

Abstract
A key step to inhibit severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is to prevent the entry of the virus into the host cells. The receptor-binding domains (RBDs) of spike proteins of SARS-CoV and other human coronaviruses utilize heparan sulfate proteoglycans (HSPGs) as the primary receptors for their accumulation on the cell surface and then scan for binding to the main entry receptor angiotensin-converting enzyme 2 (ACE2). SARS-CoV and SARS-CoV-2 share structurally similar RBDs and therefore, it is possible that SARS-COV-2 primarily binds to HSPGs followed by binding to the ACE2 receptors. A promising strategy to inhibit virus infection is to circulate exogenous bioactive moieties structurally mimicking cellular HSPG and ACE2 which act as decoy receptors binding to SARS-CoV-2 and competitively inhibit virus entry to the host cells mediated by cellular-bound HSPG and ACE2. Using a molecular docking tool, we identified carboxymethyl benzyl amide sulfonate (CMBS) and polyanetholesulfonic acid (PAS) as the suitable HSPG mimicking ligands, and Paenibacillus sp. B38-derived carboxypeptidase (B38-CAP) and Bacillus subtilis-derived carboxypeptidase (BS-CAP) as the potential ACE2-like enzymes having a strong binding affinity to the spike proteins as that of cellular HSPG and ACE2. Further, the binding stability and compactness of these moieties with SARS-CoV-2 were analyzed through molecular dynamics (MD) simulations, and the results indicated that these moieties form well-stable complexes with the RBD of spike proteins. The identified moieties could be conjugated to the surfaces of non-toxic nanoparticles to provide multiple interactions to efficiently shield SARS-CoV-2, and inhibit viral entry to the host cells.
Keywords: SARS-CoV-2, HSPG, ACE2, Mimicking moieties, Molecular docking, Molecular dynamics simulations, Inhibition
1. Introduction
Highly infectious and transmissible severe acute respiratory syndrome corona virus-2 (SARS-CoV-2) is responsible for the global pandemic outbreak of coronavirus disease (COVID-19). Being first reported in Wuhan, China in 2019, it gradually spread all over the world and created a serious threat to human health and public safety. Patients infected with SARS-CoV-2 experience mild to moderate respiratory diseases like dyspnea, dry cough, fatigue, fever, and pneumonia, while some with primary medical conditions like diabetes, chronic respiratory disease, and cancer develop serious issues like multiple organ failure or death [1], [2], [3], [4]. World Health Organization (WHO) statistics (as of October 2022) reveal that around 619 million cases and 6.5 million deaths are due to this contagious viral body [5]. Other variant forms of SARS-CoV-2 are the SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) reported in 2003 and 2012, respectively. SARS-CoV and SARS-CoV-2 strains share 73 to 76 percent similarity in their RNA [6], [7] and target angiotensin-converting enzyme Ⅱ (ACE2) receptors present on the cell membrane for their entry to the host cells with a similar binding mechanism [8], [9], [10], [11]. SARS-CoV-2 is a single-stranded positive-sense large RNA virus and the spike proteins present on its surface facilitate it's entry into the host cell [11]. The spike proteins contain functional S1 and S2 subunits, with S1 responsible for direct binding to the ACE2 through its receptor-binding domain (RBD) and S2 responsible for the membrane fusion of the virus with the host cell membrane [6], [8], [12], [13]. Spike protein is a trimer complex with 3 RBDs which helps in the interaction of the virus with the cell surface receptors through the receptor-binding motifs (RBMs) present in them [13].
ACE2 (a type I membrane protein) receptors are broadly expressed, including alveolar cells of lungs, capillary cells of organs like lungs, kidneys, gut, central nervous system, and monocytes or macrophages [13], [14]. An important function of membrane-bound ACE2 is to catalyze the degradation of the angiotensin Ⅱ to angiotensin 1 to 7 [13], [15]. Angiotensin Ⅱ is a bioactive peptide present in the renin-angiotensin system, extensively involved in the development of cardiovascular diseases (hypertension, myocardial infarction, heart failure), increased blood pressure, cytokine storm, endothelial injury, and acute respiratory distress syndrome (ARDS) [11]. However, the unfortunate scenario of SARS-CoV-2 binding to the ACE2 receptors significantly decreases the ACE2 expression at the external surfaces of the cell membrane, which results in complete loss of these receptors' catalytic activity [13]. Once SARS-CoV-2 binds to ACE2, the viral spike proteins dissociate into S1 and S2 subunits, and the RBMs present in the RBDs of S1 subunit get attached to the protease domain of ACE2 (through its interaction with alpha1 and alpha 2 helices) [11]. After binding, the S1 subunit gets detached and the cleavage sites of the S2 subunit are exposed, which then gets cleaved by the protease domain of ACE2; marking as an important process for virus infection [13].
Several studies reveal that heparin sulfate proteoglycans (HSPG) receptors (polysaccharide in nature) present on the external surfaces of the cell membrane acts as receptors (in addition to ACE2) for the binding of SARS-CoV [16], [17], [18], [19], [20], [21], [22], [23], [24], [25]. The RBDs in spike S1 proteins of SARS-CoV have heparin sulfate binding sites which help them to bind with HSPG receptors. The virus utilizes HSPGs as the primary receptors for their accumulation on the cell surface and then scans (viral surfing) for ACE2 for its entry into the cells [26], [27]. SARS-CoV and SARS-CoV-2 share structurally similar RBDs and therefore, it is possible that SARS-COV-2 primarily binds to HSPGs followed by binding to the ACE2 receptors [6], [7]. Also, it is reported that the interaction of HSPG with the spike protein of SARS-CoV-2 results in a conformational change in its RBD and facilitates its binding with ACE2 [28], [29], [30].
Many of the antiviral drugs used to treat other viral diseases were initially proclaimed to be effective against SARS-CoV-2. However, their efficacy against COVID-19 disease in terms of either reducing the mortality rate or hospitalization time of patients was later disproved. Therefore, the search for other non-toxic and broadly active agents that specifically inhibit SARS-CoV-2 infections remains urgent. The present work addresses the identification of bioactive moieties structurally mimicking cellular-bound HSPG and ACE2 receptors with a strong binding affinity to the spike proteins of SARS-CoV-2 through molecular docking studies and molecular dynamic (MD) simulations. Once the identified bioactive moieties are circulated in the soluble form, they act as decoy receptors binding to SARS-CoV-2, thus resulting a competitive inhibition for virus entry to the cells mediated by the cellular-bound HSPG and ACE2. Fig. 1 shows the schematic of the proposed SARS-CoV-2 entry inhibition mechanism in presence of a soluble form of exogenous HSPG and ACE2 mimicking moieties.
Fig. 1.

