

Abstract
Cellular homeostasis is continuously challenged by damage from reactive oxygen species (ROS) and numerous reactive electrophiles. Human cells contain various protective systems that are upregulated in response to protein damage by electrophilic or oxidative stress. In addition to the NRF2-mediated antioxidant response, ROS and reactive electrophiles also activate HSF1 and HIF1 that control heat shock response and hypoxia response, respectively. Here, we review chemical and biological mechanisms of activation of these three transcription factors by ROS/reactive toxicants and the roles of their gene expression programs in antioxidant protection. We also discuss how NRF2, HSF1, and HIF1 responses establish multilayered cellular defenses consisting of largely nonoverlapping programs, which mitigates limitations of each response. Some innate immunity links in these stress responses help eliminate damaged cells, whereas others suppress deleterious inflammation in normal tissues but inhibit immunosurveillance of cancer cells in tumors.
Introduction
Biological injury by oxidants and reactive electrophiles is causally linked to the development of major human pathologies such as neurodegeneration, atherosclerosis, diabetes, and cancer.1−5 Chronic production of these toxic molecules also contributes to declining tissue functioning in aging. Endogenous reactive oxygen species (ROS), reactive nitrogen species (RNS), reactive carbonyls, and other electrophiles produced during inflammation, normal metabolism, and lipid peroxidation are usually the major endogenous causes of chemical damage to proteins and DNA. Ambient pollution and the presence of various undesired xenobiotics in food and drinking water also lead to human exposure to a large number of exogenous toxicants. The most frequently modified/oxidized chemical group in proteins is Cys-SH, especially when cysteines are positioned next to positively charged amino acids.6,7 Other targets for electrophile adduction in proteins include side chains of histidine, lysine, and arginine. DNA is also readily oxidized and chemically modified at various sites, especially in nucleobases. Protein damage induces a range of physiological dysfunctions, whereas DNA damage leads to mutations and, ultimately, to cancerous transformation of cells. There is a growing body of evidence that stable chemical modifications in histones by electrophilic substances are also capable of causing abnormalities in the epigenome.8 Methylglyoxal, a reactive byproduct of glycolysis, was recently found to produce adducts at histone Lys and Arg side chains, which affected global transcriptome and histone acetylation.9 The threat to cellular fitness posed by biological oxidants and reactive electrophiles is so great that all organisms contain specific protein sensors and protection responses against these toxic substances.
The NRF2-induced transcription program has long been viewed as a central cellular response to oxidative stress and the overabundance of reactive electrophiles. However, this response is limited in its ability to address several important vulnerabilities in stressed cells, such as accumulation of toxic damaged proteins and generation of oxidants by damaged mitochondria. Other conservative stress-sensitive transcription factors are also induced by the same types of reactive toxicants as NRF2, indicating that the full cellular response to oxidants includes much more than the NRF2 pathway. Here, we review mechanisms of cytosolic stress sensing and complementary defense activities governed by three oxidant-activated transcription factors HSF1, HIF1, and NRF2 that control the heat shock (unfolded proteins) response, hypoxia response, and antioxidant response, respectively. Although HSF1, HIF1, and NRF2 pathways have been extensively reviewed individually, analyses of their collaborative activities have been limited to the tumor state/microenvironment such as between HIF1 and NRF2 in tumor hypoxia.10 The focus of this review is to provide an integrated view of major functions of HSF1, HIF1, and NRF2 as components of a global cellular defense against protein-reactive oxidants and electrophiles. HSF1 is an archaic stress-responsive factor with well-conserved mechanisms of activation and protective properties of its effectors between yeast to humans.11,12 HIF1 and NRF2 are evolutionarily younger systems that are best characterized in mammals, but orthologue regulatory pathways are also known to exist in lower animals. The closest homologue of NRF2 in yeast is the transcriptional factor Yap1 which directly senses oxidants via the disulfide formation blocking its nuclear export and permitting transactivation of its targets.13 The human KEAP1-NRF2 pathway is a more versatile system that senses a much greater range of Cys-reactive toxicants, including those that form adducts. HIF1 is a metazoan transcription factor.14
NRF2 Antioxidant Response and Its Limitations
NRF2 is a constitutively expressed transcription factor, commonly known as a master regulator of the oxidative/electrophilic stress-induced response in cells. Despite its ongoing production, protein levels of NRF2 in normal cells are low due to its rapid destruction by 26S proteasomes.15,16 The stability of NRF2 is primarily controlled by KEAP1, which is a substrate-binding component of the CUL3-RBX1 E3 ubiquitin ligase complex. KEAP1 also serves as a sensor of the cellular abundance of electrophiles in the activation of the NRF2 response. This electrophile-sensing potential of KEAP1 is created by the presence of numerous functionally important Cys-SH groups, which can be oxidized or adducted with the ensuing loss of the ubiquitin ligase activity toward NRF2 (Figure 1). In addition to its overall large content of Cys-SH (27 in humans, 25 in mice), KEAP1 sensitivity toward reactive toxicants is further enhanced by embedding its critical cysteines next to positively charged amino acids which lower the pKa of the SH group.15 Deprotonation of the SH group enhances its reactivity with carbonyls and the most common cellular ROS and RNS (H2O2, O2•–, CO3•–, •NO2, ONOO–, HOCl), which have higher reaction rates with thiolate (RS–) than with RSH.17 KEAP1 inactivation by damage to its sensory Cys-SH groups results in the accumulation of NRF2, its nuclear translocation, heterodimerization with bZip domain-containing proteins such as small MAFs, and transactivation of genes whose promoters contain the antioxidant response element sequence. In addition to its canonical regulation by inactivation of KEAP1 during electrophilic stress, NRF2 stability can be affected by other mechanisms which either diminish NRF2-KEAP1 interactions or engage an alternative degradation pathway of NRF2 involving its phosphorylation by GSK3β and then ubiquitination by the β-TrCP E3 ligase.15,16 NRF2-induced genes are primarily involved in ROS detoxification (glutathione reductase, glutathione peroxidase, glutaredoxin, peroxiredoxin, thioredoxin), Phase II xenobiotics inactivation (NQO1, GSTA1, GSTA3, carbonyl reductase), glutathione biosynthesis (GCLC, GCLM), and regulation of heme and Fe metabolism (HMOX1, ferritin chains).15,18,19 NRF2 also regulates expression of a Phase III detoxification protein MRP2 which excretes conjugates of electrophilic compounds with glutathione, glucuronate, or sulfate.20
Figure 1.
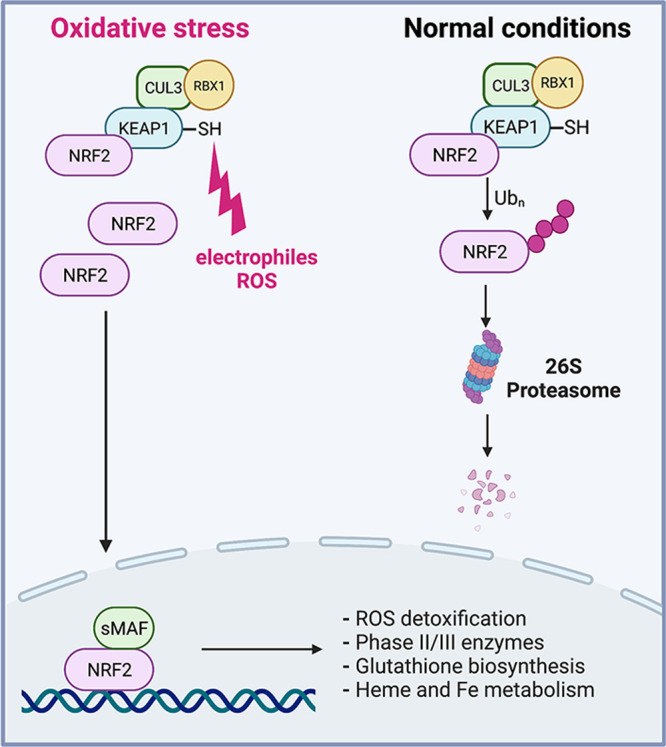
NRF2 regulation under normal and stress conditions. The transcription factor NRF2 is constitutively expressed, but its protein levels are low in normal cells due to its rapid destruction by 26S proteasomes. Constitutive polyubiquitination of NRF2 is performed by an E3 ubiquitin ligase complex containing SH-rich KEAP1 which serves as a sensor of cellular ROS and electrophiles in the activation of the NRF2 response. Damage to sentinel SH groups in KEAP1 causes its inactivation leading to the stabilization and accumulation of NRF2, its nuclear translocation, heterodimerization with bZip domain-containing proteins such as small MAFs, and transactivation of genes involved in many direct antioxidant activities such as ROS detoxification, Phase II/III detoxification reactions, glutathione biosynthesis, and iron/heme metabolism.
