

Abstract
This study aimed to evaluate the pharmacokinetics (PKs), safety, and immunogenicity of the biosimilar (RD12014) compared to reference liraglutide (Victoza) in healthy Chinese male subjects, so as to provide the basis for the similarity evaluation of the two drugs. Eligible subjects were randomized 1:1 to two sequences (RD12014‐Victoza or Victoza‐RD12014). Subjects received a single 0.6 mg dose of Victoza or RD12014 by abdominal subcutaneous injection during the first period. After a 7‐day washout period, subjects received the alternative drug during the second period. Blood samples were collected at predefined timepoints for PKs and immunogenicity assessment. The primary PK end points were maximum plasma concentration (C max) and area under the concentration‐time curve from time zero to the time of the last quantifiable concentration (AUC0−last). PK bioequivalence was achieved, if the 90% confidence intervals (CIs) of the geometric mean ratio (GMR) of C max and AUC0−last were within the range of 80.00–125.00%. Safety was assessed throughout the study. The 90% CIs of the GMR of RD12014 to Victoza for C max and AUC0−last were completely within the range of 80.00–125.00%. Thirteen treatment‐related adverse events (TRAEs) were reported in 11 subjects (22.4%) in the RD12014 group, compared to 12 TRAEs reported in 12 subjects (24.5%) in the Victoza group. The blood samples of 49 subjects were negative for anti‐drug antibody and the neutralizing antibody was not further detected. This study demonstrated PK similarity of RD12014 to Victoza in healthy Chinese male subjects. Safety and immunogenicity profiles were comparable between the two groups.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
RD12014, the biosimilar to liraglutide (Victoza), is indicated for the treatment of type 2 diabetes mellitus (T2DM), and is to be administered by subcutaneous injection once daily. At the time of this study, RD12014 was not approved for the treatment of T2DM in China, and there are no liraglutide biosimilar products in the Chinese market.
WHAT QUESTION DID THIS STUDY ADDRESS?
The primary objective of this phase I study was to evaluate the pharmacokinetic (PK) similarity between RD12014 and reference liraglutide (Victoza), and the secondary objective was to assess the similarity of safety and immunogenicity.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study indicated PK similarity of RD12014 to Victoza in healthy Chinese male subjects. The safety of the two drugs was good and similar. Neither of the two drugs showed immunogenicity.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Findings from this investigation were used to support a new drug application in China.
INTRODUCTION
Diabetes mellitus (DM) is a chronic metabolic disease characterized by hyperglycemia caused by insulin secretion deficiency and insulin resistance. The global diabetes map (2021) released by the International Diabetes Federation (IDF) shows that about 537 million (10.5%) of adults aged 20–79 years worldwide are diabetic. 1 Type 2 DM (T2DM) accounts for ~90% of all cases of diabetes and its prevalence is increasing yearly. T2DM increases the risk of cardiovascular disorders, blindness, renal failure, amputation, cognitive decline, cancer, and cognitive decline. There are some deficiencies in traditional antidiabetic drugs, such as insulin and sulfonylurea, which may cause hypoglycemia. Therefore, it is necessary to develop new drugs of new mechanisms with a good efficacy and low side effects to effectively control the blood sugar of patients so as to reduce the occurrence of complications.
Glucagon‐like peptide‐1 (GLP‐1), an incretin hormone, 2 is known to lower glucose levels, suppress glucagon secretion, protect pancreatic beta‐Cells, 3 decrease gastric emptying, 4 and increase reduction in bodyweight. 5 These properties make GLP‐1 an ideal target in treating T2DM. But endogenous GLP‐1 has a very short elimination half‐life (<1.5 min after intravenous) due to rapid degradation by dipeptidyl peptidase IV (DPP‐IV) and neutral endopeptidase (NEP), so native GLP‐1 is not a practical option for treatment and it is necessary to develop GLP‐1 analogue with a longer half‐life.
Liraglutide is an acylated GLP‐1 analogue with 97% amino acid homology with human GLP‐1 and greatly protracted action. 6 It can stimulate insulin secretion and reduce excessive glucagon secretion in a glucose concentration‐dependent mode. It is widely used for the treatment of T2DM, and administered by subcutaneous injection once daily. 7 The mechanism of prolonging the action time includes: self‐association in the pharmaceutical solution; binding to albumin; high enzyme stability for DPP‐IV and NEP. 8
Liraglutide (Victoza) was approved as an adjunct therapy to diet and exercise for treatment of T2DM in adults by the European Medicines Agency in 2009 and by the US Food and Drug Administration (FDA) in 2010. In addition, Victoza has been approved to prevent cardiovascular events in adults with T2DM by the FDA in 2017 and China in 2020. Despite the significant improvement in the treatment of T2DM in recent years, the high cost of biotherapy limits the treatment of patients. New biosimilar products with low cost can overcome cost barriers to a certain extent and improve their use worldwide. Several liraglutide biosimilars are still in clinical trials. Some of them are in phase I (www.chinadrugtrials.org.cn, register number: CTR20202 001, CTR20210493, CTR20210968, and CTR20211463; https://clinicaltrials.gov, register number: NCT05029076) or phase III clinical trials (www.chinadrugtrials.org.cn, register number: CTR20192168, CTR20190791, CTR20200348, CTR 20200400, CTR20201453, CTR20210173, and CTR20211086). But there are no liraglutide biosimilar products in the Chinese market so far. These proposed liraglutide biosimilars are mainly intended for the treatment of T2DM Mai et al. 9 However, there are few published literatures available at present.
