

Abstract
In recent years, rapid detection methods such as polymerase chain reaction (PCR) and quantitative real-time PCR (qPCR) have been continuously developed to improve the detection of food-borne pathogens in food samples. The recent developments of PCR and qPCR in the detection and identification of these food-borne pathogens are described and elaborated throughout this review. Specifically, further developments and improvements of qPCR are discussed in detecting Salmonella and norovirus. Promising advances in these molecular detection methods have been widely used to prevent human food-borne illnesses and death caused by the food-borne pathogens. In addition, this review presents the limitations and challenges of the detection methods which include conventional culture method and conventional PCR method in detecting Salmonella and norovirus. Furthermore, several advances of qPCR such as viability PCR (vPCR) and digital PCR (dPCR) have been discussed in the detection of Salmonella and norovirus. Good practice of analysis of the food-borne pathogens and other contaminants in the food industry as well as the advancement of molecular detection methods will help improve and ensure food safety and food quality.
Keywords: Polymerase chain reaction, Real-time PCR, Food-borne bacteria, Food-borne viruses, Salmonella, Norovirus, Food safety
Introduction
Food-borne pathogens which include bacteria, viruses, fungi and parasites, are causing human illnesses and outbreaks which can lead to death. Most food-borne illnesses are caused by pathogens such Salmonella and norovirus (NoV) (Koopmans and Duizer 2004). Other common food-borne illnesses are caused by toxins that are released by pathogens, toxic chemicals, natural contaminants and some unspecified agents. Common symptoms of these illnesses include diarrhea, vomiting, abdominal pain and fever. However, life-threatening food-borne illnesses can cause neurologic, hepatic and renal diseases and some, if not treated, can lead to death (Mead et al. 1999). The typical route for most food-borne illnesses is fecal–oral transmission through consumption of contaminated food and/or water. Other routes include person-to-person contact and contaminated surfaces. The growing concern of food-borne illnesses and outbreaks has implemented for better control of food safety and supply by preventing food-borne illnesses, such as salmonellosis, in processes of food handling, processing, storage arrangements, packaging and distribution of the produce.
Globalization, population movement and supply chain introduce pathogens to different regions causing emerging infections. Approximately 600 million people—1 in 10 people worldwide, have food-borne illness due to the consumption of contaminated food and 420,000 people die annually (WHO 2020). Food-borne bacteria and viruses, such as Salmonella and NoV, respectively, are some of the most common food-borne pathogens that are known to cause food-borne illnesses whereby Salmonella nontyphoidal spp. and NoV cause an estimated 1,027,561 and 5,461,731 number of illnesses, respectively (Scallan et al. 2011). The severity and incidence rate have increased for food-borne illnesses despite well-established preventive measures and improved food safety to prevent food-borne illnesses. Thus, rapid and sensitive detection methods for food-borne pathogens in food products are of great importance to the food industry as well as in preventing and containing outbreaks and cases of bioterrorism.
The introduction of PCR has aided the reduction of food-borne illness throughout the years with improved early detection and better sensitivity. These improvements of the molecular detection methods are critical to make the detection of the food-borne pathogens more reliable and sensitive by reducing the time consumption for rapid data acquisition. Other detection methods such as recombinase polymerase amplification (RPA) (Rostron et al. 2019), loop-mediated isothermal amplification (LAMP) (Zou et al. 2020) and electrochemiluminescence (ECL)-based detection (J. Shen et al. 2019) have also been used to detect pathogens. Real-time PCR (rt-PCR), also known as quantitative real-time PCR (qPCR), is the advanced method of the conventional PCR in which targeted DNA is amplified and detected in real-time with quantification of the targeted genomic sequence. This detection method has been widely used in many diagnostic areas which includes analysis of food-borne microbes. In this review, the use of PCR-based methods for the detection and identification of Salmonella and NoV will be focused on.
Salmonella
Salmonella, a Gram-negative and flagellated anaerobic bacillus, is one of the most common food-borne bacteria which causes salmonellosis. The structure of Salmonella can be seen in Fig. 1 represented in a schematic diagram. Initially, sample collection is obtained and sample will be prepared for the real-time PCR method. The sample preparation of the food-borne pathogens, such as Salmonella, will undergo extraction in which the genetic material, DNA, found in the cytoplasm of the bacteria is being isolated for PCR amplification using specific thermal cycler that will amplify and make millions of copies of the DNA (Fig. 1). Salmonellosis is found in the gastrointestinal tract of an individual infected with Salmonella. Most infections are caused by the ingestion of contaminated food and water as Salmonella can be found frequently in fresh produce, raw food products and unpasteurized milk. Most infected individuals would usually recover from diarrhea, vomiting and abdominal cramps without treatment; however, infected individuals with an impaired immune system, infants and elderly require medical treatment as they are more prone to infection. Salmonellosis has a number of syndromes whereby different serovars show a specific syndrome which include gastroenteritis, septicemia, enteric fevers, focal infections and asymptomatic carrier state (Giannella 1996). In order for the Salmonella bacteria to be fully pathogenic, In order for the Salmonella are virulence factors that the bacteria must possess. These virulence factors include (1) complete lipopolysaccharide coat, (2) ability of cell invasion, (3) ability of intracellular replication and (4) toxin(s) production (Giannella 1996). Firstly, the bacteria colonise the ileum and colon after entering the body by binding on to a specific receptor on the surface of the epithelial cell membrane. The bacteria will then invade the epithelial cells of the ileum and a series of acute inflammatory response will occur which induces proliferation of the Salmonella bacteria within the follicles of the epithelium and lymphoid cells (Giannella 1996). Moreover, Salmonella has the ability to secrete one or more enterotoxin-like substances in which could influence Salmonella pathogenesis and diarrhea (Giannella 1996). Salmonella genus includes 2 species—Salmonella enterica and Salmonella bongori. There are 6 subspecies for S. enterica—Enterica, Salamae, Arizonae, Diarizonae, Houtenae and Indica. S. bongori and S. enterica subsp. Enterica were found to be pathogenic to humans in reported cases of salmonellosis. Moreover, Salmonella has more than 2500 serovars that are characterised according to O, H and Vi antigens found on the somatic and flagella regions of Salmonella. The diversity of Salmonella serotypes has been an issue as a causative factor of salmonellosis in humans (Merino et al. 2017) which may vary geographically and with time.
Fig. 1.
