a solution of (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)piperidin-1-yl)pyrimidine-4-carboxylate ((±)-61, 0.100 g, 0.244 mmol) in CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (58.4 mg, 2.44 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous mixture was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3. The acidified mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)piperidin-1-yl)pyrimidine-4-carboxylic acid ((±)-62) as a white solid (96.4 mg, 66%): 1H NMR (400 MHz, DMSO-d6) δ 7.60−7.56 (m, 2H), 7.23 (d, J = 8.8 Hz, 1H), 7.06 (t, J = 8.0 Hz, 1H), 6.91 (s, 1H), 4.77 (d, J = 10.4 Hz, 1H), 4.57 (d, J = 13.2 Hz, 1H), 4.02−4.00 (m, 2H), 2.92−2.84 (m, 3H), 2.29 (s, 3H), 1.93−1.71 (m, 4H), 1.41−1.39 (m, 2H); ESI MS m/z 396 [M + H]+; HPLC >99% (AUC), tR = 16.2 min.
(±)-6-Methyl-2-(7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro-[4.4]nonan-2-yl)pyrimidine-4-carboxylic Acid ((±)-67).
Step A: A mixture of (±)-tert-butyl 2,7-diazaspiro[4.4]nonane-2-carboxylate ((±)-63, 0.250 g, 1.11 mmol) and 1-bromo-2-(trifluoromethyl)-benzene (0.273 g, 1.22 mmol) in 1,4-dioxane was degassed with N2 for 5 min followed by the addition of Cs2CO3 (1.08 g, 3.31 mmol), XPhos (0.105 mg, 0.223 mmol), and Pd2(dba)3 (0.101 g, 0.112 mmol). The reaction mixture was stirred at 110 °C for 16 h in a sealed vessel. The mixture was then allowed to cool to rt and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–30% EtOAc in hexane) to give (±)-methyl tert-butyl 7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonane-2-carboxylate ((±)-64) as an off-white amorphous solid (0.230 g, 56%): ESI MS m/z 371 [M + H]+.
Step B: To a 0 °C solution of (±)-methyl tert-butyl 7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonane-2-carboxylate ((±)-64, 0.100 g, 2.69 mmol) in CH2Cl2 (5 mL) was added TFA (0.20 mL, 2.61 mmol), and the resulting solution was stirred for 8 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-2-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonane ((±)-65) as a white solid (65.0 mg, 90%): ESI MS m/z 271 [M + H]+.
Step C: To a solution of (±)-2-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonane ((±)-65, 0.150 g, 0.554 mmol) in THF (5 mL) were added i-Pr2NEt (0.29 mL, 1.66 mmol) and methyl 2-chloro-6-methylpyrimidine-4-carboxylate (0.103 g, 0.554 mmol), and the resulting mixture was stirred at reflux for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonan-2-yl)-pyrimidine-4-carboxylate ((±)-66) as a white solid (0.205 g, 88%): 1HNMR (400 MHz, CDCl3) δ 7.55 (d, J = 6.4 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.00 (s, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.87 (t, J = 7.6 Hz, 1H), 3.91 (s, 3H), 3.74−3.61 (m, 4H), 3.47−3.44 (m, 2H), 3.30 (s, 2H), 2.39 (s, 3H), 2.08−1.93 (m, 4H); ESI MS m/z 421 [M + H]+.
Step D: To a solution of (±)-methyl 6-methyl-2-(7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonan-2-yl)pyrimidine-4-carboxylate ((±)-66, 0.100 g, 0.237 mmol) in CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (56.9 mg, 2.37 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous mixture was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3. The acidified mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-6-methyl-2-(7-(2-(trifluoromethyl)phenyl)-2,7-diazaspiro[4.4]nonan-2-yl)-pyrimidine-4-carboxylic acid ((±)-67) as a white solid (65.5 mg, 68%):1H NMR (400 MHz, CDCl3) δ 7.56 (d, J = 8.0 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.13 (s, 1H), 7.01−6.99 (m, 1H), 6.93−6.88 (m, 1H), 3.67−3.55 (m, 4H), 3.46 (t, J = 6.8 Hz, 2H), 3.31−3.26 (m, 2H), 2.44 (s, 3H), 2.10−1.97 (m, 4H); ESI MS m/z 407 [M + H]+; HPLC 98.4% (AUC), tR = 15.5 min.
(±)-6-Methyl-2-(3-(((2-(trifluoromethyl)phenyl)thio)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-71).
Compound (±)-71 was prepared from (±)-tert-butyl 3-((tosyloxy)methyl)-pyrrolidine-1-carboxylate ((±)-40) and 2-(trifluoromethyl)-benzenethiol according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, acetone-d6) δ 7.72 (t, J = 7.2Hz, 2H), 7.61 (t, J = 7.2 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.05 (s, 1H), 3.89−3.70 (m, 2H), 3.59−3.55 (m, 1H), 3.42−3.74 (m, 1H), 3.25 (d, J = 7.2 Hz, 2H), 2.64−2.62 (m, 1H), 2.40 (s, 3H), 2.27−2.24 (m, 1H), 1.92−1.87 (m, 1H); ESI MS m/z 398 [M + H]+; HPLC 97.1% (AUC), tR = 14.9 min.
(±)-6-Methyl-2-(3-(((2-(trifluoromethyl)phenyl)amino)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-76).
Step A: To a solution of (±)-tert-butyl 3-formylpyrrolidine-1-carboxylate ((±)-72, 0.300 g, 1.51 mmol) and 2-(trifluoromethyl)aniline (0.242 g, 1.51 mmol) in CH2Cl2 (10 mL) was added NaBH(OAc)3 (0.960 g, 4.53 mmol), and the mixture was stirred at rt for 16 h. The mixture was washed with saturated aqueous NaHCO3 solution (5 mL) and brine (5 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give crude (±)-tert-butyl 3-(((2-(trifluoromethyl)phenyl)-amino)methyl)pyrrolidine-1-carboxylate ((±)-73) as an oil, which was used as is in the next step (0.400 g, 77% crude yield): ESI MS m/z 345 [M + H]+.
Step B: To a 0 °C solution of (±)-tert-butyl 3-(((2-(trifluoromethyl)phenyl)amino)methyl)pyrrolidine-1-carboxylate ((±)-73, 0.400 g, 1.16 mmol) in CH2Cl2 (5 mL) was added TFA (0.89 mL, 11.6 mmol), and the resulting solution was stirred for 12 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into a solution of saturated aqueous NaHCO3 (10 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-N-(pyrrolidin-3-ylmethyl)-2-(trifluoromethyl)aniline ((±)-74) as a yellow oil and used as is in the next step (0.360 g): ESI MS m/z 245 [M + H]+.
Step C: To a solution of ((±)-N-(pyrrolidin-3-ylmethyl)-2-(trifluoromethyl)aniline ((±)-74), 0.360 g, 1.16 mmol) in THF (5 mL) were added i-Pr2NEt (0.61 mL, 3.48 mmol) and methyl 2-chloro-6-methylpyrimidine-4-carboxylate (0.216 g, 1.16 mmol), and the resulting mixture was stirred at reflux for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(3-(((2-(trifluoromethyl)phenyl)amino)methyl)pyrrolidin-1-yl)-pyrimidine-4-carboxylate ((±)-75) as a white solid (0.290 g, 63%): 1HNMR (400 MHz, CDCl3) δ 7.43−7.40 (m, 1H), 7.35−7.31 (m, 1H), 7.00 (s, 1H), 6.71−6.68 (m, 2H), 4.40 (br, 1H), 3.91 (s, 3H), 3.90−3.86 (m, 1H), 3.79−3.58 (m, 1H), 3.41−3.19 (m, 4H), 2.68−2.62 (m, 1H), 2.40 (s, 3H), 2.22−2.16 (m, 1H), 1.82−1.77 (m, 1H); ESI MS m/z 395 [M + H]+.
Step D: To a solution of (±)-methyl 6-methyl-2-(3-(((2-(trifluoromethyl)phenyl)amino)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-75, 0.110 g, 0.278 mmol) in CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (56.9 mg, 2.37 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous mixture was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3. The acidified mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-6-methyl-2-(3-(((2-(trifluoromethyl)phenyl)amino)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-76) as a white solid (90.0 mg, 68%): 1H NMR (400 MHz, DMSO-d6) δ 7.36−7.32 (m, 2H), 6.84−6.79 (m, 2H), 6.20 (t, J = 7.6 Hz, 1H), 5.53 (br, 1H), 3.63−3.58 (m, 2H), 3.41−3.39 (m, 1H), 3.25−3.17 (m, 3H), 2.58−2.57 (m, 1H), 2.23 (s, 3H), 1.98−1.95 (m, 1H), 1.70−1.67 (m, 1H); ESI MS m/z 381 [M + H]+; HPLC >99% (AUC), tR = 14.5 min.