According to the biosimilar guidelines from relevant regulatory agencies, biosimilars are therapeutic biological products that are similar in quality, safety, and efficacy to approved reference biological drugs. 10 , 11 , 12 , 13 , 14 Based on Victoza (Nordisk Pharmaceutical, Denmark) as the reference drug, RD12014 was independently developed as the biosimilar to Victoza by Sunshine Lake Pharma Co., Ltd. (Guangdong, China). The preparation systems of the two drugs were different: the original expression system was saccharomyces cerevisiae; the self‐developed was E. coli. Preclinical studies had confirmed that RD12014 was similar to Victoza in pharmacodynamics (PDs), pharmacokinetics (PKs), and toxicology.
According to an additional directive guidance for liraglutide biosimilars by China National Medical Products Administration (NMPA) recently and current global regulatory guidelines on biosimilars, 10 , 11 , 12 , 13 , 14 , 15 , 16 further clinical comparative studies could be designed to investigate the similarity of PKs, safety, and immunogenicity between RD12014 and Victoza in healthy subjects.
SUBJECTS AND METHODS
Subjects
Subjects who met all of the following inclusion criteria were eligible for this trial: each subject was required to provide a written informed consent at the beginning of screening; body mass index from 19 to 26 kg/m2 and weight greater than or equal to 50 kg; healthy male subjects aged 18–45 years; healthy subjects must not have any clinically significant abnormalities in vital signs, physical examinations, chest radiography, abdominal B‐scan ultrasonography, 12‐lead electrocardiograms (ECGs), and clinical laboratory tests during the screening period; had no history of respiratory, circulatory, digestive, urinary, blood, endocrine, nervous system diseases, and metabolic abnormalities with clinical significance.
Subjects were excluded if they met the following exclusion criteria: taking any prescription drug, Chinese herbal medicine, over‐the‐counter drug, and health product (except routine vitamin supplements) within 2 weeks before the first administration; previously received liraglutide or any other GLP‐1 analogue; blood donation greater than 400 ml within 3 months before the first administration; hepatitis B surface antigen, hepatitis C virus antibody, human immunodeficiency virus (HIV) antibody, treponema pallidum antibody, or anti‐liraglutide antibody were positive; drug allergy or specific allergic diseases (asthma and urticaria), or known allergy to test drugs or their excipients; had a history or family history of medullary thyroid cancer or multiple endocrine neoplasia type 2; genetic diseases that are easy to induce medullary thyroid cancer; any drug abuse before screening; positive for urine drug screening test; more than 10 cigarettes per day within 3 months before the first administration; alcoholics or regular drinkers within 3 months before the first administration, or positive alcohol breath test.
The study was registered on https://clinicaltrials.gov (register no: NCT05294536). The study protocol was in accordance with Good Clinical Practice guidelines, the principles in the Declaration of Helsinki, 17 and was approved by the Ethics Committee of Shanghai Xuhui Central Hospital. Written informed consents were obtained from all subjects before any study‐related procedures were performed.
Study drug
The test drug was RD12014 (batch number: L01201910001), the specification was 3 ml: 18 mg (refill), provided by Sunshine Lake Pharma Co., Ltd. (Guangdong, China). The reference drug Victoza (Nordisk Pharmaceutical, Denmark), batch number JVGT863‐1, has been approved in China and the specification was 3 ml: 18 mg (pre‐filled injection pen). Both RD12014 and Victoza used a single batch of drugs and stored below 30°C or in a refrigerated unit at 2–8°C avoiding freezing. The validity period after the first use is 1 month. Be sure to keep the pen cap on when not in use and protect it from sunlight. Each subject was administered subcutaneously in the abdomen once with RD12014 or Victoza®, and the dosage was 0.6 mg.
Study design
This was a randomized, open‐label, two‐period crossover study with single dose abdominal subcutaneous of liraglutide in healthy Chinese male subjects from June to July 2020, conducted in phase I Clinical Research Unit of Shanghai Xuhui Central Hospital (Shanghai, China).
Using block randomization, eligible subjects were randomized at a 1:1 ratio into two sequences (RD12014‐Victoza or Victoza‐RD12014). The duration of the study was about 25 days, which was divided into three periods: the screening period (14 days), two administration periods (4 days, respectively), and a washout period between two administrations (7 days).
