

Abstract
Microbiota are a malleable part of ecosystems, including the human ecosystem. Microorganisms not only affect the chemistry of their specific niche, such as the human gut but also the chemistry of distant environments, such as other parts of the body. Mass spectrometry-based metabolomics is one of the key technologies to detect and identify the small molecules produced by the human microbiome, and to understand the functional role of these microbial metabolites. This Review aims to provide a foundational introduction to common forms of untargeted mass spectrometry and the types of data that can be obtained in the context of microbiome analysis. Data analysis remains an obstacle, therefore, the emphasis is placed on data analysis approaches and integrative analysis, including the integration of microbiome sequencing data.
Table of content:
Mass spectrometry-based metabolomics is one of the key technologies to detect and identify the small molecules produced by the human microbiota, and to understand the functional role of these microbial metabolites. In this Review, Dorrestein and colleagues review common forms of untargeted mass spectrometry and the types of data that can be obtained in the context of microbiome analysis.
Introduction
Some of the key findings from early investigations of the human microbiome were thathealthy individuals carry different microbiota and that the composition of microbial communities is different across body sites1. Humans are composed of more microbial cells than human cells (estimated to be ~1.3 times the number of human cells)2, and perturbing the human-associated microbiota is postulated, and in some cases has been demonstrated, to have substantial health implications3–5. This is also true for animals, insects, plants, and environments such as terrestrial (for example, soil), aqueous (for example, ocean) and built environments (for example, houses or offices)6–8.
Most microbiome projects start with an inventory of organisms and/or genes using DNA or RNA sequencing methods (analysis of microbial community composition), and such efforts will continue to have an important role in the field. There is also an ever-increasing emphasis in the microbiome field towards a mechanistic understanding of how chemical environments shape microbial communities and a deeper interest in the function of the microbial-derived molecules on ecosystems. Whereas sequencing provides insights into the microorganism that are present and the metabolic capacity, metabolomics is a direct readout of the function of a system. The metabolome is considered the closest representation of phenotype and, therefore, metabolomics can provide insights into the cellular processes in response to some stimuli or interactions. Theoretically, a metabolomics experiment detects all small molecules, more specifically, chemicals with molecular weights of <2000 Da; however, in practice, it is a partial picture limited by the extent to which molecules can be extracted, ionized and detected. The tools to study the metabolome are not limited to endogenous molecules, which represent only a subset of all chemicals in a biological system, but they can also detect exogenous substances (for example, xenobiotics). Mass spectrometry (MS) is often used for metabolomics analysis, especially because of its good sensitivity and its capacity to detect and quantify a large diversity of molecules in complex biological samples 9, 10 (Box 1). Data analysis remains challenging; however, in the past few years there has been a vast increase in the development of tools to analyze MS metabolomics data, including improvements in feature extraction, annotation and data analysis to improve biological contextualization, including the integration with other omics data.
Box 1. The basics of mass spectrometry.
Mass spectrometry (MS) is an analytical technique to measure molecules (metabolomics focus on small molecules), detected as ions. In cases in which the investigator already knows what molecules they want to look at ahead of time or potentially observe, a targeted MS approach should be used. Targeting specific molecules improves sensitivity and specificity as one can directly tune the workflow and instrument for the detection of those specific molecules (for example, short-chain fatty acids or specific bile acids) and will have improved quantitative abilities. However, targeted workflows limit the potential for the discovery of unexpected molecules119. Untargeted MS, the focus of this Review, enables discovery at the cost of more difficult data analysis. MS detects chemicals by measuring the ionized form as their mass-to-charge ratio (m/z) and relative abundance or abundances (MS1). By acquiring data with high-resolution mass spectrometers such as TOF, Orbitrap or FTICR, the elemental composition can be calculated. The elemental composition in combination with an experiment called tandem MS, can be used to further reduce the possible molecular formula. In this tandem MS experiment (also described as MS/MS or MS2), energy is imparted via multiple collisions with an inert gas (for example, nitrogen or helium), which causes the ion to break apart157. This form of tandem MS is called collision-induced dissociation (CID). The fragments (that is, product ions) which result from the ionized molecule breaking apart are measured (MS/MS). This results in interpretable fragmentation patterns that are related to the chemical structure156. Although the details are much more complex, one of the simplest ways to think about this type of fragmentation is that the isolated molecule is heated until it falls apart. There are other forms of fragmentation do not use thermal activation, such as electron capture dissociation or electron impact158. In gas chromatography MS (GC-MS), used for detection of volatile molecules or non-volatile molecules that can become volatile using derivatization strategies, the electron impact (EI) ionization source allows neutral molecules to be ionized using an electron beam, and instantaneously fragment them as they enter the instrument44, 45. The result of any of these methods is fragmented molecular ions. The resulting fragmentation spectrum, the measurement of the m/z of the broken pieces of the ion of the molecule is equivalent to a short sequence read in terms of what can be achieved with subsequent data analysis. As microbiome samples are inherently complex, it is often necessary to separate molecules before the sample is introduced into the mass spectrometer, techniques such as liquid chromatography (LC), GC159, and increasingly ion mobility (IMS)160, have been applied. Particularly, IMS has received much attention over the past decade to tackle the challenge of separating unresolved or co-eluting isomers and isobars160. However, the software to leverage IMS data for microbiome studies are limited. This generates a wealth of different ways to produce ions and measure microbial molecules via mass spectrometry.
In general, some preprocessing of the metabolomics data is performed before statistical and annotation analysis, including some steps such as removing noise, recognizing adducts, and finding and quantifying features161. This is a very challenging step in metabolomics and will inevitably include the detection of false, split or missing features. Software such as MZmine49, MS-DIAL47, MShub159, openMS162 and XCMS163 can be used for such data preprocessing. The metabolites in untargeted metabolomics can be compared in relative quantitative fashion due to the large diversity of physico-chemical properties present in such biological samples, whereas for absolute quantification other steps should be taken, including evaluation of extraction yields, ionization profile and peak shape; ion suppression and matrix effects can have an impact the quantification164. When true and accurate quantities are desired for target molecules then 13C, 15N isotopically labeled internal standards and a targeted MS platform could be performed. For most scenarios such labeled compounds are not available for all the molecules detected in an untargeted metabolomics experiment. Most compounds are detected as more than one ion form (for example, in-source fragments, different adducts such as protons, sodium or multimers)165, 166, and might present different fragmentation patterns. Those ions can be found with tools such as RamClust 166 (particularly for data independent analysis), or CAMERA165, that perform MS1 based peak shape analysis and rule-based discovery of co-migrating species. To recognize some of those ion forms, ion identity molecular networking leverages both peak shape analysis and molecular networking167. Some compounds, such as sugars, cannot be easily ionized and may need derivatization before they can be detected168. Other compounds, such as less polar or non-polar compounds, need specific ionization sources to be ionized, such as atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI). Ionization does not occur equally for chemicals, and hence the use of internal standards is important in metabolomics. When it is not possible to use such internal standards as is the case for most microbial metabolites, the co-elution of chemicals can influence) the ionization of each other (for example, suppress through competition of the ‘charge’, complicating accurate quantification169. For example, peptides generally fragment in predictable patterns using relatively low energies (for example, ‘b-y’ fragmentation by CID). By contrast, some polyketides and alkaloids need higher levels of energy to induce fragmentation, and frequently generate fewer peaks in their fragmentation spectra, which can be challenging to interpret. All these issues can also be optimized by choosing the correct ionization source, solvent choices, sample or chromatography additives, instrument settings and parameters. This would contribute to inter-laboratory reproducibility of the data. In addition to metabolomics analysis, the quality of the data can substantially influence data analysis. Poor quality spectra, high noise level, contaminants, stability of the molecules, baseline drifts, among other issues, can lead to data misinterpretation or prevent a discovery. In some of these cases, filtering steps can improve the data analysis, but the proper use of background and quality controls and adoption of reproducible analysis workflows is essential to avoid removing important features.
Many advances in the analysis of microbiome MS data are coming from investigators that are developing or applying computational approaches to better interpret their sequence data11. Such laboratories are introducing to the mass spectrometry community ecological concepts such as alpha diversity [G] and beta diversity [G]12, 13 and other terms such as rarefaction [G], Procrustes analysis [G]14, mmvec [G]15, and principal coordinates analysis [G] (PcoA). In addition, MS and microbiome analysis infrastructures are in the early stages of being linked to leverage the understanding of the molecular underpinnings of the microbiome16,17,18, 19,20,11, 21,22,23.
In general, it has not been established when one should use one approach over another or how to synergistically use multiple approaches, different methods usually provide complementary results. This Review does not aim to provide a comprehensive account of metabolomics, computational tools for MS data analysis, metabolomics analysis of the microbiome or statistical methods as this is beyond the intended scope of this single review article (readers are referred to reviews on these topics9, 24–26). This Review aims to provide a starting point for readers who are entering the field of microbial- and microbiome-related mass spectrometry. The annotation of metabolites and attribution to a specific producer or producers, as well as the correlation of microbial metabolites with phenotypes, are of key interest in this field and, therefore, will be emphasized. The approaches presented in this review are summarized in Table 1.
Table 1.
Currently available metabolomics analysis to investigate and interpret microbiome data.