Schematic of the SARS-CoV-2 virus entry inhibition mechanism in the presence of a soluble form of exogenous heparin sulfate proteoglycan (HSPG) and angiotensin-converting enzyme II (ACE2) mimicking moieties. (A) Conventional entry of virus to the cells via binding with HSPG and ACE2 receptors of the host cell membrane (B) Proposed mechanism of inhibition of virus entry to the cell by circulating bioactive chemical moieties structurally mimicking cellular HSPG and ACE2.
In this work, we first performed molecular docking of RBD of spike protein with cellular-bound HSPG and ACE2 and determined their binding affinities. Secondly, the binding affinities of HSPG mimicking ligands and ACE2-like enzymes were predicted by docking them with the RBD of the spike protein. Comparison of the docking results led us to identify appropriate HSPG mimicking ligands and ACE2-like enzymes exhibiting strong binding affinity towards the viral spike protein as that of cellular HSPG and ACE2. To understand how well these identified ligands and enzymes form stable complexes with the spike proteins, we performed molecular dynamics (MD) simulations and investigated their binding stability, flexibility, structure compactness, the free energy of binding (for protein–protein complex), and interaction energy (for protein–protein complex). Thus, the present in silico studies is aimed toward the identification of potential structurally mimicking bioactive moieties of HSPG and ACE2 showing a stable and stronger binding with the spike proteins of SARS-CoV-2 which helps in competitive inhibition of the virus entry to the host cells.
2. Methodology
2.1. Screening of HSPG mimicking ligands and ACE2-like enzymes
To identify and recognize the suitable HSPG and ACE2 mimicking moieties resulting strong and stable complex formation with the RBD of SARS-CoV-2 spike protein, it is necessary to understand the factors leading to the close interaction between the moieties and the virus. It is reported that the sulphonic acid, R-S(=O)2-OH functional group of cellular HSPG helps in binding to the RBD of SARS-CoV-2 spike protein [32], [33], [34], [35]. We have screened 12 such chemical moieties with sulphonic acid and other functional groups (Table 1 ) carrying similar molecular structures of cellular HSPG and studied their binding affinity with the RBD of the spike protein of SARS-CoV-2. Similarly, we have screened three bacterial-derived carboxypeptidases B38-CAP, BS-CAP, and BA-CAP as ACE2-like enzymes (Fig. 2 ) having similar protein sequences and functional features as that of cellular ACE2. Previous studies revealed that bacterial-derived carboxypeptidases have a close resemblance to the protein structures and enzyme sequences of ACE2 receptors, supporting the possibility for a similar substrate binding preferences [31]. The sequence similarity among these protein structures was evaluated using the pairwise sequence alignment analysis tool, Emboss Needle (refer to Figs. S1, S2, and S3 of the supporting information file).
Table 1.
List of screened cellular heparin sulfate proteoglycan (HSPG) mimicking ligands and their functional groups responsible for binding with RBD of spike proteins of SARS-CoV-2.
| S. No | Ligand Name | Functional Group |
|---|---|---|
|
1 |
CMBS (Carboxymethyl benzyl amide sulfonate) |
 |
|
2 |
Polyanetholesulfonic acid (PAS) | 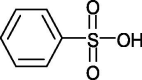 |
|
3 |
6-((3,4-dihydroxy phenethyl) amino)-6-isohexane-1-sulfonate (DOS) |  |
|
4 |
2-acrylamide 2-methyl propane sulfonic acid (PAMPS) | 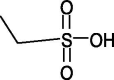 |
|
5 |
N-Sulfo-Glucosamine (SGA) |  |
|
6 |
Vinyl sulfonic acid (PVS) |  |
|
7 |
11-Mercapto-1-undecanesulfonate (MUS) | 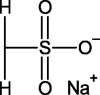 |
|
8 |
Hexamethylene diisocyanate (HIC) |  |
|
9 |
4-styrene sulfonic acid (PSS) | 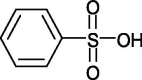 |
|
10 |
12-Mercapto-undecyl phosphoric acid (MUP) |  |
|
11 |
Oligo(ethylene glycol) (EG2-oh) | 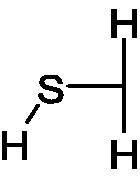 |
|
12 |
2-Mercaptoethanesulfonate (MES) | 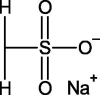 |
Fig. 2.

Crystallographic images of the protein structures studied in this work. (A) RBD domain of spike protein of SARS-CoV-2 (PDB Id: 6M0J) (B) cellular angiotensin-converting enzyme 2 (ACE2) (PDB ID: 1R4L) (C) B38-CAP (GenBank, LC406946) (D) BA-CAP (GenBank, LC417450) (E) BS-CAP (PDB ID: 3HQ2).
2.2. Molecular docking studies
Molecular docking is an important tool used to identify favorable interactions between protein–ligand or protein–protein complexes based on their binding affinity [36]. It also helps to predict the types of interactions (van der Waal’s, hydrogen bond, amide, pi-bonds, etc.), protein residues (amino acids) responsible for binding, and the distance between those binding residues. We performed docking of RBD of the SARS-CoV-2 spike protein and all the HSPG mimicking ligands (listed in Table 1) and predicted their binding affinities. Before docking, the molecular structures of all the ligands were drawn using ChemSketch software and optimized to the 3-dimensional structure. All the structures were then converted to PDB forms using Open Babel software. We used AutoDock software-defined with the standard set of input parameters and performed standard docking calculations. Before docking, the structure of the protein was edited to remove water, and extra groups such as ligands and other inhibitors and added with polar hydrogen. The charges were computed by adding Kollman and Gasteiger charges before creating the PDBQT files or autodocking form of the protein and ligand molecules. The roots for the ligand additions were detected using MGL tools and the binding positions through Discovery studio. The PDB structure of the spike protein of SARS-CoV-2 was obtained from the RCSB Protein Data Bank (PDB Id: 6M0J) and assigned to molecular docking form (PDBQT) using MGLTools, AutoDock, and Discovery studio visualizer 2021. The PDBQT files were then run on AutoDock Vina to determine the binding affinity (in kcal/mol) of each ligand to the viral spike protein domain. From the docking output, we also accessed the docked poses, types of interactions, and amino acids involved in the binding of ligands with the spike protein with the help of Discovery Studio Visualizer 2021.
For the spike protein–protein docking, the crystallographic PDB images and the sequences of the protein structures were downloaded from RCSB Protein Data Bank and GenBank. These include SARS-CoV-2 (PDB Id: 6M0J), ACE2 (PDB Id: 1R4L), and bacterial derived carboxypeptidases (ACE2-like enzymes), B38-CAP (LC406946, GenBank), BS-CAP (PDB Id: 3HQ2), BA-CAP (LC417450, GenBank). Protein-protein docking was performed using the PRODIGY and ClusPro platform to identify suitable ACE2-like enzyme(s) resulting stronger affinity to the RBD of spike protein as that of cellular ACE2. From docking, we obtained the binding affinity values (in kcal/mol), dissociation constant (M), number of clusters and the resulting members, and number of interfacial contacts present in the protein–protein complexes. We also analyzed the amino acid residues present at the interface of protein–protein complexes.
