

Abstract
Commonly overexpressed in many cancers and associated with tumor growth, metastasis, drug resistance, and poor overall survival, Axl has emerged as a promising target for cancer therapy. However, the availability of new chemical forms for Axl inhibition is limited. Herein, we present the development and characterization of novel Axl inhibitors, including the design, synthesis, and structure–activity relationships (SARs) of a series of diphenylpyrimidine–diamine derivatives. Most of these compounds exhibited remarkable activity against the Axl kinase. In particular, the promising compound m16 showed the highest enzymatic inhibitory potency (IC50 = 5 nM) and blocked multiple tumor cells' proliferation potencies (the CC50 of 4 out of 42 cancer cell lines <100 nM). Furthermore, compound m16 also possessed preferable pharmacokinetic profiles and liver microsome stability. All these favorable results make m16 a good leading therapeutic candidate for further development.
Commonly overexpressed in many cancers and associated with tumor growth, metastasis, drug resistance, and poor overall survival, Axl has emerged as a promising target for cancer therapy.
Introduction
Axl is a member of the TAM (Tyro3, Axl, Mer) subfamily of receptor tyrosine kinases (RTKs), and was first identified in chronic myeloid leukemia (CML).1,2 The most common activation mechanism of Axl signaling is binding to its endogenous ligand growth arrest-specific protein 6 (Gas6).3 Other activation mechanisms can also occur,4,5 including (1) ligand-independent homologous dimerization activation, (2) heterodimerization activation with Mer or Tyro3, (3) heterodimerization activation with non-TAM kinases (such as Met), and (4) transcellular ligand-independent activation. Dimerization induces autophosphorylation of tyrosine residues in the intracellular region of Axl, resulting in the activation of its downstream signaling pathways such as PI3K/AKT, SRC/FAK, GRB2/RAS/RAF, etc.6–8 Gas6/Axl signaling is closely correlated with various biological processes, such as proliferation, differentiation, adhesion, migration, and immune response.9,10 Collective evidence suggests that overexpression of Axl is a key inducer of epithelial to mesenchymal transition (EMT) that promotes tumor invasiveness/metastasis and drug resistance.11–13
With the widespread application of targeted therapy and immune checkpoint inhibitors, drug resistance has been a major cause of the failure in anticancer treatment.14 Understanding resistance mechanisms and identifying the molecules that drive resistance will help eradicate drug-resistant cancers. Due to its considerable clinical success, the combination therapy with tyrosine kinase inhibitors (TKIs) and immune checkpoint blockers (PD-1, PD-L1) is being reinvigorated.15 The distribution and overexpression of Axl in a variety of leukemia and solid tumors indicate that it may have an essential function in the tumorigenicity and resistance mechanism.16 If combined with PD-1 or PD-L1 blockers, Axl small molecule inhibitors may induce potent synergistic antitumor efficacy to eradicate tumors.17,18
Many inhibitors that target Axl with different selectivity profiles have entered clinical trials.19 Bemcentinib (also named: BGB-324, R428), which was disclosed as the first selective Axl inhibitor, exhibited potent activity (IC50 = 14 nM) and 50-fold higher affinity over subfamily kinases Mer and Tyro3 in vitro.20 And the FDA has granted Fast Track designation for bemcentinib in combination with an anti-PD-(L)1 agent for the treatment of patients with STK11-mutated advanced/metastatic non-small cell lung cancer (NSCLC). DS-1205b combined with osimertinib or gefitinib in the treatment of NSCLC is undergoing a phase I clinical study (NCT03255083, NCT03599518).21 Besides, numerous selective TAM family small molecule kinase inhibitors, such as ONO-7475, RXDX-106, INCB081776, SLC-391, etc., have been designed and investigated.22 Therefore, selective inhibition of Axl is a potential method to eliminate drug resistance and improve the treatment outcomes of immune or targeted therapy.
Indeed, the currently marketed Axl small-molecule inhibitors are mainly multi-target inhibitors. Gilteritinib, mainly targeting Flt3 and Axl, has been approved for the treatment of relapsed or refractory AML with FLT3 mutation.23 Cabozantinib inhibits multiple RTKs, including VEGFR2, RET, KIT, c-MET, ROS1, Flt1/3/4, Tie2, and Axl, and has been approved by the FDA for the treatment of advanced RCC and metastatic medullary thyroid carcinoma.24 Clinical studies on other indications, such as NSCLC, AML, and castration-resistant prostate cancer, are also underway. The success of these two drugs has sparked the enthusiasm of medicinal chemists to develop multi-target inhibitors for Axl, TAM, and non-TAM RTKs. Of note, the permutations and combinations of non-TAM RTKs and Axl have led to significant progress in various clinical indications.25 Among these compounds, TP-0903 was known to induce dose-dependent CLL cell death in vitro, and was first synthesized by David J. Bearss in 2011.26,27 As an Axl inhibitor, TP-0903 is currently in phase I clinical trials for the treatment of CLL and refractory solid tumors (NCT03572634, and NCT02729298) (Fig. 1).
Fig. 1. Chemical structures of the above-mentioned Axl inhibitors.

Targeting Axl, whether highly selective or multi-target inhibitors, has shown the advantages of overcoming drug resistance and improving the survival of many cancer patients. Thus, Axl small-molecule inhibitors are finally coming of age. However, the availability of new chemical forms for Axl inhibition is limited.28 Herein, we describe our efforts to discover a novel series of diphenylpyrimidine–diamine derivatives as Axl small molecule inhibitors (Fig. 2). The design and synthesis of these compounds are discussed in this manuscript.
Fig. 2. (A) Schematic illustration of the proposed binding mode of 5a with the Axl and the main interactions between Axl and 5a (PDB ID: 5U6B). (B) Structure-based discovery of potent Axl inhibitors. X = oxygen or nitrogen.

Results and discussion
Chemistry
The synthetic routes of compounds listed in Table 1 are shown in Scheme 1. In short, commercially obtained reagent 2 was treated with 2,4,5-trichloropyrimidine in the presence of DIPEA to prepare intermediate 4. Then coupling of intermediate 4 with four aniline derivatives under classic Buchwald–Hartwig coupling reaction conditions afforded 5a–c. The synthesis of 5d was catalyzed by hydrochloric acid under microwave conditions.
Structure–activity relationship of the R1 substituent on the four compounds.
| ||||||
|---|---|---|---|---|---|---|
| Cpd. | R1 | Axl IC50 (nM) | Human | Rat | ||
| T 1/2 min | Clint mL min−1 kg−1 | T 1/2 min | Clint mL min−1 kg−1 | |||
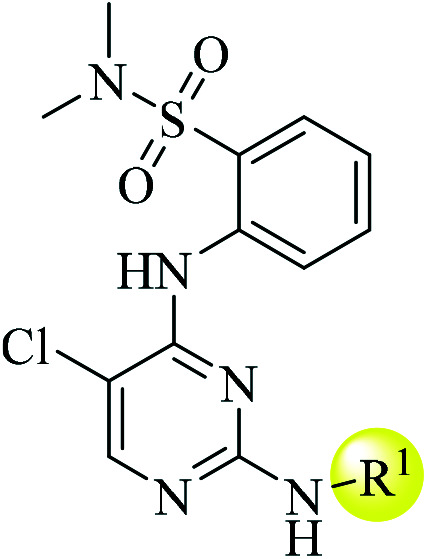
| 5a |

|
14 | 2.4 | CLhep = 720.4 | 2.4 | CLhep = 1041.0 |
| CLin vivo = 20.1 | CLin vivo = 52.4 | |||||
| 5b |
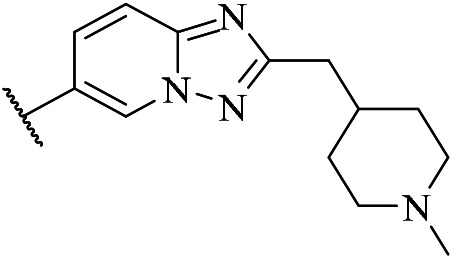
|
49 | ND. | ND. | ND. | ND. |
| 5c |

|
17 | 4.4 | CLhep = 392.8 | 7.3 | CLhep = 339.1 |
| CLin vivo = 19.6 | CLin vivo = 47.5 | |||||
| 5d |
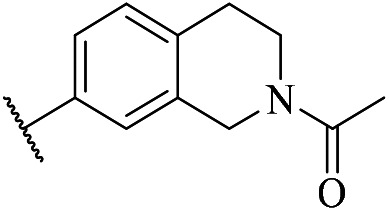
|
ND. | 1.82 | CLhep = 953.0 | 2.3 | CLhep = 1088.3 |
| CLin vivo = 20.3 | CLin vivo = 52.5 | |||||
Scheme 1. Preparation of sulfonamide derivatives 5a–d. Reagents and conditions: (a) DIPEA, THF, reflux, 16 h; (b) Pd2(dba)3, t-butyl XPhos or Xantphos, tBuONa, 1,4-dioxane, reflux 16–24 h; (c) cat. HCl, 1-butanol, microwave, 130 °C, 3 h.

The synthetic routes of compounds listed in Table 2 are shown in Scheme 2. After acyl chlorination with SOCl2, intermediate 7 was synthesized from reagent 6 in the presence of N-hydroxyacetamidine. Cyclization of compound 7 with K2CO3 generated 8. Reduction of the nitro group produced 9 in the presence of FeCl3 and hydrazine hydrate. Simultaneously, 1,2,4-oxadiazole was opened and reconstituted to 1H-1,2,4-triazole. Intermediate 9 was treated with 2,4,5-trichloropyrimidine in the presence of DIPEA to prepare compound 10, and then coupling of 10 with fourteen aniline derivatives under classic Buchwald–Hartwig coupling reaction conditions afforded w1 and 2, w4, w7, w10–17, and w19. The other seven title compounds w3, w5 and 6, w8 and 9, w18, and w20 were synthesized by a hydrochloric acid-catalyzed method under microwave conditions.
Effects of selected substitution of R2 on the kinase inhibitory activity of 1H-1,2,4-triazole derivatives.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Cpd. | R2 | Axl IC50 (nM) | Cpd. | R2 | Axl IC50 (nM) | Cpd. | R2 | Axl IC50 (nM) |
| w1 |
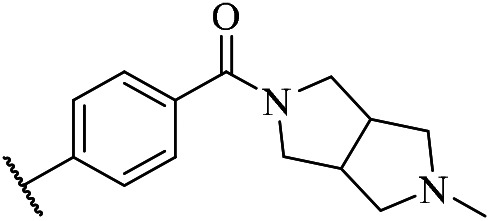
|
15 | w8 |

|
46 | w15 |

|
30 |
| w2 |
|
12 | w9 |

|
555 | w16 |

|
101 |
| w3 |

|
9 | w10 |

|
14 | w17 |

|
100 |
| w4 |

|
30 | w11 |

|
28 | w18 |

|
15 |
| w5 |

|
16 | w12 |

|
185 | w19 |

|
10 |
| w6 |

|
50 | w13 |

|
48 | w20 |

|
12 |
| w7 |

|
51 | w14 |

|
11 | |||
Scheme 2. Preparation of 1H-1,2,4-triazole derivatives w1–20. Reagents and conditions: (c) i, SOCl2, THF, 60 °C, 3 h; ii, N-hydroxy acetamidine, triethylamine, DCM, −5 °C, 16 h; (d) K2CO3, 1,4-dioxane, reflux, 16 h; (e) hydrazine hydrate, FeCl3, ethanol, 80 °C, 5.5 h; (f) 2,4,5-trichloropyrimidine, DIPEA, IPA, reflux, 6 h; (g) Pd2(dba)3, BINAP, t-butyl XPhos, tBuOH, 90 °C, 16 h; (h) cat. HCl, 1-butanol, microwave, 130 °C, 3 h.

