

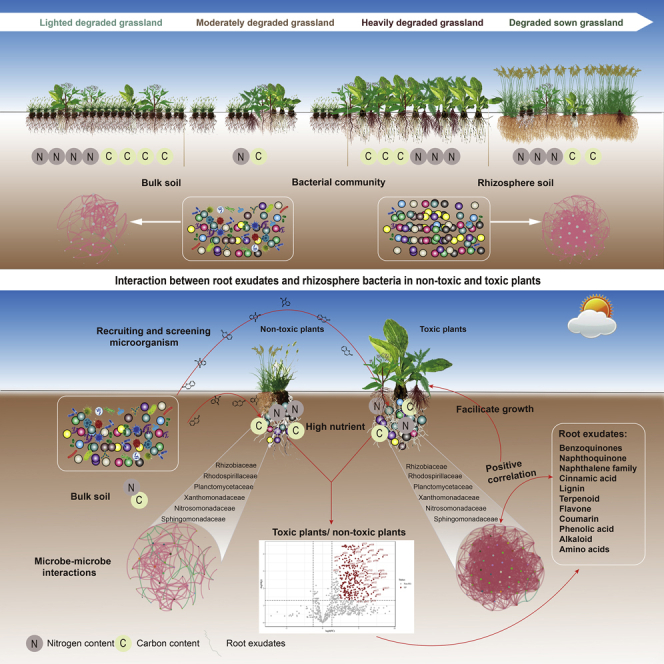
Summary
Root exudates and rhizosphere microorganisms play key roles in the colonization of toxic plants under climate change and land degradation. However, how root exudates affect the rhizosphere microorganisms and soil nutrients of toxic plants in degraded grasslands remains unknown. We compared the interaction of soil microbial communities, root exudates, microbial carbon metabolism, and environmental factors in the rhizosphere of toxic and non-toxic plants. Deterministic processes had a greater effect on toxic than non-toxic plants, as root exudates affected rhizosphere microorganisms directly. The 328 up-regulated compounds in root exudates of toxic plants affected the diversity of rhizosphere microorganisms. Rhizosphere bacteria-enriched enzymes were involved in the phenylpropanoid biosynthesis pathway. Root exudates of toxic plants form complex networks of rhizosphere microorganisms, provide high rhizosphere nutrients, and increase microbial carbon metabolism. The interaction between root exudates and rhizosphere microorganisms is the key mechanism that enables toxic plants to spread in degraded grassland habitats.
Subject areas: Biological sciences, interaction of plants with organisms, plant biology
Graphical abstract
Highlights
-
•
Root exudates are the main drivers for the spread of toxic plants in degraded grassland
-
•
Rhizosphere bacteria-enriched enzymes are involved in phenylpropanoid biosynthesis
-
•
Root exudates of toxic plants alter microbial network complexity
-
•
Root exudates provide nutrients and increase microbial carbon metabolism
Biological sciences; Interaction of plants with organisms; Plant biology.
Introduction
The expansion of plants that are toxic to grazing animals has become a global concern (Colautti and Barrett, 2013; Savary et al., 2019), as it is causing a substantial loss in grassland ecosystem services under climate change and anthropogenic activities (Humphries et al., 2021). Identifying the driving forces that enable the infestation of toxic plants would be beneficial in grassland management. Recent research on the spread of toxic plants has focused mainly on factors such as grazing livestock and climate change (Ricciardi et al., 2017; Liu et al., 2020). However, how toxic plants adapt to the poor soil conditions of degraded grassland is unknown.
The self-reinforcement ability of toxic plants is closely associated with the root system in degraded land (Wagg et al., 2011; Liu et al., 2018), as root exudates play a key role in plant growth and in the structure, diversity, and functioning of the rhizosphere microbial community (Koprivova et al., 2019; Zhou et al., 2021; Gross, 2022). For example, the tryptophan-derived molecule, camalexin, inhibited fungal pathogens and specific bacteria in Arabidopsis, thereby, altering the root microbiota (Koprivova et al., 2019). Umbelliferone in Stellera chamaejasme influenced cell division and induced membrane lipid peroxidation, which inhibited the growth of plants (Yan et al., 2016). Toxic plants produce a broad range of secondary metabolites (Shang et al., 2012; Yan et al., 2016), which have a substantial impact on the root environment (Goldschmidt et al., 2018).
Root exudates are the main driving forces regulating the diversity and metabolic activities of rhizosphere microorganisms during plant growth (De Vries et al., 2019). It was reported that wild oat (Avena fatua) had a more complex network in the rhizosphere than bulk soil, and microbial diversity decreased as network size decreased (Shi et al., 2016). Rhizosphere with a highly selective environment could promote species taxonomic co-occurrence, which could be indicative of increased metabolic specifics (Fan et al., 2018; Wang et al., 2020). It has been reported that microbial communities in the rhizosphere are shaped by deterministic processes (niche) and in bulk soil by stochastic processes (neutral) (Dumbrell et al., 2010; Mendes et al., 2014; Lima-Mendez et al., 2015). As habitat heterogeneity declines at smaller scales, a more apparent contribution of stochastic over deterministic processes is evident (Legendre et al., 2009).
Toxic plants are capable of spreading and dominating degraded grasslands with nutrient-poor soil, unlike non-toxic plants (Figure S1) (Sui et al., 2015; Yao et al., 2019; Humphries et al., 2021). We hypothesized that the interaction between rhizosphere microorganisms and root exudates facilitates the spread of toxic plants in degraded grasslands. To test this hypothesis, we measured microbial carbon metabolic activity, compounds, and functions of root exudates, and integrated taxonomic and functional data to describe the soil microbial communities of toxic and non-toxic plants in degraded alpine grasslands. The study was guided by the following predictions: (1) specific root exudates mediate the composition, network and carbon metabolic activity of rhizosphere microorganisms of toxic plants; (2) deterministic processes could explain the assembly of microorganisms in the rhizosphere of toxic plants better than stochastic processes; (3) the network complexity of the microbial community in toxic plants is greater than in non-toxic plants; and (4) the interaction of root exudates and rhizosphere microorganisms is the key factor that enables the toxic plant to spread in degraded grasslands.
Results
Community assembly and composition of the microbial community
For all plant species, the assemblies of microorganisms in different habitats and in rhizospheres of toxic or non-toxic plants were better described (R2 > 0.6) by the neutral theory than the niche theory (Figure 1), and R2 > 0.6 indicated that the assemblies were affected strongly by stochastic processes. In 2017, stochastic processes in the rhizosphere of toxic plants (R2 = 0.858, Figure 1B) were more evident when compared with bulk soil (R2 = 0.779, Figure 1A). Stochastic processes in the rhizosphere of toxic plants (R2 = 0.769, Figure 1D) were also more evident when compared with non-toxic plants (R2 = 0.690, Figure 1C), but Nm, meta-community size (N) times immigration rate (m), was lower (Figure 1).
Figure 1.

Fit of the neutral community model of community assemblies
(A) the bulk soil microorganisms in 2017 (n = 12); (B) the rhizosphere microorganisms of toxic plants in 2017 (n = 51); (C) non-toxic plants rhizosphere microorganisms in 2018 (n = 6); (D) rhizosphere microorganisms of toxic plants in 2018 (n = 21). The solid blue lines represent the best fit, and the dashed blue lines represent 0.95 confidence intervals around the model prediction. Different colors represent the frequency of OTUs. R2 = the coefficient of determination, Nm = the meta-community size times immigration rate.
The AIC value for toxic plants was lower than for non-toxic plants, indicating that the niche theory was more appropriate for the assembly of microorganisms in toxic plants. The Nm value in low density (135,334) Ligularia virgaurea was lower than in high (248,244) and medium densities (186,542) (Table S1). Because the number of sequences in the sample was 103,157, the m value in the high, medium, and low densities were 2.116, 1.808, and 1.312, respectively (Table S1), which indicated that as the density of L. virgaurea decreased, the niche theory was more applicable and species dispersal was more restricted. Compared with bulk soil, both theories existed in the rhizosphere of plants.
Distribution of microorganisms and influencing factors
A total of 12,282 OTUs were identified in 2017 (5923, 8031, 3110, and 6877 in degraded sown grassland (S), heavily degraded grassland (H), moderately degraded grassland (M), and lightly degraded grassland (L), respectively) and 5447 OTUs were identified in 2018 (2730, 2898, 1992 and 2525 in S, H, M, and L, respectively). The OTUs were classified into 44 microbial phyla in 2017 and 41 microbial phyla in 2018, with Proteobacteria, Actinobacteria, Acidobacteria, and Planctomycetes the four most abundant in all soil groups (Figure S2). The bulk soil of L grassland had the lowest α-diversity indices of bacterial and fungal communities across all samples (Kruskal-Wallis with Kruskal test, p < 0.05) (Figure 2A).
