

Abstract
Carotid stenosis is an important contributor to ischemic stroke risk with resultant significant impact on neurological disability and death in adults and with worldwide implications. Management of carotid stenosis is impacted by whether there are associated symptoms along with the degree of stenosis. Understanding of the pathogenesis of carotid atherosclerosis or stenosis is important in management of carotid stenosis. Atherosclerotic plaque formation is a chronic insidious process with a number of potential contributors to the formation of such a plaque. The definition of atherosclerosis is not simply limited to abnormal deposition of lipid but also includes a chronic, complex, inflammatory process. Molecularly, in atherosclerosis, there is decreasing nitric oxide (NO) bioavailability, activity and/or expression of endothelial NO synthase, or increasing degradation of NO secondary to enhanced superoxide production. These above changes cause endothelial dysfunction leading to formation of foam cell followed by formation on lipid plaque. After lipid plaque formation, stable or unstable atherosclerotic plaque is formed depending on the calcium deposition over the lipid plaque. It continues to be clearly established that carotid intervention for symptomatic high-grade carotid stenosis is best managed with intervention either by carotid endarterectomy or carotid stenting. However, asymptomatic carotid stenosis is the subject of considerable controversy in terms of optimal management. This review of carotid atherosclerosis is an attempt to incorporate the information provided by more recent studies on pathogenesis and management which may help in the decision-making process for optimal management for protection against stroke.
Keywords: Carotid stenosis, management, pathogenesis
Introduction
The most important aspect of addressing the evaluation and management of carotid stenosis is the assessment of risk of stroke and determining if the proposed treatment's benefits justify the potential risk of serious complication. Obviously, clinical features help to define the stroke risk profile.[1] Coexistent factors can have a major impact on optimal management.[2] For example, one has to question the advisability of carotid intervention in a patient with coexistent atrial fibrillation who is best managed with chronic anticoagulant therapy. It is also well recognized that a patient presenting with vertebrobasilar TIA will not necessarily have protection against stroke with carotid intervention for coexistent carotid disease.
Pathophysiology of Carotid Atherosclerotic Plaque
The layers of the arterial walls, and their functions, are important in understanding the pathophysiology of carotid artery stenosis. The normal structure of the carotid artery, or arteries in general, is composed of three layers: from internal to external, the tunica intima, tunica media, and tunica adventitia.[3] Each of the layers has a specific function to act as a nonthrombotic conduit to maintain blood flow in the brain. By nature, the carotid artery is a highly adaptable structure to ensure adequate blood flow to the respective cerebral hemisphere. The intima is the innermost and most dynamic layer of arterial wall and consists of endothelial cells. This layer plays a key role in preventing platelet aggregation and thrombus formation. The internal elastic lamina (IEM) lies outside the intima and this is a single layer of elastic lamina. Adjacent to IEM is the tunica media, which is the thickest layer of the arterial wall and consists of inner circumferential smooth muscles, and an outer longitudinal layer of smooth muscle surrounded by matrix protein, elastin, and proteoglycans.[3,4] The tunica media supports the arterial wall and alters the diameter of the vessel to maintain blood flow, tone, and pressure. Outside the tunica media lies the external elastic lamina. The outermost layer of the arterial wall is the tunica adventitia, which consists of collagen fibers. This is the strongest layer and prevents overexpansion of the arterial wall.[4]
Atherosclerotic plaque formation is a chronic insidious process with a number of potential contributors to the formation of such plaques. The definition of atherosclerosis is not only limited to abnormal deposition of lipid but also includes a chronic, complex, inflammatory process. Of particular note, alteration of shear stress is an important initiating factor in atherosclerosis. The process is initiated at the abluminal surface, where metabolic, mechanical, or physical injury causes disruption of endothelial integrity. The resultant endothelial dysfunction can lead to intimal stress associated with an abnormal biochemical signal. Over time, disruption of the tunica media also occurs.[5,6]
Endothelial Dysfunction and the Cascade of the Atherosclerosis Process
Endothelial cells are a key predictor of vascular health, in addition to being a barrier between luminal contents and the vessel wall. Normal functions of the endothelium include production of nitric oxide (NO), control of volume, regulation of platelet adhesion and coagulation, immune function, and electrolyte balance of the extravascular and intravascular spaces. Endothelial dysfunction is often related to impaired NO-mediated relaxation. The risk factors associated with atherosclerosis, which include diabetes mellitus, hypertension, hyperlipidemia, smoking, as well as genetic predisposition, often potentiate the endothelial dysfunction.[4,7] Previous in vivo and in vitro studies have demonstrated that low shear stress and disturbed flow, including flow separation and turbulent flow, predispose to endothelial dysfunction.[4,7] Endothelial dysfunction subsequently triggers atherosclerosis formation and extension.[8]
Shear stress directly impacts endothelial cell pathophysiology and is associated with the change in cellular orientation, local concentration of different cytokines, growth factors, receptors and adhesion molecules as well as gene expression. In general, high-to-medium shear stress causes release of NO and prostacyclin which maintain the vascular tone. In addition, NO is recognized as an atheroprotective factor of the endothelium. Turbulent flow and low shear, particularly in branch points of arteries, have the opposite effects of high shear including decreasing NO bioavailability, activity and/or expression of endothelial NO synthase, or increasing degradation of NO secondary to enhanced superoxide production.[9] In addition, hyperglycemia associated with diabetes mellitus, atherosclerosis-associated lipid components in hyperlipidemia, excessive homocysteine, and superoxide formation, associated with smoking, are toxic to the endothelium. In one study, elevation of serum level of inflammatory markers was associated with carotid stenosis leading to induction of oxidative stress in the wall of carotid arteries.[10] Different cytokines, growth factors, and their receptors, as well as adhesion molecules, are released by dysfunctional endothelium. Release of vascular cell adhesion molecule (VCAM), intercellular adhesion molecule, and P-selectin triggers the adhesion of monocyte, platelets, and lymphocytes and increases cell permeability. This process further enhances the inflammatory process.[11]
The increased endothelial permeability facilitates monocytes to migrate through endothelium and allows low-density lipoprotein (LDL)-cholesterol to enter into the intimal layer, where the LDL is oxidized by the superoxide molecules. The monocytes are then transformed to macrophages and ingest the oxidized LDL particles by phagocytosis. These cells are referred as “foam cells.” The foam cells, platelets, and T-lymphocytes release into the bloodstream the pro-inflammatory cytokines which include monocyte colony-stimulating factor (M-CSF) and platelet-derived epithelial growth factor.[12] Oxidized LDL also promotes the recruitment and retention of monocytes and lymphocytes.[13] The cytokines, growth factors, and adhesion molecules released by the above cells cause further recruitment and migration of the leukocytes and macrophages which potentiate the inflammatory reactions.[14] In addition, the inflammatory cascade is further magnified by the local release of interleukin (IL)-1, tumor necrosis factor (TNF)-α, and M-CSF, which facilitates the adhesion, activation, and proliferation of leukocytes.[11]
This cascade potently activates the subtypes of mitogen-activated protein kinase including extracellular signal-regulated kinase and P38 mitogen-activated protein kinase (MAPK) which induces cellular adhesion, migration, proliferation, differentiation, apoptosis, and autophagy.[15] Over time, the intimal layer becomes more permeable to circulating cells and triggers a pro-inflammatory condition with release of cytokines. These cytokines further attract monocytes, macrophages, and T-cells to potentiate the plaque formation[16] [Figure 1].