| Process | Application | Tools and methods | Requirements | Attributes |
|---|---|---|---|---|
| Data acquisition | Metabolite detection | MALDI-MS, REIMS, DESI | -Matrix to induce ionization (MALDI) | -Detection of metabolites (m/z) from surfaces (for example, histological sections or microorganisms grown in petri-dishes)27,28 -Microbial identification (biotyping)30 |
| LC-MS, LC-MS/MS | -Sample soluble in compatible organic solvents | -Chromatographic separation enables a better overview of metabolites in mixtures -MS/MS experiments provide structural information to improve annotation |
||
| GC-MS, 2D GC-MS | -Volatile sample -Deconvolution |
-Detection of less polar and non-polar, volatile and thermal stable molecules44,45. | ||
| Data preprocessing | Feature finding | AMDIS46, MS-DIAL47, XCMS48, MZmine49/ADAP50, OpenMS162 | -Most are based on MS1 detecting -If MS2 is available can be used as well |
-Provides more robust data (quantitative information) -It is not necessary for most annotation tools, but strongly encouraged for statistical analysis |
| Structural determination | Structure annotation | Spectral library | -MS1 -MS2 -Spectral databases |
-Annotation of microbial metabolites, usually level 2 or level 3 (MSI)51 |
| Combinatorial fragmentation (MetFrag)71, Competitive Fragmentation Modeling (CFM-ID)72, Fingerprint prediction (SIRIUS73, ZODIAC74, CANOPUS96) Hydrogen rearrangements (MS-Finder)75 |
-Most are based on MS1and MS2 -Structure databases |
-Provides candidate structures for the microbial metabolites -Usually these annotations are level 3 or level 4 (MSI)51 |
||
| Propagation of annotation (NAP)76 | -MS1 and MS2 -Structure databases |
-Provides candidate annotations for microbial metabolites -Enables analogs annotation by integration with molecular networking |
||
| DEREPLICATOR83 | -MS1 and MS2 -Structure databases |
-Annotation of microbial peptides, or at least a part of the amino acids sequence | ||
| Pep2Path85, Glycogenomics81, iSNAP86, RiPPquest87, NRPQuest88, DeepRIPP89, MiBIG79 | -MS1 and MS2 -Structure databases -Genomic sequence data |
-Annotation of microbial metabolites can be improved if genomics data are available | ||
| Substructure annotation and classification | MS2LDA93, MAGMA98, mzCloud99 | -MS1 and MS2 -MSn (mzCloud) |
-Annotation of chemical functions present in the dataset (for example, amino acids or glycosides) | |
| MolNetEnhancer94, Qemistree97 | -MS1 -MS2 -ClassyFire |
-Survey of the chemical classes present in the samples | ||
| Other structural characterization methods59 | NMR | -Deuterated solvents | -Has a key role in structure characterization -The entire chemical structure can be characterized, including stereochemistry |
|
| UV-Vis | -Metabolites need to absorb in the ultraviolet-visible spectrum | -Detection of metabolites that present chromophore groups -Provides qualitative and quantitative information of a given metabolite |
||
| IR | -Sample must be completely dry | -Shows the presence of specific chemical groups in a chemical structure, such as carbonyl, hydroxyl, amine, among others | ||
| Making connections | Spotting data trends | Unsupervised analysis (PCoA, PCA)102 | -MS1 | -Spots trends in the microbiome related to intrinsic or extrinsic effects -Quantitative analysis is strongly encouraged and data should be normalized to improve data integrity |
| Supervised analysis (PLS-DA)103 | -MS1 -Metadata (effect) |
-Shows the relationship of data with any perturbation the microbiome is facing (metadata) | ||
| Regression analyses104,105 | -MS1 | -Identification of metabolites that are modified in response to a particular effect | ||
| Connecting metabolites to microorganisms | Correlation methods (Pearson, Spearman, Kendall)109–111 | -MS1 | -Classification of metabolites that are most correlated to any perturbation the microbiome is facing | |
| Procrustes14 | -MS1 -16S rRNA or Metagenomics |
-Identification of metabolites and microorganisms that correlate | ||
| mmvec15 | -MS1 -16S rRNA or Metagenomics |
-Highlights metabolites and microorganism relationships by recognition of their co-occurrence | ||
| Songbird118 | -MS1 -16S rRNA or Metagenomics |
-Improves the false-positive rates of the metabolites, proteins or microorganisms loaded from the sample | ||
| Metabolic pathway-based analysis | Reverse metabolic ecology131 | -16S rRNA or Metagenomics -Metabolic knowledge |
-Prediction of an ecological relationship between the microorganisms (synergism, competition) in a microbiome | |
| MelonnPan135 | -Genomic sequencing data -Biological knowledge |
-Prediction of metabolic profiles from genomic data | ||
| PALS140 | -MS1 -BGCs knowledge |
-Highlights the metabolite set that changes under an ecological effect | ||
| MIMOSA (uses taxonomy)143, IPA (uses proteome)142, AMON (uses metagenome)141 | -MS1 -BGCs knowledge |
-Create connections between metabolites and biological information using BGC knowledge -Can indicate the origin (microorganism, host) of a metabolite |
||
| ChemRich144 | -Identified metabolites -Metabolic knowledge |
-Mapping of known metabolites by using structure similarity and chemical ontology | ||
| Data visualization | Spectral similarity | Molecular Networking16 | -MS1 and MS2 -Cosine score algorithm |
-Visualization of molecular families -Metadata can highlight metabolites related to microbial activities in the microbiome |
| Mass spectrometry Imaging | METASPACE41, MSiReader,42 ‘ili40 | -Spatial distribution (x, y, z) and MS1 -Pictures, map or 3D images can improve the visualization |
-Spatial visualization of metabolites on the microbiome-The spatial distribution can highlight interactions or the functional role of metabolites in a community |
BGC: biosynthetic gene cluster; GC-MS: gas chromatography coupled to mass spectrometry; IR: infra-red, LC-MS: liquid chromatography coupled to mass spectrometry; LC-MS/MS: untargeted analysis with acquisition of MS2 spectra; MALDI-MS: matrix assisted laser desorption ionization mass spectrometry; metadata: information about the sample (e.g. source, extraction procedure, disease association, etc.); MSI: metabolomics standards initiative; MS1: mass spectrum of the intact molecular ion (precursor ion); MS2: mas spectrum of the fragment ions; NMR: nuclear magnetic resonance; UV: ultra-violet to visible spectrum.
Detecting microbial metabolites
MS-based approaches have enabled the analysis of an immense amount of chemicals with diverse structures. MS has been used for detecting metabolites, or even quantifying them, from different types of samples, from solids (for example, directly from the surfaces)27, 28 to volatiles that the microbiome releases (for example, from the environment or a wet dog)29 (Fig. 1). One important strategy is the biotyping, which is currently used in clinics for microbial identification. This is accomplished by MS1 [G] profiling of ribosomal proteins (mass-to-charge ratio (m/z) 2000–15000) of bacterial colonies by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS. The resulting data are searched against a library of MS1 patterns obtained from well-characterized clinically relevant microorganisms to identify the taxon of the clinical culture30. This biotyping strategy has been adapted in a research setting to perform chemotyping of environmental microorganisms by collecting data in the lower m/z region; the region in which the metabolites are detected (m/z 200–2000) (Fig. 1a). This mass range shows many specialized metabolites or peptides that enable chemotyping of individual strains31, 32. Signals below m/z 200 are often excluded due to matrix interference in spite of many microbial metabolites being present in this m/z range.
Fig. 1: Mass spectrometry metabolomics approaches for studying the microbiome.
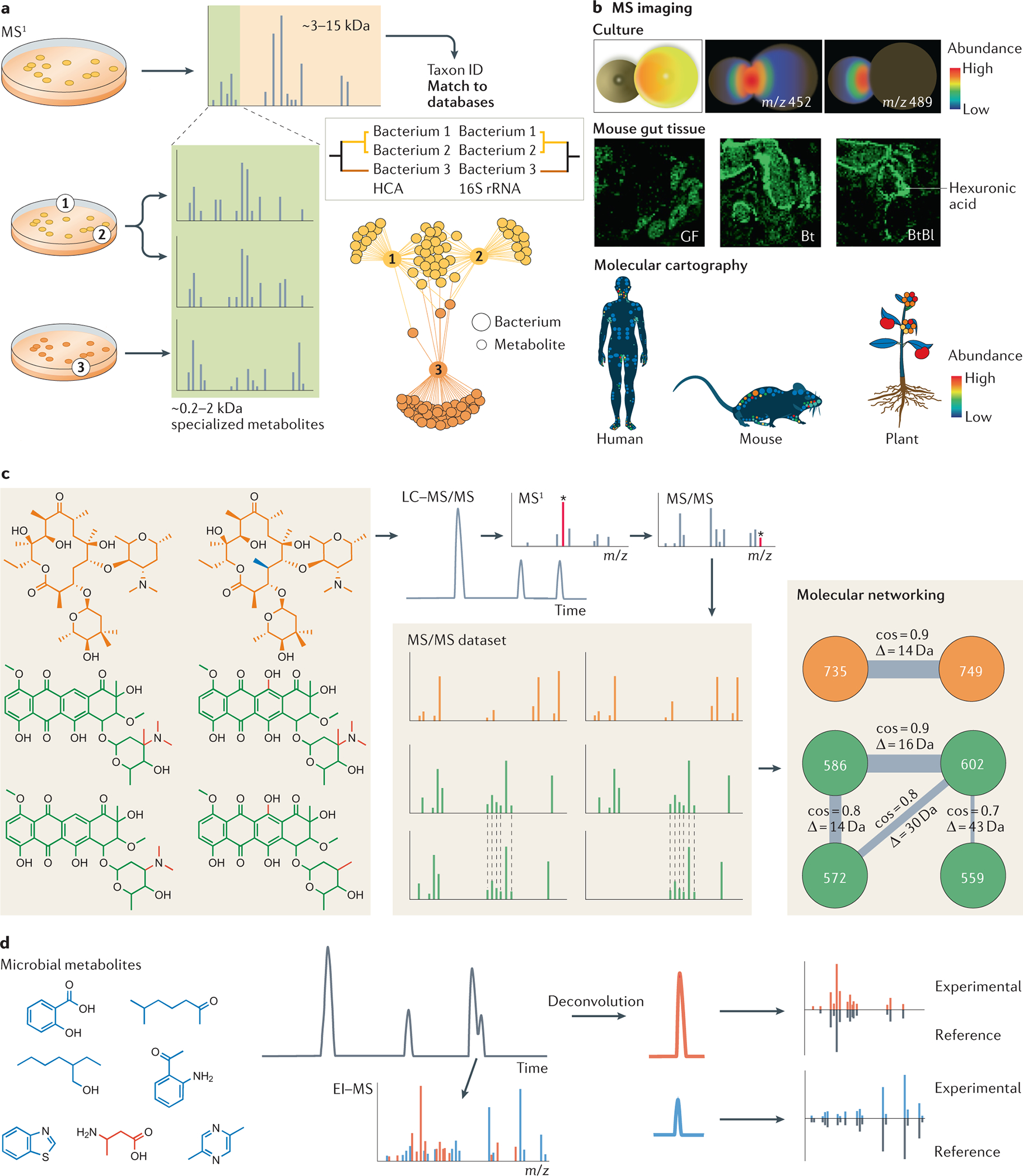
a) MS1 acquired by matrix-assisted laser desorption-ionization mass spectrometry (MALDI-MS) enables bacterial taxon identification. The range of ribosomal proteins (3–15 kDa) is used to search for a match in spectral libraries, and the hierarchical clustering generated with these data strongly correlates with 16S rRNA. The range between 0,2–2 kDa shows specialized metabolites (molecular association network)31, 32. b) Illustrative examples of imaging MS. Interactions between microorganisms can be observed by co-culture experiments (top panel). Spatial distribution of hexuronic acid in the gut of different mice can be investigated (middle panel). The examples shown are from germ-free (GT) mice, mice mono-colonized with Bacteroides thetaiotaomicron (Bt), and mice bi-colonized with Bt and Bifidobacterium longum (Bl). and molecular cartography can reveal the 3D distribution of specific ions in humans, mice and plants (bottom panel). c) Microbial metabolites can be analyzed by liquid chromatography–tandem MS (LC-MS/MS)156. The precursor mass is selected in MS1 to be fragmented, generating the MS/MS spectra. Thousands of MS/MS spectra are generated in an untargeted analysis, which can be organized by molecular networking by spectral similarities. Spectral similarity is represented by cosine score (cos), the higher the cosine the higher the similarity. D is the mass difference between two nodes (precursor ions) d) Microbial small metabolites analyzed by electron ionization (EI-MS). Deconvolution is essential to separate spectra from co-eluting compounds. The spectra can be searched for a match in spectral libraries to annotate known compounds. Images in part b adapted from Ref 36.
MS1-based microbial analysis can be performed in a spatial manner (imaging) by different MS-techniques, including MALDI, REIMS, DESI, and nanoDESI, which was reviewed27, 28. MS imaging (MSI) can be used to understand the metabolic exchange of two or more microorganisms, from a simple microbial culture to a histological slice, in two or three dimensions33, 34. It can be combined with fluorescence in situ hybridization (FISH) to observe the distributions of microorganisms35 and applied to understand the molecular distributions of a living host via 3D cartography36 (Fig. 1b). In 3D cartography, samples are taken, analyzed by MS, and the data are mapped onto the 3D surface or volume37–39. Tools such as ìli40, METASPACE41, Scils [https://scils.de/] and MSiReader42 can be used for visualization of the data. MS-based imaging in combination with other data types such as metagenomics, 16S inventories or transcriptomics can be used to establish the relationships of the spatial patterns of molecules to microbial communities.
One challenge for untargeted MS, specifically methods that only acquire MS1 data, is the annotation (identification) of the signals observed in the data. Annotation is commonly performed using a combination of exact mass, isotope patterns, retention times (if chemical separation performed), collisional cross section, or structural information provided by MS/MS [G] (also known as tandem MS or MSn). Commonly, expert MS users perform identification by analyzing one spectrum at a time; however, this is impracticable at the untargeted MS scale. One way to interpret MS/MS at a large scale is to compare the MS/MS spectra to reference MS/MS of known compounds25, 43 (Supplementary Box 1). This process is akin to matching a short sequence to a sequence in DNA, RNA, protein knowledgebases or repositories that already has an annotation or function assigned to that specific sequence. In the case of liquid chromatography [G] (LC)-MS (Fig. 1c) data can be collected via data-dependent acquisition, which functions by acquiring a survey scan after which the intact ions (also known as charged molecules) are isolated, often based on signal intensity, and subsequently fragmented, and the MS/MS spectrum is collected. This process occurs repeatedly during the duration of the experiment. Following the acquisition, the MS/MS spectra are searched against an MS/MS spectral reference library.