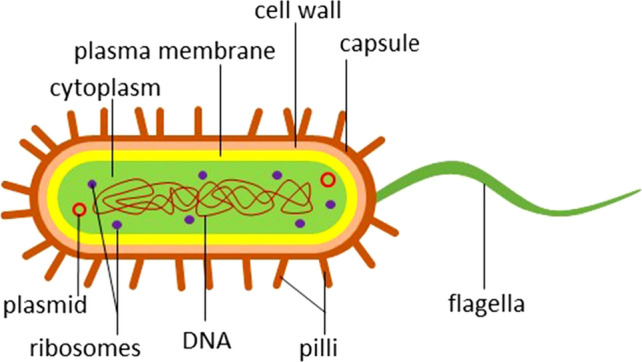
Schematic diagram representation of Salmonella particle
Norovirus
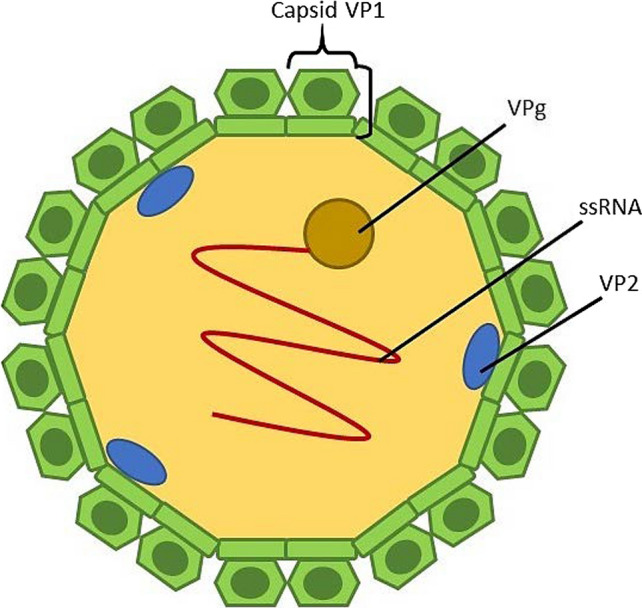
Norovirus, a non-enveloped, positive-sense single-stranded RNA (ssRNA) virus, is an enteric pathogen with the leading cause of sporadic gastroenteritis in most developing and developed countries (Koopmans and Duizer 2004). United States has an estimation of 60% of acute gastroenteritis caused by NoV annually (Robilotti et al. 2015). NoV genome is a linear ssRNA of approximately 7.5–7.7 kb in length with viral protein genome (VPg) attached at the 5’-end by covalent bonds and poly-(A)-tail at the 3’-end (Robilotti et al. 2015). The genome is organised into three open reading frames (ORFs), ORF1 to ORF3 (Fig. 2). ORF1 encodes a large nonstructural polyprotein that is cleaved into six nonstructural proteins by proteolysis. These six nonstructural proteins include N-terminal p48 protein, NTPase, 3A-like p22 protein, viral genome-linked VPg, 3C-like protease 3CL and RNA-dependent RNA polymerase RdRp (Cotten et al. 2014). ORF2 encodes major capsid protein VP1 which overlaps ORF1, and ORF3 encodes minor capsid protein VP2 and is located at the 3’-end of the viral genome. NoVs are classified into seven genogroups (GI–GVII) with each genogroup being characterised into genotypes based on the phylogenetic clusters of VP1 amino acid sequence (Vinjé 2015). NoV GI, GII and GIV are known to cause human outbreaks, GIV NoVs mostly cause epidemic and sporadic disease (Vinjé 2015). Risk assessment analysis that is related to the food-borne and waterborne viruses is required for the food industry to prevent outbreaks of these illnesses and improve food safety.
Fig. 2.

Schematic representation of the genome organization of human NoV. The NoV genome is organised into three open reading frames
The main mode of transmission of NoV is through fecal–oral transmission by the consumption of contaminated food or water. NoV can also be transmitted through aerolised particles from infected person, contaminated surfaces and environmental surroundings (Matthews et al. 2012). Furthermore, NoV can be transmitted at a low infectious dose and with environmental persistence (Rooney et al. 2014), causing symptoms which include diarrhea, vomiting, stomach pain and nausea. NoV infections are caused by the binding of the virus to histo-blood group antigens (HBGAs) in human host cells and these NoVs will bind to specific HBGAs with particular specificities according to their genotypes and genogroups (Donaldson et al. 2010). After the viral attachment, the virus will enter the human cell and uncoating of the viral particle will occur in which the positive-sense RNA will be released into the cell cytoplasm. Translation of the viral RNA will then occur and followed by post-translation cleavage of the large polyprotein ORF1 into smaller viral proteins by the virus-encoded protease (Pro). Genome replication of the viral positive-sense RNA into negative-sense RNA will occur and this will be used as a template for synthesizing new viral positive-sense RNA. Viral capsids and proteins will be assembled to form new virus particles which will be released out of the cell.
Challenges to the detection and identification of Salmonella and Noroviruses
Conventional culture method of detection
The conventional culture method is a traditional microbiological isolation technique used to detect pathogenic bacteria. The process of enrichment and confirmation of positive pathogen colonies by biochemical and serological tests is time-consuming and labour-intensive. Specifically, for Salmonella, would take up to nine days to produce results (Andrews and Hammack 2020). Thus, conventional culture method is not rapidly available to give results in a food-borne illness outbreak or bioterrorism events (K. M. Lee et al. 2015). Thus, due to the time consumption to generate results, conventional PCR was then used widely, which produced rapid and more reliable results within 24 h with higher sensitivity and specificity. Conventional culture methods are less sensitive than PCR-based methods (Table 1) in addition with pre-enrichment step such as with buffered peptone water (BPW) and lactose broth (LB), which have been found to successfully increase the sensitivity of detection of food-borne bacteria such as Salmonella spp. (Kumar et al. 2008). The ratio of positive reactions and sensitivity (%) of the samples were tested and then compared between conventional culture method and qPCR method whereby most samples show a higher sensitivity when using the qPCR method compared to the conventional method (Table 1). However, results for squid, cuttlefish and octopus (Kumar et al. 2008) have shown that conventional culture method has higher sensitivity by twofold than qPCR method and the sensitivity for fish meat (Gwida and Al-Ashmawy 2014) as well as drag swab (Eyigor et al. 2002) were equal for both methods.
Table 1.
The sensitivity of conventional culture and PCR-based methods, such as qPCR, in the detection of Salmonella spp.
| Sample type (no. of samples) | Positivity for detection of Salmonella | |||
|---|---|---|---|---|
| Target gene | Conventional culture method | qPCR method | Reference | |
| Cooked food (n = 150) | invA gene | 0/150 (0%) | 32/150 (21.3%) | Siala et al. 2017 |
| Milk (n = 93) | 6/93 (6.4%) | 31/93 (33.3%) | ||
| Fresh fruit and vegetables (n = 70) | 1/70 (1.4%) | 9/70 (12.8%) | ||
| Seafood (n = 46) | 6/46 (13%) | 11/46 (23.9%) | ||
| Raw poultry meat (n = 45) | 8/45 (17.8%) | 27/45 (60%) | ||
| Cakes (n = 41) | 0/41 (0%) | 11/41 (26.8%) | ||
| Dairy products (n = 22) | 0/22 (0%) | 5/22 (22.7%) | ||
| Charcuterie products (n = 13) | 0/20 (0%) | 5/20(25%) | ||
| Raw red meat (n = 13) | 4/13 (30.7%) | 5/13 (38.5%) | ||
| Total (n = 500) | 25/500 (5%) | 136/500 (27.2%) | ||
| Fish (n = 83) | invA gene | 20/83 (24%) | 30/83 (36.1%) | Kumar et al. 2008 |
| Shrimp (n = 58) | 11/58 (18.9%) | 20/58 (34.4%) | ||
| Crab, clam, mussel, oyster (n = 42) | 9/42 (21.4%) | 15/42 (35.7%) | ||
| Squid, cuttlefish, octopus (n = 32) | 6/32 (18.7%) | 3/32 (9.3%) | ||
| Total (n = 215) | 46/215 (21.3%) | 68/215 (31.6%) | ||
| Milk and dairy products (n = 200) | ttr gene | 24/200 (12%) | 42/200 (21%) | Gwida and Al-Ashmawy 2014 |
| Beef meat (n = 64) | invA gene | 15/64 (23.4%) | 16/64 (25%) | |
| Chicken Meat (n = 80) | 15/80 (18.6%) | 17/80 (21.3%) | ||
| Fish Meat (n = 6) | 2/6 (33.3%) | 2/6 (33.3%) | ||
| Total (n = 150) | 32/150 (21.3%) | 35/150 (33.3%) | ||
| Intestinal homogenate (n = 492) | invA gene | 32/492 (6.5%) | 62/492 (12.6%) | Eyigor et al. 2002 |
| Drag swab (n = 27) | 3/27 (11.1%) | 3/27 (11.1%) | ||
| Total (n = 519) | 35/519 (6.8%) | 65/519 (12.5%) | ||
On the other hand, the culture of NoV has been attempted throughout the years, however, there is still lack of research development due to the inadequacy of propagating the virus in-vitro (Elizaquível et al. 2014). Some studies have included B cells which were used as cell lines for the human NoV cultivation and the studies have successfully replicated and increased the number of viral genome (Jones et al. 2015). The cultivation of human NoV has to be studied more as there are still challenges in generating good results which is caused by the lack of viral source. Thus, due to the fact that human NoVs are not able to be cultivated, the traditional detection methods are electron microscopy and immunological tests such as enzyme immunoassay (EIA) and lateral-flow immunochromatographic assay (Stals et al. 2012) (Robilotti et al. 2015). However, electron microscopy could cause false negative or false positives when not properly analysed and the method could be time-consuming as well as labour-intensive (Louten 2016).