Figure 1.
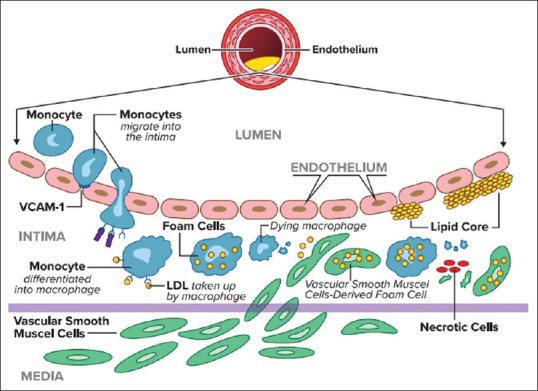
Cellular activation and conformation as well as molecules involved in the process of atherosclerosis
Besides the above factors, angiotensin II is also involved in atherosclerosis. Angiotensin II stimulates atherosclerosis by formation of reactive oxygen species (ROS) in inframammary cells, which oxidizes the LDL and promotes pro-atherogenic, pro-inflammatory, and pro-coagulant activity of platelets and monocytes.[15]
Smooth Muscle Cell Dysregulation
Smooth muscle in the tunica media migrates and proliferates to the intimal layer and results in a neointima by platelet derived growth factor (PDGF), endothelial growth factor, and transforming growth factor-beta.[17,18] High shear also decreases the release of endothelin-1, which exerts both constricting and mitogenic effects of vascular smooth muscles. Reduced bioavailability of NO is associated with increased vascular tone and increased platelet activation, as well as intimal proliferation.[19] The usual functional state of media, including contractility, is altered to a synthetic state. Smooth muscle cells (SMCs) also ingest the oxidized LDL particles and become foam cells. Endothelin released by dysfunctional endothelium causes mitogenic activity to the SMCs and vasoconstriction. Angiotensin II promotes the vascular SMC proliferation by formation of ROS and enhances the activity of membrane-bound nicotinamide adenine dehydrogenase/nicotinamide adenine dinucleotide phosphate (NADH/NADPH) oxidase.[20]
In response to alteration in function of the intima, the media responds with proliferation of SMCs, as well as further promoting the migration of leukocytes and monocytes into this layer. MAPK pathways are associated with migration, proliferation, and differentiation of the SMCs.[15] Various inflammatory mediators are released by SMCs including IL-1β, IL-8, TNF-α and β, IL-6, M-CSF, monocyte chemoattractant factor-1 (MCP-1), and CD-40 ligand. These mediators diversely stimulate mitogenesis and intracellular matrix proliferation.[15] Within the media layer, the derangements of cells and extracellular matrix initiate formation of the carotid plaque. Both genetic and environmental risk factors contribute to the progression of lipid plaque [Figure 2].
Figure 2.
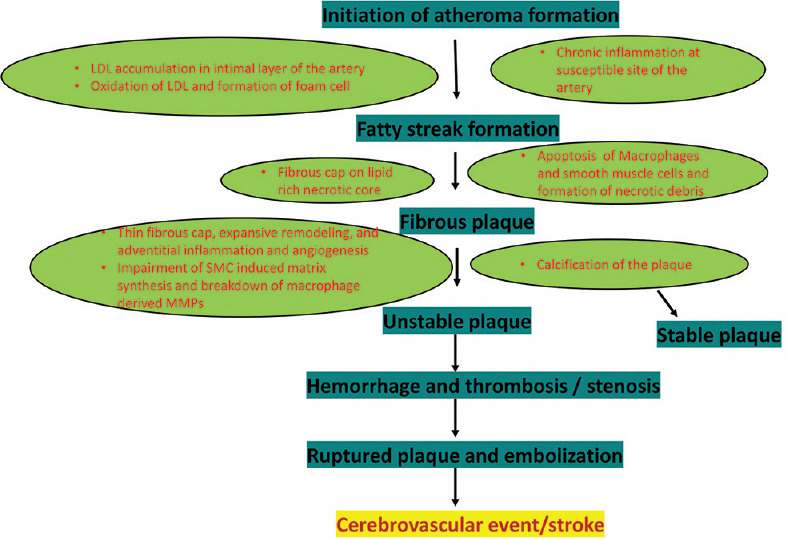
Stages of atherosclerosis plaque formation with cellular events
Histopathological Features of Carotid Atherosclerotic Plaque
The pathology of carotid stenosis begins with fatty streak formation, followed by production of atheromatous fibrous plaque. Accumulation of foam cells and SMCs in the intimal layer and production of extracellular matrix by the foam cells creates a fatty streak, elevates the endothelium, and initiates the formation of atherosclerotic plaque. Detection of active inflammation in the lipid core is an indicator of unstable plaque. When the foam cell dies, it leaves an inert necrotic, atheromatous core within the plaque which is soft and unstable. Some atherogenic LDL may be retained and accumulates within the intimal layer.[21] The lipid-rich core is soft, avascular, hypocellular, and devoid of collagen. Apoptosis of foam cells and SMCs results in the formation of an unstable lipid core and the fragile and rupture-prone fibrous cap. The above process also stimulates high tissue factor activity and thrombogenicity within the lipid core. Advanced atherosclerosis plaque is composed of 68% fibrous tissue, 16% inert necrotic core, 1% foam cells, 7% inflammatory cells, and 8% calcium.[15,22,23]
Calcification
Calcification of the lipid core is an important prognostic indicator. Plaque calcification is active and controlled and resembles bony calcification. Clinical observations suggest that calcified atheroma is stable, while the less calcified atheroma is more prone to rupture and can be associated with thromboembolism.[24,25]
Remodeling of Vascular Wall
Remodeling of the vessel wall in the atherosclerotic process triggers formation of rupture-prone inflamed thin-cap fibroatheroma. Leukocyte-induced matrix metalloproteinase (MMP)-mediated remodeling of the vascular wall is also noted in the development of carotid atherosclerosis.[15,26]
Angiogenesis
Neovascularization and inflammation coexist and mediate plaque progression. Angiogenesis starts in the vasa vasorum of the adventitia with proliferation and extension into the base of the lipid plaque. Microvessels from angiogenesis are very fragile and leaky. VCAM-1 is expressed in this process and promotes local extravasation of proteins, inflammatory cells, and erythrocytes.[15,27]
Thrombosis and Hemorrhage
The fibrous cap usually prevents the exposure of the lipid-rich core to flowing blood. However, any gap in the fibrous cap or significant injury exposes the athermanous core.[28] Exposure of the subendothelial tissue of the plaque allows platelets to adhere, resulting in the formation of fibrin. Platelet aggregation causes thrombus formation associated with luminal obstruction. Fibrinous material can help to stabilize the platelet-induced thrombus. However, this process can also allow accumulation of hemorrhage into the plaque resulting in silent, episodic progression of the atherosclerotic lesion.[28]
Ulceration of Carotid Plaque
Imaging can identify carotid plaques as being either smooth, irregular, or ulcerated and can help in determining what has been termed “vulnerable plaque” in terms of stroke risk.[29] Ulcerated plaque represents the excavated necrotic core with discontinuous fibrous cap.[30,31] Ulcerated plaques are associated with a higher risk of rupture and can lead to thrombus formation and subsequent embolization causing stroke.[32]
Stable versus Unstable Carotid Plaque
There are certain pathological features of rupture-prone plaque. Stratification of risk includes (1) structural features: large lipid reach necrotic core, thin fibrous cap, expansive remodeling, and adventitial inflammation and angiogenesis; (2) cellular features: lack of SMCs and accumulation of macrophages in the plaque; and (3) functional features: impairment of SMC-induced matrix synthesis and breakdown of macrophage-derived MMPs.[21]
Grossly complicated plaques are classified as follows: (1) plaque rupture with intraplaque hemorrhage (IPH), (2) plaque rupture plus thrombosis, (3) plaque rupture with IPH and thrombosis, and (4) IPH without plaque rupture[21] [Figure 2]. Plaque analysis with sophisticated ultrasound (US), including adding transcranial Doppler (TCD) detection of microemboli potentially associated with the unstable plaque can help to identify high-risk plaques[33] and this can include densitometric analysis of carotid plaque composition.[34] Conversely, uncomplicated, i.e., “stable,” plaques tend to be smooth and calcified.[21]
Progression of Atherosclerosis Plaque