Compounds m1–4 and m9 and 10 were synthesized in two steps starting from intermediate 10 (details are described in the ESI†). Initially, alkylation of N–H on the triazole produced methyl or ethyl substituted intermediate 10. The subsequent Buchwald–Hartwig coupling between reactant 11 and various para-substituted aniline derivatives yielded these six final compounds. The other compounds listed in Table 4 were obtained by the sequence of reactions shown in Scheme 3.
Effects of various five-membered heteroaromatic rings on the kinase inhibitory activity of diphenylpyrimidine–diamine derivatives.

| |||||||
|---|---|---|---|---|---|---|---|
| Cpd. | R3 | R4 | Axl IC50 (nM) | Cpd. | R3 | R4 | Axl IC50 (nM) |
| m1 |

|

|
8 | m9 |

|

|
10 |
| m2 |

|

|
16 | m10 |
|
|
18 |
| m3 |

|

|
30 | m11 |

|

|
25 |
| m4 |
|

|
11 | m12 |

|

|
12 |
| m5 |

|

|
22 | m13 |

|

|
22 |
| m6 |

|

|
5 | m14 |

|

|
36 |
| m7 |
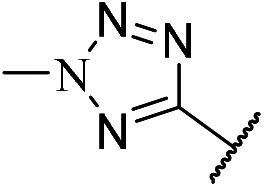
|

|
49 | m15 |

|

|
169 |
| m8 |

|

|
23 | m16 |
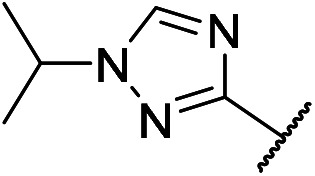
|

|
5 |
Scheme 3. Preparation of diphenylpyrimidine–diamine derivatives m5–8 and m11–16. Reagents and conditions: (h) trimethoxymethane, CF3COOH, reflux, 1.5 h; (i) methylhydrazine or ethylhydrazine, DCM, rt, 12 h; (j) iodomethane, NaH, DMF, 0 °C–rt, 3 h. (k) Pd/C, ethanol, H2, rt, 12 h. (l) i, SOCl2, THF, 60 °C, 3 h; ii, acetyl hydrazide, triethylamine, DCM, 0 °C, 16 h; (m) POCl3, toluene, 90 °C, 24 h; (n) hydrazine hydrate, FeCl3, ethanol, 80 °C, 5.5 h; (o) i, thionyl chloride, 80 °C, 5 h; ii, methylamine hydrochloride, Na2CO3, MeCN, 80 °C, 4 h; (p) NaN3, trifluoromethanesulfonic anhydride, MeCN, 0 °C, 7 h; (q) Pd/C, ethanol, H2, rt, 12 h; (r) 2,4,5-trichloropyrimidine, DIPEA, IPA, reflux, 6 h or NaH, DMF, 0 °C–rt, 12 h; (s) cat. HCl, 1-butanol, microwave, 140 °C, 3 h; (t) Pd2(dba)3, XPhos, tBuOH, 90 °C, 16 h.

The reagent methyl (E)-N-(2-cyanophenyl)formimidate was prepared by reaction of 2-aminobenzonitrile with trimethoxymethane under the catalysis of CF3COOH. Treatment of 12 with methylhydrazine or ethylhydrazine at room temperature provided intermediate 14. Through SN2 nucleophilic substitution with 2,4,5-trichloropyrimidine the corresponding intermediate 24 was obtained. The synthesis of final compounds m5 and 6, m12 and 13, m14, and m16 was performed by microwave reactions in the presence of a catalytic amount of HCl with different aniline derivatives. N-Methylation of commercially available reactant 15, followed by Pd/C catalyzed reduction of compound 16 with H2, afforded key intermediate 17. After acyl chlorination with SOCl2, 18 was synthesized from 6 in the presence of N-hydroxyacetamidine. Cyclization of 18 with POCl3 generated 19. The reduction of 19 yielded another key intermediate 20 in the presence of FeCl3 and hydrazine hydrate. 6 was treated with thionyl chloride and methylamine to obtain 21. Subsequently, cyclization of 21 with NaN3 in the presence of trifluoromethanesulfonic anhydride produced 22. Pd/C catalyzed reduction of it with H2 yielded the third key intermediate 23. The corresponding compound 24 was synthesized via SN2 nucleophilic substitution between 2,4,5-trichloropyrimidine and three key intermediates 17, 20, and 23. Finally, the synthesis of final compounds m7 and 8 and m11 was performed by microwave reactions with different aniline derivatives in the presence of a catalytic amount of HCl. Pd2(dba)3 catalyzed coupling with 4-((4-methylpiperazin-1-yl)methyl)aniline yielded the final compound m15.
Evaluation of the inhibitory activity for the Axl kinase and SAR analysis
The kinase inhibitory activities of the new derivatives were determined by KinaseProfiler™ of Eurofins. TP-0903 served as a positive control and displayed an IC50 value of 15 nM. The liver microsome assay and the pharmacokinetic assay in rats were used to evaluate the metabolic stability of our derivatives. The antiproliferative effect of the promising compound was evaluated in 42 cancer cell lines. And the cell lines were purchased from Cobioer Biosciences, Inc. The docking studies were carried out with Schrödinger suites 2018.1 using the X-ray crystal structure of Axl (PDB ID: 5U6B).
Three compounds, gilteritinib, bemcentinib, and TP-0903, were classified as type I Axl inhibitors, which target the ATP-binding domain of the Axl kinase adopting a “U shape” to generate an active conformation. Based on this information, we initially designed and synthesized compound 5a (Fig. 2), which features a bicycle moiety inserted in the solvent-accessible pocket and binds to the active conformation of Axl. The scarcity of the crystal structure of the Axl kinase domain hindered the computer-aided drug design of Axl inhibitors. In 2017, Ketan S. Gajiwala et al. first characterized the structure of the Axl kinase domain in a complex with a macrocyclic compound.29 As shown in Fig. 2A, the 2-aminopyridine segment mainly formed bidentate hydrogen bonds with the backbone peptide residue Met623 within the Axl kinase domain, although the oxy in the sulfonamide could not form a hydrogen bond with Asp690 because of the distance. Biochemical assays showed that compound 5a displayed excellent activity with an IC50 value of 14 nM, which was comparable to TP-0903. This result provided a beneficial direction for further rational design: 1) there was a large unfilled space between the bicycle core of 5a and the solvent-accessible pocket, we envisioned that the potency could be improved by introducing more heterocycles to fill the space. 2) As the sulfonamide fragment of 5a did not interact strongly with the binding pocket, shifting this moiety to a five-membered ring might allow the inhibitor to become closer to the pocket surface to enhance van der Waals interactions. With that in mind, we designed two series of diphenylpyrimidine–diamine derivatives as novel Axl inhibitors (Fig. 2B).
We initially designed four novel 2,4,5-trisubstituted pyrimidine compounds as Axl inhibitors. Early lead 5c was achieved through the introduction of a variety of aniline derivatives at the 2-position of pyrimidine, and this is depicted in Table 1. In addition to Axl kinase inhibition assessments, we also evaluated the stability of the four compounds in human and rat liver microsomes.
As shown in Table 1, compound 5a showed excellent kinase inhibitory activity (Table 1, entry 1). But its stability in human and rat liver microsomes was insufficient. The replacement of methyl with acetyl (5d) unexpectedly decreased the metabolic stability. Besides, due to its poor solubility, the kinase inhibitory activity of 5d was not investigated. Replacing phenyl with triazolopyridine, along with the introduction of N-methyl piperidine, led to a 4-fold loss in Axl inhibitory activity (5b). This was likely due to altered π-electron interactions with the hinge region of the Axl kinase. Opening of the tetrahydroisoquinoline core and construction of a rigid linker to connect the benzene with bicyclopyrrolo[3,4-c]pyrrole (5c) led to comparable Axl inhibitory activity to 5a. Fortunately, significant improvement of the liver microsome stability in humans and rats (5c) was observed. Therefore, the inhibitory activity and metabolic stability profiles of compound 5c were similar to those of the control drug TP-0903 (these data are shown in Table 5).
Liver microsomal stability of four compounds.

