

Abstract
Retinitis pigmentosa (RP) is a neurodegenerative disorder that causes irreversible vision loss in over 1.5 million individuals world-wide. The genetic heterogeneity of RP necessitates a broad therapy that is able to provide treatment in a gene- and mutation- non-specific manner. In this study, we identify the therapeutic benefits of metabolic reprogramming by targeting pyruvate kinase M2 (PKM2) in a Pde6β preclinical model of RP. The genetic contributions of PKM2 inhibition in retinal degeneration were evaluated through histology and electroretinogram (ERG) followed by a statistical analysis using a linear regression model. Notably, PKM2 ablation resulted in thicker retinal layers in Pde6β-mutated mice as compared to the controls, suggesting greater photoreceptor survival. Consistent with these anatomical findings, ERG analyses revealed that the maximum b-wave is on average greater in Pkm2 knockout mice than in mice with intact Pkm2, indicating enhanced photoreceptor function. These rescue phenotypes from Pkm2 ablation in a preclinical model of RP indicate that a metabolome reprogramming may be useful in treating RP.
Background
Retinal degenerative diseases afect a total of nine million Americans (Friedman et al. 2004; Schmier et al. 2006). Among these conditions, retinitis pigmentosa (RP) is the most devastating in terms of severity and prevalence, making it one of the most common cause of irreversible vision loss. Associated with more than 80 genes, RP is a heterogeneous condition that is characterized by rod photoreceptor degeneration followed by secondary cone death (Campochiaro and Mir 2018; Ferrari et al. 2011; Punzo et al. 2009), leading to night blindness, tunnel vision, and eventually, complete blindness. Current treatment options include the use of retinal prostheses (da Cruz et al. 2013, 2016) or gene therapy in the case of Lebers congenital amaurosis caused by mutations in RPE65 (Bainbridge et al. 2008; Maguire et al. 2008; Russell et al. 2017). However, these strategies are only applicable for patients with end-stage RP or those carrying RPE65 mutations (Duncan et al. 2018), respectively. Currently, clinical gene therapy trials for RP primarily involve augmentation or repair of a single gene, which even if successful, would only be applicable to patients with a specific mutation or gene etiology (Takahashi et al. 2018). More recently, insight into the genetic etiologies of retinal degeneration has found that photoreceptor death may be linked to apoptotic factors (Chinskey et al. 2014; Zadro-Lamoureux et al. 2009), metabolic deregulation (Punzo et al. 2009, 2012), and oxidative stress (Usui et al. 2009; Xiong et al. 2015). Targeting any one of these pathways, in turn, may offer an alternative gene non-specific therapy. As of yet, however, there is no effective therapeutic option for RP and other retinal degenerative diseases.
Punzo et al. studied gene expression changes in RP mouse models by linking mutations in rod-specific genes to their corresponding cellular processes (Punzo et al. 2009). Microarray analysis of samples collected at the onset of cone death revealed that 35% of annotated genes were involved in cellular metabolism, implicating metabolic processes in disease pathogenesis. To follow-up, Punzo et al. and Zhang et al. focused on the insulin/mTOR pathway and found that enhanced anabolic flux and lactate levels indeed slowed rodcone degeneration (Iadevaia et al. 2012; Zhang et al. 2016b). Consistent with these findings, rod-specific knockout of sirtuin 6, a transcriptional repressor of glycolytic enzymes, enhanced glycolysis, rod preservation, and photoreceptor function (Martinez-Pastor and Mostoslavsky 2012). Augmenting anabolism in rod photoreceptors could therefore serve as a potential treatment for RP.
One identified key regulator of cellular metabolism is pyruvate kinase M2 (PKM2), a critical catalyst for the final step of glycolysis that aids in the transfer of a phosphate group from phosphoenolpyruvate (PEP) to adenosine diphosphate (ADP). PKM2 is allosterically regulated so that it exists in two forms: an inactive dimer with low binding affinity to PEP and an active tetramer form with high affinity for PEP (Dong et al. 2016). Phosphorylation of PKM2 (P-PKM2) inhibits the formation of tetrameric PKM2, reducing its activity and leading to PEP accumulation. This has been found to activate the pentose phosphate pathway (PPP), a process that produces glycolytic intermediates for nucleotide and lipid synthesis. As a result, when PKM2 is inhibited, PEP accumulates, which promotes the PPP (Rajala et al. 2016). PKM2′s ability to regulate the production of glycolytic intermediates, which is essential for photoreceptor cells, makes it an attractive target to elucidate the role of glycolysis in rod and cone health. Previous studies have already found that PKM2 ablation enhances long-term photoreceptor survival in retinal detachment models, suggesting that PKM2 ablation may offer photoreceptor protection against acute external retinal stress (Wubben et al. 2017).
More recently, PKM2 has been implicated in the regulation of phosphodiesterase-6 (PDE6) (Rajala et al. 2018), a phototransduction enzyme complex that blocks Ca+2 influx by closing cyclic nucleotide-gated (CNG) channels through cGMP hydrolysis. PDE6 is a heterotetramer with two catalytic subunits (PDE6α and PDE6β) and mutations in either can similarly disrupt function and yield identical downstream effects (Bird 1995; Daiger et al. 2007; Davis et al. 2008; Hartong et al. 2006; Janisch et al. 2009; McLaughlin et al. 1995; Sancho-Pelluz et al. 2019; Tosi et al. 2011a, b; Tsang et al. 2008; Wert et al. 2013; Woodruff et al. 2008; Yang et al. 2012). One in 10 Americans carries a recessive RP allele (Hartong et al. 2006; Sohocki et al. 2001), and 10% of those alleles confer mutations in one of the two subunits of phosphodiesterase-6, accounting for 5–8% of all RP cases (Daiger et al. 2007; Hartong et al. 2006). As such, PKM2 may very well also play a role in preserving photoreceptors from inherited retinal diseases, which represent internal sources of retinal stress.
In this report, we expand on previous findings by investigating the functional and morphological contributions of rod-specific Pkm2 ablation in inherited retinal degeneration with a tamoxifen-inducible Pkm2 knockout Pde6βH620Q/H620Q RP model. We hypothesized that Pkm2 knockout in the rods would promote photoreceptor anabolism and, in doing so, preserve rod and cone survival and function, delaying retinal degeneration.
