

ABSTRACT
Enterococci are opportunistic pathogens that can cause severe bacterial infections. Treatment of these infections is challenging because enterococci possess intrinsic and acquired mechanisms of resistance to commonly used antibiotics, including cephalosporins. The transmembrane serine/threonine PASTA kinase, IreK, is an important determinant of enterococcal cephalosporin resistance. Upon exposure to cephalosporins, IreK becomes autophosphorylated, which stimulates its kinase activity to phosphorylate downstream substrates and drive cephalosporin resistance. However, the molecular mechanisms that modulate IreK autophosphorylation in response to cell wall stress, such as that induced by cephalosporins, remain unknown. A cytoplasmic protein, GpsB, promotes signaling by PASTA kinase homologs in other bacterial species, but the function of enterococcal GpsB has not been previously investigated. We used in vitro and in vivo approaches to test the hypothesis that enterococcal GpsB promotes IreK signaling in response to cephalosporins to drive cephalosporin resistance. We found that GpsB promotes IreK activity both in vivo and in vitro. This effect is required for cephalosporins to trigger IreK autophosphorylation and activation of an IreK-dependent signaling pathway, and thereby is also required for enterococcal intrinsic cephalosporin resistance. Moreover, analyses of GpsB mutants and a ΔireK gpsB double mutant suggest that GpsB has an additional function, beyond regulation of IreK activity, which is required for optimal growth and full cephalosporin resistance. Collectively, our data provide new insights into the mechanism of signal transduction by the PASTA kinase IreK and the mechanism of enterococcal intrinsic cephalosporin resistance.
IMPORTANCE Enterococci are opportunistic pathogens that can cause severe bacterial infections. Treatment of these infections is challenging because enterococci possess intrinsic and acquired resistance to commonly used antibiotics. In particular, enterococci are intrinsically resistant to cephalosporin antibiotics, a trait that requires the activity of a transmembrane serine/threonine kinase, IreK, which belongs to the bacterial PASTA kinase family. The mechanisms by which PASTA kinases are regulated in cells are poorly understood. Here, we report that the cytoplasmic protein GpsB directly promotes IreK signaling in enterococci to drive cephalosporin resistance. Thus, we provide new insights into PASTA kinase regulation and control of enterococcal cephalosporin resistance, and suggest that GpsB could be a promising target for new therapeutics to disable cephalosporin resistance.
KEYWORDS: PASTA kinase, cephalosporin resistance, enterococcus, signal transduction
INTRODUCTION
Enterococci are Gram-positive bacteria and normal commensals of the human gastrointestinal tract. However, enterococci can transition to become pathogenic under certain conditions, often leading to serious bacterial infections, including urinary tract infections, endocarditis, and bacteremia (1). Treatment of these infections can be difficult because enterococci possess intrinsic and acquired mechanisms of resistance to various commonly used antibiotics (2, 3). In particular, enterococci are intrinsically resistant to cephalosporins, a class of β-lactam antibiotics, but the mechanism of this resistance is incompletely understood. Cephalosporin therapy is a known risk factor for developing an enterococcal infection (4), likely because the intrinsic cephalosporin resistance of enterococci enables them to proliferate to abnormally high numbers in the intestinal tract during cephalosporin therapy and readily disseminate to other sites (5, 6). Therefore, elucidating the mechanisms of cephalosporin resistance can aid in the development of novel therapeutics to disable resistance and treat or prevent enterococcal infections.
One factor known to be important for cephalosporin resistance in enterococci is IreK, a transmembrane serine/threonine kinase that belongs to the bacterial “PASTA kinase” family due to the presence of multiple extracellular PASTA domains (7). An Enterococcus faecalis strain lacking IreK activity exhibits decreased cephalosporin resistance compared to wild-type cells, and conversely a strain lacking the cognate phosphatase IreP—in which IreK is hyperactivated—exhibits increased cephalosporin resistance (8, 9). IreK phosphorylates substrates to initiate a signaling cascade in response to cell wall stress, such as that induced by cephalosporins (10). Robust phosphorylation of downstream substrates requires initial activation of the kinase through autophosphorylation (9, 10). Upon exposure to cell wall stressors or other signals indicative of growth, the kinase becomes autophosphorylated at three threonine residues on a structural element known as the “activation loop” located in the intracellular kinase domain (10, 11). Mutation of these activation loop threonines to alanines to prevent their phosphorylation results in decreased cephalosporin resistance, comparable to that of a ΔireK mutant, indicating how important this autophosphorylation is for activation of IreK to phosphorylate substrates and thereby initiate a signaling cascade to drive cephalosporin resistance (9, 10). In other words, autophosphorylation of IreK “licenses” it to phosphorylate substrates at a high level, and full analysis of the activation state of the IreK signaling pathway in cells therefore requires assessment of both IreK phosphorylation (is IreK licensed to phosphorylate substrates?) and substrate phosphorylation (is IreK actually phosphorylating substrates?). To turn off the signaling pathway, the cognate phosphatase, IreP, dephosphorylates IreK (9). Accordingly, a ΔireP mutant exhibits increased levels of IreK autophosphorylation, substrate phosphorylation, and resistance to cephalosporin antibiotics (9, 10, 12). Additional mechanisms which regulate IreK activity and signaling in enterococci remain unknown.
Previous work identified two proteins important for enterococcal cephalosporin resistance, CroS and IreB, which become phosphorylated in an IreK-dependent manner (12, 13). To identify additional proteins that become phosphorylated through IreK signaling which may be important for cephalosporin resistance, a phosphoproteomic study was performed (14). Among other putative substrates, the cytoplasmic protein GpsB was identified and confirmed to be phosphorylated by IreK (14). Enterococcal GpsB has not been previously investigated, but GpsB homologs have been studied in other bacterial species. GpsB homologs have been implicated in cell division and elongation, as evidenced by localization to the septa of dividing cells; interaction with cell division and elongation proteins EzrA, MreC, and several penicillin-binding proteins (PBPs; transmembrane enzymes which catalyze the final steps of peptidoglycan cell wall synthesis); and the fact that cells lacking gpsB are either not viable or exhibit an elongated cell phenotype with multiple, non-constricted septa (15–20). The GpsB homolog in Bacillus subtilis was identified as a substrate of PrkC, the PASTA kinase (IreK) homolog (21), but whether GpsB becomes phosphorylated in other bacterial species (besides E. faecalis) has not been determined. While only one residue becomes phosphorylated on the B. subtilis GpsB homolog (21), our phosphoproteomic study identified eight putative phosphorylation sites for GpsB in E. faecalis (14). The importance of each of these predicted sites on protein phosphorylation and the role of phosphorylation at these sites in enterococci remain to be determined.
GpsB influences signaling by PASTA kinases in B. subtilis and Streptococcus pneumoniae. Specifically, B. subtilis and S. pneumoniae cells lacking gpsB exhibit decreased PASTA kinase phosphorylation (18, 21, 22). Accordingly, PASTA kinase substrate phosphorylation has been shown to be decreased in S. pneumoniae cells lacking gpsB, but has not been investigated in vivo for B. subtilis (18, 22). Incubation of the purified intracellular kinase domain of the B. subtilis PASTA kinase homolog, PrkC, with purified B. subtilis GpsB in vitro resulted in increased substrate phosphorylation compared to reaction mixtures lacking GpsB, suggesting that GpsB acts directly on the kinase (21). Additionally, a GpsB phosphomimetic mutant exhibited reduced ability to promote PrkC activity, suggesting that phosphorylation of GpsB impairs the ability of GpsB to promote PrkC signaling (21). In principle, GpsB could also influence signaling through PASTA kinase pathways by altering the activity of the cognate phosphatase for PASTA kinases (known as PrpC, Stp, or IreP in various bacterial species), although such an effect has not been reported. The importance of GpsB phosphorylation and the role of GpsB as it relates to PASTA kinase signaling has not been thoroughly investigated in other bacterial species.
Here, we investigated the function of GpsB in E. faecalis and in the context of cephalosporin resistance. We hypothesized that GpsB would be required for enterococcal cephalosporin resistance by specifically promoting IreK signaling. Using in vivo and in vitro approaches, we found that GpsB does not influence IreP phosphatase activity, but instead acts directly on IreK to promote IreK autophosphorylation and signaling in E. faecalis upon exposure to physiologically relevant stimuli, thereby promoting cephalosporin resistance.