| |||||
|---|---|---|---|---|---|
| Cpd. | Axla IC50 (nM) | Human | Rat | ||
| T 1/2 min | Clint mL min−1 kg−1 | T 1/2 min | Clint mL min−1 kg−1 | ||
| m6 | 5 | 155.9 | CLhep = 11.2 | 153.1 | CLhep = 16.2 |
| CLin vivo = 7.2 | CLin vivo = 12.5 | ||||
| m12 | 12 | 36.2 | CLhep = 48.0 | 6.4 | CLhep = 388.9 |
| CLin vivo = 14.5 | CLin vivo = 48.3 | ||||
| m13 | 22 | 74.2 | CLhep = 23.4 | 15.4 | CLhep = 161.8 |
| CLin vivo = 11.0 | CLin vivo = 41.2 | ||||
| m16 | 5 | 60.6 | CLhep = 28.7 | 28.3 | CLhep = 87.7 |
| CLin vivo = 12.0 | CLin vivo = 33.9 | ||||
| TP-0903 | 15 | 2.7 | CLhep = 645.7 | 4.3 | CLhep = 581.9 |
| CLin vivo = 20.1 | CLin vivo = 50.4 | ||||
These data were used from Table 4.
Many small-molecule inhibitors with pyrimidine scaffolds are typically utilized with various substitutions at the 2-position. However, guided by the above-mentioned tabular data, we considered that the sulfonamide segment in the trisubstituted pyrimidine derivatives may be a destabilizing factor in liver microsomes. Continuing to optimize the 2-position of pyrimidine was not a good choice. Besides, a better understanding of the binding mode of 5a with Axl has provided another way for further rational design. Therefore, a series of 2,4,5-trisubstituted pyrimidine derivatives w1–19 with a 1H-1,2,4-triazole scaffold were designed and synthesized. And SAR information for series I compounds is summarized in Table 2.
Similar to sulfonamide, the 1H-1,2,4-triazole motif could occupy the allosteric binding site, and a five-membered ring containing a hydrogen bond donor might form a hydrogen bond with residue Asp690. Compared to early lead compound 5c, a very slight improvement in activity was achieved with w1. Reassuringly, our strategy has been proven successful. And with the same substituted fragment at the 2-position of pyrimidine, the introduction of 1H-1,2,4-triazole reliably enhanced the stability of the compound in human liver microsomes (w10vs.5a, ESI† Table S3). Compounds w1–3 showed excellent potencies against the Axl kinase (IC50 = 9–15 nM). Variation from pyrrolo[3,4-c]pyrrole to the piperazine ring suggested that steric and hydrophobicity affect the inhibitory activity, which might evidence that substituted scaffolds on the para-benzene of R2 reached the solvent pocket. Although the stability of the three compounds was slightly improved, their pharmacokinetic properties were poor.
Swapping positions of piperazine analogs and carbonyl led to a nearly 2-fold loss in potency. However, significant improvements in the stability and pharmacokinetic properties of w4 and 5 were observed, further supporting the instability of the amide bond between benzene and piperazine in compounds w1–3. We next screened commonly used para-substituent phenylamines among other small molecule inhibitors. Unfortunately, the kinase inhibition results were not good enough (w6–9). N-Methyl and acetyl substituted tetrahydroisoquinolines were applied in this series. Two compounds all obtained good inhibitory activity for Axl (w10 and w11). We were encouraged by the fact that deletion of the carbonyl group did not eliminate the inhibitory activity (w14vs.w3). And the inhibitory activities of these three compounds suggested that the carbonyl linker connecting benzene with piperazine might be unnecessary. In contrast, the upward position of carbonyl is better than the downward (w12vs.w13). Replacing piperazine with cycloalkyl amines all led to significant improvement in pharmacokinetic properties, but a marked loss in potency (w15vs.w17). Otherwise, the change in electron cloud density caused a significant decrease in potency (w16vs.w15).
We next explored the effect of linkers between benzene and piperidine fragments. The oxygen-linked benzene ring with piperidine was the best (w18–20). Finally, the introduction of a fluorine atom had a weak effect on the inhibitory activity but did not contribute to the pharmacokinetic properties (w20). These initial studies established 1H-1,2,4-triazole as a suitable fragment to replace sulfonamide.
As indicated in the docking studies, our compound w14 formed bidentate hydrogen bonds with the backbone peptide residue Met623 of the Axl kinase domain. And the N–H within the triazole scaffold formed a hydrogen bond with the residue Asp690, also indicating that the replacement of the sulfonamide fragment with the triazole segment was successful. In addition, a water bridge between the nitrogen atom of the piperazine ring and His625 was observed. Moreover, there was a large unfilled space between the methyl of 1H-1,2,4-triazole and Met598 (>5.0 Å). And the methyl group was involved in hydrophobic van der Waals interactions with the DFG loop. Therefore, we envisioned that filling the space with larger alkyl might improve the potency. In contrast to TP-0903, no significant improvement in the inhibitory activity of compound w14 was observed. But the triazole scaffold improved the kinase selection for Axl, displaying more than 12, 10, or 45-fold over JAK2, IGF-1R, and ALK (Table 3).
Kinase selectivity profile of compound w14.
| Cpd. | Kinase inhibitory IC50 (nM) | ||||
|---|---|---|---|---|---|
| Axl | Aurora B | JAK2 | ALK | IGF-1R | |
| w14 | 11 | 4 | 130 | 500 | 103 |
Next, we turned our attention to modifying the triazole segment. To validate our docking hypothesis, an N-methylated 1H-1,2,4-triazole derivative m1 was synthesized. This modification led to a weak improvement in potency compared to compound w1 (IC50 = 8 nM), which encouraged our efforts toward exploring novel pharmacophores without a hydrogen bond donor in 1H-1,2,4-triazole. As shown in the fragment growth optimization in Table 2, we selected four para-substituted anilines located at the 2-position of pyrimidine due to the high binding energy interaction with the hinge region. The inhibitory activity of 2,4,5-trisubstituted pyrimidine derivatives with different five-membered heteroaromatic rings m1–m16 for Axl is summarized in Table 4.
The inhibitory activity of compounds m2 and m4 decreased slightly. And N-ethylation slightly enhanced the potency. Replacing the piperazine with piperidine resulted in decreased activity against Axl due to the decreased water solubility (m3vs.m2). When N-acetyl substituted piperazine located at the para-position of benzene, both triazole and tetrazole without hydrogen bond donors exhibited moderate inhibitory activity (m5 and m7). Upon studying the effect of alkyl substituents on the 1,2,4-triazole motif, compounds with more hydrophobic groups, such as isopropyl, were generally found to be more potent (m6vs.m5). In contrast, the N-methyl of tetrazole toward the downward position was preferred (m8vs.m7).
The interaction between the two different substituents (N-acetyl piperazine and N-methyl piperidine) with the solvent-accessible region of the kinase in 1H-1,2,4-triazole derivatives (Table 2, w18 and 19vs.w5) is similar. And this phenomenon is also reflected in the inhibitory activity of these compounds for Axl (m9vs.m2 and m10vs.m4). Likewise, compounds m11 and m8 showed similar potencies to inhibit Axl. Pleasingly, compound m12 exhibited stronger potency against Axl compared to m5. Although the activity of compound m13 with methylated N-link in R4 is insufficient, its stability and bioavailability were remarkably more pleasing (m13vs.m12). It can be rationalized that 4-(methyl(1-methylpiperidin-4-yl)amino)phenyl was chosen as a substituent at the 2-position of pyrimidine. Meanwhile, methylene-linked compound m14 exhibited moderate inhibitory activity with an IC50 value of 36 nM. As expected, with the variation from methyl to isopropyl on the triazole, the enzymatic potency and metabolic stability of compound m16 demonstrated significant improvement (IC50 = 5 nM). Replacing triazole with oxadiazol led to a loss in potency and further supported a critical role in the triazole interactions with the catalytic domain of the Axl kinase (m15).
As indicated in the docking studies, compound m16 also formed bidentate hydrogen bonds with Met623 in the hinge region of the Axl protein. And it is a structurally unique ATP-competitive small-molecule inhibitor that binds to the Axl protein in a DFG-in conformation and is designed to sequester Asp690. In addition, the ortho-N–H of pyrimidine forms an intramolecular hydrogen bond with the nitrogen of triazole. This might help the isopropyl group to extend into a hydrophobic cavity possibly composed of Leu620, Met598, Ala689, and Met598. All of these results demonstrated the rationality of our structure-based drug design.
Liver microsome stability of four diphenylpyrimidine–diamine derivatives
The metabolic stability properties of the four compounds were selected as representative examples. As shown in Table 5, m6 had a favorable half-life of 155.9 min in human liver microsomes, whereas compound m16 only exhibited a moderate half-life of 60.6 min. Compared to m12, the metabolic stability of compound m13 was preferred, especially in the human liver microsomes. And the oral bioavailability was also higher (ESI† Table S4). This is the reason that no oxy-linked compound with isopropyl substituted triazole was synthesized. Of note, all four compounds exhibited excellent metabolic stability compared to the control drug. Besides, the enzymatic inhibitory activity of m6 and m16 displayed a 3-fold improvement compared to TP-0903. Consequently, on account of their enzymatic potencies and metabolic stability properties, compounds m6 and m16 were identified as optimal diphenylpyrimidine–diamine derivatives.
In vivo pharmacokinetic study
Owing to their excellent Axl enzymatic inhibitory activity and metabolic stability, we explored the pharmacokinetic properties of compounds m6 and m16 further. The pharmacokinetic evaluations of the two compounds were conducted in Sprague–Dawley (SD) rats at a dose of 5 mg kg−1. And the results are summarized in Table 6. Compound m16 displayed extremely good oral exposure and bioavailability. In contrast, compound m6 continued to show poor exposure despite better metabolic stability. And the pharmacokinetic properties of the two compounds exhibited significant improvement compared to TP-0903. Based on the pharmacokinetic properties, together with the same Axl enzymatic inhibitory activity, compound m16 was further profiled.
Pharmacokinetic data of compounds m6, m16, and TP-0903 in rats.
| Parameter | m6 | m16 | TP-0903 | |||
|---|---|---|---|---|---|---|
| PO 5 mg kg−1 | IV 1 mg kg−1 | PO 5 mg kg−1 | IV 1 mg kg−1 | PO 5 mg kg−1 | IV 1 mg kg−1 | |
| AUClast (ng h−1 mL−1) | 661 | 497 | 1180 | 422 | 158 | 187 |
| C max (ng mL−1) | 169 | 337 | 83.9 | 56.3 | 46.8 | 89.8 |
| CL (mL h−1 kg−1) | NA | 1.95 | NA | 2.20 | NA | 4.97 |
| T 1/2 (h) | 1.32 | 1.49 | 6.45 | 6.6 | 2.94 | 1.77 |
| F% | 26.6 | 56.1 | 21.5 | |||
Biochemical kinase selectivity study
The kinase selectivity of m16 was further evaluated over a panel of 75 kinases at a single dose concentration of 1 μM (detailed information shown in Table S1†). As shown in Fig. 5, an excellent inhibition potency of more than 90% was observed across 4 kinases including Axl at this concentration. In addition to the TAM subfamily member Mer, compound m16 also significantly inhibited the kinase activity of Aurora A and Flt3. The potency of this compound against Aurora A was equal to that against Axl. In contrast to its high potency against Axl and Mer, m16 exhibited moderate inhibition against an additional 4 kinases, displaying greater than 70% (Fig. 5, KDR, Met, Ret, and Txk). Weak inhibition was observed in 6 kinases. No obvious inhibition effect was observed in the other 63 kinases (below 50%). In biochemical assays, TP-0903 significantly inhibited 10 of the kinases, including Axl and Mer. However, stronger activity against non-TAM kinases, such as Aurora A, JAK2, ALK, and ABL1, was observed.26 In contrast, compound m16 was a potent small-molecule inhibitor targeting Axl, Mer, Aurora A, and Flt3 kinases.
Fig. 5. The kinase inhibition of compound m16 was tested over a panel of 75 kinases at 1 μM. And m16 exhibited different inhibition potencies in 54 of 75 kinases.