PCR-based methods
PCR-based methods, such as conventional PCR, qPCR and dPCR, offer fast, highly specific and sensitive detection of food-borne pathogens. PCR-based methods have the ability to amplify a specific target sequence of DNA or RNA in-vitro from a small amount of target sequence. Over the years, combined techniques, such as PCR-ELISA (Sue et al. 2014), have also been developed to lower the number of food safety cases and thus, further development of these novel detection techniques should include (1) detection of multiple food-borne pathogens simultaneously in real-time, (2) good and improved sensitivity, specificity, reproducibility and convenience and (3) high potential in application for future commercial marketing in which can be used worldwide (Hu et al. 2018).
One of the challenges for PCR-based method is the detection of new and unreported pathogens due to no sequence information available in the database, GenBank (Rodríguez-Lázaro and Hernández 2013). Moreover, improving and developing a more sensitive and reliable molecular detection method is required to help overcome the dynamic nature of NoV genetic diversity. The evolutionary pattern of NoV strains which causes genetic diversity is caused by the emergence of new GII.4 NoV strains every 2–3 years, thus, the new genomic NoV strain is being replaced by the previous circulating NoV strain that have caused the outbreak (Cotten et al. 2014) (Chen et al. 2015). Moreover, the genetic diversity of NoV have caused primer designing to be more challenging as PCR requires specific tools and factors to successfully amplify specific target sequence with good quality and quantity.
Conventional PCR in detecting Salmonella and Norovirus
Conventional PCR or end-point PCR is a traditional molecular method that has been widely used to detect food-borne pathogens. A study has compared the use of conventional culture method with PCR for the isolation and identification of Salmonella in food samples such as beef and chicken meat in which the culture method has resulted in 21.3% of the total number of samples in a total of seven days to obtain results while PCR showed 23.3% in which the sensitivity and specificity were 100% and 97.5%, respectively, within a total of 12 h detection time from receiving the samples (Ahmed 2014). PCR amplification by conventional PCR is the amplification of a single specific target sequence to produce an exponential increase of the target sequence that uses relative quantification with intercalating dyes as it is inexpensive and simple to perform.
PCR amplification method involves repeated cycles of heating and cooling, and each PCR cycle will go through three phases—denaturation, annealing and extension. Initially, the hydrogen bonds of the double-stranded DNA (dsDNA) break at 94–97 °C, resulting in dsDNA to uncoil and separate, followed by annealing of primer pairs to complementary target sequence on the new single-stranded DNA (ssDNA) at a lower temperature. Free deoxynucleoside triphosphates (dNTPs) will then bind to the 3′-end of the primer by DNA polymerase at a temperature of 72–78 °C. After the amplification of the target sequence, the PCR product, will be tested by gel electrophoresis in 2% agarose gel. This method is to confirm the presence of the targeted food-borne pathogen in the sample.
Conventional PCR has also been used to detect NoVs in food, water and environmental matrices successfully with the use of conventional reverse transcriptase PCR (RT-PCR). There are a number of assays that have been developed for NoV detection using conventional RT-PCR in which first-generation assays have used primers that were designed on the first described NoV genome, the Norwalk virus genome (Robilotti et al. 2015). First-generation assay occurs when complementary DNA (cDNA) is generated from the RNA template by reverse transcriptase. The first-generation assay that uses conventional PCR, also known as one-step RT-PCR, is more commonly used for clinical screening due to decreased contamination risks and less experimental variation (Adams 2020). This method involves reverse transcriptase in a single tube preparation. Meanwhile, second-generation assay, or two-step RT-PCR, is able to perform with more reactions due to the options of wide variety of primers. Moreover, second-generation PCR assay is often used in a gene expression analysis that involves a wide variety of target sequences. Two-step method involves the process of total RNA being reversely transcribed to cDNA by reverse transcriptase. Thus, cDNA will be amplified to generate amplicons.
Throughout the years, NoV genomic strains have evolved, which results in a wider NoV genetic diversity. With this, RT-PCR assays would require improvements in order to detect a wider range of NoV genome. Two conventional RT-PCR assays and RT-qPCR assay were used to detect NoV genotypes in 186 stool samples whereby results of RT-PCR assays had poor accuracy due to lower sensitivity and were not able to detect certain NoV genotypes, while analytical sensitivity and specificity of the RT-qPCR assay have shown that the assay was able to detect all NoV genotypes with higher sensitivity and no false positives (Rooney et al. 2014). Thus, to monitor NoV epidemiology, genotyping of the NoV sequence is required. On the other hand, second-generation assays of the conventional RT-PCR have been reported to perform better compared to the first generation assays due to the use of other additional NoV strain sequences when designing primers to accommodate NoV genetic diversity (Robilotti et al. 2015).
Real-time PCR in detecting Salmonella and Norovirus
The qPCR method was first commercialized in 1997 for microbiology research purposes (DeFrancesco 2003) and over the years, qPCR has emerged as a universal method which has become the subject of focus for the detection of food-borne pathogens after the use of conventional PCR. Among the molecular detection methods, qPCR is more rapid and has better sensitivity due to the capability of targeting very low concentrations of the target sample, thus, has become the method of choice, the new “gold standard” to detect and quantify food-borne pathogens for food analysis and microbial population studies with absolute and relative quantification. Absolute quantification requires the absolute number of known standards unlike conventional PCR in which absolute standard curve is plotted for the gene of interest (Gunstream et al. 2012).
The use of qPCR compared with conventional PCR, do not require the use of gel electrophoresis, which is a post-amplification analysis to detect positive PCR products. Thus, qPCR is less labour intensive and less prone to contamination since qPCR can be used as one-step instrument without needing post-PCR analysis. Sensitivity analysis of the method requires serial dilution concentrations of the target sequence as the lowest amount is detected to check for the assay which is called limit of detection (LOD). The LOD is the lowest quantity of the target sequence that can be distinguished with a 99% confidence level. Moreover, the cross-reactivity analysis with other species is critical, in which the range of species of the food-borne pathogens are included in the PCR assay to determine and analyse whether the PCR assay amplifies other non-targeted species.