2.3. Molecular dynamics (MD) simulation studies
The binding stability, compactness, and atomic fluctuations of all the protein–ligand and protein–protein complexes were analyzed using MD simulations. Further, the binding free energy for the protein–ligand complexes and the interaction energy for the protein–protein complexes were calculated based on the MD simulation results. For protein–ligand and protein–protein systems, the simulations were performed using GROMACS 5.1.1 for 50 ns time, using the GROMOS force field. The protein topology was prepared using the GROMOS 87/96 force field and TIP3P water model for all the protein molecules, and the ligand topology was developed with the help of an external tool, PRODRG 2.5 which uses the GROMOS 87/96 force field. The protein–protein complexes were solvated by the spc216 system and neutralized by adding 26 mol of sodium ions in the spike protein-ACE2 complex, 18 mol of sodium ions in spike protein-B38-CAP, and 54 mol of sodium ions in spike protein-BS-CAP systems. Both the protein–ligand and protein–protein complexes were added to a cubic box (15 × 15 × 15 nm) and centered before the solvation step. Protein-ligand (cellular HSPG, CMBS, and PAS) complexes were neutralized by adding 2 mol of chlorine. Further, the energy minimization of the simulation system was achieved through 1000 kJ/mol steepest descents in 143 steps. To equilibrate all protein–protein and protein–ligand complexes, a two-phase equilibration process was carried out by NVT and NPT ensemble for 100 ps at a constant temperature of 300 K and 1 atm pressure conditions.
Root mean square deviation (RMSD) plots were developed to analyze the structural deviations of both HSPG mimicking ligands and ACE2-like enzymes with the RBD of the spike protein during the simulation. The details of atomic fluctuations and the compactness of the complexes were determined using root mean square fluctuation (RMSF) and radius of gyration (Rg) plots, respectively. Further, the interaction energies of the protein–protein complexes from the trajectory files obtained from the simulation, and the binding free energy of the protein–ligand complex formed using the free energy perturbation method were calculated. The binding free energy of the protein–ligand complex was calculated by considering two phases of the protein–ligand system in which one phase consists of the ligand in a de-coupled state (A) from the protein, and in the other phase, the ligand in a coupled state (B) to the protein molecule. The free energy difference between the two phases was calculated based on the concept of energy required by the system to drag the ligand away from the protein, and a higher energy (negative) indicates stronger affinity and higher interaction stability. The calculated free energy difference by integrating the potential mean force (PMF) (i.e., the average force required to pull the ligand molecule away from the protein) was then used to determine the binding free energy (). Bennet Acceptance Ratio (BAR) [37] method was used to calculate . Considering the states A and B, then
where, and are the relative probabilities of the ligand in a de-coupled and coupled state, respectively. is the free energy difference, and is the Boltzmann’s constant, and is the temperature. For free energy difference calculations, we integrated the interaction strength between the ligand and the rest of the system by a variable λ which ranges from 0 to 1. At λ = 0, the ligand is dragged away from the protein to a vacuum state, where there is no interaction between the ligand and the rest of the system. At λ = 1, the ligand is bound to the protein molecule. If the free energy for de-coupling of the ligand from the rest of the system is and the free energy for coupling the ligand to the system is , then .
3. Results and discussion
3.1. Protein-ligand docking
All the screened HSPG mimicking ligands (listed in Table 1) and cellular HSPG were docked with the RBD of SARS-CoV-2 spike protein and their respective binding affinities were predicted to choose appropriate ligands showing a stronger binding affinity for the spike protein as that of cellular HSPG. Binding affinity is expressed in kcal/mol, and a more negative affinity value is a measure of stronger interaction between a protein and a ligand. Table 2 shows the ligands ranked based on their binding affinity values, and the top two ligands showing the binding affinity values closer to that of cellular HSPG were identified as the best possible HSPG mimicking ligands that can strongly bind to the spike protein. For all the protein–ligand molecular docking analyses, a total of three runs have been performed and the average values are reported. Table S4 (of Supplementary file) shows the binding affinity values of all the protein–ligand complexes recorded from the three runs, and also the number of conformers present. Based on the rank of the conformers and the highest binding affinity values appropriate the top two ligands were chosen for MD simulation studies. From Table 2 and also from Table S4 (of Supplementary file), we find that carboxymethyl benzyl amide sulfonate (CMBS) and polyanetholesulfonic acid (PAS) ligands exhibit the respective binding affinity value of −7.3 and −7.1 kcal/mol; which are the closest to the binding affinity value of cellular HSPG (-7.7 kcal/mol). Thus, the identified CMBS and PAS ligands could be used as potential moieties which can strongly bind to the RBD of spike protein and effectively inhibit SARS-CoV-2 entry to the host cells.
Table 2.
Binding affinity values obtained from the molecular docking of RBD of SARS-CoV-2 spike protein and HSPG ligand complexes using Autodock Software.
| S. No | Ligands | Binding affinity (kcal/mol) |
|---|---|---|
| 1 | Heparin sulfate proteoglycans (HSPG) | −7.7 |
| 2 | CMBS (Carboxymethyl benzyl amide sulfonate) | −7.3 |
| 3 | Polyanetholesulfonic acid (PAS) | −7.1 |
| 4 | 6-((3,4-dihydroxy phenethyl) amino)-6-oxohexane-1-sulfonate (DOS) | −6.6 |
| 5 | 2-acrylamide 2-methyl propane sulfonic acid (PSA) | −6.4 |
| 6 | N-Sulfo-Glucosamine (SGA) | −5.7 |
| 7 | Vinyl sulfonic acid (PVS) | −5.5 |
| 8 | 11-Mercapto-1-undecanesulfonate (MUS) | −5.5 |
| 9 | Hexamethylene diisocyanate (HIC) | −5.3 |
| 10 | 4-styrene sulfonic acid (PSS) | −5.1 |
| 11 | 12-Mercapto-undecyl phosphoric acid (MUP) | −5.1 |
| 12 | Oligo(ethylene glycol) (EG2-oh) | −4.8 |
| 13 | 2-Mercaptoethanesulfonate (MES) | −4.4 |
Fig. 3 shows the docked poses of the cellular HSPG, CMBS, and PAS during their interaction with the active sites of the spike protein. These ligands form van der Waal’s, hydrogen, pi-alkyl, pi-cation/anion, and amide pi-stacked bonds with the RBD of spike protein, and the amino acid residues of spike proteins taking part during the interaction are shown in Table 3 . Further, the 2-dimensional structures depicting the spike protein–ligand interactions, amino acids of protein, and ligand atoms participating in the formation of a protein–ligand complex labeled by bond length and nature of bonds are illustrated in Fig. 4 .