Screening was completed within 14 days prior to dosing. On the day before administration (day‐1), all eligible subjects entered the research center where they were provided uniform accommodation and a standardized diet, and were randomized 1:1 into two sequences. In the first period, subjects were administered a single abdominal subcutaneous injection of 0.6 mg RD12014 or Victoza at about 8:00 (± 1 h) AM on day 1, then collected PK blood samples for 0–72 h according to the timepoint specified in the scheme and completed the safety observation. After a 7‐day washout period, another drug was cross administered for the second period. The administration method, time, dose, and PK sampling timepoint of the two periods were consistent. The safety and immunogenicity specified in the protocol were assessed throughout the study.
Subjects were required to adhere to certain restrictions. Food was forbidden for greater than or equal to 10 h before dosing. All participants were requested to maintain a light and balanced diet. Smoking or diet containing alcohol, tea, coffee, grapefruit juice, chocolate, and coca cola were not allowed. Strenuous physical activities were avoided within 24 h before administration and during the whole study. The subjects were under close medical monitoring during confinement to the center for a 72‐h period after administration.
Pharmacokinetic assessment
Blood samples were drawn through a heparin‐locked catheter (B. Braun Co., Penang, Malaysia) containing 0.5 ml of 0.4% heparin sodium. A total of 15 blood samples in a period were collected from each subject, with ~4 ml at each timepoint. Blood samples for PK assessment were collected at the following timepoints: predose (within 30 minutes before administration), and 2, 4, 6, 8, 9, 10, 11, 12, 13, 14, 24, 36, 48, and 72 h after administration. The collected blood samples were placed upright on the tube rack at room temperature, and the plasma was separated through centrifugation at 1500 g for 10 min, aliquoted into two polypropylene tubes (detection tube: 0.8 ml; backup tube: the rest) and stored at −80°C within 1 h after centrifugation until further analysis. Analysis was performed by Shanghai Xihua Scientific Co., Ltd., using a validated liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) method. Following protein precipitation and solid phase extraction, the plasma supernatant produced was injected into the LC‐MS/MS system to determine the concentration of liraglutide. The analysis was performed with an analytical column (Waters ACQUITY UPLC Peptide BEH C18, 50 × 2.1 mm, 1.7 μm) and a gradient elution consisting of mobile phase A (0.1% formic acid in 100% water) and mobile phase B (0.1% formic acid in 100% acetonitrile). The flow rate was 0.6 ml/min, and the injection volume was 15 μl. The mass spectrometer was set at the mode of positive ion electrospray and multiple reaction monitoring.
Lower limit of quantification (LLOQ) was 0.5 ng/ml and the upper limit of quantitation was 80.0 ng/ml. All plasma concentrations of subjects with less than the LLOQ were recorded as below quantification limit (BQL). The intra‐run and inter‐run precision were less than or equal to 9.2% (percent coefficient of variation [CV%]), whereas the intra‐run and inter‐run accuracy were between −7.8% and 12.8%. The sensitivity and selectivity of this method were good, and the method is suitable for the analysis of liraglutide in plasma.
Immunogenicity assessment
Blood samples for anti‐drug antibody (ADA) and neutralizing antibody (Nab) assessment were detected to evaluate immunogenicity with ~4 ml at each timepoint. During the screening period, only blood samples were collected to detect ADA. The ADA and Nab were detected on day 1 (predose in the second period), day 4 (in the second period), and early withdrawal (≥7 days after the first dose). If ADA was positive, a further analysis was used to detect the Nab. The collected blood samples were placed at room temperature for at least 30 min, then the plasma was separated through centrifugation at 1500 g for 10 min, aliquoted into two polypropylene tubes (detection tube: 1 ml; and the backup tube: the rest) and stored at −80°C until further analysis. According to the current global regulatory guidelines of the biological sample analysis, 18 a validated electro‐chemiluminescence technology based on the verified Meso Scale Discovery (MSD) platform was used to qualitatively detect ADA in human serum. The analysis method consists of three parts: screening assay, confirmation assay, and titration assay. ADA present in the samples were detected through formation of the complex of ADA and the labeled drug molecules. At first, the 1:10 diluted samples were added to the master mix containing biotin‐labeled and ruthenium‐labeled RD12014, and then incubated on the dilution plate to make the bridging reaction between the drug and antibody to form the bridging complex. After the reaction was completed, the samples were transferred to the MSD plate for incubation, and then the plate was washed to remove the unbound substance. At last, the plate buffer was added to the samples and the values on the MSD were read. The quantitative range of this method was 70–4000 ng/ml. The screening sensitivity of ADA in human serum was 56.70 ng/ml, the confirmation sensitivity was 73.65 ng/ml, and the sample volume was 20 μl.
Safety evaluation
Safety similarity was assessed primarily by comparing the type, frequency, timing, severity, and relevancy of adverse events (AEs). Safety evaluations included clinical laboratory tests (hematology, blood biochemistry, and urinalysis), vital signs (blood pressure, pulse, respiratory frequency, and body temperature), physical examinations, 12‐lead ECGs, and injection site reactions. After sitting for at least 5 min, vital signs were monitored within 1 h predose and at 2, 8, 12, 24, 48, 72 h postdose. The 12‐lead ECGs were conducted on day −1 (first period), and day 4 (both of the two periods). Clinical laboratory tests were performed at day 4 (both of the two periods). Physical examinations were performed at day 4 of the second period.
