

Abstract
Pathogenic mitochondrial DNA (mtDNA) mutations can cause a variety of human diseases. The recent development of genome-editing technologies to manipulate mtDNA, such as mitochondria-targeted DNA nucleases and base editors, offer a promising way for curing mitochondrial diseases caused by mtDNA mutations. The CRISPR-Cas9 system is a widely used tool for genome editing; however, its application in mtDNA editing is still under debate. In this study, we developed a mito-Cas9 system by adding the mitochondria-targeted sequences and 3′ untranslated region of nuclear-encoded mitochondrial genes upstream and downstream of the Cas9 gene, respectively. We confirmed that the mito-Cas9 system was transported into mitochondria and enabled knockin of exogenous single-stranded DNA oligonucleotides (ssODNs) into mtDNA based on proteinase and DNase protection assays. Successful knockin of exogenous ssODNs into mtDNA was further validated using polymerase chain reaction-free third-generation sequencing technology. We also demonstrated that RS-1, an agonist of RAD51, significantly increased knockin efficiency of the mito-Cas9 system. Collectively, we provide direct evidence that mtDNA can be edited using the CRISPR-Cas9 system. The mito-Cas9 system could be optimized as a promising approach for the treatment of mitochondrial diseases caused by pathogenic mtDNA mutations, especially those with homoplasmic mtDNA mutations.
Key words: mitochondrial disease; mtDNA editing, mito-Cas9; third-generation sequencing
Graphical abstract
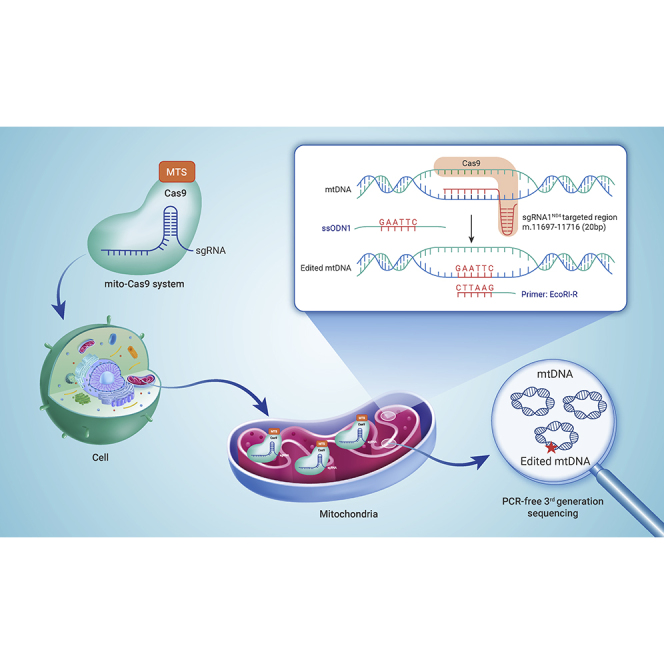
Public summary
-
•
We designed a mitochondria-targeted Cas9 system for successful mtDNA editing
-
•
Cas9-edited mtDNA was confirmed by the PCR-free third-generation sequencing
-
•
RAD51 agonist RS-1 significantly enhanced mtDNA knockin efficiency
Introduction
Mitochondria play essential roles in cell metabolism, energy production, apoptosis, calcium homeostasis, and immunity.1,2 Mammalian mitochondria are double-membrane organelles with their own genome (mitochondrial DNA [mtDNA]). Human mtDNA is a double-stranded circular molecule composed of 16,569 base pairs (bp), containing 37 genes encoding 13 respiratory chain subunits, 22 transfer RNAs, and 2 ribosomal RNAs (rRNAs).3 Mitochondrial dysfunction resulting from mtDNA mutations can cause a variety of human diseases.1,2 There are 100–100,000 copies of the mtDNA genome in a cell, depending on the cell type.3,4 Mutant and wild-type mtDNA can co-exist in one cell, known as heteroplasmy.5 The level of heteroplasmy in pathogenic mtDNA mutations can affect disease onset and clinical phenotype, with a general threshold of 60%–95% of mutant mtDNA causing biochemical and clinical defects.6, 7, 8
Currently, curing mitochondrial diseases remains a daunting task. Gene therapy through allotopic expression of mitochondrial genes shows promise for the treatment of Leber hereditary optic neuropathy9 caused by mtDNA mutations.10 Another approach is mitochondrial replacement therapy, which transfers the patient’s spindle, pronuclear, or polar body genome into healthy enucleated donor oocytes or embryos to circumvent mother-to-child mtDNA disease transmission.4,11,12 With the rapid development of genome-editing technology, several approaches for manipulating mtDNA in vitro and in vivo have been established in recent years,13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 which exploit the rapid degradation of damaged mtDNA with double-strand breaks (DSBs) via mtDNA replisome components.24,25 For instance, mitochondria-targeted DNA nucleases specific to mutant mtDNA, including mitochondrial endonucleases,14,16,20 mitochondrial transcription activator-like effector nucleases (mito-TALENs),18, 19, 20,22 mitochondrial zinc-finger nucleases (mito-ZFNs),13,15,17,21 and mitochondrial meganucleases,23 can specifically induce DSBs and promote the degradation of mutant mtDNA. These approaches can effectively shift the heteroplasmic level of mutant mtDNA13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 but are limited by the unavailability of homoplasmic pathogenic mtDNA mutations. A bacterial cytidine deaminase fused with mito-TALEN (DdCBE) was recently established to induce base editing for C > T transition in mtDNA,26 with successful application in mice, rats, zebrafish, plants, and human embryos.27, 28, 29, 30, 31 More recently, small-sized zinc-finger deaminases (ZFDs) were engineered for precise C > T base editing of nuclear and mitochondrial genes,32 offering several advantages in therapeutic application. The same research team also developed an A > G base editor for mtDNA (TALED, transcription activator-like effector-linked deaminases), providing a broader scope for mtDNA editing.33 However, although base editors are promising tools for mtDNA editing, their substantial off-target effects on nuclear genes remain poorly resolved.34,35 Moreover, no current tools are able to induce or edit other types of mutations such as insertions, deletions, and transversions in mtDNA molecules.
The CRISPR-Cas9 system is a widely used tool for genome editing.36 The core principle is to induce DSBs in the targeted genomic region, then complete genome editing via DSB repair pathways, including the non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR) pathways.37 The CRISPR-Cas9 system is user friendly and flexible, and can circumvent the limitations of TALENs and ZFNs.38 However, the suitability and efficiency of mtDNA editing by CRISPR-Cas9 remains controversial.39 First, DSB repair is inefficient in mammalian mitochondria24,25 but is essential for successful editing by CRISPR-Cas9. Second, delivery of small-guide RNA (sgRNA) and Cas9 protein complexes into mitochondria is challenging. Several recent studies have attempted to manipulate mtDNA using the CRISPR-Cas9 system,40, 41, 42, 43, 44, 45 which confirmed that the Cas9 protein can be transported into mitochondria, and that sgRNAs show mitochondrial localization.40,43 Most of these studies assessed successful manipulation of mtDNA meditated by CRISPR-Cas9 based on a decrease in mtDNA copy number40, 41, 42, 43 and positive signals of allele-specific polymerase chain reaction (PCR) and/or PCR-based sequencing.42,44 Although these studies suggest the possibility of using CRISPR-Cas9 to manipulate mtDNA, PCR-based methods may be limited in their detection of edited mtDNA due to “template switching” or other technical artifacts.46, 47, 48 Furthermore, evidence showing successful mtDNA editing by CRISPR-Cas9 remains inconclusive, which is why Gammage et al. have argued that the mitochondrial genome may not be “CRISPR-Ized.”39
In this study, we constructed a mito-Cas9 system, and confirmed that the system enabled successful knockin of exogenous single-stranded DNA oligonucleotides (ssODNs) into mtDNA using PCR-free third-generation sequencing technology. Furthermore, overexpression or activation of RAD51 significantly increased the knockin efficiency of the mito-Cas9 system. These results provide direct evidence of successful CRISPR-Cas9-mediated mitochondrial genome editing.
