

Summary
Mitochondria are major organelles responsible for cellular energy and metabolism, and their dysfunction is tightly linked to cancer. The mitochondrial ribosome (mitoribosome) is a protein complex consisting of 82 mitoribosomal proteins (MRPs) encoded by nuclear genes and is essential for mitochondrial protein synthesis. However, their roles in tumorigenesis remain poorly understood. We performed pan-cancer analyses of 18,177 tumors representing 28 cancer types to determine somatic alterations of MRP genes as a genetic basis for tumorigenesis. We identified a set of 20 altered MRPs known to be involved in early assembly of the mitoribosome complex. We found that tumors with affected MRPs were associated with impaired mitochondrial functions and TP53 mutations accompanied by increased genomic instability and intra-tumor heterogeneity. MRP deletions were associated with poor survival. Our results reveal a key role for mitochondrial ribosome biogenesis in tumor malignancy across cancer types.
Subject areas: Biological sciences, Bioinformatics, Medical informatics, Biological database, Cancer
Graphical abstract
Highlights
-
•
A widespread deletion of mitoribosomal genes in multiple tumor types
-
•
Altered mitoribosomal genes are mainly involved in early mitoribosomal assembly
-
•
Altered mitoribosomal genes are linked to mitochondrial functions and TP53 mutations
-
•
Tumors with altered mitoribosomal genes are linked to tumor stemness and malignancy
Introduction
Protein complexes are important for most functions of the cell (Alberts, 1998; Hartwell et al., 1999). Recent studies suggest that complexes are considered constitutively active and essential in cancer (Ryan et al., 2017); for example, the deregulation of cytoribosome protein (CRPs) of the complexes promotes breast cancer metastasis (Ebright et al., 2020). Furthermore, deletion of co-regulated protein complex was frequently associated with a reduction in expression of additional complex members (Ryan et al., 2017). Therefore, there is necessary to characterize the protein complexes in cancer.
Mitochondria are membrane-bound cell organelles that play a vital role in bioenergetic, biosynthetic, and signaling processes for mammalian cells. They possess a separate protein synthesis machinery, different from cytoplasmic protein synthesis, composed of the mitochondrial ribosome complexes, also known as mitoribosomes (Beckmann and Herrmann, 2015). Mitoribosomes decode the genetic information provided by mtDNA to synthesize 13 protein subunits of the oxidative phosphorylation (OXPHOS) system, all of which are integral membrane proteins (Sylvester et al., 2004). Mitoribosomes are composed of two mitochondrial ribosomal RNAs (12S and 16S rRNAs) and 82 mitoribosomal proteins (MRPs) encoded by nuclear genes (Koc et al., 2013). Recent studies indicate that the structure of mitoribosomes may vary depending on different stress conditions during protein synthesis (Janska and Kwasniak, 2014; Tomal et al., 2019). It has been shown that heterogeneity in MRP composition may create many different variants of mitoribosomes resulting in altered OXPHOS activity (Waltz and Giege, 2020). Individual MRPs may provide distinct biological functions either in the assembly and structure of mitoribosomes or at the three major steps of mitochondrial translation (initiation, elongation, and termination phases) for protein synthesis or regulation in mammalian mitochondria (De Silva et al., 2015). Therefore, the functions of mitoribosomes may be differentially affected by the silencing of individual MRPs during its assembly (Tomal et al., 2019).
We hypothesized that somatic alterations of MRPs contribute to mitochondrial dysfunctions and promote tumorigenesis. In our study, we carried out a pan-cancer analysis of 18,177 human cancer genomes to systematically identify mitochondrial ribosomal protein dysregulation. Our results demonstrated that a widespread deletion of MRP loci across multiple cancers is associated with poor patient survival. Our results provide clues as to how certain types of exogenous mutagens may contribute to selecting the loss of MRPs and TP53 mutations associated with genomic instability that drives tumor malignancy. Furthermore, single-cell transcriptome analysis suggested that loss of MRPs may contribute to intra-tumor heterogeneity (ITH). This study provides a genetic basis for the role of mitochondrial ribosome biogenesis in tumor malignancy and increases our understanding of cancer pathogenesis.
Results
MRP genes are commonly deleted in human cancers
To enable the characterization of an MRP complex (Table S1) that is dysregulated in cancer, we performed multiple functional analyses (Figure 1A). We first performed pan-cancer genomic analysis of all known MRP loci in 18,177 primary and metastatic tumor specimens across 28 cancer types from The Cancer Genome Atlas (TCGA) (International Cancer Genome et al., 2010) and the Memorial Sloan Kettering Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) (Zehir et al., 2017) cohorts (Figure 1B, upper panel). The GISTIC method was used to define genetic alterations (Korkut et al., 2018) and found many MRP genes showing a shallow deletion (equivalent to heterozygous deletions), with a few additional MRP genes having deep deletions (equivalent to homozygous deletions) (Figure 1B [lower panel] and S1A), as well as few different types of point mutations (Figure S2) across multiple cancer types. Consistent with the TCGA and MSK-IMPACT cohorts, heterozygous deletions of multiple MRP genes were also found at a high frequency in 1,043 cancer cell lines across 24 different cancer types from Cancer Cell Line Encyclopedia (CCLE) (Barretina et al., 2012) cohorts (Figure S1A). Deletions of the same MRP genes were consistently found in the top-ranked list among MRPs with heterozygous deletions across multiple cancer types (Figures 1B, S1A, and Table S2). We identified a subset of 20 MRPs that showed significantly higher frequencies of heterozygous deletions across various cancer types (Table S2). These results indicate that a subset of MRPs (referred to as affected MRPs) tends to be deleted across different cancer types. To further demonstrate if deletions of affected MRPs occur nonrandomly, we performed additional analyses by comparing the copy number distribution of affected MRPs with randomly selected genes of the same affected size across cancer types. We found that deletions of affected MRPs are much more frequent than deletions of random gene sets of the same size (p < 0.005) (Figures S1B–S1E). In addition, sequence alignment and phylogenetic analyses of affected MRPs revealed no sequence similarity among them (Figure S3).
Figure 1.

Landscape of MRP deletion in cancer genome
(A) Workflow of mitoribosomal protein complex landscape characterized through multiple analysis.
(B) Distribution of 28 cancer types in the TCGA (n = 10,201) and MSK-IMPACT (n = 7976) cohorts, including histological subtypes (upper panel). Heatmaps representing a deep (purple) and shallow (pink) deletion on different MRP genes (rows) for individual samples (columns) across 28 different tumor types in the TCGA cohort (lower panel); different colors define the cancer types as in upper panel. Binary clinical annotations are provided for survival status (dead/live).
(C) Ribbon structure graph of the human mitochondrial ribosome complex (PDB code 5AJ4). Different proteins are highlighted by different colors (affected MRP: pink, unaffected MRP: blue, mito-rRNA: green, mitoribosomal RNA: gray).
(D) Schematic diagram of the mitoribosomal assembly process occurs at the mtDNA nucleoid driven by a subset of early MRPs. Different individual MRPs can be distinguished from the early and late bind at different stages during mitoribosomal assembly.
(E) Violin plots of mitoribosomal assembly priority score in affected and unaffected MRP genes. Statistical significance was tested by t test. Data are represented as mean ± SEM.
(F) Scatterplots showing the percentage of individual MRP deletions (y axis) correlated with the mitoribosomal assembly priority score (x axis). Color indicates the stages of mitoribosomal assembly. R = Spearman’s correlation coefficient, Spearman’s p < 0.0005.
Interestingly, affected MRPs were mainly located in the 60S mitochondrial ribosome subunit (Figure 1C). In addition, affected MRPs were mainly identified as early MRPs rather than late MRPs (Figure 1D), according to a classification method to define steps of mitoribosome assembly (Bogenhagen et al., 2014, 2018). The order of different MRP proteins for the assembly of mitoribosomes was determined using stable isotope labeling with amino acids in cell culture (SILAC) and defining the ratio of the P4C10/P4 (Bogenhagen et al., 2018). A high the P4C10/P4 ratio indicates that MRP protein is assembled at early stage in mitoribosome assembly (Bogenhagen et al., 2018). We then defined the P4C10/P4 ratio as the mitoribosome assembly priority score. We found that affected MRPs had a significantly higher mitoribosome assembly priority score than unaffected MRPs (p < 0.005) (Figure 1E). Moreover, the percent of individual MRP deletions showed a positive correlation with a mitoribosome assembly priority score (p < 0.005) (Figure 1F), indicating that affected MRPs associated with early binding during mitoribosome assembly.
To further determine if MRP deletions may have a cooperative functional consequence as a complex, we analyzed the shinyDepMap database based on functional screening of cellular genes using a combined CRISPR and short hairpin RNA (shRNA) system (Shimada et al., 2021). We searched co-dependent genes based on the similarity of combined CRISPR-shRNA gene dependency score of 15,847 genes in 423 cell lines (Figure S4A). This approach was based on ensemble clustering with hierarchy over DBSCAN on t-Distributed Stochastic Neighbor Embedding (t-SNE) with Spearman distance matrix (ECHODOTS) algorithm (Shimada et al., 2021), which assigned genes into 879 small, 608 medium, and 338 large functional clusters. We found that the MRPs and mitochondrial genes are classified in the same L4 large cluster (Figure S4B), suggesting that MRP and mitochondrial genes are functionally related in cancer cell lines. The L4 large cluster comprises 24 small clusters. To determine whether affected and unaffected MRPs may have a different functional relationship, we detected the affected or unaffected MRP gene silencing cluster among the 879 small clusters based on the similarity of the dependency scores for the combined CRISPR-shRNA system. Interestingly, we found that the affected MRPs (76%) are mainly associated with the same small S2 cluster (Figure S4C). In contrast, only 41% of the unaffected MRPs are clustering in the small S2 cluster (Figure S4C). These results suggest that affected MRPs may share a similar functional relationship much more closely related than unaffected MRP genes in cancer cells.
