

Abstract
Cribriform appearance of the brain in Canavan disease is a rare finding. The two presented cases broaden the magnetic resonance imaging (MRI) phenotype wherein numerous oval, cystic structures, a few resembling dilated Virchow-Robin (VR) spaces, were noted in the centrum semiovale, periventricular, and lobar white matter producing a cribriform pattern. Besides, discrete round to oval cysts were present at the gray–white matter junctions in the second case, which were larger and appeared morphologically distinct from the VR spaces. These cysts did not elongate in any plane on imaging and were more representative of giant intramyelinic vacuoles. Genetic analysis revealed novel mutations in the aspartoacylase or ASPA gene that possibly accounts for the severe form of Canavan disease, which probably explains the imaging findings. The multicystic appearance of the white matter in Canavan disease is unusual and possibly represents two different histopathological substrates.
Keywords: Canavan disease, cribriform, novel mutation
Introduction
Canavan disease (CD) is an inborn error of metabolism caused due to the deficiency of the aspartoacylase (ASPA) enzyme. Deficiency of ASPA leads to accumulation of N -acetylaspartate (NAA), resulting in spongy degeneration of the brain. 1 Characteristic brain magnetic resonance imaging (MRI) features have been described in CD, which plays a major role in the diagnosis of the disorder. Recently three studies have described a multicystic appearance of the brain on MRI in patients with CD. 2 3 4 We describe here unusual brain imaging findings in two patients with CD with novel mutations of the ASPA gene and review the findings in the light of the previous reports.
Clinical Report
Case 1
A 14-month-old male child born of second-degree consanguineous parentage presented with delayed motor and language milestones. At presentation, the child had no neck holding, babbling, or response to light and sound. The two older siblings were apparently normal. Neurological examination revealed macrocephaly with a head circumference of 53 cm and low set ears. There was spasticity in bilateral lower limbs; however, his limb movements appeared unimpaired. The deep tendon reflexes were brisk in all the extremities, and bilateral plantar responses were extensor. Startle myoclonus was present. Abdominal examination revealed the presence of mild hepatosplenomegaly. In view of macrocephaly and organomegaly, a diagnosis of storage disorder was considered, and the patient was evaluated for the same. The routine biochemical and hematological tests were normal. Urine test for mucopolysaccharidosis was negative. The serum levels of hexosaminidase A and B levels did not reveal any abnormality.
Case 2
A 7-month-old girl born to healthy parents of a third-degree consanguineous marriage presented with developmental delay, especially in the motor domain. The maternal history was significant for first-trimester abortion in two instances. At presentation, the child had only partial head control. The child had not developed stranger anxiety or cooing. Clinical examination showed no evidence of dysmorphism, or neurocutaneous markers, or organomegaly. Neurological examination revealed a head circumference of 46 cm (85th percentile), increased tone in all four limbs, and spasticity. The child was not able to localize to sound and could track objects briefly. The plantar responses were extensor. Routine biochemical investigations were unremarkable. Blood examination with tandem mass spectrometry was negative.
Materials and Methods
Imaging
MRI brain was performed on a 3T Achieva, clinical scanner (Philips Medical Systems, The Netherlands) in case 1 and 1.5T Magnetom Aera scanner (Siemens Healthineers, Erlangen Germany) Axial T1WI, sagittal, coronal, and axial T2WI, axial fluid attenuated inversion recovery (FLAIR), diffusion-weighted imaging, magnetic resonance spectroscopy (MRS), and three-dimensional gradient-echo sequences were acquired in both cases. In addition, post-contrast magnetization prepared rapid gradient-echo (MPRAGE) images were acquired in the second case.
Mutation Analysis
Case 1
Genetic analysis of Case 1 DNA sample (extracted using peripheral venous blood by salting out method) was performed by Sanger sequencing of the coding region of ASPA gene using six primer pairs for exons (Exon1-Exon6) in both directions. Primers were designed by Primer3 software ( http://primer3.ut.ee/ ). All except Exon2 could be sequenced successfully. A healthy control DNA sample was also analyzed for comparison.
