

Abstract
Pharmacomicrobiomic studies investigate drug‐microbiome interactions, such as the effect of microbial variation on drug response and disposition. Studying and understanding the interactions between the gut microbiome and drugs is becoming increasingly relevant to clinical practice due to its potential for avoiding adverse drug reactions or predicting variability in drug response. The highly variable nature of the human microbiome presents significant challenges to assessing microbes’ influence. Studies aiming to explore drug‐microbiome interactions should be well‐designed to account for variation in the microbiome over time and collect data on confounders such as diet, disease, concomitant drugs, and other environmental factors. Here, we assemble a set of important considerations and recommendations for the methodological features required for performing a pharmacomicrobiomic study in humans with a focus on the gut microbiome. Consideration of these factors enable discovery, reproducibility, and more accurate characterization of the relationships between a given drug and the microbiome. Furthermore, appropriate interpretation and dissemination of results from well‐designed studies will push the field closer to clinical relevance and implementation.
INTRODUCTION
An understanding of how human microbiomes interact with therapeutics is likely critical to the future of precision medicine to tailor therapies to an individual patient. 1 , 2 Variability in therapeutic responses in a population can lead to inadequate dosing and the potential for adverse drug reactions for many individuals taking many drugs. 3 Decades of pharmacogenetic research into drug efficacy and toxicity have uncovered genetic mechanisms that influence a patient’s variability in response to many therapeutics. 4 , 5 , 6 However, genetic factors themselves are insufficient to explain all the interindividual variability seen in therapeutic response to a drug treatment. 6 Human microbiomes, the collective microbes in and on the human body, are likely to exert effects on many drugs and also to be affected by many drugs. Thus, understanding these relationships between therapies and microbiomes are likely to be fruitful in uncovering additional variability in drug response and thus, delivering precision medicine 2 (Figure 1). For example, indices of the microbiome, such as diversity of species 7 or presence of specific taxa or microbial genes, 8 have the potential for utilization as an indicator of efficacy 9 or adverse drug reaction risk prior to drug therapy. 10 Pharmacomicrobiomics, a term first coined in 2010, refers to the investigation of how variation in a patient’s microbiomes relates to a patient’s response to therapeutics 11 and has the potential to shed light on uncharacterized inter‐patient observed variability in both efficacy and toxicity of drug therapy.
FIGURE 1.
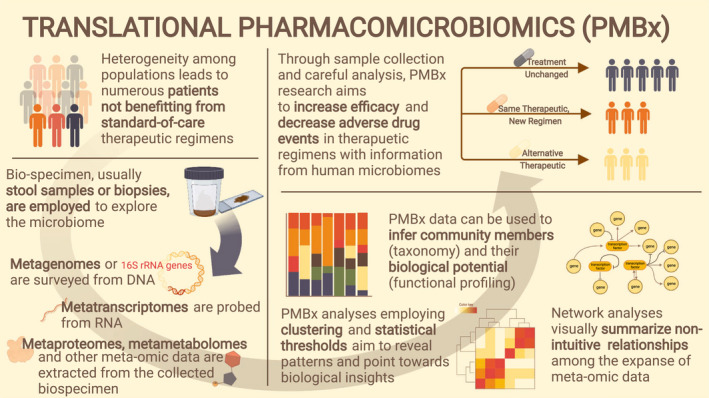
Translational pharmacomicrobiomics. With careful study design, pharmacomicrobiomics datasets will be invaluable in uncovering drug response heterogeneity that may or may not be useful in clinical prediction. Through biopsy or stool sample collection, the microbiome can be surveyed through the metagenome (DNA), metatranscriptome (RNA), metametabolome (metabolites), metaproteome (proteins), and an ever‐increasing amount of biological data. Typical analyses include profiling communities for their species and functional potential, and creating networks to uncover relationships hidden in the expansive data.
Investigating drug‐microbiome relationships in humans poses many technical challenges that must be thoughtfully considered to collect the most clinically relevant, generalizable data, and capture the inter‐ and intra‐patient variability of the microbiome in question. For example, human microbiomes’ influence on drug response is a burdensome research question due to variation in a microbiome’s composition both temporally and between individuals. A microbiome experiences fluctuations in response to genetic influences, 12 lifestyle choices, 13 and environmental forces, 14 and all these factors make up a unique microbial signature for an individual. In this review, we will focus on pharmacomicrobiomics of the human gut microbiome, first introducing previously described gut pharmacomicrobiomic relationships. We then discuss methods and considerations for designing a human pharmacomicrobiomics study, investigating the relationship between the gut microbiome and drug response variability, with a focus on the oral route of administration. We focus on the gut microbiome because it has a high propensity to affect oral drug therapy during metabolism and is a logical starting point to uncovering the utility of investigating a microbiome for drug response variability. We consider the myriad of decisions an investigator must make, from inclusion criteria to statistical analysis, to design a rigorous pharmacomicrobiomic clinical study that can adequately account for potential confounders.
THE GUT MICROBIOME HAS DIRECT AND INDIRECT EFFECTS ON DRUG THERAPY
Through mechanisms, such as biodegradation, activation, potentiation, and competition, microbes can directly affect drug pharmacokinetics. 15 Microbes can participate directly in drug activation and inactivation by biotransforming drugs into secondary metabolites. 15 For example, the chemotherapeutic irinotecan is reactivated by intestinal microorganisms back into a toxic metabolite contributing to dose‐limiting gastrointestinal side effects of the drug. 10
Indirectly, microbes can influence drug response by producing microbial metabolites that interfere with host signaling pathways and gene expression. For example, paracetamol (acetaminophen) is indirectly affected by the microbial metabolite p‐Cresol. Individuals with high levels of p‐Cresol are at an elevated risk of hepatotoxicity due to poor metabolism of the drug. 16 Further, changes in the composition of the gut microbiome can lead to a variation in drug response. 17 , 18 A typical example of this is seen with the drug digoxin, which will produce an unwanted side effect if the gut microbiome contains a specific gene encoded by Eggerthella lenta. 8 These examples of microbes, directly and indirectly, affecting drug therapy demonstrate a need for studies to understand drug‐microbiome interactions in order to safely deliver therapeutics.