Subunit defects of MRP complex have highly correlated RNA and protein abundance across tumor samples
To further examine if MRP deletions may be functional, we performed correlation analyses of MRP genes between copy number variation (CNV) and transcriptome, as well as CNV and proteome, across tumor types. We first divided samples into wild type (WT) (0%–5% deletion), lower frequency of deletion (LD, 5%–15% deletion), medium frequency of deletion (MD, 15%–40% deletion), and higher frequency of deletion (HD, 40%–65%) according to the percentage of deletion in 82 MRP genes per tumor as the MRP deletion load. The frequency of genomic alteration of MRP genes varied differently across tumor types: thyroid cancer and acute myeloid leukemia (THCA and AML) showed the lowest frequency of MRP deletion, whereas lung squamous cell carcinoma, esophageal carcinoma, and ovarian cancer (LUSC, ESAD, and OV) showed the highest frequency of MRP deletion (Figure 2A). Interestingly, we found that the ranking of cancer types with MRP deletion is highly correlated between MRP gene expression and copy number alterations (CNAs) (Figures 2A–2C). Furthermore, we found a strong correlation between CNV and transcriptome, as well as between CNV and proteome on MRP genes, especially on affected MRPs, across tumor types (Figures 2D–2F). CRP genes did not show a strong correlation between CNV and transcriptome or proteome across tumor types (Figures 2D–2F), consistent with the previous findings of Ajore et al. (2017). Even though these affected MRPs were distributed across the genome (Figure 2G), they still showed a high concordance between CNAs and gene expressions (Figures 2G and 2H). To further demonstrate if MRP loss may affect mitochondrial functions by altering the content of mtDNA and its encoded proteins, we performed additional analyses based on the approach by Yuan et al. (2020). We found that tumors with MRP deletions tend to have elevated mtDNA content, especially in tumor with high frequency of MRP deletion (HMD: 15%–65%, p < 0.005) (Figure 2I). In addition, the expressions of affected MRPs were highly correlated with the mtDNA-encoded proteins in individual cancer types compared with the expression of CRP deletions (p < 0.05) (Figure S5). Collectively, these results indicate that affected MRPs are likely to be functional in cancer.
Figure 2.

MRP loss was associated with its expression
(A) The percentage of samples in each cancer type with different MRP gene deletion status.
(B) A relative dot plot chart shows the correlation between copy number and mRNA expression level of the MRP genes for each cancer type.
(C) Scatterplot showing the percentage of samples in each cancer type with MRP gene deletion (patient with 15%–65% percentage of MRP deletions from Figure 2A) (y axis) and the averaged correlation coefficient between copy number and RNA levels from Figure 2B (x axis). Color and fit, as in Figure 2B. R = Pearson’s correlation coefficient, Pearson’s p < 0.005.
(D) The curves show the Pearson’s correlation coefficients of DNA copy number and gene expression from 8,896 tumor tissues in TCGA cohorts. The pink line represents the correlation coefficients for the affected MRP genes. The purple line represents the correlation coefficients for the MRP genes; The blue line represents CRP genes. The gray line represents the density distribution of the 1,000 random permutations of the genes. Statistical significance was tested by Wilcoxon rank-sun test. ∗∗∗p <0.005.
(E) The curves show the Pearson’s correlation coefficients of DNA copy number and protein levels in CCLE cohorts. The pink line represents the correlation coefficients for the affected MRP genes; the purple line represents the correlation coefficients for the MRP genes; the blue line represents CRP genes. The gray line represents the density distribution of the 1,000 random permutations of the genes. Statistical significance was tested by Wilcoxon rank-sun test. ∗∗∗p <0.005.
(F) The density curves show the distribution of the Pearson’s correlation coefficients of gene expression and DNA copy number from the TCGA-LIHC, ICGC-HCC, and LCI-HCC cohorts. The pink line represents the correlation coefficients for the affected MRP genes; the purple line represents the correlation coefficients for the MRP genes; the blue line represents CRP genes. ∗∗∗p <0.005. (Affected MRP genes versus CRP: ∗∗∗; MRP genes versus CRP: ∗∗∗).
(G) A relative circle plot of affected MRP genes and chromosomes is shown clockwise in the outermost circle. The two innermost circles represent the correlation between gene expression and CNV and the deletion frequencies of affected MRP genes in each cancer type.
(H) Scatterplots showing the percentage of patients with affected (left panel) and unaffected (right panel) MRP genes deletion (y axis) correlated with the correlation coefficient between copy number and RNA levels (x axis). Color and fit, as in Figure 2B. R = Pearson’s correlation coefficient, Pearson’s p < 0.005.
(I) Boxplots of mtDNA copy number in the different MRP deletions status. (HMD: high and medium frequencies of MRP deletions [n = 341]; LD: low frequencies of MRP deletions [n = 253]; WT: wild type of MRP genes [n = 345]). Statistical analyses were performed with Student’s t test for two distinct groups. ANOVA test was used to compare the mtDNA copy number between three groups, ANOVA p value <0.05. Data are represented as mean ± SEM
Subunit of MRP complex co-regulated in pan-cancer
To assess whether MRP complexes were co-regulated at mRNA levels across cancer, we performed correlation analyses between 82 MRP expressions across normal tissues and tumors. We found a positive correlation between all MRPs in normal tissues (Figures S6A and S6B). Interestingly, only some subunits of MRPs were co-regulated in tumors (Figures S6C and S6D). As proliferative cells require cellular protein synthesis, a major ATP-consuming process, via both efficient cytosolic and mitochondrial ribosomal biogenesis, one would expect a co-regulated expression between CRPs and MRPs in a normal physiological condition. Although we observed an expected positive correlation between MRPs and CRPs in normal tissues, especially in organs rich in mitochondrial activities such as the liver, a negative correlation between MRPs and CRPs in tumors was observed (Figures S6A–S6D). These data might further support the functional impact of MRP deletions in cancer.
Affected signaling pathways in tumors with MRP deletions
To determine the critically affected cellular signaling based on differentially expressed genes in tumors with MRP deletions, we performed gene ontology analysis of 7,350 curated pathways. As expected, the top-10 downregulated pathways in tumors with altered MRPs across multiple cancers were mitochondria metabolism-related pathways involving amino acid and fatty acid oxidation and tricarboxylic acid (TCA) cycle pathways (Figure S7A). Consistently, mitochondria-associated metabolic signaling was significantly depleted in tumors with altered MRPs in most cancer types (Figure S7B). Noticeably, a majority of the top-10 upregulated pathways in tumors with altered MRPs were cell cycle related (Figures S7A and S7B). We also performed a pathway cross talk analysis to determine the relationship among affected pathways. We found that different signaling pathways have cross talk, either in downregulated or upregulated pathways (Figures S7C and S7D).
To examine the correlation of the tumor cell community with affected MRPs, we reanalyzed single-cell RNA sequencing data from 3 different cancer types, i.e., head and neck squamous cell carcinoma (HNSCC), low-grade glioma (LGG), and hepatocellular carcinoma (HCC) (Table S3). As shown in Figures 3A and S8A, t-SNE dimensionality reduction analysis divided all malignant cells into different clusters according to cellular transcriptional states for each patient. We found that malignant cells formed tumor-specific subpopulations, but each subpopulation could be further divided into several subclusters that appeared to be linked to the expression of affected MRPs in cancer cells. In contrast, we did not observe subcluster-associated expression patterns for CRP genes (Figure S8B).
Figure 3.

Characterization of pathway affected by MRP deletions by single-cell analysis
(A) A t-SNE plot of malignant single cells from the HNSCC dataset. Each dot represents the tumor from which the cell comes from. Cells are colored according to the average expression of affected MRP genes.
(B) Pseudotime trajectory of malignant cells of HNS6 tumor constructed on a reversed graph embedding method. Cells were colored according to trajectory states divided into four main clusters (left) or progression of pseudotime (right).
(C) Single-cell trajectory analysis of HNS6 tumor colored by the average expression of affected MRP genes.
(D) The t-SNE dimension is reduction divided into four main clusters according to trajectory analysis from Figure 3B.
(E) Boxplots of Inferred copy number (log2) of affected MRP genes in four clusters according to trajectory analysis of HNS6 from Figure 3B. Data are represented as mean ± SEM.
(F) Violin plots of inferred copy number (log2) of affected MRP genes in four clusters according to trajectory analysis of HNSCC cells from 10 tumors. Statistical significance was tested by one-way ANOVA with Tukey’s correction for multiple testing. ∗p <0.05; ∗∗∗p <0.005; ∗∗∗∗p <0.0005; ns indicates p ≥ 0.05. Data are represented as mean ± SEM.
(G) Violin plots of inferred copy number (log2) of unaffected MRP genes in four clusters according to trajectory analysis of HNSCC cells from 10 tumors. ns indicates p ≥ 0.05. Data are represented as mean ± SEM.
(H) Gene set enrichment analysis of four clusters whose distribution is significantly altered across affected MRP genes progression and pseudotime. Density of pathways that were significantly enriched in either cluster is listed. The x axis represents a normalized enrichment score (NES). The ordinate indicates the top-ranked signal pathways enriched by KEGG in different clusters. The box on the right summarizes the types of signal pathways.
(I) Schematic diagram of the four stages across affected MRP genes progression and pseudotime.
(J) Single-cell trajectory analysis of HNS6 tumor colored by the gene set variation analysis (GSVA) score of stemness genes.