One specific noteworthy case is gas chromatography [G] (GC) coupled to an MS with an electron ionization (EI) source44, 45 (Box 1). To obtain MS/MS spectra from GC-EI-MS data, the data have to be deconvoluted (Fig. 1d). There are many packages to deconvolute, and representative examples include AMDIS46, MS-DIAL47, XCMS48, MZmine49/ADAP50, and GNPS [https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp] which enables deconvolution online using MSHub16. Once deconvolution is accomplished, the ~1.2 million public and commercial libraries can be searched via spectral similarity searches 9. As there is usually no precursor mass information with EI-MS, multiple matches to the spectral libraries are common. In principle, a Kovats index, a form or retention time comparison, could be used to help narrow down candidates. Unfortunately, only a few percent of the reference libraries have this value available. Most GC-MS instruments are low-mass resolution instruments, although high-resolution instruments are becoming available. In principle, a high resolution GC-MS instrument should provide improved annotations, but the lack of high resolution MS/MS libraries means that this improved data quality cannot yet be readily leveraged. The metabolomics community has come up with different levels of annotation confidence: four levels for the metabolomics standards initiative [G] (MSI)51, and five levels for Schymanski [G]52. In general, GC-MS spectral matching is a level 3, or molecular family level match, as the co-injection of an authentic standard, ideally with labeled isotopes, is necessary to get to a level 1 annotation (both for MSI and for Schymanski)51, 52. 2D GC-MS is another GC-MS technology that is becoming more widely available, and it refers to additional chromatographic separations that improves the distinction between similar molecules prior to MS analysis.
There is also a growing interest to monitor the microbiome at the individual cell level and the flux of metabolites. Mass spectrometry can be used to monitor the microbiome in both of those dimensions. Dynamics of metabolism can be monitored using unsteady-state flux balance analysis (uFBA), which measures the flux balance in dynamic systems such as the microbiome53. Flux is modeled using isotopically labeled reagents and is generally reserved for non-human studies54. There is also more and more interest in single-cell analysis. Single-cell metabolomics analysis has been possible for cultured microorganisms55, 56 but there is an emerging interest to apply single-cell methods in microbiome-based investigations. So far this has been largely limited to sequencing- and imaging-based technologies but single-cell metabolomics could provide an important complementary view57, 58.
Overall, MS has shown to have a great potential in providing answers for important questions raised about microorganisms in microbiomes. The interpretation of untargeted MS experiments needs to be treated with care as the results are affected by the collection, extraction, and sample preparation protocols in addition to the data acquisition and data analysis tools9, 25. There are numerous challenges (Box 1). All these options are reasons why untargeted MS and the interpretation of the data are not trivial9, 25. As an alternative or complementary approach to MS analysis, X-ray crystallography, ultra-violet, infrared or nuclear magnetic resonance (NMR) spectroscopy can be used. Each of these techniques have pros and cons, the discussion of which is outside the scope of this review. For example, NMR, despite having lesser sensitivity then MS, presents unique strengths in metabolomics and is thus a great complementary technique (reviewed in Ref. 59).
Microbial metabolite annotation
An initial step to tease apart the complexity of interactions in microbiomes is the annotation of detectable metabolites. In the past few years, many new algorithms and computational tools for improving this step in MS-based metabolomics have been introduced. MS/MS is frequently used (in conjunction with MS1 data) as it provides more structural information. The levels of confidence concerning the annotation are variable with the highest level of identification being a direct comparison to chemical standards, or via the isolation and complete structural characterization (MSI level 151 or Schymanski level [G] 152). In this section, we present some of the computational tools that can assist in metabolite annotation when chemical standards are not available (Fig. 2), along with a brief discussion of their characteristics, pros and cons, and the contextualization of how they can be used for investigations of microbial metabolites. It should be noted that although these tools speed up the process and can guide annotation, the results should be interpreted with caution. These advanced annotation tools can be invaluable in microbiome studies, and when used properly, errors are minimized.
Fig. 2: Computational tools for metabolite annotation, substructure assessment and chemical classification.
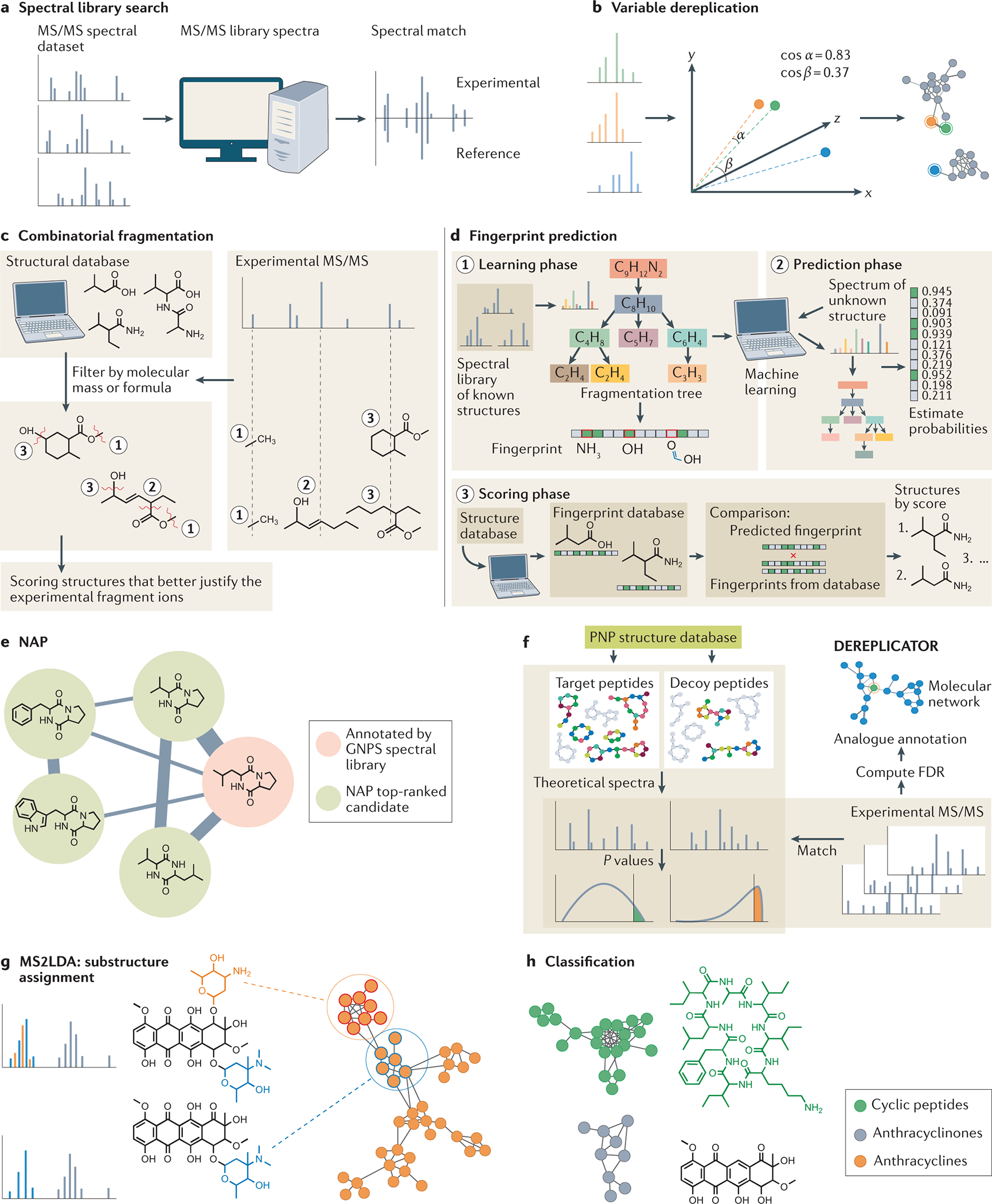
a) Tandem mass spectrometry (MS/MS) spectra can be searched against the MS/MS spectral library (for example, GNPS [https://gnps.ucsd.edu/ProteoSAFe/static/gnps-splash.jsp]) and matched based on the number of product ion matches and cosine score. b) Variable dereplication (GNPS) allows the search of structurally related metabolites (analogs) with similar MS/MS spectral data by employing the cosine similarity method. c) MetFrag71 is a combinatorial fragmentation method that focuses on the explanation of the fragment peaks from an MS/MS spectrum based on substructures generated by disconnecting the bonds of the structures from structure databases. d) SIRIUS473 and ZODIAC74 use fingerprint prediction, a fragmentation tree method to predict fingerprints (substructure properties), to score possible structures by fingerprint similarity. e) Network annotation propagation (NAP) integrates variable dereplication and combinatorial fragmentation for annotation of analogs in molecular networks. f) DEREPLICATOR annotates nonribosomal peptides and ribosomally synthesized and post-translationally modified peptides based on hypothetical spectral fragments generated from peptide natural product (PNP) structures present in structural databases, considering the false discovery rate (FDR). In addition, this tool can be used to annotate analogs by variable dereplication and also to calculate statistical significance computing false discovery rates [G]. g) MS2LDA recognizes substructures and their co-occurrence in an MS/MS dataset h) MolNetEnhancer uses such substructure information, along with ClassyFire algorithm, to classify the chemical groups present in the dataset.
Spectral libraries.
Spectral library search (Fig. 2a), mentioned above, is the most common method used for annotation of known compounds. Each experimental MS/MS spectrum is compared to reference spectra of known compounds stored in MS/MS spectral libraries, such as GNPS spectral libraries16, MassBank (Japan, EU and North America)60, NIST61, and METLIN62 and many others. The availability of chemical standards limits the scope of spectral libraries and is the reason that the majority of MS/MS spectra in these databases are from synthetic and commercially available chemicals. One key limit of a spectral library matching approach for studying microbial molecules is that most microbial molecules are not commercially available and thus are not well represented in spectral libraries. To address this limitation, GNPS16 enables contributions to its MS/MS spectral library directly from the community and data acquired from samples in addition to chemical standards. Due to a large community effort, the amount of MS/MS reference spectra for microbial molecules is growing rapidly. For known molecules only to be produced by microorganisms the library expanded from ~200 reference spectra in 2014 to thousands in 2021 in the GNPS infrastructure of the ~25,000 known microbial metabolites. Although the current MS/MS reference spectra only cover a fraction of microbial molecules, untargeted metabolomics can provide direction in studying which microorganism or microorganisms produce metabolites of interest.
It is crucial to realize that more than one annotation is often possible when comparing data to spectral libraries, such as for GC-EI-MS, as there is generally no precursor mass to filter the library with, and thus many related molecules match. High (mass) resolution combats multiple matches based on the exclusion of particular molecular formulae and isotopic patterns, hence TOF, Orbitrap and FT-ICR mass spectrometers are preferred analyzers for many metabolomics applications. Further, the extent to which a precursor ion fragments and the number of product ions that match reduces the possible number of spectral matches. Although not in all cases, it is commonplace that MS/MS spectral matching meets the requirements for an identification level 2 according to the 2007 standards initiative51. At the same time, lipids or fatty acids annotation are limited to level 3 given the number of regio- and stereoisomers. The goal of spectral matching is to narrow down the candidate molecules represented by the MS/MS signal. Thus, when one has spectral matches against the library, additional knowledge about the sample or orthogonal measurements such as co-migration with a chemical standard is needed to increase the confidence of the annotation. To complement the manual interpretation of spectral library matching accuracy, methods of controlling false discovery rate (FDR) for spectral matching are actively being developed but are not yet widely utilized in metabolomics63, 64, unlike in proteomics, for instance, where FDR methods based on the target-decoy strategy is already well-established63, 65. A study proposed empirical Bayes and target-decoy based methods to estimate the FDR in metabolomics63, 64. Assessing the FDR based on the target-decoy strategy for 70 public metabolomics data sets, it was observed that the scoring thresholds have to be adjusted for each dataset because there is a strong dependency on the number of fragmented ions in an MS/MS spectrum.