Fig. 3.

Docked poses of (A) heparin sulfate proteoglycan (HSPG) (B) carboxymethyl benzyl amide sulfonate (CMBS) (C) polyanetholesulfonic acid (PAS) ligands during their interaction with the RBD of the spike protein of SARS-CoV-2, obtained from Discovery Studio Visualizer 2021. Hydrogen and other several bonds were involved in the interaction between RBD and ligands.
Table 3.
List of amino acid residues of RBD of spike proteins of SARS-CoV-2 interacting with the cellular heparin sulfate proteoglycan (HSPG), and HSPG mimicking ligands (CMBS, PAS) classified based on the type of bonds formed.
| Types of bonds | Amino acids interacting with heparin sulfate proteoglycan (HSPG) |
Amino acids interacting with carboxymethyl benzyl amide sulfonate (CMBS) | Amino acids interacting with poly anethole sulfonic acid (PAS) |
|---|---|---|---|
| van der Waal’s Bond |
Valine 433, Isoleucine 410, Lysine 378, Threonine 376, Valine 503, Proline 412, Glutamine 414, Glycine 404 |
Tyrosine 421, Arginine 457, Tyrosine 489, Phenylalanine 456, Tyrosine 453, Arginine 403, Tyrosine 495, Tyrosine 505, Serine 494 | Glycine 431, Phenylalanine 429, Phenylalanine 515, Serine 514, Tyrosine 396, Arginine 355 |
| Hydrogen Bond |
Tyrosine 380, Tyrosine 508, Arginine 408 |
Tyrosine 473, Glutamine 493, Tyrosine 449, Glycine 496, Asparagine 501, Glutamine 498 | Threonine 430 |
| Amide- Pi Stacked Bond | Valine 407 | – | – |
| Pi- Alkyl Bond | Alanine 411 | Leucine 455, Alanine 475 | Leucine 518, Proline 426 |
| Pi-Cation Bond | – | Lysine 417 | – |
| Pi-Anion Bond | – | – | Glutamic Acid 516 |
Fig. 4.

2-dimensional structures showing the amino acid residues binding with heparin sulfate proteoglycan (HSPG) ligand indicating the type of bond and distance at which the bonds are formed in (A) heparin sulfate proteoglycans (HSPG) (B) carboxymethyl benzyl amide sulfonate (CMBS) (C) poly anethole sulfonic acid (PAS) during docking with the RBD of the spike protein of SARS-CoV-2.
3.2. Protein-protein docking
The RBD of SARS-CoV-2 spike protein was docked with cellular ACE2 and bacterial-derived carboxypeptidases B38-CAP, BS-CAP, and BA-CAP (ACE2-like enzymes) (shown in Fig. 2), and their binding affinity values, dissociation constant, and interfacial contacts directly obtained from PRODIGY platform are shown in Table 4 .
Table 4.
Protein-protein docking results obtained from the PRODIGY platform.
| S. No | Protein-Protein Complex |
Binding affinity (kcal/mol) |
Dissociation constant (M) |
Interfacial Contacts |
|---|---|---|---|---|
| 1 | S Protein-ACE2 | −12.6 | 0.85 × 10−4 | 137 |
| 2 | S Protein- BS-CAP | −11.8 | 2.59 × 10−4 | 113 |
| 3 | S Protein-B38-CAP | −11.5 | 2.73 × 10−4 | 94 |
| 4 | S Protein- BA-CAP | −10.8 | 2.96 × 10−4 | 79 |
The dissociation constant is a measure of affinity, and a smaller value of the dissociation constant indicates a more tightly binding or higher affinity between two or more interacting molecules [38]. A higher number of interfacial contacts represent the formation of more stable complexes. All the ACE2-like enzymes were ranked based on the highest negative binding affinity, lowest dissociation constant, and higher number of interfacial contacts. Though all these enzymes show closer binding affinity values for RBD of spike protein as that of cellular ACE2, the number of interfacial contacts for the spike protein-BA-CAP complex is very much lesser than the spike protein-cellular ACE2 complex. Therefore, we identified BS-CAP and B38-CAP as the most suitable ACE2-like enzymes that can be used as decoy receptors to bind to the spike protein and competitively inhibit SARS-CoV-2 entry to the host cells.
The binding energy values for all the protein–protein complexes were also obtained using ClusPro software. Table 5 shows the number of cluster members, the lowest binding energy values obtained in five conformers, and the average binding energy value for each of them. These results also confirm that BS-CAP and B38-CAP protein have a higher binding affinity towards the RBD of the spike protein, and thus considered further for MD simulations studies. Fig. 5 shows the amino acid residues present at the interface, participating in the protein–protein complex formations. The list of amino acid residues present at the interface of spike protein-cellular ACE2 complex, spike protein-BS-CAP complex, and spike protein- B38-CAP complex are shown in Tables S1, S2, and S3 of the supplementary material file.
Table 5.
Protein-Protein docking results obtained from ClusPro Software.
| S. No | Protein-Protein Complex | Number of clusters | Number of cluster members | Lowest binding energy (kJ/mol) | Average binding energy (kJ/mol) |
|---|---|---|---|---|---|
| 1 | RBD-ACE2 | 1 | 59 | −769.7 | −716.5 |
| 2 | 46 | −727.2 | |||
| 3 | 42 | −745.8 | |||
| 4 | 40 | −679.0 | |||
| 5 | 37 | −661.0 | |||
| 2 | RBD-BS-CAP | 1 2 3 4 5 |
54 54 43 41 39 |
−682.5 −746.1 −790.1 −625.2 −777.8 |
−718.3 |
| 2 | RBD-B38-CAP | 2 | 68 | −634.9 | −656.8 |
| 3 | 63 | −639.9 | |||
| 4 | 56 | −671.3 | |||
| 5 | 54 | −667.5 | |||
| 5 | 40 | −650.4 | |||
| 4 | RBD-BA-CAP | 1 | 34 | −742.9 | −645.2 |
| 2 | 34 | −614.9 | |||
| 3 | 35 | −636.7 | |||
| 4 | 33 | −622.1 | |||
| 5 | 29 | −609.6 |
Fig. 5.

Amino acid interaction at the protein–protein interfaces obtained from the docking results. (A) RBD and cellular angiotensin-converting enzyme 2 (ACE2) (B) RBD and BS-CAP (C) RBD and B-38 CAP. The maroon color indicates the RBD of the spike protein.
