

Abstract
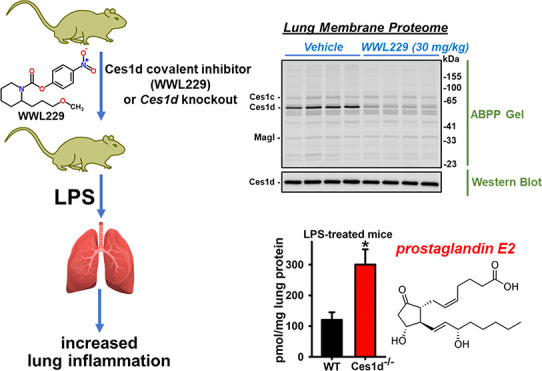
Carboxylesterases are members of the serine hydrolase superfamily and metabolize drugs, pesticides, and lipids. Previous research showed that inhibition of carboxylesterase 1 (CES1) in human macrophages altered the immunomodulatory effects of lipid mediators called prostaglandin glyceryl esters, which are produced by cyclooxygenase-catalyzed oxygenation of the endocannabinoid 2-arachidonoylglycerol (2-AG). Ces1d – the mouse ortholog of human CES1 – is the most abundant Ces isoform in murine lung tissues and alveolar macrophages and a major target of organophosphate poisons. Monoacylglycerol lipase (Magl) is also expressed in murine lung and is the main enzyme responsible for 2-AG catabolism. Several metabolic benefits are observed in Ces1d−/− mice fed a high-fat diet; thus, we wondered whether pharmacological and genetic inactivation of Ces1d in vivo might also ameliorate the acute inflammatory response to lipopolysaccharide (LPS). C57BL/6 mice were treated with WWL229 (Ces1d inhibitor) or JZL184 (Magl inhibitor), followed 30 min later by either LPS or saline. Wild-type (WT) and Ces1d−/− mice were also administered LPS to determine the effect of Ces1d knockout. Mice were sacrificed at 6 and 24 h, and cytokines were assessed in serum, lung, liver, and adipose tissues. Lipid mediators were quantified in lung tissues, while activity-based protein profiling and enzyme assays determined the extent of lung serine hydrolase inactivation by the inhibitors. WWL229 was shown to augment LPS-induced lung inflammation in a female-specific manner, as measured by enhanced neutrophil infiltration and Il1b mRNA. The marked Ces inhibition in female lung by 4 h after drug treatment might explain this sex difference, although the degree of Ces inhibition in female and male lungs was similar at 6 h. In addition, induction of lung Il6 mRNA and prostaglandin E2 by LPS was more pronounced in Ces1d−/− mice than in WT mice. Thus, WWL229 inhibited lung Ces1d activity and augmented the female lung innate immune response, an effect observed in part in Ces1d−/− mice and Ces1d/CES1-deficient murine and human macrophages. In contrast, JZL184 attenuated LPS-induced Il1b and Il6 mRNA levels in female lung, suggesting that Ces1d and Magl have opposing effects. Mapping the immunomodulatory molecules/pathways that are regulated by Ces1d in the context of lung inflammation will require further research.
Keywords: carboxylesterases, cytokines, inflammation, monoacylglycerol lipase
Carboxylesterases are members of the serine hydrolase superfamily and have metabolic functions in vivo.1 Their gene annotations are denoted Ces and CES for murine and human species, respectively.2 Ces/CES hydrolyze xenobiotics and lipids that contain carboxylic ester bonds, with pesticides and fatty acylglycerols representing important exogenous and endogenous substrates, respectively.2,3 The Ces/CES genes encode a number of unique isoforms that arose in evolution due to gene duplication events, especially in rodents.2 Consequently, mice have a larger number of carboxylesterase-encoding genes than humans (20 total in mice compared to 6 in humans). The murine genome contains eight different Ces1 genes, and each exhibits a high degree of sequence homology to the human CES1 gene.4 The Ces1d protein is the mouse ortholog of human CES1 protein in terms of both amino acid sequence homology and enzyme function.2 The Ces/CES isoforms are distributed in several mammalian tissues; for example, Ces1d/CES1 is abundant in liver, lung, and adipose in mice and humans.4−6 Further, CES1 is expressed in human monocytes and macrophages,7,8 including human alveolar macrophages,9 whereas Ces1d mRNA is detected in mouse alveolar macrophages but not in either thioglycolate (TG)-elicited peritoneal macrophages5 or bone marrow-derived macrophages (our unpublished data). Together, these findings suggest that Ces1d/CES1 enzymes have a role in innate immune responses, particularly in lung tissues.
Using activity-based protein profiling (ABPP) methods developed by Cravatt and co-workers,10 Ces1d was established to be the most active serine hydrolase in C57BL/6 mouse lung, followed by Ces1c.11 Previous research also suggested that the Ces/CES enzymes in alveolar macrophages and parenchymal lung tissues are involved in pesticide detoxication.9,11−13 For example, exposures of mice to chlorpyrifos (CPF), an organophosphate pesticide, inhibited both Ces1c and Ces1d in neonate lung and Ces1c in adult lung due to stoichiometric covalent binding and degradation of the active metabolite chlorpyrifos oxon (CPO).11 Although the lipopolysaccharide (LPS)-evoked pro-inflammatory cytokine levels in adult mouse lung were not altered by CPF exposure, CPF-treated females had augmented levels of LPS-induced eicosanoids (PGE2, PGD2, and 12-HETE). Interestingly, inflammation can also influence the extent of Ces expression. For example, treatment of isolated mouse alveolar macrophages with LPS caused Ces1d mRNA levels to be decreased,11 suggesting that inflammatory pathways can modulate Ces1d expression. Similar attenuating effects of LPS on Ces2g mRNA and Ces2g activity levels in mouse spleen were observed in vivo.14 Therefore, because the lung is exposed to noxious chemicals and pathogens that are present in the environment, further studies that examine the role of pulmonary Ces/CES enzymes during the lung’s response to inflammatory stimuli are warranted.
Diseases driven by inflammation often exhibit hallmarks related to deregulated lipid signaling. Two types of lipid mediators involved in cell signaling that are degraded by CES1 in human monocytes/macrophages are 2-arachidonoylglycerol (2-AG),15 an endocannabinoid produced on demand in activated cells,16 and prostaglandin glyceryl esters (PG-Gs),17 which are cyclooxygenase-derived oxygenated metabolites of 2-AG.18 Although inhibition of CES1 was shown to increase the levels of anti-inflammatory 2-AG in human THP-1 monocytes and macrophages,15 monoacylglycerol lipase (Magl) has a more important role than Ces1d in 2-AG turnover in murine lung.11 In addition, small-molecule inhibitors that target CES1 have only a minor effect on 2-AG hydrolytic activity in human peripheral blood mononuclear cells, whereas Magl inhibitors have a major one.8,19,20 (Patho)physiological conditions that augment 2-AG levels in vivo, which is often referred to as increased ‘endocannabinoid tone,’ can modulate the degree of inflammation in tissue injury due to autocrine and paracrine signaling evoked by 2-AG.21 For example, the drug JZL184, a small-molecule inhibitor of Magl that elevates the local concentration of 2-AG, reduced bronchoalveolar lavage (BAL) cytokine levels and decreased the extent of immune cell infiltration in an LPS-induced acute lung injury model.22 The PG-Gs, on the other hand, exhibit more complex behavior in that they can exert both anti- and pro-inflammatory effects.17,23−25 For example, PGE2-G can augment pro-inflammatory cytokine production by M1 polarized macrophages, whereas PGD2-G attenuate their levels.24 Similarly, the inhibition of α,β-hydrolase domain 6 (ABHD6), which is the primary 2-AG metabolizing enzyme in TG-elicited mouse peritoneal macrophages, was shown to attenuate the effects of LPS, putatively due to the increased conversion of 2-AG to PGD2-G. Furthermore, inactivation of CES1 in human THP-1 monocytes/macrophages promoted the anti-inflammatory effects of PGD2-G, while it diminished the pro-inflammatory effects of PGE2-G.17 Although these in vitro studies suggested that CES1 inactivation can exert anti-inflammatory effects, the role of Ces1d in murine lung and other tissues in settings of in vivo inflammation is largely unexplored. Moreover, studies that directly compare the differential effects of Ces1d and Magl inhibition in settings of inflammation have not been performed.
Inflammation and lipid metabolism are tightly linked together, and enzymes that regulate the levels of bioactive lipids can be targeted by pharmacological agents. In this study, we examined the role of Ces1d in a murine model of LPS-induced inflammation in lung, liver, and adipose tissues. WWL229, a small-molecule mechanism-based covalent inhibitor of Ces1d,26 was used to treat adult mice that were subsequently challenged with LPS. In addition, WT and Ces1d–/– mice were challenged with LPS to determine whether genotype-dependent effects were apparent. The degree of systemic inflammation was assessed by serum cytokine/chemokine levels, and tissue inflammation was determined by the levels of pro-inflammatory cytokines (lung, liver, adipose), bioactive lipids (lung), neutrophil infiltration (lung), and histopathology (liver). We determined whether Ces1d or Magl inactivation (via pharmacological or genetic disruption) altered any of these inflammatory readouts. In addition, activity-based assays established the enzyme target occupancy of the pharmacological inhibitors in the lung, both in vitro and in vivo. The main findings are that WWL229 augmented LPS-induced lung inflammation in a female-specific manner and Ces1d knockout also promoted inflammation in lung.