The MSI levels [G]51 (as well as the Schymanski)52 are not failsafe, and often annotations fall between levels. For example, an MS/MS spectral match to cis-2,3-hexenoic acid, the MS/MS spectrum within the context of a typical collision-induced dissociation (CID)-based untargeted MS experiment can differentiate neither the stereoisomers (cis- versus trans-) nor the position of the double bonds, and, therefore, considered a molecular family match or MSI level 3. Even with a standard with the correct m/z and matching retention time, one cannot rule out that other isomers do not have the same retention time. Level 1 annotation for this chemical requires additional orthogonal approaches and/or co-migration with authentic standards.
On average, using reference libraries, only 2–20% of MS/MS spectra are annotated in an untargeted MS experiment66, 67. One strategy of expanding the utility of spectral libraries, and increasing the number of candidate annotations, is through leveraging the modified cosine score as used in molecular networking [G]16, variable dereplication38, or hybrid searches68. In molecular networking (Fig. 1c), MS/MS spectra are aligned, in a process that is very similar to finding related genetic sequences by alignment. The spectral alignment in these instances is defined by using a modified cosine score, and variants with structural modifications can be detected (Fig. 2b). When visualized as networks, one can infer structural similarity. And just like sequence alignments enable the discovery of mutations and alternative splice forms, spectral alignment enables the discovery of analogs of molecules that match MS/MS reference libraries. When combined with metadata from a study (for example, germ-free versus microbiome-colonized, or healthy versus disease) it is possible to discover specific molecules associated with phenotypes37, 38, 69, which can be a powerful strategy for microbiome investigations. Molecular networking is one such approach for analyzing LC-MS/MS data and has been used to understand the chemistry of microorganisms and microbial communities. Recently, molecular networking via GNPS has been developed for analyzing GC-MS17 data which was successfully applied in an investigation of quorum sensing during fungal–bacterial interactions70. The remaining data that cannot be annotated by direct matches or similarity matches need alternative methods for annotating, and in silico annotation platforms are continuously improving.
In silico tools to improve metabolite annotation.
Several in silico annotation tools [G] have been developed to overcome the limitations of spectral library searches. Reference spectral libraries are incomplete compared to molecular structure databases, such as Pubchem. Combinatorial fragmentation methods (Fig. 2c), such as MetFrag71 and Competitive Fragmentation Modeling (CFM-ID)72, explain the fragment peaks in a given MS/MS spectrum based on substructures [G] generated by disconnecting the bonds from the known structures. Fingerprint prediction methods (Fig. 2d), such as SIRIUS473 and ZODIAC74, leverage fingerprints based on fragmentation trees for experimental spectra using machine learning [G] trained fingerprints from known structures. Hydrogen rearrangement rules during bond cleavages in low-energy fragmentation are used in tools, such as MS-Finder75. In all cases, in silico approaches, create a list of candidate structural matches from the MS data using structural databases. As with spectral matching, it is rare to obtain a unique match within acceptable scoring cutoff values, multiple matches should be cautiously trusted and interpreted.
In silico tools have been integrated into molecular networking via network annotation propagation (NAP)76 (Fig. 2e). NAP compares the structural candidates that are assigned to a specific MS/MS spectrum (node in the molecular network) and then, following the assumption that connected nodes in molecular networking correspond to similar molecular structures, NAP re-ranks the structures when a neighboring node has a related structure in the molecular network. An alternative strategy of using networking to propagate annotations, especially when some structural knowledge resulting in a more defined scope of candidate molecules is available to the user, is through either prediction of candidate-related molecules that might be present in the sample or using biotransformation logic to predict candidate molecules77, 78.
The above in silico approaches use structure databases; however, MS data can be linked with biosynthetic logic that is responsible for their production. MiBIG (minimum information about a biosynthetic gene cluster (BGC)) is currently the only repository, known to the authors, that links natural product structures directly to microbial gene clusters79. During the writing of this Review, a method that uses multiple link-scoring functions to link gene clusters, molecules and mass spectral data was reported80.
It is worth mentioning that BGC-based analysis is largely limited to protozoan microbiota and does not apply to viral or phage as they, generally, do not encode for many genes that make or modify metabolites. For some classes of bacterial molecules, it is easier to establish a link between BGC and metabolites than others81, 82. The analysis of bacterial genome databases revealed that approximately 70% of gene clusters that encode bacterial molecules contain some domain to produce nonribosomal peptides (NRPs) or ribosomally synthesized and post-translationally modified peptides (RiPPs)82. Although new methods continue to be developed for NRP and RiPP discovery within genomic and MS data, they are among the easiest to find even though NRPs and RiPPs can be extensively post-translationally modified or include hundreds of different amino acids generated by dedicated biosynthetic machineries and decorated appendages (fatty acids, halogenations, oxidations, cyclizations, among other). DEREPLICATOR83 (Fig. 2f) was designed to find such molecules. DEREPLICATOR constructs fragmentation graphs from natural product libraries such as ‘dictionary of natural products’ and AntiMarin to statistically compare the experimental spectra. DEREPLICATOR+ (Ref. 83) expands this approach to non-peptidic molecules, whereas VarQuest84 was developed to work independently from molecular networking to also enable the annotation of candidate structural analogs. Other tools that provide structural insight into detected MS/MS spectra by leveraging biosynthetic logic are Pep2Path85, Glycogenomics81, iSNAP86, RiPPquest87, NRPquest88, and DeepRIPP89. Each of these can be used to discover microbial metabolites from MS/MS data by leveraging genome sequence data. There are also metabolic models that leverage genomics data to discover microbial metabolism and will be discussed below.
As the annotations that result from in silico tools are computational matches, the confidence of the annotation is not the same level of the spectral matching, there is no level of accuracy proposed for in silico matches and the interpretation of the data has to be carefully checked. Some of these tools are based on the disconnection of chemical bonds to predict the spectra or the fragmentation; however, there are many fragmentation pathways based on rearrangements of parts of the chemical structure, which cannot yet be easily predicted. Moreover, future machine-learning approaches in combination with ever growing spectral reference libraries will improve the overall understanding of fragmentation pathways. Most often in silico annotations should be considered to be at the molecular family level or level 3 according to the metabolomics standards initiative51; however, even the assignment at the family level still needs additional validation, which can be achieved through manual inspection of the data as well as the inspection of substructure assignments. The other opportunity is that there are databases rapidly evolving that are dedicated to microbial metabolites, one of them is NPAtlas90, or databases that are based on text mining strategies that construct many loose associations91 but also have the ability to mine data from emerging metabolomics databases with strategies that enable the search for mass spectral features across the entire database, such as MASST92, Thus, there is a need for the development of improved microbial structural databases and metabolomics search engines related to the microbiome.
Substructure assignment.
The recognition of molecular substructures, such as glycosyl moieties or carboxyl groups, can provide relevant biochemical information to understand processes occurring in microbiome ecology. Although often it is not possible to completely assign a structure, it may be possible to recognize parts of molecules in fragmentation spectra through understanding specific fragments and/or neutral losses. These substructures can be annotated by MS2LDA through the recognition of the co-occurrence of patterns in MS/MS data 93 (Fig. 2g). MolNetEnhancer 94 (Fig. 2h) combines outputs from MS2LDA, NAP, DEREPLICATOR and molecular networking, along with the automated chemical classification from ClassyFire [G]95, to assign structural features to chemical classes. Canopus96 and Qemistree97 are also tools for chemical classification. The former uses neural networks [G] to improve the annotation of spectra that are not in the library, whereas the latter organizes and classifies MS data in a tree. Both tools can be combined with metadata to overlay biological or microbiome context to the chemical patterns observed. Multistage MSn spectral trees can also be used to assign substructures by recognition of hierarchical fragmentation patterns (for example, MAGMA)98 to characterize a molecular structure and get insights into the fragmentation pathway; such spectral trees are the foundation for substructure analysis with the commercial tool, such as mzCloud99. All of these tools together can provide invaluable information about the chemical content from an MS data set and have only recently been starting to be applied to investigate the chemistry of the microbiome100.
Making connections
The previous sections focus on the annotation of the specific molecules associated with an untargeted MS data set; however, many microbiome investigations aim to understand the global relationships of the molecules that are generated by microorganisms. To date, just few tools are available for this purpose (Fig. 3). Most of them are difficult to understand, requiring knowledge of very complex algorithms and statistical concepts. In this section, we present and discuss how some of these tools can be applied to study microbial interactions in microbiomes. Although it is still challenging to connect metabolites to specific microorganisms, molecular networking has been used to draw comparisons between samples and reference datasets, including data from isolated microbial cultures38, 69. A representative example is shown in Fig. 3a, in which lung samples from patients with cystic fibrosis were collected and split into two parts, one directly analyzed by LC-MS/MS and the other part was streaked onto Petri dishes for microbial isolation, and then both were analyzed by the same LC-MS/MS method. Molecular networking of both datasets enabled the discovery of metabolites in the host data that were produced by microorganisms even when cultured microorganisms produce slightly different versions of the molecules (for example, different fatty acids available that are used in the biosynthesis). Furthermore, as metabolites in culture are often slightly different (for example, different alkyl chain lengths due to promiscuous acyl-CoA loadings), molecular networking can contribute to finding connections between metabolites and microorganisms. The strategy enabled the association of Pseudomonas aeruginosa with end-stage cystic fibrosis disease by connecting the bacteria with the following metabolites: quinolones (2-heptyl-4-quinolone (HHQ), 2-nonyl-4-quinolone (NHQ) and 2-nonyl-4-quinolone-N-oxide (NQNO))69. Molecular networking can highlight molecules that are uniquely produced by microorganisms. It is much more complex to tease apart shared primary metabolite attribution, as such shared metabolites can be both produced by the host or the microorganisms (or come from diet directly). In such cases, feeding studies with labeled substrates and careful quantitation will need to be performed. Currently, no metabolomics methods exist that enables researchers to readily understand microbial contributions of shared metabolism that produce the same metabolite; however, high spatial resolution flux analysis101 may hold the key to separating each respective contribution. If a reliable low-cost method was available, it would be transformational for the functional understanding of the microbiome.
Fig. 3: Data analysis tools to uncover microbiome-derived molecules.