AEs, which were encoded using the Medical Dictionary for Regulatory Activities (MedDRA, version 23.0), were managed and recorded promptly by qualified investigators during the whole study. The severity was graded according to the common terminology criteria for adverse events (CTCAE) version 5.0. Subjects with AEs were required to follow up until the symptoms or the corresponding physical and/or clinical examinations returned to normal or baseline. The prevalence of AEs was calculated by the number of subjects who experienced at least one AE divided by the total number of subjects.
Statistical methods
Statistical analysis software SAS version 9.4 was used for statistical analysis. Based on previous studies in healthy volunteers, the CV of maximum plasma concentration (C max) is about 30%, which is higher than that of area under the concentration‐time curve (AUC; about 22%), α = 0.05, 1‐β = 80%, the 90% confidence interval (CI) of the geometric mean ratio (GMR) is 80.00–125.00%, thus the initial calculated sample size was 40. Assuming a 20% dropout rate, the final sample size of 50 subjects (25 per sequence) was planned. Charts are summarized in drug groups (RD12014/Victoza group) or sequences (RD12014‐Victoza/Victoza‐RD12014). According to the administration group, the mean plasma concentration‐time curve was drawn according to the blood collection time point and blood drug concentration (including average and individual), which was displayed on linear and semilogarithmic scales.
Phoenix WinNonlin software (version 8.2) was used to calculate PK parameters using the noncompartmental analysis model. Plasma concentrations of BQL values were represented as zero if the samples were taken before the time to maximum plasma concentration (T max) and the missing value for samples taken after T max. The calculated PK parameters based on the concentration data of each subject included: C max, T max, AUC0−last, AUC0−inf, elimination half‐life time (t 1/2), apparent total oral clearance (CL/F), elimination rate constant (λ z), and apparent volume of distribution based on the terminal phase (V d/F). AUC0−last and AUC0−inf were calculated using a linear trapezoidal rule method.
The primary PK end point parameters of this trial were C max and AUC0−last. The secondary PK end point parameters included AUC0−inf, T max, t 1/2, CL/F, V d/F, and λ z. The GMR and bilateral 90% CIs of the main PK parameters (C max and AUC0−last) of the test and the reference drug were calculated. PK bioequivalence was established, if the 90% CI of the GMR was completely within the range of 80.00–125.00%. Wilcoxon signed‐rank test was carried out for nonparametric analysis of T max. In the case where p > 0.05, it could be considered that the difference in T max between the two drug groups was not statistically significant.
Descriptive analysis was applied to summarize the demographic and baseline data. Frequency and percentage were calculated to summarize categorical variables.
RESULTS
Demographics and disposition
A total of 167 subjects were screened in this trial, of which 117 subjects failed. Fifty subjects were randomly 1:1 into two sequences (25 subjects in each group). Except for one subject (Victoza‐RD12014 sequence) that withdrew before the first dose of study drug due to the unqualified blood pressure, all other subjects received study drugs as allocated and were included in the PK and safety analysis. All subjects were men. The baseline characteristics of the subjects were generally comparable between two sequences and summarized in Table 1.
TABLE 1.
Summary of the baseline characteristics of all subjects in the study
| Parameters | RD12014‐Victoza | Victoza‐RD12014 | Total |
|---|---|---|---|
| (n = 25) | (n = 24) | (n = 49) | |
| Ethnics | |||
| Han, n (%) | 24 (96.0) | 24 (100) | 48 (98.0) |
| Other, n (%) | 1 (4.0) | 0 (0) | 1 (2.0) |
| Male, n (%) | 25 (100) | 25 (100) | 49 (100) |
| Mean age (SD), years | 27.7 (5.01) | 26.2 (5.79) | 26.9 (5.41) |
| Mean height (SD), cm | 168.40 (5.845) | 170.13 (5.005) | 169.24 (5.463) |
| Mean weight (SD), kg | 63.44 (6.874) | 65.01 (7.012) | 64.21 (6.915) |
| Mean BMI (SD), kg/m2 | 22.34 (1.882) | 22.42 (1.600) | 22.38 (1.732) |
Abbreviations: BMI, body mass index; cm, centimeter; kg, kilogram; m, meter; SD, standard deviation.