Materials and Methods
Chemicals
WWL229, (4-nitrophenyl) 2-(3-methoxypropyl)piperidine-1-carboxylate; WWL113, ethyl 4-[4-[methyl-[(3-pyridin-4-ylphenyl)methyl]carbamoyl]oxyphenyl]benzoate; JZL184, (4-nitrophenyl) 4-[bis(2H-1,3-benzodioxol-5-yl) (hydroxy)methyl]piperidine-1-carboxylate; pNPVa, p-nitrophenyl valerate; and LPS (E. coli O 111:B4) were obtained from Sigma (St. Louis, MO). Authentic standards of lipid mediators were from Cayman (Ann Arbor, MI). LC–MS solvents and the activity probe fluorophosphonate-TAMRA (FP-TAMRA) were from Thermo Fisher.
Animals
Adult C57BL/6 wild-type (WT) mice were from a breeding colony at MSU or from Jackson Laboratories (Bar Harbor, ME). Global Ces1d−/− mice on a C57BL/6 background were established as previously described.27 Mice were housed in temperature- and humidity-controlled AAALAC-approved facilities (20–25 °C and 40–60% humidity) under a 12 h light cycle and used in accordance with the Mississippi State University Institutional Animal Care and Use Committee.
Drug Treatments and Intraperitoneal LPS Challenge
Adult female and male mice (n = 5/sex/group) were injected with WWL229 (30 mg/kg, i.p.) or the drug vehicle (15:1:1 v/v/v saline/ethanol/Brij93) in a volume of 100 μL/mouse. Thirty minutes after drug injection, the mice were systemically challenged with LPS (1.25 mg/kg, i.p.) or saline vehicle in a volume of 100 μL/mouse. Six or twenty-four hours after LPS injection, mice were sacrificed by exsanguination under isoflurane anesthesia (Figure 1A depicts the experimental protocol). The LPS dose and time points were the same as those in our previous studies.11,14 Blood was collected from the inferior vena cava prior to exsanguination, and serum was prepared by centrifuging the clotted blood (4 °C, 10 min, 3,000g). Tissues (lung, liver, and adipose) were immediately flash-frozen in liquid nitrogen and stored at −80 °C until use. In some studies, JZL184 (30 mg/kg, i.p), a Magl inhibitor, was used instead of WWL229, and 0.5% w/v hydroxypropylmethylcellulose in saline was used as the vehicle. In studies involving Ces1d−/− mice (female and male), WT and Ces1d−/− mice (n = 5–6 mice/group) were injected with LPS (1.25 mg/kg, i.p.) or saline vehicle in a volume of 100 μL/mouse and sacrificed 6 h later, as described above. In studies to assess the enzyme target occupancy by inhibitors, mice were injected with drugs only (WWL229, WWL113, and JZL184; 30 mg/kg, i.p.) or the vehicle (15:1:1 v/v/v saline/ethanol/Brij93 for WWL229, 0.5% w/v hydroxypropylmethylcellulose in saline for WWL113 and JZL184) in a volume of 100 μL/mouse and sacrificed after 4 or 6 h. Mice treated for 6 h with WWL229 were subjected to a BAL procedure immediately before harvesting the lungs. Male and female mice were used in most in vivo experiments.
Figure 1.

Inflammation was augmented by WWL229 in female WT mouse lung, but not in male WT lung, 6 h after intraperitoneal LPS challenge. (A) Experimental design for the WWL229 study is depicted. (B) Serum MCP-1 levels in female and male WT mice treated with WWL229 and LPS. (C) LPS-induced MPO activity in lung was enhanced by WWL229 in a female-specific manner. LPS-induced Il1b cytokine mRNA levels were augmented by WWL229 in female lung (D) but not in male lung (E). (F) LPS-induced Il6 mRNA levels were enhanced in female Ces1d−/− mouse lung relative to WT controls. (G) LPS-induced cytokine mRNA levels were attenuated by JZL184 in female WT mouse lung. The Magl study design was the same as the WWL229 study except that mice were sacrificed at 6 h only. Data are expressed as the mean ± SD (n = 5 mice/group). One-way ANOVA (D,E,G) or two-way ANOVA (B,C,F) assessed significant differences between groups. *p < 0.05, ***p < 0.001, comparisons are indicated; ns, not significant. In panel 1B, *p < 0.05 for treatments vs vehicle control; #p < 0.05 for indicated comparison.
Intranasal LPS Challenge
LPS (in saline) or saline vehicle was administered to anesthetized male WT and Ces1d−/− mice by the intranasal (i.n.) route (1.25 mg/kg, n = 5/group; <50 μL per mouse). Mice were sacrificed 6 h later, and BAL was collected in 3 mL of phosphate-buffered saline (PBS) containing 1 mM EDTA, followed by removal of the lungs.
Quantification of Serum Cytokines by Multiplex Assay
A custom premixed Luminex xMAP multiplex assay kit (Millipore Sigma) was used to measure cytokines and inflammatory markers in mouse serum following the manufacturer’s instructions on a Luminex 200 system.
Preparation of Lung Total Membrane Fraction
Lungs were rinsed in cold PBS and Dounce-homogenized in sucrose buffer (50 mM Tris–HCl, 0.32 M sucrose, pH 7.4) and then centrifuged at a low speed (4 °C, 5 min, 1000g) to remove tissue debris, followed by centrifugation of the supernatant at high speed (4 °C, 60 min, 100,000g). The resulting pellet was washed by sonication in the sucrose buffer, followed again by centrifugation (4 °C, 30 min, 100,000g). The washed pellet (total membrane fraction) was resuspended by sonication in 200–400 μL of sucrose buffer and stored at −80 °C. Protein concentrations were determined using the BCA reagent (Thermo Pierce) with bovine serum albumin standards following the manufacturer’s instructions.
Lung Myeloperoxidase Activity
Aliquots of lung total membranes from male and female mice treated with WWL229 and LPS were assayed for myeloperoxidase (MPO) activity. In a 96-well microtiter plate, the following assay components (final concentrations indicated) were combined in 200 μL of 5 mM sodium acetate (pH 5.0) containing 75 mM NaCl and 0.005% v/v Triton X-100: H2O2 (1 mM), lung membranes (∼10–25 μg protein), and tetramethyl benzidine (500 μM; added last to begin reactions). The absorbance (λ = 650 nm) in each well was measured for 5 min in a Molecular Devices plate reader (at 37 °C) to assess the rates of MPO-catalyzed oxidation of tetramethyl benzidine.
Reverse Transcriptase Quantitative Polymerase Chain Reaction Analysis
Total RNA from adipose, liver, and lung tissues was extracted with an RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s instructions. A NanoDrop ND-1000 spectrophotometer was utilized to quantify RNA. cDNA was prepared with a RevertAid First Strand cDNA synthesis kit (Thermo Scientific). Reverese transcriptase quantitative polymerase chain reaction (RT-qPCR) was performed on a Stratagene Mx3005P thermal cycler with a QuantiFast SYBR Green PCR master mix (Qiagen) using QuantiTect primers (Qiagen) or custom oligonucleotides (Invitrogen or Eurofins) (see Table 1). The comparative cycle threshold (ΔΔCT) was used to determine gene expression changes using GADPH as the reference gene, with the results compared to the control condition [(2–ΔΔCT)] as previously described.28
Table 1. Quantitect Primers and Primer Sequences Used for RT-qPCR Analysis.
| quantitect primers | mouse | |
|---|---|---|
| Gapdh | Mm_Gapdh 3_SG QuantiTect Primer Assay—QT01658692 | |
| Il1b | Mm_Il1b_2_SG QuantiTect Primer Assay—QT01048355 | |
| Il6 | Mm_Il6_1_SG QuantiTect Primer Assay—QT00098875 | |
| Tnfa | Mm_Tnf_1_SG QuantiTect Primer Assay—QT00104006 | |
| mouse | ||
| Gene | 5′-Forward Sequence-3′ | 5′-Reverse Sequence-3′ |
| Gapdh | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
| Il1b | GCAACTGTTCCTGAACTCAACT | ATCTTTTGGGGTCCGTCAACT |
| Il6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| Tnfa | CCCTCACACTCAGATCATCTTCT | GCTACGACGTGGGCTACAG |
| Ces1c | CTCAGATATGTTCAGCACCGAAA | GCTTTTTGTCAAATCGGCAGG |
| Ces1d | ATATGGCTTTCTCTTGCTGCG | CCCAGGACTTTGCCTTTAACAGT |
| Ces2g | TCTCTGAGGTGGTTTACCAAACG | CCTCTCAGACAGCGCACCAG |
| human | ||
| Gene | 5′-Forward Sequence-3′ | 5′-Reverse Sequence-3′ |
| IL6 | AGCCACTCACCTCTTCAGAAC | GCCTCTTTGCTGCTTTCACAC |
| TNFa | CCCATGTTGTAGCAAACCCT | TGAGGTACAGGCCCTCTGAT |
| CCL3 | TTCCGTCACCTGCTCAGAAT | CAGCAGCAAGTGATGCAGAGA |
| GAPDH | GAPDH_1_SG QuantiTect Primer Assay—QT00079247 | |
Quantitation of Ces Activity in Lung Membranes
Carboxylesterase activity in lung membranes was evaluated by two methods. The first used a continuous kinetic assay with pNPVa substrate (λ = 405 nm) in a Molecular Devices plate reader.29 Enzyme activities were normalized on protein to give specific activities. The second employed gel-based activity-based protein profiling (ABPP) of the membranes with FP-TAMRA to evaluate the activities of multiple serine hydrolases in parallel, including Ces, as previously described.17 In some cases, following fluorescent visualization of in-gel proteins that had been labeled by FP-TAMRA, proteins were transferred to a polyvinylidene difluoride (PVDF) membrane and probed with a rabbit monoclonal anti-human CES1 antibody (Abcam Cat# 2312–1, RRID/AB 1266968; the antibody cross-reacts with mouse Ces1d isoform).