(±)-6-Methyl-2-(3-((2-(trifluoromethyl)benzyl)oxy)pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-80).
Step A: To a 0 °C cooled solution of tert-butyl 3-hydroxypyrrolidine-1-carboxylate ((±)-22, 0.500 g, 2.67 mmol) in DMF (5 mL) was added NaH (0.267 g, 6.68 mmol). The mixture was stirred at 0 °C under an atmosphere of N2 for 30 min and then 1-(bromomethyl)-2-(trifluoromethyl)benzene (0.766 g, 3.20 mmol) was added, and the resulting mixture was stirred for 16 h while gradually warming to rt. The mixture was cooled back to 0 °C and carefully quenched via dilution with H2O (20 mL). The aqueous mixture was extracted with EtOAc (3 × 30 mL), and the combined organic extracts were washed with H2O (3 × 30 mL) and brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-tert-butyl 3-((2-(trifluoromethyl)benzyl)oxy)-pyrrolidine-1-carboxylate ((±)-77) as an off-white solid (0.900 g, 97% crude yield), which was used as is in the next step: ESI MS m/z 346 [M + H]+.
Step B: To a 0 °C cooled solution of (±)-tert-butyl 3-((2-(trifluoromethyl)benzyl)oxy)pyrrolidine-1-carboxylate ((±)-77, 0.900 g 2.61 mmol) in CH2Cl2 (5 mL) was added TFA (2.0 mL, 26.0 mmol), and the resulting solution was stirred for 8 h while gradually warming to rt. The mixture was neutralized by carefully pouring it into aqueous saturated NaHCO3 solution (30 mL). The biphasic mixture was separated, and the aqueous layer was further extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-3-((2-(trifluoromethyl)benzyl)oxy)pyrrolidine ((±)-78) as a white solid (0.450 g, 70% crude yield): ESI MS m/z 246[M + H]+.
Step C: To a solution of (±)-3-((2-(trifluoromethyl)benzyl)oxy)-pyrrolidine ((±)-78, 0.100 g, 0.407 mmol) in THF (5 mL) were added i-Pr2NEt (0.25 mL, 1.23 mmol) and methyl 2-chloro-6-methylpyrimidine-4-carboxylate (76.2 mg, 0.408 mmol), and the resulting solution was stirred at 80 °C for 16 h under an atmosphere of N2. The mixture was allowed to cool to rt and then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)benzyl)oxy)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-79) as a white solid (0.110 g, 68%): 1H NMR (400 MHz, acetone-d6) δ 7.75−7.68 (m, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.47 (t, J = 7.6 Hz, 1H), 6.97 (s, 1H), 4.78−4.76 (m, 2H), 4.41−4.39 (m, 1H), 3.85 (s, 3H), 3.74−3.66 (m, 4H), 2.36 (s, 3H), 2.31−2.04 (m, 2H); ESI MS m/z 396 [M + H]+.
Step D: To a solution of (±)-methyl 6-methyl-2-(3-((2-(trifluoromethyl)benzyl)oxy)pyrrolidin-1-yl)pyrimidine-4-carboxylate ((±)-79, 90.0 mg, 0.228 mmol) in CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (54.5 mg, 2.28 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous layer was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3. The mixture was extracted with EtOAc (3 × 30 mL), and the combined organic extracts were washed with brine (30 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-6-methyl-2-(3-((2-(trifluoromethyl)benzyl)oxy)-pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-80) as a white solid (60.0 mg, 85%): 1H NMR (400 MHz, DMSO-d6) δ 7.67−7.59 (m, 3H), 7.47−7.45 (m,1H), 6.90 (s, 1H), 4.64−4.58 (m, 2H), 4.28−4.23 (m, 1H), 3.66−3.46 (m, 4H), 2.25 (s, 3H), 2.05−1.98 (m, 2H); ESI MS m/z 382 [M + H]+; HPLC 98.1% (AUC), tR = 14.4 min.
(±)-2-(3-((2-(tert-Butyl)phenoxy)methyl)pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-83).
Compound (±)-83 was prepared from 2-(tert-butyl)phenol and (±)-tert-butyl 3-((tosyloxy)-methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.0 Hz, 1H), 7.17−7.12 (m, 2H), 6.91−6.82 (m, 2H), 4.06−4.00 (m, 3H), 3.93−3.82 (s, 1H), 3.63−3.51 (m, 2H), 2.93−2.90 (m, 1H), 2.44 (s, 3H), 2.31−2.28 (m, 1H), 2.00−1.98 (m, 1H), 1.38 (s, 9H); ESI MS m/z 370 [M + H]+; HPLC 96.4% (AUC), tR = 16.2 min.
(±)-2-(3-((2-Cyclopentylphenoxy)methyl)pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-84).
Compound (±)-84 was prepared from 2-cyclopentylphenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.16−7.07 (m, J = 8.0 Hz, 2H), 6.94 (s, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.83 (t, J = 7.2 Hz, 1H), 3.98 (d, J = 6.4 Hz, 2H), 3.76−3.63 (m, 2H), 3.53−3.39 (m, 2H), 3.22−3.15 (m, 1H), 2.77−2.71 (m, 1H), 2.32 (s, 3H), 2.17−2.11 (m, 1H), 1.90−1.83 (m, 3H), 1.68−1.66 (m, 2H), 1.61−1.44 (m, 4H); ESI MS m/z 382 [M + H]+; HPLC 95.2% (AUC), tR = 16.4 min.
(±)-2-(3-((2-Cyclohexylphenoxy)methyl)pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-85).
Compound (±)-85 was prepared from 2-cyclohexylphenol and (±)-tert-butyl 3-((tosyloxy)-methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) 1H NMR (400 MHz, CDCl3) δ 7.17−7.15 (m, 1H), 7.13−7.11 (m, 1H), 7.12 (s, 1H), 6.92−6.88 (m, 1H), 6.82 (d, J = 8.4 Hz, 1H), 4.01−3.93 (m, 2H), 3.87−3.80 (m, 2H), 3.69−3.48 (m, 2H), 2.89−2.84 (m, 2H), 2.43 (s, 3H), 2.26−2.15 (m, 1H), 1.99−1.96 (m, 1H), 1.83−1.69 (m, 5H), 1.41−1.22 (m, 5H); ESI MS m/z 396 [M + H]+; HPLC 98.3% (AUC), tR = 17.1 min.
(±)-2-(3-((3-Chloro-2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-86).
Compound (±)-86 was prepared from 3-chloro-2-(trifluoromethyl)-phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.56 (d, J = 8.4Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 6.94 (s, 1H), 4.19−4.10 (m, 2H), 3.76−3.63 (m, 2H), 3.51−3.44 (m, 1H), 3.33−3.32 (m, 1H), 2.77−2.70 (m, 1H), 2.32 (s, 3H), 2.15−2.07 (m, 1H), 1.87−1.79 (m, 1H); ESI MS m/z 416 [M + H]+; HPLC 99.0% (AUC), tR = 15.1 min.
(±)-2-(3-((4-Fluoro-2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-87).
Compound (±)-87 was prepared from 4-fluoro-2-(trifluoromethyl)-phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.48−7.44 (m, 2H), 7.28−7.25 (m, 1H), 6.92 (s, 1H), 4.12−4.08 (m, 2H), 3.72−3.68 (m, 1H), 3.64−3.60 (m, 1H), 3.47−3.42 (m, 1H), 3.33−3.29 (m, 1H), 2.71−2.68 (m, 1H), 2.32 (s, 3H), 2.10−2.06 (m, 1H), 1.83−1.78 (m, 1H); ESI MS m/z 400 [M + H]+; HPLC 98.7% (AUC), tR = 14.8 min.
(±)-2-(3-((5-Fluoro-2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-88).
Compound (±)-88 was prepared from 5-fluoro-2-(trifluoromethyl)-phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.66−7.62 (m, 1H), 7.20 (d, J = 11.2 Hz, 1H), 6.93 (s, 1H), 6.91−6.86 (m, 1H), 4.18−4.10 (m, 2H), 3.73−3.63 (m, 2H), 3.51−3.44 (m, 1H), 3.35−3.32 (m, 1H), 2.76−2.69 (m, 1H), 2.31 (s, 3H), 2.14−2.06 (m, 1H), 1.87−1.79 (m, 1H); ESI MS m/z 400 [M + H]+; HPLC 98.0% (AUC), tR = 14.7 min.