Results
Mito-Cas9 system targeted mitochondria and decreased mtDNA content
We established the mito-Cas9 system by replacing the nuclear localization sequence (NLS) at the N terminus of Cas9 in the px330-mCherry vector with the MTS of COX8A, and the NLS at the C terminus of Cas9 with the 3′ untranslated region (UTR) of SOD2 (Figure S1). The MTSs and 3′ UTR of the respective mitochondrial genes efficiently mediated the mitochondrial localization of mRNA.49,50 Small-guide RNA, sgRNA1ND4 (Table S1), was designed to target m.11 697-11 716 in the MT-ND4 gene, with a G added to the 5′-position of the 20-bp guide sequence to obtain efficient U6 transcription of sgRNA and overall efficiency of the CRISPR-Cas9 system,37,51,52 and was cloned to create the mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 (Figure S1). Previous studies have shown that unmodified sgRNAs can co-fractionate with mitochondria,40,42,43 therefore we did not further modify the sgRNAs. We constructed other mito-Cas9 constructs with sgRNA2ND4 and MTSs of mitochondrial genes COX10 and SOD2 using a similar strategy (Figure S1).
As expected, HEK293T cells transfected with the Cas9 construct containing the MTS of COX8A (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2) contained more Cas9 protein in the cytoplasmic components than cells transfected with the same amount of px330-mCherry vector or sgRNAAPP-NLS-Cas9 vector (Figures 1A and 1B). We performed protease protection assays and confirmed the successful translocation of the Cas9 protein into the mitochondria of cells transfected with the mito-Cas9 system, albeit with low efficiency (Figure 1C). As shown in Figure 1C, the Cas9 proteins showed multiple bands (around 80–160 kDa) in samples with or without proteinase K treatment. A small fraction of the Cas9 protein with MTSs, together with COXIV (positive control for mitochondrial inner membrane proteins), was protected from proteinase K digestion by the mitochondrial membrane. In contrast, the cytoplasmic protein β-actin and mitochondrial outer membrane protein MFN2 (used as negative controls) were completely digested by proteinase K (Figure 1C). Unlike COXIV, which was completely localized in the mitochondria, Cas9 (with or without MTSs) was partially localized in the mitochondria and its protein level decreased significantly upon protease K treatment. In the immunofluorescence assays, the Cas9 protein with MTSs showed preferred co-localization with mitochondrial green fluorescent protein (Figure 1D). Notably, despite the lack of NLS or MTS, overexpression of the Cas9 protein was still observed in the nucleus and crude mitochondria (Figures 1A–1D). However, the reason for this Cas9 distribution pattern remains unknown.
Figure 1.

Mito-Cas9 system transported Cas9 protein into mitochondria and decreased mtDNA copy number
(A) Distribution of Cas9 protein in different cellular fractions. Nuclear and cytoplasmic fractions from HEK293T cells transfected with different Cas9 constructs were extracted for western blotting. Blot for H3 protein refers to nuclear fraction, whereas ATP5A and GAPDH refer to cytoplasmic fraction. Overexpression of Cas9 was determined using antibodies for Cas9 and Flag, respectively.
(B) Quantification of the relative protein level of Cas9-Flag in (A). Levels of Cas9-Flag protein in cytoplasmic and nuclear fractions were normalized to GAPDH and H3, respectively. Bars represent mean ± standard deviation (SD) of three independent tests.
(C) Proteinase protection assay and western blot analysis showing successful expression and translocation of Cas9 into mitochondria. HEK293T cells were transfected with different mito-Cas9 constructs. Crude mitochondria were isolated at 48 h after transfection, and 20 μg of crude mitochondrial fraction was treated with 50 μg/mL proteinase K for 30 min on ice, followed by western blotting, with 10 μg of crude mitochondrial fraction as a control.
(D) Immunofluorescence assay showing mitochondrial localization of Cas9. HEK293T cells were co-transfected with mito-GFP vector (expressing mitochondrial-targeted green fluorescent protein) and Cas9 constructs (NLS-Cas9: construct sgRNAAPP-NLS-Cas9 with removal of mCherry; MTS-Cas9: construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 with removal of mCherry). Cells were imaged at 488 nm (for mito-GFP) and 594 nm (for Cas9), respectively. Line graphs on right indicate fluorescence intensity in the rectangle in left images.
(E) Mitochondrial localization of FAM-labeled sgRNA1ND4. Crude mitochondria were isolated from HEK293T cells co-transfected with FAM-labeled sgRNA1 ND4 and pDsRed2-mito vector (expressing mitochondria-targeted red fluorescent protein, mito-RFP) for 48 h.
(F) Quantification of mtDNA copy number in HEK293T cells transfected with nuclear-targeted Cas9 vector (sgRNAAPP-NLS-Cas9), mitochondria-targeted Cas9 without sgRNA (MTSCOX8A-Cas9-UTRSOD2), and mito-Cas9 construct (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2). Two pairs of primers, L394/H475 and L11718/H11944, were used for cross-validation, and mtDNA content was normalized to single-copy nuclear gene β-globin. Bars are mean ± SD. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; one-way analysis of variance (ANOVA) test adjusted by Tukey’s multiple comparisons test.
We extracted mitochondria from HEK293T cells co-transfected with 6-carboxyfluorescein (FAM)-labeled sgRNA1ND4 (100 bp) and pDsRed2-mito vector (expressing mitochondria-targeted red fluorescent protein [mito-RFP]). We observed significant co-localization of FAM-labeled sgRNA1ND4 and mito-RFP (Figure 1E). We further used flow cytometry to quantify the proportion of labeled mitochondria in the total mitochondrial extract. FAM-labeled sgRNA1ND4 signals were detected in the mitochondria labeled with mito-RFP, albeit at a relatively low frequency (Figure S2A). We observed similar results using MitoTracker to label mitochondria (Figure S2B). These results suggest that Cas9 with MTSs and sgRNA can be (partially) transported into mitochondria.
Within cells, mtDNA with DSBs is rapidly degraded,24 therefore, a reduction in mtDNA copy number can be used as a marker for mtDNA manipulation.40, 41, 42 Consistent with previous studies,40, 41, 42, 43 we observed a significant reduction in mtDNA copy number in HEK293T cells overexpressing the mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 compared with cells overexpressing nuclear-targeted Cas9 (sgRNAAPP-NLS-Cas9) or non-targeted controls (MTSCOX8A-Cas9-UTRSOD2, mito-Cas9 without sgRNA) (Figure 1F). We further assessed the effect of the mito-Cas9 system on mitochondrial function. Cells transfected with mitochondria-targeted Cas9 showed significantly increased levels of reactive oxygen species (ROS) and significantly decreased levels of adenosine triphosphate compared with cells transfected with nuclear-targeted Cas9 or Cas9 without any targeting sequence (Figure S3A), indicating decreased mitochondrial function upon mito-Cas9 expression. To investigate whether the reduction in mtDNA copy number in mito-Cas9-transfected cells was caused by increased ROS, we determined the mtDNA copy number in cells treated with vitamin K3 (vitK3, reported to increase cellular ROS level53) and melatonin (reported to decrease cellular ROS54). Compared with the other groups, cells transfected with mito-Cas9 showed a significant decrease in mtDNA copy number, regardless of treatments (Figure S3C).