Quantitation of Lipid Mediators in Lung Tissues and Bronchoalveolar Lavage Fluid
Eicosanoids were extracted from lungs as previously described.30 One-hundred microliters of methanol was added to the dried extracts and transferred to LC vials with volume-reducing inserts for analysis by targeted LC–MS/MS as previously described.31 Bronchoalveolar lavage fluid (BALF) (1.5 mL volume) was spiked with deuterated internal standards and extracted twice with an equal volume hexane/ethyl acetate (1:1, v/v) containing 0.2% acetic acid. The pooled organics were dried down under nitrogen and reconstituted in methanol (100 μL) for LC–MS/MS. Aliquots of BALF (25 μL) were taken to determine their total protein concentration for normalization purposes. There was no significant difference in the BALF protein concentrations due to genotype or LPS treatment (2-way ANOVA).
In Vitro Treatments of Murine and Human Macrophages with Inflammatory Stimuli
Alveolar macrophages were harvested from male and female WT and Ces1d−/− mice lungs in cold PBS and 1 mM EDTA and pooled according to the genotype. The cells were washed and counted with a hemocytometer and then plated overnight in Dulbecco’s modified Eagle’s medium containing antibiotics and 10% FBS in an atmosphere of 5% CO2 in air (37 °C). The next day, the attached cells were gently washed and overlaid with the serum-free medium containing LPS (100 ng/mL) and incubated for 24 h. Total RNA was extracted from cells for RT-qPCR, and PGE2 levels in the culture media were quantified by LC–MS/MS. In addition, human THP-1 macrophage lines with normal and attenuated CES1 protein expression32 – termed control macrophages and CES1KD macrophages, respectively – were also treated with LPS (1 μg/mL, 6 h) or TNFα/IFNγ (100 ng/mL and 20 ng/mL, respectively, 12 h) and the RNA was extracted for RT-qPCR.
Histochemistry of Liver Sections
A portion of the liver tissue was fixed in 10% neutral-buffered formalin (10% NBF). Fixed liver was processed and embedded in paraffin using standard histologic techniques. Paraffin-embedded liver was cut into 5 μm sections and stained with hematoxylin and eosin on an automatic stainer (Leica). The number of immune cells within 4 layers of hepatocytes surrounding 20 portal zones in each mouse were quantified and averaged per portal zone.
Statistical Analysis
GraphPad Prism (Version 7, San Diego, CA) or SigmaPlot (Version 11.0, San Jose, CA) was used for statistical analyses. The results are presented as the mean ± standard deviation (SD). Data from RT-qPCR were transformed to linearized ΔΔCT values as previously described.28 Differences between groups were assessed with either an unpaired Student t-test, one-way analysis of variance (ANOVA), or two-way ANOVA as appropriate. Post-hoc testing was performed using the Student–Newman–Keuls method. Outliers were identified by Grubb’s outlier test. p-values <0.05 were considered significant.
Results
WWL229 had Minimal Effect on Serum Cytokines/Chemokines Following LPS Challenge Except for MCP-1
Cytokines and chemokines in WT mouse serum were evaluated by a Luminex assay at 6 h (male and female) and 24 h (female only). LPS challenge induced a robust increase in these molecules regardless of sex (Figure S1). WWL229 enhanced the amount of serum monocyte chemoattractant protein-1 (MCP-1) induced by LPS in females, but not in males, at 6 h (Figure 1B), although this effect was not apparent at 24 h (p > 0.05) (Figure S1). In general, the levels of the other serum cytokines/chemokines induced by LPS were not significantly altered by WWL229 (Figure S1). In addition, WWL229 treatment in the absence of LPS challenge had no impact on serum cytokines/chemokines; thus, it did not cause overt inflammation.
WWL229 Augmented Inflammation in Female Lung Following LPS Challenge
Elevated MPO activity is a surrogate measure of tissue neutrophil infiltration during inflammation.33 MPO activity in WT mouse lungs was determined at 6 h to assess the degree of inflammation induced by either LPS alone or LPS combined with WWL229 (Figure 1C). LPS caused a significant increase in MPO activity in both females and males, indicative of inflammation-dependent neutrophil infiltration into the lung. Interestingly, WWL229 augmented the LPS-induced MPO activity in females but not in males, suggesting that WWL229 enhanced female lung inflammation (Figure 1C).
We next evaluated cytokine expression in lungs by RT-qPCR. Six hours after LPS treatment, a robust increase in the expression of Il1b, Il6, and Tnfa mRNA was noted in both females and males (Figure 1D,E). Interestingly, the Ces1d inhibitor WWL229 augmented the levels of Il1b in female lungs (Figure 1D) but not in male lungs (Figure 1E). Based on gene expression and ABPP data, it is apparent that Ces1d is robustly expressed and enzymatically active in WT murine lung (Figure S2A-C). To extend these findings, we challenged female Ces1d−/− mice with LPS to assess the effect of Ces1d knockout. After 6 h, LPS-induced lung Il6 mRNA levels in this model were significantly higher than those in WT lung (Figure 1F), whereas no differences in LPS-induced Il1b and Tnfa levels were noted between WT and Ces1d−/− lungs (Figure S3). Thus, pharmacologic and genetic inactivation of Ces1d appears to modulate the degree of lung inflammation that is induced by LPS in female mice.
Next, we examined the effects of the Magl inhibitor JZL184 on lung inflammation. The LPS-induced expression of Il1b and Il6 mRNA after 6 h was significantly attenuated by JZL184 (Figure 1G). JZL184 also decreased Tnfa levels, although this did not reach statistical significance (p > 0.05). Thus, the Magl inhibitor exerted effects on lung inflammation that were opposite to those of the Ces1d inhibitor.
Levels of Lung Tissue Lipid Mediators in Ces1d−/− Mice Following LPS Challenge Were Dependent on Their Sex
Ces enzymes have roles in the regulation of certain lipid mediators,15,17 so we next evaluated eicosanoid levels in lung tissues of male and female WT and Ces1d−/− mice in the context of LPS-induced inflammation. In male mice, there was no genotype-dependent difference in PGD2, PGE2, and PGF2α levels during baseline and LPS-induced conditions (Figure S4, top), whereas AA, 12-HETE, and HODEs were generally higher in Ces1d–/– lungs than in WT lungs under either condition (Figure S4, bottom). In addition, the 12-HETE levels were lower in LPS-challenged Ces1d−/− mice than in unchallenged Ces1d−/− mice, an effect not observed in WT mice. In contrast, there was a genotype-dependent difference in the magnitude of lung lipid mediators in female mice that were induced by LPS challenge (Figure 2). For example, 12-HETE, HODEs, and PGs were markedly higher in Ces1d−/− mice than in WT mice following LPS treatment. Thus, with respect to eicosanoid production, the female Ces1d−/− lung appears to be more active than female WT lung in settings of inflammation. These results also correlate with the higher Il6 mRNA levels that were observed in female Ces1d−/− lung. Increased eicosanoid levels were also observed in female WT mice treated with both WWL229 and LPS for 24 h (Figure S5), again suggesting the augmentation of inflammatory molecules when Ces1d is inactivated.
Figure 2.

Eicosanoids were quantified in lungs of female WT and Ces1d−/− mice 6 h after intraperitoneal treatment with saline (Veh) or LPS. Ces1d-/- mice were more responsive to the effects of LPS than WT mice. HODEs represent the combined levels of 9-HODE and 13-HODE because they were not chromatographically resolved. Data are expressed as mean ± SD (n = 5 mice/group). Two-way ANOVA assessed significant differences between groups. #p < 0.05 and ***p < 0.001, comparisons are indicated; nd, not detected.