Figure 2.

Factors shaping the structure of microbial communities in 66 soil samples and 4 habitats
(A) Bray-Curtis similarity-based dendrogram showing average bacterial (left) and fungal (right) community compositions. OTUs with relative abundances >1% were considered. Levels of degradation are represented by colored squares (pink: lightly degraded grassland (L); gray: moderately degraded grassland (M); green: degraded sown grassland (S); and black: heavily degraded grassland (H)). For each sample, the community composition of bacteria and fungi at the class level is indicated by bar plots, and microbial α-diversity is represented by gray bars according to the number of observed OTUs.
(B) effect of compartment (n = 2), habitat (n = 4), plant species (n = 11) and habitat × plant species on bacterial and fungal community compositions. The explained variance for each factor is shown for bacteria and fungi, based on PERMANOVA (p < 0.001 in bacteria and p > 0.05 in fungi), habitat × plant species (Adonis: degrees of freedom (d.f.) = 21; bacterial: coefficient of determination (R2) = 0.53, p < 0.001; fungal: R2 = 0.32, p = 0.59), compartment (d.f. = 1; bacterial: R2 = 0.04, p < 0.001; fungal: R2 = 0.01, p = 0.62), degradation level (d.f. = 3; bacterial: R2 = 0.16, p < 0.001; fungal: R2 = 0.05, p = 0.91), plant species (d.f. = 10; bacterial: R2 = 0.27, p < 0.001; fungal: R2 = 0.15, p = 0.29). Compartments include bulk soil and rhizosphere.
(C) PCoA based on Bray-Curtis distances between samples across 66 samples in 4 levels of degradation. Nr, bulk soil, that is, non-rhizosphere soil. Al, Ajuga lupulina; Ef, Euphorbia fischeriana; Sg, Sphallerocarpus gracilis; Oo, Oxytropis ochrocephala; Mk, Morina kokonorica; Pk, Pedicularis kansuensis; An, Artemisia nanschanica; Ad, Artemisia dubia; Ap, Aconitum pendulum; Lv, Ligularia virgaurea.
When roots establish stable associations with microbial communities across different degradation levels, a strong host-filtering effect on degradation level is expected. Analysis of microbial community structure, based on average Bray-Curtis distances across the four degraded grasslands (Figure 2A) revealed that bacterial and fungal communities in bulk soil and in rhizosphere clustered by degradation level (Figure 2C). This pattern was corroborated by permutational multivariate analysis (PERMANOVA), which indicated that the interactions between plant species and habitat explained more of the variation in the microbial community than either compartment, degradation level, or plant species (Figure 2B). Bacterial communities were affected to a greater extent than fungal communities by degradation level and plant species.
Principal coordinate analysis (PCoA) revealed marked differences in soil bacterial and fungal communities in the 4 degraded grasslands. The bacterial community in M and the fungal community in S, in particular, were separated clearly from the others. These differences appeared in the same plant species at different degradation levels, suggesting divergence in the microbial community composition in the plant rhizosphere (Figure 2C). Nitrate nitrogen (NO3-N) in L grassland was lower (p < 0.05) than in the other three degradation levels, while ammonium nitrogen (NH4-N) exhibited an opposite trend (Figure S3A). The concentrations of TN, AP, and TOC in bulk soil of the four degraded grasslands were lower than in the rhizosphere (Tables S2 and S3). Redundancy analysis showed that soil physico-chemical properties explained 28.3% (p < 0.05) of the variation in rhizosphere bacterial communities (Figure S3B), but had no effect on fungal communities. Bacterial communities were correlated positively with TP, pH, and AP, but negatively with TN, TOC, NO3, NH4, and WC (Figure S3B).
Key-stone ecological cluster link to soil functions and root exudates
Ecological networks were used to identify clusters of microbial taxa highly correlated with each other, which could share environmental preferences and functional potentials. Six main ecological clusters (Module #0–5) were generated from the microbial network (Figures 3A and 3B). The gene network displayed that the relative abundances of microorganisms in key-stone ecological clusters were greater in most rhizospheres than in bulk soil (Table S4). The positive correlations among microbial taxa (density: 0.316–0.389) in rhizosphere were higher than in bulk soil (Figure 3D). Module #0–1 was correlated significantly and positively with soil chemical properties, and negatively with Module #2 (Figure S4).
Figure 3.

Ecological clusters based on microbial networks
(A) microbial network graph with nodes colored according to each of the six main ecological clusters (Module #0–5); (B) OTU number properties of the dominant phylotypes in the main ecological clusters; (C) regression relationships between soil biodiversity of key-stone ecological clusters (Module #0–2) and soil multi-functionality. Gray indicates confidence intervals; (D) dominant phylotypes from Module #0–2, the connection stands for a strong (Spearman’s r > 0.6) and significant (p < 0.05) correlation. The size of each node is proportional to the degree, and the thickness of edge is proportional to the value of Spearman’s correlation coefficient. A red edge indicates a positive correlation, while a green edge indicates a negative correlation.
The richness of bacteria and fungi was correlated positively with soil multi-functionality in Module #0 and Module #1, but negatively in Module #2 (Figure 3C). Given the soil multi-functionality of the three modules, Modules #0–2 were referred to as the key-stone ecological cluster and were dominated by the bacteria Alphaproteobacteria: Rhizobiales and Rhodospirillales; Betaproteobacteria: Nitrosomonadales; Deltaproteobacteria: Myxococcales, Planctomycetes: Tepidisphaeraceae, Gemmatimonadetes, Chloroflexi, Actinobacteria; and Nitrospirae; Acidobacteria (Figure 3B) and the fungi Dothideomycetes and Sordariomycetes. A complete list of the microbial taxa in the three modules is presented in Table S5. There were many positive correlations among the key-stone phylotypes (Figure 3D).
Correlations between root exudates and microorganisms
A total of 864 compounds were screened for analysis from 1092 compounds. The significantly up-regulated 328 compounds (Figure 4A) were selected through volcano analysis. The up-regulated compounds included benzoquinones, naphthoquinone (NQ), cinnamic acid (7), lignin (8), terpenoids (46), flavones (8), amino acids (11), coumarin (11), naphthalene family (17), phenolic acids (21), and alkaloids (20) (Figure 4B). Root exudates were species specific and the similarity of root exudates between different toxic plants was higher than with non-toxic plants (Figure S5).
Figure 4.

The distribution of root exudates and microbial taxa in rhizospheres of toxic and non-toxic plants
(A) The volcano plot of root exudates. Root exudates were selected with fold change threshold (x) 5 and t-test threshold (y) 0.05. The red circles represent features above the threshold.
(B) The correlation heatmap between microbial module eigengene and root exudates in toxic plants.
(C) The relative abundance of microorganisms in the rhizosphere of toxic and non-toxic plants in ME12. Values are mean ± SEM of non-toxic (n = 6) and toxic plants (n = 21).The Wilcoxon rank-sum test compared the difference between toxic and non-toxic plants.
(D) Microbial network structure of toxic and non-toxic plants in ME12. Node represent individual OTU. Node color denotes microbial phyla. A red edge indicates a positive correlation, while a green edge indicates a negative correlation.
(E) Metabolic pathways associated with toxic plants. Phenylpropanoid metabolism was significantly greater in toxic than non-toxic plants. All enzymes and enzyme nomenclature (EC) numbers were obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. The red and blue arrows indicate the compound or pathway showing a significant increase/decrease.
In total, 9852 microbial OTUs (bacteria: 5447; fungi: 4405) were generated and 15 modules (ME1-15). The microbial module eigengene was correlated significantly and positively with the up-regulated compounds in toxic plants (Figure 4B) and negatively in non-toxic plants (Figure S6). The ME12, which was correlated significantly with soil multi-functionality (Figure S7), was selected and analyzed for the network structure composition of microorganisms. The relative abundances of microorganisms in ME12 in toxic plants were greater than in non-toxic plants (Figure 4C). ME12 had 67 OTUs (Bacteria: 51; Fungi: 16) belonging to 10 phyla, with Proteobacteria (31%) and Planctomycetes dominant in bacteria, and Ascomycota and Basidiomycota dominant in fungi. The main classes of bacteria were Alphaproteobacteria and Planctomycetacia (Table S6).