RESULTS
GpsB promotes IreK signaling.
To test the hypothesis that GpsB promotes IreK signaling in enterococci, we constructed a markerless, in-frame deletion of gpsB in an otherwise wild-type E. faecalis strain. The ΔgpsB mutant grew as well as wild-type at 37°C but exhibited a growth defect at 45°C, consistent with the temperature-sensitive phenotype of the Listeria monocytogenes ΔgpsB mutant (23) (Fig. S1 and S2 in the supplemental material). The ΔgpsB mutant also exhibited an abnormal chained morphology compared to wild-type cells (Fig. S3). Both the growth and cellular morphology phenotypes of the ΔgpsB mutant mimicked the phenotypes of strains with a catalytically impaired IreK or no IreK at all (ireK K41R mutant and ΔireK mutant, respectively), consistent with the hypothesis that GpsB promotes IreK signaling. We analyzed IreK phosphorylation and activation of the IreK signaling pathway (reflected by substrate phosphorylation) in the ΔgpsB mutant by monitoring phosphorylation of IreK and MltG (a transmembrane protein phosphorylated by IreK [14]) by Phos-tag sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), as previously described (10, 14). During Phos-tag SDS-PAGE, phosphorylated proteoforms migrate more slowly than unphosphorylated proteoforms, enabling separation and quantitation of unphosphorylated versus phosphorylated proteoforms. As previously reported (10, 14), both IreK and MltG were partially phosphorylated in untreated wild-type cells (Fig. 1A), and exposure to the cephalosporin ceftriaxone significantly increased phosphorylation of both IreK and MltG (Fig. 1A). These results confirm that IreK had been licensed for signaling (via autophosphorylation), and that an IreK-dependent signaling pathway was activated in response to the cell wall stress caused by cephalosporins (10). However, both IreK and MltG phosphorylation were significantly decreased in the ΔgpsB mutant (Fig. 1B) and did not change upon exposure to ceftriaxone (Fig. 1C). Furthermore, the lack of MltG phosphorylation in the ΔgpsB mutant was comparable to that of the ΔireK mutant (Fig. S4). Together, these data indicate that IreK is unable to sense ceftriaxone-mediated cell wall stress to become activated or phosphorylate substrates in the absence of GpsB. Ectopic expression of gpsB in the ΔgpsB mutant restored IreK and MltG phosphorylation as well as the response to ceftriaxone-mediated cell wall stress (Fig. 1D and E), confirming that this defect was due to loss of GpsB. We noted that ectopic expression of GpsB from a plasmid led to a significant increase in IreK and MltG phosphorylation compared to that in wild-type cells, which may be because gpsB is overexpressed from this plasmid compared to chromosomally expressed gpsB (Fig. S5). Collectively, these data support our hypothesis that GpsB promotes both IreK phosphorylation and activation of an IreK-dependent signaling pathway in E. faecalis cells.
FIG 1.

GpsB promotes IreK phosphorylation and signaling in vivo. Exponentially growing cells were treated (or not) with ceftriaxone (Cftx, 128 μg/mL). Total protein lysates were prepared, and pairwise comparisons were subjected to Phos-tag SDS-PAGE followed by immunoblot analysis using antiserum to detect IreK or MltG. (A) WT/vector (wild-type harboring empty vector control), untreated (–) and ceftriaxone-treated (+). (B) WT/vector and ΔgpsB/vector, both untreated. (C) ΔgpsB/vector untreated and ceftriaxone-treated. (D) WT/vector and ΔgpsB/p-gpsB-His6, both untreated. (E) ΔgpsB/p-gpsB-His6, untreated and ceftriaxone-treated. Immunoblot images are representative of at least 3 biological replicates per strain or condition. Bar graph data show the average % of IreK or MltG phosphorylation (% IreK-P or % MltG-P, respectively) of the 3 replicates. Error bars represent one standard deviation. *, P < 0.05; **, P < 0.01; ns, not significant; Student’s t test (heteroscedastic, two-tailed). Strains used were WT/vector, OG1/pJRG9; ΔgpsB/vector, JL635/pJRG9; ΔgpsB/p-gpsB-His6, JL635/pNEM4; ΔireK/vector, JL206/pJRG9; and ΔmltG/vector, JL650/pJRG9.
GpsB associates with IreK in vivo.
To explore the possibility that GpsB promotes IreK signaling through an interaction with IreK in vivo, we ectopically overexpressed IreK-His6 in cells lacking ireK and used immobilized metal affinity chromatography to enrich for IreK-His6 from cell lysates along with any associated proteins. Immunoblotting verified that IreK-His6 was successfully enriched from cells that were overexpressing IreK-His6, but not in the vector-only controls (Fig. 2). The cytoplasmic GpsB was also present in the IreK-His6 containing elution fraction, but not in the vector-only controls, indicating that GpsB directly or indirectly associated with IreK in vivo. In contrast, CroR, another cytoplasmic protein important for cephalosporin resistance (12), was not present in any of the elution fractions, indicating that the pulldown of GpsB with IreK-His6 was specific.
FIG 2.
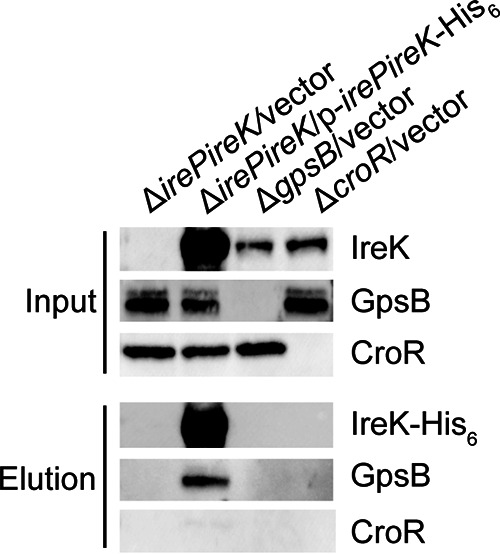
GpsB associates with IreK in vivo. Ectopically expressed IreK-His6 was enriched from exponentially growing Enterococcus faecalis cell lysates using immobilized metal affinity chromatography. Immunoblotting was performed on the input (lysate) and elution fractions using antisera to detect IreK (IreK and IreK-His6), GpsB, and CroR. Data are representative of at least 3 independent experiments. Strains were ΔirePireK/vector, CK125/pJRG9; ΔirePireK/p-irePireK-His6, CK125/pJLL47; ΔgpsB/vector, JL635/pJRG9; and ΔcroR/vector, SB23/pJRG9.
GpsB acts directly on IreK.
Having found that GpsB associated with IreK in vivo, we reasoned that GpsB could act directly on IreK to promote signaling and investigated this possibility using in vitro assays with purified proteins. To test the hypothesis that GpsB increases the rate of IreK autophosphorylation, the dephosphorylated, cytoplasmic kinase domain of IreK (His6-IreK-n) was incubated in the presence or absence of GpsB and ATP. After separating the proteins by SDS-PAGE, Pro-Q Diamond phosphoprotein gel stain was used to visualize phosphorylated His6-IreK-n and SYPRO Ruby protein gel stain was used to visualize total His6-IreK-n. Without ATP, His6-IreK-n did not become phosphorylated after incubation for 120 min (Fig. 3A). The low level of Pro-Q Diamond signal observed for these samples could be because His6-IreK-n was not completely dephosphorylated prior to the experiment, or because the stain nonspecifically interacted with His6-IreK-n at some low level. Regardless, after adding ATP, the Pro-Q Diamond signal increased over time, indicative of increasing phosphorylation of His6-IreK-n. The emergence of an additional faint upper band in the gels stained with Pro-Q Diamond represents heavily phosphorylated His6-IreK-n (IreK is capable of being phosphorylated at multiple sites) (9, 11). This upper band was not seen as clearly in the SYPRO Ruby-stained gels (total His6-IreK-n) presumably because the SYPRO stain was not sensitive enough to detect the small amount of heavily phosphorylated proteoforms present. Addition of GpsB significantly increased the relative His6-IreK-n autophosphorylation rate by 2.2-fold (Fig. 3B and C). These results indicate that GpsB acts directly on IreK to enhance IreK autophosphorylation in vitro.