As mentioned previously, formation of foam cells and SMCs in intimal layer and associated production of extracellular matrix creates fatty streaks, elevates the endothelium, and initiates the formation of atherosclerotic plaque. Over time, the repeated process of endothelial injury and alteration of shear stress, immune response, and smooth muscle function trigger the formation of a fibrous cap on lipid-laden plaque. After forming a fibrous cap, the lipid plaque begins to encroach on the lumen of carotid artery and can reduce the luminal diameter of the artery.[35] The active atheroma or plaque may continuously grow with chronic inflammation, which is triggered by alteration of the arterial geometry by the atheroma This is followed by modification of the local shear stress direction, oscillation, and magnitude. Chronic inflammation and remodeling can result in progressive focal carotid stenosis.[35]
The carotid plaque can become susceptible to rupture if the fibrous cap is associated with MMP-induced remodeling of the membrane.[36,37] Chronic ongoing inflammation in a noncalcified plaque is the key element of atherosclerotic plaque vulnerability and disruption. On the other hand, plaque calcification is an indicator of a stable plaque less likely to be associated with thromboembolism.[38] Decreased NO bioavailability induced by dysfunctional endothelial cells is associated with decreased anticoagulant properties, dysregulation of vascular tone, and increased platelet aggregation. The presence of CD40L in atherosclerosis plaque stimulates the expression of tissue factor and potentiates thrombus formation. In addition, exposure of a ruptured plaque to thrombotic material initiates formation of thrombus by activating platelets and the clotting cascade. This cascade of events eventually results in atheroembolism and resultant cerebral, or retinal, ischemia[4] [Figures 1, 2 and 3].
Figure 3.

Consequences of ruptured lipid plague
Genetic Basis of Carotid Atherosclerosis
Alteration in certain genes promotes the atherosclerotic process. Mutation in the gene-encoding LDL receptor induces hypercholesterolemia and premature atherosclerosis.[39] Studies have shown that epsilon polymorphism of the apolipoprotein E protein gene and angiotensin-converting enzyme insertion/deletion polymorphism are associated with carotid atherosclerosis.[40,41,42] Genetic factors, other than those for lipid metabolism, have also been implicated in atherosclerosis and thrombogenesis. In addition, genetic variation in coagulation factors and fibrinogen have been identified as promotors of thrombogenesis.[41] Moreover, certain genetic variants of endothelial NO synthase have been also associated with atherosclerosis.[40]
Diagnostic Approaches to Carotid Stenosis
Early detection of stenosis of carotid artery is crucial to prevent potential stroke associated with internal carotid artery (ICA) stenosis. Fortunately, there has been a significant advancement in the detection capability of ICA stenosis which includes carotid US, TCD, computed tomographic angiogram (CTA), magnetic resonance angiogram (MRA), and conventional four-vessel cerebral digital subtraction angiography (DSA).[43,44,45]
Carotid Ultrasound
Carotid duplex imaging, combining B-mode anatomical imaging with Doppler flow velocity characteristics, can be used to determine the presence of atherosclerosis and flow status of carotid artery. Grayscale US is utilized to assess the status of vessel wall including the presence and characteristics of plaque. Calcification (hyperechoic) and noncalcified portions of the plaque, as well as intimal and medial thickness of the wall, can also be determined by grayscale US.[46,47] Color Doppler US is used to determine the vascular flow direction and velocity. Continuous Doppler waveform assesses the flow characteristics, and pulsed Doppler determines the velocity in a specific field depth of the specific vascular region.[48] Peak systolic velocity is typically used to determine the degree of stenosis. However, end-diastolic velocity and carotid index are also useful. US is 89% sensitive and 84% specific determining 70%–90% stenosis while it is 36% sensitive and 91% specific to assess 50%–69% stenosis.[49] US remains a noninvasive and relatively cost-effective procedure for initial assessment of carotid stenosis.
Transcranial Doppler
TCD is another noninvasive method to determine the hemodynamic effect of extracranial ICA stenosis on intracranial circulation.[50,51,52] The Doppler signal can be obtained through transtemporal and transorbital windows. Flow direction measurement of the ophthalmic, middle cerebral, and anterior cerebral arteries can allow indirect assessment of potential collateral circulation adjustments to ipsilateral severe ICA stenosis or occlusion.[53,54]
Computed Tomographic Angiography
Studies have demonstrated that CTA is highly sensitive and specific to diagnose ICA stenosis when compared with standard DSA.[55,56] CTA (R = 0.95) was comparable to DSA (R = 0.89) to detect stenosis. It is now routinely used in rooms during stroke codes to detect extra- and intracranial carotid stenosis. CTA has higher sensitivity (98% vs. 70%) and positive predictive value (93% vs. 65%) than MRA, respectively, to detect ICA stenosis.[43] CTA is also generally more accurate in assessing the posterior circulation compared to DSA in the presence of poor flow.[57] However, the exact degree of ICA stenosis is more accurately measured by DSA. CTA has a tendency to underestimate the higher grade (CTA, 78% vs. DSA, 86%) and moderate grade stenosis (CTA, 57% vs. DSA, 63%).[58]
Magnetic Resonance Angiography
MRA can be done with or without contrast to determine carotid stenosis with the time of flight MRA without contrast and gadolinium contrast-enhanced (CE) MRA available with the contrast-enhanced study viewed as being more accurate for extracranial ICA stenosis. MRA is not generally viewed as accurate as CTA as noncontrast MRA is not as sensitive to calcification resulting in underestimation of the carotid stenosis.[46] On the other hand, contrast-enhanced MRA is more impacted by artifacts which can overestimate the stenosis.[46] With either CTA or DSA, North American Symptomatic Carotid Endarterectomy Trial (NASCET) or European carotid surgery trial (ECST) criteria can be used to calculate the degree of ICA stenosis. Of note in acknowledgment of potential iodine-based contrast contraindication, contrast-enhanced MRA is 94% sensitive and 93% specific for identifying 70%–90% ICA stenosis.[46,49] However, for 50%–69% stenosis, CE MRA is 77% and 97% sensitive and specific, respectively.[46] MRA can thus be a reasonable option for relatively noninvasive evaluation of the carotid circulation, especially in those having a contraindication to iodine-based contrast.