3.3. Molecular dynamics (MD) simulation
To get insight into the dynamic perspective of the interactions between RBD of spike protein with each ligand molecule (cellular HSPG, CMBS, and PAS) and RBD of spike protein with protein (cellular ACE2, BS-CAP, and B38-CAP) complexes, MD simulations were performed. These ligands and proteins were chosen as they exhibited the highest negative binding affinity values (towards the RBD of spike protein) in the docking analysis, and are much closer to the binding affinity values shown by RBD of spike protein-cellular HSPG and ACE2 protein. Using MD simulations, we aim to understand the self-conformational perturbations exhibited by the spike protein to attain stability and compactness with these ligands and proteins. The simulation experiments gave us the trajectories of RMSD and RMSF of Cα atoms of RBD of spike protein with respect to the initial structure, and Rg of Cα atoms of RBD of spike protein during its interaction with each of these ligands and proteins. RMSD plots were developed to analyze the structural deviations of HSPG mimicking ligands and ACE2-like enzymes upon their interaction with the RBD of spike protein during the simulation. The details of atomic fluctuations and the compactness of these complexes were also determined using RMSF and Rg plots, respectively. Further, the free energy of binding of protein–ligand complexes and interaction energies of spike protein–protein complexes were analyzed.
The initial two complexes RBD of spike protein-cellular HSPG and RBD of spike protein-cellular ACE2 were set as the standards to analyze the binding stability (based on RMSD and RMSF plots) and compactness of the complexes (based on Rg plots) formed by the mimicking ligands and proteins. Fig. 6 (A-C) shows the RMSD plots of Cα atoms of RBD of spike protein (with respect to its initial structure) during its complex formation with cellular HSPG, CMBS, and PAS ligands, respectively. It is critical to analyze the RMSD of spike protein Cα atoms which gives the binding or conformal stability with these ligands during the simulation. Low levels of RMSD indicate high stability of the complex formed, or higher fluctuations indicate lower binding stability [39]. The RBD of the spike protein in complex with cellular HSPG shows (Fig. 6 A) a maximum and minimum RMSD with very low levels of magnitudes ranging from 0.1 to 0.5 nm, indicating conformational stability achieved during simulation. The RMSD patterns of RBD of spike protein in complex with CMBS (Fig. 6 B) and PAS (Fig. 6 C) also show similar deviation values as that of cellular HSPG (below 0.5 nm), delineating strong conformational stability achieved by the spike protein with these ligands. The fluctuations observed in the simulation trajectories indicate that the spike protein experienced minor structural perturbations during its stable conformations with these ligands. Also, the RMSD values of RBD in all three ligand complexes are lower than the free RBDs, indicating that the RBD of spike protein achieves conformational stability upon its complex formation with the ligands. Further, all the three ligands (cellular HSPG, CMBS, and PAS) in their free state show (in Fig. 6 A-C) very low deviations than the free RBDs, and RBD in complex with these ligands; suggesting that they could be used as potential moieties for effective SARS-CoV-2 inhibition. A few major fluctuations in the trajectory of CMBS ligand can be observed at 10 ns and 40 ns, however, the deviations are lower than 0.35 nm and less significant as compared to the spike protein. A recent study showed an acceptable RMSD range of 0.1 nm to 0.4 nm for SARS-CoV-2 protein and other ligand complexes [40], which is consistent with the range observed in Fig. 6. Thus, the RMSD observations indicate that the spike protein attains stable complexes with cellular HSPG, CMBS, and PAS ligands during the simulations with minor structural perturbations.
Fig. 6.

RMSD plots of Cα atoms of RBD of SARS-CoV-2 during its complex formation with (A) cellular HSPG (B) CMBS and (C) PAS, in comparison to the RMSD of the respective free RBDs and free ligands.
The atoms (amino acids) of RBD of spike protein play a major role in attaining stable conformation with these ligands. We used RMSF to analyze the fluctuations (flexibility) of the atoms of spike protein in attaining complex formation with the cellular HSPG, CMBS, and PAS ligands (Fig. 7 ). RMSF gives a measure of the average movement of the atoms with respect to time calculated by the positional displacement of atoms between each conformation and averaged by many such conformations [41]. Atoms showing higher levels of RMSF signify increased flexibility and a high potential for complex formation [42]. The presence of a significant number of peaks in all three trajectories (in Fig. 7A) indicates that the atoms of RBD of spike proteins underwent significant fluctuations during complex formation. The RMSF values of all three ligands are below 0.7 nm and indicate a similar interaction potentials; suggesting that cellular HSPG, CMBS, and PAS ligands effectively attained stable conformation within the binding pockets of the spike protein. The peaks observed for the spike protein- cellular HSPG complex are due to the fluctuations in the spike protein amino acid residues Ala 475, Gly 476, Ser 477, Thr 478, Pro 479, Cys 480, Asn 481, Gly 482, Val 483, Glu 484, Gly 485. Among these, in spike protein- CMBS complex, Ala 475, Ser 477, Thr 478, Pro 479, Cys 480, Asn 481, Gly 482, Val 483, Glu 484, and Gly 485 are common amino acid residues that fluctuated; whereas in spike protein- PAS complex, Gly 476 and Ser 477 are the common amino acid residues showed fluctuations.
Fig. 7.

RMSF (A) and Rg (B) plots of Cα atoms of RBD of SARS-CoV-2 during its complex formation with cellular HSPG, CMBS, and PAS ligands.
Fig. 7 (B) shows the Rg of Cα atoms of RBD of spike protein during its complex formation with the cellular HSPG, CMBS, and PAS ligands, respectively. These plots provide insight into the distribution of atoms in a protein molecule and indicate the compactness of structures. A steady range of Rg over the simulation time indicates the compactness of the structures formed [43]. Also, a high value of Rg depicts less tight packing of atoms, whereas, a low value depicts tight packing [44]. For all protein–ligand complexes, we can observe reasonably steady Rg values indicating that the Cα atoms of RBD of spike protein are uniformly distributed and no loss of protein function occurred during complex formation. All three ligands exhibited closer Rg levels ranging from 1.7 nm to 1.8 nm indicating tight packing of atoms and are consistent with the acceptable range reported in a recent work [45]. Thus, the Rg analysis confirms that cellular HSPG, CMBS, and PAS ligands and the spike proteins of SARS-CoV-2 attain compact structures during their complex formation.