Therapeutics effect the gut microbiome
Orally ingested drugs will pass through the gastrointestinal tract where they encounter the gut microbiome and its inhabitants. Some drugs modify bacterial growth or metabolism, thereby changing the gut microbiome’s overall composition and function. For example, the effects of antibiotic therapy on the gut microbiome are well‐studied: antibiotics deplete bacteria, often selectively, and reduce important bacterial metabolites, such as short‐chain fatty acids and secondary bile acids. 19 , 20 These antibiotic‐induced changes, called dysbiosis, in the gut microbiome can increase infection susceptibility, compromise immune homeostasis, and deregulate metabolism. 20
Outside of antibiotics, studies are beginning to report the pharmacodynamic relationships of commonly used drugs with aspects of the gut microbiome, mainly focusing on how drugs impact the growth of bacterial species. 21 , 22 , 23 , 24 A high‐throughput drug screening study found that nearly 25% of drugs inhibited the growth of at least one representative gut bacterial strain in vitro. 22 Beyond depletion of the microbiome, drugs can influence the diversity of species present in a microbiome. Two extensively described examples of the impact of drugs on the diversity of the gut microbiome are proton pump inhibitors (PPIs) 23 and metformin. 24 Weakened function of the gastric mucosal barrier leaves PPI users at high risk of gastroenteritis infection and bacterial overgrowth in the small intestine. 25 Metformin treatment significantly benefits microbial environments by increasing the abundance of Escherichia spp. and decreasing the abundance of Intestinibacter. 24
Because drugs have been shown to disturb the gut microbiome through multiple pathways, more thorough investigations into gut‐microbiome associations will be necessary to determine mechanisms of these pharmacomicrobiomic interactions. Additionally, future research may uncover drugs responsible for microbiome perturbations and drugs exhibiting response variability due to microbiomic indices that remain unknown.
CLINICAL STUDY DESIGNS FOR PHARMACOMICROBIOMIC INVESTIGATIONS
Ideal studies in human systems biology longitudinally collect information on participants, such as diet, lifestyle, presence of disease, and use of drugs, while incorporating multiple “omics” data sources, such as information on the genome, transcriptome, proteome, and metabolome. The incorporation of “meta’omics” allows investigators to visualize a comprehensive picture of the host and the microbiome in question. A benchmark feat in microbiome research is the Human Microbiome Project (HMP), which collected stool samples longitudinally from healthy and diseased individuals. 26 Additional large biobanks that capture gut microbiome data have been established, such as the LifeLines‐DEEP cohort 27 and UK Biobank. 28 These large‐scale collaborative projects have provided a foundation for our understanding of host–microbe interactions in health, disease, and drug therapy. Extensive funding requirements make comprehensive multi‐institutional research endeavors not particularly feasible for many research groups. Thus, we discuss the advantages and disadvantages of study designs that can be implemented in discrete hypothesis testing clinical pharmacomicrobiomics studies. Because pharmacomicrobiomic data have not yet been collected in most cases, we focus our attention on prospective studies in this review.
Accounting for temporal changes in the gut microbiome
To examine inter‐ and intra‐patient variability in the relationship between the gut microbiome and clinical drug outcomes, longitudinally observing patients is ideal. For example, warfarin doses, in some patients, change continually and capturing this variability along with microbiomic variability at multiple timepoints is likely to provide additional insights over a cross‐sectional approach. However, cross‐sectional studies may still capture microbiomic variability, especially given stark differences across groups of prescription medicine users. Cross‐sectional studies investigating microbiome‐drug interactions have one major limitation: they do not account for the natural variance of the gut microbiome over time. Some studies have reported that higher levels of bacterial taxa (i.e., phyla and class) are stable over time, 29 , 30 thus, cross‐sectional studies may be sufficient to capture the between‐participant differences in the microbiome if species level variance is less important.
Longitudinal study designs, on the other hand, are capable of investigating temporal relationships and provide a way to study not only variability seen between individuals (inter‐) 31 but also variability within a single person (intra‐), 32 both of which exhibit high variability in species level composition. The number of repeated measurements needed for your longitudinal analysis will largely depend on the necessary power and changing environmental influences, whereas the length of time needed between measurements depends largely on the variability in the therapy being studied. 33 Ideally, the time between samples will be uniform among participants, but this will be primarily determined by the drug and outcome of interest. In longitudinal studies, dropouts will need to be pre‐emptively accounted for during recruitment and a plan will need to be implemented for missing data in the analysis phase. Both cross‐sectional studies and longitudinal studies can be used to investigate the association of the gut microbiome with drug response; longitudinal studies may answer more questions but costs may limit the scope of the investigation.
To intervene or observe?
Along with choosing whether a longitudinal or cross‐sectional approach will be applied, deciding if an interventional or observational study design will be used is a big step in designing a pharmacomicrobiomic study. Interventional studies are useful for evaluating the effectiveness of an intervention, such as drug therapy. Randomized control trials (RCTs) remain the gold standard of causation studies and an important step in implementation of new drugs and clinical approaches. Unlike genetic variation, pharmacomicrobiomic variation may be perturbed by drug therapy, dietary interventions, probiotic replacement, fecal transplants, and many other clinical interventions. As a corollary, a myriad of RCTs have been performed evaluating the effect of interventions directed at changing microbial composition in efforts to improve disease. 34 , 35 , 36 , 37 However, RCTs are often exclusive and not a reflection of real‐world situations where a patient’s daily activities are not in controlled conditions. 38 , 39 For instance, a controlled diet among participants would be desired in an RCT in order to avoid confounding effects of diet on the microbiome. Because of these controlled conditions, RCTs may overestimate the effect of an intervention in the general population. Importantly, in pharmaceutical studies, RCTs may be confronted with ethical issues as we may not be able to administer the drug‐of‐interest in a healthy population and, conversely, may not be able to restrict the drug of interest in a population. Although RCTs may not be a requirement prior to clinical translation of pharmacomicrobiomics, they remain essential to assigning causation to our findings. Observational studies are often the principal avenue for observing patients long‐term, when investigating rare effects, or in cases where RCTs would be unethical or unfeasible. As opposed to interventional studies, observational studies are generally less expensive and typically require less time to be carried out. Observational studies are more likely to reflect real‐world situations that can be generalized to broader populations with available data. Case–control studies are able to control for a multitude of confounders and the assessment of multiple exposures. However, as an individual’s microbiome is highly specific, it can be challenging to match case and control individuals, leading to a lower power to detect true effects. 40 Crossover study designs may also be implemented, where a patient is exposed to both conditions and may serve as their own control. Whereas a crossover study design is a robust way to account for confounders, it is not always feasible if patients have a clinical indication for the drug of interest.