Intra-arterial Cerebral Angiography (Digital Subtraction Angiography)
DSA is the gold standard, but invasive, method to determine the extent of carotid atherosclerotic occlusive disease in an accurate fashion. In addition to diagnostic assessment, this procedure is used during carotid stenting and angioplasty.[45] There are generally two views including lateral and anterior–posterior with DSA. Rotational view is also performed for assessing severe carotid stenosis and better visualization of cerebral aneurysm. The sensitivity, specificity, and accuracy of DSA for detection of carotid stenosis are 95%, 99%, and 97%, respectively.[59] In the NASCET[60] and European Carotid Surgery Trial (ECST),[61] DSA procedure was utilized. The NASCET criteria to calculate ICA stenosis is A − B/A × 100 (%) and ECST criteria to calculate ICA stenosis is C − B/C × 100 (%) [Figure 4]. In the NASCET criteria, severe ICA stenosis is defined as 70%–99% stenosis and moderate stenosis as 50%–69% stenosis. In ECST criteria, severe stenosis is defined as 80%–99% stenosis and moderate stenosis is 70%–79% stenosis.[45,62] The disadvantage of this procedure is that it is invasive with the potential for embolization resulting in stroke although the incidence of this should be <1%.
Figure 4.
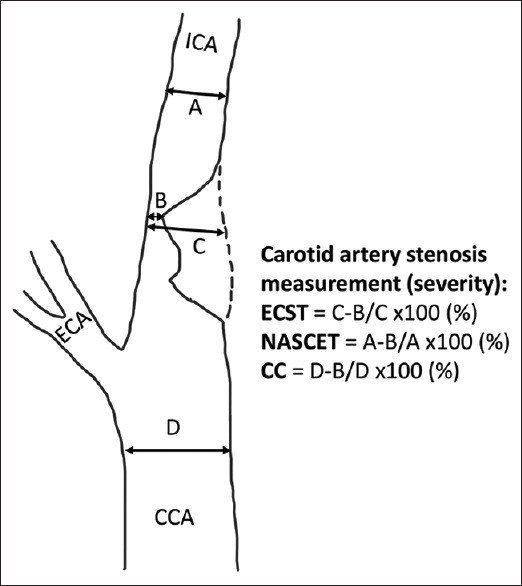
Methods of measurement of severity of carotid artery stenosis. NASCET: North American Symptomatic Carotid Endarterectomy Trial, ECST: European Carotid Surgery Trial, CC: Common carotid
As of now, a general approach is to determine carotid stenosis with carotid US as the initial screening procedure. If there is >50% ICA stenosis detected by this modality, the next step is obtained either CTA or MRA. If the ICA stenosis is <50%, serial carotid duplex scan is recommended to rule out disease progression. The DSA now tends to be reserved for those cases where the CTA or MRA results are questionable as well as for those patients who are undergoing carotid angioplasty with stenting.
Assessment and Treatment of Stroke Risk with Symptomatic Carotid Stenosis
One must first define symptomatic carotid stenosis in terms of its presentation. The presentation can be a cerebral hemispheric TIA, a cerebral hemispheric infarct, or a retinal TIA, termed amaurosis fugax or transient monocular blindness, or retinal infarction. The risk of stroke or TIA with carotid stenosis is related to the degree of stenosis along with plaque characteristics, including thrombus formation, in association with the carotid plaque. The term TIA has undergone some recent revision. Traditionally, TIA represents temporary neurological dysfunction, on a vascular basis in a single artery territory, that completely remits within 24 h.[63,64] However, because of the potential presence of tissue infarction on diffusion-weighted MRI brain scan, the tissue-based definition of TIA is now a transient episode of neurological impairment associated with either focal brain, spinal cord, or retinal ischemia without acute infarction.[65] Ischemic stroke and TIA can be ascribed to a number of mechanisms related to the diligence of the diagnostic assessment.
In a study by Cheng et al.,[66] significant carotid stenosis, of at least 50%, is seen in roughly 20% of patients presenting with TIA or stroke. The presence of such a degree of stenosis was associated with hypertension, dyslipidemia, diabetes mellitus, and ischemic cardiac disease as risk factors. Age is also a contributing factor with significant carotid stenosis affecting 7.5% of all men and 5% of all women >80 years of age.[67] In terms of association with “high-risk” plaques, which included AHA type 4, 5, or 6, ulcerations, IPH, echolucency, irregularity, mural thrombus, neovascularization, lipid-rich necrotic core, microembolic signals, and ruptured or thin fibrous cap, the prevalence of symptomatic carotid stenosis was 43.3% versus 19.9% in patients with asymptomatic stenosis.[68] IPH is viewed as being of particular risk. In one study, IPH was observed in 30%–50% of patients with symptomatic carotid stenosis compared to 20%–30% in asymptomatic carotid stenosis.[69] In a meta-analysis, using NASCET criteria for degree of stenosis,[70] even mild stenosis, <50%, the annualized ipsilateral stroke event rate, among patients with symptomatic carotid stenosis, was 9% for those with IPH versus 0.7% for those without IPH. Overall, IPH was present in 51.69% of patients with symptomatic carotid stenosis versus 29.4% in patients with asymptomatic carotid stenosis.