Results
Deletion of Pkm2 in Pde6βH620Q/H620Q mice
To evaluate whether PKM2 plays a role in rod metabolome reprogramming, we used the tamoxifen-inducible Cre recombinase system to ablate Pkm2 in a slow progressing Pde6β RP model: Pde6βH620Q/H620Q (Davis et al. 2008). To achieve a photoreceptor-specific conditional ablation of PKM2, mice were crossed with the Pde6gCreERT2 strain. Experimental and control mice were treated with tamoxifen and ethanol, respectively, at P8, P9, and P11. PKM2 protein levels were evaluated at five weeks (P36-P39). Rod selective ablation of Pkm2 significantly reduced total PKM2 levels in Pde6βH620Q/H620Q Pkm2−/− mice compared to wild-type mice (Fig. 1). Accordingly, there was also a reduction in phosphorylated PKM2 in Pde6βH620Q/H620Q Pkm2−/− mice, suggesting a decrease in the catalytic activity of PKM2. It is important to note, however, that experimental mice retained detectable levels of PKM2 and P-PKM2, shown in Fig. 1, due to the conditional nature of the rod-specific Pkm2 knockout.
Fig. 1.
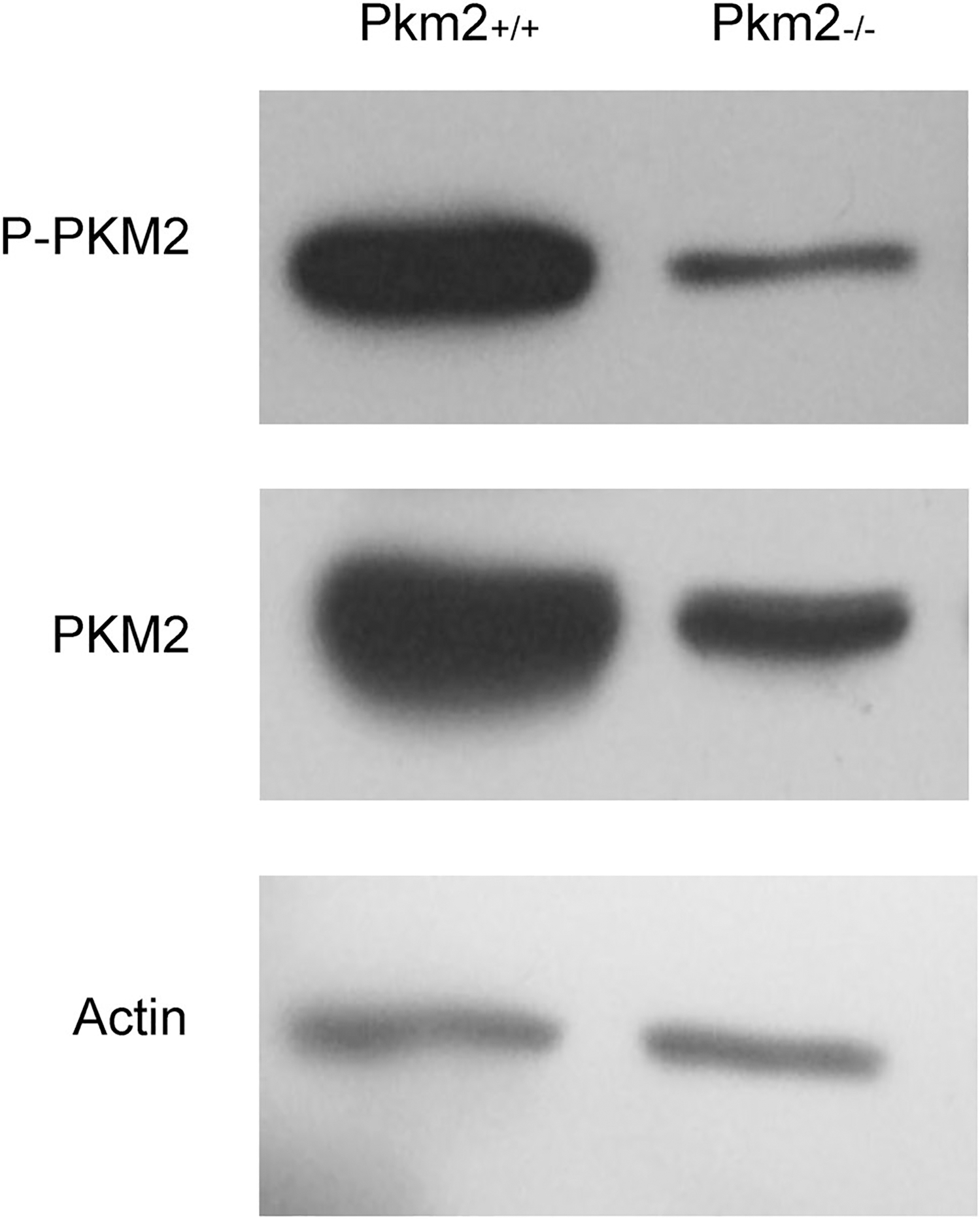
Deletion of Pkm2 in Pde6βH620Q/H620Q mice. Representative PKM2 and P-PKM2 protein levels at five weeks (P36–39). Experimental and control mice were injected with tamoxifen and ethanol, respectively at P8, P9, and P11. Selective Pkm2 deletion in photoreceptors reduced Pkm2 and P-Pkm2 expression (n = 3 for both groups). Actin served as loading control
Deletion of Pkm2 in Pde6βH620Q/H620Q mice improved retinal morphology
Subsequently, we gauged the survival impact of Pkm2 ablation on retinal morphology using histology to compare the morphology of the outer nuclear layer (ONL), inner segment (IS), and outer segment (OS). Retinal samples were extracted and evaluated at P60 for Pde6gCreERT2/+ mice and at nine weeks (P64-P67) for all other mice. Pde6βH620Q/H620Q Pkm2+/+ and Pde6gCreERT2/+ mice exhibited degenerated photoreceptors within ONL while Pde6βH620Q/H620Q Pkm2−/− mice had comparatively more cells (Fig. 2A). Similar trends were observed for IS/OS layers—compared to treated mice who demonstrated a clear IS/OS junction with relatively thick IS and OS layers, isogenic controls exhibited an atrophic IS layer and absent OS layer (Fig. 2a b). Pde6gCreERT2/+ mice also showed significantly thinner IS/OS layers to the point where the IS/OS junction was not appreciable. Strikingly, all the layers in the experimental mice were still noticeably thinner than those in wild-type mice. Overall, ablation of Pkm2 may be able to preserve but not completely rescue photoreceptor survival and preserved photoreceptors.