Fig. 8(A) shows the RMSD plots of Cα atoms of RBD of spike protein during its complex formation with the cellular ACE2, BS-CAP, and B38-CAP proteins. The RMSD values of the spike protein-BS-CAP complex range from 0.12 nm to 0.55 nm, whereas, the RMSD values of the spike protein-B38-CAP complex range from 0.19 nm to 0.72 nm. The deviations in the positions of Cα atoms in these complexes are concurrent with the RMSD values of spike protein-cellular ACE2, which range from 0.21 nm to 0.62 nm. This indicates their favorable binding with the spike protein during the simulation run time. The trajectories show the maximum RMSD values for the spike protein-BS-CAP and spike protein-B38-CAP protein complexes of 0.5 nm and 0.7 nm, respectively. Comparing these values, the lower RMSD value exhibited by the BS-CAP complex suggests that it will achieve high conformational stability during its interaction with the spike protein. The RMSF trajectories of Cα atoms of RBD of spike protein during its complex formation with cellular ACE2, BS-CAP, and B38-CAP protein are shown in Fig. 8(B). The trajectories show a maximum RMSF value of 0.9 nm and 0.5 nm for the spike protein-B38-CAP complex and spike protein-BS-CAP complex, respectively. The maximum RMSF value exhibited by the spike protein-B38-CAP complex is higher than the maximum RMSF value of the spike protein-ACE2 complex (0.7 nm); suggesting that B38-CAP protein will achieve more favorable interaction during its complex formation with the spike protein as its atoms show increased flexibility during the interaction. In spike protein-ACE2 complex, the fluctuations are exhibited by the amino acid residues Asn 334, Leu 335, Val 362, Thr 478, Pro 479, Cys 480, Asn 481, Ala 522, Thr 523, Pro 521, Gly 526, His 519, and Cys 361 (of spike protein); Pro 135, Asp 136, Asn 137, Pro 138, Gln 139, Glu 140, Asp 615 (of ACE2). In the case of spike protein-BS-CAP complex, the amino acid residues of spike protein Lys 202, Ala 203, Phe 204, Lys 317, Lys 318, and amino acid residues (Ile 47, Gln 49, Leu 50) of BS-CAP showed fluctuations. Further, in the spike protein-B38-CAP complex, the spike protein amino acid residues Phe 338, Leu 492, Gln 493, Leu 335, and B38-CAP amino acid residue Gln 337 exhibited fluctuations. Fig. 8(C) shows the Rg plots of Cα atoms of RBD of spike protein–protein (cellular ACE2, BS-CAP, and B38-CAP) complexes. The stable Rg values observed for all the protein–protein complexes indicate their compactness achieved over the simulation time. The range of Rg values of spike protein-cellular ACE2 (3.0 nm to 3.2 nm) and spike protein-B38-CAP (2.7 nm to 3.0 nm) complexes are much closer to each other depicting tight packing of atoms, and a slightly higher Rg range exhibited by spike protein-BS-CAP complex (3.5 nm to 3.7 nm) depicts less tight packing of atoms compared to the other two complexes.
Fig. 8.

RMSD (A), RMSF (B), and Rg (C) plots of Cα atoms of RBD of SARS-CoV-2 during its complex formation with cellular ACE2, BS-CAP, and B38-CAP proteins.
The binding free energy () values of the RBD of spike protein-HSPG, CMBS, and PAS ligand complexes calculated using the free energy perturbation method are shown in Table 6 .
Table 6.
Calculated binding free energies of protein–ligand complexes and interaction energies of protein–protein complexes.
| Spike protein–ligand complex | Calculated free energy of binding, () (kJ/mol) |
|---|---|
|
RBD-HSPG |
−181.5 ± 20.1 |
| RBD-CMBS | −163.6 ± 10.5 |
| RBD-PAS | −142.7 ± 25.1 |
| Spike protein–protein Complex | LJ-14 Interaction energy (kJ/mol) |
|
RBD-ACE2 |
−183.5 |
| RBD-BS-CAP | −167.5 |
| RBD-B38-CAP | −130.3 |
Comparison of the values indicate that the CMBS ligandinteracts more favorably with the RBD of spike protein (163.2 ± 10.5 kJ/mol) as that of the HSPG ligand (−181.5 ± 20.1 kJ/mol) followed by PAS ligand (−142.7 ± 25.1). The interaction energy values of the RBD of spike protein–protein (ACE-2, BS-CAP, B38-CAP) complexes calculated based on Lennard-Jones 1,4 (LJ-14) amino-acid interactions using the GROMACS tool are given in Table 6. The interaction energy values indicate that BS-CAP protein more favorably interacts with the spike protein (−167.5 kJ/mol) like ACE2 (−183.5 kJ/mol) followed by B38-CAP (-130.3 kJ/mol). The calculations of binding free energy for spike protein-(CMBS and PAS) ligands and interaction energy for the spike protein-BS-CAP and B38-CAP protein further proved their strong binding behavior in complex formation.
Thus the present in silico results suggests that the identified HSPG mimicking ligands, CMBS and PAS; and the bacterial-derived carboxypeptidase ACE2-like enzymes, BS-CAP and B38-CAP exhibit a strong binding affinity for the RBD of the spike protein of SARS-COV-2 and form well stable complexes with flexibility and high structure compactness. These bioactive molecules could effectively bind to SARS-CoV-2 and result in the competitive inhibition of the virus entry mediated by the cellular bound HSPG and ACE2. However, further in-vitro and in-vivo experiments are necessary to validate these results.
4. Conclusion
The search for novel compounds to inhibit SARS-CoV-2 infection remains a priority. In this work, we used a combined molecular docking and molecular dynamics simulation approach to identify exogenous bioactive moieties structurally mimicking cellular heparan sulfate proteoglycan (HSPG) and angiotensin-converting enzyme 2 (ACE2) which can be circulated in soluble form to achieve competitive inhibition of virus entry to the host cells mediated by cellular bound HSPG and ACE2. With this aim, we screened a total of twelve structurally mimicking HSPG ligands and three ACE2-like bacterially derived carboxypeptidase enzymes and docked them against the active binding sites (RBD) of the SARS-CoV-2 spike protein. Based on their docking results, we identified carboxymethyl benzyl amide sulfonate (CMBS) and polyanetholesulfonic acid (PAS) as HSPG mimetic ligands; whereas Paenibacillus sp. B38-derived carboxypeptidase (B38-CAP) and Bacillus subtilis-derived carboxypeptidase (BS-CAP) were identified as the ACE-2 like enzymes to have a strong binding with the spike protein as that of cellular HSPG and ACE2. To further understand the conformational behavior of these moieties with the spike protein, we carried out molecular dynamics (MD) simulations of the spike protein–ligand (cellular HSPG, CMBS, and PAS) and spike protein–protein (cellular ACE2, BS-CAP, and B38-CAP) complexes, and evaluated their RMSD, RMSF, and Rg trajectories. Analysis of the trajectories revealed that these moieties not only form well stable complexes (RMSD plots) but also indicated the increased flexibility of (i) spike protein towards the ligand and (ii) both spike protein and ACE2-like enzymes for complex formation (RMSF plots) and high structure compactness (Rg plots). Calculations related to the binding free energies for all spike protein–ligand complexes and interaction energies for all spike protein–protein complexes also proved their strong complex formation. Thus the present investigation reveals strong binding between the identified moieties and the spike protein, therefore, they could be used as potential decoy receptors to bind to SARS-CoV-2 to result in competitive inhibition of virus entry to the host cells mediated by cellular bound HSPG and ACE2. In-vitro and in-vivo experimentations could be be carried out further to verify and examine SARS-CoV-2 entry inhibition to the host cells in presence of these identified molecules. For effective SARS-CoV-2 inhibition, the identified moieties could be conjugated to the surfaces of non-toxic nanoparticles to provide multiple interactions, thus can serve as potential antiviral agents against COVID-19 disease.