(±)-2-(3-((2-Fluoro-6-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-89).
Compound (±)-89 was prepared from 2-fluoro-6-(trifluoromethyl)-phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.64−7.60 (m, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.29−7.24 (m, 1H), 6.94 (s, 1H), 4.19−4.13 (m, 2H), 3.77−3.73 (m, 1H), 3.67−3.61 (m, 1H), 3.51−3.45 (m, 1H), 3.34−3.34 (m, 1H), 2.77−2.72 (m, 1H), 2.32 (s, 3H), 2.13−2.09 (m, 1H), 1.89−1.82 (m, 1H); ESI MS m/z 400 [M + H]+; HPLC 96.7% (AUC), tR = 14.8 min.
(±)-2-(3-((5-Methoxy-2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-90).
Compound (±)-90 was prepared from 5-methoxy-2-(trifluoromethyl)-phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.8 Hz, 1H), 6.94 (s, 1H), 6.75 (s, 1H), 6.58 (d, J = 8.4 Hz, 1H), 4.13−4.07 (m, 2H), 3.79 (s, 3H), 3.74−3.64 (m, 2H), 3.51−3.45(m, 1H), 3.36−3.31 (m, 1H), 2.73−2.70 (m, 1H), 2.31 (s, 3H), 2.12−2.08 (m, 1H), 1.87−1.80 (m, 1H); ESI MS m/z 412 [M + H]+; HPLC >99% (AUC), tR = 14.6 min.
(±)-2-(3-((3,5-Bis(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)-6-methylpyrimidine-4-carboxylic Acid ((±)-91).
Compound (±)-91 was prepared from 3,5-bis(trifluoromethyl)phenol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 7.61−7.59 (m, 3H), 6.94 (s, 1H), 4.23−4.14 (m, 2H), 3.76−3.71 (m, 1H), 3.69−3.63 (m, 1H), 3.53−3.47 (m, 1H), 3.42−3.38 (m, 1 H), 2.78−2.71 (m, 1H), 2.32 (s, 3H), 2.16−2.08 (m, 1H), 1.89−1.81 (m, 1H); ESI MS m/z 450 [M + H]+; HPLC 97.0% (AUC), tR = 16.0 min.
(±)-6-Methyl-2-(3-(((4-(trifluoromethyl)pyridin-3-yl)oxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-92).
Compound (±)-92 was prepared from 4-(trifluoromethyl)pyridin-3-ol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 8.37 (d, J = 4.8 Hz, 1H), 7.43 (d, J = 4.8 Hz, 1H), 7.13 (s, 1H), 4.21−4.20 (m, 2H), 3.87−3.78 (m, 2H), 3.65−3.64 (m, 1H), 3.54−3.49 (m, 1H), 2.93−2.86 (m, 1H), 2.44 (s, 3H), 2.29−2.24 (m, 1H), 2.02−1.97 (m, 1H); ESI MS m/z 383 [M + H]+; HPLC >99% (AUC), tR = 12.6 min.
(±)-6-Methyl-2-(3-(((2-(trifluoromethyl)pyridin-3-yl)oxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic Acid ((±)-93).
Compound (±)-93 was prepared from 2-(trifluoromethyl)pyridin-3-ol and (±)-tert-butyl 3-((tosyloxy)methyl)pyrrolidine-1-carboxylate ((±)-40) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 4.4 Hz, 1H), 7.45−7.42 (m, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.13 (s, 1H), 4.11−4.07 (m, 2H), 3.86−3.85 (m, 2H), 3.63−3.61 (m, 1H), 3.51−3.47 (m, 1H), 2.91−2.84 (m, 1H), 2.44 (s, 3H), 2.28−2.25 (m, 1H), 2.00−1.97 (m, 1H); ESI MS m/z 383 [M + H]+; HPLC 99.0% (AUC), tR = 12.7 min.
(±)-2-(3-((2-(Trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)-pyrimidine-4-carboxylic Acid ((±)-94).
Compound (±)-94 was prepared from methyl 2-chloropyrimidine-4-carboxylate and (±)-3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidine ((±)-42) according to a similar procedure described for the synthesis of (±)-44: 1H NMR (400 MHz, DMSO-d6) δ 8.50 (d, J = 4.8 Hz, 1H), 7.61−7.57 (m, 2H), 7.26 (d, J = 8.4 Hz, 1H), 7.06 (t, J = 7.6 Hz, 1H), 7.00 (d, J = 5.2 Hz, 1H), 4.15 (br, 2H), 3.77−3.63 (m, 2H), 3.53−3.47 (m, 1H), 3.37−3.33 (m, 1H), 2.79−2.72 (m, 1H), 2.17−2.09 (m, 1H), 1.90−1.83 (m, 1H); ESI MS m/z 368 [M + H]+; HPLC >99% (AUC), tR = 14.3 min.
(±)-4-Methyl-6-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)picolinic Acid ((±)-95).
Step A: To a mixture of (±)-3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidine ((±)-42, 0.200 g, 0.815 mmol), methyl 6-chloro-4-methylpicolinate (0.151 g, 0.815 mmol), and Cs2CO3 (0.796 g, 2.44 mmol) in N2-degassed anhydrous 1,4-dioxane (10 mL) were added XantPhos (0.153 g, 0.265 mmol) and Pd2(dba)3 (74.6 mg, 0.082 mmol). The mixture was heated at 80 °C in a sealed vessel for 16 h. The reaction mixture was allowed to cool to rt and then diluted with H2O (30 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with H2O (3 × 50 mL) and brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–50% EtOAc in hexanes) to give (±)-methyl 4-methyl-6-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)picolinate as an off-white solid (0.180 g, 56%): 1H NMR (400 MHz, CDCl3) δ7.54 (d, J = 7.6 Hz, 1H), 7.44 (t, J = 8.0 Hz, 1H), 7.23−7.20 (m, 1H), 7.00−6.92 (m, 2H), 6.34 (s, 1H), 4.08−3.99 (m, 2H), 3.89 (s, 3H), 3.76−3.72 (m, 1H), 3.69−3.63 (m, 1H), 3.55−3.49 (m, 1H), 3.40−3.36 (m, 1H), 2.89−2.82 (m, 1H), 2.27 (s, 3H), 2.25−2.18 (m, 1H), 2.00−1.93 (m, 1H); ESI MS m/z 395 [M + H]+.
Step B: To a solution of (±)-methyl 4-methyl-6-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)picolinate (0.190 g, 0.481 mmol) in CH3OH (6 mL), THF (6 mL), and H2O (3 mL) was added LiOH (0.115 g, 4.81 mmol), and the mixture was stirred at rt for 16 h. The mixture was concentrated under reduced pressure to remove the volatile solvents, and the resulting aqueous mixture was diluted with additional H2O (10 mL) and acidified with 2 N aqueous HCl to pH = 3.The acidified mixture was extracted with EtOAc (3 × 20 mL), and the combined organic extracts were washed with brine (20 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to give (±)-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)-pyrimidine-4-carboxylic acid ((±)-95) as a white solid (0.136 g, 77%):1H NMR (400 MHz, DMSO-d6) δ 7.57−7.54 (m, 2H), 7.15 (d, J = 8.4 Hz, 1H), 7.05−7.01 (m, 2H), 6.18 (s, 1H), 4.02−3.92 (m, 2H), 3.68−3.46 (m, 3H), 3.34−3.32 (m, 1H), 2.64−2.61 (m, 1H), 2.13 (s, 3H), 2.10−2.01 (m, 1H), 1.81−1.68 (m, 1H); ESI MS m/z 381 [M + H]+; HPLC >99% (AUC), tR = 12.8 min.
(±)-6-(3-((2-(Trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)-picolinic Acid ((±)-96).
Compound (±)-96 was prepared from methyl 6-chloropicolinate and (±)-3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidine ((±)-42) according to a similar procedure described for the synthesis of (±)-95: 1H NMR (400 MHz, CDCl3) δ 7.63−7.54 (m, 2H), 7.46−7.42 (m, 2H), 7.00−6.95 (m, 2H), 6.62−6.60 (m, 1H), 4.10−4.04 (m, 2H), 3.77−3.60 (m, 2H), 3.51−3.39 (m, 2H), 2.92−2.80 (m, 1H), 2.25−2.22 (m, 1H), 2.08−1.96 (m, 1H); ESI MS m/z367 [M + H]+; HPLC 95.8% (AUC), tR = 12.7 min.