Fig. 2.
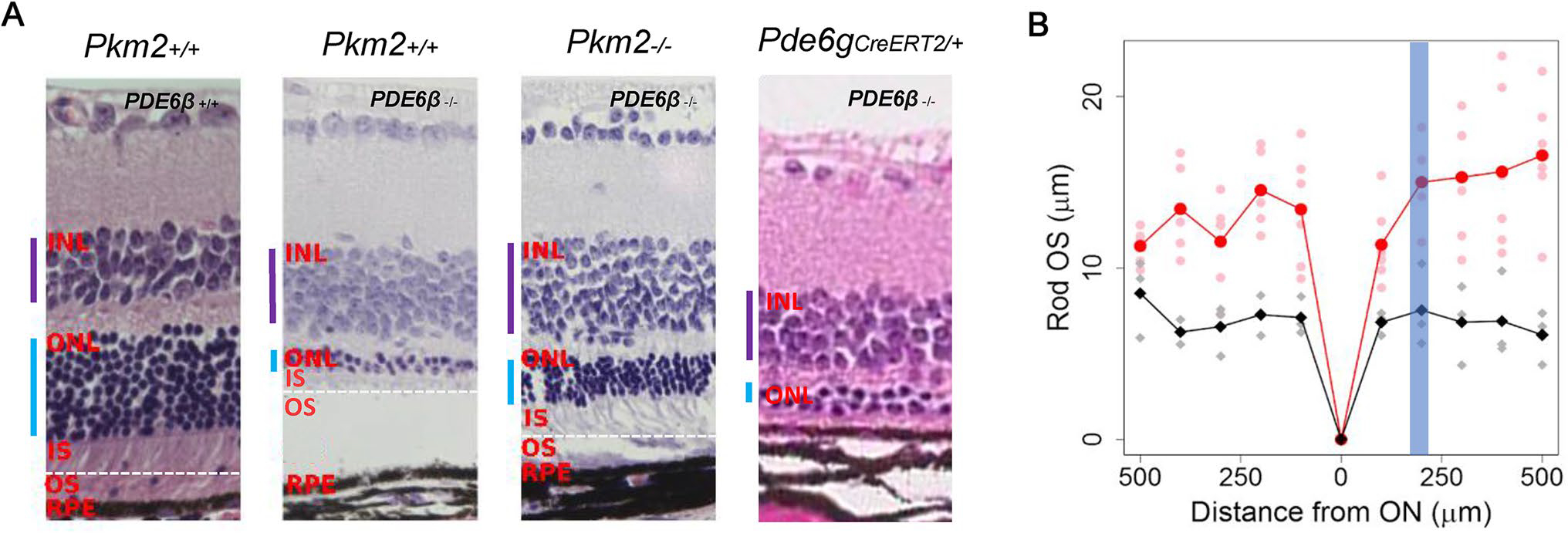
Deletion of Pkm2 in Pde6βH620Q/H620Q mice improved retinal morphology. a Representative H&E-stained retinal sections were imaged 180–220 μm from the optic nerve. Pde6gCreERT2/+ mice were imaged at P60, and all other mice were imaged at nine weeks (P64-P67). Selective deletion of Pkm2 enhanced but did not completely rescue ONL thickness in Pde6βH620Q/H620Q Pkm2−/− mice (n = 3), but not in Pkm2+/+ Pde6βH620Q/H620Q controls. At P8, P9, and P11, experimental and Pde6gCreERT2/+ mice were injected tamoxifen while wildtype and control mice were injected with oil. (Purple bar indicates INL, blue bar indicates ONL, dashed white horizontal line denotes IS/OS junction). b The difference in rod OS length was quantified at nine weeks (P64–P67). Selection deletion of PKM2 increases Rod OS thickness at every distance from the ON (n = 3). Red represents experimental mice, and black represents control mice. Highlighted line shows average Rod OS while faded dots represent individual measurements for mice. Blue band represents the area within which the histology images were taken (180–220 μm)
Deletion of Pkm2 in Pde6βH620Q/H620Q mice improved retinal function
Having established that PKM2 knockout partially rescues the number of photoreceptors, we next evaluated how it impacts the functionality of these photoreceptors. To do this, we obtained scotopic, mixed maximum, and photopic electroretinogram (ERG) recordings. At four weeks (P28), Pde6βH620Q/H620Q Pkm2−/− mice exhibited an enhanced scotopic b-wave amplitude compared to Pde6βH620Q/H620Q Pkm2+/+ (Fig. 3a), suggesting greater rod preservation. In contrast, there was no significant difference in the mixed and photopic b-wave amplitudes between the two groups, indicating that cone preservation remained approximately the same. At eight weeks (P56), all b-wave amplitudes in both groups declined considerably. Notably, Pde6βH620Q/H620Q Pkm2−/− mice maintained significantly higher scotopic, maximal, and photopic b-waves (P < 0.05) compared to Pde6βH620Q/H620Q Pkm2+/+ mice, indicating a more robust preservation in cone and rod function. Statistical analysis also revealed a significant correlation (P < 0.05) between the value of the maximal b-wave and presence of tamoxifen treatment. These results reveal that rod-specific Pkm2 ablation improved photoreceptor vitality in Pde6β-mutated mice.
Fig. 3.