To investigate whether the mito-Cas9 construct with an alternative sgRNA targeting a different mtDNA region could still be applied for mtDNA editing, we used sgRNA2ND4 (Table S1) targeting the m.11 851–11 868 (20 nt) region in the MT-ND4 gene. Consistently, upon overexpression of the sgRNA2ND4-MTSCOX8A-Cas9-UTRSOD2 construct, the mtDNA copy number decreased significantly (Figure S4A). Thus, the mito-Cas9 system established here can successfully target mitochondria and induce a decrease in mtDNA copy number.
Mito-Cas9 system-mediated knockin of exogenous ssODNs into mtDNA through homology-directed repair
Increasing evidence has demonstrated the existence of MMEJ-mediated and HR-mediated DSB repair in mammalian mitochondria.55, 56, 57, 58 Here, we investigated whether the mito-Cas9 system can mediate homology-directed repair in mtDNA. Based on previous observations that homologous arms longer than 40 bp can achieve high knockin efficiency,37,59 we designed a 96-bp ssODN1 donor template (Table S1) that contained 45-bp homologous arms (left arm: homologous to m.11 669–11 713; right arm: homologous to m.11 714–11 758) flanking a 6-bp insertion of the EcoRI site (GAATTC) (Figure 2A; Table S1). The ssODN1 donor template and Cas9 constructs were co-transfected into HEK293T cells for 48 h, after which homology-directed repair of mtDNA was analyzed using PCR and quantitative real-time PCR (qRT-PCR) with the EcoRI site-specific primer pair L11338/EcoRI-R (Figures 2A and 2B; Table S1). EcoRI site-specific PCR products were not detected in cells transfected with MTSCOX8A-Cas9-UTRSOD2 alone, but were detected in cells transfected with ssODN1 alone (Figures 2B and S5), which may be, in part, due to potential “template switching artifacts” between the ssODN1 template and mtDNA molecule.46, 47, 48 In contrast, cells transfected with a combination of sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1 showed significantly increased EcoRI site-specific PCR products compared with different controls (Figure 2B). We also constructed mito-dCas9 (dead Cas9) with a targeting sgRNA1ND4 as a control, in which the catalytic activity of the SpCas9 protein was abolished by introducing two mutations, p.D10A and p.H840A.60,61 The relative level of EcoRI site-specific PCR products of the mito-dCas9 system showed no significant differences compared with the controls transfected with ssODN1 alone or with ssODN1 + MTSCOX8A-Cas9-UTRSOD2 (no sgRNA) (Figure 2C). These findings indicated that Cas9 activity is essential for the mito-Cas9 system. To investigate whether the increase in EcoRI site-specific PCR products was caused by the potentially higher transfection efficiency/stability of ssODN1 with the mito-Cas9 system, we transfected HEK293T cells with FAM-labeled ssODN1 (which can be introduced into mitochondria after transfection, Figure S6A) and mCherry-tagged Cas9 vectors (with or without different target sequences). The proportion of fluorescently labeled cells was quantified by flow cytometry. We observed similar levels of labeled cells among the transfected cells (Figure S6B), indicating that ssODN1 exhibits a similar level of transfection efficiency in different ssODN1 and Cas9 vector combinations.
Figure 2.

Mito-Cas9 system mediated efficient knockin of exogenous ssODNs into mtDNA
(A) Design of mito-Cas9-mediated knockin system.
(B) Quantification of knockin efficiency of mito-Cas9. (Upper) Results of qRT-PCR. HEK293T cells were transfected with or without a combination of Cas9 constructs and ssODN1. Content of mtDNA with successful knockin of EcoRI site (edited mtDNA, amplified by primer pair L11338/EcoRI-R (L11338-EcoRI-R)) was normalized to whole mtDNA (total mtDNA, amplified by primer pair L394/H475 (L394-H475)), using total genomic DNA as the template. (Below) Agarose gel image showing representative PCR product for each group of cells with different transfections.
(C) Knockin efficiency of mito-Cas9 constructs with catalytically dead Cas9 (dCas9): sgRNA1ND4-MTSCOX8A-dCas9-UTRSOD2, expressing SpCas9 protein with mutations p.D10A and p.H840A. Knockin efficiency was quantified using the same qRT-PCR procedure as in (B), with total genomic DNA from respective transfected cells as the template.
(D) Sequencing chromatogram of EcoRI site-specific PCR product. Targeting region of sgRNA1ND4 is marked with a box on the mtDNA revised Cambridge reference sequence (rCRS).
(E) Knockin efficiency of different mito-Cas9 constructs with different MTSs. HEK293T cells were transfected with ssODN1 and mito-Cas9 construct fused with indicated MTSs of mitochondrial genes COX8A, COX10, or SOD2. Knockin efficiency was quantified using the same qRT-PCR procedure as in (B), with total genomic DNA from respective transfected cells as the template. NC, cells without transfection.
(F) DNase protection assay confirmed the mitochondrial source of EcoRI site-specific PCR product amplified from edited mtDNA in mitochondria. Crude mitochondria were extracted from HEK293T cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1 for 48 h, then treated with DNase I at 37°C for 1 h. PCR amplifications for nuclear APP (amplified by primer pair APP-F/APP-R), total mtDNA (amplified by primer pair L11338/H11944), and edited mtDNA (amplified by primer pair L11338/EcoRI-R) were performed using mtDNA template extracted from mitochondria with or without DNase I treatment, respectively.
(G) Quantification of knockin efficiency in mtDNAs from crude mitochondria treated with DNase I (DNase+) or without DNase I (DNase−). Bars are mean ± SD. ns, not significant; ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; one-way ANOVA test adjusted by Tukey’s multiple comparisons test for (B, C, and E); two-tailed Student's t test for (G).
We next verified insertion of the “GAATTC” sequence in the sgRNA1ND4-target site of mtDNA by direct sequencing of the EcoRI site-specific PCR product (Figure 2D). Furthermore, we found that the mito-Cas9 constructs with different MTSs of mitochondrial genes (COX8A, COX10, or SOD2) exhibited similar knockin efficiencies of exogenous ssODNs (Figure 2E). To exclude the potential effects of “template switching artifacts” during PCR, we quantified the level of ssODN1 within the transfected cells at 48, 96, and 144 h after transfection. We found that the relative amount of the EcoRI site-specific PCR product in cells transfected with ssODN1 significantly diminished with time and was barely detected at 144 h (Figure S7). In contrast, cells transfected with ssODN1 and the mito-Cas9 system contained a significantly higher level of the EcoRI site-specific PCR product relative to cells transfected with ssODN1 alone at each time point (Figure S7). Of note, a reduction in the EcoRI site-specific PCR product was observed with time (Figure S7), which might be attributed to the higher proliferation rate of cells with wild-type mtDNA compared with cells with edited mtDNA.
To further exclude potential contamination from other sources of mtDNA (e.g., leakage of mtDNA fragments in cytosol from damaged mitochondria) or mtDNA-like fragments (e.g., nuclear mitochondrial pseudogenes in the nuclear genome62) outside the mitochondria, which could potentially be recognized as a target by the mito-Cas9 system, we performed a DNase protection assay to validate the successful knockin of exogenous ssODNs into mtDNAs located within the mitochondria (Figure 2F). Upon DNase I treatment, no PCR product could be amplified for the nuclear APP gene (Figure 2F), whereas the mtDNA PCR product (amplified by primer pair L11338/H11944) and EcoRI site-specific PCR product were visible, indicating that exogenous ssODNs were inserted into mtDNA and were protected from DNase I digestion by the mitochondrial membrane (Figure 2F). Quantification of the EcoRI site-specific PCR products showed that the mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 significantly increased the knockin efficiency of ssODN1 in both crude mtDNA (DNase−) and purified mtDNA (DNase+) (Figure 2G). The EcoRI site-specific PCR product was also detected when the target and knockin sites were changed (Figures S4B–S4D). Compared with cells transfected with ssODN2 alone or with a combination of ssODN2 and MTSCOX8A-Cas9-UTRSOD2 (no sgRNA), cells transfected with a combination of sgRNA2ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN2 showed a significant increase in EcoRI site-specific PCR product (Figure S4D), indicating that mito-Cas9 system-mediated knockin can be applied to any site suitable for CRISPR-Cas9 editing. These results demonstrate that the mito-Cas9 system can mediate knockin of exogenous ssODNs into mtDNA through the HR repair pathway.