In terms of intervention, the NASCET study reported a reduction in the risk of stroke ipsilateral to >70% symptomatic stenosis from 26% to 9% after 2 years with carotid endarterectomy (CEA) compared to optimal medical management at the time of the study (P < 0.001).[71] The European Carotid Surgery Trialists (ECST) Collaborative Group reported a reduction from 20.6% to 6.8% during 3 years of follow-up for those treated with CEA (P < 0.0001). On the other hand, the benefit of intervention for moderate symptomatic carotid stenosis (50%–69%), with CEA, was much more modest in the NASCET study (22.2% vs. 15.7% after 5 years with P < 0.045).[72]
Carotid artery stenting (CAS) compared to CEA has been the subject of a number of recent studies. It is a less invasive procedure with the potential to be associated with less side effects and possibly similar efficacy to CEA. This technique is being refined in an effort to enhance its potential benefits versus risks. However, a meta-analysis of available studies, to date, reported an enhanced risk of death or any stroke within 30 days of this procedure at 7.2% compared to 4.4% for CEA (odds ratio [OR] = 1.75, P > 0.0001). This risk did diminish to nonsignificant after 30 days. However, the risk was of particular concern was in patients >70 years of age (OR = 1.11 for those <70 vs. 2.23 for those >70, P = 0.007).[73]
Assessment and Treatment of Stroke Risk with Asymptomatic Carotid Stenosis
In terms of the natural history of asymptomatic higher grade, i.e., >70% stenosis, Cheng et al.[74] reported that the 5-year risk of stroke or TIA associated with ipsilateral stenosis was 5.3%. This study was based upon serial carotid duplex scan with a progression rate of stenosis, over time, of 24.1%. With such a relatively low overall risk of stroke in asymptomatic carotid stenosis, the approach to possible intervention remains perplexing.[75] In a Veterans Administration study reported in 1993, in a male population with 50% ICA stenosis or greater, the rate of what was termed “combined ipsilateral neurological events” was 8% with CEA versus 20.6% in the medical group (P < 0.001). During follow-up, the stroke rate was 4.7% for CEA versus 9.4% for the medical group although there was no difference in combined stroke and death at 30 days.[76] There has been additional support for intervention in carefully selected patients, who have relatively low surgical risk for complication, based on additional studies.[75] However, optimization of alternative medical therapy, in this patient group, is still an evolving process, particularly with success in managing dyslipidemia with statins or other lipid-lowering agents.[77] CAS versus CEA is under investigation for asymptomatic stenosis. To date, there has been reported a borderline significant increase in stroke or death, within 30 days of the procedure, with CAS compared to CEA with OR = 1.72, P = 0.05.[73]
A recent study by Saba et al. showed that imaging techniques including MRI, CTA, and US of carotid artery can detect carotid plaque volume, maximum wall thickness, plaque inflammation, IPH, neovascularization, ulceration, lipid-rich necrotic core, fibrous cap, plaque calcification, and microembolization.[78] Moreover, Schindler et al. showed that IPH is common in both symptomatic and asymptomatic carotid stenosis, and it is a strong predictor of stroke.[70] Currently, carotid assessment depends on not only about degree of stenosis but also carotid plaque vulnerability. Therefore, MRI or CTA-based detection of carotid plaque vulnerability may be beneficial for determination of carotid revascularization.
Current Patient Selection between Carotid Artery Stenting and Carotid Endarterectomy
Carotid stenting is usually preferring in patients with restenosis after prior CEA, prior radical neck surgery, radiation therapy in the neck, young patients less than 70 years old, and concomitant heart disease. Alternatively, CEA is preferred in elderly patients older than 70 years.[79,80]
Transcarotid Artery Revascularization
Transcarotid artery revascularization (TCAR) is now available as an alternative to routine CEA or CAS for the treatment of CA stenosis. Anatomic requirements and eligibility for TCAR include diameter of carotid artery >6 mm, distance between access site and the lesion >5 cm and common carotid artery (CCA), and carotid occlusion sites free of thrombus or calcification. Recent studies[81,82,83] reported that the stroke, cranial nerve injury, or death rate is favorable for T-CAR compared to CAS. In the cases with symptomatic CA stenosis, new stroke determined within 24–48 h of procedure was 12.9% in TCAR compared to 33.3% in CAS.[81] A previous study showed that TCAR significantly reduces the rate of MI compared to CEA, and that would be choice of treatment for the particular compared to CEA or CAS as there is no head-to-head randomized control trial between the procedures.
Summary
The approach to the patient with carotid stenosis must be individualized. The medical stability of the patient and the availability of outstanding expertise in the interventional process are of utmost importance. There is increasing support for a conservative approach to management of asymptomatic carotid stenosis. However, one must factor in extenuating factors. For a relatively young and healthy patient with bilateral high-grade carotid stenosis, one might well want to consider intervention, especially if serial imaging of the carotid bifurcation shows progression of the stenosis over time despite optimal medical management. Conversely, if a patient with high-grade asymptomatic carotid stenosis, or symptomatic moderate stenosis, who is not receptive to acceptable medical management (e.g., optimal hypertension and diabetic control, optimal lipid profile, smoking cessation, dietary modification as well as use of anti-thrombotic therapy), then an interventional procedure becomes much more attractive to prevent a major cerebral infarct. Enhanced imaging of the plaque characteristics can now certainly help in the decision making process with either advanced neurosonology[84] or MR imaging of the plaque[85] with assessment of its potential vulnerability[86], as well as quantitative assessment of carotid plaque morphology with CTA.[87]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We would like to thank Mr. David Wright for the illustrations [Figures 1 and 3].
References
- 1.Sato DM, Mantovani LK, Safanelli J, Guesser V, Nagel V, Moro CH, et al. Ischemic stroke: Process perspective, clinical and profile characteristics, and external factors. J Biomed Inform. 2020;111:103582. doi: 10.1016/j.jbi.2020.103582. [DOI] [PubMed] [Google Scholar]