Based on Spearman correlations, 14 phyla in ME12 were correlated positively (p < 0.05) with up-regulated compounds in toxic plants (Figure S8A), but most were correlated negatively in non-toxic plants (Figures 4D and S8B). The network density in toxic plants (0.43) was greater than in non-toxic plants (0.07) (Figure 4D). Based on pathway analysis on root exudates and 16s function analysis, the rhizosphere microorganisms of toxic plants were involved in phenylpropanoid biosynthesis (map00940) (Figure 4E).
Relationships of microbial diversity, soil properties, carbon metabolism and root exudates
Structural equation modeling (SEM) was used to determine the drivers of changes in soil microbiota (Figure 5). Rhizosphere and plant species were the strongest direct drivers of soil properties. Soil pH and microbial α influenced microbial β diversity directly, whereas, rhizosphere, pH, microbial community, and soil properties influenced microbial C metabolism (Figure 5A). The average well-color developments (AWCDs) of rhizosphere in toxic plants were greater than in bulk soil in the four levels of degraded grasslands (Figure S9A). The first principal component in the four degraded grasslands of S, H, M, and L were 54.2%, 50.5%, 48.5%, and 47.0%, respectively, and indicated that the microorganisms of different plant rhizospheres and bulk soil used C sources differently (Figure S9B).
Figure 5.

Effects of soil properties and microbial structure on microbial carbon metabolism (A) and the complexity of microbial networks (B)
The rectangles represent variables of soil (rhizosphere and bulk soil); soil pH; species (toxic and non-toxic plants); soil property, the first axis of PCA analysis of soil total organic carbon (TOC), total nitrogen (TN), total phosphorous (TP), available phosphorous (AP), nitrate nitrogen (NO3-N), ammonium nitrogen (NH4-N), dissolved organic nitrogen (DON), available potassium (AK), available sodium (ANa) and water content (WC); microbial α diversity; microbial β diversity; compounds, the relative abundance of root exudates; AWCD, the average well-color development. Microbial network complexity, the edge of co-occurrence networks. The proportion of explained variance (R2) appears alongside variables in the model. χ2, Chi-square; df, degrees of freedom; p, probability level; RMSEA, root-mean squared error of approximation are the goodness-of-fit statistics for each model. Significance levels of each predictor were ∗p < 0.05, ∗∗p < 0.01.
Plant species, root exudates, soil pH, soil properties, and microbial diversity explained 95% of the variation of the microbial complex network (Figure 5B). Plant species (0.81), microbial α diversity (0.34), and microbial β diversity (0.34) and benzoquinones, flavones, and alkaloids in root exudates were correlated positively (p < 0.05) and soil properties were correlated negatively with the microbial complex network (Figure 5B). Plant species, soil properties, root exudates, and microbial α diversity explained 57% of the variation in microbial community compositions (Figure 5B). Naphthoquinones, terpenoids, and phthalates in root exudates affected microbial α diversity significantly; alkaloids, coumarin, cinnamic acid, and naphthoquinones affected soil pH directly, while amino acids, coumarin, and flavones affected the rhizosphere microbial community (Figure 5B).
Discussion
Assembly of rhizosphere bacterial community
The microbial community assemblies are shaped by a multitude of trophic influences, which depend upon biological diversity (Caruso et al., 2011; Lebeis et al., 2015). Neutral theory predicts that if limited dispersal and demographic stochasticity are the main drivers of community dynamics, then the random pattern in species and spatial auto-correlation of the environment should be the main factors determining community structure (Sloan et al., 2006; Mendes et al., 2014). In contrast, deterministic processes influence biodiversity and species composition, if niche partitioning interacts with environmental factors (Chase, 2007). The present study indicated that the microbial assemblies of rhizospheres were shaped mainly by niche filtering, which supported our hypothesis. The selection at the functional level in the rhizosphere was based on deterministic processes according to the niche-based theory. Furthermore, habitat and species affected the composition of microbial community structure. This trend was evident for toxic plants, which indicated that the rhizosphere of toxic plants had a strong influence in shaping the bacterial community in degraded grassland.
Three densities of L. virgaurea were identified in the present study. There was a high death rate with high density, which may have been caused by autotoxicity and/or ‘self-thinning’. Autotoxicity is allelopathy in which an individual inhibits the growth of other individuals of the same species by releasing autotoxins (Singh et al., 1999). Some autotoxins, including phenolics, omilactone B, artemisinin, phenolic acids, and cyclic hydroxamic acids (Ni et al., 2012), inhibit or delay the germination and growth of conspecific plants (Miller, 1996). Owing to overlapping and interference mechanisms, roots provide a specific micro-habitat for the proliferation of specific soil microorganisms, with new interactions developing among colonizing microbes in densely colonized rhizospheres (Petermann and Buzhdygan, 2021; Tian et al., 2022).
Microbial network complexity in rhizosphere microorganisms of toxic plants
Rhizosphere assemblages formed larger and more complex networks than bulk soil assemblages, which supported our prediction. The network modules likely resulted from microbe interactions or covariation in response to shared niches in the rhizosphere (Shi et al., 2016; Wang et al., 2021), owing to better nutrition and greater microbial C metabolism activity than in bulk soil microbes. In contrast to rhizosphere microbes, networks in bulk soil remained relatively simple, which indicated that interactions or niche sharing were minimal. The low activity of soil bacteria was another reason for the lack of networks in bulk soil (Fierer and Lennon, 2011).
Within rhizosphere modules, 15 modules were identified, which further indicated that the rhizosphere microbial networks were more complex than in bulk soil. The related properties of network modules were closely related to soil multi-functionality, suggesting that microorganisms influenced soil nutrient cycling processes (Wagg et al., 2019; Marqués-Gálvez et al., 2021). In the process of grassland degradation, soil nitrate nitrogen content increased, while ammonia nitrogen content decreased, which may be owing to enhanced nitrification and decreased denitrification by microorganisms. Studies have reported that low denitrification in soil may be related to plant interactions (Dassonville et al., 2011). The root exudates of toxic plants contained higher contents of biologically active compounds (coumarin, cinnamic acid, flavones) than in non-toxic plants, and they were correlated positively with the dominant taxa in the module. Flavonoid compounds could interfere with bacterial respiration, especially with denitrification (Bardon et al., 2014). Therefore, root exudates promoted the development of the niche occupied by dominant taxa, and greater interactions owing to the shared ecological niche, which resulted in more complex co-occurrence patterns in toxic than in non-toxic plants. This could explain the greater abundance of microorganisms in toxic than in non-toxic plants.
In degraded sown grassland in this study, high-density L. virgaurea had a related exudate module, and the middle and low densities L. virgaurea had related bacterial modules (Figure S10). It is probable that the root-associated microbial populations were determined not only by the available C sources but also by selective and inhibitory interactions (Trivedi et al., 2020). Compared with high-density L. virgaurea, the rhizosphere microbes of low-density L. virgaurea had less nutrients available. The roots of low-density L. virgaurea provided less C for their rhizosphere microorganisms, and, as a result, the rhizosphere microorganisms may have been affected by nutrient stress. Therefore, the stronger the positive interaction between microbes, the more restricted the dispersal of species (Table S1), which was consistent with the stress gradient hypothesis (David et al., 2020), and further emphasized the importance of root exudates to the assemblies of rhizosphere microorganisms.
Root exudates affect rhizosphere microorganisms
In the present study, root exudates of toxic plants were correlated positively with rhizosphere microorganisms (Figure S11), which supported the first and fourth hypotheses. This relationship has been observed in many plant species and soil types (Liu et al., 2020); however, this is not always the case. Roots of Morina kokonorica and Aconitum pendulum released unique compounds that were not correlated with rhizosphere microorganisms. This may have been caused by sampling time or plant communities, as the adjacent plants or different growth stages of plants release complex root exudates that enrich different rhizosphere microorganisms (Kong et al., 2018). When roots of Lupinus albus mature, organic acids are released that decrease the pH of the soil to inhibit bacteria (Weisskopf et al., 2006). In the current study, the pH and organic matter and nitrogen contents in the rhizosphere were higher than in bulk soil. In addition, the SEM demonstrated that pH influenced the bacterial communities directly. As reported by Hermans et al. (2020), an increase in pH affected the solubility of elements, promoted the growth of plants, and led to an increase in root exudates and soil organic matter content. Furthermore, soil surface nutrient cycling was enhanced, as plants extracted more nutrients (K and NO3), which were decomposed and mineralized at the surface (Liddicoat et al., 2019).