NRF2 plays a dual role in carcinogenesis by inhibiting its initiation but promoting the progression of already transformed cells.21,22 Consistent with its control of cellular defenses against reactive electrophiles, abrogation of NRF2 increased the susceptibility of animals to oncogene-, chemical-, and inflammation-induced cancers, as evident by the incidence and size of tumors.22−27 However, once tumors were formed, NRF2 inactivation was detrimental to tumor growth.26,27 In many human cancers, NRF2 is constitutively elevated due to its higher stability and mRNA expression.21,22 Approximately 20% of lung cancers contain mutationally inactivated KEAP1, which eliminates constitutive ubiquitination and degradation of NRF2, and another 10% have activating mutations in this transcription factor. Another, more unexpected mechanism for persistent NRF2 activation in a large subset of human tumors is a recently discovered inactivation of KEAP1 via its neomorph binding by the oncogenic mutant BRAFV600E.28 BRAF mutations are particularly common in human epidermal melanoma, colon, and thyroid cancers. An analysis of TCGA data revealed that somatic gain-of-function mutations in NRF2 are common and correlate with poor prognosis.29 Thus, cancers benefit from elevated protection against electrophile stress, which allows them to thrive in challenging growth conditions.
Activation of the NRF2 pathway does not appear to involve significant adjustments to cell cycle progression or respiratory metabolism in order to minimize consequences of the oxidant-induced damage to the genome or diminish the production and/or leakage of ROS by mitochondria, respectively. The NRF2 response also does not directly regulate the cellular capacity to stabilize unfolded damaged proteins or target them for proteasomal degradation, although it can promote another protein quality control mechanism – autophagy.30 The impact of the KEAP1-NRF2 pathway on the elimination of DNA double-stranded breaks (DSBs), the most toxic form of DNA damage by oxidants, is directed toward their repair via nonhomologous end-joining (NHEJ). NHEJ is error-prone but faster than the accurate repair of DSBs by homologous recombination, which requires a long-range resection of DSB ends and fully functional BRCA1 and BRCA2 proteins. KEAP1 inactivation leads to a stimulation of NHEJ through (i) the NRF2-mediated induction of 53BP1,31 which acts as an antagonist of the homologous recombination factor BRCA1,32 and (ii) stabilization of the second KEAP1 target EMSY, which suppresses BRCA2 functions.33 For the vast majority of cells in vivo, which are either nondividing or rarely dividing, NHEJ activation is likely beneficial as homology-based repair can operate only in S and G2 phases, and a long-range resection of DSBs in G1 phase is genetically deleterious.34 In actively proliferating cancer cells, however, mutational inactivation of KEAP1 results in the genomic instability and presents as a BRCA deficiency phenotype.33
HIF1 Response
HIF1 is the primary transcriptional mediator of the hypoxia response activated in all human cells by low O2 or chemical agents acting as hypoxia mimetics.35,36 HIF1 is a heterodimer-containing stable HIF1β and O2-regulated unstable HIF1α. Similar to NRF2, HIF1α is a constitutively expressed protein that is present in normoxic cells at very low levels due to its continuous degradation by 26S proteasomes (Figure 2). This process is initiated by hydroxylation of Pro402 and Pro564 in HIF1α that are recognized by VHL, which acts as a substrate adaptor in the Cullin 2-elongin B/C-Rbx1 E3 ubiquitin ligase. HIF1α-targeting hydroxylases PHD1-3 are Fe(II)-dependent enzymes that split O2 to hydroxylate Pro and the cosubstrate 2-oxoglutarate. In hypoxia, this Pro hydroxylation reaction is suppressed due to the lack of O2. Stabilization of HIF1α by hypoxia or by drug-induced inhibition of its Pro hydroxylases activate a large transcription program that includes upregulation of several hundreds of genes.37,38 This hypoxia adaptation response promotes cell survival by rewiring cellular energy metabolism, which includes shifting to O2-independent, glycolysis-based production of ATP and decreasing nonessential metabolic activities. The metabolic switch that directs the glucose metabolite pyruvate away from mitochondria results from the inactivation of pyruvate dehydrogenase by the HIF1 target PDK1.39,40 Other effects of HIF1 on mitochondrial metabolism include the substitution of the cytochrome c oxidase subunit COX4-1 with COX4-2 to improve efficiency of electron transfer to O2 and lower ROS production.41 HIF1 also upregulates expression of the NADH dehydrogenase NDUFA4L2 that inhibits mitochondrial Complex I activity and thereby diminishes O2 consumption and ROS formation.42 In moderate hypoxia, however, ATP production by the mitochondrial respiratory chain is not yet significantly diminished, and the primary purpose of the switch to glycolysis in these conditions is to prevent excessive formation and leakage of mitochondrial ROS.35 Consistent with this role, mitochondrial ROS are also important for the stabilization of HIF1α in hypoxia.43−46
Figure 2.
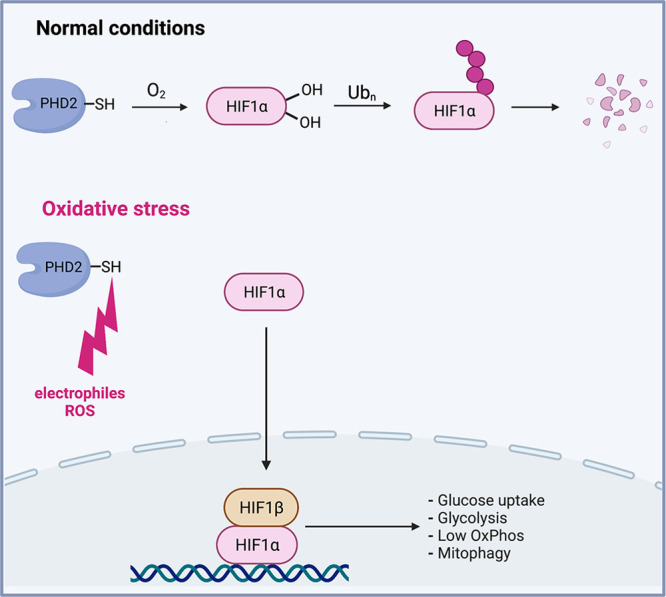
Regulation of HIF1 by O2 and oxidants. The hypoxia-inducible transcription factor HIF1 is a heterodimer composed of stable HIF1β and O2-regulated unstable HIF1α subunits. HIF1α is constitutively expressed, but it is present in normoxic cells at very low levels due to its degradation by 26S proteasomes. Hydroxylation of two Pro residues in HIF1α by the O2-dependent hydroxylases PHD1-3 (primarily PHD2) acts as a trigger for its proteolytic ubiquitination. In hypoxia, Pro hydroxylation of HIF1α is suppressed due to the lack of O2. Several Cys-SH in the catalytic domain of PHD2 (also known as EGLN1) are sensitive to oxidation or adduction by various ROS, RNS, and toxic metals, causing the loss of its Pro hydroxylation activity. Stabilization of HIF1α activates a specialized transcription program known as hypoxia response which stimulates glucose uptake, upregulates glycolysis, downregulates oxidative phosphorylation in mitochondria, and promotes mitophagy.