Case 2
DNA extraction from peripheral blood was performed using the Qiagen kit method. Polymerase chain reaction amplification of all coding exons of the ASPA gene, including exon-intron boundaries, was performed. Cycle sequencing was performed using forward and reverse primers. The amplicons were run in ABI3500xl. The electropherogram, thus generated, was analyzed.
Results
Imaging
Case1
Brain MRI revealed diffuse T2 and FLAIR hyperintensities in the subcortical and deep white matter. The posterior limbs of internal capsules and cerebellar white matter were spared. The corpus callosum was dysgenetic, and bilateral middle cerebellar peduncles were seen to exhibit abnormal signals. Symmetrical T2 hyperintensities were evident in the brainstem, bilateral globus pallidus, thalami, and dentate nuclei. Diffusion restriction was apparent in the affected white matter structures and dentate nuclei. The affected supratentorial white matter simulated a cribriform appearance with multiple interspersed oblong and round relatively T2 hyperintense lesions. The lesions were of varying sizes and demonstrated inversion on FLAIR, indicating their cystic nature( Fig. 1A , 1B ). Cysts were observed in the centrum semiovale, gray–white matter junctions, and lobar white matter of the temporal and occipital lobes. The cysts located in the centrum semiovale and lobar white matter demonstrated an elongating radiating pattern. Interestingly some of the cysts located at the gray–white matter interface were large and did not exhibit the elongated morphology ( Fig. 1C , 1D , 1E , 1F , 1G ). Multivoxel proton MRS performed at TE = 144 milliseconds revealed an elevated NAA peak with an NAA: Creatine ratio of 3.26 ( Fig. 1H ). The imaging diagnosis of CD was made, and the patient underwent urine examination for organic acids, which revealed a 27.19-fold elevation in NAA, confirming the diagnosis of CD.
Fig. 1.
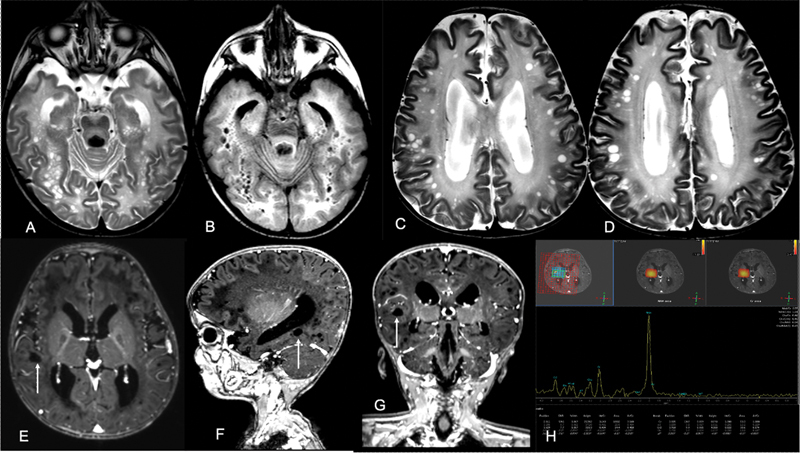
Axial T2 WI ( A ) shows multiple small cysts in the lobar white matter of bilateral cerebral hemispheres in the background of diffuse white matter hyperintensities. Axial FLAIR image ( B ) shows suppression of signals from these cystic spaces. Axial T2WI ( C, D ) showing multiple oval to round discrete cyst in the subcortical location. Axial, sagittal, and coronal T1 post contrast MPRAGE images ( E–G ) show these cystic spaces ( arrows ) as nonelongating lesions without any traversing vessels. MRS image ( H ) shows tall NAA peak within the white matter. FLAIR, Fluid attenuated inversion recovery; MPRAGE, magnetization prepared rapid gradient-echo; MRS, magnetic resonance spectroscopy; NAA, N -acetylaspartate.