Ces1d Deficiency Augmented Localized Inflammation in Lungs
Male WT and Ces1d−/− mice were treated with LPS intranasally to induce a localized inflammation in the alveolar space. Mice were sacrificed 6 h later and PGE2 was quantified in BALF and lung tissues, while Il1b mRNA expression was determined in lung tissues. The total protein concentration in BALF, a measure of capillary leakage, was unaffected by treatment or genotype (2-way ANOVA; treatment, p = 0.638; genotype, p = 0.303); thus, overt lung injury at 6 h was not yet apparent. However, the PGE2 levels in BALF from Ces1d−/− mice were ∼10-fold more than those in BALF from WT mice following the LPS challenge (Figure 3A). PGE2 levels were not induced by i.n. LPS administration in either WT or Ces1d−/− lung tissues (Figure 3B), which is similar to the situation following i.p. LPS administration in male WT and Ces1d−/− mice (Figure S4). In addition, Il1b mRNA was also significantly induced (∼5-fold) in Ces1d−/− lung by LPS, whereas no significant increase was noted in the LPS-treated WT mice (Figure 3C). These findings again suggest that Ces1d deficiency causes a more pronounced LPS-induced inflammatory response in the lung.
Figure 3.

PGE2 and Il1b mRNA levels in lungs of male WT and Ces1d−/− mice 6 h after i.n. treatment with saline or LPS. (A) PGE2 levels in BALF. (B) PGE2 levels in lung tissues. (C) Il1b mRNA levels in lung tissues. Data are expressed as mean ± SD (n = 5 mice/group). Two-way ANOVA assessed significant differences between groups. ***p < 0.001; ns, not significant; comparisons are indicated.
In Vivo WWL229 Treatment Inhibited Lung Ces Activity
Ces enzymatic activity in lung membranes was determined using the pan-Ces substrate pNPVa.29 When WT mice were treated with WWL229 for 4 h, lung Ces activity in females, but not males, was inhibited (Figure 4A). By 6 h, however, lung Ces activity was inhibited in both females and males. Treatment with LPS had no effect on the degree of inhibition caused by WWL229 in female mice (Figure 4B). Ces activity in lung was also inhibited when female mice received WWL113, another Ces1d inhibitor (Figure 4C). The extent of inhibition caused by WWL229 was slightly greater than that caused by WWL113. Although not as potent as the other inhibitors, JZL184 also decreased lung Ces activity in vivo (Figure 4D). LPS challenge also had no impact on JZL184-dependent Ces inhibition (Figure 4D). Furthermore, lung Ces activity in male and female Ces1d−/− mice was also markedly lower than that in WT mice, with Ces1d responsible for up to ∼80% of the total lung Ces activity (Figure 4E). These data again indicate that functional Ces1d enzyme is present in mouse lung and can be inactivated in vivo by covalent inhibitors.
Figure 4.

Ces activity in lung membranes from vehicle- and inhibitor-treated WT mice and Ces1d−/− mice. (A) Lung Ces activity was reduced in WT mice by WWL229 in a sex-dependent and -independent manner at 4 h and 6 h, respectively. (B) Intraperitoneal LPS challenge had no effect on lung Ces activity in female mice, and it did not alter WWL229-mediated Ces inhibition. Lung Ces activity was inhibited in female mice by WWL113 (C) and JZL184 (D). Lung Ces activity in Ces1d−/− mice were markedly lower than those in WT mice (E). Data are expressed as the mean ± SD [n = 5 mice, n = 3 male mice at 4 h, (A); n = 3 mice, (B,C); n = 5 mice, (D,E)]. One-way ANOVA (C,E) or two-way ANOVA (A,B,D) assessed significant differences between groups. *p < 0.05, ***p < 0.001 for treatments vs vehicle or Ces1d−/− vs WT.
As indicated by the ABPP gel in Figure S2B, the major 60 kDa protein detected in WT lung membranes had almost disappeared in Ces1d−/− lung membranes, enabling it to be annotated as Ces1d. Ces1c protein was also detected at ∼70 kDa, which is due to this soluble protein being more extensively glycosylated than other Ces isoforms. It should be noted that Ces1c mRNA is detected along with Ces1d and Ces2g mRNA in lung tissues; thus, it is expressed in lung too (Figure S2A). However, Ces1c protein is abundant in mouse plasma; thus, residual plasma contamination might have also contributed to its presence in lung membranes, despite the extensive washing protocol used. Therefore, the Ces1c band detected in the ABPP gels might come from both lung tissues and plasma.
To examine the potency and selectivity of the inhibitors WWL229, WWL113, and JZL184 toward the serine hydrolases found in mouse lung, we employed the gel-based ABPP approach in a competitive format (Figure 5). Each inhibitor bears an electrophilic carbamate functional group that covalently modifies the nucleophilic serine residue in the active site of its target serine hydrolase,26 thereby preventing the covalent labeling of the enzyme by the FP-TAMRA probe. Lung membranes obtained from naïve WT mice were pretreated in vitro with increasing concentrations of small-molecule inhibitors for 30 min, followed by addition of FP-TAMRA. Of the serine hydrolases detected following gel electrophoresis, the intensity of the major 60 kDa protein band represented by Ces1d was reduced by WWL229 in a concentration-dependent manner (IC50 = 2.4 μM), whereas WWL113 and JZL184 inhibited Ces1d with greater potency but with less selectivity than WWL229 (Figure 5A–C). This is evident from the protein bands besides Ces1d that were also reduced in intensity by WWL113 and JZL184 in a concentration-dependent manner.
Figure 5.

Gel-based activity-based protein profiling of lung serine hydrolases. Lung membranes from naïve WT mice were treated in vitro with the indicated concentrations of WWL229 (A), WWL113 (B), and JZL184 (C) for 30 min at 37 °C before incubation with the activity probe FP-TAMRA (0.2 μM). The IC50s for the inhibition of Ces1d are indicated. Lung membranes were also prepared from WT mice 4 h after in vivo treatments with WWL229 (D) and JZL184 (E). WWL229 selectively inhibited lung Ces1d, whereas JZL184 was less selective in that it also inhibited Ces1c, Ces1d, and Magl (its canonical target). Data are expressed as the mean ± SD (n = 4 mice, D,E). Student’s t-test assessed significant differences between groups, *p < 0.05 for treatments vs vehicle. The lanes designated as “Heat” indicate that lung membranes were heat-denatured (95 °C, 3 min) prior to adding the activity probe FP-TAMRA.
Next, we examined lung membranes from female WT mice 4 h after receiving the vehicle, WWL229 (30 mg/kg, i.p), or JZL184 (30 mg/kg, i.p.) by gel-based ABPP. Densitometry of band intensities indicated WWL229 inhibited Ces1d in vivo by ∼75%, whereas it did not inhibit either Ces1c or Magl (Figure 5D). A similar degree of Ces1d inhibition by WWL229 in both female and male lungs was seen at 6 h (Figure S6A,B), consistent with the 6 h data in Figure 4A. Western blot analysis of the 6 h female lung membranes also indicated that Ces1d protein abundance was not altered by the drug treatment (Figure S6A).
In contrast, JZL184 only slightly inhibited Ces1d activity in vivo, whereas it strongly inactivated Ces1c by >90% and inactivated the ∼35 kDa band denoted Magl by >50% (Figure 5E). JZL184 is a potent inhibitor of Magl,34 but the band intensity attributed to Magl in Figure 5E was not completely ablated as expected. In vitro treatment of naïve lung membranes with JZL184 indicated a similar situation (Figure 5C). Quantitation of this “Magl” band suggested the presence of two serine hydrolase activities that migrate in the gels with similar Rf; one is sensitive to JZL184 (IC50 = 0.46 μM), while the other is insensitive even at 100 μM JZL184 (Figure 5C, inset).
Based on these in vitro and in vivo results, we can conclude that WWL229 is the more selective covalent inhibitor of Ces1d in mouse lung and a good tool to probe Ces1d functions in vivo. Furthermore, these biochemical data indicate that Ces1d is the major carboxylesterase isoform expressed in murine lung and WWL229 can selectively inactivate lung Ces1d activity in vivo.
Macrophages Deficient in Ces1d/CES1 are More Responsive to LPS-Induced Inflammation
Ces1d is expressed in mouse alveolar macrophages.11 In vitro experiments with these cells showed that baseline and LPS-induced levels of PGE2 and cytokine mRNA (Il6 and TNFa) were higher in alveolar macrophages from Ces1d–/– mice compared to those from WT mice (Figure 6A). Consistent with these findings, when CES1 expression in human THP-1 macrophages – a cell line that strongly expresses CES1 mRNA and protein35 – was knocked down by an shRNA-containing lentivirus32 (Figure S2D), the levels of inflammatory cytokines were greater than those expressed by control THP-1 macrophages in the setting of LPS exposure (Figure 6B, left). Similar effects were also observed when the inflammatory stimulus was TNFα/IFNγ instead of LPS (Figure 6B, right). TLR4 mRNA transcript levels were also found to be slightly higher in CES1KD macrophages than in control macrophages under baseline conditions (1.6-fold, p < 0.05). Similarly, the baseline IL1b, IL6, and TNFa mRNA levels in CES1KD macrophages were 3–5-fold higher than those in control macrophages (Figure 6B, right; data not shown). Thus, these results suggested, regardless of whether an immune stimulus is present or not, that macrophages deficient in Ces1d/CES1 are more poised toward the M1 phenotype than those that express Ces1d/CES1. From another perspective, due to the higher basal cytokine levels in the enzyme-deficient cells, the fold change in cytokine levels is more pronounced in the WT/control cells than in the Ces1d/CES1-deficient cells following an immune stimulus (Figure 6).