- 2.Meschia JF, Bushnell C, Boden-Albala B, Braun LT, Bravata DM, Chaturvedi S, et al. Guidelines for the primary prevention of stroke: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2014;45:3754–832. doi: 10.1161/STR.0000000000000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charlick M, Das JM. StatPearls. Treasure Island (FL): StatPearls Publishing; 2022. Anatomy, head and neck, internal carotid arteries. [PubMed] [Google Scholar]
- 4.Milutinović A, Šuput D, Zorc-Pleskovič R. Pathogenesis of atherosclerosis in the tunica intima, media, and adventitia of coronary arteries: An updated review. Bosn J Basic Med Sci. 2020;20:21–30. doi: 10.17305/bjbms.2019.4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prasad K. Pathophysiology and medical treatment of carotid artery stenosis. Int J Angiol. 2015;24:158–72. doi: 10.1055/s-0035-1554911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingstone LL, Currie GM, Torres C. The pathogenesis, analysis, and imaging methods of atherosclerotic disease of the carotid artery: Review of the literature. J Med Imaging Radiat Sci. 2012;43:84–94. doi: 10.1016/j.jmir.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Ku DN, Giddens DP, Zarins CK, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arteriosclerosis. 1985;5:293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 8.Lind L, Andersson J, Larsson A, Sandhagen B. Shear stress in the common carotid artery is related to both intima-media thickness and echogenecity. The Prospective Investigation of the Vasculature in Uppsala Seniors study. Clin Hemorheol Microcirc. 2009;43:299–308. doi: 10.3233/CH-2009-1241. [DOI] [PubMed] [Google Scholar]
- 9.Gambillara V, Chambaz C, Montorzi G, Roy S, Stergiopulos N, Silacci P. Plaque-prone hemodynamics impair endothelial function in pig carotid arteries. Am J Physiol Heart Circ Physiol. 2006;290:H2320–8. doi: 10.1152/ajpheart.00486.2005. [DOI] [PubMed] [Google Scholar]
- 10.Alexander RW. Theodore Cooper Memorial Lecture. Hypertension and the pathogenesis of atherosclerosis. Oxidative stress and the mediation of arterial inflammatory response. A new perspective. Hypertension. 1995;25:155–61. doi: 10.1161/01.hyp.25.2.155. [DOI] [PubMed] [Google Scholar]
- 11.Crowther MA. Pathogenesis of atherosclerosis. Hematology Am Soc Hematol Educ Program. 2005;1:436–41. doi: 10.1182/asheducation-2005.1.436. [DOI] [PubMed] [Google Scholar]
- 12.Kim KJ, Jeong SW, Ryu WS, Kim DE, Saver JL, Kim JS, et al. Platelet-derived growth factor is associated with progression of symptomatic intracranial atherosclerotic stenosis. J Clin Neurol. 2021;17:70–6. doi: 10.3988/jcn.2021.17.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irani K. Oxidant signaling in vascular cell growth, death, and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ Res. 2000;87:179–83. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 14.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–14. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 15.Singh RB, Mengi SA, Xu YJ, Arneja AS, Dhalla NS. Pathogenesis of atherosclerosis: A multifactorial process. Exp Clin Cardiol. 2002;7:40–53. [PMC free article] [PubMed] [Google Scholar]
- 16.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res. 1998;82:532–9. doi: 10.1161/01.res.82.5.532. [DOI] [PubMed] [Google Scholar]
- 17.Bowen-Pope DF, Ross R, Seifert RA. Locally acting growth factors for vascular smooth muscle cells: Endogenous synthesis and release from platelets. Circulation. 1985;72:735–40. doi: 10.1161/01.cir.72.4.735. [DOI] [PubMed] [Google Scholar]
- 18.Clowes AW, Ryan GB, Breslow JL, Karnovsky MJ. Absence of enhanced intimal thickening in the response of the carotid arterial wall to endothelial injury in hypercholesterolemic rats. Lab Invest. 1976;35:6–17. [PubMed] [Google Scholar]
- 19.Abrams J. Role of endothelial dysfunction in coronary artery disease. Am J Cardiol. 1997;79:2–9. doi: 10.1016/s0002-9149(97)00379-2. [DOI] [PubMed] [Google Scholar]
- 20.Haendeler J, Ishida M, Hunyady L, Berk BC. The third cytoplasmic loop of the angiotensin II type 1 receptor exerts differential effects on extracellular signal-regulated kinase (ERK1/ERK2) and apoptosis via Ras- and Rap1-dependent pathways. Circ Res. 2000;86:729–36. doi: 10.1161/01.res.86.7.729. [DOI] [PubMed] [Google Scholar]
- 21.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–74. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 22.Pedrigi RM, de Silva R, Bovens SM, Mehta VV, Petretto E, Krams R. Thin-cap fibroatheroma rupture is associated with a fine interplay of shear and wall stress. Arterioscler Thromb Vasc Biol. 2014;34:2224–31. doi: 10.1161/ATVBAHA.114.303426. [DOI] [PubMed] [Google Scholar]
- 23.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol. 1995;15:1512–31. doi: 10.1161/01.atv.15.9.1512. [DOI] [PubMed] [Google Scholar]
- 24.Wahlgren CM, Zheng W, Shaalan W, Tang J, Bassiouny HS. Human carotid plaque calcification and vulnerability. Relationship between degree of plaque calcification, fibrous cap inflammatory gene expression and symptomatology. Cerebrovasc Dis. 2009;27:193–200. doi: 10.1159/000189204. [DOI] [PubMed] [Google Scholar]
- 25.Hunt JL, Fairman R, Mitchell ME, Carpenter JP, Golden M, Khalapyan T, et al. Bone formation in carotid plaques: A clinicopathological study. Stroke. 2002;33:1214–9. doi: 10.1161/01.str.0000013741.41309.67. [DOI] [PubMed] [Google Scholar]
- 26.Kiechl S, Willeit J. The natural course of atherosclerosis. Part II. Vascular remodeling. Bruneck Study Group. Arterioscler Thromb Vasc Biol. 1999;19:1491–8. doi: 10.1161/01.atv.19.6.1491. [DOI] [PubMed] [Google Scholar]
- 27.Munro JM. Endothelial-leukocyte adhesive interactions in inflammatory diseases. Eur Heart J. 1993;14(Suppl K):72–7. [PubMed] [Google Scholar]
- 28.Moody AR, Murphy RE, Morgan PS, Martel AL, Delay GS, Allder S, et al. Characterization of complicated carotid plaque with magnetic resonance direct thrombus imaging in patients with cerebral ischemia. Circulation. 2003;107:3047–52. doi: 10.1161/01.CIR.0000074222.61572.44. [DOI] [PubMed] [Google Scholar]
- 29.Rafailidis V, Chryssogonidis I, Tegos T, Kouskouras K, Charitanti-Kouridou A. Imaging of the ulcerated carotid atherosclerotic plaque: A review of the literature. Insights Imaging. 2017;8:213–25. doi: 10.1007/s13244-017-0543-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Homburg PJ, Rozie S, van Gils MJ, van den Bouwhuijsen QJ, Niessen WJ, Dippel DW, et al. Association between carotid artery plaque ulceration and plaque composition evaluated with multidetector CT angiography. Stroke. 2011;42:367–72. doi: 10.1161/STROKEAHA.110.597369. [DOI] [PubMed] [Google Scholar]
- 31.Kolodgie FD, Yahagi K, Mori H, Romero ME, Trout HH Rd, Finn AV, et al. High-risk carotid plaque: Lessons learned from histopathology. Semin Vasc Surg. 2017;30:31–43. doi: 10.1053/j.semvascsurg.2017.04.008. [DOI] [PubMed] [Google Scholar]
- 32.Avril G, Batt M, Guidoin R, Marois M, Hassen-Khodja R, Daune B, et al. Carotid endarterectomy plaques: Correlations of clinical and anatomic findings. Ann Vasc Surg. 1991;5:50–4. doi: 10.1007/BF02021778. [DOI] [PubMed] [Google Scholar]