Antiproliferative activities against 42 cancer cell lines
Next, the anti-proliferative activity of the selected inhibitor in various cancer cell lines was evaluated. Table 7 shows that compound m16 exhibited high potency against the proliferation of 4 out of 42 cancer cell lines (CC50 < 100 nM). Our compound significantly inhibited the proliferation of two hematological cancer cell lines MV-4-11 and RS4;11 with CC50 values of 66.0 and 137.5 nM, respectively. And the best cellular potency of m16 was a CC50 value of 35.9 nM against a human lymphoma cell line (SU-DHL-4). Besides, this compound exhibited excellent anti-proliferation activity in two solid cancer cells, Hep 3B2.1-7 and Fadu (CC50 < 100 nM). The anti-proliferative activity results, together with the biochemical kinase inhibitory profile data, confirmed that compound m16 can be used as a promising lead compound for the development of potent Axl inhibitors.
Cell line proliferation inhibitory profile of compound m16.
| Cpd. | Cell line | CC50 (nM) | Cell line | CC50 (nM) | Cell line | CC50 (nM) |
|---|---|---|---|---|---|---|
| m16 | HCC827 | 2047.5 | HT29 | 504.1 | U87 | 3010.7 |
| NCI-H226 | 1237.2 | SW620 | 349.1 | Caki-1 | 493.2 | |
| NCI-H1954 | 1214.7 | KYSE510 | 116.7 | Colo205 | 349.6 | |
| MSTO-211H | 451.3 | T.Tn | 118.7 | Namalwa | 494.1 | |
| A549 | 293.4 | ASPC-1 | 754.7 | Raji | 4557.5 | |
| Calu-3 | 107.0 | BXPC-3 | 381.8 | SU-DHL-4 | 35.9 | |
| Hepg2 | 351.9 | Panc-1 | N/A | OVCAR-3 | 223.9 | |
| Hep 3B2.1-7 | 81.2 | MGC803 | 102.7 | T24 | 933.0 | |
| NCI-H2052 | 1179.0 | SNU-5 | 356.5 | MOLM13 | 205.8 | |
| MCF-7 | 5194.0 | SUN16 | 148.6 | Hel92.1-7 | 456.8 | |
| MDA-MB-231 | N/A | Fadu | 79.8 | RS4;11 | 137.5 | |
| EVSA-T | 1147.9 | CAL27 | 106.51 | MV-4-11 | 66.0 | |
| A375 | 23895.9 | HeLa | 198.9 | KU812 | 233.1 | |
| 143B | 1336.0 | PC-3 | 5589.0 | K562 | N/A |
Conclusions
In summary, a series of diphenylpyrimidine–diamine derivatives were designed as novel potent Axl kinase inhibitors through SAR exploration and structure-based drug design (SBDD) efforts. The candidate was selected by using in vitro kinase potency, selectivity, cellular activity, and pharmacokinetics. Starting from a series of sulfonamide compounds, we described the SAR analysis of two series (1H-1,2,4-triazole and NH-free five-membered heteroaromatic series) of novel compounds. Therefore, a new chemotype for Axl inhibition has been identified. Compound m16 presented herein showed excellent enzymatic potency against the Axl kinase (IC50 = 5 nM, 3-fold improvement compared to TP-0903). And we observed that m16 remarkably suppressed the proliferation of 4 cancer cell lines (MV-4-11, SU-DHL-4, Hep 3B2.1-7, and Fadu). It displayed better pharmacokinetic parameters in rats compared to the control drug. Of note, this compound also exhibited higher liver microsomal stability in humans and rats. It may serve as an important lead compound for further antitumor drug discovery. Further biological evaluation will be reported in due course.
Experimental section
Unless otherwise noted, all starting materials, reagents, and solvents were commercially available and used without further purification. The intermediates and end-products were purified by flash column chromatography with silica gel 60 (200–300 mesh). Chemical reactions were monitored by thin-layer chromatography (TLC) or liquid chromatography (LC)-mass spectrometry (MS). LC–MS was performed on an Agilent HPLC1260-MS6120 system (column: Agilent-SB-C18, 2.5 mm × 30 mm, 3.5 μm). 1H NMR spectral data were obtained using CDCl3, CD3OD, or DMSO-d6 as a solvent on Bruker Avance III, 400 M, or 600 M frequency spectrometers. The coupling constant J is given in Hz. NMR data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br. = broad, m = multiplet), coupling constants and integration. The purities of all the final compounds were confirmed to be >95% as determined by HPLC on an Agilent Infinity 1260 HPLC system (column: Zorbax Eclipse Plus column, C18, 4.6 mm × 150 mm, 3.5 μm; detector: diode array detector). High-resolution ESI-MS was performed on an Agilent G6500 series Q-TOF spectrometer.
2-((5-chloro-2-((2-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (5a)
2-methyl-3,4-dihydro-1H-isoquinolin-7-amine (100.0 mg, 0.6 mmol), intermediate 4 (210.0 mg, 0.6 mmol), Pd2(dba)3 (56.0 mg, 0.06 mmol), t-butyl-XPhos (53.0 mg, 0.12 mmol), tBuONa (118.0 mg, 1.2 mmol) and tBuOH (6 mL) were added to a round flask. After degassing and refilling with N2, the mixture was refluxed for 16 h. The mixture was cooled down and concentrated in vacuo. The residue was purified by flash column chromatography (dichloromethane/1.2 M ammonia in methanol = 500/1) to give a yellow solid (149.0 mg, 0.3 mmol, 52.1%). 1H NMR (400 MHz, DMSO-d6): δ 9.42 (s, 1H), 9.25 (s, 1H), 8.51 (d, 1H, J = 6.8 Hz), 8.27 (s, 1 H), 7.83 (d, 1 H, J = 7.2 Hz), 7.70 (t, 1 H, J = 7.6 Hz), 7.38 (t, 1 H, J = 7.2 Hz), 7.32 (s, 1 H), 7.29 (s, 1 H), 6.97 (d, 1 H, J = 8.0 Hz), 3.33 (s, 2 H), 2.74 (t, 2 H, J = 5.2 Hz), 2.64 (s, 6 H), 2.57 (t, 2 H, J = 5.6 Hz), 2.33 (s, 3 H). MS (ESI): 473.2 [M + H]+. HRMS: [M + H]+ calcd for C22H26ClN6O2S+, 473.1418; found, 473.1416.
2-((5-chloro-2-((2-((1-methylpiperidin-4-yl)methyl)-[1,2,4]triazolo[1,5-a]pyridin-6-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (5b)
To a mixture of 5b–10 (140.0 mg, 0.55 mmol) and compound 4 (260.0 mg, 0.75 mmol) in tBuOH (8 mL) were added Pd2(dba)3 (46 mg, 0.05 mmol), Xantphos (58 mg, 0.1 mmol) and tBuONa (0.2 g, 2 mmol). After degassing and refilling with N2, the mixture was refluxed for 16 h. The mixture was cooled down and concentrated in vacuo. The residue was purified by flash column chromatography (dichloromethane/1.2 M ammonia in methanol = 100/1) to give a yellow solid (50.0 mg, 0.09 mmol, 18.1%). 1H NMR (400 MHz, DMSO-d6) δ 9.80 (s, 1H), 9.35 (s, 1 H), 9.30 (s, 1 H), 8.53 (s, 1H), 8.38 (s, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.79–7.62 (m, 3H), 7.39 (t, J = 7.6 Hz, 1H), 2.88 (d, J = 10.9 Hz, 2H), 2.72 (d, J = 6.9 Hz, 2H), 2.66 (s, 6H), 2.27 (s, 3H), 2.20–2.01 (m, 2H), 1.83 (s, 1H), 1.71 (d, J = 12.8 Hz, 2H), 1.45–1.27 (m, 2H). MS (ESI): 556.2 [M + H]+. HRMS: [M + H]+ calcd for C25H31ClN9O2S+, 556.1932; found, 556.2021.
2-((5-chloro-2-((4-(5-methyloctahydropyrrolo[3,4-c]pyrrole-2-arbonyl)phenyl)amino)-pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (5c)
To a mixture of (4-aminophenyl)-(2-methyl-1,3,3a,4,6,6a-hexahydropyrrolo[3,4-c]pyrrol-5-yl)methanone (100 mg, 0.41 mmol) and compound 4 (142.0 mg, 0.4 mmol) in tBuOH (6 mL) were added Pd2(dba)3 (38.0 mg, 0.04 mmol), t-butyl XPhos (36.0 mg, 0.08 mmol) and tBuONa (80.0 mg, 0.82 mmol). After degassing and refilling with N2, the mixture was refluxed for 24 h. The mixture was cooled down and concentrated in vacuo. The residue was purified by flash column chromatography (dichloromethane/1.2 M ammonia in methanol = 100/1) to give a yellow solid 5c (167.0 mg, 0.4 mmol, 73.7%). 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1 H), 9.32 (s, 1 H), 8.53 (d, J = 8.0 Hz, 1 H), 8.33 (s, 1 H), 7.84 (dd, J = 7.9, 1.1 Hz, 1 H), 7.77 (t, J = 7.8 Hz, 1 H), 7.67 (d, J = 8.5 Hz, 2 H), 7.41 (d, J = 7.4 Hz, 1 H), 7.37 (d, J = 8.6 Hz, 2 H), 3.71 (s, 2 H), 3.28 (s, 2 H), 2.77 (d, J = 2.0 Hz, 2 H), 2.65 (s, 5 H), 2.40 (m, 4 H), 2.22 (s, 3 H). MS (ESI): 556.3 [M + H]+. HRMS: [M + H]+ calcd for C26H31ClN7O3S+, 556.1819; found, 556.1888.
2-((5-chloro-2-((2-acetyl-1,2,3,4-tetrahydroisoquinolin-7-yl)amino)pyrimidin-4-yl)amino)-N,N-dimethylbenzenesulfonamide (5d)
To a suspension of 1-(7-amino-3,4-dihydro-1H-isoquinolin-2-yl)ethanone (90.2 mg, 0.47 mmol) and intermediate 4 (165.2 mg, 0.47 mmol) in 1-butanol (1 mL) was added cat. HCl (10.0 μL). The reaction mixture was stirred at 130 °C for 3 h under microwave conditions. After concentration, the residue was purified by flash column chromatography with eluent (MeOH/DCM = 1/30) to give the title compound 5d as a white solid (45.6 mg, 0.09 mmol, 19.2%). 1H NMR (400 MHz, DMSO-d6) δ 9.52 (d, J = 14.9 Hz, 1H), 9.26 (d, J = 7.6 Hz, 1H), 8.50 (s, 1H), 8.29 (s, 1H), 7.96–7.66 (m, 2H), 7.41 (ddd, J = 27.5, 24.1, 11.8 Hz, 3H), 7.05 (d, J = 8.3 Hz, 1H), 4.46 (d, J = 4.4 Hz, 2H), 3.64 (t, J = 5.9 Hz, 2H), 2.83–2.66 (m, 2H), 2.70–2.52 (m, 6H), 2.08 (d, J = 4.2 Hz, 3H). MS (ESI): 501.1 [M + H]+. HRMS: [M + H]+ calcd for C23H25ClN6O3S+, 501.1397; found, 501.1490.
(4-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)(5-methylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)methanone (w1)
To a solution of 10 (131.0 mg, 0.41 mmol) in 2-methylpropan-2-ol (6.0 mL) were added (4-aminophenyl)(5-methylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)methanone (100.0 mg, 0.41 mmol), Pd2(dba)3 (38.0 mg, 41.0 μmol), t-butyl XPhos (36.0 mg, 82.0 μmol) and tBuONa (80.0 mg, 0.82 mmol). The reaction was degassed with N2. After being refluxed overnight, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography with eluent (dichloromethane/1.2 M ammonia in methanol = 100/1) to give the title compound as a yellow solid (28 mg, 53.3 μmol, 13.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.16 (bs, 1H), 11.70 (s, 1H), 9.71 (s, 1H), 8.84–8.83 (d, J = 7.9 Hz, 1H), 8.28 (s, 1H), 8.14–8.12 (d, J = 7.5 Hz, 1H), 7.77–7.75 (d, J = 8.3 Hz, 2H), 7.44–7.42 (m, 3H), 7.22–7.18 (t, J = 7.4 Hz, 1H), 3.72 (m, 2H), 3.51 (m, 2H), 2.84 (s, 2H), 2.65 (m, 2H), 2.50 (m, 2H), 2.47 (s, 3H), 2.34 (s, 3H). MS (ESI): 530.2 [M + H]+. HRMS: [M + H]+ calcd for C27H29ClN9O+, 530.2105; found, 530.2111.
The same process was used for the synthesis and characterization of w2, w4, w7, w10–17, and w19.
(4-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)(5-methyl-2,5-diazabicyclo[2.2.1]heptan-2-yl)methanone (w2)
A yellow solid (150.0 mg, 288.1 μmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 14.39 (bs, 1H), 11.73 (s, 1H), 9.77 (s, 1H), 8.85–8.84 (d, J = 8.2 Hz, 1H), 8.28 (s, 1H), 8.15–8.13 (d, J = 7.8 Hz, 1H), 7.80–7.78 (d, J = 6.5 Hz, 2H), 7.52–7.50 (d, J = 6.9 Hz, 1H), 7.44–7.40 (m, 2H), 7.22–7.18 (t, J = 7.5 Hz, 1H), 3.63–3.50 (m, 4H), 3.50 (s, 3H), 2.90 (s, 2H), 2.51 (s, 3H), 2.47 (s, 3H), 1.86 (m, 1H), 1.72 (m, 1H). MS (ESI): 516.5 [M + H]+. HRMS: [M + H]+ calcd for C26H27ClN9O+, 516.1949; found, 516.1945.
1-(5-(4-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl)ethanone (w4)