(K) Violin plots of GSVA score of stemness genes in four clusters according to trajectory analysis of HNSCC cells from 10 tumors. ∗p <0.05; ∗∗∗p <0.005. Data are represented as mean ± SEM.
(L) Single-cell trajectory analysis of HNS6 tumor colored by the GSVA score of TCA cycle genes.
(M) Violin plots of GSVA score of TCA cycle genes in four clusters according to trajectory analysis of HNSCC cells from 10 tumors. ∗p < 0.05; ∗∗∗p <0.005. Data are represented as mean ± SEM.
(N) Correlation matrix plots showing the correlation between affected MRP genes expression and enrichment score of three gene sets (TCA cycle, DNA repair, and Stemness) from single cells in eight different cancer types. The size of the dots indicates the absolute value of Pearson’s correlation coefficient. The pink color stands for a negative correlation, and the purple one stands for a positive correlation.
We used a reversed graph embedding method (Tritschler et al., 2019) to further analyze functional relationships among tumor cell clusters in each tumor. Using pseudospatial trajectory reconstruction, we generated tumor cell trajectories within each tumor lesion and found that expression patterns of affected MRPs were largely correlated with the pseudotime (Figures 3B, 3C and S8C–S8F). We followed tumor clusters as cluster 1 to cluster 4 according to the pseudotime and found a similar order of tumor cell clusters based on t-SNE analysis (Figure 3D). Consistently, the expression levels of affected MRPs were dependent on the order of the clusters, with cluster 1 having the lowest expression and cluster 4 having the highest expression in cancer cells (Figure S8G). Consistently, the expression levels of CRP genes did not vary much among tumor cell clusters (Figure S8H). Furthermore, the inferred CNV profiles of affected MRPs increased along the pseudospatial trajectory either in a representative tumor or in a combined tumor data but were not observed in unaffected MRPs (Figures 3E–3G and S8I). Consistent with bulk genomic and transcriptomic data, we observed a higher correlation between gene expression and CNV in MRPs than in CRPs in HNSCC, LGG, and HCC (p < 0.005) (Figure S9A). Consistently, we found that both inferred CNVs and gene expression of shared affected MRPs were significantly lower than in unaffected MRPs in tumor cells from HNSCC and LGG (Figures S9B and S9C). In summary, these results suggested that affected MRPs were largely enriched in specific tumor clusters with similar inferred somatic copy number alterations (SCNAs) and transcriptomes.
To further determine whether tumor cells with altered MRPs had unique cell states, we performed gene set enrichment analysis (GSEA) using kyoto encyclopedia of genes and genomes (KEGG) biological process. We identified pathways enriched in each tumor cell cluster along the trajectory in malignant cells. Genes that were activated in cluster 1 (enriching cells with MRP loss) were mainly involved in stemness regulation, whereas genes that were downregulated in cluster 4 were mainly linked to mitochondria-associated pathways (Figure 3H). In contrast, genes in cluster 2 were mainly associated with the regulation of the epithelial-mesenchymal transition program, and genes in cluster 3 were mainly related to carbon metabolism and biosynthesis of amino acids (Figure 3H), possibly generating cellular metabolites needed for cell proliferation for a mesenchymal-epithelial transition (MET)-like process (Dongre and Weinberg, 2019). Interestingly, we observed a transition from cells with stemness features toward cells with differentiated features among tumor cells that correspond to pseudotime (Figures 3I and S10A). Accordingly, we focused our attention on TCA cycle and stemness-associated pathways. Further analysis of the enriched stemness pathways revealed that the top-ranking gene is MALAT1 predominantly elevated in the cluster 1-related cells with MRP loss compared with other clusters (Table S4). We demonstrated that stemness-associated genes were expressed in cells at the start of the trajectory (cluster 1 tumors) and were silenced as cells approached the patch border (cluster 4 tumors) in the cancer cells of a single patient (Figures 3J, 3K, and Table S4). Similar finding was also found in LGG and HCC cancer cells (Figures S10B and S10C). In contrast, TCA cycle genes were significantly upregulated along the trajectory from multiple patients of different cancer types (Figures 3L, 3M, S10B, S10D, Table S5).
To further validate our hypothesized link between MRP inactivation and stemness properties, and TCA cycle across cancer types, we correlated stemness and the TCA cycle features with the expression of affected MRPs. The TCA cycle features were significantly positively correlated with affected MRPs in single cells across different cancer types, but the stemness features showed a significant negative correlation with affected MRPs (Figure 3N). In addition, the expression of affected MRPs was significantly correlated with eight common features of cancer at the single-cell level (Figure S10E). These results may suggest that subpopulations of cancer stem cells (CSCs) in the tumor exhibit mitochondrial dysregulation due to the loss of MRPs.
Loss of MRP genes is associated with poorer survival in patients with cancer
To further determine a functional consequence of MRP loss, we performed survival analysis across 27 different types of solid tumors annotated with clinical information from TCGA cohorts. We corrected the patient backgrounds for SCNA in different cancer types using propensity score matching, which has been used to predict the causal relationship between factors and results (Austin, 2011). All samples were corrected from multiple dimensions, such as sex, fraction of altered genome, mutation count, and fraction of random deletion, and it was found that the deletion frequency of MRPs is significantly higher than expected by chance (Figures S11A–S11C). The difference in overall survival (OS) between MRP-deleted and non-deleted groups had the most significant increase after correction (Figure S11C). Furthermore, we noted MRP deletions to be highly correlated with the patient’s OS across 27 different cancer types (Figure 4A). Strikingly, there was a significant decreasing trend in the OS of patients from WT, LD, and MD/HD across pan-cancer (p-trend < 0.001) (Figure 4B). We found that both OS and disease-free survival (DFS) of patients in the MRP deletion group were significantly lower than those in the WT group among 20 individual cancer types from the TCGA cohort (Figures S12A and S12B). Consistently , the MRP deletions group was also associated with poor OS in patients from the MSK-IMPACT cohort (Figure S12C). To further determine if the MRP deletion-associated prognosis is independent of other variables such as clinical tumor stage, tumor type, gender, race/ethnicity, and age, we estimated the hazard ratio for patients with tumor in the MRP deletions group compared with the WT group after adjustment with multivariable Cox proportional hazards regression. The hazard ratio was significantly (p < 0.001) greater in the MRP deletions group than in the WT group in individual cancer types and all cancers combined when adjusted by other co-variables in the multivariable model, but the ranking of different cancer types was somewhat similar but not identical (Figures 4C–4F, Table S6). Noticeably, chi-square test showed that the proportion of tumors with MRP deletions was significantly associated with older age (p = 1.408 × 10−6), malignant tumor types (p = 1.668 × 10−5), and tumors with advanced stages (p = 2.348 × 10−12) in the TCGA cohorts (Figure S13). These results indicate that tumors with altered MRPs were associated with poor survival outcome.
Figure 4.

Clinical outcomes in correlation with MRP deletions
(A) Correlation between the percentage of patients with MRP deletions by cancer type (y axis) and hazard ratio (HR) (x axis) in TCGA cohorts. Linear regression lines are drawn (black line) with 95% confidence interval (gray zone); R = Spearman’s correlation coefficient; Mann-Whitney U-test p values are stated.
(B) Survival analysis of TCGA (left) and MSK-IMPACT (right) patients for subjects grouped according to the four MRP deletions status. (HD: high frequencies of MRP deletions; MD: medium frequencies of genes deletion; LD: low frequencies of MRP deletions; WT: wild type of MRP genes). Log-rank test for trend was performed to show the statistically significant trends in survival rates.
(C and D) Multivariate Cox regression analysis of clinical characteristics correlated for overall survival (OS) (C) and disease-free survival (DFS) (D) among groups of MRP deletion (LD, MD, HD, and WT) from the TCGA cohorts
(E) HR forest plot for OS and DFS endpoints in TCGA cohort. Patient samples in each dataset are stratified into the WT or deletion group based on the percentage of patients with MRP deletions. The classification of the groups of patients with MRP deletions is provided in Supplementary Data Table S6.
(F) HR forest plot for OS endpoints in MSK-IMPACT cohort. Patient samples in each dataset are stratified into the WT or deletion group based on the percentage of patients with MRP deletions. The classification of the groups of patients with MRP deletions is provided in Supplementary Data Table S6.
The landscape of driver mutations in tumors with MRP deletions
To further explore the impact of MRP deletions on cancer pathogenesis, we determined the landscape of driver mutations across 28 different cancer types stratified by MRP deletions. Commonly mutated genes were selected by the criteria of MutSig analysis q-value < 0.1. We found that TP53 mutations are enriched in 62% of the HMD tumors, almost four times higher than that in the WT group (17%) (Figure 5A) in the TCGA dataset. Similar results were found in the MSK-IMPACT dataset, in which TP53 mutations are also enriched in 58% of the HMD tumors (Figure 5A). Other frequently mutated genes, e.g., PIK3CA and PTEN, were not enriched in tumors with MRP deletions (Figure 5A). Among 1,549 somatically mutated genes, 475 genes were significantly enriched in the HMD group, and 1,047 genes were significantly enriched in the WT group from the TCGA dataset (Figure 5B; left panel). We identified the most significant enrichment of TP53 and CDKN2A mutations in tumors with altered MRPs in the TCGA cohorts, as determined by odds ratio (Figure 5B; right panel). TP53 mutations were also found to be significantly associated with the HMD group in the MSK-IMPACT cohorts (Figure 5C). In addition, TP53-mutant tumors showed higher percentage of MRP deletion compared with TP53 WT tumors, whereas low percentage of MRP deletion or no difference was detected in the other top-nine frequently mutated genes (Figure S14).
Figure 5.