Although no study design fits all potential investigations into the effects of the microbiome on human health, some important considerations can be used to inform researchers regarding the most appropriate study design. Prospective cohort studies afford the ability to control for multiple confounders, sample the microbiome at multiple timepoints, and explore various outcomes but can be time‐intensive and costly. Interventional prospective studies, such as RCTs, offer the potential benefits of assessment of causation and enabling rapid clinical translation. Cohort studies are also susceptible to selection bias, information bias, and confounding from variables that are not prospectively captured. 41 If patients are able to complete study procedures both on and off of drug treatment, a crossover design would likely be the optimal study design to account for intra‐patient variation in a pharmacomicrobiomic analysis. When choosing the type of observational study to implement, one must consider the time, effort, and financial support available to choose the best fitting design appropriately.
DETERMINING THE STUDY POPULATION FOR A PHARMACOMICROBIOMIC INVESTIGATION
Choosing the appropriate study population is vital to the success of the research in answering the question at hand. Due to the unique nature of a pharmacomicrobiomics study in its need for a medication‐taking population and access to representative gut microbiome samples, there are essential questions to consider when selecting a target population for a pharmacomicrobiomic study. Here, we consider two critical aspects of subject recruitment in a pharmacomicrobiomic study, (1) inpatient versus outpatient and (2) inclusion/exclusion criteria.
Inpatient or outpatient, does it make a difference?
Gut microbiome sampling may be more easily performed in an inpatient population due to easy access to patient bowel movements or the ability to collect an intestinal biopsy, as well as close monitoring and stability of daily diets and adherence to drug dosing schedules. Despite this benefit, observational studies of outpatient medication users may provide more generalizable information about those users rather than inpatient care, which itself can affect the gut microbiome. Additionally, outpatient studies mimic real‐world scenarios that are not necessarily apparent when conducting inpatient research, such as medication adherence. Some drugs, such as those that require therapeutic drug monitoring or are given via nonoral routes of administration, may also dictate the use of outpatient or inpatient populations, respectively.
Considerations for exclusion and inclusion criteria
Robust data will need to be collected to account for variables confounding the relationship of the microbiome and drug therapy. There are significant effects of age, disease, and medication‐use on the microbiome that should be considered when determining a study’s exclusion criteria (Table 1). Infants and children have been shown to have rapid changes to their gut microbiome, and elderly patients experiencing frailty display a marked decrease in microbial diversity, thus, including these age groups when investigating a general population may bias results. 42 Regardless, if the indication of the drug designates the age of the study population, accounting for the effects of age on the microbiome, will be important. Exclusions related to disease history should also be assessed because numerous diseases are associated with microbiomic fluctuations. Diseases with known relationships with the gut microbiome include inflammatory bowel disease, 43 , 44 psychological disorders, 45 autoimmune disorders, 46 , 47 cardiovascular diseases, 48 and various cancers. 49 , 50 , 51 When choosing which diseases to exclude from a target population, it is advisable to assess which diseases must be excluded as to still represent the target population and obtain a sufficient sample size. Finally, some drugs dramatically change an individuals’ microbiome and should be considered when deciding on exclusion criteria. Antibiotics are well‐studied and linked to the gut microbiome’s deprivation, with variability in the microbiome’s recovery time among individuals. Unless antibiotics are the drug of interest, it may be useful to delay study recruitment for those who have recently been prescribed antibiotics. It is suggested that the microbiome can return to its pre‐antibiotic state after 4 weeks, but several bacteria have been shown to be depleted for up to 6 months. 52 Apart from antibiotics, other drugs, such as PPIs 25 , 53 and metformin, 54 have been shown to affect the gut microbiome of users and may be considered as exclusion criteria when necessary for the phenotype of interest. Numerous additional confounders are likely to exist in studies of the gut microbiome, such as pets in the house 55 and mode of birth, 14 and should be evaluated when sample sizes permit.
TABLE 1.
Factors to consider in population recruitment for a pharmacomicrobiomic analysis
| Factor | Finding | Recommendation |
|---|---|---|
| Aging | ||
| Adulthood | Temporal stability 30 | Consider selecting an age range, take multiple measurements if not studying adults |
| Frailty | Reduced diversity of core microbiota 42 | Consider setting a maximum age for enrollment or studying an older population separately |
| Antibiotic use | Compositional changes (i.e., decreased Bacteroides 52 ) | Consider a minimum time since antibiotics of at least 4 weeks |
| Concomitant medications | ||
| Metformin | Compositional differences (i.e., increased Akkermansia muciniphila 24 ) | Collect medication history information |
| Proton pump inhibitors | Stark compositional differences (i.e., increased Streptococcaceae 23 ) | Collect medication history |
| Diet | The microbiome adapts to a participant’s diet 58 | Obtain dietary records/recalls or utilize standardized meal plans |
| Medical conditions | ||
| Inflammatory bowel diseases | Loss in species diversity 43 , 44 | Collect medical history, consider exclusion |
| Cancer | Dysbiosis compared to healthy controls 49 , 50 | Collect medical history, consider exclusion |
| Autoimmune disease | Dysbiosis compared to healthy controls 46 , 47 | Collect medical history, consider exclusion |
CONSIDERATIONS FOR DATA COLLECTION
Careful data collection will be pivotal to discerning a relationship between the gut microbiome and drug response. In addition to collecting gut microbiome data, we suggest prioritizing data related to the patient’s diet and concomitant drugs. Within each of these data subsets, we discuss options available to researchers and how different methods compare in the context of a pharmacomicrobiomic study.
Collecting patient dietary data
Diet has a significant effect on the composition of the gut microbiome and thus is important to capture. 56 , 57 For example, diets high in animal products (low‐fiber and high‐fat) have been shown to promote growth of bile‐tolerant microorganisms. 57 In comparison, plant‐based diets have shown an elevated abundance of polysaccharide‐digesting microorganisms. 56 Further, the microbiome can change rapidly due to the effects of diet, with a study showing that dietary alterations can induce large species‐level microbial shifts within 24 h. 58 Dietary habits can be either prescribed during study involvement or statistically accounted for by rigorous collection proximal to sampling the microbiome. If an inpatient study design is applied, controlling or standardizing diet may be useful to minimize the confounding effects of diet. In an outpatient setting, data should be collected to account for the variation in study participants’ diets. An outpatient study design may also seek to decrease diet variability by providing a predetermined diet for study participants, however, predetermined diets have also shown to decrease microbiome biodiversity, 59 which may lead to non‐generalizable results. Administering all study participants’ a controlled diet is particularly important to avoid confounding in RCTs. Because controlling for, through scheduled diets or statistical tools, variation in the gut microbiome due to diet is necessary, dietary assessments are an essential tool.