The qPCR method has the ability to detect and identify the species of food-borne pathogens phylogenetically. Moreover, qPCR also performs analysis authenticity and adulteration of food products such as the reported European horse meat scandal. The advancements made in qPCR allow the quantification of the amplified target product in which can be specific and non-specific detection chemistries (Postollec et al. 2011). Recently, the development of qPCR has been used to detect and quantify Salmonella spp. from sheep feces and tissue samples, with and without the pre-enrichment step, BPW enrichment culture method, which have resulted in a 91% sensitivity and a specificity of 100% (Parker et al. 2020). Thus, the use of molecular tools, especially qPCR, has increased over the years due to a number of reasons such as better specificity, sensitivity, dynamic range of qPCR assay methods in which will help improve advanced novel high throughput nanoliter qPCR with shorter time consumption, rapid reaction rates of heating and cooling and minimal reagent consumption. Moreover, the combination of different techniques will help to decrease the size and power usage in which will cause qPCR method to be more feasible for point-of-care diagnostics (Johnson et al. 2013).
Two common fluorescence detection assays in qPCR are widely used for the amplification of target sequences, dsDNA binding dye assay and TaqMan probe assay (Smith and Osborn 2009). Dye-based PCR methods use fluorescent dyes that bind to the target sequence which results in the production of fluorescent signals that are monitored by thermocycler during each PCR cycle and an amplification plot generated in real-time. Fluorescent dyes intercalate and bind to the minor grooves of dsDNA at an average emission range of 487 nm and 560 nm. The PCR system records the data from PCR amplification cycle and plots the data to generate an amplification curve. Moreover, the PCR amplification cycle consists of four phases which include baseline, exponential phase, linear phase and plateau phase. During the exponential phase, the increase in fluorescence generated is from the PCR products, also known as amplicons, being produced. Thus, the change in fluorescence is directly proportional to the amount of amplicons produced.
Some advantages of using dsDNA-binding dye are simplicity of amplification of target sequence without the difficulty and need to design a probe. It is less expensive and can be used for any primer set. However, the limitations of using dsDNA-binding dyes include non-specific binding of fluorescent dye to the dsDNA, as the dye is unable to discriminate the targeted dsDNA. In addition, primer-dimer formation may occur which will affect the sensitivity of the qPCR. The fluorescent non-specific dsDNA binding dye that has been widely used is SYBR Green I dye, a dsDNA intercalating dye that is able to bind to dsDNA easily compared to ethidium bromide. The next generation DNA binding dye is EvaGreen dye which provides brighter and clearer signal compared to SYBR Green I dye because EvaGreen dye has less inhibitory effects that allow the saturating dye concentration for better signal and high quality DNA melt analysis with less background fluorescence (Biotium 2010).
In addition, qPCR is able to perform singleplex and multiplex PCR whereby singleplex uses one primer set in a reaction well while multiplex PCR uses two or more primer sets in a reaction well whereby each primer set amplifies a specific target sequence (Salihah et al. 2016). The design of multiplex PCR assay is more difficult as the requirement to ensure the primer pair will not misprime with other primers for non-specific amplification and demirisation between the primers (Z. Shen et al. 2010). Probe-based assays, such as TaqMan probes, hybridisation probes and molecular beacon are more specific compared to the dsDNA-binding dye method as these probes are sequence specific and are able to discriminate the target specific dsDNA from non-specific dsDNA with a reporter and a quencher attached to the probe as well as to perform quantitative analysis (Salihah et al. 2016). The reporter is a fluorophore molecule which is attached at the 5’-end while the quencher can be found at the 3’-end of the probe and is responsible for generating fluorescence by fluorescence resonance energy transfer (FRET) mechanism when Taq polymerase has cleaved the reporter molecule from the probe. Hence, TaqMan probe assay has more specificity as the fluorescence generated is measured for the specific targeted amplicons produced. However, TaqMan probe assay is expensive compared to dye-based PCR as the probe-based assay requires specific synthesis and design of probes as well as primer pairs.
As for NoV, after the use of conventional RT-PCR to perform the detection and identification of enteric viruses such as NoV, further development and improvements of the molecular detection methods were made. The detection and identification of NoV are commonly performed by using reverse transcriptase PCR (RT-PCR) (Elizaquível et al. 2014). The detection and identification of food-borne viruses such as NoV, may use either one-step or two-step RT-PCR method as both perform the same function which is to quantify the targeted genome sequence (Adams 2020). The diversity of NoV genome, thus, requires more accurate and sensitive detection method. The use of real-time RT-PCR to monitor viruses in fresh produce and surface water which include NoV GI genogroup, adenoviruses, hepatitis A virus and rotavirus group A have been studied recently (Shaheen et al. 2019). The results of the study have shown that a wide range of enteric viruses have contaminated the fresh produce and the viruses detected by the molecular detection method may originate from contaminated irrigation water. In addition, the study has stated that irrigation water may be a vehicle of virus transmission to fresh produce such as fruits and vegetables, which was carried out by determining the presence of food-borne viruses in fresh produce and irrigation water (Shaheen et al. 2019).
Moreover, several RT-qPCR assays have been evaluated for the detection of NoV to assess the most accurate assay that detects the NoV genogroups, GI and GII (Yoo et al. 2017). This method has used both conventional RT-PCR and RT-qPCR to compare the performance, efficiency, sensitivity and specificity between the two. RT-qPCR has shown better results with regards to specificity due to high specificity of probe-based primers. Furthermore, the addition of internal quencher, such as ZEN quencher, has improved and increased the sensitivity of signal by decreasing the background fluorescence (Yoo et al. 2017). The evidence of a study in evaluating outbreaks of food-borne viral illnesses has shown that rapid molecular detection methods, RT-PCR and RT-qPCR, are more sensitive compared to conventional culture methods (Bosch et al. 2011). The presence of food-borne viruses could be due to pollution in sewage and water system that could have led to food products such as fresh produce being contaminated with the viruses.
Sample preparation for Salmonella and Norovirus
The importance of sample preparation for qPCR is to obtain good quality and quantity of pure DNA efficiently for successful and reliable results. Infected feces and contaminated food are some common examples of samples used for the detection and identification of these food-borne pathogens. Samples collected have to be aliquoted from pre-enriched cultures which will then be prepared for DNA extraction which involves three critical steps, lysis of the cell, precipitation and purification of DNA from any proteins, RNA, lipids and macromolecules (Gupta 2019). A number of components are required to make an enriched culture medium for the growth of bacteria which include sufficient nutrients such as carbon source, nitrogen source, water and mineral salts (Bonnet et al. 2020). Moreover, a selective culture medium would culture the growth of selected bacteria in which antibiotics, antiseptics, dyes, chemicals and sodium salts are added (Bonnet et al. 2020). The method of enrichment of food-borne pathogens such as Salmonella is challenging due to the number of steps required to successfully separate Salmonella from other microorganisms (Bell et al. 2016). Immunomagnetic separation (IMS) has been widely used to specifically separate and isolate the target cells, such as Salmonella, from the rest of the microorganisms found in the sample by using antibodies which will bind to paramagnetic beads (Málková et al. 1998) (Steingroewer et al. 2005) (Hsu et al. 2014).