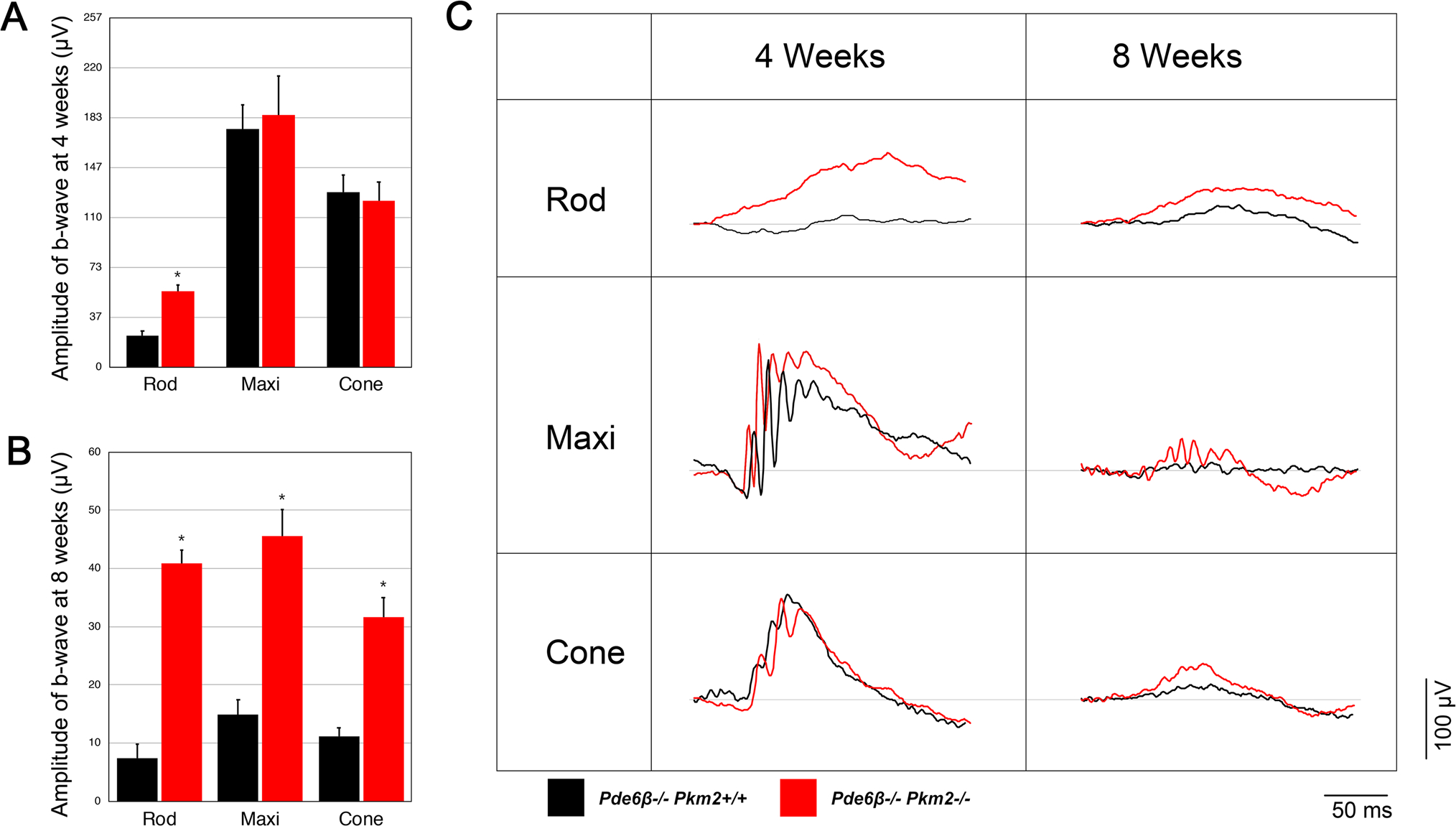
Deletion of Pkm2 in Pde6βH620Q/H620Q mice preserves rod and cone function. ERG b-wave magnitudes for scotopic, mixed maximal, and photopic response were taken and compared for control (Pkm2+/+ Pde6βH620Q/H620Q, blue bar) and experimental (Pkm2−/− Pde6βH620Q/H620Q, green bar) mice. Experimental and control mice were injected with tamoxifen and ethanol, respectively at P8, P9, and P11. a ERG recordings at four weeks (P28). Experimental mice exhibited enhanced scotopic b-wave amplitudes compared to controls. Differences in the magnitudes of mixed maximal and photopic b-waves were not statistically significant. (n = 12 for both groups; (*P < 0.05). b ERG recordings at eight weeks (P56). Experimental mice exhibited enhanced b-wave magnitudes for all groups compared to controls. (n = 6 for control group, n = 8 for experimental group; *P < 0.05). c Representative ERG tracings for scotopic, mixed maximal, and photopic response at four weeks (P28) and eight weeks (P56)
Discussion
In this report, we demonstrated that rod-specific ablation of Pkm2 can rescue both cone and rod photoreceptor degeneration. Using both structural and functional assays, we are the first to use a preclinical RP model to study the effects of Pkm2 knockout. These findings further advance the work done by Chinchore et al. who first discovered that PKM2 mediates Warburg glycolysis in healthy photoreceptors (Chinchore et al. 2017), implicating its role in photoreceptor wellbeing. Since then, various studies have sought to elucidate a direct cause-and-effect relationship. Notably, Wubben et al. observed that photoreceptor-specific ablation of Pkm2 impaired photoreceptor survival and function under baseline conditions, but improved long-term photoreceptor survival under acute outer retinal stress. To account for the rescue in the retinal detachment model, they noted the upregulation of Pkm1 and other genes involved in aerobic metabolism and proposed that accumulation of PK substrate 2-phosphoenolpyruvate was sufficient to counteract the insult from induced cell damage.
Here, we advance the current understanding of PKM2 by uncovering its genetic contributions in inherited retinal degeneration. Notably, rod-specific Pkm2 knockout noticeably increases the thickness of the ONL and IS/OS layers, indicating better photoreceptor survival. ERG functional analysis was consistent with anatomical findings and revealed that Pkm2 ablation also improved both cone and rod function significantly. These results suggest that PKM2 deficiency was indeed able to rescue retinal degeneration. As previous studies have proposed, the rescue phenotypes may be a direct result of enhancing catabolism and therefore ATP output in photoreceptors. This explanation, however, handles photoreceptors as isolated systems whose metabolism works in a cell autonomous manner. In this paper, we propose an alternative yet complementary framework that aims to consider PKM2′s role in photoreceptors in relation to the retinal pigment epithelium (RPE), offering insight into how it may regulate the delicate metabolic dynamic between the two.
One of the first evidences of a metabolic coupling was described by Punzo et al. (2009) who found that the disruptions in the insulin/mTOR signaling pathway coincided with cone death in four mouse models for RP. Strikingly, systemic supplementation of insulin was able to rescue cone death, suggesting that photoreceptor death in RP progresses in a cell non-autonomous manner. Subsequent studies characterizing the nature of this relationship have led to the emergence of the “metabolic coupling hypothesis”, which proposes that metabolic imbalance leads to cone photoreceptor death via glucose deprivation (Punzo et al. 2009; Venkatesh et al. 2015; Zhang et al. 2016b). Normally, photoreceptors take up glucose from the RPE and convert it into fat and lactate (Kanow et al. 2017), which are subsequently used to drive essential processes throughout the retina. In photoreceptors, the steady production of protein and lipids drives the daily disposal and regeneration of the OS (Hamel 2006; Molday and Moritz 2015). In contrast, the RPE consumes the generated lactate as the carbon source for the tricarboxylic acid (TCA) cycle (Barabas et al. 2010; Chinchore et al. 2017; Du et al. 2016), saving glucose for photoreceptors (Fig. 4a). In RP, however, impaired photoexcitation prevents diseased rods from generating sufficient amounts of lipid and lactate. This not only stagnates OS biogenesis but also prompts the RPE to use glucose through anabolism (Fig. 4b). The depletion of glucose by the RPE, in turn, deprives photoreceptors of nourishment and leads to prevalent rod and subsequent cone death—the hallmark of RP pathogenesis.