CRediT authorship contribution statement
Sreya Sreekumar: Investigation, Methodology, Data Curation, Formal analysis, Validation, Writing – original draft. Abhaykumar M. Kuthe: Resources, Writing – review & editing. Satyendra Chandra Tripathi: Resources, Writing – review & editing. Ganesh C. Patil: Resources, Writing – review & editing. C. Ravikumar: Conceptualization, Methodology, Supervision, Visualization, Formal analysis, Validation, Writing – review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
The authors would like to thank Visvesvaraya National Institute of Technology (VNIT) Nagpur, India for providing the infrastructural facilities to carry out the research work. The authors also thank Dr. Ravi N. Methekar, Department of Chemical Engineering, VNIT Nagpur for providing us with the computational facility to perform MD simulations.
Funding information
The authors gratefully acknowledge the Ministry of Human Resource Development (MHRD), New Delhi for financial support to carry out the research.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molliq.2022.120566.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
The corresponding author can provide data to back up the findings of this study upon reasonable request.
References
- 1.Zhang, Xiang Q., Huo R., Zhou S., Jiang Y., Wang S., Yu Q.F. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Signal Transduct. Target Ther. 2021;6:233. doi: 10.1038/s41392-021-00653-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B.C., features of patients infected with, novel coronavirus in Wuhan, China. Lancet. 2019;395(2020):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gu J., Han B., Wang J. COVID-19: Gastrointestinal manifestations and potential fecal-oral transmission. Gastroenterology. 2020;158:1518–1519. doi: 10.1053/j.gastro.2020.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiao F., Tang M., Zheng X., Liu Y., Li X., Shan H. Evidence for Gastrointestinal Infection of SARS-CoV-2. Gastroenterology. 2020;158(2020):1831–1833.e3. doi: 10.1053/j.gastro.2020.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO Coronavirus (COVID-19) Dashboard, https://covid19.who.int/.
- 6.Ali A., Vijayan R. Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020;10:14214. doi: 10.1038/s41598-020-71188-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol.. 2020;94:e00127–e220. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jawad B., Adhikari P., Podgornik R., Ching W.Y. Key Interacting Residues between RBD of SARS-CoV-2 and ACE2 Receptor: Combination of Molecular Dynamics Simulation and Density Functional Calculation. J. Chem. Inf. Model. 2021;61:4425–4441. doi: 10.1021/acs.jcim.1c00560. [DOI] [PubMed] [Google Scholar]
- 9.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 10.Shang J., Ye G., Shi K., Wan Y., Luo C., Aihara H., Geng Q., Auerbach A., Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaiswal G., Kumar V., Abel S.M. In-silico design of a potential inhibitor of SARS-CoV-2 S protein. PLoS ONE. 2020;15(10):e0240004. doi: 10.1371/journal.pone.0240004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim C., Ryu D.K., Lee J., Kim Y.I., Seo J.M., Kim Y.G., Jeong J.H., Kim M., Kim J.I., Kim P., Bae J.S., Shim E.Y., Lee M.S., Kim M.S., Noh H., Park G.S., Park J.S., Son D., An Y., Lee J.N., Kwon K.S., Lee J.Y., Lee H., Yang J.S., Kim K.C., Kim S.S., Woo H.M., Kim J.W., Park M.S., Yu K.M., Kim S.M., Kim E.H., Park S.J., Jeong S.T., Yu C.H., Song Y., Gu S.H.O., Koo H., Hong B.S., Ryu J.J., Park C.M., Oh W.B., Choi M.D., Lee Y.K., S.y. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021;12:288. doi: 10.1038/s41467-020-20602-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verdecchia P., Cavallini C., Spanevello A., Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020;76:14–20. doi: 10.1016/j.ejim.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reza-Zaldívar E.E., Hernández-Sapiéns M.A., Minjarez B., Gómez-Pinedo U., Márquez-Aguirre A.L., Mateos-Díaz J.C., Matias-Guiu J., Canales-Aguirre A.A. Infection mechanism of SARS-COV-2 and its implication on the nervous system. Front. Immunol. 2021;11 doi: 10.3389/fimmu.2020.621735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrario C.M. Role of angiotensin II in cardiovascular disease therapeutic implications of more than a century of research. J. Renin. Angiotensin. Aldosterone. Syst. 2006;7:3–14. doi: 10.3317/jraas.2006.003. [DOI] [PubMed] [Google Scholar]
- 16.Milewska A., Zarebski M., Nowak P., Stozek K., Potempa J., Pyrc K., Perlman S. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014;88(22):13221–13230. doi: 10.1128/JVI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guimond S.E., Mycroft-West C.J., Gandhi N.S., Tree J.A., Le T.T., Spalluto C.M., Humbert M.V., Buttigieg K.R., Coombes N., Elmore M.J., Wand M., Nyström K., Said J., Setoh Y.X., Amarilla A.A., Modhiran N., Sng J.D.J., Chhabra M., Young P.R., Rawle D.J., Lima M.A., Yates E.A., Karlsson R., Miller R.L., Chen Y.-H., Bagdonaite I., Yang Z., Stewart J., Nguyen D., Laidlaw S., Hammond E., Dredge K., Wilkinson T.M.A., Watterson D., Khromykh A.A., Suhrbier A., Carroll M.W., Trybala E., Bergström T., Ferro V., Skidmore M.A., Turnbull J.E. Synthetic Heparan Sulfate Mimetic Pixatimod (PG545) Potently Inhibits SARS-CoV-2 by Disrupting the Spike–ACE2 Interaction. ACS Cent. Sci. 2022;8(5):527–545. doi: 10.1021/acscentsci.1c01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Pasquale V., Quiccione M.S., Tafuri S., Avallone L., Pavone L.M. Heparan sulfate proteoglycans in viral infection and treatment: Aspecial focus on SARS-CoV-2. Int. J. Mol. Sci. 2021;22:6574. doi: 10.3390/ijms22126574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madavaraju K., Koganti R., Volety I., Yadavalli T., Shukla D. Herpes simplex virus cell entry mechanisms: An update. Front. Cell. Infect. Microbiol. 2020;10 doi: 10.3389/fcimb.2020.617578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schowalter R.M., Pastrana D.V., Buck C.B., Imperiale M. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011;7(7):e1002161. doi: 10.1371/journal.ppat.1002161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feldman S.A., Audet S., Beeler J.A. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 2000;74:6442–6447. doi: 10.1128/JVI.74.14.6442-6447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiwari V., Clement C., Xu D.V., Yue N.T., Liu B.Y.J.T., Shukla J., D. Role for 3-O-sulfated heparan sulfate as the receptor for herpes simplex virus type 1 entry into primary human corneal fibroblasts. J. Virol. 2006;80:8970–8980. doi: 10.1128/JVI.00296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choudhary, S. Marquez, M. Alencastro, F. Sports, F. Zhao, Y. Tiwari, V. Herpes simplex virus type-1 (HSV-1) entry into human mesenchymal stem cells is heavily dependent on heparan sulfate. J. Biomed. Biotechnol. 2011 (2011) 264350, https://doi.org/10.1155/2011/264350 [DOI] [PMC free article] [PubMed]