FIG 3.

GpsB enhances the rate of IreK autophosphorylation in vitro. In vitro kinase assays were performed with purified proteins to assess the impact of wild-type (WT) GpsB, the GpsB 8A mutant, or the GpsB ΔP114-F136 mutant on IreK autophosphorylation. Dephosphorylated, wild-type His6-IreK-n was incubated with or without a 10-fold excess of wild-type GpsB, GpsB 8A mutant, or GpsB ΔP114-F136 mutant at 37°C. Samples of each reaction were taken at intervals after the addition of excess ATP. (A) After SDS-PAGE, Pro-Q Diamond phosphoprotein stain was used to visualize phosphorylated His6-IreK-n (P-His6-IreK-n) and SYPRO Ruby protein gel stain was used to visualize total His6-IreK-n. Gel images are representative of at least three independent experiments. (B) The phosphorylated His6-IreK-n and total His6-IreK-n signals from the gel images were quantified. Relative His6-IreK-n phosphorylation at each time point was calculated by determining the ratios of phosphorylated His6-IreK-n to total His6-IreK-n for each time point. Graph is representative of results from at least 3 independent experiments. (C) Initial relative His6-IreK-n phosphorylation rates were determined by finding the slopes of the best-fit lines in graph (panel B) from time points 2 to 30 min for each reaction. Data are the mean x-fold changes in rates for each reaction relative to the His6-IreK-n-only reaction from at least 3 independent experiments. Error bars represent one standard deviation. *, P < 0.05; Student’s t test (heteroscedastic, two-tailed).
Another potential way that GpsB could impact activation of IreK-dependent signaling pathways would be to alter the activity of the cognate phosphatase, IreP, thereby modulating IreK phosphorylation indirectly. To test whether GpsB could act on the IreP phosphatase to alter its dephosphorylation activity, we monitored IreP-mediated His6-IreK-n dephosphorylation in the presence and absence of a 10-fold excess of GpsB relative to His6-IreK-n. The presence of GpsB did not alter the rate of His6-IreK-n dephosphorylation by IreP in vitro (Fig. S6), indicating that GpsB does not influence IreK-dependent signaling by altering IreP activity, at least in vitro.
GpsB is required for cephalosporin resistance.
Because GpsB promotes IreK signaling in E. faecalis, and IreK signaling is important for cephalosporin resistance (8–10), we hypothesized that GpsB was also essential for cephalosporin resistance in enterococci. To test this, we performed antibiotic susceptibility assays. The E. faecalis ΔgpsB mutant exhibited decreased resistance to the cephalosporins ceftriaxone and cefuroxime compared to the wild type (Table 1, Table S1 in the supplemental material), but exhibited little to no changes in resistance to other antibiotics with diverse targets (Table S1). Additionally, the ceftriaxone MIC value for the ΔgpsB mutant was the same as that for the ΔireK mutant (Table 1), as might be predicted if IreK signaling is defective in the ΔgpsB mutant. An E. faecium ΔgpsB mutant also exhibited decreased resistance to ceftriaxone compared to its otherwise isogenic wild-type strain (Table 1), indicating the importance of GpsB for cephalosporin resistance across clinically relevant enterococci. Ectopic overexpression of GpsB restored ceftriaxone resistance to the ΔgpsB mutant and even led to increased cephalosporin resistance compared to the wild type (Table 2), presumably due to overexpression of GpsB from the plasmid (Fig. S5). This is also unsurprising given that IreK signaling was found to be significantly increased in the strain ectopically overexpressing gpsB. Taken together, these data indicate that GpsB is required for cephalosporin resistance in enterococci.
TABLE 1.
GpsB is required for enterococcal cephalosporin resistance
| Enterococcus straina | Ceftriaxone MIC (μg/mL)b |
|---|---|
| E. faecalis | |
| WTc | 64 |
| ΔgpsB | 2 |
| ΔireK | 2 |
| ΔireK gpsB | 0.5 |
| E. faecium | |
| WT | 32 |
| ΔgpsB | 2 |
E. faecalis strains: WT, OG1; ΔgpsB, JL635; ΔireK, JL206; and ΔireK gpsB, NM6. E. faecium strains: WT, 1141733; and ΔgpsB, JL638.
Data represent the median minimal inhibitory concentration (MIC) from 3 independent biological replicates.
WT, wild type.
TABLE 2.
Median MICs of E. faecalis strains ectopically expressing GpsB mutants
| E. faecalis straina | Ceftriaxone MIC (μg/mL)b |
|---|---|
| WT/vectorc | 64 |
| ΔgpsB/vector | 2 |
| ΔgpsB/p-gpsB-His6 | 512 |
| ΔgpsB/p-gpsB 8A-His6 | 8 |
| ΔgpsB/p-gpsB ΔP114-F136-His6 | 32 |
WT/vector, OG1/pJRG9; ΔgpsB/vector, JL635/pJRG9; ΔgpsB/p-gpsB-His6, JL635/pNEM4; ΔgpsB/p-gpsB 8A-His6, JL635/pNEM29; and ΔgpsB/p-gpsB ΔP114-F136-His6, JL635/pNEM6.
Data represent the median MIC from 3 independent biological replicates.
WT, wild type.
In other bacterial species, GpsB homologs have been found to interact with and are important for the localization and function of penicillin-binding proteins (PBPs) (15, 18, 19, 23). To test whether E. faecalis GpsB might have some additional function beyond promoting activation of IreK, we constructed an ΔireK gpsB double-deletion mutant and analyzed its growth and cephalosporin resistance properties. The ΔireK gpsB double mutant exhibited a modestly reduced MIC for ceftriaxone compared to the ΔireK strain (Table 1). Additionally, the ΔireK gpsB mutant exhibits a marked growth defect at 37°C compared to the ΔireK strain (Fig. S1). Complementation of the ΔireK gpsB double mutant with gpsB from a plasmid rescued the growth defect (Fig. S7) and modestly increased cephalosporin resistance (Table S2). Together, these data demonstrating the phenotypic effects of GpsB deletion in the absence of IreK indicate that GpsB has some IreK-independent function in E. faecalis.
Unphosphorylated GpsB enhances IreK autophosphorylation in vitro.
GpsB is itself phosphorylated by IreK, with 8 putative sites of phosphorylation identified via phosphoproteomics: S80, T84, T107, S109, T110, T113, T120, and T133 (14) (Fig. S8). Accordingly, incubation of purified wild-type GpsB with wild-type His6-IreK-n, but not a catalytically impaired mutant His6-IreK-n (K41R), resulted in phosphorylation of GpsB (Fig. 4), as previously observed (14). To determine if GpsB phosphorylation impacts its ability to promote IreK signaling, we created a GpsB phosphoablative mutant in which all 8 putative phosphorylation sites were simultaneously mutated to alanine (here referred to as the GpsB 8A mutant). Analysis of the purified GpsB 8A mutant by in vitro phosphorylation assays revealed that it did not become phosphorylated by IreK (Fig. 4), confirming that one or more of those 8 sites are critical for GpsB phosphorylation. To determine if unphosphorylated GpsB can promote IreK autophosphorylation, we performed in vitro IreK autophosphorylation assays as previously in the presence and absence of the GpsB 8A mutant. As in the case of wild-type GpsB, addition of the GpsB 8A mutant significantly increased the relative phosphorylation rate of His6-IreK-n to levels comparable to wild-type GpsB (Fig. 3) indicating that phosphorylation of GpsB is not required to enhance IreK-n autophosphorylation in vitro.
FIG 4.