(±)-2-(3-((2-(Trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)-nicotinic Acid ((±)-97).
Compound (±)-97 was prepared from methyl 2-chloronicotinate and (±)-3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidine ((±)-42) according to a similar procedure described for the synthesis of (±)-95: 1H NMR (400 MHz, CDCl3) δ 8.40 (d, J = 4.4 Hz, 1H), 8.23 (d, J = 7.6 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.47−7.43 (m, 1H), 7.00−6.91 (m, 3H), 4.10−4.02 (m, 2H), 3.61−3.37 (m, 4H), 2.93−2.86 (m, 1H), 2.29−2.21 (m, 1H), 1.98−1.89 (m, 1 H); ESI MS m/z 367 [M + H]+; HPLC 98.3% (AUC), tR = 12.0 min.
(±)-4-Fluoro-3-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)benzoic Acid ((±)-98).
Step A: To a mixture of (±)-3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidine ((±)-42, 0.300 g, 1.22 mmol) and methyl 3-bromo-4-fluorobenzoate (0.342 g, 1.47 mmol) in N2-degassed 1,4-dioxane were added Cs2CO3 (1.2 g, 3.66 mmol), XPhos (58.1 mg, 0.12 mmol), and Pd2(dba)3 (37.9 mg, 0.037 mmol). The mixture was stirred at 110 °C in a sealed vessel for 16 h and then allowed to cool to rt. The mixture was concentrated under reduced pressure, and the resulting residue was chromatographed over silica gel (0–40% EtOAc in hexanes) to give methyl 4-fluoro-3-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)benzoate as a white solid (0.198 mg, 41%): 1H NMR (400 MHz, CDCl3) δ 7.54 (d, J = 7.6Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.37−7.32 (m, 2H), 7.00−6.95 (m, 3H), 4.04 (d, J = 7.2 Hz, 2H), 3.85 (s, 3H), 3.64−3.59 (m, 1H), 3.52−3.46 (m, 2H), 3.40−3.35 (m, 1H), 2.85−2.81 (m, 1H), 2.22−2.17 (m, 1H), 1.93−1.88 (m, 1H); ESI MS m/z 398 [M + H]+.
Step B: To a solution of methyl 4-fluoro-3-(3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidin-1-yl)benzoate (0.120 g, 0.63 mmol) in a mixture of CH3OH (4 mL), THF (4 mL), and H2O (2 mL) was added LiOH (0.144 g, 6.04 mmol). The mixture was stirred at rt for 16 h and was concentrated under reduced pressure to remove the volatile solvents. The resulting aqueous layer was diluted with H2O (50 mL) and acidified with 2 N aqueous HCl to pH = 3. The aqueous mixture was extracted with EtOAc (3 × 50 mL), and the combined organic extracts were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure to give 4-fluoro-3-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)benzoic acid ((±)-98) as a white solid (78.0 mg, 67%): δ 7.59−7.55 (d, J = 7.6 Hz, 2H), 7.24−7.20 (m, 3H), 7.13−7.02 (m, 2H), 4.13−4.07 (m, 2H), 3.52−3.48 (m, 1H), 3.39 (br, 2H), 3.29−3.25 (m, 1H), 2.73−2.70 (m, 1H), 2.11−2.08 (m, 1H), 1.81−1.76 (m, 1H); ESI MS m/z 384 [M + H]+; HPLC 98.5% (AUC), tR = 16.1 min.
(±)-6-Methyl-N-(methylsulfonyl)-2-(3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-99).
Step A: To a mixture of 6-methyl-2-(3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-44, 50.0 mg, 0.131 mmol), HBTU (74.5 mg, 0.197 mmol), and i-Pr2NEt (0.08 mL, 0.393 mmol) in DMF (4 mL) was added methane sulfonamide (19.1 mg, 0.197 mmol). The resulting solution was stirred at rt for 18 h under an atmosphere of N2. The mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with H2O (3 × 20 mL) and brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude residue was chromatographed over silica gel (0–80% EtOAc in hexanes) to give 6-methyl-N-(methylsulfonyl)-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-99) as a white solid (30.0 mg, 50%): 1H NMR (400 MHz, acetone-d6) δ 10.36 (br, 1H), 7.61−7.57 (m, 2H), 7.27−7.25 (m, 1H), 7.10−7.06 (m, 2H), 4.23 (br, 2H), 3.92−3.82 (m, 2H), 3.65−3.49 (m, 2H), 3.34 (s, 3H), 2.92−2.82 (m, 2H), 2.42 (s, 3H), 2.25−2.22 (m, 1H); ESI MS m/z 459 [M + H]+; HPLC 98.4% (AUC), tR = 15.9 min.
(±)-6-Methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-100).
Compound (±)-100 was prepared from NH4Cl and 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-44) according to a similar procedure described for the synthesis of (±)-99: 1H NMR (400 MHz, CDCl3) δ 7.71 (br, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.14 (s, 1H), 7.00−6.94 (m, 2H), 5.57 (br, 1H), 4.06−4.02 (m, 2H), 3.88−3.83 (m, 1H), 3.79−3.73 (m, 1H), 3.63−3.57 (m, 1H), 3.51−3.47 (m, 1H), 2.89−2.84 (m, 1H), 2.40 (s, 3H), 2.26−2.19 (m, 1H), 1.99−1.94 (m, 1H); ESI MS m/z 381 [M + H]+; HPLC >99% (AUC), tR = 14.3 min.
(±)-N,6-Dimethyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-101).
Step A: To a solution of 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-44, 50.0 mg, 0.131 mmol), T3P (50% w/w in CH2Cl2) (83.4 mg, 0.262 mmol), and i-Pr2NEt (0.2 mL, 1.05 mmol) in CH2Cl2 (3 mL) was added methyl amine hydrochloride (44.0 mg, 0.393 mmol). The mixture stirred at an ambient temperature for 18 h and was then concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–60% EtOAc in hexane) to give N,6-dimethyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-101) as a white solid (40.0 mg, 77%): 1H NMR (400 MHz, acetone-d6) δ 8.25 (br, 1H), 7.62−7.59 (m, 2H), 7.26 (d, J = 8.0Hz, 1H), 7.08−7.05 (m, 2H), 4.23−4.19 (m, 2H), 3.88−3.83 (m, 1H), 3.79−3.74 (m, 1H), 3.60−3.58 (m, 1H), 3.50−3.48 (m, 1H), 2.88 (s, 3H), 2.87−2.82 (m, 1H), 2.36 (s, 3H), 2.22−2.19 (m, 1H), 2.02−2.01 (m, 1H); ESI MS m/z 395 [M + H]+; HPLC >99% (AUC), tR = 14.7 min.
(±)-N-Cyclopropyl-6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)-methyl)pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-102).
Compound (±)-102 was prepared from cyclopropylamine and 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine-4-carboxylic acid ((±)-44) according to a similar procedure described for the synthesis of (±)-100: 1H NMR (400 MHz, acetone-d6) δ 8.17 (br, 1H), 7.62−7.57 (m, 2H), 7.26 (d, J = 8.4 Hz, 1H), 7.10−7.04 (m, 2H), 4.21 (br, 2H), 3.87−3.82 (m, 1H), 3.76−3.72 (m, 1H), 3.58−3.55 (m, 1H), 3.49−3.44 (m, 1H), 2.88−2.78 (m, 2H), 2.36 (s, 3H), 2.23−2.13 (m, 1H), 2.02−1.95 (m, 1H), 0.75−0.73 (m, 2H), 0.58 (br, 2H); ESI MS m/z 421 [M + H]+; HPLC >99% (AUC), tR = 15.3 min.
(±)-4-Methyl-6-(2H-tetrazol-5-yl)-2-(3-((2-(trifluoromethyl)-phenoxy)methyl)pyrrolidin-1-yl)pyrimidine ((±)-103).