Case 2
Brain imaging revealed a diffuse T2/FLAIR hyperintense signal in the subcortical and deep white matter. A relative sparing of the bilateral posterior limb of the internal capsule was seen, while the cerebellar white matter was involved. Symmetric involvement of the bilateral deep gray nuclei and diffuse involvement of the brainstem and the corpus callosum was observed. These regions appeared hypointense on T1-weighted imaging with no abnormal post-contrast enhancement. Diffusion restriction was evident in the supratentorial lobar white matter. No foci of blooming/hemorrhage were identified on susceptibility-weighted imaging. The affected periventricular and lobar white matter had a characteristic appearance rendered by variable-sized, ovoid-cystic structures that were differentially T2 hyperintense (compared with the lobar white matter) and demonstrated inversion on FLAIR ( Fig. 2A , 2B , 2C ). These abnormal structures exhibited a radiating, elongated morphology, and a few of the larger cysts showed a linear enhancing structure in the center, consistent with a vessel ( Fig. 2D , 2E ). The resultant cribriform pattern was akin to the distribution of dilated Virchow-Robin (VR) spaces. Confluence and coalescence of these structures were striking in the occipital lobes. Multivoxel proton spectroscopy performed at intermediate echo time (TE = 144 milliseconds) showed an elevated NAA resonance with an NAA: Creatine ratio of 3.89 ( Fig. 2F ). An imaging diagnosis of CD was offered, following which the patient underwent urine examination for organic acids, that showed a 22.6-fold elevation in NAA, confirming the diagnosis of CD. The patient thereafter underwent genetic testing.
Fig. 2.
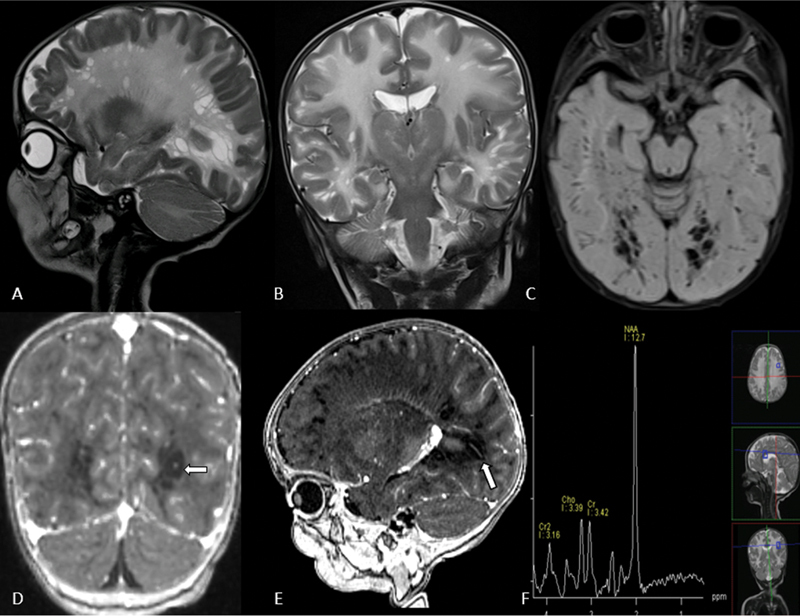
Sagittal and coronal T2 WI ( A,B ) showing multiple enlarged Virchow-Robin spaces in the subcortical and periventricular white matter of bilateral cerebral hemispheres along with diffuse white matter signal changes. Axial FLAIR image ( C ) shows suppression of signals of these cystic spaces. Coronal and sagittal T1 post-contrast MPRAGE images ( D, E ) show vessels traversing ( arrows ) these cystic spaces. MRS image ( F ) shows significantly elevated NAA peak within the white matter. FLAIR, fluid attenuated inversion recovery; MPRAGE, magnetization prepared rapid gradient-echo; MRS, magnetic resonance spectroscopy; NAA, N -acetylaspartate.
Mutation Analysis
Case 1
The analysis of sequencing traces of the exonic region showed a novel 8 base pair deletion c.446_453delCACTACCC in Exon3 of ASPA in the homozygous state in Case 1 ( Fig. 3 ). None of the family members were available for the genetic analysis; however, the variation has not been reported in any public database of 1000 Genome project data and ExAC (Exome Aggregation Consortium). In addition, the variation was not observed in 100 in-house exome data of healthy controls. The deletion is predicted to cause premature truncation of ASPA protein by frameshift of open reading frame; p.P149Lfs*7 and generation of stop codon after 154 amino acid residue thus shortening the protein length by 50% (full-length protein of 313 amino acid).