Figure 6.

Mouse alveolar macrophages and human THP-1 macrophages are more responsive to inflammatory stimuli when Ces1d and CES1 expression are ablated. (A) Alveolar macrophages from WT and Ces1d−/− mice (male and female) were isolated and pooled to obtain enough cells, which were then treated with LPS. The levels of PGE2, the most abundant prostaglandin in lung tissues, were determined in culture supernatants and normalized on cell protein content, left. The extent of murine Il6 and Tnfa mRNA expression was also determined, right. (B) Human THP-1 macrophages with ablated CES1 expression were treated with either LPS (left) or TNFα/IFNγ (right), and their inflammatory responses were compared to control THP-1 macrophages (no CES1 knockdown). The expression levels of human IL1b, IL6, TNFa, and CCL3 (MIP-1α) mRNA are shown. Data are expressed as mean ± SD (n = 3–4 replicate wells), and the results are representative of two independent experiments (alveolar macrophages) or three independent experiments (THP-1 macrophages). Two-way ANOVA assessed significant differences between groups. #p < 0.05, ##p < 0.01, ###p < 0.001, **p < 0.01, and ***p < 0.001; comparisons are indicated. In panel B, ***p < 0.001 for TNFα/IFNγ treatment vs corresponding untreated controls.
Ces1d Knockout Augmented IL-6 Expression in Female Liver Following LPS Challenge
Because Ces1d is also expressed in other metabolically active tissues such as liver and adipose, inflammation was evaluated in these tissues in female WT mice treated with WWL229 and in female Ces1d–/– mice (Figures S7 and 7). Systemically administered LPS significantly induced Il1b, Il6, and Tnfa mRNA in WT liver (Figure S7A), as expected, but their expression was not altered by WWL229. However, although female WT and Ces1d−/− mouse liver expressed similar levels of Il1b and Tnfa following LPS challenge (Figure S7B), Il6 was significantly higher in Ces1d−/− liver than in WT liver (Figure 7A). Histology of liver portal zones from female WT and Ces1d−/− mice showed that LPS increased the number of total immune cells per portal zone regardless of the genotype (Figure 7B); however, no genotype × LPS treatment interaction was noted (p > 0.05). When further stratified by immune cell type, baseline lymphocyte numbers were significantly increased in Ces1d−/− liver compared to those in WT liver (Figure 7B), suggesting that the Ces1d−/− liver is poised toward higher numbers of T- and/or B-cells under homeostatic conditions than the WT liver.
Figure 7.

LPS-induced Il6 mRNA levels were enhanced in female Ces1d−/− mouse liver compared to WT mouse liver at 6 h following intraperitoneal LPS challenge (A). Hematoxylin and eosin (H&E) histology and immune cell quantification of female WT and Ces1d−/− mouse livers 6 h following LPS challenge (B). Representative H&E-stained images from each treatment group are shown. Total immune cells were quantified within 4 layers of hepatocytes surrounding 20 portal zones from each mouse. The average number of cells per portal zone is represented in the bar graphs. Immune cells were further distinguished by cell type, and the lymphocyte numbers are shown. Data are expressed as mean ± SD (n = 5–6 mice/group). Two-way ANOVA assessed significant differences between groups. In panel A, *p < 0.05, comparisons are indicated. In panel B, #p < 0.05, comparison is indicated; *p < 0.05 for LPS vs saline.
LPS challenge also significantly induced Il1b, Il6, and Tnfa mRNA in WT adipose tissues, but WWL229 did not modulate their levels (Figure S7C). Although the baseline expression of Il1b in Ces1d−/− adipose tissues was lower than that in WT adipose tissues, no significant differences were found in the inflammatory cytokines in adipose tissues from WT and Ces1d−/− mice following LPS challenge (Figure S7D). Finally, the effects of JZL184 were also examined in female WT liver and adipose tissues. However, JZL184 also did not alter the levels of LPS-induced cytokines in either tissue (Figure S8); thus, its immune modulating effects appeared to be confined to the lung in our animal model.
Discussion
Carboxylesterases play an important role in the metabolism of xenobiotics and lipids. In addition to their abundant expression in mammalian liver and lung, CES1 is found in human monocytes and macrophages, including alveolar macrophages, and Ces1d in murine alveolar macrophages.5,7,8,15 Our previous work identified Ces1d as the most abundant serine hydrolase in mouse lung, although Magl was more important in the catabolism of 2-AG.11 Here, we investigated the roles of lung Ces1d and Magl in an LPS model of inflammation. The main findings are as follows: 1) inhibition of Ces1d by WWL229 augmented LPS-induced inflammation in female lung, whereas it did not alter the degree of inflammation in male lung, suggesting a sex-specific effect for this compound in mice; 2) the levels of LPS-induced Il6 mRNA in female Ces1d−/− lung and liver were higher than those in WT lung and liver; 3) in contrast to the potentiating effects of Ces1d inactivation, inhibition of Magl by JZL184 attenuated LPS-induced inflammatory cytokines in female lungs, suggesting that Ces1d and Magl exert opposing effects in the setting of lung inflammation.
The inflammatory effects made more potent by the Ces1d inhibitor WWL229 in female lungs were observed in part in Ces1d−/− mice, which was evident from the higher expression of Il6 mRNA and eicosanoids in Ces1d−/− lung compared to WT lung following LPS challenge. In support of this, the LPS-induced PGE2 and Il6 mRNA response was stronger in Ces1d−/− alveolar macrophages than in WT alveolar macrophages. In addition, CES1 knockdown in a human macrophage cell line also resulted in a stronger response to immune stimuli. Our previous studies using cultured macrophages have also identified immunomodulatory effects caused by the inhibition of CES1.15,17,32 For example, CES1 inactivation by either reactive pesticide metabolites or lentivirus-mediated knockdown resulted in an increase in cellular levels of endogenous 2-AG and PG-Gs due to the more limited degradation of these lipid mediators.15,32 These bioactive lipids can modulate inflammation.36−38 In addition, we showed that CES1 knockdown decreased the production of the oxysterol 27-hydroxycholesterol (27-OHC) in cholesterol-loaded macrophages, which was due to marked downregulation of the 27-OHC biosynthetic enzyme CYP27A1.32 27-OHC is another lipid mediator that exerts immune effects and is an endogenous ligand for LXRα.39 Furthermore, combined treatment of M1 polarized macrophages with WWL113 and PGD2-G attenuated IL-6 levels more strongly than that of PGD2-G alone,17 which suggested that blocking CES1 hydrolysis of PGD2-G potentiated the anti-inflammatory effect of this lipid mediator. Because of the instability and transient nature of PG-Gs, they are difficult to detect in vivo. We attempted to measure these endogenous compounds in lung extracts by LC–MS/MS but to no avail. Indeed, it was previously shown that PG-Gs could only be detected in tissues of genetically engineered animal models under specific conditions.40 Specifically, they were identified in brains of transgenic COX2-overexpressing mice treated with LPS, but only when MAGL was concomitantly inactivated. Therefore, it remains technically challenging to detect the PG-Gs in (patho)physiological settings.
Similar effects to those observed in the current study were seen with the lipolytic enzyme acyloxyacyl hydrolase (AOAH) in alveolar macrophages.41 AOAH can disarm LPS by removing the secondary acyl chains from it, and AOAH inactivation was shown to augment the expression of LPS-induced Il1b and Tnfa. Thus, an explanation for the in vivo effects in our study is that Ces1d functions (directly or indirectly) to reduce the levels of inflammatory molecules; therefore, its enzymatic inhibition may stabilize those compounds, whether they are host- or pathogen-derived, that increase the production of inflammatory cytokines. Along these lines, lipid mediators, such as PGE2, help shape the course of inflammation by stimulating immune cells to synthesize cytokines. For example, we previously showed that the potentiating effect of PGE2 on IL-6 production by M1 macrophages was strongly augmented by a CES1/Ces1d inhibitor.17 In the current study, within the setting of LPS-induced inflammation, PGE2 levels were elevated in female Ces1d−/− lungs compared to WT lungs, while Ces1d−/− alveolar macrophages produced more PGE2 than their WT counterparts. Thus, the elevated tissue levels of PGE2 might be a mechanistic driver of the augmented inflammation that results following Ces1d inactivation. Increased levels of PGE2 were also seen in male Ces1d−/− mice after i.n. LPS administration, where alveolar macrophages in the lung lumen are directly activated by the endotoxin. The higher levels of LPS-induced PGE2 in the BALF of Ces1d−/− mice compared to that of WT mice also correlated with the higher levels of Il1b mRNA in Ces1d−/− lungs.