Similar to the oxidant-sensing function of cysteines in KEAP1, several Cys-SH in the catalytic domain in PHD2 (the main HIF1α prolyl hydroxylase, also known as EGLN1) act as targets for inactivation by various ROS, RNS and toxic metals.47−50 PHDs also sense the depletion of cellular antioxidants such as ascorbate,51 which is important for the reduction of Fe(III) to Fe(II) in the catalytic center of these enzymes.52 In addition to its accumulation due to inactivation of PHDs, human HIF1α is also stabilized by oxidants-induced covalent modifications at its Cys520 residue (Cys533 in mouse HIFα) such as glutathionylation (protein-glutathione disulfide) and S-nitrosylation which inhibit ubiquitination by VHL at the nearby Lys532 in the oxygen degradation domain.53,54 Mitochondria can be damaged by a large number of endogenous and exogenous toxic agents, and the resulting distortions in the respiratory metabolism produce excessive amounts of ROS.55,56 Thus, it is reasonable to view HIF1-driven suppression of the entry of pyruvate into the Krebs cycle35 and, consequently, a decreased flow of electrons through the respiratory chain as a general protective response from ROS overproduction by diseased mitochondria, including in normoxic conditions. Upregulation of BNIP3 and BNIP3L by HIF1 also stimulates mitophagy which eliminates severely damaged mitochondria that generate especially large amounts of ROS.35 In addition to ROS, mitochondrial stress also leads to cytosolic release of the Krebs cycle intermediate fumarate which activates HIF1 through inhibition of PHD257 and NRF2 via inactivation of KEAP1.58,59 Fumarate is an α,β-unsaturated carbonyl that nonenzymatically reacts with proteins generating cysteine-S-succination adducts.60 High levels of fumarate are also present in individuals with inactivating mutations in fumarate hydratase, where it acts as an oncometabolite causing leiomyomatosis and renal cell carcinoma.61
Increased expression of glycolytic factors by activated HIF1 is also important to counteract losses of the essential glycolytic enzyme GAPDH which is susceptible to oxidative inactivation, resulting in the collapse of ATP production during strong oxidative stress.62 HIF1-induced routing of pyruvate into the production of lactic acid results in the increased acidification of cells, which together with increased utilization of NAD+ leads to the global slowdown of cell cycle,63 a beneficial response, preserving energy and genome integrity in stressed cells. Lowering cellular pH is also likely protective by globally decreasing oxidation of protein-SH groups due to their increased protonation. As discussed above, the protonated SH group is much less reactive with various oxidants and electrophiles than the thiolate anion (RS–). Consistent with its importance in antioxidant protection, HIF1 depletion led to hyperactivation of NRF2 in cells experiencing moderate oxidative stress.47 HIF1 is known to promote malignant characteristics in established cancers,64 which already acquired metabolic adaptation neutralizing growth inhibitory effects of high HIF1.63 In contrast, HIF1 activity in normal tissues is anticarcinogenic as evidenced by its role as a tumor suppressor gene in the development of human kidney cancer65 and animal models of leukemia and squamous cell carcinoma.66,67
HSF1 Response
HSF1 is the primary transcriptional mediator of a rapid cellular response to proteotoxic stress known as the heat shock response. The main gene targets and effectors of HSF1 are heat shock proteins which are molecular chaperones acting to prevent aggregation of misfolded proteins, to promote their refolding or when proteins are damaged beyond repair, and to stimulate their degradation by proteasomes and via autophagy.11,12,68 HSF1 also contributes to cytoprotection against toxic xenobiotics by promoting their efflux by MDR1 (ABCB1). MDR1 is a plasma membrane-based pump removing a range of toxic substances from cells. The promoter of the MDR1 gene is stress responsive and contains several HSF1-binding heat shock elements.69 The introduction of a constitutively active HSF1 into cells increased MDR1 expression, which was dependent on HSF1 binding to the heat shock elements in the MDR1 promoter.70
In unstressed cells, HSF1 is an inactive monomer localized in the cytoplasm and bound by HSP70 and HSP90 protein chaperones (Figure 3). In stress conditions, HSF1 undergoes a multistep activation process which involves dissociation of both HSPs, trimerization, translocation to the nucleus and binding to the target promoters.11,12 HSPs expression and HSF1 activity are higher in cancers due to dysregulated metabolism producing proteotoxic conditions in malignant cells.71 Although HSF1 was initially characterized as a regulator of cellular responses to hyperthermia, its activation is also well-established for many other protein-damaging agents, including cellular oxidants and electrophilic lipid peroxidation products.72−74 For 4-hydroxynonenal, one of the most toxic products of lipid oxidation, HSF1 was more important for cytoprotection than NRF2.75 Sulforaphane, a chemopreventive isothiocyanate compound from cruciferous vegetables and one of the most widely studied NRF2 activators, also acts as a strong inducer of the heat shock response.76 Stimulation of anti-inflammatory and antiapoptotic responses by electrophilic nitro-fatty acids is mediated by both NRF2 and, even more significantly, HSF1.77 A side-by-side comparison of several structurally distinct chemical NRF2 activators, all of which react with Cys-SH groups in KEAP1, found a strong upregulation of the canonical HSF1 target gene HSP70.78 HSP70 induction was HSF1-dependent but NRF2-independent. Analysis of the concentration dependence showed that activation of HSF1 closely followed that of NRF2, which is remarkable considering the evolutionarily selected structure of KEAP1 designed to act as a sensor of SH-reactive chemicals. The basal expression of NRF2 targets was higher in HSF1-knockout MEFs,78 suggesting that the diminished expression of molecular chaperones in these cells increased the abundance of reactive species sensed by the KEAP1-NRF2 pathway.
Figure 3.
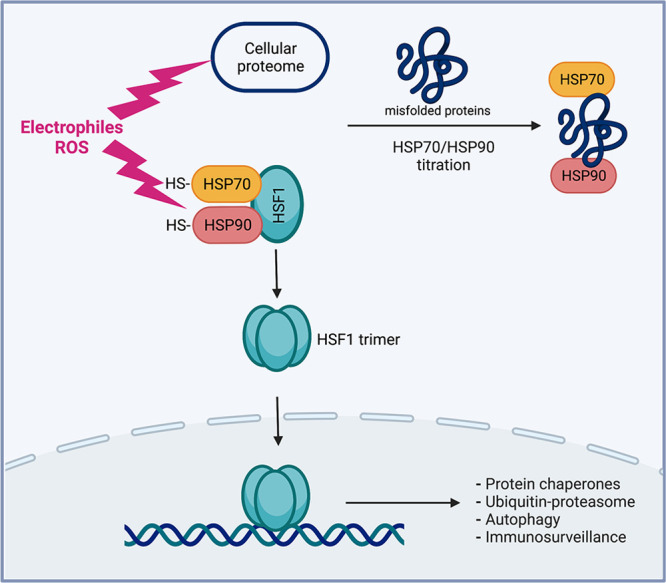
HSF1 activation by oxidants and other protein-damaging stressors. In unstressed cells, HSF1 is present in the cytoplasm as an inactive monomer that is repressed by HSP70 and HSP90 chaperones. In proteotoxic conditions causing dissociation of both HSPs, HSF1 forms a trimer which translocates to the nucleus and binds to the target promoters. HSF1 activation by oxidants and other reactive electrophiles results from dissociation of inhibitory HSP70 and HSP90 by direct damage to their SH groups and titration away of these chaperones by unfolded protein substrates. The HSF1-driven transcription program upregulates protein quality mechanisms (through increased abundance of protein chaperones, components of ubiquitin-proteasome systems, and autophagy factors) and enhances immunosurveillance of damaged cells (through expression of stress markers recruiting innate immune cells).
Activation of HSF1 by heat shock and by oxidants such as H2O2 was initially linked to the disulfide cross-linking of HSF1 molecules to form active trimers.79,80 Two out of five cysteines in human HSF1 were found to mediate intermolecular cross-linking that was needed for HSF1 trimerization and DNA binding. The disulfide bridging of HSF1 into trimers by heat stress and proteasome inactivation has been recently attributed to the protein disulfide isomerase activity of transglutaminase 2.81 This study found that heat shock and proteasome inhibition induced the trimerization of HSF1 only in Tg2+/+ MEFs and these multimeric products were present exclusively in the nuclear pool of HSF1. However, the multimers constituted only a small fraction of the total nuclear HSF1, and structural studies of DNA-bound HSF1 at a single site in the heat shock element82 do not support the importance of the earlier reported Cys36-Cys103 cross-linking.79 However, cross-linking between HSF1 trimers bound to the heat shock element containing multiple binding sites could not be excluded. Irrespective of its significance for H2O2, it seems unlikely that Cys-Cys cross-linking or other forms of Cys damage in HSF1 could represent a general route to its activation by numerous other electrophilic chemicals. Chemically diverse forms of Cys-SH modifications can serve as a sensitive and effective mechanism for the inactivation of proteins (KEAP1 as an example) but not for the formation of a specific active conformation.
A generally accepted mechanism for activation of HSF1 by the overabundance of misfolded proteins in the absence of chemical protein damage (for example, after heat shock or proteasome inhibition) involves dissociation of HSP70 and HSP90 from monomeric HSF1, permitting the formation of active trimers and gene expression.11,12 This process is fine-tuned by numerous posttranslational modifications, especially by phosphorylation.83,84 The loss of HSF1-bound inhibitory HSP70 and HSP90 is believed to result from their titration by unfolded proteins (higher affinity targets) that compete for a limited pool of these chaperones in unstressed cells. Recent studies provided strong evidence that the main source of these unfolded proteins triggering HSF1 activation during moderate proteotoxic stress are nascent polypeptides that are more vulnerable to heat-induced unfolding and aggregation due to their immature, unstable structures.85,86 In comparison to mature proteins, the unfolded structure of newly translated proteins is also expected to make them more vulnerable to chemical damage due to greater steric accessibility of reactive amino acids and higher abundance of Cys-SH groups prior to the formation of physiological disulfide bridges stabilizing specific protein folds. Therefore, it seems likely that a major source of unfolded proteins induced by oxidants and other electrophiles and sensed by HSF1 also originates from nascent polypeptides. Consistent with this notion, newly synthesized proteins were the major pools of degraded proteins after both heat shock and oxidative damage.87 There is also strong evidence that HSP70 and HSP90 can act as direct sensors of electrophilic stressors, leading to the dissociation of these chaperones from HSF1 and allowing its activation. HSP70 has been found to undergo different modifications of Cys-SH groups, such as conjugation with 4-hydroxynonenal,75,88 glutathione,89 and other compounds.73,90,91 HSP90 was also chemically modified by reactive electrophiles, predominantly targeting Cys-SH groups but also histidine residues.73,78,92 Overall, it is likely that HSF1 activation by oxidants and other reactive electrophiles reflects a combined effect of derepression from HSP70 and HSP90 by direct damage to these chaperones and their titration away from HSF1 by the buildup of unfolded substrates, predominantly originating from nascent polypeptides and intrinsically unstable proteins (Figure 3).