Clinical impact of co-occurring heterozygous deletion in MRP genes and TP53 mutation in pan-cancer
(A) The landscape of somatic mutations in all cancer types from TCGA according to the MRP deletions subgroups (HMD, LD, and WT). The top panel shows 10 genes identified using MutSigCV (q value < 0.1). The right side of the columns shows the fraction of mutated samples according to the MRP subgroup from TCGA and MSK-IMPACT cohorts (T: TCGA; M: MSK-IMPACT). Clinical annotations for cancer type and survival status across subjects are shown at the bottom.
(B) (Left panel) A volcano plot of the negative log of the p value on the y axis; the x axis shows the magnitude of association (odds ratio (log)). The red line represents the threshold of significance determined by multiple hypothesis testing. Dot colors indicate the gene was enriched in HMD groups (pink) or WT groups (blue). (Right panel) Forest plots show the top five genes highly mutated in HMD groups (pink) or WT groups (blue) from TCGA cohorts. The x axis shows the log odds ratio with error bars.
(C) Forest plots show the top five genes highly mutated in HMD groups (pink) or WT groups (blue) from MSK-IMPACT cohorts. The x axis shows the log odds ratio with error bars.
(D) Effects of MRP deletions group detectable gene-level alterations on overall survival. A volcano plot was constructed by plotting the negative log of the p value on the y axis; the x axis shows the hazard ratio (log2) of the different genes in the HMD group. p values calculated by using the Cox proportional HR with 95% confidence interval are shown.
(E) Survival analysis of TCGA and MSK-IMPACT patients from the wild type or MRP deletion group (LD and HMD) alone or co-occurring with P53 mutations.
(F and G) (F) Multivariate Cox regression analysis of clinical characteristics correlated for overall survival among groups of MRP deletion (LD, HMD, and WT) alone or co-occurring with P53 mutations across multiple cancers from the TCGA cohorts. (G) Multivariate Cox regression analysis of clinical characteristics correlated for overall survival among groups of MRPs deletion (LD, HMD, and WT) alone or co-occurring with P53 mutations in ACC from the TCGA cohorts.
To investigate whether the clinical outcomes of patients with MRP deletions can be affected by co-occurring mutations, we analyzed the combined impact of different mutations and loss of MRPs on patient survival across multiple cancers. Interestingly, among known cancer drivers, TP53 mutations in the HMD genes group were the only gene mutations showing a significantly positive association with survival outcomes as determined by the log rank test (Figure 5D). Moreover, patients with tumors without altered MRPs and TP53 had the best survival, whereas patients with tumors having both TP53 mutations and altered MRP had the worst survival, as determined by the log rank test for trend (Figure 5E). To further determine if the MRP deletion-associated survival is influenced by TP53 mutations, we performed multivariable Cox proportional hazards regression analysis by adjusting the TP53 mutation status. We found that MRP loss is significantly associated with poor survival independent of TP53 mutations in most individual cancer types as well as all cancers combined, although a combination of MRP loss and TP53 mutations further contributes to a worse prognosis in both TCGA and MSK-IMPACT cohorts (Figures 5F, 5G, and S15). These findings indicated that the co-occurrence of altered MRPs and TP53 mutations were common in pan-cancer and were linked to cancer malignancy.
Mutational signatures associated with MRP loss
Deciphering mutational signatures provides insight into the mutational processes caused by exogenous carcinogenesis and endogenous somatic mutagenesis. Previous studies of multiple mutational types have identified 30 signatures from the 96 mutational trinucleotides that may involve enzymatic modification of DNA, defective DNA repair and replication, or exogenous or endogenous mutagen exposures, and some with unknown etiologies (Maura et al., 2019). Accordingly, we analyzed mutational signatures that consist of frequency patterns along 96 trinucleotides specific for each group with WT, P53-mut, HMD, or P53-mut/HMD. We found that signatures 2, 4, 7, 11, 13, 22, 24, and 29, which relate to exogenous mutagens, were much more prevalent in the HMD group as well as the P53-mut/HMD group across multiple cancers from both TCGA and MSK-IMPACT cohorts (Figure 6A). In contrast, the signatures 1, 6, 9, 10, 15, 20, 21, and 26, which are linked to endogenous mutagens, were much more prevalent in the WT group in pan-cancer as well as individual cancer types from TCGA datasets (Figures 6A and S16A). Statistical analysis revealed that signatures significantly associated with HMD or P53-mut/HMD include mutational patterns linked to UV exposure, alkylating agents (such as temozolomide), tobacco smoking, viral infection, and DNA double-strand breaks (Figure 6B). However, WT was differentially associated with defective DNA mismatch repair, spontaneous deamination of 5-methylcytosine, and error-prone polymerase POLE (Figure 6B). Tumors with HMD exhibited increased signature 7 in TCGA and MSK-IMPACT dataset (Figure 6C), characterized primarily by the presence of C>T mutations predominantly induced by UV light exposure (Trucco et al., 2019). In contrast, the frequency distribution of single-nucleotide substitutions of P53-mut/HMD tumors was mainly enriched in C>A, and C>G single nucleotide (Figure S16B). HMD also exhibited increased signature 13, which is associated with the activation of the APOBEC family cytidine deaminases. Indeed, activation of APOBEC cytidine deaminases is associated with viral infection (Zhu et al., 2020) and tobacco smoke (Alexandrov et al., 2016). In contrast, P53-mut/HMD mainly expressed two signatures, including signature 4, whose display is correlated with tobacco smoking-induced C>A mutations (Alexandrov et al., 2016), and signature 7 (Figure 6C). These results suggest that the MRP deletions are mainly linked to increasing exogenous environmental exposures and p53 mutations contributed to additional mutational processes to drive tumorigenesis.
Figure 6.

Profiles of MRP deletions causing mutational signatures
(A) The average relative contribution for 30 COSMIC signatures from the mutation spectrum of each tumor sample across multiple cancers from TCGA (left) and MSK-IMPACT (right) cohorts. Signatures have been ordered according to their characteristic of mutation types arising from specific mutagenesis processes, including exogenous, endogenous, and unknown.
(B) Cause analysis of different mutation signatures. The mutation signatures are summarized into different causes (exogenous, endogenous, and unknown). (Left) Bar plot represents the fold change of mutation signature enrichment in WT and HMD of MRP subgroups. (Right) Statistically significant differences in mutation signature are indicated by the heatmap (p value <0.05, one-tailed hypergeometric test).
(C) Mutation signature analysis of TCGA and MSK-IMPACT patients subgrouped by different types of MRP deletion (HMD and WT) alone or co-occurring with P53 mutations. Y axis shows the enrichment level of different types of nucleotide substitution (mutation type probability %).
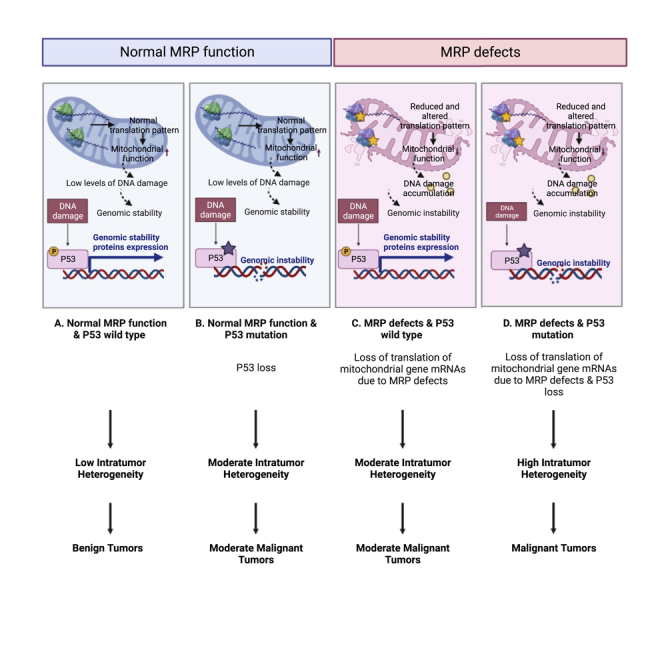
MRP loss and TP53 mutations are associated with intra-tumor heterogeneity
Genomic instability is a major contributing factor to ITH, a tumor feature responsible for treatment resistance and therapeutic failure (Andor et al., 2017; Ma et al., 2019; Wolf et al., 2019). Several studies have demonstrated the important role of p53 in maintaining genomic stability (Donehower et al., 2019). We hypothesized that deletions of multiple MRP loci distributed across the genome in pan-cancer is due to p53-induced genomic instability. Our observation revealing that p53 mutations are tightly associated with MRP deletion in multiple human cancer types is consistent with this hypothesis. To further determine the relationship between MRP deletions and p53 mutations, we measured the Mutant-Allele Tumor Heterogeneity (MATH) score, a method to monitor ITH linked to tumor prognosis and metastasis with the use of bulk tumor whole-exome sequencing data (Noorbakhsh et al., 2018). We found that MRP loss is significantly associated with MATH values in most individual cancer types as well as all cancers combined (Figures 7A and S17A). We also determined the tumor mutational burden (TMB) score, a method to calculate the number of somatic mutations per megabase in the genomes (Angus et al., 2019). Indeed, increased TMB scores were evident in P53-mut/HMD tumors in individual cancer types and all cancers combined (Figures 7B and S1B). We also used the entropy-based diversity score across different cancer types to determine the link between transcriptomic diversity and MRP deletions in the context of p53 mutations (Park et al., 2016). We found a positive correlation between MRP deletions and transcriptomic diversity across pan-cancer (Figure 7C). Noticeably, the transcriptomic diversity scores were also significantly higher in P53-mut/HMD tumors than in tumors with other types in most cancer types (Figure S18).
Figure 7.