Fig. 4.
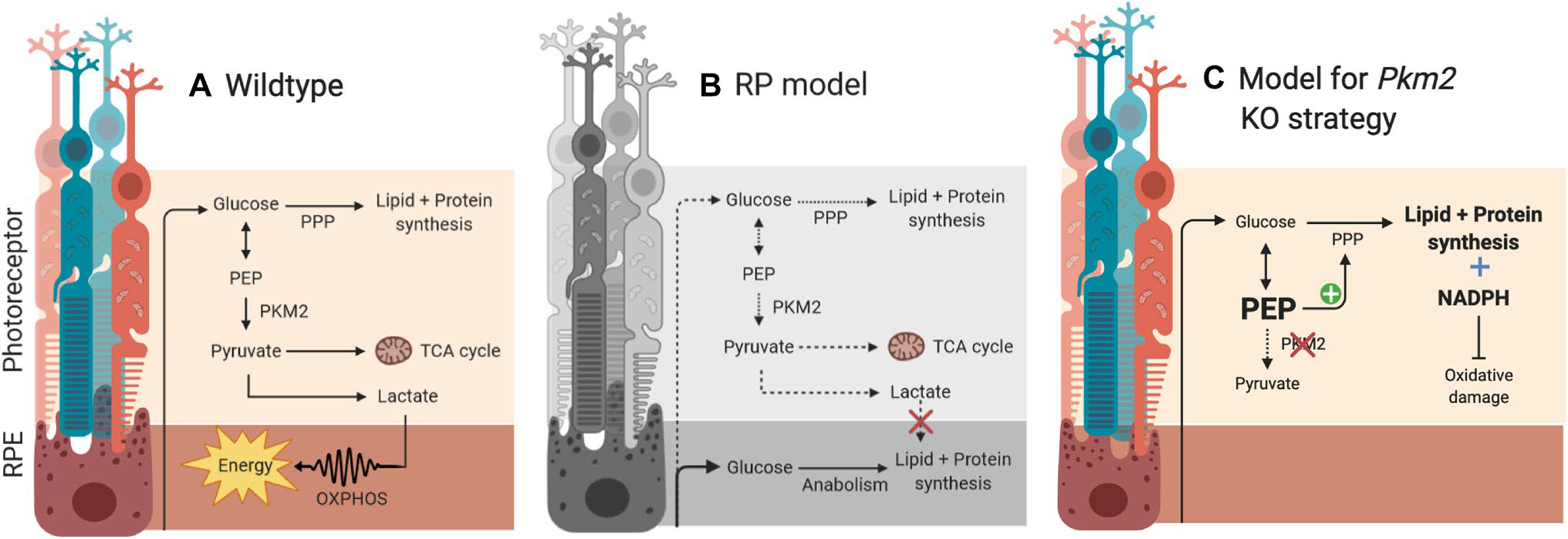
Schematic of metabolic pathways and the role of PKM2 in a wildtype, b RP model, and c RP model with Pkm2 KO
In light of this framework, the rescued phenotypes indicate a rejuvenation of the disrupted metabolic dynamic within the diseased retina. Previous studies have shown that phosphorylated PKM2 leads to the accumulation of 2-phosphoenolpyruvate and the subsequent activation of the anabolic PPP pathway (Rajala et al. 2018). As such, PKM2 deficiency may elevate anabolism and lead to recovery of normal cellular processes. This metabolic reprogramming, in turn, restores a steady supply of glucose to the diseased photoreceptors, precluding the need to optimize metabolism. As a result, the photoreceptors return to a preferred anabolic state and employ aerobic glycolysis to satisfy their ATP needs, promoting photoreceptor health and function (Fig. 4c). It is important to note that this model is not necessarily incompatible with the notion of catabolism-mediated photoreceptor survival as described by Wubben et al. Roughly 80%–90% of the glucose in photoreceptors is metabolized through aerobic glycolysis, indicating that a small, but significant, fraction still undergoes catabolism. While we highlight that different anabolic processes can provide photoreceptors with both the raw materials and energy to survive and function, it is possible that a certain level of catabolism is needed to sufficiently meet the high energy demands of a healthy photoreceptor and counteract the insult from metabolic imbalance.
Another notable finding is that despite the significant improvements in both retinal morphology and function, PKM2 ablation was not able to induce a complete rescue in either case. Pde6βH620Q/H62Q Pkm2−/− mice exhibited thinner ONLs than that of wild-type mice, indicating increased photoreceptor death. Moreover, the retinal function of Pde6βH620Q/ H62Q Pkm2−/− mice decreased after the initial improvement. Both these findings are likely a result of the short-lived increase in anabolism from Pkm2 ablation. It has been suggested that PKM1 mediates ATP synthesis by promoting catabolism in the absence of PKM2 (Rajala et al. 2018; Wubben et al. 2017). However, Rajala et al. recently demonstrated that the upregulation of PKM1 from Pkm2 deletion is unable to match the catalytic activity of endogenous PKM2, suggesting that PKM1 cannot compensate for PKM2 deficiency (Rajala et al. 2018). Together, these findings suggest the possibility that PKM1 upregulation interferes with the anabolic influx from PKM2 inhibition and promotes catabolism. The resultant inhibition of anabolism deprives photoreceptors of essential building blocks, leading to the observed decrease in ONL thickness. At the same time, PKM1 is unable to generate sufficient energy to sustain photoreceptor function, resulting in the decrease in ERG response over time. Alternatively, it is possible that retinal degeneration impacts a wide range of independent metabolic pathways. As such, PKM2 ablation is only able to affect PKM2-dependent processes, leading to a partial but not complete rescue of the deleterious phenotypes.