Pharmacokinetics
Comparing the mean plasma concentration‐time curves between RD12014 and Victoza, PK similarity was presented clearly (Figure 1). The mean values of PK parameters (including C max, AUC0−last, AUC0−inf, T max, t 1/2, CL/F, V d /F, and λ z) were similar between the two groups (data are shown in Table 2). Based on the overall data analysis of all evaluable subjects, the PK parameter similarity analysis was carried out. As displayed in Table 3 and Figure 2, The GMR of the test to reference for C max and AUC0−last were 112.07% and 111.52%, respectively. The 90% CIs of the GMR for C max and AUC0−last were 106.29–118.18% and 106.73–116.53%, respectively, which were completely within the predefined equivalence margin of 80.00–125.00%, and the bioequivalence of these PK parameters was demonstrated. Additionally, the GMR value of AUC0−inf is almost equal to that of AUC0−last (AUC0−last/AUC0−inf ≈ 98%), therefore 90% CI of the GMR for AUC0−inf (106.37–115.57%) was also within the equivalence margin of 80.00–125.00%, which also support the bioequivalence.
FIGURE 1.
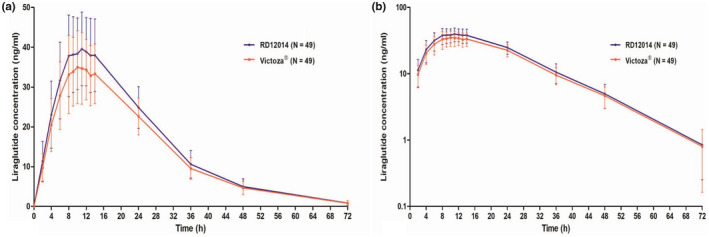
Mean plasma concentration‐time profiles of liraglutide after a single 0.6 mg subcutaneous administration of Victoza or RD12014. (a) Linear scale; (b) semilogarithmic scale. The error bars are SD.
TABLE 2.
Summary of PK parameters of RD12014 and Victoza
| PK parameter arithmetic mean (SD) [CV%] | RD12014 (n = 49) | Victoza (n = 49) |
|---|---|---|
| C max (ng/ml) | 42.11 (9.868) [23.43] | 37.58 (9.025) [24.01] |
| AUC0−last (h*ng/ml) | 1084.8130 (225.2798) [20.77] | 967.8264 (183.0252) [18.91] |
| AUC0−inf (h*ng/ml) | 1103.7647 (225.1409) [20.40] | 990.1840 (181.6512) [18.35] |
| T max (h) | 11 (8–14.017) | 11 (8–24) |
| t 1/2 (h) | 9.8677 (1.2213) [12.38] | 10.3209 (1.8196) [17.63] |
| CL/F (ml/h) | 567.7798 (126.3296) [22.25] | 626.3771 (116.5173) [18.60] |
| V d/F (ml) | 8060.3928 (1978.2818) [24.54] | 9363.2055 (2624.9996) [28.04] |
| λ z (1/h) | 0.0713 (0.0090) [12.63] | 0.0689 (0.0103) [14.94] |
Note: Data are presented as arithmetic mean (SD) [CV%] except T max are presented as median (minimum‐maximum).
Abbreviations: AUC0−last, area under the concentration‐time curve from time 0 to the time of the last quantifiable concentration; AUC0−inf, area under the concentration‐time curve from time 0 to infinity; CL/F, apparent total oral clearance; C max, maximum plasma concentration; h, hour; ml, milliliter; ng, nanogram; PK, pharmacokinetic; SD, standard deviation; t 1/2, elimination half‐life time; T max, time to maximum plasma concentration; V d/F, apparent volume of distribution based on the terminal phase; λ z, elimination rate constant.
TABLE 3.
Similarity analysis results of primary PK parameters
| PK parameter geometric mean | GM | GMR a (%) | 90% CI (%) | CVw (%) | |
|---|---|---|---|---|---|
| RD12014 | Victoza | ||||
| C max (ng/ml) | 41.00 | 36.58 | 112.07 | (106.29, 118.18) | 15.7 |
| AUC0−last (h*ng/ml) | 1061.06 | 951.44 | 111.52 | (106.73, 116.53) | 13.0 |
| AUC0−inf (h*ng/ml) | 1080.49 | 974.52 | 110.87 | (106.37, 115.57) | 12.3 |
Note: Bioequivalence was declared if the 90% CIs were within the range of 80.00–125.00%. Test group: RD12014; reference group: Victoza.
Abbreviations: AUC0−last, area under the concentration‐time curve from time 0 to the time of the last quantifiable concentration; AUC0−inf, area under the concentration‐time curve from time 0 to infinity; CI, confidence interval; C max, maximum plasma concentration; CVw, within‐subject coefficient of variation; GM, geometric mean; GMR, geometric mean ratio; h, hour; ml, milliliter; ng, nanogram; PK, pharmacokinetic; SD, standard deviation.
Test‐to‐reference ratio of geometric means.
FIGURE 2.