Dietary records, food frequency questionnaires, and 24‐h dietary recalls are open‐ended surveys that collect a variety of detailed information about food consumed over a predetermined period of time. 60 In the dietary record approach, the respondent records foods and beverages and the amounts of each consumed over 1 or more days at the time of consumption, thus not relying on the participant’s memory. The dietary record method can provide quantitatively accurate information on food consumed during the recording period. In a 24‐h dietary recall, each respondent is asked to remember and report all consumed foods and beverages in the preceding 24 h or on a preceding day. Validated 24‐h dietary recall surveys are useful for generating high‐quality dietary data and are considered the preferred tool for studying diet‐related associations. The National Cancer Institute has developed a free, web‐based tool that enables multiple, automatically coded, self‐administered 24‐h diet recalls and single or multiday food records. The Automated Self‐Administered 24‐h food recall survey (ASA24) is a reliable method to complete a dietary assessment that accurately approximates dietary food intake. 61 To assess long‐term dietary intake, food frequency questionnaires are often used as they can be developed uniquely for each study. A food frequency approach asks respondents to report their usual frequency of food consumption from a list of foods for a specific period. The purpose of a food frequency questionnaire is to obtain a crude estimate of the usual total daily intakes over a designated time period. When deciding to use a food frequency or 24‐h food recall survey, barriers to participation need to be assessed. The ASA24, for example, which relies heavily on access to and knowledge of technology, may be prohibitive for some individuals. Any option, unfortunately, is prone to missing data and incomplete records, however, minimal research has been done into appropriate handling of this missingness. 62 Because a study participants’ diet is the fuel for the microbes in their gut, understanding what the participants eat is essential.
Data related to medication use and adherence
Both medication adherence and concomitant medications have the potential to confound a pharmacomicrobiomic analysis. Not only do many medications have an effect on the microbiome, but there are also drug–drug interactions that should be considered. Further, a participant on multiple medications makes drawing conclusions on the microbiome’s relationship with one drug difficult, particularly if therapy fluctuates. As discussed above under determining the study population, many medications have been shown to interact with the gut microbiome through direct and indirect mechanisms. It will be valuable to collect information on concomitant drug use and statistically control for use of these medications as they may skew study results related to the microbiome.
Drug nonadherence will also skew results or lead to an inability to find true relationships in the data during analysis. 1 Direct measures of drug adherence (e.g., measuring drug metabolites in blood) can be the best route for tracking a patient’s medication adherence but have a downside of being costly to implement and not always feasible in outpatient studies. Subjective or indirect adherence measures, which include patient‐clinician reports and self‐reported adherence surveys, are simple and less costly than direct measure. We refer the reader to a review of self‐report measures of drug aherence. 63 Surveys of both concomitant drug use and target medication adherence will allow control over drug‐related confounding of pharmacomicrobiomic results.
CAPTURING THE MICROBIOME
Appropriate sampling of the microbiome will be central to any pharmacomicrobiomic study. Both sampling methods and DNA isolation have been shown to affect bacterial composition. 64 Further, anaerobic bacteria cannot survive outside of the gut making capturing the true state of a microbiome difficult. 65 Thus, sampling the gut microbiome presents a significant challenge. Although we will not discuss the methods in DNA isolation here, below we discuss sampling and sequencing methods of the gut microbiome in‐depth. Please see Lim et al. for an evaluation of popular DNA extraction protocols. 66
Sampling methods
There are two popular methods in which gut microbial information can be collected: biopsy or stool sample. 67 A biopsy may represent the most accurate picture of the gut microbiome and can more easily be performed in inpatient studies and via routine colonoscopy. However, because colonoscopies typically use high potency laxatives prior to the procedure, the gut microbiome can become disrupted and depleted. 68 Additionally, biopsies are invasive, expensive, and require a medical professional, thus, this section will focus on the benefits and drawbacks of stool sample collection and major stool sample collection methods. No matter which sample collection method is used, the golden rule of all methods is to avoid temperature fluctuations.
Stool samples are often used as representative proxy samples from the gut microbiome. Whereas stool samples are abundantly available for all patients, sample collection can be challenging due to sample degradation as well as patient comfort and compliance. One study found that stool collections pose high barriers to patient participation due to concerns related to hygiene, contamination, discretion, and lack of information. 69 Providing sterile gloves and sufficient patient education, and choosing a collection method that allows for discrete sampling will help reduce barriers to stool sample collection. 69 There are several options for collecting stool samples that involve immediate cooling, the use of a stabilizing mechanism, or minimal sample collection (Table 2) and are discussed below.
TABLE 2.
Stool sample collection methods
| Method of collection | Stability | Advantages | Disadvantages | Modalities that can be assessed |
|---|---|---|---|---|
| Immediate cooling | ||||
| Flash‐frozen | Freeze immediately | Preserves microbial signature |
Cold chain management necessary Need for freezer Batch processing not possible |
Metagenomics, 16S Amplicon sequencing, metatranscriptomics, metaproteomics, metabolomics |
| Refrigeration (4°C) | 2–24 h |
Slowed bacterial growth Slowed down fermentation |
Cold chain management necessary Need for refrigerator Deviation from the original sample Batch processing not possible |
Metagenomics, 16S Amplicon sequencing, metatranscriptomics, metabolomics (may be affected) |
| Stabilizing solution (room temperature) | ||||
| Omnigene GUT | 60 days |
It can be done at home and shipped back Immediate homogenization No PCR inhibition No preprocessing before DNA extraction High DNA/RNA recovery |
Expensive (compared to freezing) |
Metagenomics, 16S Amplicon sequencing, metatranscriptomics (may be skewed), metabolomics (may be affected) |
| RNAlater | Seven days |
It can be done at home and shipped back High DNA/RNA recovery |
Large container needed A large volume of liquid needed (5:1 ratio) Preprocessing needed for DNA extraction Expensive (compared to freezing) |
Metagenomics, 16S Amplicon sequencing, metatranscriptomics |
| Ethanol (95–99%) | Two days |
It can be done at home and shipped back Inexpensive |
Large container needed Flammable liquid Preprocessing needed for DNA extraction A large volume of liquid needed (5:1 ratio) Quality of DNA affected |
Metagenomics, 16S Amplicon sequencing |
| Minimal collection | ||||
| Swab | Variable by solution |
It can be done at home and shipped back Inexpensive Reduce deviations from the original sample |
Overgrowth of gram‐negative bacteria Highly diluted sample DNA yield poor A small amount of sample Modified DNA extraction method needed |
Metagenomics, 16S Amplicon sequencing |
| Card | Stable once dry |
It can be done at home and shipped back Reduce deviations from the original sample |
DNA yield poor A small amount of sample Modified DNA extraction method needed Increased contamination risk during the drying process |
Metagenomics, 16S Amplicon sequencing |
Abbreviation: PCR, polymerase chain reaction.