Deletion of MRP-induced intra-tumor heterogeneity in pan-cancer
(A) Ridgeline plots showing the distribution of MRP deletions, Mutant-Allele Tumor Heterogeneity (MATH) in different types of MRP deletion (HMD, LD, and WT) alone and co-occurring with P53 mutations. The dotted lines represent the median of the P53-mut/HMD group under the x axis transformation.
(B) Ridgeline plots showing the distribution of mutation burden in different MRP deletion (HMD, LD, and WT) alone and co-occurring with P53 mutations. The dotted lines represent the median of the P53-mut/HMD group under the x axis transformation.
(C) Scatterplots showing the percentage of patients with MRP deletions by cancer type (y axis) and the diversity score (x axis). Color and fit, as in Figure 2B. R = Spearman’s correlation coefficient, Spearman’s p < 0.0005.
(D) Violin plot of expression (log2) of affected MRP genes in WT and P53 mutation patients from HNSCC single-cell datasets. The upper part is the distribution of diversity scores. ∗p <0.05. Data are represented as mean ± SEM.
(E) Validation of affected MRP expression and tumor biodiversity using the HNSCC malignant single-cell dataset. The red line represents the linear regression fit, with the gray band representing the 95% confidence interval.
(F) Validation of affected MRP expression and tumor biodiversity using the HNSCC non-malignant single-cell dataset. The red line represents the linear regression fit, with the gray band representing the 95% confidence interval.
We further determined an association between transcriptomic diversity and the co-occurrence of MRP deletions and TP53 mutations at the single-cell level using a method described recently (Ma et al., 2019). In HNSCC tumor samples with available single cell transcriptomic data, we found that transcriptomic diversity was higher in P53-mut tumors, with the lower expression of affected MRPs, than in P53-WT tumors (Figure 7D). There was an inverse correlation between transcriptomic diversity and the expressions of affected MRPs only in malignant single cells (Figure 7E) but not in non-malignant cells (Figure 7F), suggesting MRP loss is an important contributor to tumor heterogeneity. Taken together, our results are consistent with the hypothesis that TP53 mutations drive ITH, which further selectively enriches MRP deletions to promote tumor progression.
Discussion
Tumor heterogeneity continues to affect poor prognosis thereby reducing therapy responses in various cancer types (Andor et al., 2016). Recent studies suggest that cellular metabolic heterogeneity may contribute to tumor heterogeneity by allowing cells to adapt to environmental exposures for maintaining homeostasis and growth (Kim and DeBerardinis, 2019). For example, metabolic heterogeneity may promote malignant cell growth and stemness properties and may render malignant cells resistant to therapeutics (Kim and DeBerardinis, 2019). Several studies have reported the relationships between cell metabolism and gain or loss of mutation in a particular gene linked to metabolism (Ferreira et al., 2012). Although the roles of mitochondrial dysfunction in metabolic heterogeneity of tumors have been studied (Xiao et al., 2019), genetic basis for the role of mitochondrial ribosome biogenesis in cancer types has not been explored. As metabolic heterogeneity in malignant cells may be the consequence of OXPHOS-mediated activities (Xiao et al., 2019), it is reasonable to speculate that the OXPHOS function, in turn, may be tightly linked to metabolic heterogeneity, which contributes to tumor cell plasticity, tumor cell evolution, and survival. Our study confirms high variation of MRPs in different tumor types at both tissue bulk and single-cell levels. For example, we found that the degree of MRP deletions can be linked to prognosis. We suggest that the degree of MRP deletions may be closely related to OXPHOS-mediated activities and may serve as an alternative mechanism to determine metabolic heterogeneity.
Chromosomal instability may contribute to ITH observed in tumors and may drive phenotypic plasticity during tumor evolution (Raynaud et al., 2018). TP53 is commonly referred to as the “guardian of the genome” to prevent chromosomal instability. TP53 mutations lead to the loss of proper cell cycle checkpoint control, resulting in enhanced chromosomal instability (Raynaud et al., 2018). In this study, we found that tumors with loss of affected MRPs exhibited chromosomal instability and ITH. We also revealed that MRP defects frequently accompanied TP53 mutations. However, this feature does not suggest that TP53 mutations are required to drive observed MRP deletions because molecular and clinical features associated with MRP loss can be independent of TP53 mutations. Rather, it is plausible that dysfunctions of mitoribosomal biogenesis in some tumors may be the consequence of TP53 mutation-induced chromosomal instability and, subsequently, metabolic heterogeneity, collectively leading to malignant transformation. As genomic instability-induced tumor heterogeneity accelerates the development of multidrug resistance, leading to treatment failure and tumor recurrence (Andor et al., 2017), identifying novel strategies to prevent tumor heterogeneity is, therefore, of paramount importance for cancer treatment. Together, our findings provide important new insights into the key role of mitochondrial ribosome biogenesis in genomic instability and ITH with implications for our understanding of cancer pathogenesis and therapy.
It is noted that malignant properties enabled by p53 inactivation are acquired through a pattern of genome evolution that involves four sequential phases—TP53 loss of heterozygosity, accumulation of deletions, genome doubling, and the emergence of gains and amplifications (Baslan et al., 2022), indicating that accumulation of deletions is an important feature of cancer evolution. Recent studies suggest that heterozygous genetic alterations may cause genetic instability in a yeast model, which may mimic genetic instability during cancer evolution in mammals. Interestingly, inactivation of genes identified by selecting for genetic instability in yeast model includes several MRPs (e.g., MRPL1), which is consistent with the findings of MRP mutations in human cancer (Coelho et al., 2019). Our work suggests that affected MRPs were clonally evolved using single-cell analysis, a phenomenon linked to metabolic heterogeneity and tumor stemness. However, single-cell data are mainly from inferring CNAs using transcriptome data, which mainly links to their cellular states. Future studies will be required to analyze single-cell DNA sequencing data to study the relationship between affected MRPs and p53 mutations during cancer evolution.
Our pan-cancer analysis of 18,177 tumors from 28 tumor types suggests that MRP deletions were functionally important in driving tumorigenesis. This plausible interpretation is supported by the following observations. First, heterozygous deletions of certain MRPs are frequently found in most human solid malignancies and this non-random feature is stable across cancer types. Second, loss of the MRP loci is accompanied by reduced expression of affected MRPs at both mRNA and protein levels. Third, tumors with MRP deletions frequently show malignant features including an association with poor survival independent of tumor staging and TP53 mutations. Fourth, tumors with MRP deletions carry unique driver mutations and unique mutational signatures that reflect an exposure to environmental mutagens rather than endogenous mutagens across tumor types. Fifth, tumors with MRP deletions display elevated mtDNA content, a sign of dysregulated mitochondrial functions, accompanied by an enrichment of affected mitochondrial signaling pathways. Sixth, a functional screening of co-dependent cellular genes using the CRISPR-shRNA system-generated shinyDepMap database further supports a direct consequence of affected MRPs on mitochondrial functions. Collectively, these results support the hypothesis that dysregulation of mitoribosomal biogenesis by somatic deletions of certain MRPs may drive tumor malignancy.
Limitations of the study
Although this study may provide new insights into a potential mechanism as to how genetically altered MRPs might be cancer drivers, there are several limitations regarding the causal relationship between MRP defects and cancer progression. First, although the strength of association between MRP defects and tumor malignancy is strong, its causal role will need to be further determined using both in vitro and in vivo model systems. We also do not know the mechanism that leads to altered MRPs. Second, our single-cell RNA sequencing analysis revealed that affected MRPs may be clonally evolved and are associated with metabolic heterogeneity and tumor stemness. However, single-cell analysis was mainly from inferring CNAs using transcriptome data, and this feature is mainly linked to cellular states rather than somatically evolved clonality. Future studies using single-cell DNA sequencing together with single-cell transcriptome analysis may be needed to study tumor cell evolution in the context of MRP alteration and p53 mutations during cancer progression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| TCGA genomic, transcriptomic, and clinical data | NIH Genomic Data Commons (GDC) | https://portal.gdc.cancer.gov/; https://gdc.cancer.gov/about-data/publications/pancanatlas; cBioPotal: https://www.cbioportal.org/ |
| TCGA mutation annotation file | NIH Genomic Data Commons (GDC) | cBioPotal: https://www.cbioportal.org/ |
| MSK-IMPACT genomic, mutation, and clinical data | Memorial Sloan Kettering cancer center | cBioPotal: https://www.cbioportal.org/ |
| CCLE genomic, mutation, and clinical data | Broad Institute Cancer Cell Line Encyclopedia (CCLE) | https://portals.broadinstitute.org/ccle |
| Mitoribosome structure | This paper | PDB: 5AJ4 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| HNSCC Single Cell RNA expression | (Puram et al., 2017) | GSE103322 |
| Gliomas Single Cell RNA expression | (Filbin et al., 2018) | GSE102130 |
| HCC Single Cell RNA expression | (Ma et al., 2019) | GEO125449 |
| AML Single Cell RNA expression | (Fan et al., 2018) | GSE110499 |
| BRCA Single Cell RNA expression | (Chung et al., 2017) | GSE75688 |
| COAD Single Cell RNA expression | (Li et al., 2017) | GSE81861 |
| KIRP Single Cell RNA expression | (Kim et al., 2016) | GSE73121 |
| LUSC Single Cell RNA expression | (Lambrechts et al., 2018) | E-MTAB-6149 |
| Software and algorithms | ||
| Significance of copy-number changes – GISTIC 2.0 | (Mermel et al., 2011) | GenePattern: https://cloud.genepattern.org/gp/pages/index.jsf |
| R version 3.6.2 | The R Project for Statistical Computing | https://www.r-project.org/ |
| UCSF Chimera | Resource for Biocomputing, Visualization, and Informatics (RBVI) | https://www.cgl.ucsf.edu/chimera/ |
| Maftools version 2.4.05 | Mayakonda Lab | https://www.bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html |
| DeconstructSigs version 1.8.0 | Swanton Lab | https://github.com/raerose01/deconstructSigs |
| GSEA version 4.0 | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| Monocle version 2.6.3 | Trapnell Lab | http://cole-trapnell-lab.github.io/monocle-release/ |
| InferCNV | GitHub | https://github.com/broadinstitute/inferCNV |
| Fgsea version 1.10.1 | Sergushichev Lab | http://bioconductor.org/packages/release/bioc/html/fgsea.html |
Resource availability
Lead contact
Further information and requests for resources data analysis results should be directed to and will be fulfilled by the Lead Contact, Xin Wei Wang (xw3u@nih.gov).