Although this study identifies PKM2 as one of the major players in slowing down retinal degeneration, future studies identifying specific molecules and pathways would be beneficial in understanding the mechanism of this relationship. Wubben et al. offered critical insight into the specific genes impacted by PKM2 ablation in the retina (Wubben et al. 2017). Expanding their analysis to include gene expression changes in the RPE offers a quantitative way to not only support PKM2′s role in regulating metabolic coupling, but also track the metabolic changes over time. Another promising direction for research is to characterize the role of PKM1 and whether its interaction with PKM2 is conserved in inherited retinal disease. This question is important as it would clarify whether PKM1 indeed acts as an antagonistic regulator of anabolic influx that may limit the impact of metabolic reprogramming. Other potential directions include tuberous sclerosis complex 1 (TSC1), an mTOR inhibitor, and hypoxia inducible factor-1α (HIF-1α), a transcription factor responsible for oxygen consumption regulation. In our earlier studies, we showed that both of these molecules are linked to photoreceptor rescue from metabolic reprogramming (Zhang et al. 2016a, b). Moreover, separate groups have found that PKM2 interacts directly with mTOR and HIF-1α, hinting at possible mechanistic pathways (Anastasiou et al. 2011; Sun et al. 2011; Zhang et al. 2019).
One important consideration is that Rajala et al. have shown in vitro evidence that PKM2 may regulate transcriptional activity through the PDE6β promoter (Rajala et al. 2018). This finding suggests that PKM2 knockout may have promoted photoreceptor health by working directly with Pde6β, which would indicate that the beneficial effects are specific to Pde6β deficiency. As such, we highlight the need to use a different RP model to evaluate whether the rescue phenotypes from PKM2 ablation are in fact a result of its role in a gene non-specific metabolic pathway.
Conclusions
Given that over 80 genes have been implicated in the development of RP, current CRISPR/Cas9-based gene therapy strategies are impractical as they are not amenable to large-scale production or treatment. Here, we have demonstrated that PKM2 ablation offers 6 equivalent years of improved eyesight in our RP model (Dutta and Sengupta 2016), suggesting that elevations in rod-specific anabolism can offer protection against degeneration in a gene non-specific manner. Importantly, the metabolic pathways highlighted in this study are common to not only retinal degeneration, but also neurodegenerative conditions including Alzheimer’s Disease (Demetrius and Driver 2013; Demetrius and Simon 2012). As such, reprogramming the metabolism of a terminally differentiated neuron towards a more anabolic state may offer a universal, gene non-specific treatment for multiple neurodegenerative conditions. The function of PKM2 as a catalyst for the final step in glycolysis lies at the heart of metabolic regulation. Therefore, a deeper understanding of its role in coupling the dynamic metabolisms of the photoreceptor and RPE may offer critical insight into and advancement towards the development of gene non-specific therapeutics. Such a strategy can then be used synergistically with gene supplementation therapy to optimize efficacy.
Materials and methods
Animals
All mice were maintained in the Columbia University Pathogen-free Eye Institute Annex Animal Care Services Facility under a 12-h light/12-h dark cycle. PKM2flox/flox mice (B6;129S-Pkmtm1.1Mgvh/J) were purchased from The Jackson Laboratory while Pde6βH620Q/H620Q mice were re-derived via oviduct transfer from morulae provided by the European Mouse Mutant Archive (EMMA). Our lab generated Pde6gCreERT2 mice (Koch et al. 2015).
The inducible Pde6gCreERT2 driver was used to exert both temporal and spatial control over PKM2 ablation in Pde6βH620Q/H620Q mice. PKM2 ablation was activated during the mid-stage of degeneration for clinical relevance and limited to the rod photoreceptors to prevent embryonic lethality. To generate this conditional PKM2 knockout in a Pde6βH620Q/H620Q mouse model, Pde6βH620Q/+ mice were crossed first with Pde6gCreERT2 mice and then Pkm2flox/flox mice. The resultant progeny were then backcrossed four generations to generate heterozygous Pde6βH620Q/+ Pkm2flox/flox Pde6gCreERT2. A total of six generations of backcrosses were performed to produce the desired homozygous genotype of Pde6βH620Q/H620Q Pkm2flox/flox Pde6gCreERT2.
The experimental mice were injected intraperitoneally with tamoxifen (amount: 100 μg/g body weight; concentration: 100 mg/ml in ethanol; dilution: 10 mg/ml in corn oil at 42 °C; catalog T5648; Sigma-Aldrich) to generate a genotype of Pde6βH620Q/H620Q Pkm2−/−. Isogenic control and wild-type mice were injected with ethanol (amount: 100 μg/g body weight; dilution: 10% in corn oil) to generate a genotype of Pde6βH620Q/H620Q Pkm2+/+. All mice were treated with their respective injections at three time points: P8, P9, and P11. They were not discriminated based on sex, and different mice were used for each of the experiments performed.
Immunoblotting
Following the protocol described by Tsang et al. (1998, 1996), retina samples were collected from mice at five weeks (P36-P39). To evaluate PKM2 expression levels, the samples were homogenized using a solution consisting of M-PER Mammalian Protein Extraction Reagent (Prod #78501 Thermo Scientific), phosphatase inhibitor cocktail (catalog P2850–5 mL; Sigma), and protease inhibitor cocktail (catalog P8340–1 mL; Sigma). After transfer onto nitrocellulose membrane (BioRad), samples were blocked in 5% skim milk 902887 MP Biomedicals, LLC). The membrane was then immersed overnight at 4 °C in the following primary antibodies: rabbit Anti-PKM2 (1:1000; #3198S; Cell Signaling Technology), mouse anti–β-actin (1:1000; ab125248; Abcam), and rabbit anti-phospho-PKM2 (1:1000; #3827S; Cell Signaling Technology). The following day, the membrane was washed in 0.5% PBST (500 μl Tween-20 in 1000 ml PBS), and subsequently incubated in mouse monoclonal anti-rabbit IgG-HRP antibody (1:2000; ab99697; Abcam) and rabbit anti-mouse IgG-HRP antibody (1:2,000; sc-358914; Santa Cruz Biotechnology Inc.). Chemiluminescence (EMD Millipore) was detected using Biomax film (Kodak), and PKM2, P-PKM2, and actin levels were then quantified.
Histology
As described by Velez et al. (2017), eyes were enucleated from euthanized mice, by following along the limbus to create a puncture at the 12 o’clock position. Each eye was then placed in a solution of 1:2 × Karnovsky fixative for 24 h. Following fixation, eyes underwent a saline wash and then dehydration to be embedded in paraffin. Afterwards, hematoxylin and eosin were used to stain paraffin sections, which were then examined by light microscopy. Pde6gCreERT2/+ mice were imaged at P60, and all other mice were imaged at nine weeks (P64–P67). Imaging was taken 180–220 μm from the optic nerve.