A yellow solid (70.0 mg, 0.13 mmol, 32.0%). 1H NMR (400 MHz, DMSO-d6) δ δ 14.09 (s, 1H), 11.58 (s, 1H), 9.09 (s, 1H), 8.89 (s, 1H), 8.14–8.09 (m, 2H), 7.43–7.41 (d, J = 8.5 Hz, 2H), 7.36–7.32 (m, 1H), 7.16–7.12 (m, 1H), 6.55–6.52 (d, J = 8.8 Hz, 2H), 3.77–3.72 (m, 1H), 3.62–3.57 (m, 1H), 3.46–3.42 (m, 3H), 3.31–3.25 (m, 1H), 3.19–3.12 (m, 2H), 3.09–3.05 (m, 1H), 3.02–2.97 (m, 1H), 2.47 (s, 3H), 1.95 (s, 3H). MS (ESI): 530.2 [M + H]+. HRMS: [M + H]+ calcd for C27H29ClN9O+, 530.2105; found, 530.2131.
5-Chloro-N2-(4-(6,7-dihydropyrazolo[1,5-a]pyrazin-5(4H)-yl)phenyl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w7)
A yellow solid (101.0 mg, 0.2 mmol, 43.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.09 (s, 1H), 11.59 (s, 1H), 9.26 (s, 1H), 8.85–8.84 (d, J = 7.1 Hz, 1H), 8.19 (s, 1H), 8.15–8.13 (d, J = 7.4 Hz, 1H), 7.55–7.53 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 1.2 Hz, 1H), 7.36–7.32 (t, J = 7.4 Hz, 1H), 7.17–7.13 (t, J = 7.6 Hz, 1H), 7.05–7.03 (d, J = 8.9 Hz, 2H), 6.15 (s, 1H), 4.42 (s, 2H), 4.20–4.17 (t, J = 5.4 Hz, 2H), 3.74–3.72 (t, J = 5.4 Hz, 2H), 2.49 (s, 3H). MS (ESI): 499.2 [M + H]+. HRMS: [M + H]+ calcd for C25H24ClN10+, 499.1796; found, 499.1798.
5-Chloro-N2-(2-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w10)
A yellow solid (106.0 mg, 0.23 mmol, 39.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.14 (s, 1H), 11.67 (s, 1H), 9.36 (s, 1H), 8.83 (s, 1H), 8.22 (s, 1H), 8.13–8.12 (d, J = 7.6 Hz, 1H), 7.39–7.37 (m, 3H), 7.19–7.17 (t, J = 7.5 Hz, 1H), 7.02–7.01 (d, J = 8.1 Hz, 1H), 3.42 (s, 2H), 2.78–2.76 (t, J = 5.7 Hz, 2H), 2.60–2.58 (t, J = 5.8 Hz, 2H), 2.47 (s, 3H), 2.34 (s, 3H). MS (ESI): 447.3 [M + H]+. HRMS: [M + H]+ calcd for C23H24ClN8+, 447.1734; found, 447.1759.
5-Chloro-N2-(2-methyl-1,2,3,4-tetrahydroisoquinolin-7-yl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w11)
A beige solid (384.1 mg, 1.1 mmol, 82.5%). 1H NMR (400 MHz, DMSO-d6) δ 14.09 (s, 1H), 11.59 (d, J = 8.0 Hz, 1H), 9.45 (d, J = 12.5 Hz, 1H), 8.79 (t, J = 8.1 Hz, 1H), 8.24 (s, 1H), 8.16 (d, J = 7.5 Hz, 1H), 7.59 (d, J = 16.8 Hz, 1H), 7.42 (dd, J = 14.2, 6.5 Hz, 2H), 7.09 (d, J = 8.3 Hz, 1H), 4.55 (d, J = 7.2 Hz, 2H), 3.66 (t, J = 5.8 Hz, 2H), 2.82 (t, J = 5.6 Hz, 1H), 2.71 (t, J = 5.5 Hz, 1H), 2.49 (s, 3H), 2.09 (d, J = 3.7 Hz, 3H).
MS (ESI): 475.2 [M + H]+. HRMS: [M + H]+ calcd for C24H24ClN8O+, 475.1683; found, 475.3069.
6-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)-2-cyclopropylisoindolin-1-one (w12)
A yellow solid w12 (10.6 mg, 0.02 mmol, 8.3%). 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 9.67 (s, 1H), 8.91 (d, J = 8.3 Hz, 1H), 8.30 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 8.05 (s, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 8.1 Hz, 2H), 7.18 (t, J = 7.3 Hz, 2H), 4.34 (s, 2H), 2.48 (s, 3H), 2.05–1.94 (m, 2H), 0.88–0.85 (m, 2H), 0.81–0.77 (m, 2H). MS (ESI): 473.2 [M + H]+. HRMS: [M + H]+ calcd for C24H22ClN8O+, 473.1527; found, 473.1617.
5-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)-2-cyclopropylisoindolin-1-one (w13)
A yellow solid (10.0 mg, 0.02 mmol, 4.0%). 1H NMR (400 MHz, DMSO-d6) δ 11.62 (s, 1H), 9.81 (s, 1H), 8.76–8.74 (d, J = 8.2 Hz, 1H), 8.28 (s, 1H), 8.12–8.10 (d, J = 7.3 Hz, 1H), 7.99 (s, 1H), 7.69–7.66 (d, J = 8.4 Hz, 1H), 7.55–7.53 (d, J = 8.3 Hz, 1H), 7.48–7.44 (t, J = 7.7 Hz, 1H), 7.24–7.21 (t, J = 7.5 Hz, 1H), 4.32 (s, 2H), 2.92–2.86 (m, 1H), 2.46 (s, 3H), 0.81–0.78 (m, 4H). MS (ESI): 473.1 [M + H]+. HRMS: [M + H]+ calcd for C24H22ClN8O+, 473.1527; found, 473.1569.
5-Chloro-N4-(2-(3-methyl-1H-1,2,4-triazol-5-yl)phenyl)-N2-(4-((4-methylpiperazin-1-yl)methyl)phenyl)pyrimidine-2,4-diamine (w14)
A yellow solid (38.4 mg, 76.8 μmol, 2.5%). 1H NMR (400 MHz, DMSO-d6) δ 11.70 (s, 1H), 9.64 (s, 1H), 8.83 (d, J = 7.5 Hz, 1H), 8.27 (s, 1H), 8.13 (d, J = 8.1 Hz, 1H), 7.74 (d, J = 8.0 Hz, 2H), 7.51–7.15 (m, 5H), 3.98 (s, 2H), 3.28–3.09 (m, 4H), 2.80 (s, 3H), 2.50–2.43 (m, 7H). MS (ESI): 490.4 [M + H]+. HRMS: [M + H]+ calcd for C25H29ClN9+, 490.2156; found, 490.2233.
4-((5-chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)-N-cyclopropylbenzamide (w15)
A yellow solid (101.0 mg, 0.22 mmol, 39.0%). 1H NMR (400 MHz, DMSO) δ 14.11 (s, 1H), 11.66 (s, 1H), 9.74 (s, 1H), 8.85–8.84 (d, J = 6.6 Hz, 1H), 8.30 (s, 1H), 8.27–8.26 (d, J = 3.8 Hz, 1H), 8.16 (s, 1H), 7.79–7.74 (m, 3H), 7.48–7.44 (t, J = 7.6 Hz, 1H), 7.24–7.20(t, J = 7.3 Hz, 1H), 2.86–2.80 (m, 1H), 2.48 (s, 3H), 0.72–0.67 (m, 2H), 0.59–0.55 (m, 2H). MS (ESI): 461.3 [M + H]+. HRMS: [M + H]+ calcd for C23H22ClN8O+, 461.1527; found, 461.1597.
5-((5-Chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)-N-cyclopropylpicolinamide (w16)
A yellow solid (68.0 mg, 33.6 μmol, 6.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.11 (s, 1H), 11.71 (s, 1H), 9.98 (s, 1H), 8.82 (d, J = 2.1 Hz, 2H), 8.53–8.52 (d, J = 4.8 Hz, 1H), 8.44–8.42 (dd, J = 8.5, 1.9 Hz, 1H), 8.34 (s, 1H), 8.18–8.16 (d, J = 7.4 Hz, 1H), 7.95–7.92 (d, J = 8.6 Hz, 1H), 7.48–7.44 (t, J = 8.0 Hz, 1H), 7.23–7.20 (t, J = 7.4 Hz, 1H), 2.92–2.86 (m, 1H), 2.50 (m, 3H), 0.70–0.66 (m, 4H). MS (ESI): 462.1 [M + H]+. HRMS: [M + H]+ calcd for C22H21ClN9O+, 462.1479; found, 462.1574.
4-((5-Chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)-N-cyclopentylbenzamide (w17)
A yellow solid (632.0 mg, 1.22 mmol, 21.7%). 1H NMR (400 MHz, DMSO-d6) δ 11.73 (s, 1H), 9.74 (s, 1H), 8.85 (d, J = 8.2 Hz, 1H), 8.30 (s, 1H), 8.13 (d, J = 7.6 Hz, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.84–7.72 (m, 4H), 7.46 (t, J = 7.8 Hz, 1H), 7.22 (t, J = 7.4 Hz, 1H), 4.22 (dd, J = 14.1, 7.1 Hz, 1H), 2.47 (s, 3H), 1.87 (dd, J = 12.9, 7.4 Hz, 2H), 1.76–1.63 (m, 2H), 1.60–1.46 (m, 4H). MS (ESI): 489.1 [M + H]+. HRMS: [M + H]+ calcd for C26H26ClN8O+, 489.1840; found, 489.1846.
5-Chloro-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyrimidine-2,4-diamine (w19)
A yellow solid (1.02 g, 2.08 mmol, 45.2%). 1H NMR (400 MHz, DMSO-d6) 14.13 (s, 1H), 11.67 (s, 1H), 9.28 (s, 1H), 8.86–8.84 (d, J = 5.7 Hz, 1H), 8.19 (s, 1H), 8.12–8.10 (d, J = 7.4 Hz, 1H), 7.54–7.52 (d, J = 8.8 Hz, 2H), 7.39–7.35 (t, J = 7.7 Hz, 1H), 7.18–7.14 (t, J = 7.3 Hz, 1H), 6.91–6.89 (d, J = 8.9 Hz, 2H), 4.33–4.26 (m, 1H), 2.64–2.61 (m, 2H), 2.47 (s, 3H), 2.19 (s, 3H), 2.15 (m, 2H), 1.94–1.91 (m, 2H), 1.67–1.59 (m, 2H). MS (ESI): 491.1 [M + H]+. HRMS: [M + H]+ calcd for C25H28ClN8O+, 491.1996; found, 491.2062.
(4-((5-Chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)(4-methylpiperazin-1-yl)methanone (w3)
To a solution of 10 (139 mg, 0.43 mmol) in isopropyl alcohol (4.0 mL) were added (4-aminophenyl)-(4-methylpiperazin-1-yl)methanone (100.0 mg, 0.43 mmol) and HCl/dioxane (4.0 mol L−1, 0.1 mL, 0.4 mmol). After being stirred at 130 °C in a microwave reactor for 4 h, the reaction mixture was diluted with methanol (10.0 mL) and neutralized with a saturated aqueous solution of NaHCO3 (6.0 mL). The crude was collected through filtration. The crude was purified by silica gel column chromatography with eluent (dichloromethane/1.2 M ammonia in methanol = 200/1) to give the title compound as a yellow solid (51.0 mg, 98.9 μmol, 23.0%). 1H NMR (400 MHz, DMSO-d6) δ 12.15 (s, 1H), 9.69 (s, 1H), 8.84–8.82 (d, J = 8.3 Hz, 1H), 8.27 (s, 1H), 8.14–8.12 (d, J = 8.0 Hz, 1H), 7.79–7.77 (d, J = 8.4 Hz, 2H), 7.40–7.32 (m, 3H), 7.19–7.15 (t, J = 7.6 Hz, 1H), 3.50 (m, 4H), 2.43 (s, 3H), 2.33 (s, 4H), 2.21 (s, 3H). MS (ESI): 504.3 [M + H]+. HRMS: [M + H]+ calcd for C25H27ClN9O+, 504.1949; found, 504.2020.
The same process was used for the synthesis and characterization of w5 and 6, w8 and 9, w18, and w20.
1-(4-(4-((5-Chloro-4-((2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethanone (w5)
A yellow solid (17.0 mg, 34.4 μmol, 8.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.13 (s, 1H), 11.65 (s, 1H), 9.25 (s, 1H), 8.88 (s, 1H), 8.18 (s, 1H), 8.12 (s, 1H), 7.53–7.51 (d, J = 7.0 Hz, 2H), 7.39 (m, 1H), 7.17 (m, 1H), 6.94–6.92 (d, J = 7.1 Hz, 2H), 3.59 (s, 4H), 3.10 (s, 2H), 3.03 (s, 2H), 2.47 (s, 3H), 2.05 (s, 3H). MS (ESI): 504.1 [M + H]+. HRMS: [M + H]+ calcd for C25H27ClN9O+, 504.1949; found, 504.2003.
5-Chloro-N2-(4-(4-(dimethylamino)piperidin-1-yl)phenyl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w6)
A yellow solid (20.0 mg, 40.5 μmol, 9.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.09 (s, 1H), 11.70 (s, 1H), 9.20 (s, 1H), 8.89–8.87 (m, 1H), 8.17 (s, 1H), 8.12–8.10 (d, J = 7.1 Hz, 1H), 7.48–7.46 (d, J = 8.7 Hz, 2H), 7.38–7.35 (t, J = 7.4 Hz, 1H), 7.17–7.14 (t, J = 7.6 Hz, 1H), 6.91–6.89 (d, J = 8.9 Hz, 2H), 3.65–3.62 (d, J = 12.1 Hz, 2H), 3.43–3.40 (m, 1H), 2.64–2.59 (t, J = 11.3 Hz, 3H), 2.47 (s, 3H), 2.20 (s, 6H), 1.85–1.83 (d, J = 10.9 Hz, 2H), 1.54–1.45 (m, 2H). MS (ESI): 504.1 [M + H]+. HRMS: [M + H]+ calcd for C26H31ClN9+, 504.2313; found, 504.2375.
5-Chloro-N4-(2-(3-methyl-1H-1,2,4-triazol-5-yl)phenyl)-N2-(4-morpholinophenyl)pyrimidine-2,4-diamine (w8)
A yellow solid (30.4 mg, 64.5 μmol, 8.3%). 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H), 9.53 (d, J = 0.6 Hz, 1H), 8.81 (s, 2 H), 8.22 (s, 1 H), 8.11 (d, J = 7.6 Hz, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.38 (t, J = 7.6 Hz, 1 H), 7.19 (t, J = 7.4 Hz, 1 H), 7.08–6.97 (m, 2 H), 3.87–3.69 (m, 4 H), 3.14 (s, 4H), 2.47 (s, 3 H). MS (ESI): 463.1 [M + H]+. HRMS: [M + H]+ calcd for C23H24ClN8O+, 463.1683; found, 463.1770.
5-Chloro-N2-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w9)
A yellow solid (10 mg, 38.7 μmol, 9.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.12 (s, 1H), 11.66 (s, 1H), 8.70 (s, 1H), 8.16 (s, 1H), 8.11 (s, 1H), 8.08–8.06 (d, J = 6.9 Hz, 1H), 7.42–7.40 (d, J = 8.5 Hz, 1H), 7.24–7.20 (m, 1H), 7.12–7.08 (t, J = 7.6 Hz, 1H), 6.66 (d, J = 1.6 Hz, 1H), 6.52–6.49 (dd, J = 8.8, 2.0 Hz, 1H), 3.77 (s, 3H), 3.17 (s, 4H), 2.51 (m, 4H), 2.46 (s, 3H), 2.25 (s, 3H). MS (ESI): 506.3 [M + H]+. HRMS: [M + H]+ calcd for C25H29ClN9O+, 506.2105; found, 506.2169.
5-Chloro-N2-(4-(methyl(1-methylpiperidin-4-yl)amino)phenyl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w18)
A yellow solid (61.1 mg, 0.12 mmol, 18.0%). 1H NMR (400 MHz, DMSO-d6) δ 14.12 (s, 1H), 11.66 (s, 1H), 9.11 (s, 1H), 8.89 (s, 1H), 8.15 (s, 1H), 8.11–8.09 (d, J = 7.6 Hz, 1H), 7.43–7.40 (d, J = 8.6 Hz, 2H), 7.34–7.31 (t, J = 7.7 Hz, 1H), 7.16–7.12 (t, J = 7.5 Hz, 1H), 6.80–6.78 (d, J = 9.0 Hz, 2H), 3.54–3.48 (m, 1H), 2.87–2.85 (d, J = 10.4 Hz, 2H), 2.70 (s, 3H), 2.47 (s, 3H), 2.20 (s, 3H), 2.06–2.00 (t, J = 11.4 Hz, 2H), 1.74–1.67 (m, 2H), 1.60–1.57 (m, 2H). MS (ESI): 504.6 [M + H]+. HRMS: [M + H]+ calcd for C26H31ClN9+, 504.2313; found, 504.2395.
5-Chloro-N2-(3-fluoro-4-((1-methylpiperidin-4-yl)oxy)phenyl)-N4-(2-(5-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (w20)