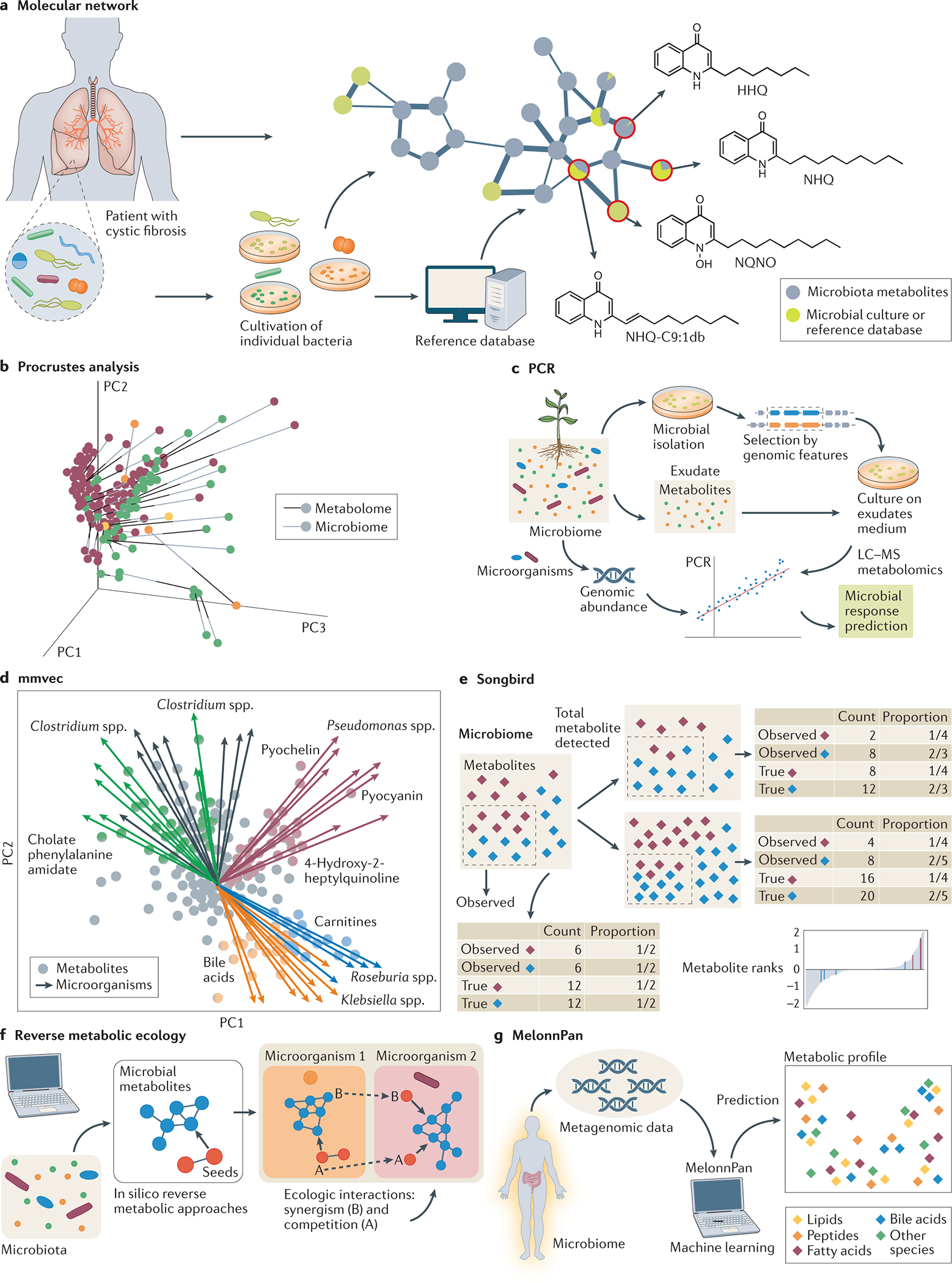
a) Molecular networks can attribute the producer of specific metabolites detected in microbiome, from cultured systems or reference databases. b) Procrustes analysis allows integration of omics data based on canonical correlation. The results are summarized in a low-dimensional space representation known as principal components (PC1, PC2 and PC3) c) Principal component regression (PCR) is a statistical method based on regression analysis and principal component (analysis. In the example, metabolomics and metagenomics data were integrated to investigate the microbial response to plant growth. d) mmvec uses co-occurrence probabilities to predict microorganism–metabolite interactions from metabolomic data and is visualized with a biplot. The results are shown in three-dimension space and the illustration shows two principal components (PC1 and PC2). e) Songbird introduced ‘reference frames’ by using ratios to compute the abundance of compositional data overcoming common pitfalls in comparing relative abundances across samples. f) Ecological interactions, such as competition or synergistic (for example, symbiosis), can be predicted by reverse metabolic ecology. Seeds are known as specific metabolites used to evaluate the interaction. g) MelonnPan is a machine learning method, trained with metabolomics and metagenomics data, aiming to predict the metabolome of microbial communities, including those metabolites usually not observed by common analytical techniques.
Spotting data trends.
Spotting patterns in untargeted MS data is challenging given the number of variables detected, thus multivariate analysis is immensely helpful. There are tens to hundreds of different methods to uncover data trends, including many unsupervised multivariate statistical methods (for example clustering analyses, PCoA102, and PCA102, principal component analysis)) and supervised multivariate statistical methods (for example, partial least squares regression discriminant analysis (PLS-DA)103). PCA is widely used in metabolomics and creates uncorrelated variables to maximize variances in the data102; it is usually used to understand the chemical similarity of samples holistically and interpreted with metadata (post-computation) to reveal the rationale for the separation of samples. Further, the PCA loadings, interpreted as vector quantities, indicate the variables which contribute to the separation. Similarly, PCoA can be used to analyze untargeted MS data with different distance metrics than that of the Euclidean distance used in PCA102. Contrasting unsupervised multivariate methods, supervised multivariate methods of data analysis, such as PLS-DA103, use class labels (metadata) in calculations, viz. healthy versus unhealthy. Although supervised methods are useful in extracting the variables that contribute to the separation, they are fallible (overfitting is a primary concern) and all important variables (chemicals or microorganisms) should be evaluated carefully.
Further, PCA can be combined with linear regression (for example, to model the relationship between independent and dependent variables), which is called principal component regression. Linear component regression can be applied to define features (principal components) that are modified in response to a particular phenotype104, 105. Principal component regression was used to accurately predict the microbial response to changing nutrients 105 (Fig. 3c). The microbial response was evaluated by metabolomic analyses of the culture of isolated microorganisms, in which the consumption or production of specific substrates related to the abundance dynamics was observed. In this example, the integration of the data by principal component regression enabled the prediction of bacterial behavior. The combination of these experiments provides evidence of plant–microbial interactions, in which the plant regulates the molecular composition of its rhizosphere to manipulate the microbial community for its own benefit. It is likely that this approach can be leveraged to study other microorganism–host systems.
It should be noted that all omics data not only contain immediate response to specific evaluated challenges or effects, but also inform a multitude of other factors, such as age, sex, diet, medications, lifestyle, genetic background, among others. Chemicals derived from such factors can often be obtained from untargeted metabolomics, and the discovery of such potential confounders that can be reused as metadata to a microbiome project is currently underutilized. A well-curated metadata can be leveraged to correct errors. Additional methods such as regression analysis can also be used to identify and perhaps remove the confounders106. There are other resources that are emerging and are becoming available that enable researchers to discover metabolites from diets, exposome or other sources67, 107, 108. However, they have not been used to link microbial metabolism or to leverage microbiome linkages, and these are good opportunities to provide additional context to microbiome studies.
Connecting metabolites and microorganism.
Although commonly of interest, drawing connections from chemical–chemical, microorganism–microorganism or chemical–microorganism interactions remains immensely challenging. Correlation analyses, in general, are statistical methods to evaluate and predict possible connections between two or more variables that can be either quantitative or categorical. Pearson109, Spearman110 and Kendall111 correlations generate correlation coefficients that measure the strength of the relationship, which can vary from −1 to +1, representing a perfect negative or positive correlation, respectively. These correlation methods can be integrated with molecular networks for visualization112, and are commonly performed in microbiome studies to find, for instance, which metabolite is positively or negatively related to a specific microorganism or event. These methods tend to work well with infrequent observations (for example, an observation in less than 5% of the samples) but are also very prone to incorrect correlations, especially when false discovery rate is not controlled15, 113. Data compositionality (that is; data that are naturally described as proportions or probabilities or with a constant or irrelevant sum) is problematic for correlation methods as accuracy of correlation methods strongly depend on the on the variance of the data. Data are compositional114 in sequencing techniques, and although MS data are not inherently compositional, certain normalization, standardization or transforms can introduce compositionality. It cannot be overstated, that these connections are merely associations (correlations) and do not indicate causation. An understanding of causation requires further experimentation, analysis and interpretation. For integration analysis, the reader should be aware of the nuances associated with each of the technologies, regarding sample acquisition and processing; please see Box 1 for metabolomics and these reviews for genomics115, 116.
Data analysis tools based on co-occurrence (that is, co-occurrence networks) focus on the frequency of a specific feature occurring between different datasets117. Microorganism–metabolite vectors (mmvec)15 (Fig. 3d) uses the probability of co-occurrence instead of correlation methods, in which the presence of a specific metabolite is conditioned to the observation of a particular microorganism in a microbiome. As it is based on neural networks, this approach tends to work best with large data sets. In mmvec, and neural networks in general, the learning rate is an important parameter to be adjusted as it is responsible for determining how fast the model adapts to the datasets entered. In this context, Songbird (Fig. 3e) introduces ‘reference frames’, to overcome false-positive rates when comparing relative abundances118. The idea is similar to a well-known concept in physics, in which the velocity of one object can be measured relative to another moving object. In this sense, the microbial population can be measured as a reference frame to another microbial population and can also be applied to chemical-to-microorganism relationships. Thus, strategies are emerging to infer microbial-metabolite relationships regarding both the absolute and relative abundances106. It is crucial for the field to continue to develop new correlation approaches. It is also likely that there are specific data collection and processing circumstances where one works better than others, but such guidelines are not yet clearly delineated, even for the existing approaches.
Procrustes analysis (Fig. 3b) is one method by which to integrate different data types and visualize paired -omics data based on the correlations (canonical correlation) between their loadings14. In other words, this method shapes the distribution of two or more datasets or matrices with different representations of the same system (for example, genomics, proteomics, exposomics or metabolomics). When the microbiome data and untargeted MS data, represented as separate points, are more similar, they are closer to one another and reflect a stronger correlation. There are now a few examples when Procrustes have been used to understand the relationship of the metabolome to microbiome sequencing data119–121.
As sequencing has become more affordable, more emphasis is being placed on obtaining functional insights. 16SrRNA and often, shallow shotgun sequencing, fail to point to the functional metabolome or fail to identify the functional biosynthons. In such cases metabolomics analysis can be a complementary approach or even provide the sole data on function122, 123. Even though is difficult to connect function from 16S inventories, the community has developed other approaches to overcome such limitations. One of them is PICRUSt (phylogenetic investigation of communities by reconstruction of unobserved states)124, 125, which predicts the functional composition of a metagenome using marker gene data and a database of reference genomes. Another approach is Mummichog126 which predicts the functional activity of a metabolite by organization of metabolic networks and pathways. These prediction tools can be used to give additional support to the connections drawn.
Pathway analysis.
Reconstruction of the metabolome and pathways based on genomic information127, such as KEGG128, WikiPathways129 and MetaCyc130, iis one way to predict the molecules that might be present in metabolomics experiments. This approach has been expanded to predict the causality of microbial community dynamics. Following an observation in ecology, the cause of a specific ecological phenomenon is established. This concept was integrated into the algorithms used in the reverse metabolic ecology131 (Fig. 3f) approach, in which the metabolic profile of a community (the biochemical environment) is inferred from genomic or metagenomic data, and the metabolic information is leveraged to predict the ecological phenomenon. Different relationships can be observed in a community or microbiome, such as competition for the same nutrient or synergism. In the case of nutrient dependency, such as essential amino acids, lipids or vitamins, one organism tends to co-occur with another (that is, one species depends on some nutrient provided by other species). Based on these concepts, the first step in reverse metabolic ecology is to define the ‘seed set framework’ (the characterizations of the biochemical composition of an organism’s habitat based on its genome), in which seeds mean metabolites considered important for relationships (that is, competition or synergism)132. Subsequently, the method calculates the interface or connections between the seed set and the organisms in the environment133, 134. The metabolic network is represented as a directed graph in which nodes denote metabolites and directed edges connect substrates to products. Therefore, reverse metabolic ecology uses genomic or metagenomic data and metabolic information to raise hypotheses about possible microbial interactions in a given microbiome. This approach was used to find the most promising prebiotic combinations aiming to promote the development of the infant immune system131.