Step A: A mixture of 6-methyl-2-(3-((2-(trifluoromethyl)phenoxy)methyl)-pyrrolidin-1-yl)pyrimidine-4-carboxamide ((±)-100, 0.200 g, 0.526 mmol), NaN3 (0.142 g, 0.375 mmol), and tetrachlorosilane (98.5 mg, 0.579 mmol) in CH3CN (4 mL) was stirred at 80 °C for 18 h in a sealed vessel. The reaction mixture was allowed to cool to rt and diluted with saturated NaHCO3 (5 mL). The aqueous mixture was extracted with CHCl3 (3 × 50 mL), and the combined organic extracts were washed with brine (50 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was chromatographed over silica gel (0–10% CH3OH in CH2Cl2) to give 4-methyl-6-(2H-tetrazol-5-yl)-2-(3-((2-(trifluoromethyl)phenoxy)methyl)pyrrolidin-1-yl)pyrimidine ((±)-103) as a white solid (66.0 mg, 30%): 1H NMR (400 MHz, acetone-d6) δ 7.62−7.58 (m, 2H), 7.28−7.25 (m, 2H), 7.08 (t, J = 7.6 Hz, 1H), 4.28−4.21 (m, 3H), 3.91−3.86 (m, 1H), 3.80−3.74 (m, 1H), 3.653.58 (m, 1H), 3.52−3.47 (m, 1H), 2.92−2.85 (m, 1H), 2.42 (s, 3H), 2.27−2.21 (m, 1H); ESI MS m/z 406 [M + H]+; HPLC 97.4% (AUC), tR = 14.6 min.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Eye Institute of the National Institutes of Health under award number R01EY028549. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This project has been funded in whole or in part with Federal funds from the National Eye Institute, the National Institutes of Health, and the Department of Health and Human Services, NIH Grant R01EY028549 (to K.P. and C.L.C.). This study was supported by NIH Grant P30 EY019007 (Core Support for Vision Research) and unrestricted funds from the Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University. The authors thank The Burch Family Foundation, the Mary Jaharis-John Catsimatidis Scholarship Fund, the Kaplen Foundation, and the Eye Surgery Fund for gifts supporting this study.
ABBREVIATIONS
- Aβ
β-amyloid
- Abca4
ATP-binding cassette, subfamily A (ABC1), member 4
- ADME
absorption, distribution, metabolism, elimination
- AMD
age-related macular degeneration
- aq
aqueous
- Arg
arginine
- ATTR
transthyretin amyloidosis
- ATTR-CM
transthyretin amyloidosis cardiomyopathy
- ATTR-PN
transthyretin amyloidosis polyneuropathy
- AUC
area under the curve
- A2E
N-retinide-N-retinylidene ethanolamine
- Boc2O
di-tert-butyl dicarbonate
- CH2Cl2
dichloromethane
- CH3CN
acetonitrile
- CH3OH
methyl alcohol
- CL
clearance
- CLint
intrinsic clearance
- Cs2CO3
cesium carbonate
- CF3
trifluoromethyl
- CSF
cerebral spinal fluid
- CYP
cytochrome P450
- CYP2C9
cytochrome P450 2C9
- CYP2C19
cytochrome P450 2C19
- CYP2D6
cytochrome P450 2D6
- CYP3A4
cytochrome P450 3A4
- DMAP
1,4-dimethylaminopyridine
- DME
dimethoxyethane
- DMF
N,N-dimethylformamide
- Et3N
triethylamine
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- EtOH
ethyl alcohol
- %F
% oral bioavailability
- FITC
fluorescein isothiocyanate
- FP
fluorescence polarization assay
- Gln
glutamine
- Glu
glutamic acid
- Gly
glycine
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- HBP
halogen binding pocket
- HBTU
(2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HCl
hydrochloric acid
- hERG
human ether-a-go-go channel
- HLM
human liver microsomes
- HOAc
acetic acid
- HTRF
homogeneous time-resolved fluorescence assay
- HRMS
high-resolution mass spectrometry
- i-Pr2NEt
N,N-diisopropylethylamine
- IV
intravenous
- LiOH
lithium hydroxide
- Leu
leucine
- Lys
lysine
- Met
methionine
- MLM
mouse liver microsomes
- NAFLD
nonalcoholic fatty liver disease
- NaBH4
sodium borohydride
- NaBH(OAc)3
sodium triacetoxyborohydride
- NaN3
sodium azide
- NH4Cl
ammonium chloride
- PBR
phosphate-buffered saline
- PD
pharmacodynamics
- PDB
Protein Data Bank
- Pd2(dba)3
Tris(dibenzylideneacetone)-dipalladium(0)
- Phe
phenylalanine
- PK
pharmacokinetics
- PO
oral
- PPARγ
nuclear peroxisome proliferator-activated receptor-γ
- %PPB
% plasma protein binding
- RBP4
retinol binding protein-4
- RLM
rat liver microsomes
- RPE
retinal pigment epithelium
- SAR
structure–activity relationship
- Ser
serine
- SN2
bimolecular nucleophilic displacement
- SPA
scintillation proximity assay
- SSA
systemic senile amyloidosis
- STD-NMR
saturation transfer difference-NMR
- TBG
thyroxine binding globulin
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Thr
threonine
- TTR
transthyretin
- TsCl
tosyl chloride
- Tyr
tyrosine
- T3P
propanephosphonic acid anhydride
- T4
thyroxine
- Val
valine
- Vss
volume of distribution at steady state
- XantPhos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
- XPhos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00996.
RBP4 in vitro assay protocols, TTR in vitro assay protocols, mouse PK study protocols, serum RBP4 collection and measurement protocols, in vitro ADME assay protocols, general chemistry information, and spectroscopic and analytical data for compound (±)-44 (1H NMR, 13C NMR, 19F NMR, MS, and HPLC) (PDF) Molecular formula strings for biologically tested compounds (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00996
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Steinmetz AC; Renaud JP; Moras D Binding of ligands and activation of transcription by nuclear receptors. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 329–359. [DOI] [PubMed] [Google Scholar]; (b) Clagett-Dame M; DeLuca HF The role of vitamin A in mammalian reproduction and embryonic development. Annu. Rev. Nutr 2002, 22, 347–381. [DOI] [PubMed] [Google Scholar]; (c) Wolf G Multiple functions of vitamin A. Physiol. Rev 1984, 64, 873–937. [DOI] [PubMed] [Google Scholar]
- (2).(a) Kiser PD; Golczak M; Palczewski K Chemistry of the retinoid (visual) cycle. Chem. Rev 2014, 114, 194–232. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tsin A; Betts-Obregon B; Grigsby J Visual cycle proteins: structure, function, and roles in human retinal disease. J. Biol. Chem 2018, 293, 13016–13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Kanai M; Raz A; Goodman DS Retinol-binding protein: the transport protein for vitamin A in human plasma. J. Clin. Invest 1968, 47, 2025–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schlehuber S; Skerra A Lipocalins in drug discovery: from natural ligand-binding proteins to ″anticalins″. Drug Discovery Today 2005, 10, 23–33. [DOI] [PubMed] [Google Scholar]
- (4).(a) Bobbert T; Raila J; Schwarz F; Mai K; Henze A; Pfeiffer AFH; Schweigert FJ; Spranger J Relation between retinol, retinol-binding protein 4, transthyretin and carotid intima media thickness. Atherosclerosis 2010, 213, 549–551. [DOI] [PubMed] [Google Scholar]; (b) Kawaguchi R; Zhong M; Kassai M; Ter-Stepanian M; Sun H Vitamin A transport mechanism of the multitransmembrane cell-surface receptor STRA6. Membranes 2015, 5, 425–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Blake CC; Geisow MJ; Oatley SJ; Rerat B; Rerat C Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 Å. J. Mol. Biol 1978, 121, 339–356. [DOI] [PubMed] [Google Scholar]; (b) Nilsson SF; Rask L; Peterson PA Studies on thyroid hormone-binding proteins. II. Binding of thyroid hormones, retinol-binding protein, and fluorescent probes to prealbumin and effects of thyroxine on prealbumin subunit self-association. J. Biol. Chem 1975, 250, 8554–8563. [PubMed] [Google Scholar]; (c) Wojtczak A; Cody V; Luft JR; Pangborn W Structures of human transthyretin complexed with thyroxine at 2.0 Å resolution and 3′,5′-dinitro-N-acetyl-L-thyronine at 2.2 Å resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr 1996, 52, 758–765. [DOI] [PubMed] [Google Scholar]; (d) Hamilton JA; Benson MD Transthyretin: a review from a structural perspective. Cell. Mol. Life Sci 2001, 58, 1491–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Monaco HL; Rizzi M; Coda A Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [DOI] [PubMed] [Google Scholar]