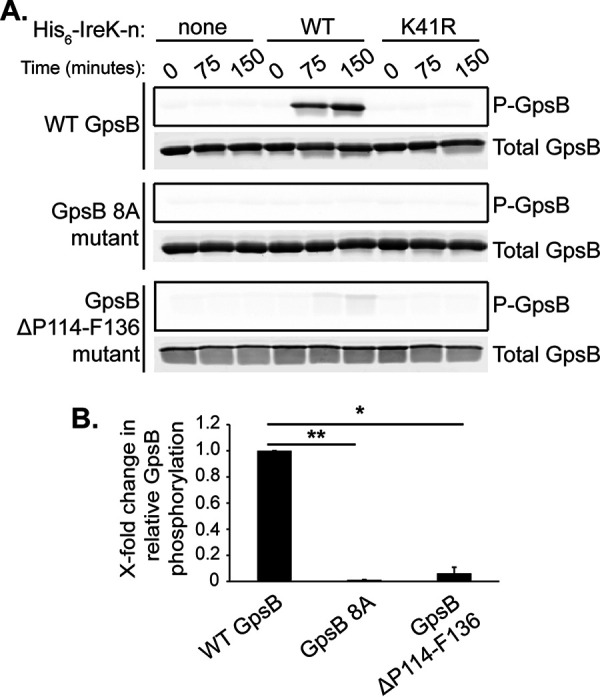
Mutations impair GpsB phosphorylation by IreK in vitro. In vitro phosphorylation assays were performed with purified proteins to assess phosphorylation of GpsB by IreK. Reactions contained GpsB (wild-type [WT], 8A mutant, or ΔP114-F136 mutant) and either no kinase (none), wild-type His6-IreK-n (WT), or a catalytically impaired His6-IreK-n K41R mutant (K41R), and were incubated at 37°C. Samples of the reactions were taken at intervals after the addition of excess ATP. (A) After SDS-PAGE, GpsB phosphorylation (P-GpsB) was visualized using Pro-Q Diamond phosphoprotein gel stain and total GpsB was visualized using SYPRO Ruby protein gel stain. Data are representative of at least 2 independent experiments. (B) The x-fold change in relative GpsB phosphorylation was determined by calculating the ratio of P-GpsB to total GpsB at 150 min in the reactions containing wild-type His6-IreK-n. Data are the mean x-fold changes in phosphorylation for each GpsB mutant relative to wild-type GpsB from 2 independent experiments. Error bars represent one standard deviation. *, P < 0.05; **, P < 0.01; Student’s t test (heteroscedastic, two-tailed).
Unphosphorylated GpsB impairs cephalosporin resistance and IreK phosphorylation in vivo.
Because the GpsB 8A mutant increased the rate of IreK-n autophosphorylation like wild-type GpsB in vitro, and wild-type GpsB is required for IreK signaling in vivo, we hypothesized that the GpsB 8A mutant would promote cephalosporin resistance and IreK signaling in vivo as well. Immunoblotting confirmed that the GpsB 8A mutant was ectopically expressed at levels comparable to ectopically expressed wild-type GpsB (Fig. S5). Expression of the GpsB 8A mutant led to a modest increase in ceftriaxone resistance of the ΔgpsB mutant, but unlike wild-type GpsB, the GpsB 8A mutant did not promote robust ceftriaxone resistance (Table 2), suggesting that phosphorylation of GpsB is critical for some aspect of its function in driving cephalosporin resistance. Phos-tag SDS-PAGE and immunoblotting revealed that the strain ectopically expressing the GpsB 8A mutant exhibited decreased IreK phosphorylation in untreated, growing cells compared to the strain ectopically expressing wild-type GpsB (Fig. 5A). However, there was no significant difference in MltG phosphorylation between the two strains (Fig. 5A), suggesting that the (relatively) low level of IreK signaling occurring in unstressed growing cells was unaffected by expression of the GpsB 8A mutant. When exposed to ceftriaxone, both IreK and MltG phosphorylation significantly increased in the strain ectopically expressing the GpsB 8A mutant (Fig. 5B). Therefore, because IreK signaling (as reflected by MltG phosphorylation) is unchanged and is responsive to cell wall stress induced by cephalosporins, we conclude that phosphorylation of GpsB is not required for the ability of GpsB to promote IreK signaling in vivo.
FIG 5.
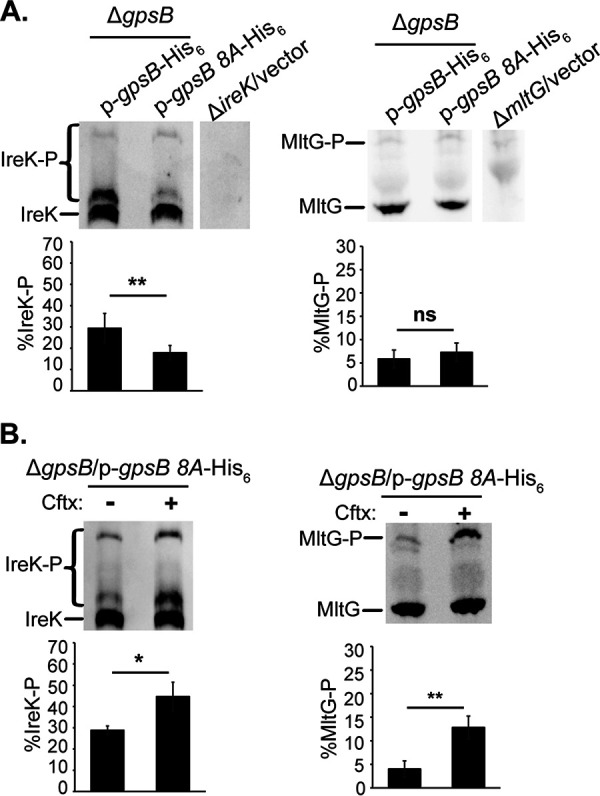
The GpsB 8A mutant impairs IreK autophosphorylation, but not IreK signaling, in unstressed, growing cells in vivo. Exponentially growing cells were treated (or not) with ceftriaxone (Cftx, 128 μg/ml)″ as was done in the legend to Fig. 1A. Total protein lysates were prepared, and pairwise comparisons were subjected to Phos-tag SDS-PAGE followed by immunoblot analysis using antiserum to detect IreK or MltG. (A) ΔgpsB/p-gpsB-His6 and ΔgpsB/p-gpsB 8A-His6, both untreated. (B) ΔgpsB/p-gpsB 8A-His6, untreated (–) and ceftriaxone-treated (+). Immunoblot images are representative of at least 3 biological replicates per strain or condition. Bar graph data show the average % of IreK or MltG phosphorylation (% IreK-P or % MltG-P, respectively) of the 3 replicates. Error bars represent one standard deviation. *, P < 0.05; **, P < 0.01; ns, not significant; Student’s t test (heteroscedastic, two-tailed). Strains were ΔgpsB/p-gpsB-His6, JL635/pNEM4; ΔgpsB/p-gpsB 8A-His6, JL635/pNEM29; ΔireK/vector, JL206/pJRG9; and ΔmltG/vector, JL650/pJRG9.
The C-terminal tail of GpsB promotes cephalosporin resistance and GpsB phosphorylation.
Enterococcal GpsB possesses an extension of ~25 amino acids of unknown function at its C terminus relative to GpsB homologs in other bacterial species (Fig. S8). To determine if this “C-terminal tail” is functionally important for GpsB to promote IreK signaling and cephalosporin resistance, we deleted the codons which encode amino acids P114 to F136 from E. faecalis gpsB (here referred to as GpsB ΔP114-F136) and ectopically expressed this truncated protein in the ΔgpsB mutant. Antibiotic susceptibility assays indicated that the GpsB ΔP114-F136 mutant slightly increased the cephalosporin resistance of the ΔgpsB mutant, but not to the same extent as wild-type GpsB (Table 2). However, immunoblotting revealed that the GpsB ΔP114-F136 mutant exhibited decreased abundance compared to ectopically expressed wild-type GpsB (Fig. S5), which may account for the difference in cephalosporin resistance between the two strains. Analysis of the GpsB ΔP114-F136 mutant by in vitro phosphorylation assays revealed that the mutant is minimally phosphorylated after 150 min (Fig. 4), indicating that the C-terminal tail is important for phosphorylation of GpsB by IreK.
The GpsB C-terminal tail modulates IreK signaling.