A pink solid (146.0 mg, 0.26 mmol, 83.7%). 1H NMR (400 MHz, CDCl3) δ 13.47 (s, 1H), 11.44(s, 1 H), 8.58 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 7.5 Hz, 1 H), 7.93 (s, 1 H), 7.74 (s, 1 H), 7.54 (dd, J = 13.6, 2.0 Hz, 1 H), 7.23 (d, J = 7.7 Hz, 1 H), 6.99 (t, J = 7.5 Hz, 2 H), 6.79 (t, J = 9.0 Hz, 1 H), 4.07 (s, 1 H), 2.63 (m, 2 H), 2.38 (s, 3 H), 2.19(s, 5 H), 1.93–1.85 (m, 2 H), 1.74 (m, 2 H). 19F NMR (CDCl3) δ 130.82; MS (ESI): 509.2 [M + H]+. HRMS: [M + H]+ calcd for C25H27ClFN8O+, 509.1902; found, 502.1988.
2,5-Dichloro-N-(2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidin-4-amine (m1–1)
To a suspension of 10 (2.0 g, 6.2 mmol) in THF (30 mL) was added potassium tert-butoxide (1.14 g, 10.0 mmol) and the mixture was stirred at rt for 0.5 h. To the reaction mixture was added iodomethane (0.7 mL, 10.0 mmol). After being stirred at rt for 4 h, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography with eluent (petroleum ether/ethyl acetate = 3/1) to give the title compound as a yellow solid (1.14 g, 3.4 mmol, 55.1%). 1H NMR (400 MHz, CDCl3) δ 11.83 (s, 1H), 8.80–8.78 (d, J = 8.5 Hz, 1H), 8.22–8.20 (m, 2H), 7.50–7.46 (t, J = 7.9 Hz, 1H), 7.22–7.19 (t, J = 7.6 Hz, 1H), 3.92 (s, 3H), 2.55 (s, 3H). MS (ESI): 355.2 [M + H]+.
(4-((5-Chloro-4-((2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)(4-methylpiperazin-1-yl)methanone (m1)
To a solution of m1–1 (145.0 mg, 0.43 mmol) in 2-methylpropan-2-ol (6.0 mL) were added (4-aminophenyl)-(4-methylpiperazin-1-yl)methanone (100.0 mg, 0.43 mmol), Pd2(dba)3 (40.0 mg, 0.043 mmol), t-butyl XPhos (38.0 mg, 0.087 mmol) and tBuONa (85.0 mg, 0.87 mmol). The reaction was degassed with nitrogen. After being stirred at reflux overnight, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography with eluent (dichloromethane/methanol = 100/1) to give the title compound as a yellow solid (118.0 mg, 0.23 mmol, 53.0%). 1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 1H), 9.70 (s, 1H), 8.83–8.81 (d, J = 8.3 Hz, 1H), 8.28 (s, 1H), 8.13–8.11 (m, 1H), 7.77–7.75 (d, J = 8.5 Hz, 2H), 7.44–7.40 (t, J = 7.8 Hz, 1H), 7.34–7.32 (d, J = 8.5 Hz, 2H), 7.21–7.17 (t, J = 7.5 Hz, 1H), 3.89 (s, 3H), 3.51–3.48 (m, 4H), 2.51 (s, 3H), 2.32 (m, 4H), 2.20 (s, 3H). MS (ESI): 518.5 [M + H]+. HRMS: [M + H]+ calcd for C26H29ClN9O+, 518.2105; found, 518.2139.
The similar Buchwald–Hartwig coupling process was used for the synthesis and characterization of m2–4, m9 and 10, and m14 and 15.
1-(4-(4-((5-Chloro-4-((2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethanone (m2)
A yellow solid (66.0 mg, 124.7 μmol, 29.0%). 1H NMR (400 MHz, DMSO-d6) δ 11.43 (s, 1H), 9.24 (s, 1H), 8.87–8.85 (d, J = 5.9 Hz, 1H), 8.18 (s, 1H), 8.11–8.09 (d, J = 7.8 Hz, 1H), 7.52–7.50 (d, J = 8.5 Hz, 2H), 7.39–7.35 (t, J = 7.7 Hz, 1H), 7.17–7.13 (t, J = 7.3 Hz, 1H), 6.94–6.92 (d, J = 8.8 Hz, 2H), 3.89 (s, 3H), 3.60–3.59 (m, 4H), 3.10 (m, 2H), 3.04–3.02 (m, 2H), 2.51 (s, 3H), 2.05 (s, 3H). MS (ESI): 518.2 [M + H]+. HRMS: [M + H]+ calcd for C26H29ClN9O+, 518.2105; found, 518.2127.
1-(4-(4-((5-Chloro-4-((2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperidin-1-yl)ethan-1-one (m3)
A yellow solid (8.1 mg, 16.0 μmol, 4.1%). 1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 1H), 9.41 (s, 1H), 8.87–8.85 (d, J = 8.4 Hz, 1H), 8.23 (s, 1H), 8.12–8.10 (d, J = 7.8 Hz, 1H), 7.61–7.58 (d, J = 8.4 Hz, 2H), 7.41–7.37 (t, J = 7.4 Hz, 1H), 7.19–7.16 (t, J = 6.6 Hz, 3H), 4.55–4.52 (d, J = 12.4 Hz, 1H), 3.94–3.89 (m, 1H), 3.89 (s, 3H), 3.16–3.09 (m, 1H), 2.76–2.68 (m, 1H), 2.62–2.51 (m, 1H), 2.51 (s, 3H), 2.04 (s, 3H), 1.82–1.75 (m, 2H), 1.63–1.56 (m, 1H), 1.48–1.41 (m, 1H). MS (ESI): 517.3 [M + H]+. HRMS: [M + H]+ calcd for C27H30ClN8O+, 517.2153; found, 517.2151.
1-(4-(4-((5-Chloro-4-((2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethanone (m4)
A yellow solid (103.1 mg, 0.19 mmol, 28.9%). 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.25 (s, 1H), 8.86–8.84 (d, J = 7.5 Hz, 1H), 8.19 (s, 1H), 8.14–8.12 (d, J = 7.8 Hz, 1H), 7.53–7.50 (d, J = 8.8 Hz, 2H), 7.39–7.36 (t, J = 7.6 Hz, 1H), 7.17–7.13 (t, J = 7.7 Hz, 1H), 6.94–6.92 (d, J = 8.9 Hz, 2H), 4.26–4.21 (dd, J = 14.4, 7.2 Hz, 2H), 3.60–3.59 (m, 4H), 3.11–3.08 (m, 2H), 3.04–3.02 (m, 2H), 2.53 (s, 3H), 2.05 (s, 3H), 1.42 (t, J = 7.4 Hz, 3H). MS (ESI): 532.3 [M + H]+. HRMS: [M + H]+ calcd for C27H31ClN9O+, 532.2262; found, 532.2274.
5-Chloro-N4-(2-(1,5-dimethyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyrimidine-2,4-diamine (m9)
A yellow solid (95.1 mg, 0.19 mmol, 41.1%). 1H NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H), 9.31 (s, 1H), 8.85–8.83 (d, J = 6.6 Hz, 1H), 8.19 (s, 1H), 8.11–8.09 (d, J = 7.8 Hz, 1H), 7.56–7.54 (d, J = 8.7 Hz, 2H), 7.38–7.34 (t, J = 7.8 Hz, 1H), 7.17–7.13 (t, J = 7.5 Hz, 1H), 6.95–6.93 (d, J = 8.8 Hz, 2H), 4.45 (s, 1H), 3.88 (s, 3H), 2.97 (m, 2H), 2.70 (s, 2H), 2.51 (s, 3H), 2.48 (s, 3H), 2.07–2.03 (m, 2H), 1.82–1.80 (m, 2H). MS (ESI): 505.2 [M + H]+. HRMS: [M + H]+ calcd for C26H30ClN8O+, 505.2153; found, 505.2254.
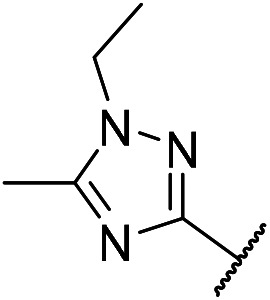
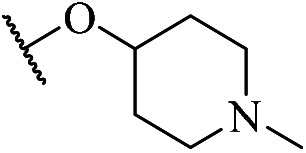
5-Chloro-N4-(2-(1-ethyl-5-methyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-((1-methylpiperidin-4-l)oxy)phenyl)pyrimidine-2,4-diamine (m10)
A yellow solid (85.2 mg, 166.3 μmol, 24.1%). 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.28 (s, 1H), 8.83–8.82 (d, J = 7.0 Hz, 1H), 8.20 (s, 1H), 8.14–8.12 (dd, J = 7.8, 1.1 Hz, 1H), 7.54–7.52 (d, J = 8.8 Hz, 2H), 7.38–7.34 (t, J = 7.7 Hz, 1H), 7.17–7.14 (t, J = 7.5 Hz, 1H), 6.92–6.90 (d, J = 8.9 Hz, 2H), 4.37–4.31 (m, 1H), 4.26–4.21 (q, J = 7.2 Hz, 2H), 2.76–2.73 (m, 2H), 2.53 (s, 3H), 2.37–2.331 (m, 2H), 2.29 (s, 3H), 1.98–1.94 (m, 2H), 1.72–1.65 (m, 2H), 1.44–1.40 (t, J = 7.2 Hz, 3H). MS (ESI): 519.3 [M + H]+. HRMS: [M + H]+ calcd for C27H32ClN8O+, 519.2309; found, 519.2403.
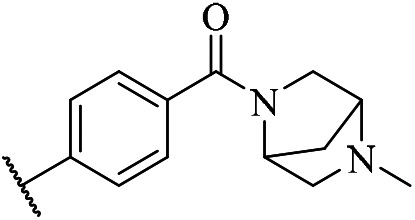
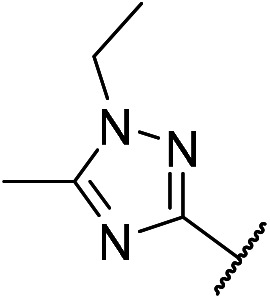
5-Chloro-N4-(2-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-((1-methylpiperidin-4-yl)methyl)phenyl)pyrimidine-2,4-diamine (m14)
A yellow solid (135.6 mg, 0.28 mmol, 28.0%). 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.49 (s, 1H), 8.84 (d, J = 8.5 Hz, 1H), 8.75 (s, 1H), 8.24 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.62 (d, J = 8.3 Hz, 2H), 7.40 (t, J = 7.8 Hz, 1H), 7.20 (t, J = 7.7 Hz, 3H), 4.00 (s, 3H), 3.45 (s, 2H), 3.31–3.18 (m, 4H), 2.38 (ddd, J = 18.9, 14.2, 8.3 Hz, 4H), 2.29 (s, 3H). MS (ESI): 490.3 [M + H]+. HRMS: [M + H]+ calcd for C25H29ClN9+, 490.2156; found, 490.2238.
5-Chloro-N4-(2-(5-methyl-1,3,4-oxadiazol-2-yl)phenyl)-N2-(4-((4-methylpiperazin-1-yl)methyl)phenyl)pyrimidine-2,4-diamine (m15)
A white solid m15 (7.9 mg, 15.0 μmol, 6.1%). 1H NMR (400 MHz, CDCl3) δ 8.88 (d, J = 8.5 Hz, 1H), 8.15 (s, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.58–7.45 (m, 3H), 7.30 (dd, J = 8.2, 1.1 Hz, 1H), 7.19 (dd, J = 16.4, 9.1 Hz, 2H), 3.60 (s, 2H), 2.66 (s, 3H), 2.58 (s, 3H), 2.39–2.16 (m, 4H), 2.09–1.95 (m, 4H). MS (ESI): 491.3 [M + H]+. HRMS: [M + H]+ calcd for C25H28ClN8O+, 491.1960; found, 491.1966.
1-(4-(4-((5-Chloro-4-((2-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethan-1-one (m5)
An aqueous solution of HCl (0.01 mL, 6 mol L−1, 0.06 mmol) was added into a solution of 24–m5 (400 mg, 1.245 mmol) and 1-[4-(4-aminophenyl)piperazin-1-yl]-ethanone (275 mg, 1.2541 mmol) in nBuOH (6.0 mL). The mixture was stirred under microwave conditions for 6 h at 120 °C. The solvent was removed in vacuo and purified by flash column chromatography with eluent (dichloromethane/methanol = 100/1–10/1) to give a yellow solid m5 (326.1 mg, 0.64 mmol, 51.4%). 1H NMR (600 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.26 (s, 1H), 8.86 (s, 1H), 8.73 (s, 1H), 8.20 (s, 1H), 8.16 (dd, J = 7.8, 1.5 Hz, 1H), 7.52 (d, J = 8.8 Hz, 2H), 7.41 (t, J = 7.4 Hz, 1H), 7.22–7.15 (m, 1H), 4.00 (s, 3H), 3.60 (dd, J = 10.1, 8.2 Hz, 4H), 3.14–3.08 (m, 2H), 3.06–3.00 (m, 2H), 2.06 (s, 3H). MS (ESI): 504.1 [M + H]+. HRMS: [M + H]+ calcd for C25H27ClN9O+, 504.1949; found, 504.2028.
The synthesis of compounds m6–8, m11–13, and m16 were catalyzed by hydrochloric acid under microwave conditions.
1-(4-(4-((5-Chloro-4-((2-(1-isopropyl-1H-1,2,4-triazol-3-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethan-1-one (m6)
A pink solid (0.2 g, 0.38 mmol, 53.0%). 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 1H), 9.27 (s, 1H), 8.80 (s, 2H), 8.20 (q, J = 1.9 Hz, 2H), 7.52 (d, J = 8.8 Hz, 2H), 7.40 (t, J = 7.4 Hz, 1H), 7.21–7.17 (m, 1H), 6.93 (d, J = 9.0 Hz, 2H), 4.83–4.71 (m, 1H), 3.59 (dd, J = 10.5, 7.4 Hz, 4H), 3.11–3.08 (m, 2H), 3.05–3.01 (m, 2H), 2.05 (s, 3H), 1.55 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ (ppm) 168.77, 160.44, 158.47, 155.82, 155.26, 146.79, 143.21, 137.29, 133.20, 129.77, 128.42, 123.03, 122.01, 121.71, 118.82, 116.91, 52.47, 50.09, 49.69, 46.07, 41.25, 22.57, 21.66. MS (ESI): 532.2 [M + H]+. HRMS: [M + H]+ calcd for C27H31ClN9O+, 532.2340; found, 532.2327.
1-(4-(4-((5-Chloro-4-((2-(2-methyl-2H-tetrazol-5-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethan-1-one (m7)
A yellow solid (86.5 mg, 0.17 mmol, 50.1%). 1H NMR (600 MHz, DMSO-d6) δ 10.20 (s, 1H), 9.28 (s, 1H), 8.74 (s, 1H), 8.22 (s, 1H), 8.17 (d, J = 7.7 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 8.2 Hz, 2H), 7.30 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 4.49 (s, 3H), 3.59 (d, J = 3.0 Hz, 5H), 3.13–3.07 (m, 2H), 3.06–3.00 (m, 2H), 2.05 (s, 3H). MS (ESI): 505.2 [M + H]+. HRMS: [M + H]+ calcd for C24H26ClN10O+, 505.1901; found, 505.1970.
1-(4-(4-((5-Chloro-4-((2-(1-methyl-1H-tetrazol-5-yl)phenyl)amino)pyrimidin-2-yl)amino)phenyl)piperazin-1-yl)ethan-1-one (m8)
A yellow solid (98.5 mg, 0.2 mmol, 41.8%). 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 9.07 (s, 1H), 8.26–7.91 (m, 2H), 7.83–7.62 (m, 2H), 7.57–7.29 (m, 3H), 6.81 (d, J = 7.8 Hz, 2H), 4.03 (s, 3H), 3.57 (s, 4H), 3.01 (d, J = 24.4 Hz, 4H), 2.04 (s, 3H). MS (ESI): 505.3 [M + H]+. HRMS: [M + H]+ calcd for C24H26ClN10O+, 505.1901; found, 505.1897.
5-Chloro-N4-(2-(1-methyl-1H-tetrazol-5-yl)phenyl)-N2-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyrimidine-2,4-diamine (m11)
A yellow solid (91.6 mg, 185.4 μmol, 33.7%). 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 9.15 (s, 1H), 8.07 (d, J = 6.5 Hz, 2H), 7.76 (d, J = 7.5 Hz, 1H), 7.67 (t, J = 7.6 Hz, 1H), 7.55–7.30 (m, 3H), 6.83 (d, J = 8.8 Hz, 2H), 4.47 (s, 1H), 4.03 (s, 3H), 3.10 (s, 2H), 2.92 (s, 2H), 2.60 (s, 3H), 2.08 (d, J = 4.1 Hz, 2H), 1.89 (d, J = 13.8 Hz, 2H). MS (ESI): 492.2 [M + H]+. HRMS: [M + H]+ calcd for C24H26ClN9O+, 492.1914; found, 492.2024.