Third-generation sequencing verified knockin of exogenous ssODNs into mtDNA through mito-Cas9 system
To exclude the possibility that the EcoRI site-specific PCR products were caused by PCR artifacts, such as template-switching artifacts46, 47, 48 between exogenous ssODNs and mtDNA, we performed PCR-free third-generation sequencing to verify the knockin of exogenous ssODNs into mtDNA (Figure 3A). The ssODN1 donor template and mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 were co-transfected into HEK293T cells for 48 h. Cells transfected with ssODN1 only or with a combination of ssODN1 and MTSCOX8A-Cas9-UTRSOD2 (no sgRNA) were considered as controls. BamHI digestion was used to linearize purified mtDNA isolated from DNase I-treated mitochondria (Figure 3A). Total genomic DNA was completely digested by BamHI and showed no clear bands on the gel, whereas successfully linearized mtDNA was evidenced by a single band of mtDNA after BamHI digestion (Figure S8A). We also measured the efficiency of linearization by quantifying the mtDNA fragment flanking the BamHI site using qRT-PCR with the primer pair L14054/H14573 (Table S1; linearized mtDNA could not be amplified by L14054/H14573) and observed a significant decrease in the PCR product for purified mtDNA subjected to BamHI compared with undigested samples (Figure S8B), indicating that mtDNA was thoroughly linearized. Direct sequencing of linearized mtDNA using PCR-free PacBio sequencing technology yielded a distinct sequence peak at ∼16 kb (Figure 3B, left), indicating full-length capture and sequencing of the mtDNA genome. Reads Mapping using the mtDNA reference sequence63 and nuclear mitochondrial (NUMTs) reference sequences64 showed that most reads mapped to NUMTs were <16 kb (Figure 3C, left). Therefore, we only used reads >16 kb in length, and with higher mapping percentage and concordance to mtDNA than to NUMTs, for the following analyses, thus reliably excluding the potential effects of NUMTs and other artifacts. We identified precise insertion of GAATTC at the sgRNA1ND4 target site in mtDNA reads from the entire mitochondrial genome (Figure 3D). Of note, a higher frequency of GAATTC insertion at the sgRNA1ND4 targeting site was observed for cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN1(3/11,484, 0.026%) than for control cells transfected with MTSCOX8A-Cas9-UTRSOD2 (no sgRNA) + ssODN1 (3/22,995, 0.0087%) (Figure 3E, top). In both samples, we observed randomly distributed GAATTC insertions across the mtDNA genome at extremely low frequencies (Figure 3E, top), which may be caused by potential errors in the third-generation sequencing technology.65
Figure 3.

Third-generation sequencing identified mtDNA editing introduced by mito-Cas9 system
(A) Flowchart of PCR-free third-generation sequencing of purified mtDNA.
(B) Length distribution patterns of sequencing reads generated by PacBio (left) and Nanopore (right) sequencing. All reads were mapped to mtDNA.
(C) Length distributions of sequencing reads mapped to mtDNA (pink) and potential NUMTs (blue).
(D) Sequencing reads (>16 kb) showing successful knockin of EcoRI site GAATTC at sgRNA1ND4-targeted site.
(E) Distribution of GAATTC insertion across the entire mtDNA genome based on sequencing reads >16 kb in length.
(F) Counts (left) and frequencies (right) of mtDNA sequencing reads with targeted GAATTC insertion in all reads grouped by sequence length.
To exclude the possibility that these targeted insertions were introduced by sequencing errors in PacBio Sequel II, we used another PCR-free sequencing method, Nanopore sequencing, to sequence mtDNAs extracted from cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN1. In the captured reads across the entire mtDNA genome (peak near 16,569 bp; Figures 3B and 3C, right), we confirmed a higher rate of targeted GAATTC insertion in cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN1 (2/411,780) compared with cells transfected with ssODN1 alone (1/314,840) (Figure 3E, bottom). We observed several untargeted GAATTC insertions across the mtDNA genome in the PacBio data (Figure 3E, top), but these insertions were not replicated in the Nanopore sequencing data (Figure 3E, bottom), suggesting that most untargeted insertions were introduced by PacBio sequencing errors.65 We further analyzed the number of reads with targeted insertions in the mtDNA sequencing data of different lengths. We identified a higher frequency of precise GAATTC insertions in mtDNA sequences larger than 10 kb at the sgRNA1ND4 target region in cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN1 (n = 6) than in cells transfected with ssODN1 alone (n = 2) (Figure 3F). Overall, the total numbers of targeted insertions in the mtDNA sequences with different length cutoffs (from 10 to 16 kb) were consistently higher in cells transfected with the mito-Cas9 system than cells transfected with ssODN1 alone (Figure 3F). Therefore, the higher rate of targeted insertions in cells transfected with the mito-Cas9 system is unlikely to be due to random sequencing errors, but may be indicative of the editing capability of the mito-Cas9 system. The occurrence of targeted insertions in cells transfected with ssODN1 alone, even at a very low frequency, deserves further attention and focused study in the future, which may suggest an unexpected role of ssODN1 in the initiation of mtDNA replication in the absence of the mito-Cas9 system (Bi et al., unpublished data).
RAD51 activation enhanced mito-Cas9 system knockin efficiency
We further tested whether modulating those factors involved in genome stability maintenance and repair pathways could improve the knockin efficiency of the mito-Cas9 system. HR is essential for maintaining mitochondrial genome integrity.55,56,58 Key factors in the HR pathway, such as RAD51 and XRCC3, can be recruited to mitochondria and participate in mtDNA maintenance under DNA damage stress.58,66,67 We confirmed the presence of RAD51 in mitochondria using a proteinase protection assay (Figure 4A). As our mito-Cas9 system was based on HR-mediated effects, we hypothesized that enhancing RAD51 function would increase the knockin efficiency of ssODNs into mtDNA, as shown by the Cas9-mediated knockin efficiency for nuclear DNA.68, 69, 70 We treated HEK293T cells transfected with the mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and/or ssODN1 with RAD51 agonist RS-1 (10 μM) for 42 h, then quantified the level of edited mtDNA (Figure 4B). Stimulation of RAD51 with RS-1 significantly increased the knockin efficiency of the mito-Cas9 system (Figure 4B), whereas RS-1 had no significant effect on cells transfected with ssODN1 alone Figures 4B and S9A) or on the other three controls (mito-Cas9 without ssODN1, mito-Cas9 without sgRNA, and mito-Cas9 with sgRNA2ND4 targeting another region) (Figures 4B and S9A). Overexpression of the RAD51 protein resulted in a significant increase in the knockin efficiency of the mito-Cas9 system, similar to the treatment of the RAD51 agonist (Figures 4C and S9B). However, knockdown of RAD51 or inhibition of RAD51 by RI-171 had no or a weak effect on the knockin efficiency of the mito-Cas9 system (Figures 4C, 4D, S9C, and S9D), suggesting a potential compensatory effect in cells with RAD51 knockdown or inhibition during the maintenance of genome stability. Interestingly, an increase in the EcoRI site-specific PCR product signal was observed in cells transfected with sgRNA1ND4-NLS-Cas9 and ssODN1 compared with cells transfected with ssODN1 alone (Figure 4B). We speculated that this may be caused by a minimum level of sgRNA1ND4 guiding Cas9 and ssODN1 or penetrance of Cas9 and ssODN1 into the mitochondria, similar to that observed for nuclear-imported Cas9 in the absence of NLS (Figure 1A).