The concentrations of coumarin derivatives, naphthoquinones, salicylic acid, cinnamic acid, and terpenoids were greater in the root exudate of toxic than non-toxic plants in the present study. This is consistent with the report that toxic plants have improved chemical defense substances (terpenoids and phenols), potentially contributing to their reproductive success. Mendes et al. (2011) reported that Proteobacteria and Actinobacteria were associated with disease suppression, and Proterobacteria was correlated positively with bio-active compounds of root exudates in toxic plants (Figure S8). In addition, the relative abundances of Bradyrhizobium, Pseudomonas, Rhizobium, and Sphingomonas, mainly plant growth promoting rhizobacteria (PGPR), were greater in toxic than non-toxic plants.
The significantly up-regulated compounds in this study were correlated positively with bacterial phyla (Figure S8). Root exudates have been shown to affect the composition of the rhizosphere microbial community. Salicylic acid plays an important role in plant resistance to pathogens (Leibold and McPeek, 2006), and coumarin is beneficial to the interaction among probiotics (Voges et al., 2019; Harbort et al., 2020). Huang et al. (2019) reported that Arabidopsis produced specialized triterpenes that shaped and customized the microorganisms within and around its roots and maintained specific microbiota. Naphthoquinones possess antimicrobial activity (Brigham et al., 1999), are derived from phenylpropanoid and isoprenoid precursors (Gaisser and Heide, 1996), and enhance the self-growth and defense of the next-generation of plants (Hu et al., 2018). These compounds may play a similar role in toxic plants by inhibiting the growth of nearby plants, enriching beneficial bacteria, and helping plants resist pathogens. The effects of bio-active compounds in root exudates of toxic plants on rhizosphere microorganisms warrant further studies.
Life cycle influenced the adaptation of toxic plants
According to our survey, L. virgaurea has a long pre-flowering vegetative growth stage, generally 3–6 years, and produces rhizomes, which store nutrients for clonal growth. In the current study, L. virgaurea had a wider niche than other toxic plants; perhaps the spatial colonization mechanisms of clonal plants are driven by the growth of the rhizome. The rhizosphere bacterial communities had higher positive correlations with root exudates in L. virgaurea and Pedicularis kansuensis than with other toxic plants. The samples were taken in August, which was the peak time for the development of plants and L. virgaurea, a perennial, was at the stage of asexual reproduction. In addition, P. kansuensis, an annual, parasitizes the host during growth and reproduction (Sui et al., 2015). As root exudates could be affected by the developmental stage of plants (Zhalnina et al., 2018), further studies are needed to determine the effect of the growth stage on microbial communities.
Conclusions
Overall, the convergence of root functions in toxic plants could improve their adaptability in different degraded grasslands. Toxic plants secrete more bio-active compounds beneficial to plant growth than non-toxic plants. These compounds include naphthoquinones, terpenoids, phthalates, amino acids, coumarin, and flavones, which alter microbial diversity. The assembly of rhizosphere microorganisms was mainly a stochastic process. The deterministic process (niche theory) also contributed and had a greater effect on toxic than non-toxic plants, as root exudates directly affected the assembly of rhizosphere microorganisms. Compared with bulk soil, the rhizosphere had higher nutrients and greater microbial C metabolism activity. In addition, the network of the microbial community in the rhizosphere of toxic plants was more complex and had higher positive correlations among microorganisms than bulk soil and rhizosphere of non-toxic plants, indicating greater interaction and niche sharing potential among microorganisms in the rhizosphere of toxic plants. Bacteria in the rhizosphere of toxic plants were enriched in the phenylpropanoid biosynthesis pathway. It is concluded that the strong interaction between root exudates and microorganisms is the key mechanism for the divergent habitat adaptation of toxic plants in different degraded grasslands, supporting above-ground plant growth and reproduction.
Limitations of the study
Climate warming may affect the metabolism of plants, increase the defense substances and promote the invasion of weeds (Rice et al., 2021). In the present study, increased content of defensive substances was present in the roots of toxic plants in a degraded alpine grassland on the Qinghai-Tibetan Plateau. These substances have a direct effect on the assembly process of rhizosphere microorganisms, which may have a positive effect on the reproduction of toxic plants. Studies that support this argument directly are few. In addition, since toxic weeds are usually perennial plants, it is not clear whether responses are similar at different stages of growth. Further studies on different growth stages of toxic weeds are needed, in particular in the later stages. Whether rhizosphere microorganisms and root exudates are similar in different growth stages also requires further research.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Plant and soil samples | Maqin County, Guoluo Tibetan Autonomous Prefecture, Qinghai Province | 3740 m above sea level |
| Software and algorithms | ||
| R (v3.6.1) | r-project | https://www.r-project.org/ |
| RStudio (V1.2.1335) | RStudio, Inc. | rstudio.com |
| R package vegan | CRAN | https://cran.r-project.org/web/packages/vegan/vegan.pdf |
| R package WGCNA | CRAN | https://cran.r-project.org/web/packages/WGCNA/WGCNA.pdf |
| R package car | CRAN | https://cran.r-project.org/web/packages/car/car.pdf |
| R package pheatmap | CRAN | https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf |
| Cytoscape (V 3.7.0) | https://cytoscape.org | https://cytoscape.org |
| Gephi (0.9.2) | https://gephi.org/ | https://gephi.org/ |
| IBM SPSS Statistics 25.0 | IBM® SPSS® software platform | RRID:SCR_002865 |
| MetaboAnalyst 5.0 | https://www.metaboanalyst.ca/ | https://www.metaboanalyst.ca/ |
| Deposited data | ||
| Datasets and statistical analyses | This paper | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhanhuan Shang (Shangzh@lzu.edu.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Neutral and niche theories
Stochastic (birth/death, speciation/extinction and immigration) and deterministic [biotic (e.g., competition and predation) and abiotic (e.g., temperature and nutrients) environmental factors] processes of the plant rhizosphere microbial assembly could explain the self-adjustment response ability of plant rhizosphere to changes in the soil environmental (Mendes et al., 2014). The neutral and niche theoretical models of microbial assembly tested the adaptive mechanisms of plant roots to the soil environment (Jiao et al., 2020). When the assembly of rhizosphere microorganisms conforms to the neutral theoretical model, then the root exudates have little impact on rhizosphere microorganisms. When the rhizosphere microbial assembly conforms to the niche theoretical model, then the root exudates strongly influence the rhizosphere microbial assembly to improve the survival and growth of plants. We calculated the rank abundance distribution and migration rate of each toxic and non-toxic plant in different habitats. The abundance of each was fitted into two theoretical assembly models: the neutral community model (NCM) and niche-based model (log-normal) (Dumbrell et al., 2010) for both the rhizosphere and bulk soil microorganisms. In this model, there were NT species in the community. For a community to change, a species must die or leave the community at a speed (δ) independent of the species. The dead species is replaced immediately by an immigrant species from the community (with probability m), or by the regeneration of a member of the local community (with probability 1-m). Assuming that deaths are distributed evenly over time, it is expected that 1/δ individuals will die within a period of time, and the i-th species with initial absolute abundance Ni will either increase by 1, remain unchanged, or decrease by 1. The probabilities are presented by the Equations 1, 2, and 3 (Sloan et al., 2006):
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
where pi is the relative abundance of the ith species in the community. If αi is positive, the probability of obtaining an individual for the ith species increases, but, if αi is negative, the probability decreases. Ni/NT is the relative abundance of i species. In the NCM, Nm is an estimate of dispersal between communities, with N the community size and m the immigration rate. It represents the link between occurrence frequency and regional relative abundance. The coefficient of determination (R2) represented the overall fit to the NCM (Chen et al., 2019), while the 95% confidence intervals around all fitting statistics were done by 1000 bootstrap replicates. Log-normal model assumes that the logarithmic deformation of the number of individuals conforms to normal distribution, that is, Z = ln(N) is distributed normally. The model was compared based on the Akaike Information Criterion (AIC) — the lower the value, the better the microbial community fitted to a specific model. AIC values were calculated based on the equation AIC = - 2log-likehood + 2 × n, where n represents the number of parameters in the fitted model (Feinstein and Blackwood, 2012), and ut was generated using the function “radfit” from the Vegan package. All statistical analyses were done in R (version 3.6.1).
Method details
Study sites and design
The study was done on 100 ha at the headwater region of the Yellow River in the eastern Tibetan plateau in Maqin County, Guoluo Tibetan Autonomous Prefecture, Qinghai Province (34°28′N, 100°12′E; 3740 m above sea level) (Figure S1). The site is characterized by a highland continental climate and no absolute frost-free days, with an annual average temperature range between −0.1 and 1.2°C, precipitation between 463 and 602 mm, total sunshine between 2272 and 2632 h, and evaporation of approximately 1460 mm. The grassland type was alpine meadow (Shang et al., 2013, 2016), with the dominant plant species consisting of Kobresia pygmaea, Stipa spp. and Kobresia humilis. Traditionally, the site is used for winter grazing of yaks and sheep from December to March.