- (6).(a) Johnson SM; Wiseman RL; Sekijima Y; Green NS; Adamski-Werner SL; Kelly JW Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc. Chem. Res 2005, 38, 911–921. [DOI] [PubMed] [Google Scholar]; (b) Foss TR; Wiseman RL; Kelly JW The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry 2005, 44, 15525–15533. [DOI] [PubMed] [Google Scholar]
- (7).(a) Newcomer ME; Jones TA; Aqvist J; Sundelin J; Eriksson U; Rask L; Peterson PA The three-dimensional structure of retinol-binding protein. EMBO J 1984, 3, 1451–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cowan SW; Newcomer ME; Jones TA Crystallographic refinement of human serum retinol binding protein at 2 Å resolution. Proteins 1990, 8, 44–61. [DOI] [PubMed] [Google Scholar]
- (8).Motani A; Wang Z; Conn M; Siegler K; Zhang Y; Liu Q; Johnstone S; Xu H; Thibault S; Wang Y; Fan P; Connors R; Le H; Xu G; Walker N; Shan B; Coward P Identification and characterization of a non-retinoid ligand for retinol-binding protein 4 which lowers serum retinol-binding protein 4 levels in vivo. J. Biol. Chem 2009, 284, 7673–7680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Zanotti G; Folli C; Cendron L; Alfieri B; Nishida SK; Gliubich F; Pasquato N; Negro A; Berni R Structural and mutational analyses of protein–protein interactions between transthyretin and retinol-binding protein. FEBS J 2008, 275, 5841–5854. [DOI] [PubMed] [Google Scholar]; (b) Noy N; Slosberg E; Scarlata S Interactions of retinol with binding proteins: studies with retinol-binding protein and with transthyretin. Biochemistry 1992, 31, 11118–11124. [DOI] [PubMed] [Google Scholar]; (c) Noy N; Xu ZJ Interactions of retinol with binding proteins: implications for the mechanism of uptake by cells. Biochemistry 1990, 29, 3878–3883. [DOI] [PubMed] [Google Scholar]; (d) Monaco HL The transthyretin-retinol-binding protein complex. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 2000, 1482, 65–72. [DOI] [PubMed] [Google Scholar]; (e) Monaco HL Three-dimensional structure of the transthyretinretinol-binding protein complex. Clin. Chem. Lab. Med 2002, 40, 1229–1236. [DOI] [PubMed] [Google Scholar]; (f) Monaco HL; Rizzi M; Coda A Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science 1995, 268, 1039–1041. [DOI] [PubMed] [Google Scholar]
- (10).Naylor HM; Newcomer ME The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry 1999, 38, 2647–2653. [DOI] [PubMed] [Google Scholar]
- (11).(a) Radu RA; Han Y; Bui TV; Nusinowitz S; Bok D; Lichter J; Widder K; Travis GH; Mata NL Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest. Ophthalmol. Visual Sci 2005, 46, 4393–4401. [DOI] [PubMed] [Google Scholar]; (b) Palczewski K Retinoids for treatment of retinal diseases. Trends Pharmacol. Sci 2010, 31, 284–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Petrukhin K Pharmacological inhibition of lipofuscin accumulation in the retina as a therapeutic strategy for dry AMD treatment. Drug Discovery Today: Ther. Strategies 2013, 10, e11–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Petrukhin K New therapeutic targets in atrophic age-related macular degeneration. Expert Opin. Ther. Targets 2007, 11, 625–639. [DOI] [PubMed] [Google Scholar]
- (13).Dobri N; Qin Q; Kong J; Yamamoto K; Liu Z; Moiseyev G; Ma J-X; Allikmets R; Sparrow JR; Petrukhin K A1120, a nonretinoid RBP4 antagonist, inhibits formation of cytotoxic bisretinoids in the animal model of enhanced retinal lipofuscinogenesis. Invest. Ophthalmol. Visual Sci 2013, 54, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Racz B; Varadi A; Kong J; Allikmets R; Pearson PG; Johnson G; Cioffi CL; Petrukhin K A non-retinoid antagonist of retinol-binding protein 4 rescues phenotype in a model of Stargardt disease without inhibiting the visual cycle. J. Biol. Chem 2018, 293, 11574–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Young RW Pathophysiology of age-related macular degeneration. Surv. Ophthalmol 1987, 31, 291–306. [DOI] [PubMed] [Google Scholar]; (b) Dorey CK; Wu G; Ebenstein D; Garsd A; Weiter JJ Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest. Ophthalmol. Visual Sci 1989, 30, 1691–1699. [PubMed] [Google Scholar]; (c) Holz FG; Bellman C; Staudt S; Schutt F; Volcker HE Fundus autofluorescence and development of geographic atrophy in age-related macular degeneration. Invest. Ophthalmol. Visual Sci 2001, 42, 1051–1056. [PubMed] [Google Scholar]; (d) Holz FG; Bellmann C; Margaritidis M; Schutt F; Otto TP; Volcker HE Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol 1999, 237, 145–152. [DOI] [PubMed] [Google Scholar]; (e) Holz FG; Bindewald-Wittich A; Fleckenstein M; Dreyhaupt J; Scholl HP; Schmitz-Valckenberg S Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am. J. Ophthalmol 2007, 143, 463–472. [DOI] [PubMed] [Google Scholar]; (f) Schmitz-Valckenberg S; Fleckenstein M; Scholl HP; Holz FG Fundus autofluorescence and progression of age-related macular degeneration. Surv. Ophthalmol 2009, 54, 96–117. [DOI] [PubMed] [Google Scholar]; (g) Finnemann SC; Leung LW; Rodriguez-Boulan E The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 3842–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Suter M; Reme C; Grimm C; Wenzel A; Jaattela M; Esser P; Kociok N; Leist M; Richter C Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J. Biol. Chem 2000, 275, 39625–39630. [DOI] [PubMed] [Google Scholar]; (i) Sparrow JR; Fishkin N; Zhou J; Cai B; Jang YP; Krane S; Itagaki Y; Nakanishi K A2E, a byproduct of the visual cycle. Vision Res 2003, 43, 2983–2990. [DOI] [PubMed] [Google Scholar]; (j) Sparrow JR; Gregory-Roberts E; Yamamoto K; Blonska A; Ghosh SK; Ueda K; Zhou J The bisretinoids of retinal pigment epithelium. Prog. Retinal Eye Res 2012, 31, 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Delori FC RPE Lipofuscin in Ageing and Age-Related Macular Degeneration. In Retinal Pigment Epithelium and Macular Disease (Documenta Ophthalmologica); Coscas G; Piccolino FC, Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Vol. 62, pp 37–45. [Google Scholar]
- (16).(a) Weng J; Mata NL; Azarian SM; Tzekov RT; Birch DG; Travis GH Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell 1999, 98, 13–23. [DOI] [PubMed] [Google Scholar]; (b) Sparrow JR; Cai B Blue light-induced apoptosis of A2E-containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest. Ophthalmol. Visual Sci 2001, 42, 1356–1362. [PubMed] [Google Scholar]; (c) Bergmann M; Schutt F; Holz FG; Kopitz J Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J 2004, 18, 562–564. [DOI] [PubMed] [Google Scholar]; (d) Sparrow JR; Parish CA; Hashimoto M; Nakanishi K A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest. Ophthalmol. Visual Sci 1999, 40, 2988–2995. [PubMed] [Google Scholar]; (e) De S; Sakmar TP Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol 2002, 120, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Vives-Bauza C; Anand M; Shirazi AK; Magrane J; Gao J; Vollmer-Snarr HR; Manfredi G; Finnemann SC The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem 2008, 283, 24770–24780. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhou J; Jang YP; Kim SR; Sparrow JR Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. U.S.A 2006, 103, 16182–16187. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Radu RA; Hu J; Yuan Q; Welch DL; Makshanoff J; Lloyd M; McMullen S; Travis GH; Bok D Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem 2011, 286, 18593–18601. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ben-Shabat S; Parish CA; Vollmer HR; Itagaki Y; Fishkin N; Nakanishi K; Sparrow JR Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J. Biol. Chem 2002, 277, 7183–7190. [DOI] [PubMed] [Google Scholar]; (j) Rózanowska M; Jarvis-Evans J; Korytowski W; Boulton ME; Burke JM; Sarna T Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J. Biol. Chem 1995, 270, 18825–18830. [DOI] [PubMed] [Google Scholar]; (k) Sparrow JR; Zhou J; Ben-Shabat S; Vollmer H; Itagaki Y; Nakanishi K Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Invest. Ophthalmol. Visual Sci 2002, 43, 1222–1227. [PubMed] [Google Scholar]; (l) Dontsov AE; Sakina NL; Golubkov AM; Ostrovsky MA Light-induced release of A2E photooxidation toxic products from lipofuscin granules of human retinal pigment epithelium. Dokl. Biochem. Biophys 2009, 425, 98–101. [DOI] [PubMed] [Google Scholar]