Rod OS Measurement
Following euthanasia, the eye was enucleated followed by cornea and lens dissection and vitreous removal. At every 100 microns from the optic nerve, rod OS was measured using a Image J (NIH, Bethesda). Measurements were taken for segments 500 microns away from the optic nerve on both eyes. All procedures were done in accordance with previously established IACUC guidelines.
ERG
Scotopic, mixed maximum, and photopic full-field electroretinogram (ERG) recordings were obtained for experimental and control mice at two timepoints: four weeks (P28) and eight weeks (P56). Both eyes of each mouse (20 eyes) were analyzed and averaged due to the correlation between mouse eyes. Following a 12 h overnight dark adaptation, mice were anesthetized using the protocol described by Sancho-Pelluz et al. (2019). All procedures performed on the mice were completed under dim lighting. Mice were dilated using a solution composed of phenylephrine hydrochloride (2.5%, Paragon) and tropicamide (1%, Akorn) that was administered topically. Mouse body temperature was maintained during all experiments using a heating pad. Three different experiments were performed—scotopic recording (pulse intensity: 0.001 Cd/m2), mixed maximal recording (pulse intensity: 3 Cd/m2), and photopic recording (pulse intensity: 30 Cd/m2). All ERG data were collected using Espion E2 Electroretinography System (Diagnosys LLC).
Statistical Analysis
Using the ERG results, the maximal b-wave wave of each eye was taken and used to fit a linear model using generalized least squares. This method of regression was chosen because of the correlation between the eyes of each mouse and to account for unequal variance between treated and untreated mice.
Acknowledgements
Jonas Children’s Vision Care and Bernard & Shirlee Brown Glaucoma Laboratory are supported by the National Institutes of Health [P30EY019007, R01EY018213, R01EY026682, R24EY027285, R24 EY28758-U01EY030580, National Cancer Institute Core [5P30CA013696], Foundation Fighting Blindness [TA-NMT-0116–0692-COLU], the Research to Prevent Blindness (RPB) Physician-Scientist Award, and unrestricted funds from RPB, New York, NY, USA. S.H.T. is a member of the RD-CURE Consortium and is supported by Kobi and Nancy Karp, the Crowley Family Fund, the Rosenbaum Family Foundation, the Tistou and Charlotte Kerstan Foundation, the Schneeweiss Stem Cell Fund, New York State [C029572], and the Gebroe Family Foundation.
Funding
Funding for this research was supported by the Global Ophthalmology Awards Program (GOAP), a Bayer-sponsored initiative committed to supporting ophthalmic research across the world.
Footnotes
Conflict of interest There are no conflicts of interest to disclose for any author.
Compliance with ethical standards
Ethical approval All experiments were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC) prior to the start of the study. All mice were used and maintained in accordance with the Statement for the Use of Animals in Ophthalmic and Vision Research of the Association for Research in Vision and Ophthalmology and the Policy for the Use of Animals in Neuroscience Research of the Society for Neuroscience.
Data availability
All data in this manuscript are available for share. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Anastasiou D (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283 10.1126/science.1211485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge JW (2008) Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358:2231–2239 10.1056/NEJMoa0802268 [DOI] [PubMed] [Google Scholar]
- Barabas P, Cutler Peck C, Krizaj D (2010) Do calcium channel blockers rescue dying photoreceptors in the Pde6b (rd1) mouse? Adv Exp Med Biol 664:491–499 10.1007/978-1-4419-1399-9_56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird AC (1995) Retinal photoreceptor dystrophies: the LI Edward Jackson Memorial Lecture. Am J Opthalmol 119:543–562 [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Mir TA (2018) The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res 62:24–37. 10.1016/j.preteyeres.2017.08.004 [DOI] [PubMed] [Google Scholar]
- Chinchore Y, Begaj T, Wu D, Drokhlyansky E, Cepko CL (2017) Glycolytic reliance promotes anabolism in photoreceptors. Elife. 10.7554/eLife.25946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinskey ND, Besirli CG, Zacks DN (2014) Retinal cell death and current strategies in retinal neuroprotection. Curr Opin Ophthalmol 25:228–233. 10.1097/ICU.0000000000000043 [DOI] [PubMed] [Google Scholar]
- Cruz da L (2013) The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br J Ophthalmol 97:632–636. 10.1136/bjophthalmol-2012-301525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz da L (2016) Five-year safety and performance results from the argus II retinal prosthesis system clinical trial. Ophthalmology 123:2248–2254. 10.1016/j.ophtha.2016.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Bowne SJ, Sullivan LS (2007) Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 125:151–158. 10.1001/archopht.125.2.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (2008) Functional rescue of degenerating photoreceptors in mice homozygous for a hypomorphic cGMP phosphodiesterase 6 b allele (Pde6bH620Q). Investig Ophthalmol Vis Sci 49:5067–5076. 10.1167/iovs.07-1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrius LA, Driver J (2013) Alzheimer’s as a metabolic disease. Biogerontology 14:641–649. 10.1007/s10522-013-9479-7 [DOI] [PubMed] [Google Scholar]
- Demetrius LA, Simon DK (2012) An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 13:583–594. 10.1007/s10522-012-9403-6 [DOI] [PubMed] [Google Scholar]
- Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, Jiang F (2016) PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol Lett 11:1980–1986. 10.3892/ol.2016.4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J (2016) Reductive carboxylation is a major metabolic pathway in the retinal pigment epithelium. Proc Natl Acad Sci USA 113:14710–14715. 10.1073/pnas.1604572113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JL (2018) Inherited retinal degenerations: current landscape and knowledge gaps. Transl Vis Sci Technol 7:6. 10.1167/tvst.7.4.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta S, Sengupta P (2016) Men and mice: relating their ages. Life Sci 152:244–248. 10.1016/j.lfs.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Ferrari S, Iorio Di E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F (2011) Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics 12:238–249. 10.2174/138920211795860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DS (2004) Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 122:564–572. 10.1001/archopht.122.4.564 [DOI] [PubMed] [Google Scholar]
- Hamel C (2006) Retinitis pigmentosa. Orphanet J Rare Dis 1:40. 10.1186/1750-1172-1-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP (2006) Retinitis pigmentosa. Lancet 368:1795–1809 10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Iadevaia V, Huo Y, Zhang Z, Foster LJ, Proud CG (2012) Roles of the mammalian target of rapamycin, mTOR, in controlling ribosome biogenesis and protein synthesis. Biochem Soc Trans 40:168–172 10.1042/BST20110682 [DOI] [PubMed] [Google Scholar]