Stress-Regulated Immunosurveillance of Damaged Cells
Recent successes in the treatment of human malignancies by derepression of anticancer immune responses provided clear evidence for the central role of the immune system in the maintenance and growth of highly immunogenic tumors.93 However, the functions of the immune system, especially components of its innate immunity branch, are also critical for the prevention of cancer. Mouse models with systemic losses of NK cells, NK/T cell perforin deletion, or ablation of epidermal γ/δ T cells all showed increased susceptibility to carcinogen-induced tumorigenesis.94−96 In humans, drug-induced immunosuppression for the prevention of organ transplant rejection is associated with several-fold higher risks of melanoma and other skin cancers.96 For the innate immune system to identify and eliminate damaged cells, its targets need to emit activation/recruitment signals and express distress markers. Two key groups of immunostimulatory danger markers expressed by damaged cells are the MIC and RAET1 families. These proteins serve as ligands for NKG2D receptors expressed by NK cells, subpopulations of γ/δ T cells, and by all human CD8+ T cells.97 Promoter regions of NKG2D receptors-engaging MICA and MICB contain heat shock elements that are bound by activated HSF1 to upregulate transcription.98 Promoters of the ULBP1-6 members of the RAET1 family of NKG2D ligands also contain heat shock elements and are likely to be upregulated by HSF1 during proteotoxic stress.71
Another cellular response stimulating the innate immune system is the production of interferon type I (IFN-I).99 IFN-I operates in a paracrine manner to stimulate macrophages and NK cells and in an autocrine manner to activate cellular production of a number of immunostimulatory cytokines. The binding of IFN-I to the plasma membrane receptors triggers activation of STAT1 by JAK kinase. Tyrosine-phosphorylated STAT1 dimerizes and translocates to the nucleus, leading to the induction of IFN-responsive genes. Although it has been initially characterized in pathogen-infected cells, activation of IFN-I is now a well-established cellular response to damage-associated molecular patterns, which are abnormal molecules in the cytosolic compartment. These abnormal molecules include leaked fragments of nuclear DNA and mitochondrial DNA and dsRNA.100,101 A recent report by Marzio et al. (2022)33 provided strong evidence from experimental models and human clinical studies that INF-I response is regulated by KEAP1 as a separate function from its control of NRF2. Similar to its regulation of NRF2 in unstressed cells, KEAP1 was found to target a proto-oncogene EMSY for ubiquitin-mediated proteasomal degradation. EMSY acts as a negative regulator of IFN-stimulated genes, and consequently, its build-up as a result of KEAP1 inactivation limits the production of proinflammatory cytokines under the conditions of oxidative/electrophilic stress in cells, which restricts recruitment of inflammatory immune cells and their release of oxidants in pathogen-free tissue. Prolonged or overly robust inflammation in the absence of pathogen infections is undesirable and associated with higher cancer risks.102,103 In tumors, proinflammatory cytokines are necessary for efficient recruitment of immune cells and cancer immunosurveillance, and consequently, KEAP1 inactivation/EMSY upregulation is protumorigenic due to suppression of IFN-I response.33 Inflammatory responses in sterile tissues are also regulated by the transcription factor NF-κB which modulates the strength and duration of inflammation.104,105 It was reported that IKKβ, a positive regulator of NF-κB, was a ubiquitination target of KEAP1 and its stabilization upon KEAP1 inactivation led to NF-κB activation.106 However, this function of KEAP1 was not confirmed in a more recent study28 and is inconsistent with suppression of proinflammatory cytokines production in cells with inactive KEAP1.33 Although hypoxic tumor microenvironment is known to be immunosuppressive, this state is induced by low O2 and not by HIF1/HIF2,107 indicating that the oxidant-activated hypoxia response in normoxic cells would not interfere with immunomodulatory actions of HSF1 or KEAP1.
Oxidant-Induced HSF1, HIF1, and NRF2 Responses: Integrative View
Proteotoxicity and the resulting cell injury by oxidants and other reactive electrophiles arise from damage to different amino acids, but Cys-SH is by far the most vulnerable group. Protein-Cys is approximately 1000-times more nucleophilic than the next two most reactive amino acids in proteins: histidine and lysine.108 This sensitivity of Cys-SH was evolutionarily incorporated into the design of specific protein sensors of electrophilic toxicants using Cys-SH as thiol-based switches for the inactivation of negative regulators of cellular stress responses. Operating on this principle are KEAP1 (sensor of oxidants/electrophiles in the NRF2 pathway), PHD2 (oxidant-sensitive regulator of HIF1), and to some extent, HSP70 and HSP90 (negative regulators of HSF1). Another trigger of the heat shock response is the accumulation of oxidant/chemically damaged proteins, which activate HSF1 in a manner similar to that by other protein-denaturing stressors such as hyperthermia. Cys-SH is susceptible to oxidation by all common ROS and RNS (H2O2, O2•–, CO3•–, •NO2, ONOO–, HOCl),17,108 which can originate from both normal metabolism and redox cycling and other prooxidant activity of numerous xenobiotics in their parental or metabolically activated forms. The SH group is a soft Lewis nucleophile (base), making it highly reactive with soft Lewis electrophiles (acids) such as several toxic metals. Numerous organic electrophiles of endogenous and exogenous origin with high Cys-SH reactivity include quinones, epoxides, aldehydes, and other reactive carbonyls, especially α,β-unsaturated aldehydes. Reactivity of these carbonyl compounds with the protein-SH groups involves a rapid Michael addition at the highly polarized β-carbon of the double bond.17
In light of its evolutionary conserved function as a defense response to the buildup of denatured proteins, activation of HSF1 by oxidants and reactive electrophiles (which all primarily damage proteins) is not surprising. As discussed above, induction of the HIF1 responses by oxidants is also well-established.43,44,47−50 To estimate HIF1 responsiveness to protein-damaging agents more generally, we compared the lists of activators of HSF1 (a marker of proteotoxic stress) and HIF1 that were identified in the currently largest screen of chemicals (∼7.5K chemical library, Tox21 program). Almost 80% (77.5%) of HIF1-activating chemicals also tested positive as activators of HSF1, indicating that chemicals with protein-damaging properties constitute a clear majority of the inducers of HIF1 pathway (Figure 4, Supporting Data File S1). As the number of chemicals screened for HSF1 response was smaller, the overlap in activators of HIF1 and HSF1 in the entire library was likely somewhat underestimated. The number of inducers of HSF1 was larger than that for HIF1, which could be due to the detection of chemicals that denature proteins and trigger the heat shock response in the absence of chemical damage. These nonreactive protein denaturants are not expected to act as selective inhibitors of negative regulators of the hypoxia response.
Figure 4.
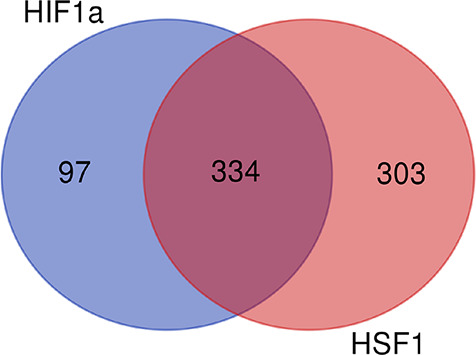
Venn diagram showing the number of chemicals activating HIF1, HSF1, or both transcription factors in a 7.5K chemical library. The diagram was generated using the results of high-throughput screens of HIF1 and HSF1 agonists (activators) performed as part of Tox21 program (https://comptox.epa.gov/dashboard/assay-endpoints; accessed on June 27, 2022). A DMSO-soluble library of stable, low-volatility chemicals was tested for activation of HIF1 (7871 chemicals) and HSF1 (7521 chemicals) in human cervical carcinoma cells. HSF1 agonists were screened in HeLa cells and HIF1 activators in ME-180 cells. HIF1 was activated by 431 and HSF1 by 637 chemicals. The same chemical library has not yet been screened for NRF2 activators.
Activation of the NRF2 pathway represents the most direct protective response to oxidants/electrophiles due to the upregulation of enzymes with a variety of antioxidant activities, including detoxification of ROS, increased glutathione biosynthesis, inactivation of reactive toxicants by Phase II enzymes, and excretion of the resulting conjugates from cells by MRP2. Stimulation of the HIF1 pathway by oxidative inactivation of PHD2, evolutionarily selected primarily as part of cellular hypoxia sensing, suppresses the activity of the electron transport chain and the release of ROS by mitochondria. Mitochondria are frequently the main source of ROS in stressed cells due to their vulnerability to damage by various toxicants. The role of HIF1 as a guardian of mitochondrial health is further supported by its involvement in the elimination of severely diseased mitochondria through activation of mitophagy by two canonical hypoxia response-activated targets BNIP and BNIP3L.35,109 Downregulation of the mitochondrial respiratory chain is accompanied by increased uptake and flow of glucose through glycolysis, establishing O2-independent generation of ATP and producing large amounts of lactic acid, which slows down cell cycle and promotes oxidant-protective protonation of protein-SH groups. Electrophile-induced upregulation of HSF1-inducible genes (such as protein chaperones and ubiquitin) acts to enhance repair and disposal of damaged/unfolded proteins. During chronic stress, all three oxidant-responsive pathways governed by NRF2, HSF1, and HIF1 are typically upregulated as observed in cancers.21,22,35,71 In these conditions, the HSF1 pathway probably deals with unfolded proteins damaged by oxidants and electrophiles escaping detoxification activities of the NRF2-regulated processes and defensive metabolism promoted by HIF1. In the acute oxidative/electrophilic stress rapidly inducing large amounts of protein damage, HSF1 activation is probably the most immediate response due to the evolutionary selected processes for a rapid transcription and translation of HSF1 targets.110 The unparalleled immediacy of the HSF1 response evolved to deal with a massive protein unfolding produced after even brief hyperthermia. Unlike gene expression and protein translation of HSF1 targets, cap-dependent mRNA translation (∼90% of total protein synthesis) and global transcription are actively repressed under severe stress conditions. Components of the cap-dependent translation machinery along with mRNA and damaged nascent polypeptides become insoluble and sequestered in the cytoplasmic granules.110 Upregulation of chaperones and other protein quality control factors accelerate repair and removal of damaged proteins (mostly nascent proteins for both heat shock and oxidative damage),87 allowing resumption of global transcription and translation, including those of NRF2 and HIF1 targets.