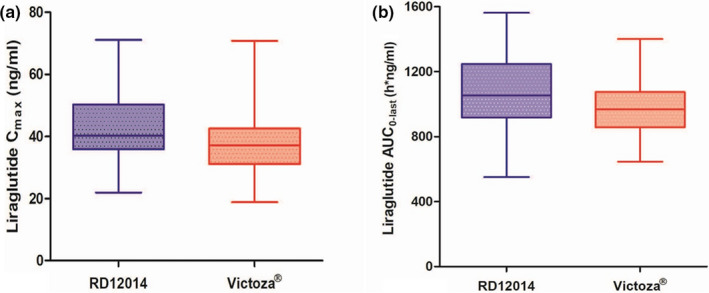
Box‐plot comparing C max (a), AUC0−last (b) of RD12014 and Victoza. The bottom and top of the box represent the first and third quartiles, respectively, and the horizontal lines inside the box represent the median, and the ends of the extension line are the minimum and maximum, respectively. AUC0−last, area under the concentration‐time curve from time 0 to the time of the last quantifiable concentration; C max, maximum plasma concentration.
Safety
The overall safety profiles for both RD12014 and Victoza were similar (Table 4). A total of 33 cases of AEs occurred in 24 subjects (49.0%) during the whole study: 14 AEs occurred in 11 subjects (22.4%) in the RD12014 group; and 19 AEs occurred in 16 subjects (32.7%) in the Victoza group. Of these, all 33 AEs were mild, and all had been resolved without medical intervention by the end of the study. There were no serious AEs or deaths that occurred during the study.
TABLE 4.
Summary of TRAEs in the RD12014 and Victoza groups
| MedDRA preferred term | RD12014 group (n = 49) | Victoza group (n = 49) | Total (n = 49) |
|---|---|---|---|
| Any AE | 11 (22.4) | 16 (32.7) | 24 (49.0) |
| Number of events | 14 | 19 | 33 |
| Any TRAE | 11 (22.4) | 12 (24.5) | 21 (42.9) |
| Number of events | 13 | 12 | 25 |
| Any SAE | 0 | 0 | 0 |
| AEs | |||
| Serum uric acid increased | 3 (6.1) | 3 (6.1) | 6 (12.2) |
| Blood bilirubin increased | 2 (4.1) | 3 (6.1) | 5 (10.2) |
| Blood triglyceride increased a | 1 (2.0) | 3 (6.1) | 4 (8.2) |
| Blood glucose decreased | 1 (2.0) | 2 (4.1) | 3 (6.1) |
| Upper respiratory tract infection b | 0 (0) | 2 (4.1) | 2 (4.1) |
| Total bile acid increased | 1 (2.0) | 1 (2.0) | 2 (4.1) |
| Conjugated bilirubin increased | 2 (4.1) | 0 (0) | 2 (4.1) |
| Alanine aminotransferase increased | 1 (2.0) | 0 (0) | 1 (2.0) |
| Neutrophil count increased | 0 (0) | 1 (2.0) | 1 (2.0) |
| Aspartate aminotransferase increased | 1 (2.0) | 0 (0) | 1 (2.0) |
| Vomit a | 0 (0) | 1 (2.0) | 1 (2.0) |
| Nausea a | 0 (0) | 1 (2.0) | 1 (2.0) |
| Diarrhea | 1 (2.0) | 0 (0) | 1 (2.0) |
| Abdominal distention | 1 (2.0) | 0 (0) | 1 (2.0) |
| Fever | 0 (0) | 1 (2.0) | 1 (2.0) |
| Hematuria a | 0 (0) | 1 (2.0) | 1 (2.0) |
Note: Data are presented as number of subjects (%).
Abbreviations: AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities; SAE, serious adverse event.
AEs were not considered treatment‐related.
One of the upper respiratory tract infections was not considered treatment‐related.
The most frequently reported AE was serum uric acid increase, experienced by three subjects (6.1%) in the RD12014 group. Serum uric acid increase, blood bilirubin increase, and blood triglyceride increase were the most frequently reported AEs in three subjects (6.1%), respectively, in the Victoza group. A total of 21 subjects (42.9%) reported 25 TRAEs: 11 subjects (22.4%) with 13 TRAEs in the RD12014 group; and 12 subjects with 12 TRAEs (24.5%) in the Victoza group. The incidence of AEs in the RD12014 group (22.4%) was slightly lower compared with that in the Victoza group (32.7%), whereas TRAEs was more similar (22.4% vs. 24.5%) in the two groups. The common TRAEs (incidence ≥10%) in this study were serum uric acid increased (12.2%) and blood bilirubin increased (10.2%), the incidence of which were comparable in the two groups (6.1% vs. 6.1% and 4.1% vs. 6.1%). No major hypoglycemic episodes occurred during the trial. Only three had a slight decrease in fasting blood glucose of clinical laboratory tests with no symptomatics were reported in the three subjects. This study results indicated that GLP‐1 analogues reduce blood glucose in a glucose‐dependent mode and may even reduce the risk of hypoglycemia. The 12‐lead ECGs and physical examinations showed no clinically significant changes.
Immunogenicity
All healthy subjects enrolled were negative for ADA before dosing. The blood samples of 49 subjects who completed the study were negative for ADA after dosing, and the Nab was not further detected.
DISCUSSION
The primary objective of this phase I study was to evaluate the PK similarity between RD12014 and reference liraglutide (Victoza), and the secondary objective was to assess the similarity of safety and immunogenicity.