The detection of NoV by RT-qPCR using monoclonal antibodies that act against the NoV has been studied (Yao et al. 2009) (Liu et al. 2015) (K. Lee et al. 2014). RNA extraction for human NoV involves the virus extraction from samples and nucleic acid extraction from the concentrated viruses by using specific extraction kits according to instructions of the manufacturer as described in a review in 2011 (Baert et al. 2011). In a study, 500 µl PCR-grade water was added to a 10% (w/v) fecal suspension which was then centrifuged for 5 min at 16.000 × g to collect the supernatant used for RNA extraction (Schmid et al. 2004). In infected individuals, NoV can be found in feces samples in larger quantity compared to vomitus and rectal swabs (Vinjé 2015). The samples obtained for the use of qPCR has to be checked for the quantity and quality by using a UV–Vis spectrophotometer such as NanoDrop (Thermo Scientific). Meanwhile, Salmonella enterica has been detected in food samples by a novel technique which involved immune-magnetic bead concentration and direct PCR with sensitivity of 91.6%, specificity of 100% and detection as low as 2–3 CFU/ml (Vinayaka et al. 2019). In addition, it has been stated that gene expression analysis would help the sample preparation method to have lesser influence on de novo gene expression (Postollec et al. 2011).
Viability PCR in detecting Salmonella and Norovirus
Viability PCR (vPCR) is known to be an evolution of the PCR method, as it is the only PCR-based method that could amplify viable cells (Fig. 3). Viability dyes, propidium monoazide (PMA) and ethidium monoazide (EMA), are used as nucleic acid intercalating dyes in both conventional PCR and qPCR (Elizaquível et al. 2014). These dyes are cell membrane impermeable and dead-cell specific whereby the dye binds to DNA of the dead cell with high affinity by forming covalent bonds in the presence of visible light (Fig. 4). Initially, sample preparation of the specific pathogen is done in which the viability dye, PMA or EMA, is added into the sample which can be seen as step 1 in Fig. 4. The mixture is then incubated in the dark for 10 min at room temperature, according to the protocol (Biotium 2010). After incubation, light is then introduced to the sample whereby the viability dye will bind to the dsDNA forming a covalent bond. DNA modification will prevent PCR amplification of the dead cell. DNA extraction is then performed to quantify the DNA of viable cells by qPCR. PMA has successfully been used in qPCR for the quantification of viable Salmonella and E. coli O157:H7 (Sánchez et al. 2012). PMA has been found to be more effective than EMA in terms of discrimination between living and dead cells and there is a great effect on the activity of viability dyes due to the differences in cell envelope structure of the Gram-positive and Gram-negative bacteria (Elizaquível et al. 2014). The concept for PMA penetration is the cell structure whereby the permeability barrier for Gram-negative bacteria is the complex structure of the outer membrane while for Gram-positive bacteria would be the peptidoglycan layer (Nogva et al. 2003).
Fig. 3.
Schematic diagram representation of Norovirus particle
Fig. 4.

Viability PCR workflow involving PMA and EMA for selective detection and quantification of the viable cells. Image adapted and
modified from: Biotium website. (https://biotium.com/technology/microbiology/pma-for-viability-pcr/)
The application of vPCR to detect food-borne pathogens has been beneficial in producing results that could help provide solutions for the challenges faced in detection of food-borne pathogens. Some factors that could affect vPCR efficiency include complex matrices, pH and salt concentration, turbidity and dead cell concentration (Fittipaldi and Codony 2011). The results of vPCR have shown that there could be a change in polymerase activity due to the presence of PCR inhibitors, interference in cell lysis and inability to perform photo-activation in the presence of organic compounds (Fittipaldi and Codony 2011).
The detection of food-borne viruses does not indicate the infectivity of the viruses directly (Elizaquível et al. 2014). Several studies have attempted to use PCR to differentiate between infectious and non-infectious NoV, such as the use of RNAse for pre-treatments to digest the viral RNA of non-intact viral particles as well as the use of viability dyes such as PMA to monitor the infectivity of NoV (Topping et al. 2009) (S. Y. Kim and Ko 2012). Infectivity of food-borne viruses such as enteroviruses, NoV, hepatitis A virus, murine norovirus (MNV) and feline calicivirus have been tested with vPCR (K. Kim et al. 2011) (S. Y. Kim and Ko 2012). Viability dyes have been reported to have different effects depending on the virus type (Elizaquível et al. 2014) which has been investigated in a study whereby PMA was used in RT-qPCR to differentiate the infectious and non-infectious bacteriophage MS2, however, MNV was not able to be differentiated by the RT-qPCR (S. Y. Kim and Ko 2012).
Digital PCR in detecting Salmonella and Norovirus
Digital PCR (dPCR) is the newest advancement method which has improved the limitations of the qPCR method in which the sample is partitioned into many individual PCR reaction wells (Fig. 5). Moreover, unlike qPCR which collects the data in real-time, dPCR collects data at end point and involves absolute quantification. The dPCR method uses the traditional end-point PCR and fluorescent-probe-based method for amplification in order to generate extremely sensitive quantification of the targeted sequences without the use of standard curve (Salipante and Jerome 2020). In addition, dPCR is able to perform very low-target quantitation from different contaminated samples without standard curves in which will be able to generate more accurate and reproducible data while in qPCR, Taq polymerase can be inhibited when sample contaminants are present (Taylor et al. 2017). The effects of two rapid detection methods, qPCR and dPCR were compared and studied to detect S. typhimurium which have shown that dPCR has produced more sensitive results with less pre-culturing and enrichment time compared to qPCR (Wang et al. 2018). The less pre-enrichment time, the more efficient measurement of food-borne bacteria with a 2 h difference compared to qPCR, as the start-up time to generate a standard curve is removed. Furthermore, the detection of S. typhimurium by dPCR has stronger resistance to PCR inhibitors and lower LOD than qPCR, with dPCR being more stable compared to qPCR due to stronger resistance to PCR inhibitors when comparing with different DNA concentrations (ng/µl). Thus, resistance to PCR inhibitors has shown that the dPCR methods are able to generate reliable results with different varieties of DNA samples.
Fig. 5.

Digital PCR method showing a single sample with the target sequence which will be separated and then amplified by end-point to generate copies of the target sequence. Reference: Nyaruaba R. et al. 2019
ATP bioluminescence assay
The use of biosensors to detect food-borne bacteria is also known for years. The application of biosensors to detect bacteria has been developed to help improve the use of conventional culture methods. One of the methods in biosensor is the Adenosine triphosphate (ATP) bioluminescence which is known to be one of the highly effective biosensors (Ali et al. 2020). This method has been used in the early days whereby firefly luciferase was used for ATP detection to monitor the growth of microorganisms. The bioluminescence mechanism involves the emission of light from the organisms which will be converted from energy into light (Ali et al. 2020). Since ATP is an energy source that is significantly found in all living microorganisms which includes living microbes, the ATP bioluminescence assay test is used for the detection of ATP in these living or viable microorganisms as this shows that the living microorganisms are present (Jayan et al. 2020). This method is an advantage as is able to detect the presence of living food-borne bacteria at a cost-effective rate as well as rapid and high sensitivity (Eed et al. 2016). Thus, the ATP bioluminescence assay test can be studied and used as an alternative way and to detect the living food-borne bacteria.