Additional methods based on genomic or metagenomic data135 recognize biosynthetic gene clusters [G] (BGCs) or use machine-learning predictions of metabolites that are most likely to be produced in a given microbial community. MelonnPan 135 (Fig. 3g), for example, is an algorithm developed to predict the metabolites produced by the microbial community based on metabolomic scoring of a combination of genetic sequencing along with biological knowledge. This can then be leveraged to predict the response of a metabolic profile, but also suggests what data need to be collected to validate the predictions. Even though the predicted metabolic profiles still do not present high levels of similarity to the empirically measured metabolome136, such predictions can be very useful to guide MS analysis as there are many different types of experiments that are possible (due to the many combinations of sample preparation, separation, and detection strategies). Strategies such as metabolic pathway-based approaches137 can be used to link metabolites to biochemical pathways (largely consisting of primary metabolites) and gain insight into the function of microbial communities138, 139. But the production of many specialized metabolites, secondary metabolites or natural products are generally not mapped out on biochemical schemas. Metabolites can be connected to BGCs by pathway activity level scoring (PALS)140 that ranks changing metabolite sets over different experimental conditions. The valuable knowledge on metabolic pathways enabled the annotation of metabolite origins via networks (AMON)141, which annotates which metabolite is produced by microorganisms in a microbiome; the ingenuity pathway analysis (IPA)142, which elucidates underlying relationships of metabolites and differentially expressed proteins; and model-based integration of metabolite observations and species abundances (MIMOSA)143, twhich unravel ecological links with metabolic changes. Enrichment strategies based on biochemical knowledge enable the interpretation of the metabolic regulation from the metabolomics dataset by recognizing sets of connected metabolites. These methods are usually applied to primary metabolites, such as sugar and lipids, but fail for compounds that comprise multiple structural components in a single structure. However, tools such as ChemRich can overcome some of these limitations144, once it recognizes these molecules in a non-overlapping way by chemical similarity (spectral similarity networks) instead of fixed definitions of metabolic pathways. One key opportunity for improving metabolic models is to also model microbial molecules that are not part of common and/or primary metabolites. A bottleneck for the inclusion of such metabolites as part of metabolic reconstructions is that they are generally not yet part of existing biochemical pathway maps. It is therefore rare that metabolic models, including those used to understand metabolic flux53, consider specialized metabolites, but this is an important consideration, as such natural products are often produced in very large quantities and must therefore dictate large portions of flux.
Perspective
It is remarkable to think that we are surrounded by an entirely invisible ecosystem (at least to the naked eye) that can be shaped and is shaping us. The magnitude and grandeur of these microbial communities have been revealed by the advancement of more affordable and faster sequencing. However, there is much to learn, for example each microbial genome encodes for the ability to generate hundreds if not thousands of molecules. Further, a functional understanding of most of such molecules is strikingly absent from our knowledge, although insights into the host–microbiota interactions are emerging (see some representative examples in Box 2). We learned that a single chemical can substantially influence the microbiome (Box 2; Supplementary Box 2) when penicillin, vancomycin and many other drugs145, 146 were introduced in the 20th and 21st centuries. However, a much deeper understanding is required to predictably control the microbiome that the scientific community aspires to achieve.
Box 2. How do microbial metabolites shape the hosthealth and behavior?
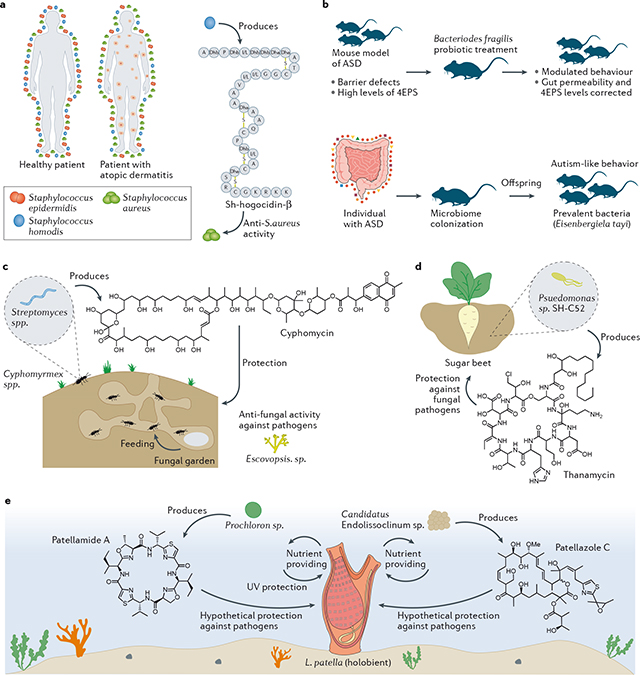
Several human diseases and conditions are associated with microbial dysbiosis, which can even impact human behavior and the proper functioning of organs. For instance, bacteria from the human gut can exert effects on the host immune system, from systemic autoimmunity170 to the evolution of cancer171. It is also known that external cues such as diet can influence the gut microbiome, and a shift in the microbiome may be related to metabolic syndromes, such as type 2 diabetes and nonalcoholic fatty liver disease172. Dysbiosis has also been observed to affect other environments, such as the soil, important for agriculture and production of food; the ocean, which affects biodiversity; and in the insects community; among several others examples not further specified here. Some examples from different organisms and environments are discussed that highlight the efforts in studying microbial-derived molecules and their role in microbiomes and hosthealth and behavior. Staphylococcus hominis has an important role in maintaining the equilibrium of the human skin microbiome. The commensal produces the metabolite Sh-hogocidin-β, which inhibits the growth of S. aureus. Dysbiosis of the skin microbiota (that is, a decreased abundance of S. hominis and thus increased abundance of S. aureus) can contribute to atopic dermatitis 173 (see the figure, part a). In addition, several studies report on the possible link between the gut microbiota and symptoms of nervous systems diseases, such as autism disorder 3. In a mouse model of autism spectrum disorder (ASD) it was shown that treatment with probiotics containing Bacteroides fragilis restored barrier integrity, decreased serum levels of the metabolite 4-ethylphenyl sulfate (4EPS) and modulated behaviour (see the figure, part b, top panel)174. The same group3 transplanted gut microbiota from individuals with ASD into mice and reported that the offspring presented autism-like behavior (see the figure part b, bottom pane), further suggesting a role of the gut microbiota in the development of neurological disorders.
In social insects, Streptomyces spp. colonize the cuticles of ants and produce cyphomycin, a compound that protects the ants’ fungal garden against fungal pathogens 175 see the figure, part c). In the rhizosphere, Pseudomonas sp. SH-C52 produces thannamycin to protect the sugar beet from fungal pathogens 6 (see the figure, part d). In the marine environment, an interesting example is the ascidian Lissoclinum patella, in which the colonizing bacteria produce compounds that may protect the host. In return, the ascidian provides essential nutrients for bacterial survival 8 (see the figure, part e).
Therefore, it is imperative that a large inventory of microbial-derived metabolites and their functions is established. Further, this includes the understanding of their interconnectivity as well as database and knowledgebase resources of how microorganisms process different metabolites, including natural products, medications147–149 and not only the active ingredients), and available nutrients150–152. The interaction of chemicals with ecosystems, and how chemicals influence drivers of ecosystems such as pH, salinity, temperature, oxygen, natural defenses should be investigated153–155. A major obstacle to advancement in these areas is the lack of machine-readable data and information in centralized resources. The sequencing field has adopted data sharing and data repositories, whereas the MS field lags behind, but can benefit from learned lessons. The first steps towards the transition from snapshots of microbial or chemical inventories to contextualized and deep understanding of function, effect, and meaning are near.
How do we advance? We must improve our understanding of the chemistry of individual organisms at a functional level, the chemistry of the microbial interactions with other microorganisms and host cell types, the effect of chemical exposures to the microbiome, and more. MS, specifically untargeted metabolomics, and sequencing are naturally complementary, and the wealth of information, when used in combination, is largely untapped. It will not be one laboratory that will solve these puzzles, but it will require the community to share knowledge, as well as systematic and reproducible analysis pipelines and collaborations.
Supplementary Material
Acknowledgments:
A.B. acknowledges the FAPESP (2018/24865-4), H.M.-R. the Brazilian Fulbright Commission and CNPq (142014/2018-4), L.V.C.-L. the FAPESP (2015/17177-6, 2018/07098-0) and CNPQ (443281/2019-0, 306913/2017-8), and P.C.D. the NIH (1 U19 AG063744), The Gordon and Betty Moore Foundation and Crohn’s and Colitis foundations for financial support.
Glossary:
- Alpha diversity
A metric that summarizes how many taxonomic groups or unique molecular features (species richness) and the evenness or balance of those microorganisms (species diversity) or molecular features that can be detected in the sample
- Beta diversity
This is the ratio between the diversity of molecules or organisms in the entire data set divided by the diversity of the specific sample. This metric represents the diversity of microbial communities across different environments, also referred to compositional heterogeneity
- Rarefaction
It is a strategy whereby the summed number of unique data points (for example, OTU’s in microbiome data or MS/MS in mass spectrometry) are inventoried with each sample that is added to the sample set. It is often used to standardize the samples of different sizes and determine whether a sample has been sequenced enough in order to represent its true diversity
- Procrustes analysis
A statistical model based on canonical correlation to shape the distribution of two or more groups of features from different omics datasets
- mmvec
A simple one-layer neural networking strategy using bi-loglinear multinominal regression to predict the probability for co-occurrence relative to metadata
- MS1
Precursor mass of the intact molecular ion
- MS/MS
Also known as MS2. The fragment ion spectrum
- Molecular networking
A computational algorithm that organizes fragmentation spectra into a dataset by spectral similarities from which structural similarity is inferred. It is a neutral mass difference network obtained via spectral alignment
- Metabolomics Standards Initiative (MSI)
A formal definition of metabolite annotation and identification of the metabolomics standard initiative. It comprises four levels: Level 1 represent the identified metabolites; level 2 represents the putatively annotated compounds; level 3 – represents the putatively characterized chemical classes or molecular family; level 4 represents unknown but real mass spectrometry signal
- Schymanski
A system for metabolites annotation. Level 1 represents the confirmed structure; Level 2 represents the probable structure; Level 3 represents the tentative candidate of compound class; Level 4 and 5 correspond to an unequivocal molecular formula assignment, and exact mass of interest that still lacks molecular formula assignment
- Substructures
A small part or a functional group in a chemical entity
- ClassyFire
A hierarchical chemical classification of chemical entities
- Principal Coordinates Analysis (PCoA)
Unsupervised multivariate analysis used to calculate the interrelationships of a data set, and is often used to reduce the dimensionality of large data sets
- Machine learning
Application of artificial intelligence that is able to learn, adapt and improve their accuracy over time without being explicitly programmed
- Neural networks
A set of algorithms designed to recognize patterns and used to classify entities or make predictions. Modeled around the concept of neurons
- Biosynthetic gene clusters (BGCs)
Group of two or more genes in a particular genome that together encode a biosynthetic pathway
- Gas chromatography
A technique to separate compounds in a complex sample which occurs between a stationary phase and a gaseous mobile phase, usually an inert gas such as helium
- Liquid chromatography
A technique to separate two or more compounds present in a sample by exploiting the affinity balance between a stationary phase placed inside the chromatographic column and a mobile phase that flows through
- In silico annotation tools
Computational methods to improve compound annotation by exploring other approaches besides spectral libraries, such as machine learning
- False discovery rates
A method to calculate the proportion of events that falsely seem to be significant
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
References:
- 1.Consortium THMP & The Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sender R, Fuchs S & Milo R Are We Really Vastly Out numbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 164, 337–340 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Sharon G et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 177, 1600–1618.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Claesen J et al. Cutibacterium acnes antibiotic production shapes niche competition in the human skin microbiome.
- 5.Garg N et al. Natural products as mediators of disease. Nat. Prod. Rep. 34, 194–219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mendes R et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332, 1097–1100 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Douglas AE Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morita M & Schmidt EW Parallel lives of symbionts and hosts: chemical mutualism in marine animals. Nat. Prod. Rep. 35, 357–378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aksenov AA, da Silva R, Knight R, Lopes NP & Dorrestein PC Global chemical analysis of biology by mass spectrometry. Nat. Rev. Chem. 1, 0054 (2017). [Google Scholar]
- 10.Misra BB The Connection and Disconnection Between Microbiome and Metabolome: A Critical Appraisal in Clinical Research. Biol. Res. Nurs. 22, 561–576 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Dhariwal A et al. Microbiome Analyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceccarani C et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 9, 14095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilmanski T et al. Blood metabolome predictsgut microbiome α-diversity in humans. Nat. Biotechnol. 37, 1217–1228 (2019). [DOI] [PubMed] [Google Scholar]
- 14. Gower JC Generalized procrustes analysis. Psychometrika 40, 33–51 (1975). A canonical correlation method to shape the distribution of multi-omics datasets.