The assessment of bioequivalence needs to take a stepwise approach, starting with in vitro studies (structural/physicochemical and functional analysis) and proceeding to in vivo animal (PKs, safety, and immunogenicity comparison) and clinical studies (PKs, safety, and efficacy assessment). 10 , 11 , 12 , 13 , 14 The reference liraglutide (Victoza) has been approved for registration in China. According to the preclinical study of Victoza and previous Liraglutide PK studies, 9 , 19 , 20 , 21 the PK comparative study of single administration and cross design was used to compare the drug exposure characteristics, and the design of the washout period was reasonable. According to the drug instruction, Victoza is dosed once daily using a prefilled pen. Treatment is initiated at 0.6 mg per day for 1 week. This initial dose is intended to reduce gastrointestinal symptoms. After 1 week, the dose is increased to 1.2 mg, and can be further increased to 1.8 mg based on glycemic control. 6 In order to protect healthy subjects and meet the sufficient exposure to accurately assess PKs, this study was designed to choose a relatively sensitive low‐dose 0.6 mg abdominal subcutaneous injection in a unified injection site. To avoid more variation factors of PKs in this study, healthy subjects were selected as the most sensitive population for the single‐dose PK study.
For the PK similarity comparison of C max, AUC0−last, and AUC0−inf, 90% CIs of the GMR for the test to reference were completely within the predefined equivalence margin of 80.00–125.00%. These data indicated that the PKs of RD12014 and reference Victoza were equivalent in healthy Chinese male subjects.
In the original research instructions, the common adverse reactions of liraglutide during the clinical trial (phase III and after marketing) include nausea, diarrhea, vomiting, constipation, abdominal pain, abdominal distention, dyspepsia, hypoglycemia, upper respiratory tract infection, nasopharyngitis, headache, etc. The common adverse reactions in laboratory examinations are increased lipase and amylase. A previous study reported that the most frequent AEs were of gastrointestinal origin after multiple Victoza administration in healthy Chinese male subjects, by Jiang et al. 21 The most frequent AEs were hyperidrosis, dizziness, and malaise after a single proposed liraglutide biosimilar administration in healthy Chinese male subjects Mai et al. 9 The safety was comparable for both the two groups and it is basically consistent with the original research data. The incidence of gastrointestinal origin (mainly nausea, diarrhea, and abdominal pain) in this study was not high, possibly due to the low dose. 9 , 21 However, the incidence of various clinical laboratory abnormalities in this study is high, but were mild and transient, and had recovered without any medical treatment.
In this study, there was no difference in immunogenicity between the two groups (no subject tested positive for ADA, and the Nab was not further detected). The results might indicate that the incidence of the positive ADA response after a single dose was relatively low in the short term. However, in the subsequent clinical trials, the observation of long‐term immunogenicity should be taken into account.
This study had some limitations. Only healthy male volunteers aged 18–45 years were selected in this study. However, male, female, elderly patients, and even adolescents will use liraglutide to treat T2DM in clinical practice. Although no major hypoglycemic events occurred, finger‐stick blood glucose monitoring was not included in the safety assessment. Future research will continue to focus on gender and age factors and PDs on blood glucose and insulin.
In conclusion, the PKs, safety, and immunogenicity were similar between RD12014 and reference liraglutide (Victoza) in this study. In addition to the preclinical results, these results could support further clinical trials in patients, and provide an experimental support and data basis for clinical evaluation of biosimilars in China.
AUTHOR CONTRIBUTIONS
All authors: Wrote manuscript. RZ, LG, YZ, YL, GL: Designed Research. RZ, YW, WX, YZ, YL, GL: Performed Research. All authors: Analyzed Data.
FUNDING INFORMATION
This study was supported by Sunshine Lake Pharma Co., Ltd. (Guangdong, China).
CONFLICTS OF INTEREST
Linfeng Guo, Xianglei Gao, Wenjia Li, and Yulei Zhuang are employees of Sunshine Lake Pharma Co., Ltd. All other authors declared no competing interests for this work.
ACKNOWLEDGMENTS
The authors would like to extend thanks to all enrolled participants, investigators, and people who contributed to this study. The authors also acknowledge Janus Clinical Research Institude Co., Ltd (Tianjin, China) for data statistics. The authors acknowledge Shanghai Xihua Scientific Co., Ltd (Shanghai, China) for bioanalytical method validation and sample detection.
Zhou R, Guo L, Gao X, et al. A phase I study comparing the pharmacokinetics of the biosimilar (RD12014) with liraglutide (Victoza) in healthy Chinese male subjects. Clin Transl Sci. 2022;15:2458‐2467. doi: 10.1111/cts.13374
Ruirui Zhou and Linfeng Guo contributed equally to this work.
Clinicaltrials.gov identifier: United States National Library of Medicine (grant number NCT05294536).
Contributor Information
Gangyi Liu, Email: gyliu@shxh-centerlab.com.