Other negative impacts of Ces1d inactivation have been reported in the context of metabolic syndrome.42 Ces1d ablation in adipose tissues led to a local unhealthy microenvironment in metabolically active tissues. Specifically, an adipose tissue-specific Ces1d knockout mouse, placed on a high-fat diet, exhibited a more pronounced lipotoxicity and proinflammatory phenotype in adipose and liver tissues as compared to control mice, resulting in greater metabolic dysregulation and systemic insulin resistance. These effects were linked to the absence of Ces1d-dependent triacylglycerol lipase activity in adipose tissues, causing ectopic accumulation of triacylglycerols in peripheral tissues. Whether Ces1d has a role in triacylglycerol metabolism within lung tissues, however, will require further study.
Another important issue is that the WWL229-dependent augmentation of lung inflammation seen in female mice was not observed in male mice. This is congruent with the female-specific inactivation of lung Ces activity by WWL229 observed at 4 h in vivo. However, lung Ces activity was decreased by WWL229 equally in females and males by 6 h (Figures 4A and S6), suggesting a differential rate of drug uptake into the lung. Because the levels of Ces activity in male and female lungs are comparable, and Ces1d accounts for most of this enzymatic activity, this implies that lung Ces1d concentrations are similar in both sexes. Indeed, this appears to be borne out by gel-based ABPP of male and female mouse lung membranes (Figure S2C). It is possible that the time-dependent effects on Ces inhibition in females and males might be related to differences in the pharmacokinetic behavior of WWL229, that is, it may be inferred from the enzyme activity data that the biologically effective concentration of WWL229 required to inhibit Ces1d was achieved earlier in female lung than in male lung. It is known that sex-related differences in drug metabolism exist, especially in the liver;43,44 males often have higher levels of xenobiotic metabolism enzymes (e.g., CYPs and Ces) than females.11,45 Therefore, it is possible that WWL229 is inactivated in vivo more efficiently in males by either hepatic oxidative metabolism or covalent adduction of Ces enzymes, altering the rate of compound uptake into the lung. This also could be the reason that WWL113, a more potent in vitro inhibitor of Ces1d, did not inhibit Ces activity to the same extent as WWL229 in female lung in vivo.
The observed in vivo effects caused by Ces inhibition might also be specific to mice. For instance, recent work showed that the inhibition of Ces in porcine alveolar macrophages by bis(4-nitrophenyl) phosphate (BNPP) reduced the expression of Il1b, Il6, and Tnfa mRNA,46 which is opposite to our findings. Furthermore, combined treatment of pigs with BNPP and LPS in vivo led to decreased levels of AA and other pro-inflammatory lipid mediators, which was attributed to inhibited Ces-dependent endocannabinoid hydrolysis. We had previously suggested that the LPS-dependent reductions of Ces1d and Ces2g activity in murine alveolar macrophages, lung, and spleen might be part of a negative feedback mechanism to control inflammation.11,14 However, our current data seem to suggest that Ces inactivation in lung induces a pro-inflammatory response rather than an anti-inflammatory one. The identification of those endogenous substrates that are possibly metabolized by Ces1d in the murine lung will help to unravel the species-related differences observed in these different models. In addition, as shown by the significantly altered levels of CYP27A1 and its metabolite 27-OHC in human CES1KD macrophages,32 the inactivation of CES1 (and its mouse counterpart Ces1d) may indirectly regulate the levels of immune-modulating lipid mediators, such as PGE2 and other eicosanoids. Further research on the signaling pathways that are involved is needed.
Magl is the primary hydrolytic enzyme of the endocannabinoid 2-AG in murine lung despite the fact that the catalytic efficiencies of recombinant human CES1 and MAGL enzymes toward 2-AG are equally comparable.11,14,15,47 Furthermore, the in vitro hydrolysis rates of 2-AG by WT and Ces1d–/– lung membranes were not significantly different (data not shown), indicating that other enzymes, notably Magl, are responsible. Therefore, we treated mice with JZL184, a Magl inhibitor with some known off-targets, for example, Ces/CES isoforms48 (Figure 5C,E), and found that it could attenuate lung cytokines induced by LPS challenge. These findings are opposite to those of WWL229 but are in line with others that showed JZL184 could exert anti-inflammatory effects.22,49−52 Two of these studies examined the murine lung. The first showed that JZL184 reduced IL-6 and TNF-α levels in BAL fluid after i.n. LPS challenge.22 JZL184 also reduced MCP-1, an effect opposite to that in our study when female mice were treated with WWL229. The authors attributed the JZL184-mediated effects to increased 2-AG levels. The second study showed that JZL184 reduced TNF-α and IL-1β in BAL.49 Lung 2-AG was also found to be significantly increased by JZL184. Thus, our JZL184 findings are consistent with other reports that indicate that Magl inactivation in settings of lung inflammation has benefits. One caveat to this is that Ces1c can also be potently inhibited by JZL184 in vivo, but whether this contributes to the immune-dampening effects of JZL184 in the lung is not currently known. Another study has demonstrated that the ABHD6 inhibitor WWL70 decreased LPS-induced murine lung inflammation to an even greater extent than JZL184 did, which was attributed to increases in 2-AG levels and some lysoglycerophospholipids following ABHD6 inactivation.53 ABHD6 adds another node to the intricate lipid biochemical pathways that can be targeted to alleviate inflammation during lung injury.
Finally, the effects observed in our study also have implications regarding mechanisms of immunotoxicity. Our previous work showed that lung Ces1d had a protective role against CPF-mediated lung immune effects.11 For example, in the setting of inflammation, a CPF-dependent increase in TNFa mRNA was revealed in adult Ces1d−/− mouse lung, an effect not seen in WT mice. In addition, Ces1c expression in male Ces1d−/− LPS-treated lungs was lower than that in LPS-treated WT lungs, which could have compounded the CPF-dependent immune effects in Ces1d−/− mice because Ces1c can also detoxify the bioactive metabolite of CPF (i.e., CPO). These findings, which were obtained with a chemical toxicant instead of a drug, also suggested that the inactivation or downregulation of Ces functions in lungs can exaggerate immune effects;11 thus, they are consistent with the changes seen in the present study.
Conclusions
Here, we showed that opposing effects on inflammation can occur when Ces1d and Magl are inhibited in mouse lungs. Although the effects of the Magl inhibitor are most likely due to increased levels of inflammation-dampening 2-AG, the identification of the effector molecules responsible for the Ces1d-dependent effects will require further research. For example, it will be of interest to examine the connections between the transcription factors (e.g., NF-κB and AP-1) activated by LPS and the signaling pathways that are unmasked when Ces1d is inhibited or knocked out. Overall, our study further supports the benefits that Magl inhibitors might offer in inflammatory settings. Moreover, it suggests that functional Ces1d has a role, albeit undefined, in regulating the degree of acute inflammation in the lung. The mechanisms that explain this regulation will be of interest to explore further..
Acknowledgments
The authors thank Shirley Guo-Ross for assistance with animal experiments and Daniel Young for help with ABPP experiments. We are grateful to Katie Webb and Hudson Chenault for their assistance with the human THP-1 cell experiments. Funding was provided by NIH R15GM128206. C.B.W. was supported by NIH T35OD010432-21.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.2c00098.
Mouse serum cytokine/chemokine levels; Ces1d expression/activity in WT mouse lung; Il1b and Tnfa mRNA levels in female WT and Ces1d−/− mouse lungs; eicosanoids in lungs from male WT and Ces1d−/− mice treated with saline and LPS; eicosanoids in lungs from female WT mice treated with WWL229 and LPS; cytokine mRNA levels in female mouse liver and adipose resulting from Ces1d inhibition or Ces1d genetic knockout; and cytokine mRNA levels in female mouse liver and adipose after Magl inhibition (PDF)
Author Contributions
B.N.S. and A.B. contributed equally. B.N.S., A.B., B.L.F.K., and M.K.R. designed the study. B.N.S., A.B., H.S., A.M.M., J.E.C., A.O., C.B.W., and M.K.R. carried out experiments, data analyses, and interpretation. R.L. provided Ces1d–/– mice and interpretation. The manuscript was drafted by B.N.S. and M.K.R., and revised by B.L.F.K. and M.K.R. All authors approved the final manuscript.
The authors declare no competing financial interest.
This paper was published ASAP on September 12, 2022, with an error in the Abstract and also the Macrophages Deficient in Ces1d/CES1 are More Responsive to LPS-Induced Inflammation section. The corrected version was reposted on September 14, 2022.