Materials availability
This study did not generate new unique reagents and animal strain.
Experimental model and subject details
Clinical and genomic data were obtained from TCGA, MSK-IMPACT, and CCLE projects. The TCGA (phs000178.v10. p8) copy number data from GISTIC2, and log2 copy-number values are consist of 10,201 cancer and sample metadata from the NIH Genomic Data Commons (GDC). The MSK-IMPACT copy number data from GISTIC2 and log2 copy-number values consist of 7,976 cancer and sample metadata from Memorial Sloan Kettering Cancer Center. The CCLE copy number data from GISTIC2 and log2 copy-number values consist of 1,657 cancer and sample metadata. The scRNA-seq of eight individual different cancer types in humans collected from ArrayExpress and Gene Expression Omnibus (GEO). The accession number of the datasets were listed in key resources table. All single cells from individual different cancer types were analyzed using gene expression quantification and characterization of indicated functional states.
Method details
Availability of data
TCGA genomic, transcriptomic, mutation, and clinical data from NIH Genomic Data Commons (GDC) are available for download at the cBioPotal datasets (https://www.cbioportal.org/). ICGC/TCGA PCAWG genomic and clinical data are available for download at the PCAWG data portal (https://dcc.icgc.org/releases/PCAWG). MSK-IMPACT genomic, mutation and clinical data from Memorial Sloan Kettering cancer center are available for download at the cBioPotal database (https://www.cbioportal.org/). CCLE genomic, mutation, and clinical data are available for download at the Broad Institute Cancer Cell Line Encyclopedia (CCLE) datasets. The scRNA-seq of eight individual different cancer types in humans was collected from ArrayExpress and Gene Expression Omnibus (GEO). All single-cell data were provided from the expression matrix data of the original papers. The accession number of the datasets used in this study are listed in method. The mtDNA copy number was estimated using the TCMA data portal (http://bioinformatics.mdanderson.org/main/TCMA:Overview).
Identification and analysis of somatic MRPs copy number alterations
Clinical and genomic data were obtained from TCGA, MSK-IMPACT, and CCLE projects. The TCGA (phs000178.v10. p8) copy number data from GISTIC2, and log2 copy-number values are consist of 10,201 cancer and sample metadata from the NIH Genomic Data Commons (GDC). The MSK-IMPACT copy number data from GISTIC2 and log2 copy-number values consist of 7,976 cancer and sample metadata from Memorial Sloan Kettering Cancer Center. The CCLE copy number data from GISTIC2 and log2 copy-number values consist of 1,657 cancer and sample metadata. We determine copy number alteration thresholds according to the set of discrete copy number calls provided by GISTIC analysis: −2, deep loss/homozygous deletion; −1, shallow loss/heterozygous deletion; 0, diploid; 1, one copy gain; 2, high-level amplification. We considered as deletion altered only samples with either deep loss (−2) or shallow loss (−1) of genes located in regions with copy number alterations.
Percentage of deleted MRP genes in each patient is expressed as Equation 1
| Equation 1 |
where, = number of total MRP protein in the human genome, where is a constant = 82; = indicator function that returns 1 if the GISTIC number of such MRP gene is −1 or −2, otherwise returns 0; A = a MRP gene list.
The distribution of shallow deletions of affected MRPs gene per tumor
We determined shallow deletions according to the set of discrete copy number calls provided by GISTIC analysis: −1, shallow loss/heterozygous deletion. We employed the Equation 1 formula to calculate the percentage of shallow deletions of affected MRPs gene per tumor and the percentage of 1,000 equally sized random gene sets. Using R with coin package, we employed the permutation test to compare the shallow deletions of affected MRPs and 1,000 equally sized random gene sets (background). To find the difference in the percentage of defect of MRPs and random groups, we calculated the difference in means for each group. We then perform a two-sided t-test computing the percentage of shallow deletions of affected MRPs that exceeded equally sized random gene.
Identification and analysis of somatic MRPs mutation alterations
Clinical and mutation data were obtained from TCGA projects. Differences in MRPs mutation in multiple cancers were obtained from the Gene Set Cancer Analysis (GSCA) platform (http://bioinfo.life.hust.edu.cn/GSCA/#/) (Liu et al., 2018). For the analyses in the manuscript, missense mutation, nonsense mutation, splice site, frameshift mutation, and in-frame mutation were included.
Molecular structure analysis
The molecular model of the mitochondrial ribosome is based on the structural data (5AJ4) published in the PDB database (https://www.rcsb.org/). The structure analysis and visualization adopt the software UCSF Chimera, and the affected and unaffected MRPs are marked with different colors to form a 3D view with shadows.
Correlation analyses
To determine the correlation of MRP loss in different cancer types, we performed a correlation analysis between MRP loss and gene expression across different cancer types. We first identified the Pearson correlation between gene expression and copy number alteration from the TCGA cohort using R with cor.test () function in different cancer types. We then evaluated the correlation coefficients (r) and corresponding p value between percentage of samples in each cancer type with MRP gene deletion (patient with 15%–65% percentage of MRP deletions) and the averaged correlation coefficient between copy number and RNA levels. To determine the functional consequences of MRP loss by showing changes at the protein level in addition to mRNA, we performed correlation analysis of MRP genes between CNV and transcriptome, as well as CNV and proteome, across tumor types. We identified patients from the TCGA cohort with available information on transcriptome and copy number. The Pearson correlation coefficients (r) and corresponding p value between MRP gene expression and copy number alteration calculate by R with cor.test () function. We also evaluated the Pearson correlation between MRP proteome and copy number alteration from the CCLE cohort. For each gene sets (Affected MRP, Total MRP and Total CRP) or 1000 times randomly permuted 82 genes, the distribution graphs were obtained by the Pearson correlation coefficients calculated for each gene sets between the CNV and transcriptome, as well as CNV and proteome, across tumor types. To further demonstrate if MRP loss may affect mitochondrial functions by altering the levels of mtDNA encoded proteins, we also evaluated the Pearson correlation coefficients (r) and corresponding p value between the levels of mtDNA encoded proteins and expression levels of MRP genes from the CCLE cohort in different gene sets including affected MRP, total MRP and total CRP.
mtDNA copy number in different degree of MRPs defect
The mtDNA copy number was estimated based on the approach of the study by Yuan et al. (Yuan et al., 2020). We then calculated the percentage of MRP defects per tumor from the PCAWG cohort and matched it with the mtDNA copy number. To assess the correlation of deficiency of MRPs with mtDNA copy-number, we used t-test and ANOVA(if there are more than two groups).
Survival analyses
Clinical information of the TCGA and MSK-IMPACT cohorts was obtained from the cBioPortal (https://www.cbioportal.org/). Survival analyses based on MRPs defects were performed using R (v3.6.2) with survival package. Different stratified groups were compared using Kaplan-Meier curves and using the Log rank test for trend to calculate p values. The Cox Proportional Hazard model includes the different status of MRPs defect, age, gender, race/ethnicity, stage, and tumor type were performed using R (v3.6.2) with survival and survcomp package. The multivariate model was compared using the permutation test.
Propensity score matching of clinical parameters
To correct the selection bias of patients caused by the SCNA backgrounds and clinical parameters, we performed propensity score matching to predict the causal relationship between factors and results. The statistical quantity is the causal effect of defected of MRPs (5–60%) compared to the Wild type of MRPs (0–5%) on tumors from the MSK-IMPACT cohort. The clinical parameters including the mutation count, fraction genome altered, and gender provided by the MSK-IMPACT cohort. The SCNA backgrounds, including the percentage of 82 CRPs defects per tumor and 1,000 equally sized random gene sets defects, were calculated by the Equation 1 formula. The SCNA backgrounds and clinical parameters were fit into the nearest neighbor algorithm with a multivariate logistic model. Next, we set one to four matches in the logistic model. Propensity score matching for the calculation algorithm was performed by R version 3.6.2, primarily utilizing the packages MatchIt (version 4.2.0).
Gene set enrichment analysis
Pathway analyses in bulk data were performed on Gene Ontology (GO) biological process gene sets from the Molecular Signatures Database (MSigDB v6.2) by using GSEA (version 4.0). The detailed parameters of GSEA are set according to the recommendations from the Broad (Subramanian et al., 2005). Pre-ranked GSEA was performed with 1,000 permutations for wild type and a high degree of MRPs defect tumor. Pathways were selected with both p value and q-value < 0.05 ranks of the 20 top-ranked genes. The pre-ranked gene list is generated by the p value for differential analysis. Pathway analyses in single-cell data were performed on the KEGG biological process from the Molecular Signatures Database (MSigDB v6.2) using GSEA (version 4.0). Pathways were selected with the 5 top-ranked genes for either different cluster. The enrichment of each pathway was ranked by the normalized enrichment score (NES).