To determine if the GpsB ΔP114-F136 mutant impacts the rate of IreK autophosphorylation in vitro, we performed an in vitro IreK autophosphorylation assay in the presence and absence of the purified GpsB ΔP114-F136 mutant. The presence of GpsB ΔP114-F136 significantly increased the rate of IreK autophosphorylation by 2.8-fold compared to a reaction that did not contain the mutant (Fig. 3), indicating that the C-terminal tail is not required for GpsB to enhance IreK autophosphorylation in vitro. Phos-tag SDS-PAGE analysis revealed that the strain ectopically expressing GpsB ΔP114-F136 exhibited significantly increased IreK and MltG phosphorylation in untreated, growing cells compared to the strain ectopically expressing wild-type GpsB (Fig. 6A), suggesting that the C-terminal tail normally impairs the ability of GpsB to activate IreK signaling in unstressed cells. When exposed to ceftriaxone, the distribution of phosphorylated IreK proteoforms shifted in the strain ectopically expressing GpsB ΔP114-F136 (Fig. 6B; note the decrease in the “middle” phosphorylated band and increase in the “upper” phosphorylated band), indicating ceftriaxone-responsive changes in IreK phosphorylation, although the overall level of IreK phosphorylation was not statistically different upon ceftriaxone exposure (possibly because IreK was already so highly phosphorylated in the unstressed condition). MltG phosphorylation was significantly increased upon ceftriaxone exposure (Fig. 6B), confirming that activation of IreK-dependent signaling pathways can occur in the absence of the GpsB C-terminal tail. Taken together, these data suggest that one role of the GpsB C-terminal tail may be to impair the ability of GpsB to promote IreK phosphorylation in unstressed cells when low levels of IreK signaling are sufficient.
FIG 6.
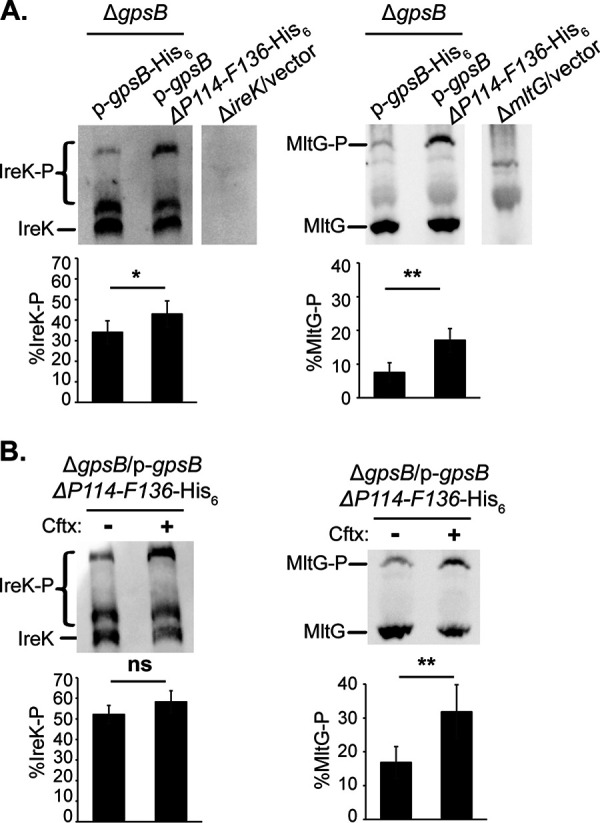
The GpsB ΔP114-F136 mutant enhances IreK autophosphorylation and signaling in vivo. Exponentially growing cells were treated with ceftriaxone (Cftx, 128 μg/mL) or untreated. Total protein lysates were prepared, and pairwise comparisons were subjected to Phos-tag SDS-PAGE followed by immunoblot analysis using antiserum to detect IreK or MltG. (A) ΔgpsB/p-gpsB-His6 and ΔgpsB/p-gpsB ΔP114-F136-His6, both untreated. (B) ΔgpsB/p-gpsB ΔP114-F136-His6, untreated (–) and ceftriaxone-treated (+). Immunoblot images are representative of at least 3 biological replicates per strain or condition. Bar graph data show the average % of IreK or MltG phosphorylation (% IreK-P or % MltG-P, respectively) of the 3 replicates. Error bars represent one standard deviation. *, P < 0.05; **, P < 0.01; ns, not significant; Student’s t test (heteroscedastic, two-tailed). Strains were ΔgpsB/p-gpsB-His6, JL635/pNEM4; ΔgpsB/p-gpsB ΔP114-F136-His6, JL635/pNEM6; ΔireK/vector, JL206/pJRG9; and ΔmltG/vector, JL650/pJRG9.
DISCUSSION
GpsB has been shown to be important for PASTA kinase phosphorylation and/or signaling in B. subtilis and S. pneumoniae (18, 21, 22), but whether this occurs in other bacterial species, and the mechanism by which GpsB promotes PASTA kinase signaling, are poorly understood. Here, we identified GpsB as a new determinant of cephalosporin resistance in both of the most clinically relevant species of enterococci. Using both in vitro and in vivo approaches, we determined that GpsB directly promotes autophosphorylation of the PASTA kinase IreK and subsequent activation of IreK-dependent signaling pathways to drive cephalosporin resistance. Multiple lines of evidence establish connections between the biochemical results and in vivo phenotypes to support this conclusion: (i) deletion of gpsB leads to losses in IreK phosphorylation, IreK substrate phosphorylation, and cephalosporin resistance (Fig. 1, Tables 1 and 2); (ii) overexpression of wild-type GpsB leads to elevated levels of IreK and substrate phosphorylation, and elevated cephalosporin resistance (Fig. 1, Table 2); and (iii) exposure to cephalosporins leads to enhanced levels of IreK phosphorylation and substrate phosphorylation concomitant with cephalosporin resistance (Fig. 1). The impact of GpsB on activation of IreK-dependent signaling does not appear to involve modulation of the cognate phosphatase IreP, because GpsB did not alter IreP-dependent dephosphorylation of IreK in vitro. Because the amount of GpsB added to these dephosphorylation reactions was in 10-fold excess of His6-IreK-n, and His6-IreK-n was in 100-fold excess of IreP, our results argue against the following possibilities: (i) that GpsB acts directly on IreP to inhibit phosphatase activity or (ii) that it acts on His6-IreK-n to block IreP from dephosphorylating the PASTA kinase.
This is the first report, to the best of our knowledge, to identify that the cytoplasmic protein GpsB is required for activation of PASTA kinase (IreK) signaling in response to a physiologically relevant external cell wall stress (cephalosporins). In other words, IreK appears to be “blind” to cephalosporin-induced stress without the assistance of GpsB. While the extracellular domains of PASTA kinases have traditionally been viewed as the functionally important “sensory input” modules that recognize cell wall stress to trigger kinase activation, and the extracellular module of IreK is required for IreK to respond to cell wall stress (10), our data suggest that regulation of PASTA kinase signaling is more complex in that a cytoplasmic partner (GpsB) is also required for this response. One possible model for how GpsB promotes IreK signaling in response to cell wall stress is that upon sensing cell wall stress via the PASTA domains, a conformational change occurs in the intracellular kinase domain of IreK, and GpsB is required to stabilize this conformational change to allow for efficient autophosphorylation of the activation loop and subsequent activation of IreK signaling. However, more work is required to elucidate the molecular mechanism by which GpsB acts to modulate PASTA kinase phosphorylation.
GpsB has been identified as a substrate of the PASTA kinase homolog in B. subtilis (21) and, more recently, of IreK in E. faecalis (14). Eight putative phosphorylation sites were identified for E. faecalis GpsB (14), but the importance of phosphorylation on GpsB function had not been previously determined. In B. subtilis, unphosphorylated GpsB promotes PASTA kinase autophosphorylation as well as wild-type GpsB (21). Our data agrees with the findings for B. subtilis, where unphosphorylated GpsB enhances IreK autophosphorylation as well as wild-type GpsB in vitro (Fig. 3). In contrast, unphosphorylated GpsB does not promote IreK phosphorylation as well as wild-type GpsB in unstressed, growing E. faecalis cells (Fig. 5A), although phosphorylation of both IreK and MltG was enhanced upon exposure to ceftriaxone (Fig. 5B). Our previous work (10) suggested that IreK may respond to two types of signals: one associated with growth and/or division, and another resulting from cephalosporin-mediated cell wall stress. It is clear from our results that unphosphorylated GpsB promotes the response to cephalosporins normally, but the decrease in IreK phosphorylation observed in unstressed growing cells (Fig. 5A) suggests that phosphorylation of GpsB might be required for IreK to efficiently detect the “growth signal” in vivo. Further work is required to determine whether phosphorylation at all eight GpsB residues collectively is important to regulate the role of GpsB on IreK activation and cephalosporin resistance, or whether phosphorylation at different sites has distinct functions which differentially impact IreK phosphorylation and signaling in vivo.