Conclusion
Rapid, reliable, sensitive and inexpensive detection methods for food-borne pathogens are in great demand due to the increasing burden of food-borne disease outbreaks affecting public health, welfare and economy. State-of-art molecular detection methods including PCR and qPCR, which have been widely used in clinical and research laboratories, started to play an important role in point-to-care diagnostics and help in implementing a surveillance system for rapid and precise detection of pathogens at the nucleic acid level. On the other hand, there are challenges and limitations that come with the detection methods, such as sample extraction and specific primer designing which still have to be overcome. Advanced developments and improvements of the molecular detection methods are critical to making the detection of food-borne pathogens more reliable, sensitive and time-saving, in order to provide rapid data acquisition. Thus, the recent trends and developments of molecular detection methods, specifically qPCR, to detect food-borne pathogens Salmonella and norovirus were reviewed and concluded in Table 2. The comparison of conventional culture methods and end-point PCR with qPCR were discussed as qPCR has proven to decrease the time consumption and manpower needed.
Table 2.
The molecular detection methods discussed in this study
| Method | Purpose | Improvements | References |
|---|---|---|---|
| Conventional culture | Isolation and identification | Agar culture can help distinguish pathogens morphologically | (Andrews and Hammack 2020) |
| Conventional end-point PCR | Detection and identification of gDNA of pathogens at end point | Less time consuming than conventional culture | (Ahmed 2014) |
| Real-time PCR | Detection and identification of gDNA of pathogens at real time and use of melting curve | Time-saving and does not require post-PCR amplification | (Gunstream et al. 2012) (Postollec et al. 2011) |
| Viability PCR | Detection of viable cells | Viability dye used to distinguish viable and dead cells | (Elizaquível et al. 2014) |
| Digital PCR | Detection of gDNA of pathogens at end point | Highly sensitive, standard curve not required, lower LOD than real-time PCR and stronger resistance to PCR inhibitors | (Salipante and Jerome 2020) (Taylor et al. 2017) |
| ATP bioluminescence | Measures ATP from viable cells of bacteria to monitor growth | Rapid and high sensitivity | (Eed et al. 2016) |
Acknowledgements
The author is grateful to Universiti Brunei Darussalam and the Ministry of Education for supporting and funding this research.
Abbreviations
- ATP
Adenosine triphosphate
- PCR
Polymerisation chain reaction
- qPCR
Quantitative real-time polymerisation chain reaction
- vPCR
Viability polymerisation chain reaction
- dPCR
Digital polymerisation chain reaction
- NoV
Norovirus
- rt-PCR
Real-time polymerisation chain reaction
- ssRNA
Single-stranded ribonucleic acid
- VPg
Viral protein genome-linked
- ORF
Open reading frame
- BPW
Buffered peptone water
- LB
Lactose broth
- EIA
Enzyme immunoassay
- dsDNA
Double-stranded deoxynucleic acid
- ssDNA
Single-stranded ribonucleic acid
- dNTP
Deoxynucleoside triphosphate
- RT-PCR
Reverse transcriptase polymerisation
- cDNA
Complementary deoxyribonucleic acid
- LOD
Limit of detection
- Tm
Melting temperature
- FRET
Fluorescence resonance energy transfer
- IMS
Immunomagnetic separation
- PMA
Propidium monoazide
- EMA
Ethidium monoazide bromide
- HBGA
Histo-blood group antigens
Authors’ contribution
NAC: Conceptualization, Methodology, Investigation, Writing original draft, Visualization. NTS: Resources, Writing—review & editing, PSB: Resources, Writing—review & editing. MUA: Supervision, Project administration, Resources, Writing—review & editing.
Funding
This research was funded by the Ministry of Education under Brunei Darussalam Government Scholarship.
Availability of data and material
Not Applicable.
Code availability
Not Applicable.
Declarations
Conflict of interest
The author declares no conflict of interest.
Consent for publication
All authors have agreed to publish the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Adams G. A Beginner’s Guide to RT-PCR, qPCR and RT-qPCR. Biochem. 2020;42(3):48–53. doi: 10.1042/BIO20200034. [DOI] [Google Scholar]
- Ali AA, Altemimi AB, Alhelfi N, Ibrahim SA (2020) Application of biosensors for detection of pathogenic food bacteria: a review. Biosensors 10(58). 10.3390/bios10060058 [DOI] [PMC free article] [PubMed]
- Andrews WH, Hammack TS (2020) Bacteriological analytical manual, 8th Edition, Revision A. Chapter 5 (Old version 1998, Chapter 4). https://www.fda.gov/food/laboratory-methods-food/bam-chapter-1-foodsamplingpreparation-sample-homogenate
- Baert L, Mattison K, Loisy-Hamon F, Harlow J, Martyres A, Lebeau B, Stals A, Van Coillie E, Herman L, Uyttendaele M. Review: Norovirus prevalence in Belgian, Canadian and French fresh produce: a threat to human health? Int J Food Microbiol. 2011;151(3):261–269. doi: 10.1016/j.ijfoodmicro.2011.09.013. [DOI] [PubMed] [Google Scholar]
- Bell RL, Jarvis KG, Ottesen AR, Mcfarland MA, Brown EW. Recent and emerging innovations in Salmonella detection: a food and environmental perspective. Microb Biotechnol. 2016;9(3):279–292. doi: 10.1111/1751-7915.12359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biotium (2010) Product Information: EvaGreen™ Dye, 20x in water
- Bonnet M, Lagier JC, Raoult D, Khelaifia S. Bacterial culture through selective and non-selective conditions: the evolution of culture media in clinical microbiology. New Microbes New Infect. 2020 doi: 10.1016/j.nmni.2019.100622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch A, Bidawid S, Le Guyader FS, Lees DN, Jaykus L-A. Norovirus and Hepatitis A virus in shellfish, soft fruits and water. Rapid Detect Identif Quantif Foodborne Pathog. 2011;44:466. [Google Scholar]
- Chen SY, Feng Y, Chao HC, Lai MW, Huang WL, Lin CY, Tsai CN, Chen CL, Chiu CH. Emergence in Taiwan of novel norovirus GII.4 variants causing acute gastroenteritis and intestinal haemorrhage in children. J Med Microbiol. 2015;64:544–550. doi: 10.1099/jmm.0.000046. [DOI] [PubMed] [Google Scholar]
- Cotten M, Petrova V, Phan MVT, Rabaa MA, Watson SJ, Ong SH, Kellam P, Baker S. Deep sequencing of Norovirus genomes defines evolutionary patterns in an urban tropical setting. J Virol. 2014;88(19):11056–11069. doi: 10.1128/jvi.01333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defrancesco L. Real-time PCR takes center stage. Anal Chem. 2003;75(7):175–179. doi: 10.1021/ac031280e. [DOI] [PubMed] [Google Scholar]
- Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. Viral shape-shifting: norovirus evasion ofthe human immune system. Nat Rev Microbiol. 2010;8(3):231–241. doi: 10.1038/nrmicro2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eed HR, Abdel-Kader NS, El Tahan MH, Dai T, Amin R (2016) Bioluminescence-sensing assay for microbial growth recognition. J Sens 2016. 10.1155/2016/1492467
- Elizaquível P, Aznar R, Sánchez G. Recent developments in the use of viability dyes and quantitative PCR in the food microbiology field. J Appl Microbiol. 2014;116(1):1–13. doi: 10.1111/jam.12365. [DOI] [PubMed] [Google Scholar]
- Eyigor A, Carli KT, Unal CB. Implementation of real-time PCR to tetrathionate broth enrichment step of Salmonella detection in poultry. Lett Appl Microbiol. 2002;34(1):37–41. doi: 10.1046/j.1472-765X.2002.01036.x. [DOI] [PubMed] [Google Scholar]
- Fittipaldi M, Codony F, Adrados B, Camper AK, Morató J. Viable real-time pcr in environmentalsamples: can all data be interpreted directly? Microb Ecol. 2011;61:7–12. doi: 10.1007/s00248-010-9719-1. [DOI] [PubMed] [Google Scholar]
- Giannella RA (1996) Salmonella. Medical Microbiology. 4th edition. Galveston (TX): University of Texas Medical Branch at Galveston. Available from: https://www.ncbi.nlm.nih.gov/books/NBK8435/
- Gunstream S, Hellemans J, Menezes A, Owens B, Rose S, Sander R, Vandesompele J (2012) qPCR Application Guide: Experimental Overview, Protocol, Troubleshooting
- Gupta N. DNA extraction and polymerase chain reaction. J Cytol. 2019;36(2):116–117. doi: 10.4103/JOC.JOC_110_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwida MM, Al-Ashmawy MAM. Culture versus PCR for salmonella species identification in some dairy products and dairy handlers with special concern to its zoonotic importance. Vet Med Int. 2014 doi: 10.1155/2014/502370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed OB, Asghar AH, El-Rahim IHA, AI H. Detection of salmonella in food samples by culture and polymerase chain reaction methods. J Bacteriol Parasitol. 2014;05(03):5–7. doi: 10.4172/2155-9597.1000187. [DOI] [Google Scholar]
- Hsu CY, Hsu BM, Chang TY, Hsu TK, Shen SM, Chiu YC, Wang HJ, Ji WT, Fan CW, Chen JL. Evaluation of immunomagnetic separation for the detection of Salmonella in surface waters by polymerase chain reaction. Int J Environ Res Public Health. 2014;11(9):9811–9821. doi: 10.3390/ijerph110909811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Huang R, Wang Y, Wei X, Wang Z, Geng Y, Jing J, Gao H, Sun X, Dong C, Jiang C. Development of duplex PCR-ELISA for simultaneous detection of Salmonella spp. and Escherichia coli O157: H7 in food. J Microbiol Methods. 2018;154(136):127–133. doi: 10.1016/j.mimet.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Jayan H, Pu H, Sun DW. Recent development in rapid detection techniques for microorganismactivities in food matrices using bio-recognition: a review. Trends Food Sci Technol. 2020;95:233–246. doi: 10.1016/j.tifs.2019.11.007. [DOI] [Google Scholar]
- Johnson G, Nolan T, Bustin SA. Real-time quantitative PCR, pathogen detection and MIQE. Methods Mol Biol. 2013;943:1–16. doi: 10.1007/978-1-60327-353-4_1. [DOI] [PubMed] [Google Scholar]
- Jones MK, Grau KR, Costantini V, Kolawole AO, De Graaf M, Freiden P, Graves CL, Koopmans M, Wallet SM, Tibbetts SA, Schultz-Cherry S, Wobus CE, Vinjé J, Karst SM. Human norovirus culture in B cells. Nat Protoc. 2015;10(12):1939–1947. doi: 10.1038/nprot.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Ko G. Using propidium monoazide to distinguish between viable and nonviable bacteria, MS2 and murine norovirus. Lett Appl Microb. 2012;55(3):182–188. doi: 10.1111/j.1472-765X.2012.03276.x. [DOI] [PubMed] [Google Scholar]
- Kim K, Katayama H, Kitajima M, Tohya Y, Ohgaki S. Development of a real-time RT-PCR assay combined with ethidium monoazide treatment for RNA viruses and its application to detect viral RNA after heat exposure. Water Sci Technol. 2011;63(3):502–507. doi: 10.2166/wst.2011.249. [DOI] [PubMed] [Google Scholar]
- Koopmans M, Duizer E. Foodborne viruses: an emerging problem. Int J Food Microbiol. 2004;90(1):23–41. doi: 10.1016/S0168-1605(03)00169-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Surendran PK, Thampuran N. Evaluation of culture, ELISA and PCR assays for the detection of Salmonella in seafood. Lett Appl Microbiol. 2008;46(2):221–226. doi: 10.1111/j.1472-765X.2007.02286.x. [DOI] [PubMed] [Google Scholar]
- Lee K, Park K, Seo DJ, Lee MH, Jung JY, Park GJ, Yoon D, Park KH, Choi C. Enhanced immunomagnetic separation for the detection of norovirus using the polyclonal antibody produced with human norovirus GI.I4-like particles. Food Sci Biotechnol. 2014;23(5):1569–1576. doi: 10.1007/s10068-014-0213-2. [DOI] [Google Scholar]
- Lee KM, Runyon M, Herrman TJ, Phillips R, Hsieh J. Review of Salmonella detection and identification methods: aspects of rapid emergency response and food safety. Food Control. 2015;47:264–276. doi: 10.1016/j.foodcont.2014.07.011. [DOI] [Google Scholar]
- Liu P, Kim M, Schlesinger D, Kranz C, Ha S, Ha J, Slauch J, Baek S, Moe C. Immunomagnetic separation combined with RT-qPCR for determining the efficacy of disinfectants against human noroviruses. J Infect Public Health. 2015;8(2):145–154. doi: 10.1016/j.jiph.2014.08.007. [DOI] [PubMed] [Google Scholar]
- Louten J (2016) Detection and diagnosis of viral infections. In: Essential Human Virology, pp 111–132
- Málková K, Rauch P, Wyatt GM, Morgan MRA. Combined immunomagnetic separation and detection of Salmonella enteritidis in food samples. Food Hydrocoll. 1998;10(3):271–280. doi: 10.1080/09540109809354990. [DOI] [Google Scholar]
- Matthews JE, Dickey BW, Miller RD, Felzer JR, Dawson BP, Lee AS, Rocks JJ, Kiel J, Montes JS, Moe CL, Eisenberg JNS, Leon JS. The epidemiology of published norovirus outbreaks: a systematic review of risk factors associated with attack rate and genogroup. Epidemiol Infect. 2012;140(7):1161–1172. doi: 10.1017/S0950268812000234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, Griffin PM, Tauxe RV. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5(5):607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino L, Procura F, Trejo FM, Bueno DJ, Golowczyc MA. Biofilm formation by Salmonella sp. in the poultry industry: detection, control and eradication strategies. Food Res Int. 2017 doi: 10.1016/j.foodres.2017.11.024. [DOI] [PubMed] [Google Scholar]
- Nogva HK, Drømtorp SM, Nissen H, Rudi K. Ethidium monoazide for DNA-based differentiation of viable and dead bacteria by 5′-nuclease PCR. Biotechniques. 2003;34(4):804–813. doi: 10.2144/03344rr02. [DOI] [PubMed] [Google Scholar]
- Nyaruaba R, Mwaliko C, Kering KK, Wei H. Droplet digital PCR applications in the tuberculosis world. Tuberculosis. 2019;117:85–92. doi: 10.1016/j.tube.2019.07.001. [DOI] [PubMed] [Google Scholar]