Flash‐freezing and refrigerating are recommended in any stool sample collection method in order to try to preserve the microbial signature and DNA. 70 Lower temperatures limit the proliferation of bacteria and can bridge the time between collection and processing. 71 Flash‐freezing has been shown to produce the least change in microbiomic samples, however, it is problematic in terms of batching samples, and the time from sample collection to flash freezing depends largely on patient proximity to flash freezing equipment. Refrigerated samples, alternatively, need to be processed within 24 h. However, even if the sample is kept cold, bacteria in the sample that thrive at refrigeration temperatures will proliferate. 71 Although immediate cooling methods have been shown to produce the most reliable microbiome data, stabilizing methods may reduce the researchers’ and study participants’ burden.
Stool collection methods with mechanisms for stabilizing the sample may be desired to reduce bacterial shifts and are particularly useful in outpatient studies. A stabilizing solution allows study participants to collect samples in the privacy of their own home and discreetly ship samples to the laboratory. One popular stabilizing solution system is DNAgenotek’s Omnigene GUT•200 (DNA Genoteck), which is a simple to use system that stabilizes the stool at room temperature for up to 60 days. 72 Studies have shown no significant changes in microbial composition or alpha diversity with the Omnigene system, compared to flash‐frozen samples. 71 , 73 Other stabilizing solutions include RNALater and 95% ethanol. With these solutions, samples are collected in a container and submerged into an RNA stabilization reagent and are stable for up to 7 days at room temperature with minimal shifts in diversity. 74 RNA stabilization comes with limitations, such as time stable at room temperature, need to remove RNAlater before downstream methods, and yield and quality of DNA when compared to other stabilizing methods. The 95–99% ethanol comes with its own limitations, such as stability for 48 h at refrigerated temperature and special shipping requirements because ethanol is a flammable liquid.
Methods of stool sampling that require only a minimal sample amount are fecal swabs and fecal occult blood tests. 75 Swab and card methods reduce deviations from the original sample and require little effort from the study participant. Swabs are as simple as dipping the swab into the stool and storing it into a tube with solution. Nucleic acid stabilizing cards are another minimal collection method that stabilizes DNA and RNA by trapping nucleic acids in a fiber matrix; this method yields low amounts of DNA and increases contamination risk. 75 These two minimal methods require modified DNA extraction methods due to the small amount of stool collected.
Shotgun or amplicon sequencing
Identification and characterization of the microbes inhabiting the gut is a significant focus of pharmacomicrobiomic research. Sequencing the human microbiome provides vital insights into bacterial community structure and diversity. There are two main ways to survey the microbes in the gut microbiome with DNA: (1) amplicon sequencing (16S sequencing) and (2) shotgun sequencing (metagenomics). Review papers by Ranjan et al. 76 and Jovel and Patterson et al., 77 have in‐depth summaries of the benefits and drawbacks of choosing 16S or shotgun sequencing. Briefly, amplicon sequencing uses the 16S ribosomal gene that exists in all bacteria. This gene has regions throughout that are highly variable between species. The conserved and variable regions in the 16S gene differentiate between bacterial genera, and uniquely tag bacteria such that filtering human sequences from a biosample is straightforward. The 16S sequencing is a cost‐effective solution to surveying gut bacterial communities with a fast turnaround of results (i.e., straightforward computational analysis). If researchers are interested in the entire microbiome, however, shotgun sequencing surveys the entire genome of all the organisms present in a sample, including human, viral and fungal DNA. 76 Shotgun sequencing offers the ability to detect species of organisms in the microbiome to a much higher resolution and reveal the genes that potentially influence the phenotype, whereas 16S sequencing provides information on the 16S gene alone. The appropriate sequencing methodology depends mainly on the specific research question. Questions regarding presence or absence of bacterial genera can be answered with 16S sequencing but questions regarding specific genes or species will only be answered with shotgun sequencing.
Beyond bacterial genomes
Understanding the gut microbiome and its influence on drug variability will often require a multi‐meta‐omics approach. Multi‐meta‐omics combines multiple “omes” such as the metagenome, metaproteome, metatranscriptome, and metametabolome to study a complex outcome holistically. The ability to use this type of approach is primarily defined by the method of gut microbiome sampling (Table 2). Although metagenomics is a useful tool for identifying microbes in the gut, it only provides information about genetic signatures that have the potential to be expressed, not what is actually being expressed in the cell. Applying multiple meta‐omics techniques can characterize functional pathways and how they are impacted by or impact drug therapy.
Metatranscriptomics use sequences of RNA to identify repressed or enhanced gene activity. 78 High‐quality RNA is needed for metatranscriptomic analyses, which can be difficult due to RNases in stool and general RNA instability. Highly expressed transcripts will be overinflated and could obscure the detection of rare, functionally important transcripts. Not all transcripts are translated into proteins, thus, evaluating proteins in a stool sample can provide insight into what the microbiome is doing functionally. Proteins are typically analyzed through mass spectrometry and there are reviews on the analysis of such information. Metaproteomics, analyzing all the proteins in a microbiome, may capture additional information on top of metatranscriptomics. Metaproteomics, however, is challenging due to the instability of proteins, the fact that only 10–20% of expressed proteins will be captured, and the issue of supersaturation of abundant proteins from inflated gut microbes. 79 Finally, the metabolome measures metabolites from both the host and microbiome. Pharmacometabolomic studies, in theory, could report pharmacokinetic drug responses alongside the creation of a prediction model that includes metabolite markers. 80 With the implementation of multiple meta‐“omic” approaches, genes to functional pathways can be interrogated to understand the microbiome’s role in drug response variability.
ANALYSIS OF PHARMACOMICROBIOMIC DATA
Quality control methods used and analyses conducted depends on the type of sequencing performed. The most popular tool for 16S studies is QIIME2. 81 This program untangles sequence data into discrete units called operational taxonomic units, which are then taxonomically assigned using reference databases, such as Greengenes or SILVA. 82 , 83 As previously stated, the analysis of metagenomic data is computationally intensive. Perhaps most important, shotgun metagenomic sequencing requires preprocessing to remove host DNA sequences. Shotgun data can then be compiled with tools, such as Kraken 84 and HUManN2, 85 to generate taxonomic and functional profiles through steps such as assembly, binning, and alignment. Even if all other factors are held constant, results will vary with different analytic methods and programs. Thus, it is exceeding important to create thorough documentation of your quality control and analysis methods.