One striking feature of enterococcal GpsB is that it possesses a functionally important extension of ~25 amino acids at the C terminus which is not found in the previously studied GpsB homologs. There are two putative phosphorylation sites found in the C-terminal tail: T120 and T133. Our in vitro data reveal that the GpsB ΔP114-F136 mutant gets phosphorylated at a reduced level relative to wild-type GpsB (Fig. 4). Therefore, either or both T120 and T133 residues may be important for phosphorylation of GpsB overall, although one or more other sites must also be phosphorylated at some level. Pompeo et al. (21) identified that T75 in B. subtilis GpsB (which roughly corresponds to E. faecalis GpsB T84 and is close to S80) is important for GpsB phosphorylation and may be important for regulating PrkC signaling. Phosphorylatable residues are conserved in this region of the sequence among GpsB homologs from diverse species, including enterococci (Fig. S8). Therefore, we speculate that the residual phosphorylation on the GpsB ΔP114-F136 mutant occurs on T84 and/or S80. Future work will tease apart the functional importance and relationships between the multiple sites of phosphorylation on enterococcal GpsB.
In vivo, ectopic expression of the GpsB ΔP114-F136 mutant resulted in significantly increased IreK autophosphorylation and IreK-dependent signaling in unstressed cells compared to ectopic expression of wild-type, full-length GpsB (Fig. 6). This indicates that the C-terminal tail is not required for GpsB to directly promote IreK autophosphorylation, and instead may act to autoinhibit this function in unstressed cells where (relatively) low levels of IreK signaling are needed. Notably, the GpsB ΔP114-F136 mutant exhibited decreased abundance compared to full-length GpsB, despite being expressed from the same constitutive promoter (Fig. S4). It is unknown why this mutant exhibits decreased abundance, but it is worth noting that the GpsB ΔP114-F136 mutant drives elevated IreK phosphorylation despite the reduced abundance in vivo, suggesting the GpsB ΔP114-F136 mutant is especially proficient at activating IreK.
Previous studies have correlated increased IreK signaling with hyper-resistance to cephalosporins (9, 10), consistent with the majority of results reported here. However, in our study, even though ectopic expression of the GpsB ΔP114-F136 mutant leads to increased IreK signaling, the strain exhibits decreased cephalosporin resistance compared to the strain expressing wild-type GpsB (Fig. 6; Table 2). Similarly, in the strain expressing the GpsB 8A mutant, IreK signaling appears unchanged compared to that in the strain expressing wild-type GpsB, but there is a decrease in cephalosporin resistance (Fig. 5; Table 2). These two instances represent departures from the otherwise robust correlation between IreK-dependent signaling and cephalosporin resistance noted above, and suggest that GpsB possesses an additional function that acts independently and downstream of IreK to promote cephalosporin resistance. We tested this by constructing the ΔireK gpsB double mutant and found that even in the absence of IreK, loss of GpsB resulted in additional phenotypic consequences (Table 1, Fig. S1): the ΔireK gpsB mutant exhibited reduced cephalosporin resistance compared to the ΔireK mutant, and the ΔireK gpsB mutant exhibited a growth defect at 37°C that was not present for the ΔireK mutant. Thus, our data are consistent with the hypothesis that GpsB, in addition to promoting IreK activation, also acts independently of IreK to promote normal growth and full cephalosporin resistance. In other bacterial species, GpsB homologs are important for the localization and function of PBPs, enzymes which catalyze the last steps in peptidoglycan synthesis (15, 18, 19, 23). Enterococcal PBPs have been identified as additional determinants important for cephalosporin resistance (24–26) and connections have been made between PASTA kinase signaling and peptidoglycan synthesis (27–29). Therefore, we speculate that GpsB has an additional role involving one or more of the PBPs, either by correctly localizing the enzymes in the cell or by promoting their enzymatic activity, which can drive cephalosporin resistance downstream of IreK signaling.
Given the fact that GpsB is required for IreK signaling, it is unsurprising that GpsB is also required for cephalosporin resistance in both E. faecalis and E. faecium. While IreK is an appealing target for drug development to disable cephalosporin resistance, it may also be useful to target factors that drive or support IreK signaling. Based on our study, GpsB is one such candidate, but more work is required to determine the detailed molecular mechanisms by which GpsB carries out its cellular functions.
MATERIALS AND METHODS
Bacterial strains, growth, media, and chemicals.
The bacterial strains and plasmids used in this study are listed in Table S2. E. coli strains were grown in Lysogeny broth (LB) and E. faecalis and E. faecium strains were grown in Mueller-Hinton broth (MHB) (prepared according to the manufacturer’s instructions; Difco) for all experiments. Archives of bacterial strains were kept at −80°C in MHB or LB with 30% glycerol. Antibiotics were used at the following concentrations: kanamycin, 50 μg/mL; chloramphenicol, 10 μg/mL.
Genetic manipulation of enterococci.
In-frame deletions of enterococcal genes were constructed using the temperature-sensitive, counter-selectable allelic exchange plasmid pJH086 as previously described (30). In addition to p-Cl-Phe, counterselection plates contained 20% sucrose. Flanking segments of genomic DNA from upstream and downstream of the gene of interest were amplified by PCR and introduced into pJH086 via isothermal assembly (31). Deletion alleles were in-frame and retained a variable number of codons at the beginning and end of each gene to avoid perturbing the expression of adjacent genes. Table S2 contains the specific details of each deletion allele. All mutant or complemented strains were constructed independently at least twice and analyzed to ensure that their phenotypes were concordant.
Construction of plasmids.
All recombinant plasmids were constructed using Gibson assembly (31) and sequencing of the full insert was performed to confirm absence of errors. Ectopic expression of GpsB (wild-type and mutants) in E. faecalis OG1 was accomplished using the enterococcal expression plasmid pJRG9 under the control of the constitutive P23s promoter. The native ribosome binding site for GpsB was included. Expression of wild-type and mutant GpsB in E. coli for protein purification was accomplished using the IPTG (isopropyl β-D-1-thiogalactopyranoside)-inducible expression vector pET28a-His-smt3, which encodes a His6-SUMO fusion.
Antibiotic susceptibility assays.
MICs were determined using a broth microdilution method. Briefly, bacteria from stationary-phase cultures were inoculated into wells containing 2-fold serial dilutions of antibiotics in MHB (including 10 μg/mL chloramphenicol when required for maintenance of plasmids) at a normalized optical density at 600 nm (OD600) of 4 × 10−5 (~1 × 105 CFU). Plates were incubated in a Bioscreen C plate reader at 37°C for 24 h with brief shaking before each measurement. The OD600 was determined every 15 min, and the lowest concentration of antibiotic that prevented growth was recorded as the MIC.
Whole-cell lysate preparation.
Stationary-phase cultures of bacteria were diluted to OD600 = 0.01 in MHB supplemented with 10 μg/mL chloramphenicol (for plasmid maintenance) and grown to exponential-phase at 37°C. Cells were treated with or without 128 μg/mL ceftriaxone for 20 min at 37°C before harvesting by adding an equal amount of ice-cold ethanol/acetone (1:1). The cells were collected by centrifugation, washed with water, and resuspended in lysozyme solution (10 mM Tris, 20% sucrose, 50 mM NaCl [pH = 8.0]). After normalizing based on OD600 measurements, the cells were treated with 10 mg/mL lysozyme for 30 min at 37°C and mixed with 5× Laemmli buffer. Cell lysates used for GpsB immunoblot analysis were additionally boiled for 5 min, but lysates used for Phos-tag analysis were not.
GpsB immunoblot analysis.
Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at 200 V for 45 min followed by transfer to polyvinylidene fluoride (PVDF) membranes using a Transblot Turbo system at 25 V for 7 min. After transfer, membranes were blocked in 3% milk for 1 h at room temperature. Membranes were probed with custom primary antiserum to detect GpsB, followed by horseradish peroxidase-conjugated secondary antibody, before developing with Super-Signal (Thermo Fisher Scientific, Waltham, MA) using a ChemiDoc Touch imaging system (Bio-Rad, Hercules, CA).
Phos-tag SDS-PAGE and IreK/MltG immunoblot analysis.
Immunoblot analysis for IreK and MltG Phos-tag SDS-PAGE were performed as previously described for GpsB standard immunoblot analysis with for the following alterations. Proteins were separated on SDS-PAGE gels supplemented with 50 μM Phos-tag and 100 μM Zn(NO3)2. Electrophoresis conditions were 200 V for 50 min (IreK Phos-tag gels) or 150 V for 1 h (MltG Phos-tag gels). Following electrophoresis, all Phos-tag gels were soaked in 5 mM EDTA 3 times for 10 min each. Proteins were transferred to nitrocellulose membranes using a Transblot Turbo system at 25 V for 7 to 14 min. Membranes were probed with custom primary antisera to detect IreK or MltG. Samples for specific pairwise comparisons of interest were always on the same membranes to minimize differences introduced between membranes during immunoblotting.
Copurification.
Stationary-phase cultures of E. faecalis cells harboring the vector alone or plasmid overexpressing IreK-His6 were diluted to OD600 = 0.01 in MHB supplemented with 10 μg/mL chloramphenicol and grown to exponential-phase at 37°C. The cells were treated with 1% formaldehyde for 10 min at room temperature and then with 0.5 M glycine for 5 min at room temperature. Cells were lysed by treatment with 15 mg/mL lysozyme for 30 min at 37°C followed by bead beating 30 s on, 30 s off six times each at room temperature. The lysates were incubated with binding-buffer-equilibrated Ni-charged resin for 1 h at room temperature. The resin was washed with binding buffer (50 mM Tris, 300 mM NaCl, 5 mM imidazole [pH = 8.0]) and IreK-His6 was recovered from the resin with elution buffer (50 mM Tris, 300 mM NaCl, 500 mM imidazole [pH = 8.0]). All eluates were boiled for at least 30 min. The input and elution fractions were subjected to SDS-PAGE and immunoblotting was performed using custom primary antisera to detect IreK or GpsB.
Protein purification.
Recombinant His6-SUMO-GpsB (wild-type, 8A mutant, and ΔP114-F36 mutant), wild type His6-IreK-n (intracellular domain only), a catalytically impaired His6-IreK-n K41R mutant, and His6-SUMO-IreP were each purified from E. coli BL21(DE3) or Nico21(DE3) cells. Stationary-phase cultures were diluted 1:50 into LB media supplemented with 50 μg/mL kanamycin (for plasmid maintenance) and grown for 3 h at 37°C, then induced with either 1 mM IPTG for 1 h at 37°C or 0.4 mM IPTG at 16°C overnight. Cells were pelleted by centrifugation at 17,000 × g for 10 min at 4°C and resuspended in binding buffer (50 mM Tris, 300 mM NaCl, 5 mM imidazole [pH = 7.5]), then lysed using a French press. The lysates were then centrifuged at 4,200 × g for at least 25 min at 4°C and passed through a 0.2-μM filter to remove cellular debris. Filtered lysates were applied to binding-buffer-equilibrated Ni-charged resin. The protein-bound resin was washed with wash buffer (50 mM Tris, 300 mM NaCl, 20 mM imidazole [pH = 7.5]) and bound His6-tagged proteins were subsequently recovered using elution buffer (50 mM Tris, 300 mM NaCl, 500 mM imidazole [pH = 7.5]). The elution fractions containing the His6-IreK-n proteins were each dialyzed against 1 L of 50 mM Tris, 25 mM NaCl [pH = 7.5] for 1 h, then overnight at 4°C in 1 L fresh buffer. The elution fractions containing His6-SUMO proteins (GpsB WT, GpsB 8A mutant, GpsB ΔP114-F36 mutant, or IreP) were dialyzed for 1 h at 4°C against 1 L 50 mM Tris, 150 mM NaCl [pH = 7.5], then His-GB1-Ulp1 protease and 0.1% β-mercaptoethanol were added to the dialysis bag and dialyzed overnight at 4°C in 1 L fresh buffer. After dialysis and Ulp1 cleavage, proteins were applied to binding-buffer-equilibrated Ni-charged resin, and cleaved (untagged) GpsB or IreP were collected in the flowthrough and wash fractions as verified by SDS-PAGE and GelCode Blue staining. Untagged proteins were subjected to size exclusion chromatography using a HiLoad Superdex 200-pg 16/600 column (Cytiva, Marlborough, MA) with running buffer (50 mM Tris, 25 mM NaCl [pH = 7.5]) at a flow rate of 1 mL/min. Fractions containing untagged protein were concentrated using an Amicon Ultra 15-mL 3 kDa-cutoff concentrator.
In vitro enzyme assays.
All kinase (phosphorylation) and phosphatase (dephosphorylation) assays were performed the same way unless otherwise indicated below for each reaction type. Purified proteins were pre-incubated in 50 mM Tris [pH = 7.5], 25 mM NaCl, and 5 mM MgCl2 (kinase assays) or 1 mM MnCl2 (phosphatase assays) for 5 min at 37°C. Next, 2 mM ATP (kinase assays) or 0.1 μM IreP (phosphatase assays) was added to initiate the reactions after which samples were removed at intervals. The reactions were quenched by the addition of 5 × SDS-PAGE Laemmli buffer and boiled for 5 min. The samples were subjected to SDS-PAGE at 200 V for 40 to 45 min. The gels were stained with Pro-Q Diamond Phosphoprotein Gel Stain (Invitrogen) followed by SYPRO Ruby Protein Gel Stain (Invitrogen) according to the manufacturer’s instructions to visualize phosphorylated protein and total protein, respectively. Gels were imaged using an Amersham Typhoon 5 (Cytiva, Marlborough, MA).
-
a)
GpsB phosphorylation by His6-IreK-n. Purified wild-type GpsB, GpsB 8A mutant, and GpsB ΔP114-F136 mutant (14.2 μM) were incubated at 37°C in the presence or absence of recombinant wild-type His6-IreK-n (0.33 μM) or a catalytically impaired His6-IreK-n K41R mutant (0.33 μM).
-
b)
His6-IreK-n autophosphorylation. Purified wild-type His6-IreK-n was dephosphorylated by purified IreP and then repurified before use in these assays. Dephosphorylated His6-IreK-n (1 μM) was incubated at 37°C in the presence or absence of wild-type GpsB, GpsB 8A mutant, or GpsB ΔP114-F136 mutant (10 μM).
-
c)
His6-IreK-n dephosphorylation by IreP. Phosphorylated, purified His6-IreK-n (10 μM) was pre-incubated in the presence or absence of wild-type GpsB (100 μM) at room temperature for 20 minutes. Reactions were performed at room temperature.
Image processing and statistical analysis
-
a)
Phos-tag SDS-PAGE immunoblot analysis. Band intensities of Phos-tag blots were quantified using the rectangle function on ImageLab. Total IreK was determined by taking the intensity of the entire IreK signal and phosphorylated IreK was determined by taking the intensity of the upper bands. Total MltG was determined by combining the intensities of the upper band (phosphorylated MltG) and the lower band (nonphosphorylated MltG). Data were exported to Microsoft Excel, where all calculations and graphing was performed. The percentage of phosphorylated IreK or MltG (% IreK-P or % MltG-P) was determined by calculating the ratio of phosphorylated IreK or MltG to total IreK or MltG. The ratios from at least 3 biological replicates were averaged and graphed using Microsoft Excel. Standard deviations and statistical analyses (Student’s t test [two-tailed, heteroscedastic]) were also performed using Microsoft Excel functions.