Fig. 3.
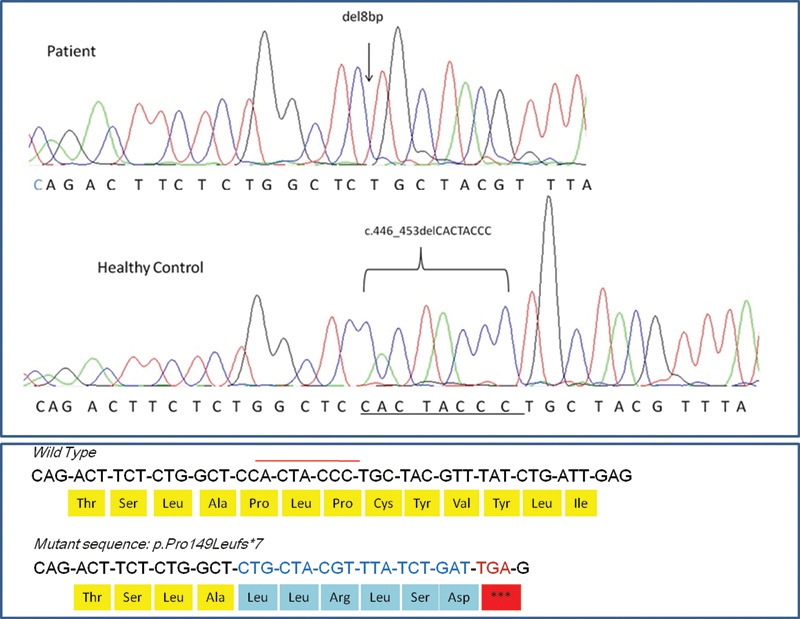
Representative electropherogram of Sanger sequencing of exon3 of ASPA gene showing 8 base-pair deletion (CACTACCC). The upper panel shows homozygous deletion in patient's DNA sample. Middle panel shows wild-type homozygous sequence from a representative healthy control. Lower panel shows the change at protein level, producing premature truncation at 155 amino acid in comparison with wild type 314 amino acid residue sequences. ASPA, aspartoacylase.
Case 2
The analysis of the Exon1 of the ASPA gene revealed c.188G > A corresponding to p.Arg63Lys at the genomic co-ordinate chr17:3379641G > A. This was a homozygous missense variation in Exon 1 that results in the amino acid substitution of Lysine for Arginine at codon 63 (p.Arg63Lys). This variant has not hitherto been reported in the GenomeAsia, gnomAD, NHLBI-ESP, and 1000Genome, and ExAC databases and has a minor allele frequency at 0.005% within the internal database. In-silico predictions of this variant were considered to be “likely-pathogenic” and probably damaging by PolyPhen-2 (HumDiv) and damaging by sorting tolerant from intolerant (SIFT), likelihood ratio test (LRT), as well as disease causing by MutationTaster2. The variant lies in the succinyl glutamate desuccinylase/aspartoacylase family domain of the ASPA protein, and the reference codon/native amino acid is conserved across species. On segregation analysis, the observed variant was noted in a heterozygous state among the unaffected parents. Collating all the evidence, the mutation was considered “likely-pathogenic”.
Discussion
CD is an autosomal recessive neurometabolic disorder characterized by swelling and spongy degeneration of the brain white matter. 5 Myelin loss with myelin splitting and intramyelinic vacuole formation are the characteristic pathological findings.
Diagnosis is supported by elevated levels of NAA in the urine. The absence of ASPA activity in cultured skin fibroblasts and mutation of the ASPA gene are confirmatory.
Imaging findings on MRI in conjunction with MRS, is virtually diagnostic. The imaging pattern in CD includes diffuse involvement of the supratentorial white matter, including subcortical and deep white matter. Globus pallidi, thalami, and dentate nuclei are commonly involved. Brain stem and cerebellar white matter affliction are less severe. 6 7 The absence of caudate nuclei and putaminal involvement is characteristic; however, there are sporadic reports of caudate and putaminal involvement in CD. 8 Proton MRS evaluation of the abnormal white matter reveals elevated NAA, a CD signature. 9 A metabolite at 2.2 ppm ( N -acetylneuraminic acid) masquerading as NAA has been described in Salla disease, a hypomyelinating disorder with routine imaging findings unlike that of CD.