Figure 4.

RAD51 was involved in mtDNA editing mediated by the mito-Cas9 system
(A) RAD51 protein was detected in mitochondria from HEK293T cells treated with or without proteinase K.
(B) RAD51 agonist RS-1 (10 μM) significantly increased knock-in efficiency of the mito-Cas9 system (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1). Total genomic DNA (total DNA, upper) and mtDNA from crude mitochondria (crude mtDNA, lower) from transfected HEK293T cells with or without RS-1 treatment were used for quantification of edited mtDNA.
(C) Overexpression of RAD51 protein significantly increased knockin efficiency of the mito-Cas9 system (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1), but RAD51 knockdown did not affect knockin efficiency. HEK293T cells were co-transfected with expression vector of RAD51 and mito-Cas9 system for 48 h before harvest for qRT-PCR.
(D) RAD51 inhibitor RI-1 (20 μM) decreased knockin efficiency of the mito-Cas9 system (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1). Bars in (B)–(D) are mean ± SD. ns, not significant; ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; one-way ANOVA adjusted by Tukey’s multiple comparisons test.
We further quantified the knockin efficiency of the mito-Cas9 system using second-generation sequencing technology. Using a primer pair outside of ssODN1, we amplified a fragment flanking the knockin site (m.11 600–11 820) in crude and purified mtDNAs (Table 1; Figure S9E), respectively, then subjected the PCR products to second-generation sequencing. Knockin of ssODNs was identified in the sequencing reads (Table 1; Figure S9E). Consistent with the qRT-PCR results, in HEK293T cells transfected with the mito-Cas9 construct sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1, RS-1 treatment resulted in a nearly 5-fold increase in knockin efficiency in the crude mtDNA (0.14%) relative to that in cells without RS-1 treatment (0.03%; p < 2.2 × 10−16, Fisher’s exact test) (Table 1). Knockin frequency further increased in the purified mtDNA (0.23%; p < 2.2 × 10−16, Fisher’s exact test) (Table 1). Of note, the relative number of sequence reads with knockin was significantly lower (p < 2.2 × 10−16, Fisher’s exact test) in cells transfected with ssODN1 alone than in cells transfected with mito-Cas9 constructs and ssODN1 (Table 1). Furthermore, no significant changes in knockin efficiency were observed in the ssODN1-transfected cells with or without RS-1 treatment, suggesting that sequence reads with GAATTC knockin in cells transfected with ssODNs alone were not HR mediated (Table 1).
Table 1.
Frequency of reads with GAATTC knockin based on second-generation sequencing
| Sample | Total no. of reads | No. of reads with GAATTC insertion | Knockin ratio |
|---|---|---|---|
| Crude mtDNA | |||
| ssODN1 | 5,028,392 | 343 | 0.00006821 |
| ssODN1 + RS-1 | 6,838,719 | 324 | 0.00004738 |
| mito-Cas9 + ssODN1 | 5,367,579 | 1608 | 0.00029958 |
| mito-Cas9 + ssODN1 + RS-1 | 6,208,098 | 8839 | 0.00142379 |
| Purified mtDNA | |||
| ssODN1 + RS-1 | 5,991,432 | 210 | 0.00003505 |
| mito-Cas9 + ssODN1 + RS-1 | 4,992,779 | 11,714 | 0.00234619 |
Counts for sequencing reads with the GAATTC insertion in a short PCR fragment (region m.11 600–11 820 in mtDNA) flanking the knockin site. Crude mtDNA and purified mtDNA were used as respective templates for PCR, and PCR products were sequenced by second-generation sequencing technology. Crude mtDNA, mtDNAs isolated from crude mitochondria in HEK293T cells transfected with ssODN1 alone, mito-Cas9 system (sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 and ssODN1), with or without RS-1 treatment; purified mtDNA, mtDNAs isolated from crude mitochondria after DNase I digestion.
Collectively, these results suggest that RAD51 is a key factor for the knockin of exogenous ssODNs into mtDNA. Small-molecule RS-1 increased knockin efficiency of the mito-Cas9 system through RAD51 activation, further supporting HR-mediated knockin of exogenous ssODNs into mtDNA. Although template switching artifacts or other potential factors may introduce some noise in regard to the interpretation of knockin frequency, this noise is unlikely to have contributed to the significant differences observed between groups.
Lack of evident off-target sites in nuclear genome by the mito-Cas9 system
We performed whole-genome sequencing (WGS) for genomic DNA isolated from HEK293T cells transfected with sgRNA1ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN1 and sgRNA2ND4-MTSCOX8A-Cas9-UTRSOD2 + ssODN2, respectively. Mean sequence depth for the two samples was about 34×. The nuclear genome was screened for potential off-target sites with “NGG” or “NAG” protospacer adjacent motif and with up to nine mismatches relative to sgRNA. No insertions or deletions were identified in the potential off-target sites of sgRNA1ND4 and sgRNA2ND4 (Table S2). We further screened the whole genome for GAATTC insertions using the WGS data. No nuclear DNA reads with the GAATTC insertion were detected, indicating that GAATTC knockin in the mtDNA reads of third-generation sequencing data was not caused by potential off-target and/or unspecific knockin in nuclear DNA or from NUMTs.72
mtDNA pathogenic mutation generation using the mito-Cas9 system
Intragenic inversion mutation m.3902_3908inv (m.3902_3908 ACCTTGC>GCAAGGT) in the MT-ND1 gene is reported to cause fatal infantile lactic acidosis and mitochondrial myopathy.73,74 At present, however, the underlying mechanism remains unclear. Here, using the same strategy, we designed the mito-Cas9 system to introduce the m.3902_3908inv mutation into HEK293T cells (Figure 5A). Successful knockin of ssODN3902 in mtDNA within the mitochondria was demonstrated using a DNase protection assay (Figure 5B). Consistent with previous observations from the ssODN1ND4 and ssODN2ND4 knockin experiments, cells co-transfected with the mito-Cas9 constructs and ssODN3902 showed a significant increase in m.3902_3908inv-specific PCR products compared with cells transfected with ssODN3902 alone or with a combination of ssODN3902 and MTSCOX8A-Cas9-UTRSOD2 (no sgRNA) (Figure 5C). In addition, mutation m.3902_3908inv was identified in the second-generation sequencing reads (Figure 5D). The knockin efficiency of mutation m.3902_3908inv was about 0.05% (2,432 reads successfully edited among 5,284,619 raw reads). These results suggest that the mito-Cas9 system may serve as a promising approach for targeted knockin in mtDNA and could be optimized as a workable way to establish cellular models for studying mtDNA pathogenic mutations.
Figure 5.

Introduction of pathogenic mutation m.3902_3908inv to mtDNA of HEK293T cells
(A) Design of sgRNA and mito-Cas9-mediated knockin of mtDNA mutation m.3902_3908inv.
(B) Site-specific PCR of mutation m.3902_3908inv. Crude mitochondria were extracted from HEK293T cells transfected with sgRNA3902-MTSCOX8A-Cas9-UTRSOD2 and ssODN3902 for 48 h, then were treated with DNase I at 37°C for 1 h. PCR amplifications for nuclear APP (amplified by primer pair APP-F/APP-R), total mtDNA (amplified by primer pair L394/H475), and edited mtDNA (amplified by primer pair 3902F/H4227) were performed using the DNA template extracted from mitochondria with or without DNase I treatment.