The study site includes grasslands at different levels of degradation. Sown grassland, with mainly Elymus nutans and Poa annua, was established more than 10 years ago to restore heavily degraded grassland, but this grassland became degraded again with the invasion and spread of toxic plants (Shang et al., 2013; Gao et al., 2019). Lightly (L), moderately (M) and heavily (H) degraded native grasslands, and degraded sown grassland (S) were selected (Table S1).
Vegetation survey and root and soil sampling
Field sampling was done in August 2017 and 2018, when alpine plants reached peak growth. Four plots, each of 5 ha were selected randomly at each level of degradation. The distance between any two plots was greater than 100 m, which exceeded the spatial dependence of microbial variables (Franklin and Mills, 2003), and, thus, the samples in each plot were independent. Three sub-plots, with more than 20 m between any two, were selected in each plot. Fifteen random quadrats (50 cm × 50 cm) in each sub-plot were selected to measure plant cover, height and composition (Wang et al., 2012). After a vegetation survey, some plants with high frequency and coverage were selected (Table S7). Toxic plants secrete secondary compounds, which are poisonous to livestock, wild herbivores and humans (James et al., 2005). We selected Aconitum pendulum, Ajuga lupulina, Artemisia dubia, Artemisia nanschanica, Euphorbia fischeriana, Ligularia virgaurea, Morina kokonorica, Oxytropis ochrocephala, Pedicularis kansuensis, and Sphallerocarpus gracilis, which were the dominant toxic plants (Gao et al., 2011; Shang et al., 2012; Zhao et al., 2013; Li et al., 2014; Xie et al., 2014; Ma et al., 2015). Kobresia pygmaea and Elymus nutans, native forages which are highly palatable with a high nutritive value for livestock (Long et al., 1999), were selected as non-toxic plants.
Rhizosphere was collected from M. kokonorica in L; A. pendulum, A. lupulina, E. fisheriana, L. virgaurea, and M. kokonorica in M; A. lupulina, E. fisheriana, S. gracilis, L. virgaurea, M. kokonorica, Artemisia dubia, Artemisia nanschanica, and A. pendulum in H; and L. virgaurea, M. kokonorica, O. ochrocephala, S. gracilis, and P. kansuensis in S (Figure S1 and Table S7). In addition, bulk (non-rhizosphere) soil was collected in each habitat as control. In total, 66 samples (3 replicates) in 4 the habitats were collected in 2017; 6 in L, 18 in M, 27 in H and 15 in S. Rhizosphere and root exudation were collected from L. virgaurea, M. kokonorica, and K. pygmaea in L; A. pendulum and L. virgaurea in M; L. virgaurea in H; and P. kansuensis, E. nutans and low (<120 plants/m2), medium (120–240 plants/m2) and high (>360 plants/m2) densities of L. virgaurea in S (Figure S1 and Table S7). In total, 33 samples (3 replicates) in the 4 grasslands were collected in 2018; 9 in L, 6 in M, 3 in H and 15 in S.
Rhizosphere was collected as described by Edwards et al. (2015). In brief, the whole plant was dug out with a shovel, the soil surrounding the root was removed manually by gentle shaking, and the soil within 2 mm of the root surface was shaken into a sterile self-sealing bag and mixed well. Then, roots and litter were removed manually, and a small amount of soil was placed into a 3 mL sterile centrifuge tube and stored in liquid nitrogen for determining the diversity of the soil microbial community. The remaining soil was divided into two parts: one part, covered by an ice pack, was brought to the laboratory and refrigerated at 4°C for determination of soil microbial C metabolism, and the other part was air-dried and passed through a 2-mm sieve. Nine soil core samples (3.5 cm in diameter) at a depth of 0–10 cm were collected at each plot, mixed and homogenized, and three core soil samples were combined into one composite sample. This sample was termed ‘bulk soil’ and was free from the influence of plants. Fifteen individuals of each toxic plant species were collected, the roots were rinsed with distilled water and then immersed in 500 mL distilled water for 10 h. The exudates were placed into five 100 mL plastic bottles and refrigerated at 4°C.
Soil chemical properties
Soil pH was measured using a pH meter (Sartorius PB-10, Goettingen, Germany) in a 1:2.5 soil:water solution (w/v). Soil total N (TN) was determined by the micro-Kjeldahl method with digestion in H2SO4 followed by steam distillation. Soil total phosphorous (TP) was digested with HF-HClO4 and ammonium and nitrate were extracted by 2 mol/L potassium chloride. TN, TP, ammonium and nitrate were measured with a flow injection analyzer (FIA star5000 Analyzer, Höganäs, Sweden). Soil total organic carbon (TOC) content was determined by wet digestion using the potassium dichromate method and available P (AP) was determined by the molybdenum-blue method using an UV spectrophotometer (Hitachi U-2910, Tokyo, Japan), after extraction with sodium bicarbonate. Soil nitrate nitrogen (NO3-N) and ammonium nitrogen (NH4-N) were extracted with a 1 M KCl solution for 1 h at 25°C. Available potassium (AK) and available sodium (ANa) were measured by flame photometry after extraction with sodium hydroxide and ammonium acetate, respectively. Dissolved organic nitrogen (DON) was measured using the alkaline hydrolysis diffusion method. Data were standardized using Z-score, and then the average Z-score was used to calculate the soil multi-functionality.
Microbial community sequencing and data processing
To extract DNA from soil samples, the E.Z.N.A. Soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) was used according to the manufacturer’s protocol. Illumina sequencing was performed by amplifying the V4/V5 region of the bacterial 16S rRNA gene using individually bar-coded forward primers 515F (5′-GTGCCAGCMGCCGCGG-3′) and reverse primers 907R (5′-CCGTCAATTCMTTTRAGTTT-3′) and ITS1 region of ITS using individually bar-coded forward primers ITS1F (5′-CTTGGTCATTTAGAGGAAGTAA-3′) and reverse primers ITS1R (5′-GCTGCGTTCTTCATCGATGC-3′). The PCR reactions were done in triplicate 20 μL mixtures containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. Amplicons were extracted from 2% agarose gels, purified according to the manufacturer’s instructions and quantified using QuantiFluor™-ST (Promega, Madison, WI, USA). The PCR conditions were as follows: 95°C for 2 min, followed by 25 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s and a final extension at 72°C for 5 min. Purified PCR products were quantified by Qubit®3.0 (Life Invitrogen Technologies, Carlsbad, CA, USA). The pooled DNA product was used to construct an Illumina Pair-End library following Illumina’s genomic DNA library preparation procedure. The amplicon library was paired-end sequenced on an Illumina HiSeq 2500 according to standard protocols.
Sequence data are deposited in the NCBI Sequence Read Archive under the accession number PRJNA693171. Raw fastq files were demultiplexed and quality-filtered using quantitative insights into microbial ecology (QIIME, version 1.17). Operational taxonomic units (OTUs) were clustered with 97% similarity cutoff using USEARCH and chimeric sequences were identified and removed by denovo. The rarefaction curves of alpha diversity index were drawn by QIIME. The phylogenetic affiliation of each 16S rRNA gene sequence was aligned against the SILVA database for 16S rRNA of bacterial sequences and against the UNITE database for fungal sequences. After classification, the OTU abundance table was obtained according to the number of sequences in each OTU, and subsequent analyses were done according to the OTU abundance table (Caporaso et al., 2010).
Microbial carbon metabolism assay
Soil microbial C source metabolism was determined using microplates (Biolog-Eco, Hayward, CA, USA). Ten g of soil were placed in a 250 mL conical bottle, 90 mL sterile saline (0.85% NaCl solution) were added, the bottle was sealed with sealing film, shaken at 200 r/min at 25°C for 30 min and left to stand for 10 min. The supernatant was brought to a 103 final dilution (Nair and Ngouajio, 2012), and then 150 μL of diluted supernatant were added to the microplate and placed in an incubator at 25°C in the dark for 9 days. Absorbance at 590 nm and 750 nm were measured every 24 h with a microplate reader, and the distribution of 31C sources were identified.
The relative absorbance indicates the ability of the microbial community to utilize the C source. The average well color development (AWCD) of the pores reflects the average metabolic capacity of the microbial community with 31C sources, indicating the overall metabolic activity of the microorganisms. To calculate AWCD, Equation 4 was used:
| (Equation 4) |
where Ci is the absorbance at 590 nm minus 750 nm of each C source, and R0 is the absorbance of the control.