-
b)
In vitro assay analysis. Band intensities of Pro-Q Diamond-stained and SYPRO Ruby-stained gel images were quantified using AzureSpot software. Lanes were defined manually, and background subtraction was defined using the rolling ball function. Bands were detected manually and intensity values were exported to Microsoft Excel, where all calculations and graphing was performed. The ratio of phosphorylated protein to total protein of interest was determined and graphed over time. To determine relative initial rates, we identified the time interval exhibiting a linear rate for each reaction type, and determined the slope from the best fit line. To determine the x-fold change in rates for each reaction compared to the control reaction, the ratio of the rate of each test reaction to the rate of the control reaction was calculated. The mean x-fold change in rate for at least 2 independent experiments was determined and graphed. Standard deviations and statistical analyses (Student’s t test [two-tailed, heteroscedastic]) were also performed using Microsoft Excel functions.
ACKNOWLEDGMENTS
This study was supported in part by grants AI134660 and AI150895 from the National Institutes of Health (NIH).
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material is available online only.
Contributor Information
Christopher J. Kristich, Email: ckristich@mcw.edu.
Michael J. Federle, University of Illinois at Chicago
REFERENCES
- 1.Higuta NIA, Huyecke MI. 2014. Enterococcal disease, epidemiology, and implication for treatment. In Gilmore MS, Clewell DB, Ike Y, Shankar N (ed), Enterococci: from commensals to leading causes of drug resistant infection. Massachusetts Eye and Ear Infirmary, Boston, MA. [PubMed] [Google Scholar]
- 2.Hollenbeck BL, Rice LB. 2012. Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3:421–433. doi: 10.4161/viru.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller WR, Munita JM, Arias CA. 2014. Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther 12:1221–1236. doi: 10.1586/14787210.2014.956092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carmeli Y, Eliopoulos G, Mozaffari E, Samore M. 2002. Health and economic outcomes of vancomycin-resistant enterococci. Arch Intern Med 162:2223–2228. doi: 10.1001/archinte.162.19.2223. [DOI] [PubMed] [Google Scholar]
- 5.Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. 2000. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343:1925–1932. doi: 10.1056/NEJM200012283432604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG. 2010. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120:4332–4341. doi: 10.1172/JCI43918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeats C, Finn R, Bateman A. 2002. The PASTA domain: a β-lactam binding domain. Trends Biochem Sci 27:438–440. doi: 10.1016/S0968-0004(02)02164-3. [DOI] [PubMed] [Google Scholar]
- 8.Kristich CJ, Wells CL, Dunny GM. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc Natl Acad Sci USA 104:3508–3513. doi: 10.1073/pnas.0608742104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kristich CJ, Little JL, Hall CL, Hoff JS. 2011. Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2:e00199-11. doi: 10.1128/mBio.00199-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Labbe BD, Kristich CJ. 2017. Growth- and stress-induced PASTA kinase phosphorylation in Enterococcus faecalis. J Bacteriol 199:e00363-17. doi: 10.1128/JB.00363-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labbe BD, Hall CL, Kellogg SL, Chen Y, Koehn O, Pickrum AM, Mirza SP, Kristich CJ. 2019. Reciprocal regulation of PASTA kinase signaling by differential modification. J Bacteriol 201:e00016-19. doi: 10.1128/JB.00016-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellogg SL, Kristich CJ. 2018. Convergence of PASTA kinase and two-component signaling in response to cell wall stress in Enterococcus faecalis. J Bacteriol 200:e00086-18. doi: 10.1128/JB.00086-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall CL, Tschannen M, Worthey EA, Kristich CJ. 2013. IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob Agents Chemother 57:6179–6186. doi: 10.1128/AAC.01472-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iannetta AA, Minton NE, Uitenbroek AA, Little JL, Stanton CR, Kristich CJ, Hicks LM. 2021. IreK-mediated, cell wall protective phosphorylation in Enterococcus faecalis. J Proteome Res 20:5131–5144. doi: 10.1021/acs.jproteome.1c00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, Edwards DH. 2008. Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Mol Microbiol 68:1029–1046. doi: 10.1111/j.1365-2958.2008.06210.x. [DOI] [PubMed] [Google Scholar]
- 16.Tavares JR, de Souza RF, Meira GL, Gueiros-Filho FJ. 2008. Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisome. J Bacteriol 190:7096–7107. doi: 10.1128/JB.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. 2013. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol 90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rued BE, Zheng JJ, Mura A, Tsui HT, Boersma MJ, Mazny JL, Corona F, Perez AJ, Fadda D, Doubravova L, Buriankova K, Branny P, Massidda O, Winkler ME. 2017. Suppression and synthetic-lethal genetic relationships of ΔgpsB mutations indicate that GpsB mediates protein phosphorylation and penicillin-binding protein interactions in Streptococcus pneumoniae D39. Mol Microbiol 103:931–957. doi: 10.1111/mmi.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleverley RM, Rutter ZJ, Rismondo J, Corona F, Tsui HT, Alatawi FA, Daniel RA, Halbedel S, Massidda O, Winkler ME, Lewis RJ. 2019. The cell cycle regulator GpsB functions as cytosolic adaptor for multiple cell wall enzymes. Nat Commun 10:261. doi: 10.1038/s41467-018-08056-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mura A, Fadda D, Perez AJ, Danforth ML, Musu D, Rico AI, Krupka M, Denapaite D, Tsui HT, Winkler ME, Branny P, Vicente M, Margolin W, Massidda O. 2017. Roles of the essential protein FtsA in cell growth and division in Streptococcus pneumoniae. J Bacteriol 199:e00608-16. doi: 10.1128/JB.00608-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pompeo F, Foulquier E, Serrano B, Grangeasse C, Galinier A. 2015. Phosphorylation of the cell division protein GpsB regulates PrkC kinase activity through a negative feedback loop in Bacillus subtilis. Mol Microbiol 97:139–150. doi: 10.1111/mmi.13015. [DOI] [PubMed] [Google Scholar]
- 22.Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, Grangeasse C. 2014. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet 10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rismondo J, Cleverley RM, Lane HV, Grosshennig S, Steglich A, Möller L, Mannala GK, Hain T, Lewis RJ, Halbedel S. 2016. Structure of the bacterial cell division determinant GpsB and its interaction with penicillin-binding proteins. Mol Microbiol 99:978–998. doi: 10.1111/mmi.13279. [DOI] [PubMed] [Google Scholar]
- 24.Djoric D, Little JL, Kristich CJ. 2020. Multiple low-reactivity class B penicillin-binding proteins are required for cephalosporin resistance in enterococci. Antimicrob Agents Chemother 64:e02273-19. doi: 10.1128/AAC.02273-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arbeloa A, Segal H, Hugonnet J, Josseaume N, Dubost L, Brouard J, Gutmann L, Mengin-Lecreulx D, Arthur M. 2004. Role of class A penicillin-binding proteins in PBP5-mediated b-lactam resistance in Enterococcus faecalis. J Bacteriol 186:1221–1228. doi: 10.1128/JB.186.5.1221-1228.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice LB, Carias LL, Rudin S, Hutton R, Marshall S, Hassan M, Josseaume N, Dubost L, Marie A, Arthur M. 2009. Role of class A penicillin-binding proteins in the expression of beta-lactam resistance in Enterococcus faecium. J Bacteriol 191:3649–3656. doi: 10.1128/JB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vesic D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob Agents Chemother 56:2443–2451. doi: 10.1128/AAC.05984-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wamp S, Rutter ZJ, Rismondo J, Jennings CE, Moller L, Lewis RJ, Halbedel S. 2020. PrkA controls peptidoglycan biosynthesis through the essential phosphorylation of ReoM. Elife 9:e56048. doi: 10.7554/eLife.56048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelliher JL, Grunenwald CM, Abrahams RR, Daanen ME, Lew CI, Rose WE, Sauer JD. 2021. PASTA kinase-dependent control of peptidoglycan synthesis via ReoM is required for cell wall stress response, cytosolic survival, and virulence in Listeria monocytogenes. PLoS Pathog 17:e1009881. doi: 10.1371/journal.ppat.1009881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kellogg SL, Little JL, Hoff JS, Kristich CJ. 2017. Requirement of the CroRS two-component system for resistance to cell wall-targeting antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61:e02461-16. doi: 10.1128/AAC.02461-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA kilobases up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S3 and Fig. S1 to S8. Download jb.00304-22-s0001.pdf, PDF file, 0.6 MB (658.1KB, pdf)