The above review of the major functional changes induced by NRF2, HSF1 and HIF1 indicated that their effects on cellular functions are largely nonoverlapping. To support this conclusion by quantitative analyses, we examined gene expression profiles for all three transcription factors to identify their unique and shared transcription targets. First, we compared HSF1 target genes from a comprehensive database described by Kovacs et al.,111 core transcription targets for NRF2 using high confidence NRF2-regulated genes from Ibrahim et al.,112 and HIF1 targets using RNA-seq data38 for the hypoxia-induced genes applying a 2-fold change as a cutoff. Analysis of these data sets showed that there were 0 genes induced by all three transcription factors and only 2 NRF2-activated genes (0.9% of NRF2 targets) were also upregulated by HIF1 (Figure 5A, Supporting Data File S2). The percentage of NRF2 transcription targets that were also induced by HSF1 was higher but still small (8.4% of NRF2 genes). Next, we analyzed the same comprehensive data set for HSF1 target genes111 together with all HIF1-upregulated genes,38 which dramatically increased the number of transcripts from 161 to >1000, and a broader set of NRF2-regulated genes (Table S3 in Ibrahim et al.112). Even in these larger sets of genes, there were only 4 genes (0.6% of NRF2 targets) induced by all three transcription factors (Figure 5B, Supporting Data File S3). The percentages of NRF2-activated genes that were also upregulated by HIF1 and HSF1 also remained small: 5.7 and 4.9%, respectively. Although transcription programs driven by HSF1, HIF1, and NRF2 are clearly very distinct, these factors do not operate completely independently, as they can directly or indirectly affect each other’s activity.47,113,114
Figure 5.
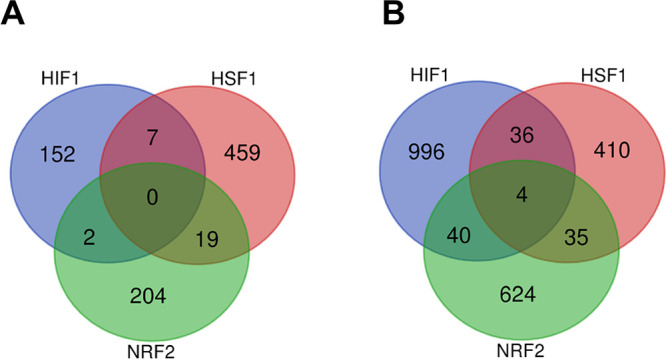
Venn diagrams depicting common and unique transcripts induced by HIF1, HSF1, and NRF2. (A) Venn diagram of overlapping and unique genes generated using a comprehensive HSF1 target genes data set,111 a high confidence NRF2 genes data set (Table S5 in Ibrahim et al.112), and hypoxia-induced genes with 2 or higher fold change.38 (B) Venn diagram of overlapping and unique genes from a comprehensive HSF1 target genes data set111 and broader sets of target transcripts for NRF2 (Table S3 in Ibrahim et al.112) and HIF1 (all hypoxia-induced genes).38
Overall, NRF2, HIF1, and HSF1 pathways represent largely nonoverlapping transcription responses to oxidative/electrophilic stress, which collectively provide a comprehensive defense inside the cell and generate activation signals for surveillance of injured cells by innate immunity (Figure 6). The relative significance of these three responses probably varies dependent on the severity and complexity of the injury, abundance of unfolded proteins (more for bulky protein adducts than for simple SH oxidation), and the extent of mitochondrial damage. Some protein-reactive toxicants can inhibit a specific stress-responsive pathway, leading to the increased reliance of cells on the remaining protective processes. For example, HSF1 activators formaldehyde, acetaldehyde, and cadmium cause impairment of the hypoxia response by damaging HIFα subunits of HIF1 and the related transcription factor HIF2.115 The importance of different oxidant-responsive pathways is also likely to vary among different types of cells. Epithelial cells in tissues that are most commonly exposed to xenobiotics (liver, intestine, lung) typically show the most robust upregulation of a broad set of defense genes by NRF2.15 In lymphoid cells, activation of NRF2 induced a more limited transcription response with many canonical targets remaining unchanged.19 Hematopoietic stem cells contain unusually high concentrations of the nonthiol antioxidant ascorbate,52 and HIF1 plays a critical role in healthy functioning of these and other stem cells.116
Figure 6.
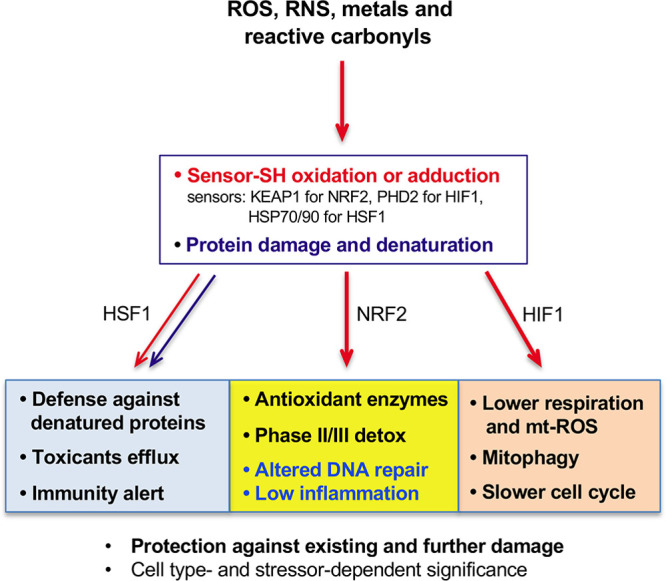
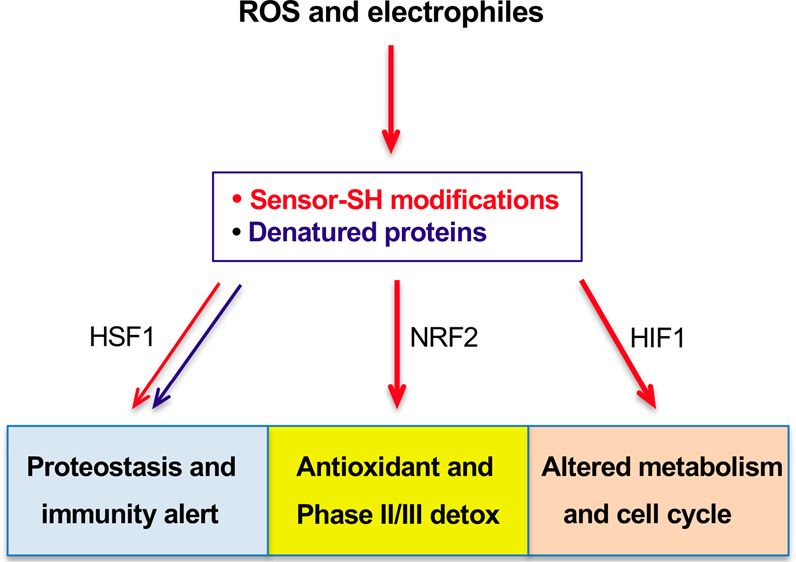
Flowchart depicting activation of HIF1, HSF1, and NRF2 and their main functions in protection against ROS and reactive electrophiles. All three factors are induced through inactivation of their negative regulators that act as sensors of ROS/reactive toxicants via modifications of their Cys-SH groups. HSF1 is also derepressed via titration of HSP70 and HSP90 by unfolded proteins which predominantly include damaged nascent polypeptides. KEAP1/NRF2-dependent promotion of NHEJ at the expense of homologous recombination represents a shift to a faster but more mutagenic repair of oxidants-induced DSBs. Suppression of sterile inflammation by KEAP1 inactivation during oxidative stress in normal tissue is beneficial by limiting a secondary damage. In tumor tissue, a constitutive loss of KEAP1 and the resulting ineffective production of proinflammatory cytokines inhibit cancer immunosurveillance.
Glossary
Abbreviations
- DSBs
DNA double-stranded breaks
- IFN-I
interferon type I
- NHEJ
nonhomologous end-joining
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
Biographies
Anna M. Cyran, MD, is a postdoctoral researcher in the Department of Pathology and Laboratory Medicine at Brown University. She received a medical degree from the Medical University of Warsaw, Poland. As a resident physician in the Department for Otolaryngology, Head and Neck Surgery at Otto-von-Guericke University in Magdeburg, Germany, she received a scholarship from the Else-Kroener Research College for clinician scientists. Her research interests include translational oncology and molecular aspects of chemical carcinogenesis.
Anatoly Zhitkovich is Professor of Medical Science in the Department of Pathology and Laboratory Medicine at Brown University. He received B.Sc. with distinction in biochemistry from Belarusian State University and Ph.D. in biochemistry from Belarusian Academy of Sciences. Prof. Zhitkovich’s research is focused on chemical and biological mechanisms of cell injury by reactive carcinogens and anticancer drugs and activation of stress signaling networks promoting cell survival and DNA repair.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.2c00131.