Root exudate determination
A liquid chromatograph mass spectrometer (Ultimate 3000LC, Q Exactive Thermo Fisher Scientific, San Jose, CA, USA) was used to measure the composition of root exudate. Five mL of exudate were freeze-dried, 300 μL of methanol and 10 μL of internal standard (3 mg/mL, dichlorophenylalanine) were added, homogenized for 1 min, centrifuged at 12,000 rpm for 15 min at 4°C, and 200 μL of supernatant were transferred into a vial. Compounds with positive and negative ion modes were determined with a Hypersil Gold C18 column (100 mm × 2.1 mm, 1.9 μm). Column temperature was 45°C, the flow rate was 0.35 mL/min and mobile phase composition A was: water +5% acetonitrile +0.1% formic acid, and B was: acetonitrile +0.1% formic acid. The injection volume was 10 μL and auto-sampler temperature was 4°C. Compound databases (Predicted Compositions, mzCloud Search, ChemSpider Search) were used for comparison.
Network construction and visualization
The scale function standardized the original data and then the network analysis of microbial communities and root exudates were completed with weighted gene co-expression network analysis (WGCNA) in R. To reduce the redundancy of data, some values with little information were removed, and the mad function in R was used to screen the OTUs/exudates (75% before the median absolute deviation and greater than 0.01). The network was constructed as an unsigned type, and the Pearson correlation coefficient was used to test for relationships between variables. For the constructed network to conform more closely to the scale-free feature, the appropriate soft threshold was calculated to complete the integration of the modules. The OTUs/exudates were classified into modules, which were represented by different colors.
Correlation analysis was employed between modules (module eigengenes, ME) and plant species after modules merged. The adjacency and colSums functions in the WGCNA package were used to calculate the connectivity of the nodes. The Pearson correlation coefficient tested the relation between root exudates and microorganisms. Significance was accepted at p < 0.05, and the Holm method was used for multiple test adjustment. The visualization of the network was completed by Gephi 0.9.2 and Cytoscape 3.7.0, which only showed the nodes and edges with significant correlations. The colors of nodes were consistent with the colors of the modules where they were located.
Quantification and statistical analysis
One-way analysis of variance (ANOVA) for three or more treatments, or t-test for two treatments were used to test for differences among or between means (SPSS 25.0, SPSS Inc., Chicago, IL, USA). Values are presented as mean ± S.D. or S.E.M. Statistical analysis and graphing were done with R 3.6.1, and principal component analysis (PCA), principal coordinates analysis (PCoA), permutational multivariate analysis (PERMANOVA) and redundancy analysis (RDA) were done in R. Structural equation modelling (SEM) used AMOS Graphics based on the site score of the first axis of PCoA for bacterial communities, and PCA for root exudates and AWCD. Bacterial communities were based on weighted UniFrac distances, and PCA of root exudates and AWCD were based on Bray-Curtis distances. Data of soil physico-chemical properties were log transformed. The initial model was simplified by a stepwise removal of uninformative paths until a suitable model was generated. In addition, modification indices were used to identify missing paths. The χ2 and the root mean square error of approximation (RMSEA) tested the overall goodness of fit, which was indicated by a low χ2, a high probability (p > 0.05), and a RMSEA near 0. Screening of root exudates was based on Kyoto Encyclopedia of Genes and Genomes (KEGG), and statistical analysis and pathway analysis used MetaboAnalyst 5.0.
Acknowledgments
We thank two reviewers for helpful suggestions on the article. This work was supported by the Second Tibetan Plateau Expedition (2019QZKK0302), the Natural Science Foundation of China (31870433, U21A20183, 31961143012, 42041005), the Fundamental Research Funds for the Central Universities (lzujbky-2021-ct10), the ‘111’ Programme 2.0 (BP0719040). We would like to thank the Central Laboratory of the School of Life Science, Lanzhou University, for providing instruments and equipment.
Author contributions
Conceptualization, Z.H.S.; Methodology, W.Y.W., T.H.J., Z.H.S.; Investigation, W.Y.W., T.H.J., J.H., Y.F.B., T.Z., S.Q., M.H., Z.H.L., and Z.H.S; Data Curation, W.Y.W., T.H.J., T.Y.Q., S.S.L., J.X.J; Writing−Original Draft, W.Y.W; Writing−Review & Editing, W.Y.W., A.A.D., and Z.H.S.; Funding Acquisition, Z.H.S.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105243.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Bardon C., Piola F., Bellvert F., Haichar F.E.Z., Comte G., Meiffren G., Pommier T., Puijalon S., Tsafack N., Poly F. Evidence for biological denitrification inhibition (BDI) by plant secondary metabolites. New Phytol. 2014;204:620–630. doi: 10.1111/nph.12944. [DOI] [PubMed] [Google Scholar]
- Brigham L.A., Michaels P.J., Flores H.E. Cell-specific production and antimicrobial activity of naphthoquinones in roots of Lithospermum erythrorhizon. Plant Physiol. 1999;119:417–428. doi: 10.1104/pp.119.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peña A.G., Goodrich J.K., Gordon J.I., et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso T., Chan Y., Lacap D.C., Lau M.C.Y., McKay C.P., Pointing S.B. Stochastic and deterministic processes interact in the assembly of desert microbial communities on a global scale. ISME J. 2011;5:1406–1413. doi: 10.1038/ismej.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase J.M. Drought mediates the importance of stochastic community assembly. Proc. Natl. Acad. Sci. USA. 2007;104:17430–17434. doi: 10.1073/pnas.0704350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Ren K., Isabwe A., Chen H., Liu M., Yang J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome. 2019;7:138. doi: 10.1186/s40168-019-0749-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colautti R.I., Barrett S.C.H. Rapid adaptation to climate facilitates range expansion of an invasive plant. Science. 2013;342:364–366. doi: 10.1126/science.1242121. [DOI] [PubMed] [Google Scholar]
- Dassonville N., Guillaumaud N., Piola F., Meerts P., Poly F. Niche construction by the invasive Asian knotweeds (species complex Fallopia): impact on activity, abundance and community structure of denitrifiers and nitrifiers. Biol. Invasions. 2011;13:1115–1133. doi: 10.1007/s10530-011-9954-5. [DOI] [Google Scholar]
- David A.S., Thapa-Magar K.B., Menges E.S., Searcy C.A., Afkhami M.E. Do plant-microbe interactions support the stress gradient hypothesis? Ecology. 2020;101:e03081. doi: 10.1002/ecy.3081. [DOI] [PubMed] [Google Scholar]
- De Vries F.T., Williams A., Stringer F., Willcocks R., McEwing R., Langridge H., Straathof A.L. Changes in root-exudate-induced respiration reveal a novel mechanism through which drought affects ecosystem carbon cycling. New Phytol. 2019;224:132–145. doi: 10.1111/nph.16001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbrell A.J., Nelson M., Helgason T., Dytham C., Fitter A.H. Relative roles of niche and neutral process in structuring a soil microbial community. ISME J. 2010;4:337–345. doi: 10.1038/ismej.2009.122. [DOI] [PubMed] [Google Scholar]
- Edwards J., Johnson C., Santos-Medellín C., Lurie E., Podishetty N.K., Bhatnagar S., Eisen J.A., Sundaresan V. Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. USA. 2015;112:911–920. doi: 10.1073/pnas.1414592112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan K., Weisenhorn P., Gilbert J.A., Shi Y., Bai Y., Chu H. Soil pH correlates with the co-occurrence and assemblage process of diazotrophic communities in rhizosphere and bulk soils of wheat fields. Soil Biol. Biochem. 2018;121:185–192. doi: 10.1016/j.soilbio.2018.03.017. [DOI] [Google Scholar]