- 33.Madani A, Beletsky V, Tamayo A, Munoz C, Spence JD. High-risk asymptomatic carotid stenosis: Ulceration on 3D ultrasound vs TCD microemboli. Neurology. 2011;77:744–50. doi: 10.1212/WNL.0b013e31822b0090. [DOI] [PubMed] [Google Scholar]
- 34.Beletsky VY, Kelley RE, Fowler M, Phifer T. Ultrasound densitometric analysis of carotid plaque composition. Pathoanatomic correlation. Stroke. 1996;27:2173–7. doi: 10.1161/01.str.27.12.2173. [DOI] [PubMed] [Google Scholar]
- 35.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 36.Welgus HG, Campbell EJ, Cury JD, Eisen AZ, Senior RM, Wilhelm SM, et al. Neutral metalloproteinases produced by human mononuclear phagocytes. Enzyme profile, regulation, and expression during cellular development. J Clin Invest. 1990;86:1496–502. doi: 10.1172/JCI114867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaalan WE, Cheng H, Gewertz B, McKinsey JF, Schwartz LB, Katz D, et al. Degree of carotid plaque calcification in relation to symptomatic outcome and plaque inflammation. J Vasc Surg. 2004;40:262–9. doi: 10.1016/j.jvs.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 39.Hegele RA. The genetic basis of atherosclerosis. Int J Clin Lab Res. 1997;27:2–13. doi: 10.1007/BF02827237. [DOI] [PubMed] [Google Scholar]
- 40.Juo SH. Genetics of carotid atherosclerosis. Front Biosci (Landmark Ed) 2009;14:4525–34. doi: 10.2741/3545. [DOI] [PubMed] [Google Scholar]
- 41.Casas JP, Hingorani AD, Bautista LE, Sharma P. Meta-analysis of genetic studies in ischemic stroke: Thirty-two genes involving approximately 18,000 cases and 58,000 controls. Arch Neurol. 2004;61:1652–61. doi: 10.1001/archneur.61.11.1652. [DOI] [PubMed] [Google Scholar]
- 42.Ariyaratnam R, Casas JP, Whittaker J, Smeeth L, Hingorani AD, Sharma P. Genetics of ischaemic stroke among persons of non-European descent: A meta-analysis of eight genes involving approximately 32,500 individuals. PLoS Med. 2007;4:e131. doi: 10.1371/journal.pmed.0040131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bash S, Villablanca JP, Jahan R, Duckwiler G, Tillis M, Kidwell C, et al. Intracranial vascular stenosis and occlusive disease: Evaluation with CT angiography, MR angiography, and digital subtraction angiography. AJNR Am J Neuroradiol. 2005;26:1012–21. [PMC free article] [PubMed] [Google Scholar]
- 44.Nemoto S. Diagnostic imaging of carotid stenosis: Ultrasound, magnetic resonance imaging, and computed tomography angiography. Nihon Geka Gakkai Zasshi. 2011;112:371–6. [PubMed] [Google Scholar]
- 45.Saxena A, Ng EY, Lim ST. Imaging modalities to diagnose carotid artery stenosis: Progress and prospect. Biomed Eng Online. 2019;18:66. doi: 10.1186/s12938-019-0685-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adla T, Adlova R. Multimodality imaging of carotid stenosis. Int J Angiol. 2015;24:179–84. doi: 10.1055/s-0035-1556056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Netuka D, Belšán T, Broulíková K, Mandys V, Charvát F, Malík J, et al. Detection of carotid artery stenosis using histological specimens: A comparison of CT angiography, magnetic resonance angiography, digital subtraction angiography and Doppler ultrasonography. Acta Neurochir (Wien) 2016;158:1505–14. doi: 10.1007/s00701-016-2842-0. [DOI] [PubMed] [Google Scholar]
- 48.Hartley CJ, Cole JS. An ultrasonic pulsed Doppler system for measuring blood flow in small vessels. J Appl Physiol. 1974;37:626–9. doi: 10.1152/jappl.1974.37.4.626. [DOI] [PubMed] [Google Scholar]
- 49.Wardlaw JM, Chappell FM, Stevenson M, De Nigris E, Thomas S, Gillard J, et al. Accurate, practical and cost-effective assessment of carotid stenosis in the UK. Health Technol Assess. 2006;10:x–182. doi: 10.3310/hta10300. [DOI] [PubMed] [Google Scholar]
- 50.Kelley RE, Namon RA, Juang SH, Lee SC, Chang JY. Transcranial Doppler ultrasonography of the middle cerebral artery in the hemodynamic assessment of internal carotid artery stenosis. Arch Neurol. 1990;47:960–4. doi: 10.1001/archneur.1990.00530090030009. [DOI] [PubMed] [Google Scholar]
- 51.Hochuli E. “Fetal monitoring” within the framework of actual obstetric monitoring. Schweiz Med Wochenschr. 1976;106:841–6. [PubMed] [Google Scholar]
- 52.Sloan MA, Alexandrov AV, Tegeler CH, Spencer MP, Caplan LR, Feldmann E, et al. Assessment: Transcranial Doppler ultrasonography: Report of the therapeutics and technology assessment subcommittee of the American Academy of Neurology. Neurology. 2004;62:1468–81. doi: 10.1212/wnl.62.9.1468. [DOI] [PubMed] [Google Scholar]
- 53.Wilterdink JL, Feldmann E, Furie KL, Bragoni M, Benavides JG. Transcranial Doppler ultrasound battery reliably identifies severe internal carotid artery stenosis. Stroke. 1997;28:133–6. doi: 10.1161/01.str.28.1.133. [DOI] [PubMed] [Google Scholar]
- 54.Molina CA, Montaner J, Abilleira S, Ibarra B, Romero F, Arenillas JF, et al. Timing of spontaneous recanalization and risk of hemorrhagic transformation in acute cardioembolic stroke. Stroke. 2001;32:1079–84. doi: 10.1161/01.str.32.5.1079. [DOI] [PubMed] [Google Scholar]
- 55.Nguyen-Huynh MN, Wintermark M, English J, Lam J, Vittinghoff E, Smith WS, et al. How accurate is CT angiography in evaluating intracranial atherosclerotic disease? Stroke. 2008;39:1184–8. doi: 10.1161/STROKEAHA.107.502906. [DOI] [PubMed] [Google Scholar]
- 56.Duffis EJ, Jethwa P, Gupta G, Bonello K, Gandhi CD, Prestigiacomo CJ. Accuracy of computed tomographic angiography compared to digital subtraction angiography in the diagnosis of intracranial stenosis and its impact on clinical decision-making. J Stroke Cerebrovasc Dis. 2013;22:1013–7. doi: 10.1016/j.jstrokecerebrovasdis.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 57.Fiaccavento W, Camardi V, Litrico V, Nanfitò L, Fiaccavento R, Petrino P, et al. The surgical therapy of alkaline esophagitis. The authors' own experience. Minerva Chir. 1992;47:579–81. [PubMed] [Google Scholar]
- 58.Silvennoinen HM, Ikonen S, Soinne L, Railo M, Valanne L. CT angiographic analysis of carotid artery stenosis: Comparison of manual assessment, semiautomatic vessel analysis, and digital subtraction angiography. AJNR Am J Neuroradiol. 2007;28:97–103. [PMC free article] [PubMed] [Google Scholar]
- 59.Chilcote WA, Modic MT, Pavlicek WA, Little JR, Furlan AJ, Duchesneau PM, et al. Digital subtraction angiography of the carotid arteries: A comparative study in 100 patients. Radiology. 1981;139:287–95. doi: 10.1148/radiology.139.2.7012921. [DOI] [PubMed] [Google Scholar]
- 60.North American Symptomatic Carotid Endarterectomy Trial. Methods, patient characteristics, and progress. Stroke. 1991;22:711–20. doi: 10.1161/01.str.22.6.711. [DOI] [PubMed] [Google Scholar]