For both sequencing methods, higher‐level analyses, such as describing diversity, are used to evaluate patterns of variation prior to downstream analyses. Diversity is a measure of both richness, how many different species are in a sample, and their abundance, how much of each species is in the sample. The microbiome can be described in terms of alpha and beta diversities. Alpha diversity can be thought of as the variance within‐subjects. There are three main indices to examine this relationship: CHAO1 index, 86 Simpson diversity index, 87 and Shannon diversity index, 88 each with its strengths and limitations. Beta diversity, on the other hand, is how patient samples differ or are similar between each other. Beta diversity can be calculated and visualized by four main methods: Bray‐Curtis dissimilarity, 89 UniFrac distances, 90 and Principal Coordinates Analysis (PCoA). 91 Both types of diversity indices consider two aspects: how many organisms are present in the sample and their abundances. Alpha and beta diversities can be inferred from both cross‐sectional and longitudinal studies; however, longitudinal analysis holds the advantage of being able to study beta diversity between samples from one patient.
Statistical analyses of community composition
Pharmacomicrobiomic studies have three main investigative components: host, drug therapy, and microbiome. We can think about testing associations between these variables as three separate testable hypotheses. First, testing an association between the host and drug therapy, which will not change from other biomedical research questions involving the host and an intervention. Second, testing the association between the microbiome and the host, which will answer questions regarding the composition of the microbiome and phenotypic variability. Third, testing the association of drug response variability on a specific microbiome composition. Here, we will focus on the latter two types of hypotheses. Although these two types of hypotheses ask different questions, they may both investigate various components of the microbiome, such as species richness or evenness, alpha diversity, total number of reads, or phylogenetic diversity. For example, if we hypothesize that warfarin has antibacterial effects, we may wish to investigate the total number of reads of treated versus untreated patients. If we hypothesize that the gut microbiome effects stable warfarin dose, we may wish to investigate alpha diversity among low and high warfarin responders.
Many common tests are appropriate for analyzing the microbiome. We may use a t‐test or the corresponding nonparametric test, for example, to investigate differences between alpha diversities of experimental groups. The analysis of the human gut microbiome is complexified, however, by numerous advanced statistical features. The data are (1) compositional (parts of a whole), (2) high dimensional (data with many columns), (3) overdispersed (highly variable in read count), and (4) sparse with many zeros (many taxa have a zero read count). 92 Thus, we may choose to use more advanced statistical models, such as a negative binomial model or zero‐inflated negative binomial to account for the excess zeros and overdispersion. Further, if longitudinal studies are used, repeated measures analyses, such as general linear mixed models or generalized estimating equations, will be required to assess changes in a response variable over time. Although classical tests may be appropriate, advanced models will account for more features of the data. 93 As statistical analysis of the gut microbiome is increasingly challenging, we refer the reader to Statistical Analysis of Microbiome Data with R, by Xia, Sun, and Chen, for an in‐depth review of analyzing microbiome data. 92
Bioinformatic pipelines and R packages are central to microbiome analyses. QIIME 94 and mothur 95 are two comprehensive bioinformatics pipelines with copious documentation for the analysis of metagenomic as well as 16S amplicon sequencing data. Additionally, R packages can be repurposed from other fields to aid in microbiome analyses. DESeq2, 96 and edgeR, 97 for example, were developed to analyze transcript data and are useful in handling the overdispersed data of the gut microbiome.
CONCLUSIONS
The microbiome is providing pharmaceutical researchers with new insights into the mechanisms of variability in drug response, which allows for increases in drug efficacy and the potential to lower drug toxicity. Although large cohorts have been assembled to investigate pharmacomicrobiomic relationships, independent studies are necessary to answer specific questions, such as how does one’s microbial signature effect the dosing of a drug of interest or how does a metabolite produced in the gut interfere with a drug’s mechanism of action. Studies investigating pharmacomicrobiomic associations are challenging due to the many confounding variables, the number of methodological options available to researchers, and the advanced statistical methods required for analysis. We recommend first deciding on which sequencing method will be appropriate to answer the research question because it may determine the sampling method that will be used and will influence analyses that can be performed. Further, we recommend choosing as many exclusion criteria as feasible to still achieve a reasonably representative study population. Finally, we recommend researchers consider the advanced features of the microbiome when performing statistical analyses to decrease bias in results. Here, we provide a resource for researchers to aid in designing a pharmacomicrobiomic study that includes multiple rigorous options for each step of the study design process, helping weigh the benefits and drawbacks of the numerous currently available approaches.
FUNDING INFORMATION
This work is supported by an institutional career development award from the University of Arizona Health Science Center (J.H.K.) and a Seed Grant to Promote Translational Research in Precision Medicine from the Flinn Foundation (J.H.K.). J.H.K. is supported by the National Heart, Lung, and Blood Institute (NHLBI) grants K01HL143137, R01 HL156993, and R01HL158686.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
Steiner HE, Patterson HK, Giles JB, Karnes JH. Bringing pharmacomicrobiomics to the clinic through well‐designed studies. Clin Transl Sci. 2022;15:2303‐2315. doi: 10.1111/cts.13381
REFERENCES
- 1. Lam KN, Alexander M, Turnbaugh PJ. Precision medicine goes microscopic: engineering the microbiome to improve drug outcomes. Cell Host Microbe. 2019;26:22‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Doestzada M, Vila AV, Zhernakova A, et al. Pharmacomicrobiomics: a novel route towards personalized medicine? Protein Cell. 2018;9:432‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Spear BB, Heath‐Chiozzi M, Huff J. Clinical application of pharmacogenetics. Trends Mol Med. 2001;7:201‐204. [DOI] [PubMed] [Google Scholar]
- 4. Johnson JA, Gong L, Whirl‐Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90:625‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roden DM, Wilke RA, Kroemer HK, Stein CM. Pharmacogenomics: the genetics of variable drug responses. Circulation. 2011;123:1661‐1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belle DJ, Singh H. Genetic factors in drug metabolism. Am Fam Physician. 2008;77:1553‐1560. [PubMed] [Google Scholar]