- Feinstein L.M., Blackwood C.B. Taxa-area relationship and neutral dynamics influence the diversity of fungal communities on senesced tree leaves. Environ. Microbiol. 2012;14:1488–1499. doi: 10.1111/j.1462-2920.2012.02737.x. [DOI] [PubMed] [Google Scholar]
- Fierer N., Lennon J.T. The generation and maintenance of diversity in microbial communities. Am. J. Bot. 2011;98:439–448. doi: 10.3732/ajb.1000498. [DOI] [PubMed] [Google Scholar]
- Franklin R.B., Mills A.L. Multi-scale variation in spatial heterogeneity for microbial community structure in an eastern Virginia agricultural field. FEMS Microbiol. Ecol. 2003;44:335–346. doi: 10.1016/S0168-6496(03)00074-6. [DOI] [PubMed] [Google Scholar]
- Gaisser S., Heide L. Inhibition and regulation of shikonin biosynthesis in suspension cultures of Lithospermum. Phytochemistry. 1996;41:1065–1072. doi: 10.1016/0031-9422(95)00633-8. [DOI] [Google Scholar]
- Gao C., Tian C., Lu Y., Xu J., Luo J., Guo X. Essential oil composition and antimicrobial activity of Sphallerocarpus gracilis seeds against selected food-related bacteria. Food Control. 2011;22:517–522. doi: 10.1016/j.foodcont.2010.09.038. [DOI] [Google Scholar]
- Gao X., Dong S., Xu Y., Wu S., Wu X., Zhang X., Zhi Y., Li S., Liu S., Li Y., et al. Resilience of revegetated grassland for restoring severely degraded alpine meadows is driven by plant and soil quality along recovery time: a case study from the Three-River Headwater area of Qinghai-Tibetan Plateau. Agric. Ecosyst. Environ. 2019;279:169–177. doi: 10.1016/j.agee.2019.01.010. [DOI] [Google Scholar]
- Goldschmidt F., Regoes R.R., Johnson D.R. Metabolite toxicity slows local diversity loss during expansion of a microbial cross-feeding community. ISME J. 2018;12:136–144. doi: 10.1038/ismej.2017.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross M. How plants grow their microbiome. Curr. Biol. 2022;32:R97–R100. doi: 10.1016/j.cub.2022.01.044. [DOI] [Google Scholar]
- Harbort C.J., Hashimoto M., Inoue H., Niu Y., Guan R., Rombolà A.D., Kopriva S., Voges M.J.E.E.E., Sattely E.S., Garrido-Oter R., et al. Root-secreted coumarins and the microbiota interact to improve iron nutrition in Arabidopsis. Cell Host Microbe. 2020;28:825–837.e6. doi: 10.1016/j.chom.2020.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans S.M., Buckley H.L., Case B.S., Curran-Cournane F., Taylor M., Lear G. Using soil bacterial communities to predict physico-chemical variables and soil quality. Microbiome. 2020;8:79. doi: 10.1186/s40168-020-00858-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Robert C.A.M., Cadot S., Zhang X., Ye M., Li B., Manzo D., Chervet N., Steinger T., van der Heijden M.G.A., et al. Root exudate metabolites drive plant-soil feedbacks on growth and defense by shaping the rhizosphere microbiota. Nat. Commun. 2018;9:2738. doi: 10.1038/s41467-018-05122-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang A.C., Jiang T., Liu Y.X., Bai Y.C., Reed J., Qu B., Goossens A., Nützmann H.W., Bai Y., Osbourn A. A specialized metabolic network selectively modulates Arabidopsis root microbiota. Science. 2019;364:eaau6389. doi: 10.1126/science.aau6389. [DOI] [PubMed] [Google Scholar]
- Humphries T., Florentine S.K., Dowling K., Turville C., Sinclair S. Weed management for landscape scale restoration of global temperate grasslands. Land Degrad. Dev. 2021;32:1090–1102. doi: 10.1002/ldr.3802. [DOI] [Google Scholar]
- James L.F., Gardner D.R., Lee S.T., Panter K.E., Pfister J.A., Ralphs M.H., Stegelmeier B.L. Important poisonous plants on rangelands. Rangelands. 2005;27:3–9. doi: 10.2111/1551-501X. [DOI] [Google Scholar]
- Jiao S., Yang Y., Xu Y., Zhang J., Lu Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020;14:202–216. doi: 10.1038/s41396-019-0522-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong C.H., Zhang S.Z., Li Y.H., Xia Z.C., Yang X.F., Meiners S.J., Wang P. Plant neighbor detection and allelochemical response are driven by root-secreted signaling chemicals. Nat. Commun. 2018;9:3867. doi: 10.1038/s41467-018-06429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprivova A., Schuck S., Jacoby R.P., Klinkhammer I., Welter B., Leson L., Martyn A., Nauen J., Grabenhorst N., Mandelkow J.F., et al. Root-specific camalexin biosynthesis controls the plant growth-promoting effects of multiple bacterial strains. Proc. Natl. Acad. Sci. USA. 2019;116:15735–15744. doi: 10.1073/pnas.1818604116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeis S.L., Paredes S.H., Lundberg D.S., Breakfield N., Gehring J., Mcdonald M., Malfatti S., Glavina del Rio T., Jones C.D., Tringe S.G., et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 2015;349:860–864. doi: 10.1126/science.aaa8764. [DOI] [PubMed] [Google Scholar]
- Legendre P., Mi X., Ren H., Ma K., Yu M., Sun I.F., He F. Partitioning beta diversity in a subtropical broad-leaved forest of China. Ecology. 2009;90:663–674. doi: 10.1890/07-1880.1. [DOI] [PubMed] [Google Scholar]
- Leibold M.A., McPeek M.A. Coexistence of the niche and neutral perspectives in community ecology. Ecology. 2006;87:1399–1410. doi: 10.1890/0012-9658. [DOI] [PubMed] [Google Scholar]
- Li Y.Y., Dong S.K., Liu S., Wang X., Wen L., Wu Y. The interaction between poisonous plants and soil quality in response to grassland degradation in the alpine region of the Qinghai-Tibetan Plateau. Plant Ecol. 2014;215:809–819. doi: 10.1007/s11258-014-0333-z. [DOI] [Google Scholar]
- Liddicoat C., Weinstein P., Bissett A., Gellie N.J.C., Mills J.G., Waycott M., Breed M.F. Can bacterial indicators of a grassy woodland restoration inform ecosystem assessment and microbiota-mediated human health? Environ. Int. 2019;129:105–117. doi: 10.1016/j.envint.2019.05.011. [DOI] [PubMed] [Google Scholar]
- Lima-Mendez G., Faust K., Henry N., Decelle J., Colin S., Carcillo F., Chaffron S., Ignacio-Espinosa J.C., Roux S., Vincent F., et al. Determinants of community structure in the global plankton interactome. Science. 2015;348:1262073. doi: 10.1126/science.1262073. [DOI] [PubMed] [Google Scholar]
- Liu H., Mi Z., Lin L., Wang Y., Zhang Z., Zhang F., Wang H., Liu L., Zhu B., Cao G., et al. Shifting plant species composition in response to climate change stabilizes grassland primary production. Proc. Natl. Acad. Sci. USA. 2018;115:4051–4056. doi: 10.1073/pnas.1700299114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Huang X., Zhang J., Cai Z., Jiang K., Chang Y. Deciphering the relative importance of soil and plant traits on the development of rhizosphere microbial communities. Soil Biol. Biochem. 2020;148:107909. doi: 10.1016/j.soilbio.2020.107909. [DOI] [Google Scholar]
- Long R.J., Apori S.O., Castro F.B., Ørskov E. Feed value of native forages of the Tibetan Plateau of China. Anim. Feed Sci. Technol. 1999;80:101–113. doi: 10.1016/S0377-8401(99)00057-7. [DOI] [Google Scholar]
- Ma L., Gu R., Tang L., Chen Z.-E., Di R., Long C. Important poisonous plants in Tibetan ethnomedicine. Toxins. 2015;7:138–155. doi: 10.3390/toxins7010138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués-Gálvez J.E., Miyauchi S., Paolocci F., Navarro-Ródenas A., Arenas F., Pérez-Gilabert M., Morin E., Auer L., Barry K.W., Kuo A., et al. Desert truffle genomes reveal their reproductive modes and new insights into plant-fungal interaction and ectendomycorrhizal lifestyle. New Phytol. 2021;229:2917–2932. doi: 10.1111/nph.17044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes L.W., Kuramae E.E., Navarrete A.A., van Veen J.A., Tsai S.M. Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J. 2014;8:1577–1587. doi: 10.1038/ismej.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes R., Kruijt M., de Bruijn I., Dekkers E., van der voort M., Schneider J.H.M., Piceno Y.M., Desantis T.Z., Andersen G.L., Bakker P.A.H.M., et al. Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science. 2011;332:1097–1100. doi: 10.1126/science.1203980. [DOI] [PubMed] [Google Scholar]