- 15. Morton JT et al. Learning representations of microbe-metabolite interactions. Nat. Methods 16, 1306–1314 (2019). Mmvec integrates metabolomics and microbiome data to estimate microbe-metabolites interaction based on their co-occurrence probabilities.
- 16. Wang M et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837 (2016). A web-based MS ecosystem created by the community for the community to share, process and annotate MS/MS data.
- 17.Aksenov AA et al. Algorithmic Learning for Auto-deconvolution of GC-MS Data to Enable Molecular Networking within GNPS. Bioinformatics (2020). [Google Scholar]
- 18.Bolyen E et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez A et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giacomoni F et al. Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics 31, 1493–1495 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong J, Liu P, Zhou G & Xia J Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Stanstrup J et al. The metaRbolomics Toolbox in Bioconduct or and beyond. Metabolites 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNally CP, Eng A, Noecker C, Gagne-Maynard WC & Borenstein E BURRITO: An Interactive Multi-Omic Tool for Visualizing Taxa-Function Relationships in Microbiome Data. Front. Microbiol. 9, 365 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X-W, Li Q-H, Xu Z-D & Dou J-J Mass spectrometry-based metabolomics in health and medical science: a systematic review. RSC Advances 10, 3092–3104 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren J-L, Zhang A-H, Kong L & Wang X-J Advances in mass spectrometry-based metabolomics for investigation of metabolites. RSC Advances 8, 22335–22350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen DH, Nguyen CH & Mamitsuka H Recent advances and prospects of computational methods for metabolite identification: a review with emphasis on machinel earning approaches. Brief. Bioinform. 20, 2028–2043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih C-J, Chen P-Y, Liaw C-C, Lai Y-M & Yang Y-L Bringing microbial interactions to light using imaging mass spect rometry. Nat. Prod. Rep. 31, 739–755 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Watrous JD & Dorrestein PC Imaging mass spectrometry in microbiology. Nat. Rev. Microbiol. 9, 683–694 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weisskopf L, Schulz S & Garbeva P Microbial volatile organic compounds in intra-kingdom and inter-kingdom interactions. Nature Reviews Microbiology (2021). [DOI] [PubMed] [Google Scholar]
- 30.Freiwald A & Sauer S Phylogenetic classification and identification of bacteria by mass spectrometry. Nature Protocols 4, 732–742 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Clark CM et al. Using the Open-Source MALDI TOF-MS IDBac Pipeline for Analysis of Microbial Protein and Specialized Metabolite Data. J. Vis. Exp. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark CM, Costa MS, Sanchez LM & Murphy BT Coupling MALDI-TOF mass spectrometry protein and specialized metabolite analyses to rapidly discriminate bacterial function. Proc. Natl. Acad. Sci. U. S. A. 115, 4981–4986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watrous JD et al. Microbial metabolic exchange in 3D. ISME J. 7, 770–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebeke M et al. Unique metabolites protect earthworms against plant polyphenols. Nat. Commun. 6, 7869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geier B et al. Spatial metabolomics of insitu host–microbe interactions at the micrometre scale. Nature Microbiology 5, 498–510 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Rath CM et al. Molecular analysis of modelgut microbiot as by imaging mass spectrometry and nanodesorption electrospray ionization reveals dietary metabolite transformations. Anal. Chem. 84, 9259–9267 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quinn RA et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouslimani A et al. Molecular cartography of the human skin surface in 3D. Proc. Natl. Acad. Sci. U. S. A. 112, E2120–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Floros DJ et al. Mass Spectrometry Based Molecular 3D-Cartography of Plant Metabolites. Front. Plant Sci. 8, 429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Protsyuk I et al. 3D molecular cartography using LC–MS facilitated by Optimus and ‘ili software. Nature Protocols 13, 134–154 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Alexandrov T et al. METASPACE: A community-populated knowledge base of spatial metabolomes in health and disease. Cold Spring Harbor Laboratory (2019). [Google Scholar]
- 42.Robichaud G, Garrard KP, Barry JA & Muddiman DC MSiReader: an open-source interface to view and analyze high resolving power MS imaging files on Matlabplatform. J. Am. Soc. Mass Spectrom. 24, 718–721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wandy J et al. In Silico Optimization of Mass Spectrometry Fragmentation Strategies in Metabolomics. Metabolites 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papadimitropoulos M-EP, Vasilopoulou CG, Maga-Nteve C & Klapa MI Untargeted GC-MS Metabolomics. Methods in Molecular Biology, 133–147 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Keppler EAH, Jenkins CL, Davis TJ & Bean HD Advances in the application of comprehensive two-dimensional gas chromatography in metabolomics. Trends Analyt. Chem 109, 275–286 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stein SE An integrated method for spectrum extraction and compound identification from gas chromatography/mass spectrometry data. Journal of the American Society for Mass Spectrometry 10, 770–781 (1999). [Google Scholar]
- 47.Tsugawa H et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tautenhahn R, Patti GJ, Rinehart D & Siuzdak G XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem. 84, 5035–5039 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pluskal T, Castillo S, Villar-Briones A & Oresic M MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smirnov A et al. ADAP-GC 4.0: Application of Clustering-Assisted Multivariate Curve Resolution to Spectral Deconvolution of Gas Chromatography-Mass Spectrometry Metabolomics Data. Anal. Chem. 91, 9069–9077 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sumner LW et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schymanski EL et al. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol. 48, 2097–2098 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Kuang E, Marney M, Cuevas D, Edwards RA & Forsberg EM Towards Predicting Gut Microbial Metabolism: Integration of Flux Balance Analysis and Untargeted Metabolomics. Metabolites 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bordbar A et al. Elucidat ing dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 7, 46249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ibáñez AJ et al. Mass spectrometry-based metabolomics of single yeast cells. Proc. Natl. Acad. Sci. U. S. A. 110, 8790–8794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z et al. Single-Cell Mass Spectrometry Analysis of Metabolites Facilitated by Cell Electro-Migration and Electroporation. Anal. Chem. 92, 10138–10144 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Chijiiwa R et al. Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota. Microbiome 8, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma PV & Thaiss CA Host-Microbiome Interactions in the Era of Single-Cell Biology. Front. Cel l. Infect. Microbiol. 10, 569070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edison AS et al. NMR: Unique Strengths That Enhance Modern Metabolomics Research. Anal. Chem. (2020). [DOI] [PubMed] [Google Scholar]
- 60.Horai H et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 45, 703–714 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Oberacher H, Whitley G & Berger B Evaluation of the sensitivity of the ‘Wiley registry of tandem mass spectral data, MSforID’ wit h MS/MS data of the ‘NIST/NIH/EPA mass spectral library’. Journal of Mass Spectrometry 48, 487–496 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Guijas C et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 90, 3156–3164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scheubert K et al. Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun. 8, 1494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li D et al. XY-Meta: A High-Efficiency Search Engine for Large-Scale Metabolome Annotation with Accurate FDR Estimation. Anal. Chem. 92, 5701–5707 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Wang X et al. Target-Decoy-Based False Discovery Rate Estimation for Large-Scale Metabolite Identification. J. Proteome Res. 17, 2328–2334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.da Silva RR, Dorrestein PC & Quinn RA Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. U. S. A. 112, 12549–12550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gauglitz JM et al. Reference data based insights expand understanding of human metabolomes. Systems Biology (2020). It introduces a reference database of food to show the potential of large scale reference data to complement the understanding of microbiome.
- 68.Moorthy AS, Wallace WE, Kearsley AJ, Tchekhovskoi DV & Stein SE Combining Fragment-Ion and Neutral-Loss Matching during Mass Spectral Library Searching: A New General Purpose Algorit hm Applicable to Illicit Drug Identification. Anal. Chem. 89, 13261–13268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Garg N et al. Three-Dimensional Microbiome and Metabolome Cartography of a Diseased Human Lung. Cell Host Microbe 22, 705–716.e4 (2017). MS-based cartography allowed the visualization of host and microbial-derived metabolites by employing molecular networking and reference data of microbial cultures.
- 70.Al barracín Orio AG et al. Fungal-bacterial interaction selects for quorum sensing mutants with increased production of natural antifungal compounds. Commun Biol 3, 670 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wolf S, Schmidt S, Müller-Hannemann M & Neumann S In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 11, 148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Allen F, Greiner R & Wishart D Competitive fragmentation modeling of ESI-MS/MS spectra for putative metabolite identification. Metabolomics 11, 98–110 (2015). [Google Scholar]
- 73. Dührkop K et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019). An insilico fragmentation method based on fragmentation trees to improve metabolite annotation in metabolomics based on MS data.
- 74.Ludwig M, Nothias LF, Dührkop K et al. Database-independent molecular formula annotation using Gibbs sampling through ZODIAC. Nat Mach Intell 2, 629–641 (2020). [Google Scholar]
- 75.Tsugawa H et al. Hydrogen Rearrangement Rules: Computational MS/MS Fragmentation and Structure Elucidation Using MS-FINDER Software. Anal. Chem. 88, 7946–7958 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.da Silva RR et al. Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 14, e1006089 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Djoumbou-Feunang Y et al. BioTransformer: a comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 11, 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Showalter MR, Cajka T & Fiehn O Epimetabolites: discovering metabolism beyond building and burning. Curr. Opin. Chem. Biol. 36, 70–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kautsar SA et al. MIBiG 2.0: a repository for biosynthetic geneclusters of known function. Nucleic Acids Res. 48, D454–D458 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eldjárn GH et al. Ranking microbial metabolomic and genomic links in the NPLinker framework using complementary scoring functions. PLoS Comput. Biol. 17, e1008920 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kersten RD et al. Glycogenomics as a mass spectrometry-guided genomemining method for microbial glycosylated molecules. Proc. Natl. Acad. Sci. U. S. A. 110, E4407–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kersten RD et al. A mass spectrometry–guided genome mining approach for natural product peptidogenomics. Nature Chemical Biology 7, 794–802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mohimani H et al. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 9, 4035 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gurevich A et al. Increased diversity of peptidic natural products revealed by modification-tolerant database search of mass spectra. Nat Microbiol 3, 319–327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Medema MH et al. Pep2Path: automated mass spectrometry-guided genome mining of peptidic natural products. PLoS Comput. Biol. 10, e1003822 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ibrahim A et al. Dereplicating nonribosomal peptides using an informatic search algorithm for natural products (iSNAP) discovery. Proc. Natl. Acad. Sci. U. S. A. 109, 19196–19201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mohimani H et al. Automated genome mining of ribosomal peptide natural products. ACS Chem. Biol. 9, 1545–1551 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mohimani H et al. NRPquest : Coupling Mass Spectrometry and Genome Mining for Nonribosomal Peptide Discovery. J. Nat. Prod. 77, 1902–1909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Merwin NJ et al. DeepRiPP integrates multiomics data to automate discovery of novel ribosomally synthesized natural products. Proc. Natl. Acad. Sci. U. S. A. 117, 371–380 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Santen JA et al. The Natural Products Atlas: An Open Access Knowledge Base for Microbial Nat ural Products Discovery. ACS Cent. Sci. (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang Q & Xu R Automatic extraction, prioritization and analysis of gut microbial metabolites from biomedical literature. Sci. Rep. 10, 9996 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang M et al. Mass spectrometry searches using MASST. Nat. Biotechnol. 38, 23–26 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. vander Hooft JJJ, Wandy J, Barrett MP, Burgess KEV & Rogers S Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. U. S. A. 113, 13738–13743 (2016). MS2LDA recognizes and annotate motifs from MS data as chemical substructures.