- 7. Artacho A, Isaac S, Nayak R, et al. The pretreatment gut microbiome is associated with lack of response to methotrexate in new‐onset rheumatoid arthritis. Arthritis Rheumatol. 2021;73:931‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science. 2013;341:295‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science. 2018;359:104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takasuna K, Hagiwara T, Hirohashi M, et al. Involvement of beta‐glucuronidase in intestinal microflora in the intestinal toxicity of the antitumor camptothecin derivative irinotecan hydrochloride (CPT‐11) in rats. Cancer Res. 1996;56:3752‐3757. [PubMed] [Google Scholar]
- 11. Aziz RK, Saad R, Rizkallah MR. PharmacoMicrobiomics or how bugs modulate drugs: an educational initiative to explore the effects of human microbiome on drugs. BMC Bioinformatics. 2011;12:A10. [Google Scholar]
- 12. Goodrich JK, Waters JL, Poole AC, et al. Human genetics shape the gut microbiome. Cell. 2014;159:789‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee SH, Yun Y, Kim SJ, et al. Association between cigarette smoking status and composition of gut microbiota: population‐based cross‐sectional study. J Clin Med. 2018;7:E282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagpal R, Tsuji H, Takahashi T, et al. Ontogenesis of the gut microbiota composition in healthy, full‐term, vaginally born and breast‐fed infants over the first 3 years of life: a quantitative bird's‐eye view. Front Microbiol. 2017;8:1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Enright EF, Gahan CGM, Joyce SA, Griffin BT. The impact of the gut microbiota on drug metabolism and clinical outcome. Yale J Biol Med. 2016;89:375‐382. [PMC free article] [PubMed] [Google Scholar]
- 16. Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009;106:14728‐14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug‐microbiota interactions in anticancer therapies. Microbiome. 2018;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoo D‐H, Kim IS, Van Le TK, Jung IH, Yoo HH, Kim DH. Gut microbiota‐mediated drug interactions between lovastatin and antibiotics. Drug Metab Dispos Biol Fate Chem. 2014;42:1508‐1513. [DOI] [PubMed] [Google Scholar]
- 19. Hildebrand F, Moitinho‐Silva L, Blasche S, et al. Antibiotics‐induced monodominance of a novel gut bacterial order. Gut. 2019;68:1781‐1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Becattini S, Taur Y, Pamer EG. Antibiotic‐induced changes in the intestinal microbiota and disease. Trends Mol Med. 2016;22:458‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hao Z, Li L, Ning Z, et al. Metaproteomics reveals growth phase‐dependent responses of an in vitro gut microbiota to metformin. J Am Soc Mass Spectrom. 2020;31:1448‐1458. [DOI] [PubMed] [Google Scholar]
- 22. Maier L, Pruteanu M, Kuhn M, et al. Extensive impact of non‐antibiotic drugs on human gut bacteria. Nature. 2018;555:623‐628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jackson MA, Goodrich JK, Maxan ME, et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut. 2016;65:749‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de la Cuesta‐Zuluaga J, Mueller NT, Corrales‐Agudelo V, et al. Metformin is associated with higher relative abundance of mucin‐degrading Akkermansia muciniphila and several short‐chain fatty acid‐producing microbiota in the gut. Diabetes Care. 2017;40:54‐62. [DOI] [PubMed] [Google Scholar]
- 25. Lázaro‐Pacheco IB, Servín‐Caamaño AI, Pérez‐Hernández JL, Rojas‐Loureiro G, Servín‐Abad L, Tijera FH. Proton pump inhibitors increase the overall risk of developing bacterial infections in patients with cirrhosis. Arq Gastroenterol. 2018;55:28‐32. [DOI] [PubMed] [Google Scholar]
- 26. Integrative HMP (iHMP) Research Network Consortium . The integrative human microbiome project: dynamic analysis of microbiome‐host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16:276‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tigchelaar EF, Zhernakova A, Dekens JAM, et al. Cohort profile: lifelines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open. 2015;5:e006772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elliott P, Peakman TC, Biobank UK. The UKBiobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int J Epidemiol. 2008;37:234‐244. [DOI] [PubMed] [Google Scholar]
- 29. Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694‐1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Faith JJ, Guruge JL, Charbonneau M, et al. The long‐term stability of the human gut microbiota. Science. 2013;341:1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Goodrich JK, Davenport ER, Beaumont M, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flores GE, Caporaso JG, Henley JB, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;15:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guo Y, Logan HL, Glueck DH, Muller KE. Selecting a sample size for studies with repeated measures. BMC Med Res Methodol. 2013;13:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Srinivasan S, Hua X, Wu MC, et al. Impact of topical interventions on the vaginal microbiota and metabolome in postmenopausal women: a secondary analysis of a randomized clinical trial. JAMA Netw Open. 2022;5:e225032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Johnstone J, Meade M, Lauzier F, et al. Effect of probiotics on incident ventilator‐associated pneumonia in critically ill patients: a randomized clinical trial. JAMA. 2021;326:1024‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen RY, Mostafa I, Hibberd MC, et al. A microbiota‐directed food intervention for undernourished children. N Engl J Med. 2021;384:1517‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim CS, Grady N, Derrick M, et al. Effect of antibiotic use within first 48 hours of life on the preterm infant microbiome: a randomized clinical trial. JAMA Pediatr. 2021;175:303‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med. 2000;342:1887‐1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kunz R, Oxman AD. The unpredictability paradox: review of empirical comparisons of randomised and non‐randomised clinical trials. BMJ. 1998;317:1185‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24:392‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hannan EL. Randomized clinical trials and observational studies: guidelines for assessing respective strengths and limitations. JACC Cardiovasc Interv. 2008;1:211‐217. [DOI] [PubMed] [Google Scholar]
- 42. O'Toole PW, Jeffery IB. Gut microbiota and aging. Science. 2015;350:1214‐1215. [DOI] [PubMed] [Google Scholar]
- 43. Morgan XC, Tickle TL, Sokol H, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Imhann F, Vich Vila A, Bonder MJ, et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut. 2018;67:108‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peirce JM, Alviña K. The role of inflammation and the gut microbiome in depression and anxiety. J Neurosci Res. 2019;97:1223‐1241. [DOI] [PubMed] [Google Scholar]
- 46. Azzouz D, Omarbekova A, Heguy A, et al. Lupus nephritis is linked to disease‐activity associated expansions and immunity to a gut commensal. Ann Rheum Dis. 2019;78:947‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valitutti F, Cucchiara S, Fasano A. Celiac disease and the microbiome. Nutrients. 2019;11:E2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peng J, Xiao X, Hu M, Zhang X. Interaction between gut microbiome and cardiovascular disease. Life Sci. 2018;214:153‐157. [DOI] [PubMed] [Google Scholar]
- 49. Zhang Y, Shen J, Shi X, et al. Gut microbiome analysis as a predictive marker for the gastric cancer patients. Appl Microbiol Biotechnol. 2021;105:803‐814. [DOI] [PubMed] [Google Scholar]
- 50. Sims TT, Colbert LE, Zheng J, et al. Gut microbial diversity and genus‐level differences identified in cervical cancer patients versus healthy controls. Gynecol Oncol. 2019;155:237‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goodman B, Gardner H. The microbiome and cancer. J Pathol. 2018;244:667‐676. [DOI] [PubMed] [Google Scholar]