5-Chloro-N4-(2-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-((1-methylpiperidin-4-yl)oxy)phenyl)pyrimidine-2,4-diamine (m12)
A yellow solid (85.0 mg, 0.17 mmol, 54.6%). 1H NMR (400 MHz, DMSO-d6) δ 11.22 (s, 1H), 9.33 (s, 1H), 8.83 (d, J = 8.1 Hz, 1H), 8.73 (s, 1H), 8.20 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.39 (t, J = 7.5 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 6.96 (d, J = 8.9 Hz, 2H), 4.63–4.41 (m, 1H), 3.99 (s, 3H), 3.21–3.12 (m, 2H), 3.07–2.90 (m, 2H), 2.65 (s, 3H), 2.18–2.05 (m, 2H), 1.97–1.83 (m, 2H). MS (ESI): 491.2 [M + H]+. HRMS: [M + H]+ calcd for C25H28ClN8O+, 491.1996; found, 492.2077.
5-Chloro-N2-(4-(methyl(1-methylpiperidin-4-yl)amino)phenyl)-N4-(2-(1-methyl-1H-1,2,4-triazol-3-yl)phenyl)pyrimidine-2,4-diamine (m13)
A yellow solid (80.2 mg, 0.16 mmol, 20.4%). 1H NMR (400 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.17 (s, 1H), 8.89 (s, 1H), 8.73 (s, 1H), 8.16 (d, J = 10.0 Hz, 2H), 7.46 (d, J = 8.7 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.9 Hz, 2H), 4.00 (s, 3H), 3.79 (d, J = 11.0 Hz, 1H), 2.93 (s, 2H), 2.70 (s, 3H), 2.63 (s, 3H), 2.03 (dd, J = 22.7, 11.0 Hz, 2H), 1.75 (d, J = 12.8 Hz, 2H), 1.23 (s, 2H). MS (ESI): 504.2 [M + H]+. HRMS: [M + H]+ calcd for C26H31ClN9+, 504.2391; found, 504.2406.
5-Chloro-N4-(2-(1-isopropyl-1H-1,2,4-triazol-3-yl)phenyl)-N2-(4-(methyl(1-methylpiperidin-4-yl)amino)phenyl)pyrimidine-2,4-diamine (m16)
A pink solid (110.3 mg, 0.2 mmol, 28.9%). 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 9.17 (s, 1H), 8.81 (s, 2H), 8.19 (d, J = 10.6 Hz, 2H), 7.45 (d, J = 8.6 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 6.83 (d, J = 8.9 Hz, 2H), 4.73–4.62 (m, 1H), 3.77 (s, 1H), 2.81 (d, J = 22.1 Hz, 2H), 2.70 (s, 3H), 2.59 (s, 3H), 1.99 (dd, J = 22.8, 11.1 Hz, 2H), 1.73 (d, J = 12.7 Hz, 2H), 1.54 (d, J = 6.6 Hz, 6H), 1.22 (s, 2H). 13C NMR (151 MHz, DMSO-d6): δ (ppm) 160.46, 158.71, 155.80, 155.27, 145.94, 143.20, 137.39, 131.22, 129.68, 128.40, 122.86, 122.45, 121.92, 118.74, 115.17, 104.50, 54.61, 53.95, 52.46, 32.42, 26.59, 22.56. MS (ESI): 532.2 [M + H]+. HRMS: [M + H]+ calcd for C28H35ClN9+, 532.2704; found, 532.2717.
Kinase inhibition assay
The compounds were tested against each of the selected kinases using the Eurofins standard KinaseProfiler™ assays and following the relevant standard operating procedures. The Compounds supplied as powders were reconstituted to a 10 mM stock in 100% DMSO before further dilution to 50×. The required volume of the 50× stock of test compound was added to the assay well before a reaction mix containing the enzyme and substrate was added. The reaction was initiated by the addition of ATP at the selected concentration. There was no pre-incubation of the compound with the enzyme/substrate mix prior to ATP addition.
Data were processed using custom in-house analysis software. Results are expressed as remaining kinase activity, as a percentage of the DMSO control. This is calculated using the following formula:![]()
For IC50 determination (the concentration of an inhibitor that reduces the level of kinase activity by 50%), data were analyzed using XLFit version 5.3 (ID Business Solutions). Sigmoidal dose–response (variable slope) curves were fitted based on the mean result for each test concentration using non-linear regression analysis. Where the top and/or bottom of the curve fall >10% outwith 100 and 0, respectively, either or both of these limits may be constrained at 100 and 0. Selected kinases were diluted in the buffer (20 mM MOPS, 1 mM EDTA, 0.01% Brij-35, 5% glycerol, 0.1% β-mercaptoethanol, 1 mg mL−1 BSA) prior to addition to the reaction mix.
Kinase selectivity assay
The test compound was used in a single dose concentration of 1 μM. % Inhibition was calculated by subtracting the % remaining kinase activity from 100. And the detailed % inhibition data are shown in Table S1.† The detailed % remaining kinase activity was listed as kinase selectivity. The compound was tested against each of the selected kinases using the Eurofins standard KinaseProfiler™ assays with a single point (n = 2). The standard deviation (SD) of the four controls (as a percentage of their mean) must be 15 or less for an automatic pass. If the SD is greater than 15, an outlier may be removed according to the set criteria. If a set of controls fails to meet any of these criteria, then the assay is deemed to have failed and must be repeated.
Cell proliferation assay
Cells were seeded in 96-well plates (detailed information is shown in Table S2†) at a low density in growth media. The day after, the positive control (staurosporine) or designated concentrations of compounds were added to each well, and the cells were incubated for 72 h. Finally, cell proliferation was determined using a cell counting kit (Cell Counting kit-8) assay. The IC50 values were calculated by concentration–response curve fitting using a curve integration method (GraphPad Prism, version 8.3.0.538). The curve integration equation uses the following formula:Y = Bottom + (Top − Bottom)/(1 + 10 ^((Log CC50 − X) × HillSlope))
Microsomal stability screening in liver microsomes
The test compounds were incubated with rat and human liver microsomes (0.5 mg protein per mL) for about 10 min at 37 °C in the reaction mixture containing 0.1 M potassium phosphate (pH 7.4). The buffer was then used to prepare NADPH at a concentration of 6 mM. The stop solution was ACN (including 100 ng mL−1 propranolol as the internal standard). The resulting supernatant was subjected to an LC–MS/MS analysis. The ratio of peak area responses relative to the internal standard was determined.
Procedures
1. Working solution: stock solution_01: dilute 5 μL test compound or positive control from the respective stock solution (10 mM) with 495 μL 50% ACN/H2O (conc.: 100 μM, 50% ACN). Working solution: dilute 60 μL test compound or positive control from the respective stock solution_01 (100 μM) with 140 μL buffer (conc.: 30 μM, 15% ACN). 2. Prepare 96-well plates, named T0, T15, T30, T60, and NCF60. 3. Dispense 18.8 μL (20 mg mL−1) liver microsomes to 456.2 μL buffer, incubate the microsome solution and test compound working solution separately at 37 °C for about 10 min. 4. Transfer 25 μL test compound or positive control working solution to the microsome solution and mix well by pipetting. Immediately remove 30 μL from the mixture, add the stop solution (150 μL per well), and then add 15 μL NADPH as T0. 5. Transfer 30 μL from the mixture solution to the corresponding wells in the plates of NCF60, T60, T30, and T15. Add 15 μL buffer to NCF60 while 15 μL NADPH to T60, T30, and T15 at the required time points to start a reaction. The final reaction system contains 0.5 mg mL−1 liver microsomes and 2 mM NADPH with ACN less than 1%. 6. At 15, 30, and 60 min, add the stop solution (150 μL per well) to terminate the reaction.
7. Centrifuge the plates at 4000 rpm for 5 min. 8. While centrifuging, load a new 96-well plate with 150 μL of 50% MeOH/H2O. Then transfer 30 μL supernatant into the corresponding well and mix thoroughly for LC/MS/MS (Table 8).
Physiological parameters of human and rat liver microsomes.
| Species | mg microsomal protein per g liver weight (mg) | g liver weight per kg body weight | Factor | Hepatic blood flow (ml min−1 kg−1) |
|---|---|---|---|---|
| Rat | 44.8 | 40 | 1792 | 55.2 |
| Human | 48.8 | 25.7 | 1254.2 | 20.7 |
Data analysis
In the determination of the in vitro elimination constant of the test compound or positive control compound, the analyte/internal standard peak area ratios were converted to percentage remaining (%remaining) with the following equation:![]() CLHep = (0.693/t1/2) × 1/(microsome Conc. (0.5 mg mL−1)) × factorCLin vivo = CLHep × Hepatic blood flow/(CLHep + Hepatic blood flow)
CLHep = (0.693/t1/2) × 1/(microsome Conc. (0.5 mg mL−1)) × factorCLin vivo = CLHep × Hepatic blood flow/(CLHep + Hepatic blood flow)
Pharmacokinetic parameters obtained in rats
Male rats (SD rats, n = 2) were administered compound 22 or TP-0903 by intragastric administration at 5 mg kg−1, and intravenous injection at 1 mg kg−1, respectively. A mixed solvent of 10% DMSO + 10% Kolliphor HS15 + 80% saline was used as the formulation. Blood samples were collected at 0.083, 0.25, 0.5, 1, 2, 5, 7, and 24 h after dosing in the intravenous injection group. Blood samples were collected at 0.25, 0.5, 1, 2, 5, 7, and 24 h after dosing in the intragastric administration group. The plasma was separated by centrifugation, and the serum was collected in vials. The serum samples were frozen and stored at −20 °C before analysis. The test sample concentrations were determined using an LC–MS/MS system. Animal procedures were performed according to the institutional ethical guidelines on animal care and approved by the Institute Animal Care and Use Committee at Dongguan Institute of Sunshine Lake Pharma Co. Ltd. (Study no. 16020-5015). The PK parameters, maximum drug concentration (Cmax), area under the concentration–time curve (AUClast) from zero to 24 h post-dose and time to reach the maximum (peak) concentration following drug administration were determined based on the mean plasma concentration.
Molecular modeling
The docking studies were carried out with Schrödinger suites 2018.1 using the X-ray crystal structure of Axl (PDB ID: 5U6B). The protein preparation, optimization, and minimization were performed in the preparation process wizard (Schrödinger, LLC, New York, NY, 2019). The compounds 5a, w14, and m16 were prepared by Ligprep. The co-crystallized inhibitor was deleted from the prepared structure. The compound was then docked into the ATP-site using Glide release with standard settings except that 10 poses were retained. Docking included the calculation of a grid of the receptor site and docking into this grid. And Glide SP and Glide XP were used for docking. The docking site was defined using a grid box size that docks ligands similar in size to the workspace ligand. And the grid center was selected as the centroid of the workspace ligand (selected in the Receptor tab). Fig. 2A–4 were obtained from PyMOL.
Fig. 3. Schematic illustration of the proposed binding mode of w14 with the Axl (generated from PDB ID: 5U6B). (A) Docking of compound w14 into the Axl kinase. (B) Proposed model of binding between the 1H-1,2,4-triazole segment and Axl kinase.