- 61.MRC European Carotid Surgery Trial: Interim results for symptomatic patients with severe (70-99%) or with mild (0-29%) carotid stenosis. European Carotid Surgery Trialists' Collaborative Group. Lancet. 1991;337:1235–43. [PubMed] [Google Scholar]
- 62.Donnan GA, Davis SM, Chambers BR, Gates PC. Surgery for prevention of stroke. Lancet. 1998;351:1372–3. doi: 10.1016/s0140-6736(98)22019-8. [DOI] [PubMed] [Google Scholar]
- 63.Thom T, Haase N, Rosamond W, Howard VJ, Rumsfeld J, Manolio T, et al. Heart disease and stroke statistics-2006 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113:e85–151. doi: 10.1161/CIRCULATIONAHA.105.171600. [DOI] [PubMed] [Google Scholar]
- 64.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics-2013 update: A report from the American Heart Association. Circulation. 2013;127:e6–245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Easton JD, Saver JL, Albers GW, Alberts MJ, Chaturvedi S, Feldmann E, et al. Definition and evaluation of transient ischemic attack: A scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. The American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke. 2009;40:2276–93. doi: 10.1161/STROKEAHA.108.192218. [DOI] [PubMed] [Google Scholar]
- 66.Cheng SF, Brown MM, Simister RJ, Richards T. Contemporary prevalence of carotid stenosis in patients presenting with ischaemic stroke. Br J Surg. 2019;106:872–8. doi: 10.1002/bjs.11136. [DOI] [PubMed] [Google Scholar]
- 67.de Weerd M, Greving JP, Hedblad B, Lorenz MW, Mathiesen EB, O'Leary DH, et al. Prevalence of asymptomatic carotid artery stenosis in the general population: An individual participant data meta-analysis. Stroke. 2010;41:1294–7. doi: 10.1161/STROKEAHA.110.581058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kamtchum-Tatuene J, Noubiap JJ, Wilman AH, Saqqur M, Shuaib A, Jickling GC. Prevalence of high-risk plaques and risk of stroke in patients with asymptomatic carotid stenosis: A meta-analysis. JAMA Neurol. 2020;77:1524–35. doi: 10.1001/jamaneurol.2020.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Teng Z, Sadat U, Brown AJ, Gillard JH. Plaque hemorrhage in carotid artery disease: Pathogenesis, clinical and biomechanical considerations. J Biomech. 2014;47:847–58. doi: 10.1016/j.jbiomech.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schindler A, Schinner R, Altaf N, Hosseini AA, Simpson RJ, Esposito-Bauer L, et al. Prediction of stroke risk by detection of hemorrhage in carotid plaques: Meta-analysis of individual patient data. JACC Cardiovasc Imaging. 2020;13:395–406. doi: 10.1016/j.jcmg.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 71.North American Symptomatic Carotid Endarterectomy Trial Collaborators; Barnett HJM, Taylor DW, Haynes RB, Sackett DL, Peerless SJ, et al. Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med. 1991;325:445–53. doi: 10.1056/NEJM199108153250701. [DOI] [PubMed] [Google Scholar]
- 72.Barnett HJ, Taylor DW, Eliasziw M, Fox AJ, Ferguson GG, Haynes RB, et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1998;339:1415–25. doi: 10.1056/NEJM199811123392002. [DOI] [PubMed] [Google Scholar]
- 73.Müller MD, Lyrer P, Brown MM, Bonati LH. Carotid artery stenting versus endarterectomy for treatment of carotid artery stenosis. Cochrane Database Syst Rev. 2020;2:CD000515. doi: 10.1002/14651858.CD000515.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng TW, Pointer KE, Gopal M, Farber A, Jones DW, Eberhardt RT, et al. Natural history of non-operative management in asymptomatic patients with 70%-80% internal carotid artery stenosis by duplex criteria. Eur J Vasc Endovasc Surg. 2020;60:339–46. doi: 10.1016/j.ejvs.2020.05.039. [DOI] [PubMed] [Google Scholar]
- 75.Gaba K, Ringleb PA, Halliday A. Asymptomatic carotid stenosis: Intervention or best medical therapy? Curr Neurol Neurosci Rep. 2018;18:80. doi: 10.1007/s11910-018-0888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hobson RW, 2nd, Weiss DG, Fields WS, Goldstone J, Moore WS, Towne JB, et al. Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. The Veterans Affairs Cooperative Study Group. N Engl J Med. 1993;328:221–7. doi: 10.1056/NEJM199301283280401. [DOI] [PubMed] [Google Scholar]
- 77.Castilla-Guerra L, Del Carmen Fernandez-Moreno M, Colmenero-Camacho MA. Statins in stroke prevention: Present and future. Curr Pharm Des. 2016;22:4638–44. doi: 10.2174/1381612822666160510125229. [DOI] [PubMed] [Google Scholar]
- 78.Saba L, Agarwal N, Cau R, Gerosa C, Sanfilippo R, Porcu M, et al. Review of imaging biomarkers for the vulnerable carotid plaque. JVS Vasc Sci. 2021;2:149–58. doi: 10.1016/j.jvssci.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Narins CR, Illig KA. Patient selection for carotid stenting versus endarterectomy: A systematic review. J Vasc Surg. 2006;44:661–72. doi: 10.1016/j.jvs.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 80.Bell IR, Edman JS, Marby DW, Satlin A, Dreier T, Liptzin B, et al. Vitamin B12 and folate status in acute geropsychiatric inpatients: Affective and cognitive characteristics of a vitamin nondeficient population. Biol Psychiatry. 1990;27:125–37. doi: 10.1016/0006-3223(90)90642-f. [DOI] [PubMed] [Google Scholar]
- 81.de Borst GJ. Transcarotid artery stenting: Hype or hope? Stroke. 2022;53:108–10. doi: 10.1161/STROKEAHA.121.036464. [DOI] [PubMed] [Google Scholar]
- 82.Schermerhorn ML, Liang P, Dakour-Aridi H, Kashyap VS, Wang GJ, Nolan BW, et al. In-hospital outcomes of transcarotid artery revascularization and carotid endarterectomy in the Society for Vascular Surgery Vascular Quality Initiative. J Vasc Surg. 2020;71:87–95. doi: 10.1016/j.jvs.2018.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kashyap VS, King AH, Foteh MI, Janko M, Jim J, Motaganahalli RL, et al. A multi-institutional analysis of transcarotid artery revascularization compared to carotid endarterectomy. J Vasc Surg. 2019;70:123–9. doi: 10.1016/j.jvs.2018.09.060. [DOI] [PubMed] [Google Scholar]
- 84.Aarli SJ, Thomassen L, Waje-Andreassen U, Logallo N, Kvistad CE, Næss H, et al. The course of carotid plaque vulnerability assessed by advanced neurosonology. Front Neurol. 2021;12:702657. doi: 10.3389/fneur.2021.702657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van den Bouwhuijsen QJ, Vernooij MW, Verhaaren BFJ, Vrooman HA, Niessen WJ, Krestin GP, et al. Carotid plaque morphology and ischemic vascular brain disease on MRI. AJNR Am J Neuroradiol. 2017;38:1776–82. doi: 10.3174/ajnr.A5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Porambo ME, DeMarco JK. MR imaging of vulnerable carotid plaque. Cardiovasc Diagn Ther. 2020;10:1019–31. doi: 10.21037/cdt.2020.03.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chrencik MT, Khan AA, Luther L, Anthony L, Yokemick J, Patel J, et al. Quantitative assessment of carotid plaque morphology (geometry and tissue composition) using computed tomography angiography. J Vasc Surg. 2019;70:858–68. doi: 10.1016/j.jvs.2018.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]