Mutation analysis in different degree of MRPs defect
The mutation data (MAF files) based on whole exome sequencing is obtained from the cBioPortal database. The individual MAF files in different cancer types were concatenated into one combined MAF file for different MRPs defect groups for downstream analysis. Driver genes with statistically significant levels of recurrent mutation were determined by Mutation Significance (MutSig). Visualization and summarization of the mutations were performed by custom scripts in R version 3.6.2, primarily utilizing the packages maftools (version 2.4.05).
Mutational signatures in different degree of MRPs defect
Mutational signatures were performed by three available algorithms: deconstructSigs, NMF, and maftools R packages. Contributions of distinct mutation signatures were investigated for each sample according to the distribution of six different possible substitutions (C>A, C>G, C>T, T>A, T>C, T>G) and base on the bases immediately 5’ and -3′ to mutated base, generating 96 possible mutation subtypes (6 types of substitution ∗ 4 types of 5′ base ∗ 4 types of 3′ base). The algorithm from deconstructSigs estimates the contribution of each signature for each sample. Each of the sample signatures is assigned a weight from previously described 30 COSMIC signatures that correspond to the mutations’ percentage explained by each extracted sample signatures. We further used the maftools and NMF package for R to implement an algorithm using the non-negative matrix factorization to break down the original matrix to the minimum set of mutation signatures. This algorithm estimates each signature contribution across the samples. The unsupervised hierarchical clustering was performed on samples to evaluate cosine similarities between the extracted sample signatures and each COSMIC signature. For the analyses in the manuscript, we focused on three main mutational processes: the endogenous mutational signatures (Tubbs and Nussenzweig, 2017) (Defective mismatch repair: Signatures 6, 15, 20, 21 and 26; POLE η and ε: Signatures 9 and 10; Spontaneous deamination of 5-methylcytosine generates an aging signature: Signatures 1), exogenous as well as virus-related mutational signatures (APOBEC: Signatures 2 and 13 (Petljak et al., 2019); Smoking: Signature 4 and 29; UV exposure: Signature 7; TMZ treatment: Signature 7; Aflatoxin exposures: Signature 24; Aristolochic acid: Signature 22), and DNA double-strand break signature (Signatures 3).
Constructing single-cell trajectories
We use the R package Monocle to build a single-cell trajectory. This package is based on the reversed graph embedding method. After the built-in standardization, PCA noise filtering and t-SNE reduce the dimensions. The data set is used to select highly variable genes based on empirical dispersion. Variable genes are used for dimensional analysis based on the DDRTree algorithm, and the tree structure is further calculated the plotted. In order to study which genes have produced key changes in the single cell developmental trajectory, the BEAM function that comes with the Monocle R package is used to study differential gene expression along the trajectory.
Functional characterizing of cancer features in MRP-deficient malignant single cells
We used the expression of the functional gene sets to detect the cancer feature for MRP-deficient malignant single cells. We detected 8 critical cancer features, including quiescence, metastasis, inflammation, hypoxia, EMT, DNA damage, differentiation, and angiogenesis (Different signature gene lists from (Yuan et al., 2019)). The signature pathway activities of 8 cancer feature across various cancer types in each single cell dataset were evaluated using the GSVA function in the GSVA package in R. As described in, the GSVA enrichment scores were summarized according to the expression-level rank statistics by the Kolmogorov-Smirnov (KS) statistic to characterize the signature activity. We then analyzed the relationship between the average expression level of MRPs and different signature pathway activities across various cancer types using Spearman’s rank correlation test.
Diversity score in bulk transcriptomic data
The gene expression diversity of each TCGA patient is calculated with Shannon entropy (), using Equation 2 modified from a previous publication (Hausser and Strimmer, 2009).
| Equation 2 |
Where, are the probabilities of each gene expression.
Diversity score in single-cell data
The diversity score was determined based on the gene expression profile of a tumor to measure the intratumoral heterogeneity. We employed t-SNE to project the original expression profiles of all malignant cells to the 2-dimential space to filter out useless information and extract information that reflects the structure of the sample itself. Furthermore, the centroid and geometric distance of all malignant cells within the tumor on the t-SNE projection are calculated based on the published method (Ma et al., 2019). where, is the total cell number of each sample, is the coordinates of each point, is centroid coordinates.
Mutation burden
Mutation burden is defined as the total number of mutations in one patient’s sample. Use the following formula for calculation.
Where, is defined as a counting function that returns the number of all mutations in sample .
Functional connections between MRP deletions and mitochondria
We determined the functional consequences of MRP deletions using the shinyDepMap database (https://labsyspharm.shinyapps.io/depmap/) (Shimada et al., 2021) based on functional screening of cellular genes using combined 40:60 mixing ratio of CRISPR and shRNA system. We searched co-dependent genes based on functional screening of 15,847 genes in 423 cell lines. We selected affected MRPs to determine, for each gene, the effect of cell viability by knockout/knockdown and the functional relationships across cell lines. Next, we selected 'Large' and ‘small’ clusters that contain the most correlated genes per cluster.
Processing of the single-cell dataset across pan-cancer
The scRNA-seq of eight individual different cancer types in humans collected from ArrayExpress and Gene Expression Omnibus (GEO). All single-cell data were provided from the expression matrix data of the original papers. The accession number of the datasets used in this study are listed in the key resources table.
We first distinguished malignant or non-malignant single cells using the following criteria: (1) We extracted the malignant cells from the original literature that provided cancer types of information. (2) We distributed malignant cells and non-malignant cells by inferred DNA copy numbers from single-cell transcriptome using the inferCNV R package (Patel et al., 2014). Malignant samples of patients that had at least 40 cells were used for further analysis. We also performed quality control on the mRNA expression for each single cell, including the expression levels are greater than 0, and in principle removed cells with less than 1000 genes expressed (Tirosh et al., 2016; Yuan et al., 2019).
For visualization, we performed the dimension reduction algorithm to extend the data into two or three dimensions to hidden structures in the data that can be depicted intuitively (Wang et al., 2017). The Rtsne () function in Rtsne package was used to perform t-SNE analysis on all genes in the filtered dataset, with default parameters. The reduced maps are displayed with ggplot R package, and the expression of different genes or the average expression of the gene sets are marked on maps (Wei et al., 2020). In order to evaluate the pathway activity of a single patient or single cell, we performed Gene Set Variation Analysis (GSVA) analysis (Hanzelmann et al., 2013) on specific gene sets. We entered the gene expression profile into the GSVA function in R to get the GSVA score, which is displayed on the maps of dimensionality reduction analysis.
Quantification and statistical analysis
Statistical analysis was performed using R (version 3.6.2). A comparison of conventional clinical parameters for subjects grouped according to the four MRP deletions status was performed using Chi-square test p values are stated. Wilcoxon rank-sum test and Student’s t test were used in this study. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.005.
Acknowledgments
We thank members of the Wang laboratory for critical discussions and Wei Tang for technical advice. We thank Man Hsin Hung and Eytan Ruppin for critical discussions of this manuscript. We also thank the NIH Fellows Editorial Board for editing the manuscript. This work was supported by grants (Z01-BC010313, Z01-BC010876, Z01-BC010877, ZIA-BC011870) from the intramural research program of the Center for Cancer Research, National Cancer Institute of the United States.
Author contributions
C.-W.C. and X.W.W. developed the study concept, directed experimental design, and interpreted data. C.-W.C. performed computational analysis. C.-W.C. and S.R.D. performed structure visualization. C.-W.C., Z.W., L.M., and M.F. conducted additional data analysis. C.-W.C. and X.W.W. wrote the manuscript. All authors read, edited, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105244.
Supplemental information
Data and code availability
The copy number, RNA-seq, proteomics, and scRNA-seq data used in this study have been published, and the specific information is in the key resources table section. We used publicly available algorithms and R packages for all bioinformatic analyses in this study, the details of which have been described in STAR methods section and were cited properly in the manuscript. This paper did not produce original code. Any other items information required for reanalyzing the reported result is available from the lead contact upon request.