(C) Quantification of knockin efficiency of mutation m.3902_3908inv. HEK293T cells were transfected with or without a combination of Cas9 constructs and ssODN3902. Proportion of mtDNA with successful m.3902_3908inv knockin (amplified by 3902F/H4227 (3902F-H4227)) was normalized to whole mtDNA (amplified by L394/H475 (L394-H475)). Bars are mean ± SD. ns, not significant; ∗∗p < 0.01, ∗∗∗p < 0.001.
(D) Detection strategy and presence of m.3902_3908inv knockin in mtDNA reads generated by second-generation sequencing.
Discussion
Currently, whether mtDNA can be edited by CRISPR-Cas9 remains controversial.4,39,40 Due to the lack of NHEJ repair, mtDNA with DSBs degrades rapidly,24 resulting in a reduction in mtDNA copy number, and thus mtDNA-edited products have not been directly detected in previous research.40, 41, 42 In this study, in addition to adding MTSs to Cas9,40, 41, 42, 43, 44, 45 we optimized the mitochondria-targeted Cas9 system by adding the 3′ UTR of the SOD2 gene to the downstream region of the Cas9 gene to facilitate efficient mitochondrial localization of Cas9 mRNA. We confirmed that the developed mito-Cas9 system transported Cas9 into the mitochondria (Figure 1C) and enabled mtDNA manipulation. Importantly, we developed a PCR-free third-generation sequencing technology that effectively avoids artifacts caused by PCR amplification (e.g., template switching artifacts) and verified the accurate knockin of exogenous ssODNs into mtDNA using the mito-Cas9 system (Figure 3). Another straightforward approach to confirm the effectiveness and efficiency of mitochondria-targeted CRISPR-Cas9 system can be engineered by introducing mtDNA-based drug resistance. Currently, we are attempting to establish a drug-resistant cell line with the mtDNA mutation m.2991T>C in the 16S rRNA of mtDNA, which could facilitate selection by chloramphenicol.75
Previous studies have shown that homologous arms longer than 40 bp exhibit higher HR efficiency than shorter arms.37,59 Based on this observation, we designed the homologous arms of three ssODNs (ssODN1, ssODN2, and ssODN3902), which were all 45 bp in length. We also investigated the knockin efficiency of ssODN50 with shorter arms (22 bp). Results showed that the knockin efficiency of ssODN50 was significantly lower than that of ssODN1 (Figure S10). Thus, the knockin efficiency of ssODN may be affected by its length, and optimizing homologous arm length to balance the stability and mitochondrial transport efficiency of ssODN could be helpful for increasing the knockin efficiency of the mito-Cas9 system.
The mechanism that maintain mitochondrial genome stability remain elusive. Notably, for the two main DNA repair pathways, NHEJ cannot be detected in mitochondria and the existence of HR in mitochondria is controversial.55,56,58,76 Consistent with previous study,58 we found that RAD51, a key nuclear factor of the HR repair pathway, was translocated into the mitochondria (Figure 4A) and its activation with agonist RS-1 enabled a 2- to 5-fold increase in knockin efficiency of ssODNs into mtDNA (Figure 4B). In addition, the effects of RAD51 activation appeared to be specific to cells transfected with the mito-Cas9 system (Figure 4, Table 1). These findings and third-generation sequencing results suggest that mtDNA can be affected by the HR pathway, and activation of the HR pathway by RAD51 stimulation may enhance CRISPR-Cas9-mediated knockin in mtDNA. One unresolved question is how ssODN is imported into mitochondria, which requires further research. Several studies have shown that DNA and RNA can be transported into mitochondria in animal cells and in plants,42,77, 78, 79, 80 and our study provides further evidence that FAM-labeled sgRNA and ssODN1 can be transported into mitochondria (Figures 1E, S2, and S6A).
Enthusiasm for mtDNA editing stems from the clinical need for the treatment of mitochondrial diseases caused by pathogenic mtDNA mutations, most of which are in a heteroplasmic state. Both mito-ZFN13,15,17,21 and mito-TALEN18, 19, 20,22 are effective in altering the heteroplasmic level of mutant mtDNA.17, 18, 19,22 For homoplasmic mtDNA mutations, base editing26,32,33 and CRISPR-Cas9-mediated HR knockin can be used to edit mutant mtDNA. However, one of the key problems with mtDNA editing is that each cell may have hundreds to thousands of mtDNA copies and the editing of each copy is unlikely. Thus, employing mtDNA-editing technology to cure mitochondrial diseases caused by mtDNA mutations remains a considerable challenge. In our study, knockin efficiency of the mito-Cas9 system was rather low (0.03%–0.23%) compared with the recently developed DdCBE method (5%–50%).26 We speculate that the low efficiency may be due to inefficient mitochondrial transport of the editing system and limited editing efficiency of the Cas9 protein to mtDNA. Therefore, mito-Cas9 system optimization, either by improving the mitochondrial transport efficiency such as mitochondrial RNA transport (although we did not achieve better mtDNA editing efficiency in cells overexpressing PNPASE, which can regulate RNA-import to mitochondria,81 compared with cells overexpressing RAD51 [data not shown]) or by using engineered Cas proteins with higher editing efficiency,82 is essential for the application of this mtDNA editing system. Furthermore, the knockin efficiency varied for different types of mutations, which may be due to different sequence features of the targeted region (i.e., GC% content) or different targeting efficiencies of the sgRNAs. Despite its limited editing efficiency, the mito-Cas9 system has the potential for wider scope of variant replacement and can introduce accurate knockin of target variants without changing other loci in the same editing window, thereby serving as an alternative strategy for manipulating mtDNA, especially considering recent findings that mitochondrial base editors may induce extensive off-target editing in the nuclear genome.34,35 In addition, inducing a small fraction of wild-type mtDNA using the mito-Cas9 system, then altering the heteroplasmic level of the wild-type mtDNA using mito-ZFN or mito-TALEN technology, offers a promising strategy for editing homoplasmic pathogenic mtDNA mutations.
In conclusion, we established an mtDNA editing system based on CRISPR-Cas9-mediated knockin via the HR pathway and found that RAD51 agonist RS-1 significantly enhanced mtDNA knockin efficiency. Using PCR-free third-generation sequencing, we provide direct evidence for mtDNA editing mediated by the CRISPR-Cas9 system. Future studies devoted to increasing editing efficiency are essential for expanding the application and safety of the mito-Cas9 system in the treatment of mitochondrial diseases caused by pathogenic mtDNA mutations.
Materials and methods
Detailed materials and methods are included in the supplemental information.
Data access
The data that support the findings of this work are available from the corresponding author upon reasonable request. The sequencing data were deposited at GSA (https://ngdc.cncb.ac.cn/gsa/) under accession number HRA001435.
Acknowledgments
We thank Prof. Ge Shan for helpful discussions, Dr. Christine Watts for language editing, and Dr. Jiankui Zhou for sharing the PST1374-Cas9 vector and pST1374-N-NLS-flag-linker-Cas9-D10A vector with mutation p.H840A. This study was supported by the National Science and Technology Innovation 2030 Major Program (2021ZD0200900), the National Natural Science Foundation of China (U1702284 and 31970560), Yunnan Province (202003AD150009 and 2019FA027), and the Strategic Priority Research Program (B) of the Chinese Academy of Sciences (CAS) (XDB32020200).
Author contributions
Y.-G.Y., R.B., X.H., H.Z., and P.Z. designed the study. R.B. and Y.L. constructed the mito-Cas9 system. M.X. performed third-generation sequencing and analyzed the data. Q.Z., B.X., and X.Z. extracted DNA and performed PCR analysis. D.-F.Z. and X.L. carried out secondary generation sequencing and analyzed the results. R.B. and G.M. performed flow cytometry. Y.-G.Y., R.B., M.X., and Y.L. wrote the first draft of the paper. All authors contributed to and approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published Online: September 27, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xinn.2022.100329.