Fig. 4. Schematic illustration of the proposed binding mode of compound m16 with the Axl (generated from PDB ID: 5U6B). (A) Compound m16 with the hinge region. Yellow dotted lines indicate the intermolecular hydrogen bonds. The red dotted line indicates the intramolecular hydrogen bond. (B) Compound m16 binds to the Axl protein in a DFG-in conformation. The yellow dotted line indicates the intramolecular hydrogen bonds. All displayed residues shown are within 4 Å distance to the ligand.

Author contributions
All authors have approval for the final version of the manuscript. Shuang Wu: data collection and analysis, the design and synthesis of the compounds, CCK-8 assay, model docking, manuscript preparation. Min Liao: synthesis, liver microsomal stability assay, manuscript preparation. Minxiong Li: manuscript preparation. Mingming Sun: pharmacokinetic assay. Ning Xi and Youlin Zeng: research design, supervision of the whole project, manuscript preparation.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Material
Acknowledgments
We would like to thank Xiaoqing Chen and Shaoyu Cai for their analytical assistance. And this research was funded by the National Major Scientific and Technological Special Project for “Significant New Drugs Development” during the Twelfth Five-year Plan Period (No. 2015ZX09101013), China.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00153e
Notes and references
- Linger R. M. Keating A. K. Graham D. K. et al., Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets. 2010;14:1073–1090. doi: 10.1517/14728222.2010.515980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Bryan J. P. Frye R. A. Cogswell P. C. et al., Axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991;11:5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunger J. Schleithoff L. Janssen J. W. G. et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14:2619–2631. doi: 10.1038/sj.onc.1201123. [DOI] [PubMed] [Google Scholar]
- Valverde P. Obin M. S. Taylor A. Role of Gas6/Axl signaling in lens epithelial cell proliferation and survival. Exp. Eye Res. 2004;78:27–37. doi: 10.1016/j.exer.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Myers S. H. Brunton V. G. Unciti-Broceta A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2016;59:3593–3608. doi: 10.1021/acs.jmedchem.5b01273. [DOI] [PubMed] [Google Scholar]
- Wu X. Liu X. Zhang Z. et al., AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–9563. doi: 10.18632/oncotarget.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linger R. M. Keating A. K. Graham D. K. et al., TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu C. et al., AXL and MET in Hepatocellular Carcinoma: A Systematic Literature Review. Liver Cancer. 2022;11:94–112. doi: 10.1159/000520501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenhoff J. Dahlbäck B. Hafizi S. Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2004;319:871–878. doi: 10.1016/j.bbrc.2004.05.070. [DOI] [PubMed] [Google Scholar]
- Shan S. Liu Z. Liu C. et al., Growth arrest-specific gene 6 transfer promotes mesenchymal stem cell survival and cardiac repair under hypoxia and ischemia via enhanced autocrine signaling and paracrine action. Arch. Biochem. Biophys. 2018;660:108–120. doi: 10.1016/j.abb.2018.10.016. [DOI] [PubMed] [Google Scholar]
- Zhu C. Wei Y. Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol. Cancer. 2019;18:153–175. doi: 10.1186/s12943-019-1090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyette M.-A. Côté J.-F. AXL Receptor Tyrosine Kinase as a Promising Therapeutic Target Directing Multiple Aspects of Cancer Progression and Metastasis. Cancers. 2022;14:466–481. doi: 10.3390/cancers14030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.-M. Jang Y. Kang B. et al., AXL/MET dual inhibitor, CB469, has activity in non-small cell lung cancer with acquired resistance to EGFR TKI with AXL or MET activation. Lung Cancer. 2020;146:70–77. doi: 10.1016/j.lungcan.2020.05.031. [DOI] [PubMed] [Google Scholar]
- Tang J. Yu J. Hubbard-Lucey V. et al., The clinical trial landscape for PD1/PDL-1 immune checkpoint inhibitors. Nat. Rev. Drug Discovery. 2018;17:854–855. doi: 10.1038/nrd.2018.210. [DOI] [PubMed] [Google Scholar]
- Gainor J. F. Shaw A. T. Yeap B. Y. Engelman J. A. Mino-Kenudson M. et al., EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non–small cell lung cancer: A retrospective analysis. Clin. Cancer Res. 2016;22:4585–4593. doi: 10.1158/1078-0432.CCR-15-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. Black J. R. M. Sharma R. et al., Gene of the month: Axl. J. Clin. Pathol. 2016;69:391–397. doi: 10.1136/jclinpath-2016-203629. [DOI] [PubMed] [Google Scholar]
- Rankin E. B. Fuh K. C. Amato G. J. et al., AXL is an Essential Factor and Therapeutic Target for Metastatic Ovarian Cancer. Cancer Res. 2010;70:7570–7579. doi: 10.1158/0008-5472.CAN-10-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wium M. Ajayi-Smith A. F. Paccez J. D. Zerbini L. F. The Role of the Receptor Tyrosine Kinase Axl in Carcinogenesis and Development of Therapeutic Resistance: An Overview of Molecular Mechanisms and Future Applications. Cancers. 2021;13:1521–1541. doi: 10.3390/cancers13071521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Y. B. Kim J.-H. Lim S. M. et al., The Development of AXL Inhibitors in Lung Cancer: Recent Progress and Challenges. Front. Oncol. 2022;12:811247. doi: 10.3389/fonc.2022.811247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felip E. Brunsvig P. Spicer J. F. et al., A phase II study of bemcentinib (BGB324), a first-in-class highly selective AXL inhibitor, with pembrolizumab in pts with advanced NSCLC: OS for stage I and preliminary stage II efficacy. J. Clin. Oncol. 2019;15:9098. [Google Scholar]
- Jimbo T. Hatanaka M. Fujiwara K. et al., DS-1205b, a novel selective inhibitor of AXL kinase, blocks resistance to EGFR-tyrosine kinase inhibitors in a non-small cell lung cancer xenograft model. Oncotarget. 2019;10:5152–5167. doi: 10.18632/oncotarget.27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnete S. T. E. Lotsberg M. L. Terry S. et al., Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front. Immunol. 2022;13:869676. doi: 10.3389/fimmu.2022.869676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Gilteritinib: First Global Approval. Drugs. 2019;79:331–339. doi: 10.1007/s40265-019-1062-3. [DOI] [PubMed] [Google Scholar]
- Tovolia F. Dadduziob V. De Lorenzoc S. et al., Real-Life Clinical Data of Cabozantinib for Unresectable Hepatocellular Carcinoma. Liver Cancer. 2021;10:370–379. doi: 10.1159/000515551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolajczyk A. Mitula F. Pieczykolan J. et al., Two-Front War on Cancer-Targeting TAM Receptors in Solid Tumour Therapy. Cancers. 2022;14:2488. doi: 10.3390/cancers14102488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollard A. Steven L. Bearss D. J. et al., Design, Synthesis, and Biological Evaluation of a Series of Novel AXL Kinase Inhibitors. ACS Med. Chem. Lett. 2011;2:907–912. doi: 10.1021/ml200198x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. Boysen J. Ghosh A. K. et al., Targeted Axl Inhibition Primes Chronic Lymphocytic Leukemia B Cells to Apoptosis and Shows Synergistic/Additive Effects in Combination with BTK Inhibitors. Clin. Cancer Res. 2015;21:2115–2126. doi: 10.1158/1078-0432.CCR-14-1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng P. S. Foo K. Hung A. W. et al., Fragment-based lead discovery of indazole-based compounds as AXL kinase inhibitors. Bioorg. Med. Chem. 2021;49:116437–116448. doi: 10.1016/j.bmc.2021.116437. [DOI] [PubMed] [Google Scholar]
- Gajiwala K. S. Grodsky N. Stewart A. et al., The Axl kinase domain in complex with a macrocyclic inhibitor offers first structural insights into an active TAM receptor kinase. J. Biol. Chem. 2017;292:15705–15716. doi: 10.1074/jbc.M116.771485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.