- 52. Jernberg C, Löfmark S, Edlund C, Jansson JK. Long‐term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56‐66. [DOI] [PubMed] [Google Scholar]
- 53. Chen Y, Liu B, Glass K, du W, Banks E, Kirk M. Use of proton pump inhibitors and the risk of hospitalization for infectious gastroenteritis. PloS One. 2016;11:e0168618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu H, Esteve E, Tremaroli V, et al. Metformin alters the gut microbiome of individuals with treatment‐naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850‐858. [DOI] [PubMed] [Google Scholar]
- 55. Kates AE, Jarrett O, Skarlupka JH, et al. Household pet ownership and the microbial diversity of the human gut microbiota. Front Cell Infect Microbiol. 2020;10:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tomova A, Bukovsky I, Rembert E, et al. The effects of vegetarian and vegan diets on gut microbiota. Front Nutr. 2019;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Singh RK, Chang HW, Yan D, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med. 2017;15:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walker AW, Ince J, Duncan SH, et al. Dominant and diet‐responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shim J‐S, Oh K, Kim HC. Dietary assessment methods in epidemiologic studies. Epidemiol Health. 2014;36:e2014009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Subar AF, Kirkpatrick SI, Mittl B, et al. The automated self‐administered 24‐hour dietary recall (ASA24): a resource for researchers, clinicians, and educators from the National Cancer Institute. J Acad Nutr Diet. 2012;112:1134‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fraser GE, Yan R, Butler TL, Jaceldo‐Siegl K, Beeson WL, Chan J. Missing data in a long food frequency questionnaire: are imputed zeroes correct? Epidemiol Camb Mass. 2009;20:289‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stirratt MJ, Dunbar‐Jacob J, Crane HM, et al. Self‐report measures of medication adherence behavior: recommendations on optimal use. Transl Behav Med. 2015;5:470‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Videnska P, Smerkova K, Zwinsova B, et al. Stool sampling and DNA isolation kits affect DNA quality and bacterial composition following 16S rRNA gene sequencing using MiSeq Illumina platform. Sci Rep. 2019;9:13837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bellali S, Lagier J‐C, Raoult D, Bou Khalil J. Among live and dead bacteria, the optimization of sample collection and processing remains essential in recovering gut microbiota components. Front Microbiol. 2019;10:1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim MY, Park Y‐S, Kim J‐H, Nam Y‐D. Evaluation of fecal DNA extraction protocols for human gut microbiome studies. BMC Microbiol. 2020;20:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qian X‐B, Chen T, Xu YP, et al. A guide to human microbiome research: study design, sample collection, and bioinformatics analysis. Chin Med J (Engl). 2020;133:1844‐1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jalanka J, Salonen A, Salojärvi J, et al. Effects of bowel cleansing on the intestinal microbiota. Gut. 2015;64:1562‐1568. [DOI] [PubMed] [Google Scholar]
- 69. Lecky DM, Hawking MKD, McNulty CAM, ESBL Steering Group . Patients’ perspectives on providing a stool sample to their GP: a qualitative study. Br J Gen Pract J R Coll Gen Pract. 2014;64:e684‐e693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu W‐K, Chen CC, Panyod S, et al. Optimization of fecal sample processing for microbiome study – the journey from bathroom to bench. J Formos Med Assoc Taiwan Yi Zhi. 2019;118:545‐555. [DOI] [PubMed] [Google Scholar]
- 71. Choo JM, Leong LEX, Rogers GB. Sample storage conditions significantly influence faecal microbiome profiles. Sci Rep. 2015;5:16350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Anderson EL, Li W, Klitgord N, et al. A robust ambient temperature collection and stabilization strategy: enabling worldwide functional studies of the human microbiome. Sci Rep. 2016;6:31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lim MY, Hong S, Kim BM, Ahn Y, Kim HJ, Nam YD. Changes in microbiome and metabolomic profiles of fecal samples stored with stabilizing solution at room temperature: a pilot study. Sci Rep. 2020;10:1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sinha R, Chen J, Amir A, et al. Collecting fecal samples for microbiome analyses in epidemiology studies. Cancer Epidemiol Biomark Prev Oncologia. 2016;25:407‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang Z, Zolnik CP, Qiu Y, et al. Comparison of fecal collection methods for microbiome and metabolomics studies. Front Cell Infect Microbiol. 2018;8:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469:967‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jovel J, Patterson J, Wang W, et al. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol. 2016;7:459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Franzosa EA, Sirota‐Madi A, Avila‐Pacheco J, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4:293‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Debyser G, Mesuere B, Clement L, et al. Faecal proteomics: a tool to investigate dysbiosis and inflammation in patients with cystic fibrosis. J Cyst Fibros off J Eur Cyst Fibros Soc. 2016;15:242‐250. [DOI] [PubMed] [Google Scholar]
- 80. Beger RD, Schmidt MA, Kaddurah‐Daouk R. Current concepts in Pharmacometabolomics, biomarker discovery, and precision medicine. Metabolites. 2020;10:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rai SN, Qian C, Pan J, et al. Microbiome data analysis with applications to pre‐clinical studies using QIIME2: statistical considerations. Genes Dis. 2021;8:215‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. McDonald D, Price MN, Goodrich J, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012;6:610‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sierra MA, Li Q, Pushalkar S, et al. The influences of bioinformatics tools and reference databases in analyzing the human oral microbial community. Genes. 2020;11:E878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Franzosa EA, McIver LJ, Rahnavard G, et al. Species‐level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962‐968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chao A. Nonparametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265‐270. [Google Scholar]
- 87. Simpson EH. Measurement of diversity. Nature. 1949;163:688. [Google Scholar]
- 88. Shannon CE. A mathematical theory of communication. Bell Syst Tech J. 1948;27:379‐423, 623‐656. [Google Scholar]
- 89. Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27:325‐349. [Google Scholar]
- 90. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228‐8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kruskal JB. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika. 1964;29:1‐27. [Google Scholar]
- 92. Xia Y, Sun J. Hypothesis testing and statistical analysis of microbiome. Genes Dis. 2017;4:138‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang H, Xia Y, Chen R, Gunzler D, Tang W, Tu X. Modeling longitudinal binomial responses: implications from two dueling paradigms. J Appl Stat. 2011;38:2373‐2390. [Google Scholar]
- 94. Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high‐throughput community sequencing data. Nat Methods. 2010;7:335‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537‐7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma Oxf Engl. 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]