Supplementary Material
References
- Long J. Z.; Cravatt B. F. The Metabolic Serine Hydrolases and Their Functions in Mammalian Physiology and Disease. Chem. Rev. 2011, 111, 6022–6063. 10.1021/CR200075Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian J.; Nelson R.; Lehner R. Carboxylesterases in Lipid Metabolism: From Mouse to Human. Protein Cell 2018, 9, 178–195. 10.1007/s13238-017-0437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross M. K.; Streit T. M.; Herring K. L.; Xie S. Carboxylesterases: Dual Roles in Lipid and Pesticide Metabolism. J. Pestic. Sci. 2010, 35, 257–264. 10.1584/jpestics.R10-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes R. S.; Wright M. W.; Laulederkind S. J.; Cox L. A.; Hosokawa M.; Imai T.; Ishibashi S.; Lehner R.; Miyazaki M.; Perkins E. J.; Potter P. M.; Redinbo M. R.; Robert J.; Satoh T.; Yamashita T.; Yan B.; Yokoi T.; Zechner R.; Maltais L. J. Recommended Nomenclature for Five Mammalian Carboxylesterase Gene Families: Human, Mouse, and Rat Genes and Proteins. Mamm Genome 2010, 21, 427–441. 10.1007/s00335-010-9284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R. D.; Taylor A. M.; Tong E. Y.; Repa J. J. Carboxylesterases Are Uniquely Expressed among Tissues and Regulated by Nuclear Hormone Receptors in the Mouse. Drug Metab. Dispos. 2013, 41, 40–49. 10.1124/dmd.112.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa T.; Fukami T.; Nakajima M. Characterization of Species Differences in Tissue Diltiazem Deacetylation Identifies Ces2a as a Rat-Specific Diltiazem Deacetylase. Drug Metab. Dispos. 2015, 43, 1218–1225. 10.1124/dmd.115.064089. [DOI] [PubMed] [Google Scholar]
- Ghosh S. Cholesteryl Ester Hydrolase in Human Monocyte/Macrophage: Cloning, Sequencing, and Expression of Full-Length CDNA. Physiol Genomics 2000, 2, 1–8. 10.1152/physiolgenomics.2000.2.1.1. [DOI] [PubMed] [Google Scholar]
- Szafran B. N.; Lee J. H.; Borazjani A.; Morrison P.; Zimmerman G.; Andrzejewski K. L.; Ross M. K.; Kaplan B. L. F. Characterization of Endocannabinoid-Metabolizing Enzymes in Human Peripheral Blood Mononuclear Cells under Inflammatory Conditions. Molecules 2018, 23, 3167. 10.3390/molecules23123167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J. S.; Shi G. P.; Mark E. A.; Chin D. T.; Gerard C.; Chapman H. A. A Serine Esterase Released by Human Alveolar Macrophages Is Closely Related to Liver Microsomal Carboxylesterases. J. Biol. Chem. 1991, 266, 18832–18838. 10.1016/s0021-9258(18)55139-5. [DOI] [PubMed] [Google Scholar]
- Niphakis M. J.; Cravatt B. F. Enzyme Inhibitor Discovery by Activity-Based Protein Profiling. Annu. Rev. Biochem. 2014, 83, 341–377. 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Szafran B. N.; Borazjani A.; Seay C. N.; Carr R. L.; Lehner R.; Kaplan B. L. F.; Ross M. K. Effects of Chlorpyrifos on Serine Hydrolase Activities, Lipid Mediators, and Immune Responses in Lungs of Neonatal and Adult Mice. Chem. Res. Toxicol. 2021, 34, 1556–1571. 10.1021/acs.chemrestox.0c00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T.; Schiller N. L.; Fukuto T. R. Malathion and Phenthoate Carboxylesterase Activities in Pulmonary Alveolar Macrophages as Indicators of Lung Injury. Toxicol. Appl. Pharmacol. 1983, 70, 140–147. 10.1016/0041-008X(83)90187-4. [DOI] [PubMed] [Google Scholar]
- Wallace T. J.; Ghosh S.; McLean Grogan W. M. L. Molecular Cloning and Expression of Rat Lung Carboxylesterase and Its Potential Role in the Detoxification of Organophosphorus Compounds. Am. J. Respir. Cell Mol. Biol. 1999, 20, 1201–1208. 10.1165/AJRCMB.20.6.3402. [DOI] [PubMed] [Google Scholar]
- Szafran B.; Borazjani A.; Lee J. H.; Ross M. K.; Kaplan B. L. Lipopolysaccharide Suppresses Carboxylesterase 2g Activity and 2-Arachidonoylglycerol Hydrolysis: A Possible Mechanism to Regulate Inflammation. Prostaglandins Other Lipid Mediat 2015, 121, 199–206. 10.1016/j.prostaglandins.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S.; Borazjani A.; Hatfield M. J.; Edwards C. C.; Potter P. M.; Ross M. K. Inactivation of Lipid Glyceryl Ester Metabolism in Human THP1 Monocytes/Macrophages by Activated Organophosphorus Insecticides: Role of Carboxylesterases 1 and 2. Chem. Res. Toxicol. 2010, 23, 1890–1904. 10.1021/tx1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V.; Bisogno T.; De Petrocellis L.; Melck D.; Orlando P.; Wagner J. A.; Kunos K. Biosynthesis and Inactivation of the Endocannabinoid 2-Arachidonoylglycerol in Circulating and Tumoral Macrophages. Eur. J. Biochem. 1999, 264, 258–267. 10.1046/J.1432-1327.1999.00631.X. [DOI] [PubMed] [Google Scholar]
- Scheaffer H. L.; Borazjani A.; Szafran B. N.; Ross M. K. Inactivation of CES1 Blocks Prostaglandin D2 Glyceryl Ester Catabolism in Monocytes/Macrophages and Enhances Its Anti-Inflammatory Effects, Whereas the Pro-Inflammatory Effects of Prostaglandin E2 Glyceryl Ester Are Attenuated. ACS Omega 2020, 5, 3961. 10.1021/acsomega.0c03961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley P. J.; Rouzer C. A.; Morgan A. J.; Patel S.; Marnett L. J. Aspects of Prostaglandin Glycerol Ester Biology. Adv. Exp. Med. Biol. 2019, 1161, 77–88. 10.1007/978-3-030-21735-8_8. [DOI] [PubMed] [Google Scholar]
- Turcotte C.; Chouinard F.; Lefebvre J. S.; Flamand N. Regulation of Inflammation by Cannabinoids, the Endocannabinoids 2-Arachidonoyl-Glycerol and Arachidonoyl-Ethanolamide, and Their Metabolites. J Leukoc Biol 2015, 97, 1049–1070. 10.1189/jlb.3RU0115-021R. [DOI] [PubMed] [Google Scholar]
- Turcotte C.; Dumais É.; Archambault A. S.; Martin C.; Blanchet M. R.; Bissonnette É.; Boulet L. P.; Laviolette M.; Di Marzo V.; Flamand N. Human Leukocytes Differentially Express Endocannabinoid-Glycerol Lipases and Hydrolyze 2-Arachidonoyl-Glycerol and Its Metabolites from the 15-Lipoxygenase and Cyclooxygenase Pathways. J. Leukoc. Biol. 2019, 106, 1337–1347. 10.1002/JLB.3A0919-049RRR. [DOI] [PubMed] [Google Scholar]
- Howlett A. C.; Reggio P. H.; Childers S. R.; Hampson R. E.; Ulloa N. M.; Deutsch D. G. Endocannabinoid Tone versus Constitutive Activity of Cannabinoid Receptors. Br. J. Pharmacol. 2011, 163, 1329–1343. 10.1111/J.1476-5381.2011.01364.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costola-de-Souza C.; Ribeiro A.; Ferraz-de-Paula V.; Calefi A. S.; Aloia T. P. A.; Gimenes-Júnior J. A.; de Almeida V. I.; Pinheiro M. L.; Palermo-Neto J. Monoacylglycerol Lipase (MAGL) Inhibition Attenuates Acute Lung Injury in Mice. PLoS One 2013, 8, 77706. 10.1371/JOURNAL.PONE.0077706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhouayek M.; Lambert D. M.; Delzenne N. M.; Cani P. D.; Muccioli G. G. Increasing Endogenous 2-Arachidonoylglycerol Levels Counteracts Colitis and Related Systemic Inflammation. FASEB J. 2011, 25, 2711–2721. 10.1096/fj.10-176602. [DOI] [PubMed] [Google Scholar]
- Alhouayek M.; Masquelier J.; Cani P. D.; Lambert D. M.; Muccioli G. G. Implication of the Anti-Inflammatory Bioactive Lipid Prostaglandin D2-Glycerol Ester in the Control of Macrophage Activation and Inflammation by ABHD6. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 17558–17563. 10.1073/pnas.1314017110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M.; Rosenberg D. W. Multifaceted Roles of PGE2 in Inflammation and Cancer. Semin Immunopathol 2013, 35, 123–137. 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez E.; Galmozzi A.; Chang J. W.; Hsu K. L.; Pawlak J.; Li W.; Godio C.; Thomas J.; Partida D.; Niessen S.; O’Brien P. E.; Russell A. P.; Watt M. J.; Nomura D. K.; Cravatt B. F.; Saez E. Integrated Phenotypic and Activity-Based Profiling Links Ces3 to Obesity and Diabetes. Nat. Chem. Biol. 2014, 10, 113–121. 10.1038/nchembio.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei E.; Ben Ali Y.; Lyon J.; Wang H.; Nelson R.; Dolinsky V. W.; Dyck J. R.; Mitchell G.; Korbutt G. S.; Lehner R. Loss of TGH/Ces3 in Mice Decreases Blood Lipids, Improves Glucose Tolerance, and Increases Energy Expenditure. Cell Metab. 2010, 11, 183–193. 10.1016/j.cmet.2010.02.005. [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D.; Livak K. J. Analyzing Real-Time PCR Data by the Comparative C(T) Method. Nat. Protoc. 2008, 3, 1101–1108. 10.1038/NPROT.2008.73. [DOI] [PubMed] [Google Scholar]