Contributor Information
Rui Bi, Email: birui@mail.kiz.ac.cn.
Yong-Gang Yao, Email: yaoyg@mail.kiz.ac.cn.
Lead contact website
http://sourcedb.kiz.cas.cn/yw/zjrc/sc/200908/t20090825_2446617.html.
Supplemental information
References
- 1.Schon E.A., DiMauro S., Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rahman J., Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560–2574. doi: 10.1016/S0140-6736(18)30727-X. [DOI] [PubMed] [Google Scholar]
- 3.Falkenberg M., Larsson N.G., Gustafsson C.M. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 2007;76:679–699. doi: 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- 4.Patananan A.N., Wu T.H., Chiou P.Y., Teitell M.A. Modifying the mitochondrial genome. Cell Metab. 2016;23:785–796. doi: 10.1016/j.cmet.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao Y.G., Kajigaya S., Young N.S. Mitochondrial DNA mutations in single human blood cells. Mutat. Res. 2015;779:68–77. doi: 10.1016/j.mrfmmm.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell O., Turnbull D. Mitochondrial DNA disease-molecular insights and potential routes to a cure. Exp. Cell Res. 2014;325:38–43. doi: 10.1016/j.yexcr.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace D.C., Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013;5:a021220. doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellouze S., Augustin S., Bouaita A., et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am. J. Hum. Genet. 2008;83:373–387. doi: 10.1016/j.ajhg.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bi R., Logan I., Yao Y.G. Leber hereditary optic neuropathy: a mitochondrial disease unique in many ways. Handb. Exp. Pharmacol. 2017;240:309–336. doi: 10.1007/164_2016_1. [DOI] [PubMed] [Google Scholar]
- 11.Herbert M., Turnbull D. Progress in mitochondrial replacement therapies. Nat. Rev. Mol. Cell Biol. 2018;19:71–72. doi: 10.1038/nrm.2018.3. [DOI] [PubMed] [Google Scholar]
- 12.Kang E., Wu J., Gutierrez N.M., et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature. 2016;540:270–275. doi: 10.1038/nature20592. [DOI] [PubMed] [Google Scholar]
- 13.Minczuk M., Papworth M.A., Miller J.C., et al. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008;36:3926–3938. doi: 10.1093/nar/gkn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka M., Borgeld H.J., Zhang J., et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002;9:534–541. doi: 10.1159/000064726. [DOI] [PubMed] [Google Scholar]
- 15.Gammage P.A., Viscomi C., Simard M.L., et al. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018;24:1691–1695. doi: 10.1038/s41591-018-0165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacman S.R., Williams S.L., Duan D., Moraes C.T. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012;19:1101–1106. doi: 10.1038/gt.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gammage P.A., Rorbach J., Vincent A.I., et al. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014;6:458–466. doi: 10.1002/emmm.201303672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bacman S.R., Kauppila J.H.K., Pereira C.V., et al. MitoTALEN reduces mutant mtDNA load and restores tRNA(Ala) levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018;24:1696–1700. doi: 10.1038/s41591-018-0166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira C.V., Bacman S.R., Arguello T., et al. mitoTev-TALE: a monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 2018;10:e8084. doi: 10.15252/emmm.201708084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reddy P., Ocampo A., Suzuki K., et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161:459–469. doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minczuk M., Papworth M.A., Kolasinska P., et al. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl. Acad. Sci. USA. 2006;103:19689–19694. doi: 10.1073/pnas.0609502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bacman S.R., Williams S.L., Pinto M., et al. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013;19:1111–1113. doi: 10.1038/nm.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zekonyte U., Bacman S.R., Smith J., et al. Mitochondrial targeted meganuclease as a platform to eliminate mutant mtDNA in vivo. Nat. Commun. 2021;12:3210. doi: 10.1038/s41467-021-23561-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peeva V., Blei D., Trombly G., et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018;9:1727. doi: 10.1038/s41467-018-04131-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moretton A., Morel F., Macao B., et al. Selective mitochondrial DNA degradation following double-strand breaks. PLoS One. 2017;12:e0176795. doi: 10.1371/journal.pone.0176795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mok B.Y., de Moraes M.H., Zeng J., et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature. 2020;583:631–637. doi: 10.1038/s41586-020-2477-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee H., Lee S., Baek G., et al. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021;12:1190. doi: 10.1038/s41467-021-21464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo J., Zhang X., Chen X., et al. Precision modeling of mitochondrial diseases in zebrafish via DdCBE-mediated mtDNA base editing. Cell Discov. 2021;7:78. doi: 10.1038/s41421-021-00307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang B.C., Bae S.J., Lee S., et al. Chloroplast and mitochondrial DNA editing in plants. Nat. Plants. 2021;7:899–905. doi: 10.1038/s41477-021-00943-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X., Liang D., Guo J., et al. DdCBE-mediated mitochondrial base editing in human 3PN embryos. Cell Discov. 2022;8:8. doi: 10.1038/s41421-021-00358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei Y., Xu C., Feng H., et al. Human cleaving embryos enable efficient mitochondrial base-editing with DdCBE. Cell Discov. 2022;8:7. doi: 10.1038/s41421-021-00372-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim K., Cho S.I., Kim J.S. Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat. Commun. 2022;13:366. doi: 10.1038/s41467-022-27962-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cho S.I., Lee S., Mok Y.G., et al. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell. 2022;185:1764–1776.e12. doi: 10.1016/j.cell.2022.03.039. [DOI] [PubMed] [Google Scholar]
- 34.Wei Y., Li Z., Xu K., et al. Mitochondrial base editor DdCBE causes substantial DNA off-target editing in nuclear genome of embryos. Cell Discov. 2022;8:27. doi: 10.1038/s41421-022-00391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei Z., Meng H., Liu L., et al. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature. 2022;606:804–811. doi: 10.1038/s41586-022-04836-5. [DOI] [PubMed] [Google Scholar]
- 36.Hsu P.D., Lander E.S., Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ran F.A., Hsu P.D., Wright J., et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gammage P.A., Moraes C.T., Minczuk M. Mitochondrial genome engineering: the revolution may not be CRISPR-Ized. Trends Genet. 2018;34:101–110. doi: 10.1016/j.tig.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loutre R., Heckel A.M., Smirnova A., et al. Can mitochondrial DNA be CRISPRized: Pro and Contra. IUBMB Life. 2018;70:1233–1239. doi: 10.1002/iub.1919. [DOI] [PubMed] [Google Scholar]
- 41.Jo A., Ham S., Lee G.H., et al. Efficient mitochondrial genome editing by CRISPR/Cas9. BioMed Res. Int. 2015;2015:305716. doi: 10.1155/2015/305716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bian W.P., Chen Y.L., Luo J.J., et al. Knock-in strategy for editing human and zebrafish mitochondrial DNA using mito-CRISPR/Cas9 system. ACS Synth. Biol. 2019;8:621–632. doi: 10.1021/acssynbio.8b00411. [DOI] [PubMed] [Google Scholar]
- 43.Antón Z., Mullally G., Ford H.C., et al. Mitochondrial import, health and mtDNA copy number variability seen when using type II and type V CRISPR effectors. J. Cell Sci. 2020;133:jcs248468. doi: 10.1242/jcs.248468. [DOI] [PubMed] [Google Scholar]
- 44.Wang B., Lv X., Wang Y., et al. CRISPR/Cas9-mediated mutagenesis at microhomologous regions of human mitochondrial genome. Sci. China Life Sci. 2021;64:1463–1472. doi: 10.1007/s11427-020-1819-8. [DOI] [PubMed] [Google Scholar]