Author Contributions
CRediT: Anna M. Cyran formal analysis, visualization, writing-review & editing; Anatoly Zhitkovich conceptualization, funding acquisition, supervision, visualization, writing-original draft, writing-review & editing.
This work was supported by grants ES008786, ES028072, and ES020689 from the National Institute of Environmental Health Sciences.
The authors declare no competing financial interest.
Supplementary Material
References
- Sies H.; Berndt C.; Jones D. P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- Dasuri K.; Zhang L.; Keller J. N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- Rains J. L.; Jain S. K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaunig J. E. Oxidative stress and cancer. Cur. Pharm. Des. 2019, 24, 4771–4778. 10.2174/1381612825666190215121712. [DOI] [PubMed] [Google Scholar]
- Hajam Y. A.; Rani R.; Ganie S. Y.; Sheikh T. A.; Javaid D.; Qadri S. S.; Pramodh S.; Alsulimani A.; Alkhanani M. F.; Harakeh S.; Hussain A.; Haque S.; Reshi M. S. Oxidative stress in human pathology and aging: molecular mechanisms and perspectives. Cells 2022, 11, 552. 10.3390/cells11030552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 2010, 43, 673–683. 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Carroll K. S.; Liebler D. C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics 2016, 15, 1–11. 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan J. J.; Marnett L. J. Histone adduction and its functional impact on epigenetics. Chem. Res. Toxicol. 2017, 30, 376–387. 10.1021/acs.chemrestox.6b00379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galligan J. J.; Wepy J. A.; Streeter M. D.; Kingsley P. J.; Mitchener M. M.; Wauchope O. R.; Beavers W. N.; Rose K. L.; Wang T.; Spiegel D. A.; Marnett L. J. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. U S A 2018, 115, 9228–9233. 10.1073/pnas.1802901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth R. K.; Warfel N. A. Strange bedfellows: nuclear factor, erythroid 2-Like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants 2017, 6, 27. 10.3390/antiox6020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anckar J.; Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011, 80, 1089–1115. 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- Gomez-Pastor R.; Burchfiel E. T.; Thiele D. J. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 4–19. 10.1038/nrm.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuse Y.; Kobayashi M. Conservation of the Keap1-Nrf2 system: an evolutionary journey through stressful space and time. Molecules 2017, 22, 436. 10.3390/molecules22030436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytkönen K. T.; Williams T. A.; Renshaw G. M.; Primmer C. R.; Nikinmaa M. Molecular evolution of the metazoan PHD-HIF oxygen-sensing system. Mol. Biol. Evol. 2011, 28, 1913–1926. 10.1093/molbev/msr012. [DOI] [PubMed] [Google Scholar]
- Baird L.; Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2022, 40, e00099-20. 10.1128/MCB.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidlin C. J.; Dodson M. B.; Madhavan L.; Zhang D. D. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. 10.1016/j.freeradbiomed.2019.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A. N-Acetylcysteine: antioxidant, aldehyde scavenger, and more. Chem. Res. Toxicol. 2019, 32, 1318–1319. 10.1021/acs.chemrestox.9b00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa R. K.; Mai K. H.; Srisuma S.; Kensler T. W.; Yamamoto M.; Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002, 62, 5196–5203. [PubMed] [Google Scholar]
- Chorley B. N.; Campbell M. R.; Wang X.; Karaca M.; Sambandan D.; Bangura F.; Xue P.; Pi J.; Kleeberger S. R.; Bell D. A. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429. 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollrath V.; Wielandt A. M.; Iruretagoyena M.; Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 2006, 395, 599–609. 10.1042/BJ20051518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon S.; Columbano A.; Giordano S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- Schmidlin C. J.; Shakya A.; Dodson M.; Chapman E.; Zhang D. D. The intricacies of NRF2 regulation in cancer. Semin. Cancer Biol. 2021, 76, 110–119. 10.1016/j.semcancer.2021.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Gomez M.; Kwak M. K.; Dolan P. M.; Itoh K.; Yamamoto M.; Talalay P.; Kensler T. W. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. U S A 2001, 98, 3410–3415. 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor T. O.; Huang M. T.; Prawan A.; Liu Y.; Hao X.; Yu S.; Cheung W. K.; Chan J. Y.; Reddy B. S.; Yang C. S.; Kong A. N. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola G. M.; Karreth F. A.; Humpton T. J.; Gopinathan A.; Wei C.; Frese K.; Mangal D.; Yu K. H.; Yeo C. J.; Calhoun E. S.; Scrimieri F.; Winter J. M.; Hruban R. H.; Iacobuzio-Donahue C.; Kern S. E.; Blair I. A.; Tuveson D. A. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H.; Moriguchi T.; Takai J.; Ebina M.; Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013, 73, 4158–4168. 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- Tao S.; Rojo de la Vega M.; Chapman E.; Ooi A.; Zhang D. D. The effects of NRF2 modulation on the initiation and progression of chemically and genetically induced lung cancer. Mol. Carcinog. 2018, 57, 182–192. 10.1002/mc.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo X.; Niu Q.; Ivanov A. A.; Tsang Y. H.; Tang C.; Shu C.; Li Q.; Qian K.; Wahafu A.; Doyle S. P.; Cicka D.; Yang X.; Fan D.; Reyna M. A.; Cooper L. A.D.; Moreno C. S.; Zhou W.; Owonikoko T. K.; Lonial S.; Khuri F. R.; Du Y.; Ramalingam S. S.; Mills G. B.; Fu H. Systematic discovery of mutation-directed neo-protein-protein interactions in cancer. Cell 2022, 185, 1974–1985. 10.1016/j.cell.2022.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerins M. J.; Ooi A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. 10.1038/s41598-018-31281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajares M.; Jiménez-Moreno N.; García-Yagüe Á. J.; Escoll M.; de Ceballos M. L.; Van Leuven F.; Rábano A.; Yamamoto M.; Rojo A. I.; Cuadrado A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. 10.1080/15548627.2016.1208889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. B.; Pandita R. K.; Eskiocak U.; Ly P.; Kaisani A.; Kumar R.; Cornelius C.; Wright W. E.; Pandita T. K.; Shay J. W. Targeting of Nrf2 induces DNA damage signaling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. U S A 2012, 109, E2949–E2955. 10.1073/pnas.1207718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A.; Jeggo P. A. Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks. DNA Repair 2020, 93, 102915. 10.1016/j.dnarep.2020.102915. [DOI] [PubMed] [Google Scholar]
- Marzio A.; Kurz E.; Sahni J. M.; Di Feo G.; Puccini J.; Jiang S.; Hirsch C. A.; Arbini A. A.; Wu W. L.; Pass H. I.; Bar-Sagi D.; Papagiannakopoulos T.; Pagano M. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell 2022, 185, 169–183. 10.1016/j.cell.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiano J.; Zolnerowich N.; Wu W.; Pavani R.; Wang C.; Li H.; Zheng L.; Shen B.; Sleckman B. P.; Chen B. R.; Nussenzweig A. Role of 53BP1 in end protection and DNA synthesis at DNA breaks. Genes Dev. 2021, 35, 1356–1367. 10.1101/gad.348667.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B.; Johnson R. S.; Simon M. C. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L.; Roth P. H.; Fang H. M.; Wang G. L. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. 10.1016/S0021-9258(17)31580-6. [DOI] [PubMed] [Google Scholar]
- Chan M. C.; Ilott N. E.; Schödel J.; Sims D.; Tumber A.; Lippl K.; Mole D. R.; Pugh C. W.; Ratcliffe P. J.; Ponting C. P.; Schofield C. J. Tuning the transcriptional response to hypoxia by Inhibiting hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases. J. Biol. Chem. 2016, 291, 20661–20673. 10.1074/jbc.M116.749291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I.; Cairns R. A.; Fontana L.; Lim A. L.; Denko N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Kim J. W.; Tchernyshyov I.; Semenza G. L.; Dang C. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Fukuda R.; Zhang H.; Kim J. W.; Shimoda L.; Dang C. V.; Semenza G. L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- Tello D.; Balsa E.; Acosta-Iborra B.; Fuertes-Yebra E.; Elorza A.; Ordóñez Á.; Corral-Escariz M.; Soro I.; López-Bernardo E.; Perales-Clemente E.; Martínez-Ruiz A.; Enríquez J. A.; Aragonés J.; Cadenas S.; Landázuri M. O. Induction of the mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Brunelle J. K.; Bell E. L.; Quesada N. M.; Vercauteren K.; Tiranti V.; Zeviani M.; Scarpulla R. C.; Chandel N. S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Niecknig H.; Tug S.; Reyes B. D.; Kirsch M.; Fandrey J.; Berchner-Pfannschmidt U. Role of reactive oxygen species in the regulation of HIF-1 by prolyl hydroxylase 2 under mild hypoxia. Free Radic. Res. 2012, 46, 705–717. 10.3109/10715762.2012.669041. [DOI] [PubMed] [Google Scholar]