- Ross M. K.; Borazjani A. Enzymatic Activity of Human Carboxylesterases. Curr. Protoc. Toxicol. 2007, 33, 41–244. 10.1002/0471140856.tx0424s33. [DOI] [PubMed] [Google Scholar]
- Szafran B. N.; Pinkston R.; Perveen Z.; Ross M. K.; Morgan T.; Paulsen D. B.; Penn A. L.; Kaplan B. L. F.; Noël A. Electronic-Cigarette Vehicles and Flavoring Affect Lung Function and Immune Responses in a Murine Model. Int. J. Mol. Sci. 2020, 21, 6022. 10.3390/ijms21176022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Borazjani A.; Matthews A. T.; Mangum L. C.; Edelmann M. J.; Ross M. K. Identification of Palmitoyl Protein Thioesterase 1 in Human THP1 Monocytes and Macrophages and Characterization of Unique Biochemical Activities for This Enzyme. Biochemistry 2013, 52, 7559–7574. 10.1021/bi401138s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangum L. C.; Hou X.; Borazjani A.; Lee J. H.; Ross M. K.; Crow J. A. Silencing Carboxylesterase 1 in Human THP-1 Macrophages Perturbs Genes Regulated by PPARgamma/RXR and RAR/RXR: Down-Regulation of CYP27A1-LXRalpha Signaling. Biochem. J. 2018, 475, 621–642. 10.1042/BCJ20180008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulli B.; Ali M.; Forghani R.; Schob S.; Hsieh K. L. C.; Wojtkiewicz G.; Linnoila J. J.; Chen J. W. Measuring Myeloperoxidase Activity in Biological Samples. PLoS One 2013, 8, e67976 10.1371/journal.pone.0067976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J. Z.; Nomura D. K.; Cravatt B. F. Characterization of Monoacylglycerol Lipase Inhibition Reveals Differences in Central and Peripheral Endocannabinoid Metabolism. Chem. Biol. 2009, 16, 744–753. 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. A.; Middleton B. L.; Borazjani A.; Hatfield M. J.; Potter P. M.; Ross M. K. Inhibition of Carboxylesterase 1 Is Associated with Cholesteryl Ester Retention in Human THP-1 Monocyte/Macrophages. Biochim. Biophys. Acta - Mol. Cell Biol. Lipids 2008, 1781, 643–654. 10.1016/j.bbalip.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallily R.; Breuer A.; Mechoulam R. 2-Arachidonylglycerol, an Endogenous Cannabinoid, Inhibits Tumor Necrosis Factor-Alpha Production in Murine Macrophages, and in Mice. Eur. J. Pharmacol. 2000, 406, R5–R7. 10.1016/s0014-2999(00)00653-1. [DOI] [PubMed] [Google Scholar]
- Lourbopoulos A.; Grigoriadis N.; Lagoudaki R.; Touloumi O.; Polyzoidou E.; Mavromatis I.; Tascos N.; Breuer A.; Ovadia H.; Karussis D.; Shohami E.; Mechoulam R.; Simeonidou C. Administration of 2-Arachidonoylglycerol Ameliorates Both Acute and Chronic Experimental Autoimmune Encephalomyelitis. Brain Res. 2011, 1390, 126–141. 10.1016/j.brainres.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Peng F.; Dong M.; Yang H. Endocannabinoid 2-Arachidonylglycerol Protects Primary Cultured Neurons against LPS-Induced Impairments in Rat Caudate Nucleus. J. Mol. Neurosci. 2014, 54, 49–58. 10.1007/S12031-014-0246-2. [DOI] [PubMed] [Google Scholar]
- Ma L.; Cho W.; Nelson E. R. Our Evolving Understanding of How 27-Hydroxycholesterol Influences Cancer. Biochem. Pharmacol. 2022, 196, 114621. 10.1016/J.BCP.2021.114621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A. J.; Kingsley P. J.; Mitchener M. M.; Altemus M.; Patrick T. A.; Gaulden A. D.; Marnett L. J.; Patel S. Detection of Cyclooxygenase-2-Derived Oxygenation Products of the Endogenous Cannabinoid 2-Arachidonoylglycerol in Mouse Brain. ACS Chem. Neurosci. 2018, 9, 1552–1559. 10.1021/acschemneuro.7b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou B.; Jiang W.; Han H.; Li J.; Mao W.; Tang Z.; Yang Q.; Qian G.; Qian J.; Zeng W.; Gu J.; Chu T.; Zhu N.; Zhang W.; Yan D.; He R.; Chu Y.; Lu M. Acyloxyacyl Hydrolase Promotes the Resolution of Lipopolysaccharide-Induced Acute Lung Injury. PLoS Pathog. 2017, 13, 6436. 10.1371/JOURNAL.PPAT.1006436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Li X.; Yang L.; Wang S.; Dai Y.; Fekry B.; Veillon L.; Tan L.; Berdeaux R.; Eckel-Mahan K.; Lorenzi P. L.; Zhao Z.; Lehner R.; Sun K. Adipose Tissue-Specific Ablation of Ces1d Causes Metabolic Dysregulation in Mice. Life Sci. Alliance 2022, 5, 1209. 10.26508/lsa.202101209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikada H.; Ida K.; Ando E.; Inagaki Y.; Sakamoto A.; Kamiya A. Establishment and Analysis of a Mouse Model That Regulates Sex-Related Differences in Liver Drug Metabolism. Lab. Invest. 2018, 98, 1500–1511. 10.1038/S41374-018-0088-6. [DOI] [PubMed] [Google Scholar]
- Rinn J. L.; Snyder M. Sexual Dimorphism in Mammalian Gene Expression. Trends Genet. 2005, 21, 298–305. 10.1016/J.TIG.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Kisui F.; Fukami T.; Nakano M.; Nakajima M. Strain and Sex Differences in Drug Hydrolase Activities in Rodent Livers. Eur. J. Pharm. Sci. 2020, 142, 105143. 10.1016/J.EJPS.2019.105143. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Yan B.; Sun W.; Chen Q.; Xiao Q.; Xiao Y.; Wang X.; Shi D. Pig Liver Esterases Hydrolyze Endocannabinoids and Promote Inflammatory Response. Front. Immunol. 2021, 12, 12. 10.3389/FIMMU.2021.670427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner G. F.; Zimmermann R.; Schicho R.; Taschler U. Monoglyceride Lipase as a Drug Target: At the Crossroads of Arachidonic Acid Metabolism and Endocannabinoid Signaling. Pharmacol. Ther. 2017, 175, 35–46. 10.1016/j.pharmthera.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow J. A.; Bittles V.; Borazjani A.; Potter P. M.; Ross M. K. Covalent Inhibition of Recombinant Human Carboxylesterase 1 and 2 and Monoacylglycerol Lipase by the Carbamates JZL184 and URB597. Biochem. Pharmacol. 2012, 84, 1215–1222. 10.1016/J.BCP.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abohalaka R.; Bozkurt T. E.; Nemutlu E.; Onder S. C.; Sahin-Erdemli I. The Effects of Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase Inhibitor Treatments on Lipopolysaccharide-Induced Airway Inflammation in Mice. Pulm. Pharmacol. Ther. 2020, 62, 101920. 10.1016/j.pupt.2020.101920. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Wise L. E.; Chen Y.; Gujjar R.; Mahadevan A.; Cravatt B. F.; Lichtman A. H. The Monoacylglycerol Lipase Inhibitor JZL184 Suppresses Inflammatory Pain in the Mouse Carrageenan Model. Life Sci. 2013, 92, 498–505. 10.1016/J.LFS.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S. K.; Zhang M.; Tian Z. L.; Wang M.; Zhao R.; Wang L. L.; Li S. S.; Liu M.; Li J. Y.; Zhang M. Z.; Guan D. W. The Monoacylglycerol Lipase Inhibitor JZL184 Decreases Inflammatory Response in Skeletal Muscle Contusion in Rats. Eur. J. Pharmacol. 2015, 761, 1–10. 10.1016/J.EJPHAR.2015.04.018. [DOI] [PubMed] [Google Scholar]
- Kerr D. M.; Harhen B.; Okine B. N.; Egan L. J.; Finn D. P.; Roche M. The Monoacylglycerol Lipase Inhibitor JZL184 Attenuates LPS-Induced Increases in Cytokine Expression in the Rat Frontal Cortex and Plasma: Differential Mechanisms of Action. Br. J. Pharmacol. 2013, 169, 808–819. 10.1111/J.1476-5381.2012.02237.X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottemanne P.; Paquot A.; Ameraoui H.; Alhouayek M.; Muccioli G. G. The α/β–Hydrolase Domain 6 Inhibitor WWL70 Decreases Endotoxin-Induced Lung Inflammation in Mice, Potential Contribution of 2-Arachidonoylglycerol, and Lysoglycerophospholipids. FASEB J. 2019, 33, 7635–7646. 10.1096/fj.201802259R. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.