- Janisch KM, Kasanuki JM, Naumann MC, Davis RJ, Lin CS, Semple-Rowland S, Tsang SH (2009) Light-dependent phosphorylation of the gamma subunit of cGMP-phophodiesterase (PDE6gamma) at residue threonine 22 in intact photoreceptor neurons. Biochem Biophys Res Commun 390:1149–1153. 10.1016/j.bbrc.2009.10.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanow MA (2017) Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife. 10.7554/eLife.28899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SF (2015) Halting progressive neurodegeneration in advanced retinitis pigmentosa. J Clin Invest 125:3704–3713. 10.1172/JCI82462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM (2008) Safety and efficacy of gene transfer for Leber’s congenital amauros is. N Engl J Med 358:224–2248. 10.1056/NEJMoa0802315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pastor B, Mostoslavsky R (2012) Sirtuins, metabolism, and cancer. Front Pharmacol 3:22. 10.3389/fphar.2012.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin ME, Ehrhart TL, Berson EL, Dryja TP (1995) Mutation spectrum of the gene encoding the beta subunit of rod phosphodiesterase among patients with autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA 92:3249–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday RS, Moritz OL (2015) Photoreceptors at a glance. J Cell Sci 128:4039–4045. 10.1242/jcs.175687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Kornacker K, Cepko CL (2009) Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci 12:44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo C, Xiong W, Cepko CL (2012) Loss of daylight vision in retinal degeneration: areoxidative stress and metabolic dysregulation to blame? J Biol Chem 287:1642–1648. 10.1074/jbc.R111.304428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala A (2018) Pyruvate kinase M2 regulates photoreceptor structure, function, and viability. Cell Death Dis 9:240. 10.1038/s41419-018-0296-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala RV, Rajala A, Kooker C, Wang Y, Anderson RE (2016) The warburg effect mediator pyruvate kinase M2 expression and regulation in the retina. Sci Rep 6:37727. 10.1038/srep37727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell S (2017) Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390:849–860. 10.1016/S0140-6736(17)31868-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Pelluz J (2019) Mechanisms of neurodegeneration in a preclinical autosomal dominant retinitis pigmentosa knock-in model with a Rho(D190N) mutation. Cell Mol Life Sci 76:3657–3665. 10.1007/s00018-019-03090-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmier JK, Jones ML, Halpern MT (2006) The burden of age-related macular degeneration. Pharmacoeconomics 24:319–334 [DOI] [PubMed] [Google Scholar]
- Sohocki MM (2001) Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 17:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q (2011) Mammalian target of rapamycin up-regulation of pyruvat kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA 108 4129–4134. 10.1073/pnas.1014769108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi VKL, Takiuti JT, Jauregui R, Tsang SH (2018) Gene therapy in inherited retinal degenerative diseases: a review. Ophthalmic Genet 39:560–568. 10.1080/13816810.2018.1495745 [DOI] [PubMed] [Google Scholar]
- Tosi J, Davis RJ, Wang NK, Naumann M, Lin CS, Tsang SH (2011a) shRNA knockdown of guanylate cyclase 2e or cyclic nucleotide gated channel alpha 1 increases photoreceptor survival in a cGMP phosphodiesterase mouse model of retinitis pigmentosa. J Cell Mol Med 15:1778–1787. 10.1111/j.1582-4934.2010.01201.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi J et al. (2011b) Lentivirus-mediated expression of cDNA and shRNA slows degeneration in retinitis pigmentosa. Exp Biol Med (Maywood). 10.1258/ebm.2011.011053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH, Burns ME, Calvert PD, Gouras P, Baylor DA, Goff SP, Arshavsky VY (1998) Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science 282:117–121. 10.1126/science.282.5386.117 [DOI] [PubMed] [Google Scholar]
- Tsang SH, Gouras P, Yamashita CK, Kjeldbye H, Fisher J, Farber DB, Goff SP (1996) Retinal degeneration in mice lacking the gamma subunit of the rod cGMP phosphodiesterase. Science 272:1026–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang SH (2008) A novel mutation and phenotypes in phosphodiesterase 6 deficiency. Am J Ophthalmol 146:780–788. 10.1016/j.ajo.2008.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui S (2009) Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol Ther 17:778–786. 10.1038/mt.2009.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velez G (2017) Gene therapy restores mfrp and corrects axial eye length. Sci Rep 7:16151. 10.1038/s41598-017-16275-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh A, Ma S, Le YZ, Hall MN, Ruegg MA, Punzo C (2015) Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J Clin Invest 125:1446–1458. 10.1172/JCI79766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wert KJ, Davis RJ, Sancho-Pelluz J, Nishina PM, Tsang SH (2013) Gene therapy provides long-term visual function in a pre-clinical model of retinitis pigmentosa. Hum Mol Genet 22:558–567. 10.1093/hmg/dds466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL (2008) Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci 28:2064–2074. 10.1523/JNEUROSCI.2973-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wubben TJ, Pawar M, Smith A, Toolan K, Hager H, Besirli CG (2017) Photoreceptor metabolic reprogramming provides survival advantage in acute stress while causing chronic degeneration. Sci Rep 7:17863. 10.1038/s41598-017-18098-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, Cepko CL (2015) NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest 125:1433–1445. 10.1172/JCI79735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J (2012) Vigabatrin-induced retinal toxicity is partially mediated by signaling in rod and cone photoreceptors. PLoS ONE 7(8):e43889. 10.1371/journal.pone.0043889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadro-Lamoureux LA, Zacks DN, Baker AN, Zheng QD, Hauswirth WW, Tsilfidis C (2009) XIAP effects on retinal detachment-induced photoreceptor apoptosis [corrected]. Invest Ophthalmol Vis Sci 50:1448–1453. 10.1167/iovs.08-2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L (2016a) Reprogramming metabolism by targeting sirtuin 6 attenuates retinal degeneration. J Clin Invest 126:4659–4673. 10.1172/JCI86905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L (2016b) Reprogramming towards anabolism impedes degeneration in a preclinical model of retinitis pigmentosa. Hum Mol Genet 25:4244–4255. 10.1093/hmg/ddw256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F (2019) PKM2, function and expression and regulation. Cell Biosci 9:52. 10.1186/s13578-019-0317-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data in this manuscript are available for share. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.