- Miller D.A. Allelopathy in forage crop systems. Agron. J. 1996;88:854–859. doi: 10.2134/agronj1996.00021962003600060003x. [DOI] [Google Scholar]
- Nair A., Ngouajio M. Soil microbial biomass, functional microbial diversity, and nematode community structure as affected by cover crops and compost in an organic vegetable production system. Appl. Soil Ecol. 2012;58:45–55. doi: 10.1016/j.apsoil.2012.03.008. [DOI] [Google Scholar]
- Ni L., Acharya K., Hao X., Li S. Isolation and identification of an anti-algal compound from Artemisia annua and mechanisms of inhibitory effect on algae. Chemosphere. 2012;88:1051–1057. doi: 10.1016/j.chemosphere.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Petermann J.S., Buzhdygan O.Y. Grassland biodiversity. Curr. Biol. 2021;31:R1195–R1201. doi: 10.1016/j.cub.2021.06.060. [DOI] [PubMed] [Google Scholar]
- Ricciardi A., Blackburn T.M., Carlton J.T., Dick J.T.A., Hulme P.E., Iacarella J.C., Jeschke J.M., Liebhold A.M., Lockwood J.L., MacIsaac H.J., et al. Invasion science: a horizon scan of emerging challenges and opportunities. Trends Ecol. Evol. 2017;32:464–474. doi: 10.1016/j.tree.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Rice C., Wolf J., Fleisher D.H., Acosta S.M., Adkins S.W., Bajwa A.A., Ziska L.H. Recent CO2 levels promote increased production of the toxin parthenin in an invasive Parthenium hysterophorus biotype. Nat. Plants. 2021;7:725–729. doi: 10.1038/s41477-021-00938-6. [DOI] [PubMed] [Google Scholar]
- Savary S., Willocquet L., Pethybridge S.J., Esker P., McRoberts N., Nelson A. The global burden of pathogens and pests on major food crops. Nat. Ecol. Evol. 2019;3:430–439. doi: 10.1038/s41559-018-0793-y. [DOI] [PubMed] [Google Scholar]
- Shang Z., Yang S., Wang Y., Shi J., Ding L., Long R. Soil seed bank and its relation with above-ground vegetation along the degraded gradients of alpine meadow. Ecol. Eng. 2016;90:268–277. doi: 10.1016/j.ecoleng.2016.01.067. [DOI] [Google Scholar]
- Shang Z.H., Deng B., Ding L.M., Ren G.H., Xin G.S., Liu Z.Y., Wang Y.L., Long R.J. The effect of three years of fencing enclosure on soil seed banks and the relationship with above-ground vegetation of degraded alpine grasslands of the Tibetan plateau. Plant Soil. 2013;364:229–244. doi: 10.1007/s11104-012-1362-9. [DOI] [Google Scholar]
- Shang Z.H., Hou Y., Long R.J. Chemical composition of essential oil of Artemisia nanschanica Krasch. from Tibetan plateau. Ind. Crops Prod. 2012;40:35–38. doi: 10.1016/j.indcrop.2012.02.027. [DOI] [Google Scholar]
- Shi S., Nuccio E.E., Shi Z.J., He Z., Zhou J., Firestone M.K. The interconnected rhizosphere: high network complexity dominates rhizosphere assemblages. Ecol. Lett. 2016;19:926–936. doi: 10.1111/ele.12630. [DOI] [PubMed] [Google Scholar]
- Singh H.P., Batish D.R., Kohli R.K. Autotoxicity: concept, organisms, and ecological significance. Crit. Rev. Plant Sci. 1999;18:757–772. doi: 10.1080/07352689991309478. [DOI] [Google Scholar]
- Sloan W.T., Lunn M., Woodcock S., Head I.M., Nee S., Curtis T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006;8:732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- Sui X.L., Huang W., Li Y.J., Guan K.Y., Li A.R. Host shoot clipping depresses the growth of weedy hemiparasitic. J. Plant Res. 2015;128:563–572. doi: 10.1007/s10265-015-0727-6. [DOI] [PubMed] [Google Scholar]
- Tian Q., Lu P., Zhai X., Zhang R., Zheng Y., Wang H., Nie B., Bai W., Niu S., Shi P., et al. An integrated belowground trait-based understanding of nitrogen driven plant diversity loss. Glob. Chang. Biol. 2022;28:3651–3664. doi: 10.1111/gcb.16147. [DOI] [PubMed] [Google Scholar]
- Trivedi P., Leach J.E., Tringe S.G., Sa T., Singh B.K. Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 2020;18:607–621. doi: 10.1038/s41579-020-0412-1. [DOI] [PubMed] [Google Scholar]
- Voges M.J.E.E.E., Bai Y., Schulze-Lefert P., Sattely E.S. Plant-derived coumarins shape the composition of an Arabidopsis synthetic root microbiome. Proc. Natl. Acad. Sci. USA. 2019;116:12558–12565. doi: 10.1073/pnas.1820691116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagg C., Jansa J., Stadler M., Schmid B., van der Heijden M.G.A. Mycorrhizal fungal identity and diversity relaxes plant-plant competition. Ecology. 2011;92:1303–1313. doi: 10.1890/10-1915.1. [DOI] [PubMed] [Google Scholar]
- Wagg C., Schlaeppi K., Banerjee S., Kuramae E.E., van der Heijden M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019;10:4841. doi: 10.1038/s41467-019-12798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Duan J., Xu G., Wang Y., Zhang Z., Rui Y., Luo C., Xu B., Zhu X., Chang X., et al. Effects of warming and grazing on soil N availability, species composition, and ANPP in an alpine meadow. Ecology. 2012;93:2365–2376. doi: 10.1890/11-1408.1. [DOI] [PubMed] [Google Scholar]
- Wang X., Feng H., Wang Y., Wang M., Xie X., Chang H., Wang L., Qu J., Sun K., He W., et al. Mycorrhizal symbiosis modulates the rhizosphere microbiota to promote rhizobia−legume symbiosis. Mol. Plant. 2021;14:503–516. doi: 10.1016/j.molp.2020.12.002. [DOI] [PubMed] [Google Scholar]
- Wang X., Wang M., Xie X., Guo S., Zhou Y., Zhang X., Yu N., Wang E. An amplification-selection model for quantified rhizosphere microbiota assembly. Sci. Bull. 2020;65:983–986. doi: 10.1016/j.scib.2020.03.005. [DOI] [PubMed] [Google Scholar]
- Weisskopf L., Abou-Mansour E., Fromin N., Tomasi N., Santelia D., Edelkott I., Neumann G., Aragno M., Tabacchi R., Martinoia E. White lupin has developed a complex strategy to limit microbial degradation of secreted citrate required for phosphate acquisition. Plant Cell Environ. 2006;29:919–927. doi: 10.1111/j.1365-3040.2005.01473.x. [DOI] [PubMed] [Google Scholar]
- Xie T.P., Zhang G.F., Zhao Z.G., Du G.Z., He G.Y. Intraspecific competition and light effect on reproduction of Ligularia virgaurea, an invasive native alpine grassland clonal herb. Ecol. Evol. 2014;4:817–825. doi: 10.1002/ece3.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Wang D., Cui H., Zhang D., Sun Y., Jin H., Li X., Yang X., Guo H., He X., et al. Phytotoxicity mechanisms of two coumarin allelochemicals from Stellera chamaejasme in lettuce seedlings. Acta Physiol. Plant. 2016;38:248. doi: 10.1007/s11738-016-2270-z. [DOI] [Google Scholar]
- Yao X., Chai Q., Chen T., Chen Z., Wei X., Bao G., Song M., Wei W., Zhang X., Li C., et al. Disturbance by grazing and the presence of rodents facilitates the dominance of the unpalatable grass Achnatherum inebrians in alpine meadows of northern China. Rangel. J. 2019;41:301–312. doi: 10.1071/RJ18096. [DOI] [Google Scholar]
- Zhalnina K., Louie K.B., Hao Z., Mansoori N., da Rocha U.N., Shi S., Cho H., Karaoz U., Loqué D., Bowen B.P., et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018;3:470–480. doi: 10.1038/s41564-018-0129-3. [DOI] [PubMed] [Google Scholar]
- Zhao M., Gao X., Wang J., He X., Han B. A review of the most economically important poisonous plants to the livestock industry on temperate grasslands of China. J. Appl. Toxicol. 2013;33:9–17. doi: 10.1002/jat.2789. [DOI] [PubMed] [Google Scholar]
- Zhou J., Li X.L., Peng F., Li C., Lai C., You Q., Xue X., Wu Y., Sun H., Chen Y., et al. Mobilization of soil phosphate after 8 years of warming is linked to plant phosphorus-acquisition strategies in an alpine meadow on the Qinghai-Tibetan Plateau. Glob. Chang. Biol. 2021;27:6578–6591. doi: 10.1111/gcb.15914. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.