The MRI features in our cases were consistent with CD but for the additional finding of multiple cysts. The supratentorial white matter cysts were diffusely distributed, though they were more numerous in the centrum semiovale, lobar white matter and gray–white matter junctions in case 1, and periventricular and lobar white matter in case 2. The cysts were of varying sizes, and those located in the centrum semiovale, periventricular regions, and lobar white matter exhibited an oblong radiating pattern. Sharp and linear enhancing structures were traversing through a few of the larger cysts in case 2, reminiscent of vessels. In view of the above findings, the location and morphology, the cysts were identified as dilated perivascular VR spaces. VR spaces are multilayered, pial-lined, fluid-filled, interstitial spaces that envelope perforating vessels that assume a linear, radiating morphology on imaging. The honeycomb appearance of the white matter due to dilated VR spaces has been reported in CD. 2 However, the appearance of the cysts described by the authors differed in certain aspects from the cysts in the index cases. The cysts described by them were present in the centrum semiovale, periventricular white matter and were more localized than the cysts in our cases. In addition, they were smaller, less numerous, and nearly identical in size. The authors hypothesized that the cystic appearance could be due to leukodystrophy or postnatal ischemic events. We believe that the cysts represent overtly dilated VR spaces and are secondary to myelin loss, as corroborated by histopathological studies. 10
In case 1, in addition to the cysts in the centrum semiovale and lobar white matter, there were large cysts at the gray − white matter interface. A few of the cysts in case 1 exhibited distinct morphology with nonelongating patterns in at least two cardinal imaging planes, possibly mirroring true giant intramyelinic vacuoles considering that the subcortical white matter and deeper layers of the cortex are the earliest and most severely affected in CD. 11 Macroscopic vacuolation is seen in the cut-section of affected brains of CD. Electron microscopy of these white matter vacuoles reveals separation of lamellae of myelin and intramyelinic vacuoles. The intramyelinic vacuoles evident on histopathology correspond to similar spatial (subcortical) distribution as observed in our cases. Castillo has hypothesized that when sufficiently large, the intramyelinic vacuoles are seen on imaging in CD. 12 The cysts' location at the gray − white matter interface and their discrete nonradiating appearance made us speculate that they could represent intramyelinic vacuoles. A proliferation of swollen astroglia is a well-described neuropathological observation in CD, especially in the cortex. Similar cells are also seen to a lesser extent in the subcortical white matter. The ascribed astrocytic population is cortically predominant; however, its contribution to the described imaging phenotype cannot be completely negated. 11
Recently a multicystic-beaded and spongy white matter on MRI has been described in two cases of CD. 3 4 The cysts described by Kamate et al are quite similar to our case 2, wherein the cysts in the periventricular white matter and centrum semiovale were numerous and in multiple layers and appearing to elongate. These are more likely dilated VR spaces. 4 The cysts described by Drenckhahn et al had a periventricular and subcortical location. 3 The larger discrete, nonelongating cysts in the subcortical junctions probably represent the MRI equivalent of giant intramyelinic vacuoles. The cysts observed in case 1 in our study were similar to those observed by Drenckhahn et al except that those in case 1 were more numerous and larger in dimension.
Genetic analysis in the index cases revealed novel mutations in CD. The deletion mutations with premature truncations like the one in case 1 have been reported to cause complete loss of ASPA activity in vitro. 13 Genetic findings in our patient are in accordance with prior reports of observation of total loss of activity of ASPA in the cultured fibroblasts, thus substantiating its deterministic pathogenic role. Case 2 represents a scenario wherein the mutation was deemed “likely pathological,” considering its novel status. The pathological evidence of the severity of the disease in case1 is demonstrated by the appearance of MRI-visible macroscopic cysts representing vacuole formation due to intramyelinic edema seen in severe variants of CD and dilated VR spaces in case 1 and 2 representing severe myelin loss. In view of the above neuroimaging and genetic findings, we propose that novel mutations may, at least in part, account for the unusual imaging phenotypes. The probability that other unidentified, extraneous exacerbating factors contribute to this novel imaging phenotype cannot be entirely ruled out.