- Chandel N. S.; McClintock D. S.; Feliciano C. E.; Wood T. M.; Melendez J. A.; Rodriguez A. M.; Schumacker P. T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- Guzy R. D.; Hoyos B.; Robin E.; Chen H.; Liu L.; Mansfield K. D.; Simon M. C.; Hammerling U.; Schumacker P. T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Briggs K. J.; Koivunen P.; Cao S.; Backus K. M.; Olenchock B. A.; Patel H.; Zhang Q.; Signoretti S.; Gerfen G. J.; Richardson A. L.; Witkiewicz A. K.; Cravatt B. F.; Clardy J.; Kaelin W. G. Jr Paracrine induction of HIF by glutamate in breast cancer: EglN1 senses cysteine. Cell 2016, 166, 126–139. 10.1016/j.cell.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.; Won H. S.; Lee Y. M.; Choi J. W.; Oh T. I.; Jang J. H.; Choi D. K.; Lim B. O.; Kim Y. J.; Park J. W.; Puigserver P.; Lim J. H. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1α activation. Sci. Rep. 2016, 6, 18928. 10.1038/srep18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Flashman E.; Mecinović J.; Kramer H. B.; Kessler B. M.; Frapart Y. M.; Boucher J. L.; Clifton I. J.; McDonough M. A.; Schofield C. J. Studies on the reaction of nitric oxide with the hypoxia-inducible factor prolyl hydroxylase domain 2 (EGLN1). J. Mol. Biol. 2011, 410, 268–279. 10.1016/j.jmb.2011.04.075. [DOI] [PubMed] [Google Scholar]
- Mecinović J.; Chowdhury R.; Liénard B. M.; Flashman E.; Buck M. R.; Oldham N. J.; Schofield C. J. ESI-MS studies on prolyl hydroxylase domain 2 reveal a new metal binding site. ChemMedChem. 2008, 3, 569–572. 10.1002/cmdc.200700233. [DOI] [PubMed] [Google Scholar]
- Salnikow K.; Donald S. P.; Bruick R. K.; Zhitkovich A.; Phang J. M.; Kasprzak K. S. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem. 2004, 279, 40337–40344. 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- Zhitkovich A. Nuclear and cytoplasmic functions of vitamin C. Chem. Res. Toxicol. 2020, 33, 2515–2526. 10.1021/acs.chemrestox.0c00348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Sonveaux P.; Rabbani Z. N.; Liu S.; Yan B.; Huang Q.; Vujaskovic Z.; Dewhirst M. W.; Li C. Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. 10.1016/j.molcel.2007.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y.; Murdoch C. E.; Sano S.; Ido Y.; Bachschmid M. M.; Cohen R. A.; Matsui R. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilize HIF-1α and improve limb revascularization. Proc. Natl. Acad. Sci. U S A 2016, 113, 6011–6016. 10.1073/pnas.1524198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavers W. N.; Rose K. L.; Galligan J. J.; Mitchener M. M.; Rouzer C. A.; Tallman K. A.; Lamberson C. R.; Wang X.; Hill S.; Ivanova P. T.; Brown H. A.; Zhang B.; Porter N. A.; Marnett L. J. Protein Modification by Endogenously Generated Lipid Electrophiles: Mitochondria as the Source and Target. ACS Chem. Biol. 2017, 12, 2062–2069. 10.1021/acschembio.7b00480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P.; Hirsilä M.; Remes A. M.; Hassinen I. E.; Kivirikko K. I.; Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 2007, 282, 4524–4532. 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- Ooi A.; Wong J. C.; Petillo D.; Roossien D.; Perrier-Trudova V.; Whitten D.; Min B. W.; Tan M. H.; Zhang Z.; Yang X. J.; Zhou M.; Gardie B.; Molinié V.; Richard S.; Tan P. H.; Teh B. T.; Furge K. A. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Adam J.; Hatipoglu E.; O’Flaherty L.; Ternette N.; Sahgal N.; Lockstone H.; Baban D.; Nye E.; Stamp G. W.; Wolhuter K.; Stevens M.; Fischer R.; Carmeliet P.; Maxwell P. H.; Pugh C. W.; Frizzell N.; Soga T.; Kessler B. M.; El-Bahrawy M.; Ratcliffe P. J.; Pollard P. J. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai R.; Brock J. W.; Blatnik M.; Baatz J. E.; Bethard J.; Walla M. D.; Thorpe S. R.; Baynes J. W.; Frizzell N. Succination of protein thiols during adipocyte maturation. J. Biol. Chem. 2007, 282, 34219–34228. 10.1074/jbc.M703551200. [DOI] [PubMed] [Google Scholar]
- Morin A.; Letouzé E.; Gimenez-Roqueplo A. P.; Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int. J. Cancer 2014, 135, 2237–2248. 10.1002/ijc.29080. [DOI] [PubMed] [Google Scholar]
- Yun J.; Mullarky E.; Lu C.; Bosch K. N.; Kavalier A.; Rivera K.; Roper J.; Chio I. I.; Giannopoulou E. G.; Rago C.; Muley A.; Asara J. M.; Paik J.; Elemento O.; Chen Z.; Pappin D. J.; Dow L. E.; Papadopoulos N.; Gross S. S.; Cantley L. C. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luczak M. W.; Krawic C.; Zhitkovich A. NAD+ metabolism controls growth inhibition by HIF1 in normoxia and determines differential sensitivity of normal and cancer cells. Cell Cycle 2021, 20, 1812–1827. 10.1080/15384101.2021.1959988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schito L.; Semenza G. L. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. 10.1016/j.trecan.2016.10.016. [DOI] [PubMed] [Google Scholar]
- Shen C.; Beroukhim R.; Schumacher S. E.; Zhou J.; Chang M.; Signoretti S.; Kaelin W. G. Jr Genetic and functional studies implicate HIF1a as a 14q kidney cancer suppressor gene. Cancer Discovery 2011, 1, 222–235. 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scortegagna M.; Martin R. J.; Kladney R. D.; Neumann R. G.; Arbeit J. M. Hypoxia-inducible factor-1α suppresses squamous carcinogenic progression and epithelial-mesenchymal transition. Cancer Res. 2009, 69, 2638–2646. 10.1158/0008-5472.CAN-08-3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco-Hernandez T.; Hyrenius-Wittsten A.; Rehn M.; Bryder D.; Cammenga J. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood 2014, 124, 3597–3607. 10.1182/blood-2014-04-567065. [DOI] [PubMed] [Google Scholar]
- Dokladny K.; Zuhl M. N.; Mandell M.; Bhattacharya D.; Schneider S.; Deretic V.; Moseley P. L. Regulatory coordination between two major intracellular homeostatic systems: heat shock response and autophagy. J. Biol. Chem. 2013, 288, 14959–14972. 10.1074/jbc.M113.462408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kioka N.; Yamano Y.; Komano T.; Ueda K. Heat-shock responsive elements in the induction of the multidrug resistance gene (MDR1). FEBS Lett. 1992, 301, 37–40. 10.1016/0014-5793(92)80205-U. [DOI] [PubMed] [Google Scholar]
- Vilaboa N. E.; Galan A.; Troyano A.; de Blas E.; Aller P. Regulation of multidrug resistance 1 (MDR1)/P-glycoprotein gene expression and activity by heat-shock transcription factor 1 (HSF1). J. Biol. Chem. 2000, 275, 24970–24976. 10.1074/jbc.M909136199. [DOI] [PubMed] [Google Scholar]
- Cyran A. M.; Zhitkovich A. Heat shock proteins and HSF1 in Cancer. Front. Oncol. 2022, 12, 860320. 10.3389/fonc.2022.860320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Systems analysis of protein modification and cellular responses induced by electrophile stress. Acc. Chem. Res. 2010, 43, 673–683. 10.1021/ar900286y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patinen T.; Adinolfi S.; Cortés C. C.; Härkönen J.; Jawahar Deen A.; Levonen A. L. Regulation of stress signaling pathways by protein lipoxidation. Redox Biol. 2019, 23, 101114. 10.1016/j.redox.2019.101114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J. D.; Wang Y.; Morano K. A. Small molecule activators of the heat shock response: Chemical properties, molecular targets, and therapeutic promise. Chem. Res. Toxicol. 2012, 25, 2036–2053. 10.1021/tx300264x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs A. T.; Marnett L. J. Heat shock factor 1 attenuates 4-hydroxynonenal-mediated apoptosis: Critical role for heat shock protein 70 induction and stabilization of Bcl-XL. J. Biol. Chem. 2007, 282, 33412–33420. 10.1074/jbc.M706799200. [DOI] [PubMed] [Google Scholar]
- Gan N.; Wu Y. C.; Brunet M.; Garrido C.; Chung F. L.; Dai C.; Mi L. Sulforaphane activates heat shock response and enhances proteasome activity through up-regulation of Hsp27. J. Biol. Chem. 2010, 285, 35528–35536. 10.1074/jbc.M110.152686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kansanen E.; Jyrkkanen H.-K.; Volger O. L.; Leinonen H.; Kivela A. M.; Hakkinen S.-K.; Woodcock S. R.; Schopfer F. J.; Horrevoets A. J.; Yla-Herttuala S.; Freeman B. A.; Levonen A.-L.; et al. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: Identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009, 284, 33233–33241. 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Ahn Y. H.; Benjamin I. J.; Honda T.; Hicks R. J.; Calabrese V.; Cole P. A.; Dinkova-Kostova A. T. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem. Biol. 2011, 18, 1355–1361. 10.1016/j.chembiol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S. G.; Thiele D. J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003, 17, 516–528. 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M.; Kim H. E.; Li C. R.; Kim S.; Kwak I. J.; Lee Y. J.; Kim S. S.; Moon J. Y.; Kim C. H.; Kim D. K.; Kang H. S.; Park J. S. Two distinct disulfide bonds formed in human heat shock transcription factor 1 act in opposition to regulate its DNA binding activity. Biochemistry 2008, 47, 6007–6015. 10.1021/bi702185u. [DOI] [PubMed] [Google Scholar]