- (17).(a) Berni R; Formelli F In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBR Lett 1992, 308, 43–45. [DOI] [PubMed] [Google Scholar]; (b) Adams WR; Smith JE; Green MH Effects of N-(4-hydroxyphenyl)retinamide on vitamin A metabolism in rats. Exp. Biol. Med 1995, 208, 178–185. [DOI] [PubMed] [Google Scholar]; (c) Mata NL; Lichter JB; Vogel R; Han Y; Bui TV; Singerman LJ Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina 2013, 33, 498–507. [DOI] [PubMed] [Google Scholar]
- (18).Cioffi CL; Dobri N; Freeman EE; Conlon MP; Chen P; Stafford DG; Schwarz DMC; Golden KC; Zhu L; Kitchen DB; Barnes KD; Racz B; Qin Q; Michelotti E; Cywin CL; Martin WH; Pearson PG; Johnson G; Petrukhin K Design, synthesis, and evaluation of nonretinoid retinol binding protein 4 antagonists for the potential treatment of atrophic age-related macular degeneration and Stargardt disease. J. Med. Chem 2014, 57, 7731–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cioffi CL; Racz B; Freeman EE; Conlon MP; Chen P; Stafford DG; Schwarz DM; Zhu L; Kitchen DB; Barnes KD; Dobri N; Michelotti E; Cywin CL; Martin WH; Pearson PG; Johnson G; Petrukhin K Bicyclic [3.3.0]-octahydrocyclopenta[c]-pyrrolo antagonists of retinol binding protein 4: potential treatment of atrophic age-related macular degeneration and Stargardt disease. J. Med. Chem 2015, 58, 5863–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Racz B; Varadi A; Pearson PG; Petrukhin K Comparative pharmacokinetics and pharmacodynamics of the advanced retinol-binding protein 4 antagonist in dog and cynomolgus monkey. PLoS One 2020, 15, No. e0228291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Graham TE; Yang Q; Bluher M; Hammarstedt A; Ciaraldi TP; Henry RR; Wason CJ; Oberbach A; Jansson PA; Smith U; Kahn BB Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med 2006, 354, 2552–2563. [DOI] [PubMed] [Google Scholar]; (b) Yang Q; Graham TE; Mody N; Preitner F; Peroni OD; Zabolotny JM; Kotani K; Quadro L; Kahn BB Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [DOI] [PubMed] [Google Scholar]
- (22).Aeberli I; Biebinger R; Lehmann R; L’Allemand D; Spinas GA; Zimmermann MB Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J. Clin. Endocrinol. Metab 2007, 92, 4359–4365. [DOI] [PubMed] [Google Scholar]
- (23).Kowalska I; Straczkowski M; Adamska A; Nikolajuk A; Karczewska-Kupczewska M; Otziomek E; Gorska M Serum retinol binding protein 4 is related to insulin resistance and nonoxidative glucose metabolism in lean and obese women with normal glucose tolerance. J. Clin. Endocrinol. Metab 2008, 93, 2786–2789. [DOI] [PubMed] [Google Scholar]
- (24).(a) Ingelsson E; Sundstrom J; Melhus H; Michaelsson K; Berne C; Vasan RS; Riserus U; Blomhoff R; Lind L; Arnlov J Circulating retinol-binding protein 4, cardiovascular risk factors and prevalent cardiovascular disease in elderly. Atherosclerosis 2009, 206, 239–244. [DOI] [PubMed] [Google Scholar]; (b) Qi Q; Yu Z; Ye X; Zhao F; Huang P; Hu FB; Franco OH; Wang J; Li H; Liu Y; Lin X Elevated retinol-binding protein 4 levels are associated with metabolic syndrome in Chinese people. J. Clin. Endocrinol. Metab 2007, 92, 4827–4834. [DOI] [PubMed] [Google Scholar]; (c) Norseen J; Hosooka T; Hammarstedt A; Yore MM; Kant S; Aryal P; Kiernan UA; Phillips DA; Maruyama H; Kraus BJ; Usheva A; Davis RJ; Smith U; Kahn BB Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol 2012, 32, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lee SA; Yuen JJ; Jiang H; Kahn BB; Blaner WS Adipocyte-specific overexpression of retinol-binding protein 4 causes hepatic steatosis in mice. Hepatology 2016, 64, 1534–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cioffi CL; Racz B; Varadi A; Freeman EE; Conlon MP; Chen P; Zhu L; Kitchen DB; Barnes KD; Martin WH; Pearson PG; Johnson G; Blaner WS; Petrukhin K Design, synthesis, and preclinical efficacy of novel nonretinoid antagonists of retinol-binding protein 4 in the mouse model of hepatic steatosis. J. Med. Chem 2019, 62, 5470–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).(a) Falk RH; Comenzo RL; Skinner M The systemic amyloidoses. N. Engl. J. Med 1997, 337, 898–909. [DOI] [PubMed] [Google Scholar]; (b) Brunjes DL; Castano A; Clemons A; Rubin J; Maurer MS Transthyretin cardiac amyloidosis in older Americans. J. Card. Failure 2016, 22, 996–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ton VK; Mukherjee M; Judge DP Transthyretin cardiac amyloidosis: pathogenesis, treatments, and emerging role in heart failure with preserved ejection fraction. Clin. Med. Insights: Cardiol 2014, 8s1, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).White JT; Kelly JW Support for the multigenic hypothesis of amyloidosis: the binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 13019–13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hyung SJ; Deroo S; Robinson CV Retinol and retinol-binding protein stabilize transthyretin via formation of retinol transport complex. ACS. Chem. Biol 2010, 5, 1137–1146. [DOI] [PubMed] [Google Scholar]
- (30).(a) Ruberg FL; Berk JL Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Connors LH; Doros G; Sam F; Badiee A; Seldin DC; Skinner M Clinical features and survival in senile systemic amyloidosis: comparison to familial transthyretin cardiomyopathy. Amyloid 2011, 18, 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Westermark P; Bergstrom J; Solomon A; Murphy C; Sletten K Transthyretin-derived senile systemic amyloidosis: clinicopathologic and structural considerations. Amyloid 2003, 10, 48–54. [PubMed] [Google Scholar]
- (31).(a) Buxbaum JN; Ruberg FL Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet. Med 2017, 19, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alexander KM; Falk RH V122I T. T. R. cardiac amyloidosis in patients of African descent: recognizing a missed disease or the dog that didn’t bark? Circ.: Heart Failure 2016, 9, No. e003489. [DOI] [PubMed] [Google Scholar]
- (32).Sekijima Y; Hammarström P; Matsumura M; Shimizu Y; Iwata M; Tokuda T; Ikeda S-I; Kelly JW Energetic Characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology Lab. Lab. Invest 2003, 83, 409–417. [DOI] [PubMed] [Google Scholar]
- (33).(a) Kerschen P; Plante-Bordeneuve V Current and future treatment approaches in transthyretin familial amyloid polyneuropathy. Curr. Treat. Options Neurol 2016, 18, No. 53. [DOI] [PubMed] [Google Scholar]; (b) Almeida MR; Gales L; Damas AM; Cardoso I; Saraiva MJ Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr. Drug Targets: CNS Neurol. Disord 2005, 4, 587–596. [DOI] [PubMed] [Google Scholar]; (c) Almeida MR; Macedo B; Cardoso I; Alves I; Valencia G; Arsequell G; Planas A; Saraiva MJ Selective binding to transthyretin and tetramer stabilization in serum from patients with familial amyloidotic polyneuropathy by an iodinated diflunisal derivative. Biochem. J 2004, 381, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Johnson SM; Wiseman RL; Sekijima Y; Green NS; Adamski-Werner SL; Kelly JW Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidosis. Acc. Chem. Res 2005, 38, 911–921. [DOI] [PubMed] [Google Scholar]; (e) Nencetti S; Orlandini E TTR fibril formation inhibitors: is there a SAR? Curr. Med. Chem 2012, 19, 2356–2379. [DOI] [PubMed] [Google Scholar]; (f) Adams D; Cauquil C; Labeyrie C; Beaudonnet G; Algalarrondo V; Theaudin M TTR kinetic stabilizers and TTR gene silencing: a new era in therapy for familial amyloidotic polyneuropathies. Expert Opin. Pharmacother 2016, 17, 791–802. [DOI] [PubMed] [Google Scholar]