- 94.Ernst M et al. MolNet Enhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Djoumbou Feunang Y et al. ClassyFire: automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 8, 61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dührkop K et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nature Biotechnology (2020). [DOI] [PubMed] [Google Scholar]
- 97.Tripathi A et al. Chemically informed analyses of metabolomics mass spectrometry data with Qemistree. Nat. Chem. Biol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ridder L et al. Substructure-based annotation of high-resolution multistage MS(n) spectral trees. Rapid Commun. Mass Spectrom. 26, 2461–2471 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Wang J, Peake DA, Mistrik R & Huang Y A platform to identify endogenous metabolites using a novel high performance Orbit rap MS and the mzCloud Library. Blood 4, 2–8 (2013). [Google Scholar]
- 100.Hulme H et al. Microbiome-derived carnitine mimics as previously unknown mediators of gut-brain axis communication. Sci Adv 6, eaax6328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sugiura Y et al. Visualization of in vivo metabolic flows reveals accelerated utilization of glucose and lactate in penumbra of ischemic heart. Scient ific Reports 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mohammadi SA & Prasanna BM Analysis of genetic diversity in crop plants—salient statistical tools and considerations. Crop Sci. 43, 1235–1248 (2003). [Google Scholar]
- 103.Lee LC, Liong C-Y & Jemain AA Partial l east squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: a review of contemporary practice strategies and knowledge gaps. Analyst 143, 3526–3539 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Jolliffe IT A Note on the Use of Principal Components in Regression. Appl. Stat. 31, 300 (1982). [Google Scholar]
- 105.Zhalnina K et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat Microbiol 3, 470–480 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Gauglitz JM et al. Metabolome-Informed Microbiome Analysis Refines Metadata Classifications and Reveals Unexpected Medication Transfer in Captive Cheetahs. mSystems 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Naveja JJ, Rico-Hidal go MP & Medina-Franco JL Analysis of a large food chemical database: chemical space, diversity, and complexity. F1000Res. 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Neveu V et al. Exposome-Explorer: a manually-curated database on biomarkers of exposure to dietary and environmental factors. Nucleic Acids Res. 45, D979–D984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pearson K Determination of the coefficient of correlation. Science 30, 23–25 (1909). [DOI] [PubMed] [Google Scholar]
- 110.Spearman C The Proof and Measurement of Association between Two Things. Am. J. Psychol. 15, 72 (1904). [PubMed] [Google Scholar]
- 111.Kendall MG & Gibbons JD Rank Correlation Methods (Oxford University Press, 1990). [Google Scholar]
- 112.Basu S et al. Sparse network modeling and metscape-based visualization methods for the analysis of large-scale metabolomics data. Bioinformatics 33, 1545–1553 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Noecker C, Chiu H-C, McNally CP & Borenstein E Defining and Evaluating Microbial Cont ributions to Metabolite Variation in Microbiome-Metabolome Association Studies. mSystems 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gloor GB, Macklaim JM, Pawlowsky-Glahn V & Egozcue JJ Microbiome Datasets Are Compositional: And This Is Not Optional. Frontiers in Microbiology 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Knight R et al. Best pract ices for anal ysing microbiomes. Nat. Rev. Microbiol. 16, 410–422 (2018). [DOI] [PubMed] [Google Scholar]
- 116.Weinstock GM Genomic approaches to studying the human microbiota. Nature 489, 250–256 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Freilich S et al. The large-scale organization of the bacterial net work of ecological co-occurrence interactions. Nucleic Acids Res. 38, 3857–3868 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Morton JT et al. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 10, 2719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Melnik AV et al. Coupling Targeted and Untargeted Mass Spectrometry for Metabolome-Microbiome-Wide Association Studies of Human Fecal Samples. Anal. Chem. 89, 7549–7559 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Ayeni FA et al. Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep. 23, 3056–3067 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Yuan C, Graham M, Staley C & Subramanian S Mucosal microbiota and metabolome along the intestinal tractsreveals location specific relationship. Genomics (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Visconti A et al. Interplay between the human gut microbiome and hostmet abolism. Nat. Commun. 10, 4505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK & Knight R Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Langille MGI et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Douglas GM et al. PICRUSt 2: An improved and cust omizable approach for metagenome inference. Cold Spring Harbor Laboratory (2020). [Google Scholar]
- 126.Li S et al. Predicting network activity from hight hroughput metabolomics. PLoS Comput. Biol. 9, e1003123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Magnúsdóttir S et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiot a. Nat. Biotechnol. 35, 81–89 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Kanehisa M & Goto S KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Slenter DN et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 46, D661–D667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Caspi R et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 36, D623–31 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Michelini S et al. A reverse metabolic approach to weaning: in silico identification of immune-beneficial infant gut bacteria, mining their metabolism for prebiotic feeds and sourcing these feeds in the natural product space. Microbiome 6, 171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moradi F, Olovsson T & Tsigas P in 2014 IEEE/ACM International Conference on Advances in Social Net works Analysis and Mining (ASONAM 2014) 1–8 (IEEE, 2014). [Google Scholar]
- 133.Carr R & Borenstein E Net Seed: a net work-based reverse-ecology tool for calculating the metabolic interface of an organism with its environment. Bioinformatics 28, 734–735 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Su Y, Wang B & Zhang X A seed-expanding method based on random walks for community detection in networks with ambiguous community structures. Sci. Rep. 7, 41830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mallick H et al. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 10, 3136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yin X et al. A Comparative Evaluation of Tools to Predict Metabolite Profiles From Microbiome Sequencing Dat a. Front. Microbiol. 11, 595910 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Karnovsky A et al. Met scape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 28, 373–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kamburov A, Cavill R, Ebbels TMD, Herwig R & Keun HC Integrated pathway-level analysis of transcript omics and metabolomics data with IMPaLA. Bioinformatics 27, 2917–2918 (2011). [DOI] [PubMed] [Google Scholar]
- 139.Hosseini R, Hassanpour N, Liu L-P & Hassoun S Pathway-Activity Likelihood Analysis and Metabolite Annotation for Untargeted Metabolomics Using Probabilistic Modeling. Metabolites 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.McLuskey K et al. Decomposing metabolite set activity levels with PALS. Bioinformatics (2020). [Google Scholar]
- 141.Shaffer M et al. AMON: annotation of metabolite origins via networks to integrate microbiome and metabolome data. BMC Bioinformatics 20, 614 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang L-N et al. Integrated Metabolomics and Proteomics Analysis Revealed Second Messenger System Disturbance in Hippocampus of Chronic Social Defeat Stress Rat. Front. Neurosci. 13, 247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Noecker C et al. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Barupal DK & Fiehn O Chemical Similarity Enrichment Analysis (ChemRICH) as alternative to biochemical pathway mapping for metabolomic datasets. Sci. Rep. 7, 14567 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bryrup T et al. Metformin-induced changes of the gut microbiot a in healthy young men: results of a non-blinded, one-armed intervention study. Diabetologia 62, 1024–1035 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Savage N The complex relationship between drugs and the microbiome. Nature 577, S10–S11 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R & Goodman AL Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570, 462–467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vich Vil a A et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 11, 362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mullard A Understanding how microbiome bugs metabolize drugs. Nat. Rev. Drug Discov. 18, 488 (2019). [DOI] [PubMed] [Google Scholar]
- 150.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hehemann J-H et al. Transfer of carbohydrate active enzymes from marine bacteria to Japanese gut microbiota. Nature 464, 908–912 (2010). [DOI] [PubMed] [Google Scholar]
- 152.Kolodziejczyk AA, Zheng D & Elinav E Diet–microbiota interactions and personalized nutrition. Nature Reviews Microbiology 17, 742–753 (2019). [DOI] [PubMed] [Google Scholar]
- 153.Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ & Balskus EP Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Delzenne NM & Bindels LB Food for thought about manipulating gut bacteria. Nature 577, 32–34 (2020). [DOI] [PubMed] [Google Scholar]
- 155.Zheng D, Liwinski T & Elinav E Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bauermeister A, Zucchi TD & Moraes LAB Mass spectrometric approaches for the identification of anthracycline analogs produced by actinobacteria. J. Mass Spectrom. 51, 437–445 (2016). [DOI] [PubMed] [Google Scholar]
- 157.Johnson AR & Carlson EE Collision-Induced Dissociation Mass Spectrometry: A Powerful Tool for Natural Product Structure Elucidation. Anal. Chem. 87, 10668–10678 (2015). [DOI] [PubMed] [Google Scholar]
- 158.Lermyte F, Valkenborg D, Loo JA & Sobott F Radical solutions: Principles and application of electron-based dissociation in mass spectrometry-based anal ysis of protein structure. Mass Spectrom. Rev. 37, 750–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Aksenov AA et al. Auto-deconvolution and molecular networking of gas chromatography–mass spectrometry data. Nat. Biotechnol, 1–5 (2020). Auto-deconvolution by machine learning and molecular networking of GC-MS data withing the GNPS platform.
- 160.Paglia G, Kliman M, Claude E, Geromanos S & Astarita G Applications of ion-mobility mass spectrometry for lipid analysis. Anal. Bioanal. Chem. 407, 4995–5007 (2015). [DOI] [PubMed] [Google Scholar]
- 161.Tian H, Li B & Shui G Untargeted LC–MS Data Preprocessing in Metabolomics. Journal of Analysis and Testing 1, 187–192 (2017). [Google Scholar]
- 162.Röst HL et al. OpenMS: a flexible open-source software plat form for mass spectrometry data analysis. Nat. Methods 13, 741–748 (2016). [DOI] [PubMed] [Google Scholar]
- 163.Tautenhahn R, Böttcher C & Neumann S Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9, 504 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kapoore RV & Vaidyanathan S Towards quantitative mass spectrometry-based metabolomics in microbial and mammalian systems. Philos. Trans. A Math. Phys. Eng. Sci 374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kuhl C, Tautenhahn R, Böttcher C, Larson TR & Neumann S CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283–289 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Broeckling CD, Afsar FA, Neumann S, Ben-Hur A & Prenni JE RAMClust: a novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 86, 6812–6817 (2014). [DOI] [PubMed] [Google Scholar]
- 167.Schmid R et al. Ion Identity Molecular Networking in the GNPS Environment. Cold Spring Harbor Laboratory (2020). [Google Scholar]
- 168.Qi B-L et al. Derivatization for liquid chromatography-mass spectrometry. Trends Analyt. Chem. 59, 121–132 (2014). [Google Scholar]
- 169.Furey A, Moriarty M, Bane V, Kinsella B & Lehane M Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta 115, 104–122 (2013). [DOI] [PubMed] [Google Scholar]
- 170.Li B, Selmi C, Tang R, Gershwin ME & Ma X The microbiome and autoimmunity: a paradigm from the gut–liver axis. Cell. Mol. Immunol. 15, 595–609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zitvogel L, Daillère R, Roberti MP, Routy B & Kroemer G Anticancer effects of the microbiome and its products. Nat. Rev. Microbiol. 15, 465–478 (2017). [DOI] [PubMed] [Google Scholar]
- 172.Wan Y et al. Effects of dietary fat on gut microbiota and faecal metabolites, and their relationship with cardiometabolic risk factors: a 6-month randomised controlled-feeding trial. Gut 68, 1417–1429 (2019). [DOI] [PubMed] [Google Scholar]
- 173. Nakatsuji T et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Science Translational Medicine 9, eaah4680 (2017). Staphylococcus hominis, a commensal bacteria from the human skin, produces an antibiotic metabolite that control the S. aureus grow, contributing to the equilibrium of the microbiota.
- 174. Hsiao EY et al. Microbiot a modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013). It reports a treatment of mouse model of ASD to restore some symptoms and modulate behavior, which support a gut-microbiome-brain connection.
- 175.Chevrette MG et al. The antimicrobial potential of Streptomyces from insect microbiomes. Nature Communications 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.