- Parker AM, Mohler VL, Gunn AA, House JK. Development of a qPCR for the detection and quantification of Salmonella spp. in sheep feces and tissues. J Vet Diagn Investig. 2020 doi: 10.1177/1040638720952359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postollec F, Falentin H, Pavan S, Combrisson J, Sohier D. Recent advances in quantitative PCR (qPCR) applications in food microbiology. Food Microbiol. 2011;28(5):848–861. doi: 10.1016/j.fm.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Robilotti E, Deresinski S, Pinsky BA. Norovirus. Clin Microbiol Rev. 2015;28(1):134–164. doi: 10.1128/CMR.00075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Lázaro D, Hernández M. Real-time PCR in food science: introduction. Current Issues Mol Biol. 2013;15(2):25–38. doi: 10.21775/cimb.015.025. [DOI] [PubMed] [Google Scholar]
- Rooney BL, Pettipas J, Grudeski E, Mykytczuk O, Pang XL, Booth TF, Hatchette TF, Leblanc JJ. Detection of circulating norovirus genotypes: hitting a moving target. Virol J. 2014;11(1):20–25. doi: 10.1186/1743-422X-11-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostron P, Pennance T, Bakar F, Rollinson D, Knopp S, Allan F, Kabole F, Ali SM, Ame SM, Webster BL (2019) Development of a recombinase polymerase amplification (RPA) fluorescence assay for thedetection of Schistosoma haematobium. Parasites Vectors 1–7. 10.1186/s13071-019-3755-6 [DOI] [PMC free article] [PubMed]
- Salihah NT, Hossain MM, Lubis H, Ahmed MU. Trends and advances in food analysis by realtimepolymerase chain reaction. J Food Sci Technol. 2016;53(5):2196–2209. doi: 10.1007/s13197-016-2205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez G, Elizaquível P, Aznar R. A single method for recovery and concentration of enteric viruses and bacteria from fresh-cut vegetables. Int J Food Microbiol. 2012;152(1–2):9–13. doi: 10.1016/j.ijfoodmicro.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. Foodborne illness acquired in the United States-Major pathogens. Emerg Infect Dis. 2011;17(1):7–15. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Oehme R, Schalasta G, Brockmann S, Kimmig P, Enders G. Fast detection of Noroviruses using a real-time PCR assay and automated sample preparation. BMC Infect Dis. 2004;4:1–8. doi: 10.1186/1471-2334-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen MNF, Elmahdy EM, Chawla-Sarkar M. Quantitative PCR-based identification of enteric viruses contaminating fresh produce and surface water used for irrigation in Egypt. Environ Sci Pollut Res. 2019;26(21):21619–21628. doi: 10.1007/s11356-019-05435-0. [DOI] [PubMed] [Google Scholar]
- Shen Z, Qu W, Wang W, Lu Y, Wu Y, Li Z, Hang X, Wang X, Zhao D, Zhang C (2010) MPprimer: a program for reliable multiplex PCR primer design. BMC Bioinf 11. 10.1186/1471-2105-11-143 [DOI] [PMC free article] [PubMed]
- Shen J, Zhou T, Huang R (2019) Recent advances in Electrochemiluminescence sensors for pathogenicbacteria detection. Micromachines 10(8). 10.3390/mi10080532 [DOI] [PMC free article] [PubMed]
- Siala M, Barbana A, Smaoui S, Hachicha S, Marouane C, Kammoun S, Gdoura R, Messadi-Akrout F. Screening and detecting Salmonella in different food matrices in Southern Tunisia using a combined enrichment/real-time PCR method: correlation with conventional culture method. Front Microbiol. 2017 doi: 10.3389/fmicb.2017.02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Osborn AM. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol Ecol. 2009;67(1):6–20. doi: 10.1111/j.1574-6941.2008.00629.x. [DOI] [PubMed] [Google Scholar]
- Stals A, Mathijs E, Baert L, Botteldoorn N, Denayer S, Mauroy A, Scipioni A, Daube G, Dierick K, Herman L, van Coillie E, Thiry E, Uyttendaele M. Molecular detection and genotyping of Noroviruses. Food Environ Virol. 2012;4(4):153–167. doi: 10.1007/s12560-012-9092-y. [DOI] [PubMed] [Google Scholar]
- Steingroewer J, Knaus H, Bley T, Boschke E. A rapid method for the pre-enrichment and detection of Salmonella typhimurium by immunomagnetic separation and subsequent fluorescence microscopical techniques. Eng Life Sci. 2005;5(3):267–272. doi: 10.1002/elsc.200420072. [DOI] [Google Scholar]
- Sue MJ, Yeap SK, Omar AR, Tan SW (2014) Application of PCR-ELISA in molecular diagnosis. BioMed Res Int 2014. 10.1155/2014/653014 [DOI] [PMC free article] [PubMed]
- Taylor SC, Laperriere G, Germain H. Droplet Digital PCR versus qPCR for gene expression analysiswith low abundant targets: from variable nonsense to publication quality data. Sci Rep. 2017;7(1):1–8. doi: 10.1038/s41598-017-02217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topping JR, Schnerr H, Haines J, Scott M, Carter MJ, Willcocks MM, Bellamy K, Brown DW, Gray JJ, Gallimore CI, Knight AI. Temperature inactivation of Feline calicivirus vaccine strain FCV F-9 in comparison with human noroviruses using an RNA exposure assay and reverse transcribed quantitative real-time polymerase chain reaction-A novel method for predicting virus infectivity. J Virol Methods. 2009;156(1–2):89–95. doi: 10.1016/j.jviromet.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Vinayaka AC, Ngo TA, Kant K, Engelsmann P, Dave VP, Shahbazi MA, Wolff A, Bang DD. Rapid detection of Salmonella enterica in food samples by a novel approach with combination of sample concentration and direct PCR. Biosens Bioelectron. 2019 doi: 10.1016/j.bios.2018.09.078. [DOI] [PubMed] [Google Scholar]
- Vinjé J. Advances in laboratory methods for detection and typing of norovirus. J Clin Microbiol. 2015;53(2):373–381. doi: 10.1128/JCM.01535-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yang J, Gai Z, Huo S, Zhu J, Li J, Wang R, Xing S, Shi G, Shi F, Zhang L. Comparison between digital PCR and real-time PCR in detection of Salmonella typhimurium in milk. Int J Food Microbiol. 2018;266:251–256. doi: 10.1016/j.ijfoodmicro.2017.12.011. [DOI] [PubMed] [Google Scholar]
- World Health Organisation (WHO) (2020) Food Safety. https://www.int/news-room/factsheets/detail/food-safety. Accessed 20 Mar 2021
- Yao L, Wu Q, Wang D, Kou X, Zhang J. Development of monoclonal antibody-coated immunomagnetic beads for separation and detection of norovirus (genogroup II) in faecal extract samples. Lett Appl Microbiol. 2009;49(2):173–178. doi: 10.1111/j.1472-765X.2009.02638.x. [DOI] [PubMed] [Google Scholar]
- Yoo JE, Lee C, Park SJ, Ko G. Evaluation of various real-time reverse transcription quantitative pcr assays for norovirus detection. J Microbiol Biotechnol. 2017;27(4):816–824. doi: 10.4014/jmb.1612.12026. [DOI] [PubMed] [Google Scholar]
- Zou Y, Mason MG, Botella JR. Evaluation and improvement of isothermal amplification methods for point-of-need plant disease diagnostics. PLoS ONE. 2020;15(6):1–19. doi: 10.1371/journal.pone.0235216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not Applicable.
Not Applicable.