- Rossin F.; Villella V. R.; D’Eletto M.; Farrace M. G.; Esposito S.; Ferrari E.; Monzani R.; Occhigrossi L.; Pagliarini V.; Sette C.; Cozza G.; Barlev N. A.; Falasca L.; Fimia G. M.; Kroemer G.; Raia V.; Maiuri L.; Piacentini M. TG2 regulates the heat-shock response by the post-translational modification of HSF1. EMBO Rep. 2018, 19, e45067. 10.15252/embr.201745067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N.; Feng H.; Wang S.; Punekar A. S.; Ladenstein R.; Wang D.-C.; Zhang Q.; Ding J.; Liu W.; et al. Structures of heat shock factor trimers bound to DNA. iScience 2021, 24, 102951. 10.1016/j.isci.2021.102951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzyński M. A.; Puustinen M. C.; Joutsen J.; Sistonen L. Uncoupling Stress-Inducible Phosphorylation of Heat Shock Factor 1 from Its Activation. Mol. Cell. Biol. 2015, 35, 2530–2540. 10.1128/MCB.00816-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X.; Krakowiak J.; Patel N.; Beyzavi A.; Ezike J.; Khalil A. S.; Pincus D. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. Elife 2016, 5, e18638. 10.7554/eLife.18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masser A. E.; Kang W.; Roy J.; Mohanakrishnan Kaimal J.; Quintana-Cordero J.; Friedländer M. R.; Andréasson C. Cytoplasmic protein misfolding titrates Hsp70 to activate nuclear Hsf1. Elife 2019, 8, e47791. 10.7554/eLife.47791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye B. W.; Churchman L. S. Hsf1 activation by proteotoxic stress requires concurrent protein synthesis. Mol. Biol. Cell 2021, 32, 1800–1806. 10.1091/mbc.E21-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B.; Goldberg A. L. Heat shock and oxygen radicals stimulate ubiquitin-dependent degradation mainly of newly synthesized proteins. J. Cell Biol. 2008, 182, 663–673. 10.1083/jcb.200803022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone D. L.; Doorn J. A.; Kiebler Z.; Sampey B. P.; Petersen D. R. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 2004, 17, 1459–1467. 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang H.; Gong W.; Liu Z.; Wu H.; Hu W.; Chen X.; Wang L.; Wu S.; Chen C.; Perrett S. S-Glutathionylation of human inducible Hsp70 reveals a regulatory mechanism involving the C-terminal α-helical lid. J. Biol. Chem. 2020, 295, 8302–8324. 10.1074/jbc.RA119.012372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Gibney P. A.; West J. D.; Morano K. A. The yeast Hsp70 Ssa1 is a sensor for activation of the heat shock response by thiol-reactive compounds. Mol. Biol. Cell 2012, 23, 3290–3298. 10.1091/mbc.e12-06-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Gong W.; Wu S.; Perrett S. Hsp70 in redox homeostasis. Cells 2022, 11, 829. 10.3390/cells11050829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R. E.; Marnett L. J.; Liebler D. C. Protein-selective capture to analyze electrophile adduction of Hsp90 by 4-hydroxynonenal. Chem. Res. Toxicol. 2011, 24, 1275–1282. 10.1021/tx200157t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A.; Wolchok J. D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi M.; Oppenheim D. E.; Steele C. R.; Lewis J. M.; Glusac E.; Filler R.; Hobby P.; Sutton B.; Tigelaar R. E.; Hayday A. C.; et al. Regulation of cutaneous malignancy by gammadelta T cells. Science 2001, 294, 605–609. 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- Ortner D.; Tripp C. H.; Komenda K.; Dubrac S.; Zelger B.; Hermann M.; Doppler W.; Tymoszuk P. Z.; Boon L.; Clausen B. E.; Stoitzner P.; et al. Langerhans cells and NK cells cooperate in the inhibition of chemical skin carcinogenesis. Oncoimmunology 2017, 6, e1260215. 10.1080/2162402X.2016.1260215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley M. J.; Thomson J.; Harwood C.; Leigh I. The role of the immune system in cutaneous squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2009. 10.3390/ijms20082009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel D.; Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front. Immunol. 2018, 9, 2040. 10.3389/fimmu.2018.02040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh V.; Bahram S.; Bauer S.; Herman A.; Beauchamp M.; Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc. Natl. Acad. Sci. U S A 1996, 93, 12445–12450. 10.1073/pnas.93.22.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton S. E.; Saleiro D.; Platanias L. C. Type I and II interferons in the anti-tumor immune response. Cancers 2021, 13, 1037. 10.3390/cancers13051037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury A.; Witte S.; Aich A. Role of mitochondrial nucleic acid sensing pathways in health and patho-physiology. Front. Cell Dev. Biol. 2022, 10, 796066. 10.3389/fcell.2022.796066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Zhao H.; Shen Y.; Chen Q. A Variety of nucleic acid species are sensed by cGAS, implications for its diverse functions. Front. Immunol. 2022, 13, 826880. 10.3389/fimmu.2022.826880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten F. R.; Grivennikov S. I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity 2019, 51, 27–41. 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerling C.; Casbon A.-J.; Werb Z. Balancing the innate immune system in tumor development. Trends Cell Biol. 2015, 25, 214–220. 10.1016/j.tcb.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonina I. S.; Zhong Z.; Karin M.; Beyaert R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. 10.1038/ni.3772. [DOI] [PubMed] [Google Scholar]
- Liu D.; Zhong Z.; Karin M. NF-κB: A double-edged sword controlling inflammation. Biomedicines 2022, 10, 1250. 10.3390/biomedicines10061250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. F.; Kuo H. P.; Liu M.; Chou C. K.; Xia W.; Du Y.; Shen J.; Chen C. T.; Huo L.; Hsu M. C.; Li C. W.; Ding Q.; Liao T. L.; Lai C. C.; Lin A. C.; Chang Y. H.; Tsai S. F.; Li L. Y.; Hung M. C. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miar A.; Arnaiz E.; Bridges E.; Beedie S.; Cribbs A. P.; Downes D. J.; Beagrie R. A.; Rehwinkel J.; Harris A. L. Hypoxia induces transcriptional and translational downregulation of the type i IFN pathway in multiple cancer cell types. Cancer Res. 2020, 80, 5245–5256. 10.1158/0008-5472.CAN-19-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvez S.; Long M. J. C.; Poganik J. R.; Aye Y. Redox signaling by reactive electrophiles and oxidants. Chem. Rev. 2018, 118, 8798–8888. 10.1021/acs.chemrev.7b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure N. M.; Pouysségur J. Hypoxia-induced autophagy: Cell death or cell survival?. Curr. Opin.Cell Biol. 2010, 22, 177–180. 10.1016/j.ceb.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Alagar Boopathy L. R.; Jacob-Tomas S.; Alecki C.; Vera M. Mechanisms tailoring the expression of heat shock proteins to proteostasis challenges. J. Biol. Chem. 2022, 298, 101796. 10.1016/j.jbc.2022.101796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács D.; Sigmond T.; Hotzi B.; Bohár B.; Fazekas D.; Deák V.; Vellai T.; Barna J. HSF1Base: A comprehensive database of HSF1 (heat shock factor 1) target genes. Int. J. Mol. Sci. 2019, 20, 5815. 10.3390/ijms20225815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L.; Mesgarzadeh J.; Xu I.; Powers E. T.; Wiseman R. L.; Bollong M. J. Defining the Functional Targets of Cap’n’collar Transcription Factors NRF1, NRF2, and NRF3. Antioxidants 2020, 9, 1025. 10.3390/antiox9101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küper A.; Baumann J.; Göpelt K.; Baumann M.; Sänger C.; Metzen E.; Kranz P.; Brockmeier U. Overcoming hypoxia-induced resistance of pancreatic and lung tumor cells by disrupting the PERK-NRF2-HIF-axis. Cell Death Dis. 2021, 12, 82. 10.1038/s41419-020-03319-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte T. L.; Talbot N. P.; Drakesmith H. NRF2 and hypoxia-inducible factors: key players in the redox control of systemic iron homeostasis. Antioxid. Redox Signal. 2021, 35, 433–452. 10.1089/ars.2020.8148. [DOI] [PubMed] [Google Scholar]
- Meyers L. M.; Luczak M. W.; Krawic C.; Zhitkovich A. Vulnerability of HIF1a and HIF2a to damage by proteotoxic stressors. Toxicol. Appl. Pharmacol. 2022, 445, 116041. 10.1016/j.taap.2022.116041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. Dynamic regulation of stem cell specification and maintenance by hypoxia-inducible factors. Mol. Aspects Med. 2016, 47–48, 15–23. 10.1016/j.mam.2015.09.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.