Conclusion
Cribriform appearance of the brain on MRI is an unusual imaging phenotype in CD. It represents either enlarged VR spaces, or a combination of dilated VR spaces and giant intramyelinic vacuoles. In the reported cases, it may result from c.446_453delCACTACCC mutation or homozygous missense variation in exon 1 (chr17: g.3379641G > A) in the ASPA gene. This warrants further genetic and histological evaluation in a larger number of subjects. CD should be included in the imaging differential diagnosis of neurometabolic disorders, demonstrating the supratentorial white matter's multicystic appearance.
Footnotes
Conflict of Interest None declared.
References
- 1.Matalon R, Michals K, Sebesta D, Deanching M, Gashkoff P, Casanova J. Aspartoacylase deficiency and N -acetylaspartic aciduria in patients with Canavan disease . Am J Med Genet. 1988;29(02):463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- 2.Pradhan S, Goyal G. Teaching neuroimages: honey-comb appearance of the brain in a patient with Canavan disease. Neurology. 2011;76:e68. doi: 10.1212/WNL.0b013e318212a887. [DOI] [PubMed] [Google Scholar]
- 3.Drenckhahn A, Schuelke M, Knierim E. Leukodystrophy with multiple beaded periventricular cysts: unusual cranial MRI results in Canavan disease. J Inherit Metab Dis. 2015;38(05):983–984. doi: 10.1007/s10545-015-9812-1. [DOI] [PubMed] [Google Scholar]
- 4.Kamate M, Kabate V, Malhotra M. Spongy white matter: a novel neuroimaging finding in Canavan disease. Pediatr Neurol. 2016;56:92–93. doi: 10.1016/j.pediatrneurol.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Matalon R M, Michals-Matalon K. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Front Biosci. 2000;5:D307–D311. doi: 10.2741/matalon. [DOI] [PubMed] [Google Scholar]
- 6.Brismar J, Brismar G, Gascon G, Ozand P. Canavan disease: CT and MR imaging of the brain. AJNR Am J Neuroradiol. 1990;11(04):805–810. [PMC free article] [PubMed] [Google Scholar]
- 7.Matalon R M, Michals-Matalon K, Kaul R, Mafee M. Spongy degeneration of the brain, Canavan disease: biochemical and molecular findings. Front Biosci. 2000;5:D307–D311. doi: 10.2741/matalon. [DOI] [PubMed] [Google Scholar]
- 8.Toft P B, Geiss-Holtorff R, Rolland M O. Magnetic resonance imaging in juvenile Canavan disease. Eur J Pediatr. 1993;152(09):750–753. doi: 10.1007/BF01953994. [DOI] [PubMed] [Google Scholar]
- 9.Karimzadeh P, Jafari N, Nejad Biglari H. The clinical features and diagnosis of Canavan's disease: a case series of Iranian patients. Iran J Child Neurol. 2014;8(04):66–71. [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo A, Suzuki K. The blood brain barrier in human leukodystrophies and allied diseases. Ultrastructural and morphometric studies on the capillaries in brain biopsies. Clin Neuropathol. 1993;12(03):169–174. [PubMed] [Google Scholar]
- 11.van der Knaap M S, Valk J. Berlin Heidelberg: Springer-Verlag; 2005. Canavan Disease. In: Magnetic resonance of myelin, myelination and myelin disorders; pp. 326–327. [Google Scholar]
- 12.Castillo M. Philadelphia: Lippincott Williams & Wilkins; 2006. Neuroradiology Companion: Methods, Guidelines, and Imaging Fundamentals; p. 177. [Google Scholar]
- 13.Kaul R, Gao G P, Matalon R. Identification and expression of eight novel mutations among non-Jewish patients with Canavan disease. Am J Hum Genet. 1996;59(01):95–102. [PMC free article] [PubMed] [Google Scholar]