Yanmei Liu, Email: ymliu@shxh-centerlab.com.
REFERENCES
- 1. IDF (International Diabetes Federation) Diabetes Atlas . 10th edn; 2021. Accessed December 18, 2021. https://diabetesatlas.org/data/en/country/42/cn.html
- 2. Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127‐136. [DOI] [PubMed] [Google Scholar]
- 3. Kim MH, Lee MK. The incretins and pancreatic beta‐cells: use of glucagon‐like peptide‐1 and glucose‐dependent insulinotropic polypeptide to cure type 2 diabetes mellitus. Korean Diabetes J. 2010;34:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horowitz M, Flint A, Jones KL. Effect of the once‐daily human GLP‐1 analogue liraglutide on appetite, energy intake, energy expenditure and gastric emptying in type 2 diabetes. Diabetes Res Clin Pract. 2012;97:258‐266. [DOI] [PubMed] [Google Scholar]
- 5. Ard J, Fitch A, Fruh S, Herman L. Weight loss and maintenance related to the mechanism of action of glucagon‐like peptide 1 receptor agonists. Adv Ther. 2021;38:2821‐2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jacobsen LV, Flint A, Olsen AK, Ingwersen SH. Liraglutide in type 2 diabetes mellitus: clinical pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2016;55:657‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sfairopoulos D, Liatis S, Tigas S, Liberopoulos E. Clinical pharmacology of glucagon‐like peptide‐1 receptor agonists. Hormones (Athens). 2018;17:333‐350. [DOI] [PubMed] [Google Scholar]
- 8. Malm‐Erjefält M, Bjørnsdottir I, Vanggaard J, et al. Metabolism and excretion of the once‐daily human glucagon‐like peptide‐1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug Metab Dispos. 2010;38:1944‐1953. [DOI] [PubMed] [Google Scholar]
- 9. Mai G, Fan L, Li M, et al. A randomized phase 1 pharmacokinetic study comparing the potential biosimilar LRG201902 with liraglutide (Victoza®) in healthy male subjects. Front Pharmacol. 2021;11:610880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. China Food and Drug Administration . Guidelines for the development and evaluation of similar biological products. China Food and Drug Administration; 2015. Accessed December 10, 2021. https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20150228155701114.html. [Google Scholar]
- 11. China Food and Drug Administration . Technical guidelines for the study of human bioavailability and bioequivalence in chemical pharmaceutical preparations. China Food and Drug Administration; 2005. Accessed December 10, 2021. https://www.nmpa.gov.cn/wwwroot/gsz05106/08.pdf. [Google Scholar]
- 12. European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: non‐clinical and clinical issues. European Medicines Agency; 2014. Accessed December 10, 2021. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐similar‐biological‐medicinal‐products‐containing‐biotechnology‐derived‐proteins‐active_en‐2.pdf [Google Scholar]
- 13. US Food and Drug Administration . Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. US Food and Drug Administration; 2015. Accessed December 18, 2021. https://www.fda.gov/media/82647/download [Google Scholar]
- 14. World Health Organization . Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs). WHO Technical Report Series; 2009: 1–134.
- 15. US Food and Drug Administration . Liraglutide clinical review. US Food and Drug Administration; 2018. Accessed November 18, 2021. https://www.fda.gov/media/130093/download [Google Scholar]
- 16. China National Medical Products Administration . Guidance for designing clinical trials of biosimilars of liraglutide injection; 2020. Accessed November 18, 2021. https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20200624092101964.html.
- 17. World Medical Association . Declaration of Helsinki. Ethical principles for medical research involving human subjects; 2001. Accessed December 18, 2021. https://www.wma.net/wp‐content/uploads/2016/11/DoH‐Oct2013‐JAMA.pdf [PMC free article] [PubMed]
- 18. US Food and Drug Administration . Immunogenicity Testing of Therapeutic Protein Products‐Developing and Validating Assays for Anti‐Drug Antibody Detection Guidance for Industry; US Food and Drug Administration; 2019. Accessed December 18, 2021. https://www.fda.gov/media/119788/download [Google Scholar]
- 19. Scott LJ. Liraglutide: a review of its use in adult patients with type 2 diabetes mellitus. Drugs. 2014;74:2161‐2174. [DOI] [PubMed] [Google Scholar]
- 20. Østoft SH, Bagger JI, Hansen T, et al. Glucose‐lowering effects and low risk of hypoglycemia in patients with maturity‐onset diabetes of the young when treated with a GLP‐1 receptor agonist: a double‐blind, randomized, crossover trial. Diabetes Care. 2014;37:1797‐1805. [DOI] [PubMed] [Google Scholar]
- 21. Jiang J, Zhang J, Jacobsen LV, Hu P. The pharmacokinetics, pharmacodynamics, and tolerability of liraglutide, a once‐daily human GLP‐1 analogue, after multiple subcutaneous administration in healthy Chinese male subjects. J Clin Pharmacol. 2011;51:1620‐1627. [DOI] [PubMed] [Google Scholar]