- 24.Tiwari V., Tarbutton M., Shukla D. Diversity of heparan sulfate and HSV entry: basic understanding and treatment strategies. Molecules. 2015;20:2707–2727. doi: 10.3390/molecules20022707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akhtar J., Shukla D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009;276:7228–7236. doi: 10.1111/j.1742-4658.2009.07402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalra R.S., Kandimalla R. Engaging the spikes: heparan sulfate facilitates SARS-CoV-2 spike protein binding to ACE2 and potentiates viral infection. Signal Transduct. Target Ther. 2021;6:39. doi: 10.1038/s41392-021-00470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clausen T.M., Sandoval D.R., Spliid C.B., Pihl J., Perrett H.R., Painter C.D., Narayanan A., Majowicz S.A., Kwong E.M., McVicar R.N., Thacker B.E., Glass C.A., Yang Z., Torres J.L., Golden G.J., Bartels P.L., Porell R.N., Garretson A.F., Laubach L., Feldman J., Yin X., Pu Y., Hauser B.M., Caradonna T.M., Kellman B.P., Martino C., Gordts P.L.S.M., Chanda S.K., Schmidt A.G., Godula K., Leibel S.L., Jose J., Corbett K.D., Ward A.B., Carlin A.F., Esko J.D. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell J. 2020;183(4):1043–1057.e15. doi: 10.1016/j.cell.2020.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li F., Goff S.P. Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J. Virol. 2015;89(4):1954–1964. doi: 10.1128/JVI.02615-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C.C., Farzan H., M. Angiotensin-converting enzyme 2 is a functional receptor for the BSCAP coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minato T., Nirasawa S., Sato T., Yamaguchi T., Hoshizaki M., Inagaki T., Nakahara K., Yoshihashi T., Ozawa R., Yokota S., Natsui M., Koyota S., Yoshiya T., Yoshizawa-Kumagaye K., Motoyama S., Gotoh T., Nakaoka Y., Penninger J.M., Watanabe H., Imai Y., Takahashi S., Kuba K. B38-CAP is a bacteria-derived ACE2-like enzyme that suppresses hypertension and cardiac dysfunction. Nat. Commun. 2020;11(1) doi: 10.1038/s41467-020-14867-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cagno V., Andreozzi P., D’Alicarnasso M., Jacob Silva P., Mueller M., Galloux M., Le Goffic R., Jones S.T., Vallino M., Hodek J., Weber J., Sen S., Janeček E.-R., Bekdemir A., Sanavio B., Martinelli C., Donalisio M., Rameix Welti M.-A., Eleouet J.-F., Han Y., Kaiser L., Vukovic L., Tapparel C., Král P., Krol S., Lembo D., Stellacci F. Broad-spectrum non-toxic antiviral nanoparticles with a virucidal inhibition mechanism. Nature Mater. 2018;17(2):195–203. doi: 10.1038/nmat5053. [DOI] [PubMed] [Google Scholar]
- 32.Lang J., Yang N., Deng J., Liu K., Yang P., Zhang G., Jiang C., Geraghty R.J. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE. 2011;6(8):e23710. doi: 10.1371/journal.pone.0023710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paluck S.J., Nguyen T.H., Maynard H.D. Heparin-mimicking polymers: Synthesis and biological applications. Biomacromolecules. 2016;17:3417–3440. doi: 10.1021/acs.biomac.6b01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bugatti A., Urbinati C., Ravelli C., Clercq E.D., Liekens S., Rusnati M. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 tat and gp120 proteins. Antimicrob. Agents Chemother. 2007;51:2337–2345. doi: 10.1128/AAC.01362-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meng X.Y., Zhang H.X., Mezei M., Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011;7:146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett J. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976;22:245–268. doi: 10.1016/0021-9991(76)90078-4. [DOI] [Google Scholar]
- 37.Jarmoskaite, I. AlSadhan, I. Vaidyanathan, P.P. Herschlag, D. How to measure and evaluate binding affinities. eLife. 9 (2020) e57264, https://doi.org/10.7554/eLife.57264 [DOI] [PMC free article] [PubMed]
- 38.Kwofie S.K., Broni E., Asiedu S.O., Kwarko G.B., Dankwa B., Enninful K.S., Tiburu E.K., Wilson M.D. Cheminformatics-based identification of potential novel anti-SARS-CoV-2 natural compounds of African origin. Molecules. 2021;26:406. doi: 10.3390/molecules26020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lokhande K.B., Doiphod S., Vyas R., Swamy K.V. Molecular docking and simulation studies on SARS-CoV-2Mpro reveals Mitoxantrone, Leucovorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn. 2021;39:7294–7305. doi: 10.1080/07391102.2020.1805019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martínez L., Kleinjung J. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE. 2015;10(3):e0119264. doi: 10.1371/journal.pone.0119264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma J., Kumar Bhardwaj V., Singh R., Rajendran V., Purohit R., Kumar S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021;346:128933. doi: 10.1016/j.foodchem.2020.128933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pandey B., Grover A., Sharma P. Molecular dynamics simulations revealed structural differences among WRKY domain-DNA interaction in barley (Hordeum vulgare) BMC Genom. 2018;19:132. doi: 10.1186/s12864-018-4506-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nazar A., Abbas G., Azam S.S. Deciphering the inhibition mechanism of under trial Hsp90 inhibitors and their analogues: A comparative molecular dynamics simulation. J. Chem. Inf. Model. 2020;60(8):3812–3830. doi: 10.1021/acs.jcim.9b01134. [DOI] [PubMed] [Google Scholar]
- 44.Muhseen Z.T., Hameed A.R., Al-Hasani H.M.H., Tahir ul Qamar M., Li G. Promising terpenes as SARS-CoV-2 spike receptor-binding domain (RBD) attachment inhibitors to the human ACE2 receptor: Integrated computational approach. J. Mol. Liq. 2020;320:114493. doi: 10.1016/j.molliq.2020.114493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The corresponding author can provide data to back up the findings of this study upon reasonable request.