References
- Ajore R., Raiser D., McConkey M., Jöud M., Boidol B., Mar B., Saksena G., Weinstock D.M., Armstrong S., Ellis S.R., et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol. Med. 2017;9:498–507. doi: 10.15252/emmm.201606660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Alexandrov L.B., Ju Y.S., Haase K., Van Loo P., Martincorena I., Nik-Zainal S., Totoki Y., Fujimoto A., Nakagawa H., Shibata T., et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618–622. doi: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N., Graham T.A., Jansen M., Xia L.C., Aktipis C.A., Petritsch C., Ji H.P., Maley C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016;22:105–113. doi: 10.1038/nm.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andor N., Maley C.C., Ji H.P. Genomic instability in cancer: teetering on the limit of tolerance. Cancer Res. 2017;77:2179–2185. doi: 10.1158/0008-5472.CAN-16-1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus L., Smid M., Wilting S.M., van Riet J., Van Hoeck A., Nguyen L., Nik-Zainal S., Steenbruggen T.G., Tjan-Heijnen V.C.G., Labots M., et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019;51:1450–1458. doi: 10.1038/s41588-019-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin P.C. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav. Res. 2011;46:399–424. doi: 10.1080/00273171.2011.568786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslan T., Morris J.P., Zhao Z., Reyes J., Ho Y.-J., Tsanov K.M., Bermeo J., Tian S., Zhang S., Askan G., et al. Ordered and deterministic cancer genome evolution after p53 loss. Nature. 2022;608:795–802. doi: 10.1038/s41586-022-05082-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann R., Herrmann J.M. Structural biology. Mitoribosome oddities. Science. 2015;348:288–289. doi: 10.1126/science.aab1054. [DOI] [PubMed] [Google Scholar]
- Bogenhagen D.F., Martin D.W., Koller A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metabol. 2014;19:618–629. doi: 10.1016/j.cmet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Bogenhagen D.F., Ostermeyer-Fay A.G., Haley J.D., Garcia-Diaz M. Kinetics and mechanism of mammalian mitochondrial ribosome assembly. Cell Rep. 2018;22:1935–1944. doi: 10.1016/j.celrep.2018.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho M.C., Pinto R.M., Murray A.W. Heterozygous mutations cause genetic instability in a yeast model of cancer evolution. Nature. 2019;566:275–278. doi: 10.1038/s41586-019-0887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva D., Tu Y.T., Amunts A., Fontanesi F., Barrientos A. Mitochondrial ribosome assembly in health and disease. Cell Cycle. 2015;14:2226–2250. doi: 10.1080/15384101.2015.1053672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower L.A., Soussi T., Korkut A., Liu Y., Schultz A., Cardenas M., Li X., Babur O., Hsu T.K., Lichtarge O., et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019;28:3010. doi: 10.1016/j.celrep.2019.08.061. [DOI] [PubMed] [Google Scholar]
- Dongre A., Weinberg R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019;20:69–84. doi: 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- Ebright R.Y., Lee S., Wittner B.S., Niederhoffer K.L., Nicholson B.T., Bardia A., Truesdell S., Wiley D.F., Wesley B., Li S., et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science. 2020;367:1468–1473. doi: 10.1126/science.aay0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira L.M.R., Hebrant A., Dumont J.E. Metabolic reprogramming of the tumor. Oncogene. 2012;31:3999–4011. doi: 10.1038/onc.2011.576. [DOI] [PubMed] [Google Scholar]
- Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell L.H., Hopfield J.J., Leibler S., Murray A.W. From molecular to modular cell biology. Nature. 1999;402:C47–C52. doi: 10.1038/35011540. [DOI] [PubMed] [Google Scholar]
- Hausser J., Strimmer K.J. Entropy inference and the james-stein estimator, with application to nonlinear gene association networks. J. Mach. Learn. Res. 2009;10 [Google Scholar]
- International Cancer Genome Consortium. Hudson T.J., Anderson W., Artez A., Barker A.D., Bell C., Guyer M., Bhan M.K., Calvo F., Eerola I., et al. International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janska H., Kwasniak M. Mitoribosomal regulation of OXPHOS biogenesis in plants. Front. Plant Sci. 2014;5:79. doi: 10.3389/fpls.2014.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., DeBerardinis R.J. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metabol. 2019;30:434–446. doi: 10.1016/j.cmet.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc E.C., Cimen H., Kumcuoglu B., Abu N., Akpinar G., Haque M.E., Spremulli L.L., Koc H. Identification and characterization of CHCHD1, AURKAIP1, and CRIF1 as new members of the mammalian mitochondrial ribosome. Front. Physiol. 2013;4:183. doi: 10.3389/fphys.2013.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkut A., Zaidi S., Kanchi R.S., Rao S., Gough N.R., Schultz A., Li X., Lorenzi P.L., Berger A.C., Robertson G., et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-beta superfamily. Cell Syst. 2018;7:422–437.e7. doi: 10.1016/j.cels.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.J., Hu F.F., Xia M.X., Han L., Zhang Q., Guo A.Y. GSCALite: a web server for gene set cancer analysis. Bioinformatics. 2018;34:3771–3772. doi: 10.1093/bioinformatics/bty411. [DOI] [PubMed] [Google Scholar]
- Ma L., Hernandez M.O., Zhao Y., Mehta M., Tran B., Kelly M., Rae Z., Hernandez J.M., Davis J.L., Martin S.P., et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell. 2019;36:418–430.e6. doi: 10.1016/j.ccell.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maura F., Degasperi A., Nadeu F., Leongamornlert D., Davies H., Moore L., Royo R., Ziccheddu B., Puente X.S., Avet-Loiseau H., et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019;10:2969. doi: 10.1038/s41467-019-11037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noorbakhsh J., Kim H., Namburi S., Chuang J.H. Distribution-based measures of tumor heterogeneity are sensitive to mutation calling and lack strong clinical predictive power. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-29154-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Lim S., Nam J.W., Kim S. Measuring intratumor heterogeneity by network entropy using RNA-seq data. Sci. Rep. 2016;6 doi: 10.1038/srep37767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C., et al. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019;176:1282–1294.e20. doi: 10.1016/j.cell.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud F., Mina M., Tavernari D., Ciriello G. Pan-cancer inference of intra-tumor heterogeneity reveals associations with different forms of genomic instability. PLoS Genet. 2018;14 doi: 10.1371/journal.pgen.1007669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C.J., Kennedy S., Bajrami I., Matallanas D., Lord C.J. A compendium of Co-regulated protein complexes in breast cancer reveals collateral loss events. Cell Syst. 2017;5:399–409.e5. doi: 10.1016/j.cels.2017.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K., Bachman J.A., Muhlich J.L., Mitchison T.J. shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife. 2021;10:e57116. doi: 10.7554/eLife.57116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U SA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvester J.E., Fischel-Ghodsian N., Mougey E.B., O'Brien T.W. Mitochondrial ribosomal proteins: candidate genes for mitochondrial disease. Genet. Med. 2004;6:73–80. doi: 10.1097/01.gim.0000117333.21213.17. [DOI] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G., et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomal A., Kwasniak-Owczarek M., Janska H. An update on mitochondrial ribosome biology: the plant mitoribosome in the spotlight. Cells. 2019;8 doi: 10.3390/cells8121562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler S., Buttner M., Fischer D.S., Lange M., Bergen V., Lickert H., Theis F.J. Concepts and Limitations for Learning Developmental Trajectories from Single Cell Genomics. Development. 2019;146 doi: 10.1242/dev.170506. [DOI] [PubMed] [Google Scholar]
- Trucco L.D., Mundra P.A., Hogan K., Garcia-Martinez P., Viros A., Mandal A.K., Macagno N., Gaudy-Marqueste C., Allan D., Baenke F., et al. Ultraviolet radiation-induced DNA damage is prognostic for outcome in melanoma. Nat. Med. 2019;25:221–224. doi: 10.1038/s41591-018-0265-6. [DOI] [PubMed] [Google Scholar]
- Tubbs A., Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168:644–656. doi: 10.1016/j.cell.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz F., Giegé P. Striking diversity of mitochondria-specific translation processes across eukaryotes. Trends Biochem. Sci. 2020;45:149–162. doi: 10.1016/j.tibs.2019.10.004. [DOI] [PubMed] [Google Scholar]
- Wang B., Zhu J., Pierson E., Ramazzotti D., Batzoglou S. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat. Methods. 2017;14:414–416. doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- Wei Z., Lei J., Shen F., Dai Y., Sun Y., Liu Y., Dai Y., Jian Z., Wang S., Chen Z., et al. Cavin1 deficiency causes disorder of hepatic glycogen metabolism and neonatal death by impacting fenestrations in liver sinusoidal endothelial cells. Adv. Sci. 2020;7:2000963. doi: 10.1002/advs.202000963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y., Bartok O., Patkar S., Eli G.B., Cohen S., Litchfield K., Levy R., Jiménez-Sánchez A., Trabish S., Lee J.S., et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell. 2019;179:219–235.e21. doi: 10.1016/j.cell.2019.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Dai Z., Locasale J.W. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat. Commun. 2019;10:3763. doi: 10.1038/s41467-019-11738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Yan M., Zhang G., Liu W., Deng C., Liao G., Xu L., Luo T., Yan H., Long Z., et al. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2019;47:D900–D908. doi: 10.1093/nar/gky939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Ju Y.S., Kim Y., Li J., Wang Y., Yoon C.J., Yang Y., Martincorena I., Creighton C.J., Weinstein J.N., et al. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020;52:342–352. doi: 10.1038/s41588-019-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A., Benayed R., Shah R.H., Syed A., Middha S., Kim H.R., Srinivasan P., Gao J., Chakravarty D., Devlin S.M., et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10, 000 patients. Nat. Med. 2017;23:703–713. doi: 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B., Xiao Y., Yeager M., Clifford G., Wentzensen N., Cullen M., Boland J.F., Bass S., Steinberg M.K., Raine-Bennett T., et al. Mutations in the HPV16 genome induced by APOBEC3 are associated with viral clearance. Nat. Commun. 2020;11:886. doi: 10.1038/s41467-020-14730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TCGA genomic, transcriptomic, mutation, and clinical data from NIH Genomic Data Commons (GDC) are available for download at the cBioPotal datasets (https://www.cbioportal.org/). ICGC/TCGA PCAWG genomic and clinical data are available for download at the PCAWG data portal (https://dcc.icgc.org/releases/PCAWG). MSK-IMPACT genomic, mutation and clinical data from Memorial Sloan Kettering cancer center are available for download at the cBioPotal database (https://www.cbioportal.org/). CCLE genomic, mutation, and clinical data are available for download at the Broad Institute Cancer Cell Line Encyclopedia (CCLE) datasets. The scRNA-seq of eight individual different cancer types in humans was collected from ArrayExpress and Gene Expression Omnibus (GEO). All single-cell data were provided from the expression matrix data of the original papers. The accession number of the datasets used in this study are listed in method. The mtDNA copy number was estimated using the TCMA data portal (http://bioinformatics.mdanderson.org/main/TCMA:Overview).
The copy number, RNA-seq, proteomics, and scRNA-seq data used in this study have been published, and the specific information is in the key resources table section. We used publicly available algorithms and R packages for all bioinformatic analyses in this study, the details of which have been described in STAR methods section and were cited properly in the manuscript. This paper did not produce original code. Any other items information required for reanalyzing the reported result is available from the lead contact upon request.