- (34).(a) Coelho T; Merlini G; Bulawa CE; Fleming JA; Judge DP; Kelly JW; Maurer MS; Plante-Bordeneuve V; Labaudinie re R; Mundayat R; Riley S; Lombardo I; Huertas P Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol. Ther 2016, 5, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nencetti S; Rossello A; Orlandini E Tafamidis (Vyndaqel): A light for FAP patients. ChemMedChem 2013, 8, 1617–1619. [DOI] [PubMed] [Google Scholar]; (c) Lamb YN; Deeks ED Tafamidis: A review in transthyretin amyloidosis with polyneuropathy. Drugs 2019, 79, 863–874. [DOI] [PubMed] [Google Scholar]; (d) Bulawa CE; Connelly S; Devit M; Wang L; Weigel C; Fleming JA; Packman J; Powers ET; Wiseman RL; Foss TR; Wilson IA; Kelly JW; Labaudiniere R Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).(a) Penchala SC; Connelly S; Wang Y; Park MS; Zhao L; Baranczak A; Rappley I; Vogel H; Liedtke M; Witteles RM; Powers ET; Reixach N; Chan WK; Wilson IA; Kelly JW; Graef IA; Alhamadsheh MM AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 9992–9997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miller M; Pal A; Albusairi W; Joo H; Pappas B; Haque Tuhin MT; Liang D; Jampala R; Liu F; Khan J; Faaij M; Park M; Chan W; Graef I; Zamboni R; Kumar N; Fox J; Sinha U; Alhamadsheh M Enthalpy-driven stabilization of transthyretin by AG10 mimics a naturally occurring genetic variant that protects from transthyretin amyloidosis. J. Med. Chem 2018, 61, 7862–7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Judge DP; Heitner SB; Falk RH; Maurer MS; Shah SJ; Witteles RM; Grogan M; Selby VN; Jacoby D; Hanna M; Nativi-Nicolau J; Patel J; Rao S; Sinha U; Turtle CW; Fox JC Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J. Am. Coll. Cardiol 2019, 74, 285–295. [DOI] [PubMed] [Google Scholar]
- (37).Berk JL; Suhr OB; Sekijima Y; Yamashita T; Heneghan M; Zeldenrust SR; Ando Y; Ikeda S; Gorevic P; Merlini G; Kelly JW; Skinner M; Bisbee AB; Dyck PJ; Obici L Familial amyloidosis consortium. The diflunisal trial: study accrual and drug tolerance. Amyloid 2012, 19, 37–38. [DOI] [PubMed] [Google Scholar]
- (38).Sant’Anna R; Gallego P; Robinson LZ; Pereira-Henriques A; Ferreira N; Pinheiro F; Esperante S; Pallares I; Huertas O; Almeida MR; Reixach N; Insa R; Velazquez-Campoy A; Reverter D; Reig N; Ventura S Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun 2016, 7, No. 10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gimeno A; Santos LM; Alemi M; Rivas J; Blasi D; Cotrina EY; Llop J; Valencia G; Cardoso I; Quintana J; Arsequell G; Jiménez-Barbero J Insights on the interaction between transthyretin and Aβ in solution. A saturation transfer difference (STD) NMR analysis of the role of iododiflunisal. J. Med. Chem 2017, 60, 5749–5758. [DOI] [PubMed] [Google Scholar]
- (40).Gales L; Macedo-Ribeiro S; Arsequell G; Valencia G; Saraiva M; Damas AM Human transthyretin in complex with iododiflunisal: structural features associated with a potent amyloid inhibitor. Biochem. J 2005, 388, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Rodrigues DA Drug–Drug Interactions, 2nd ed.; CRC Press: New York, 2008. [Google Scholar]
- (42).Ahmadian M; Suh JM; Hah N; Liddle C; Atkins AR; Downes M; Evans RM PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med 2013, 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Palaninathan SK; Mohamedmohaideen NN; Orlandini E; Ortore G; Nencetti S; Lapucci A; Rossello A; Freundlich JS; Sacchettini JC Novel transthyretin amyloid fibril formation inhibitors: synthesis, biological evaluation, and X-ray structural analysis. PLoS One 2009, 4, No. e6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Petrassi HM; Klabunde T; Sacchettini J; Kelly JW Structure-based design of N-phenyl phenoxazine transthyretin amyloid fibril inhibitors. J. Am. Chem. Soc 2000, 122, 2178–2192. [Google Scholar]
- (45).Petrassi HM; Johnson SM; Purkey HE; Chiang KP; Walkup T; Jiang X; Powers ET; Kelly JW Potent and selective structure-based dibenzofuran inhibitors of transthyretin amyloido-genesis: kinetic stabilization of the native state. J. Am. Chem. Soc 2005, 127, 6662–6671. [DOI] [PubMed] [Google Scholar]
- (46).Green NS; Foss TR; Kelly JW Genistein, a natural product from soy, is a potent inhibitor of transthyretin amyloidosis. Proc. Nat. Acad. Sci. U.S.A 2005, 102, 14545–14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hurshman AR; White JT; Powers ET; Kelly JW Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004, 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- (48).Klabunde T; Petrassi HM; Oza VB; Raman P; Kelly JW; Sacchettini JC Rational design of potent human transthyretin amyloid disease inhibitors. Nat. Struct. Biol 2000, 7, 312–321. [DOI] [PubMed] [Google Scholar]
- (49).Niemietz C; Fleischhauer L; Sandfort V; Guttmann S; Zibert A; Schmidt HH Hepatocyte-like cells reveal novel role of SERPINA1 in transthyretin amyloidosis. J. Cell Sci 2018, 131, No. jcs219824. [DOI] [PubMed] [Google Scholar]
- (50).Klein R; Klein BE; Cruickshanks KJ The prevalence of age-related maculopathy by geographic region and ethnicity. Prog. Retinal Eye Res 1999, 18, 371–389. [DOI] [PubMed] [Google Scholar]
- (51).Basaiawmoit RV; Rattan SIS Cavalotti CAP, Cerulli L (eds) Age-related Changes of the Human Eye. Biogerontology 2009, 10, 95–96. [Google Scholar]
- (52).Westermark P; Sletten K; Johansson B; Cornwell GG 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. U.S.A 1990, 87, 2843–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wong WL; Su X; Li X; Cheung CM; Klein R; Cheng CY; Wong TY Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Global Health 2014, 2, e106–e116. [DOI] [PubMed] [Google Scholar]
- (54).Wolf G Retinol transport and metabolism in transthyretin-″knockout″ mice. Nutr. Rev 1995, 53, 98–99. [DOI] [PubMed] [Google Scholar]
- (55).Vogel S; Piantedosi R; O’Byrne SM; Kako Y; Quadro L; Gottesman ME; Goldberg IJ; Blaner WS Retinol-binding protein-deficient mice: biochemical basis for impaired vision. Biochemistry 2002, 41, 15360–15368. [DOI] [PubMed] [Google Scholar]
- (56).Quadro L; Blaner WS; Salchow DJ; Vogel S; Piantedosi R; Gouras P; Freeman S; Cosma MP; Colantuoni V; Gottesman ME Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J 1999, 18, 4633–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Quadro L; Hamberger L; Colantuoni V; Gottesman ME; Blaner WS Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol. Aspects Med 2003, 24, 421–430. [DOI] [PubMed] [Google Scholar]
- (58).Thompson CL; Blaner WS; Van Gelder RN; Lai K; Quadro L; Colantuoni V; Gottesman ME; Sancar A Preservation of light signaling to the suprachiasmatic nucleus in vitamin A-deficient mice. Proc. Nat. Acad. Sci. U.S.A 2001, 98, 11708–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wolf G A case of human vitamin A deficiency caused by an inherited defect in retinol-binding protein without clinical symptoms except night blindness. Nutr. Rev 1999, 57, 258–260. [DOI] [PubMed] [Google Scholar]
- (60).Seeliger MW; Biesalski HK; Wissinger B; Gollnick H; Gielen S; Frank J; Beck S; Zrenner E Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest. Ophthalmol. Visual Sci 1999, 40, 3–11. [PubMed] [Google Scholar]
- (61).Alhamadsheh MM; Connelly S; Cho A; Reixach N; Powers ET; Pan DW; Wilson IA; Kelly JW; Graef IA Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci. Transl. Med 2011, 3, No. 97ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.