- 45.Hussain S.R.A., Yalvac M.E., Khoo B., et al. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front. Genet. 2021;12:627050. doi: 10.3389/fgene.2021.627050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hofreiter M., Jaenicke V., Serre D., et al. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29:4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu S., Thaler D.S., Libchaber A. Signal and noise in bridging PCR. BMC Biotechnol. 2002;2:13. doi: 10.1186/1472-6750-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilbert M.T.P., Hansen A.J., Willerslev E., et al. Characterization of genetic miscoding lesions caused by postmortem damage. Am. J. Hum. Genet. 2003;72:48–61. doi: 10.1086/345379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sylvestre J., Margeot A., Jacq C., et al. The role of the 3' untranslated region in mRNA sorting to the vicinity of mitochondria is conserved from yeast to human cells. Mol. Biol. Cell. 2003;14:3848–3856. doi: 10.1091/mbc.E03-02-0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginsberg M.D., Feliciello A., Jones J.K., et al. PKA-dependent binding of mRNA to the mitochondrial AKAP121 protein. J. Mol. Biol. 2003;327:885–897. doi: 10.1016/s0022-2836(03)00173-6. [DOI] [PubMed] [Google Scholar]
- 51.Gao Z., Harwig A., Berkhout B., Herrera-Carrillo E. Mutation of nucleotides around the +1 position of type 3 polymerase III promoters: the effect on transcriptional activity and start site usage. Transcription. 2017;8:275–287. doi: 10.1080/21541264.2017.1322170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang D., Zhang H., Li T., et al. Perfectly matched 20-nucleotide guide RNA sequences enable robust genome editing using high-fidelity SpCas9 nucleases. Genome Biol. 2017;18:191. doi: 10.1186/s13059-017-1325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.He T., Hatem E., Vernis L., et al. PRX1 knockdown potentiates vitamin K3 toxicity in cancer cells: a potential new therapeutic perspective for an old drug. J. Exp. Clin. Cancer Res. 2015;34:152. doi: 10.1186/s13046-015-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feng Y.M., Jia Y.F., Su L.Y., et al. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy. 2013;9:1395–1406. doi: 10.4161/auto.25468. [DOI] [PubMed] [Google Scholar]
- 55.Kajander O.A., Karhunen P.J., Holt I.J., Jacobs H.T. Prominent mitochondrial DNA recombination intermediates in human heart muscle. EMBO Rep. 2001;2:1007–1012. doi: 10.1093/embo-reports/kve233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.D'Aurelio M., Gajewski C.D., Lin M.T., et al. Heterologous mitochondrial DNA recombination in human cells. Hum. Mol. Genet. 2004;13:3171–3179. doi: 10.1093/hmg/ddh326. [DOI] [PubMed] [Google Scholar]
- 57.Tadi S.K., Sebastian R., Dahal S., et al. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell. 2016;27:223–235. doi: 10.1091/mbc.E15-05-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dahal S., Dubey S., Raghavan S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell. Mol. Life Sci. 2018;75:1641–1655. doi: 10.1007/s00018-017-2702-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X., Li T., Ou J., et al. Homology-based repair induced by CRISPR-Cas nucleases in mammalian embryo genome editing. Protein Cell. 2022;13:316–335. doi: 10.1007/s13238-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qi L.S., Larson M.H., Gilbert L.A., et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen B., Zhang W., Zhang J., et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods. 2014;11:399–402. doi: 10.1038/nmeth.2857. [DOI] [PubMed] [Google Scholar]
- 62.Parr R.L., Maki J., Reguly B., et al. The pseudo-mitochondrial genome influences mistakes in heteroplasmy interpretation. BMC Genom. 2006;7:185. doi: 10.1186/1471-2164-7-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrews R.M., Kubacka I., Chinnery P.F., et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 1999;23:147. doi: 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- 64.Tsuji J., Frith M.C., Tomii K., Horton P. Mammalian NUMT insertion is non-random. Nucleic Acids Res. 2012;40:9073–9088. doi: 10.1093/nar/gks424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ardui S., Ameur A., Vermeesch J.R., Hestand M.S. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018;46:2159–2168. doi: 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sage J.M., Gildemeister O.S., Knight K.L. Discovery of a novel function for human Rad51: maintenance of the mitochondrial genome. J. Biol. Chem. 2010;285:18984–18990. doi: 10.1074/jbc.M109.099846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mishra A., Saxena S., Kaushal A., Nagaraju G. RAD51C/XRCC3 facilitates mitochondrial DNA replication and maintains integrity of the mitochondrial genome. Mol. Cell Biol. 2018;38:e00489-17. doi: 10.1128/MCB.00489-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song J., Yang D., Xu J., et al. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun. 2016;7:10548. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pinder J., Salsman J., Dellaire G. Nuclear domain 'knock-in' screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015;43:9379–9392. doi: 10.1093/nar/gkv993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jayathilaka K., Sheridan S.D., Bold T.D., et al. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl. Acad. Sci. USA. 2008;105:15848–15853. doi: 10.1073/pnas.0808046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Budke B., Logan H.L., Kalin J.H., et al. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012;40:7347–7357. doi: 10.1093/nar/gks353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yao Y.G., Kong Q.P., Salas A., Bandelt H.J. Pseudomitochondrial genome haunts disease studies. J. Med. Genet. 2008;45:769–772. doi: 10.1136/jmg.2008.059782. [DOI] [PubMed] [Google Scholar]
- 73.Musumeci O., Andreu A.L., Shanske S., et al. Intragenic inversion of mtDNA: a new type of pathogenic mutation in a patient with mitochondrial myopathy. Am. J. Hum. Genet. 2000;66:1900–1904. doi: 10.1086/302927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blakely E.L., Rennie K.J., Jones L., et al. Sporadic intragenic inversion of the mitochondrial DNA MTND1 gene causing fatal infantile lactic acidosis. Pediatr. Res. 2006;59:440–444. doi: 10.1203/01.pdr.0000198771.78290.c4. [DOI] [PubMed] [Google Scholar]
- 75.King M.P., Attardi G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell. 1988;52:811–819. doi: 10.1016/0092-8674(88)90423-0. [DOI] [PubMed] [Google Scholar]
- 76.Hagström E., Freyer C., Battersby B.J., et al. No recombination of mtDNA after heteroplasmy for 50 generations in the mouse maternal germline. Nucleic Acids Res. 2014;42:1111–1116. doi: 10.1093/nar/gkt969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koulintchenko M., Temperley R.J., Mason P.A., et al. Natural competence of mammalian mitochondria allows the molecular investigation of mitochondrial gene expression. Hum. Mol. Genet. 2006;15:143–154. doi: 10.1093/hmg/ddi435. [DOI] [PubMed] [Google Scholar]
- 78.Tarasenko T.A., Klimenko E.S., Tarasenko V.I., et al. Plant mitochondria import DNA via alternative membrane complexes involving various VDAC isoforms. Mitochondrion. 2021;60:43–58. doi: 10.1016/j.mito.2021.07.006. [DOI] [PubMed] [Google Scholar]
- 79.Liu X., Wang X., Li J., et al. Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci China Life Sci. 2020;63:1429–1449. doi: 10.1007/s11427-020-1631-9. [DOI] [PubMed] [Google Scholar]
- 80.Boesch P., Ibrahim N., Dietrich A., Lightowlers R.N. Membrane association of mitochondrial DNA facilitates base excision repair in mammalian mitochondria. Nucleic Acids Res. 2010;38:1478–1488. doi: 10.1093/nar/gkp1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang G., Chen H.W., Oktay Y., et al. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen Y., Hu Y., Wang X., et al. Synergistic engineering of CRISPR-Cas nucleases enables robust mammalian genome editing. Innovation. 2022;3:100264. doi: 10.1016/j.xinn.2022.100264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.