

Abstract

The high mobility group box 1 (HMGB1), which is released during acute acetaminophen (APAP) overdose, is thought to mediate a subsequent immune response, particularly hepatic infiltration of macrophages. The redox behavior of HMGB1 and the proteoforms of HMGB1 present in oxidative environments has been the subject of a number of confusing and contradictory studies. Therefore, a stable isotope dilution two-dimensional nanoultrahigh-performance liquid chromatography parallel reaction monitoring/high-resolution mass spectrometry method was developed in order to characterize and quantify oxidative modifications to the cysteine (Cys) residues (Cys-23, Cys-45, and Cys-106) that are present in HMGB1. Disulfide linkages were determined using carbamidoethyl derivatization before and after reduction as well as by direct analysis of disulfide cross-linked peptides. A stable isotope labeled form of HMGB1 was used as an internal standard to correct for sample to sample differences in immunoaffinity precipitation, derivatization, and electrospray ionization. Four discrete HMGB1 proteoforms were found to be released from a hepatocarcinoma cell model of APAP overdose after 24 h. Fully reduced HMGB1 with all three Cys-residues in their free thiol state accounted for 18% of the secreted HMGB1. The proteoform with disulfide between Cys-23 and Cys-45 accounted for 24% of the HMGB1. No evidence was obtained for a disulfide cross-link between Cys-106 and the other two Cys-residues. However, 45% of the HMGB1 formed a cross-link with unidentified intracellular proteins via an intermolecular disulfide bond, and 12% was present as the terminally oxidized cysteic acid. Surprisingly, there was no evidence for the formation of HMGB1 disulfides with GSH or other low molecular weight thiols. Secreted plasma HMGB1 Cys-23/Cys45 disulfide proteoform together with the Cys-106/protein disulfide proteoforms could potentially serve as early biomarkers of hepatoxicity after APAP overdose as well as biomarkers of drug-induced liver injury.
Introduction
High mobility group box 1 (HMGB1), the most abundant nonhistone protein in the nucleus, was first isolated as a heparin binding protein (p30) with neurite outgrowth activity1 and its primary structure deduced from the nucleotide sequence.2 The 215 amino acid single-chain polypeptide is composed of A-box and B-box DNA-binding domains, an acidic C-terminal tail, and several redox-sensitive amino acids, including three highly conserved Cys residues (Scheme 1). HMGB1 is a multifunctional protein that acts as a DNA chaperone as well as a damage-associated molecular pattern with immune-stimulating effects.3
Scheme 1. Amino Acid Sequence of HMGB1 Showing the 3 Cys-Residues at 23, 45, and 106 (in brown), the 2 NLS Regions, and the Tail with 30 Acidic Amino Acids (in blue).

HMGB1 can be released in multiple contexts and bind to a variety of extracellular receptors. Release of HMGB1 occurs passively in necrotic cells,3 and it can be released during regulated apoptosis of certain cell types.4 Active secretion of HMGB1 from activated immune cells is possible, at least in part through CD14-and TNF-dependent mechanisms.5 The immunological effects exerted by extracellular HMGB1 are mediated through multiple receptors, including the receptor for advanced glycation end products6 as well as toll-like receptors 2 and 4.7,8 In addition, HMGB1 can recruit inflammatory cells to damaged tissues by forming a complex with chemokine receptors such as CXCL 12 and signaling via CXCR4.9 HMGB1 binding to extracellular receptors can induce a range of immunological effects including inflammation,3 migration of maturing dendritic cells,10 and release of cytokines like TNFα.11 The contribution of HMGB1 to pathological processes is evident from the decrease in lethality of APAP overdose resulting from the administration of anti-HMGB1 antibodies3 or heparin.12
It has been widely proposed that the HMGB1 oxidation state and extracellular function are linked in a ternary relationship through its three Cys-residues: Cys-23, Cys-45, and Cys-106 (Scheme 1). It was suggested that HMGB1 Cys residues could be fully reduced, form a disulfide between Cys-23 and Cys-45 with Cys-106 being reduced, or all three Cys-residues could be terminally oxidized to cysteic acid residues.13 These proteoforms with different oxidation states were proposed to correspond to chemokine functionality and cytokine functionality and inactivity, respectively. Sequential release of each HMGB1 oxidative proteoform from injured cells as pathological oxidative stress progressed and therefore corresponded with each stage of inflammation; specifically, leukocyte recruitment, leukocyte activation, and eventual resolution of inflammation (reviewed in Lu et al.).14 Much of the literature supporting such a relationship has been confused by retractions, temporary removal, or expressions of concern, and there is a paucity of studies providing definitive characterization of secreted oxidized HMGB1 proteoforms.15−23 The role of HMGB1 oxidation in many pathological conditions is therefore unclear despite the potential effects this might have on immune-stimulating function. Drug-induced liver injury (DILI) is a form of liver damage during which HMGB1 is released into circulation where it can alter the immunological response. Oxidized proteoforms of HMGB1 have the potential to act as biomarkers of DILI and facilitate the development of specific therapeutics, further motivating clarification of the specific oxidized proteoforms of HMGB1 that are secreted.
APAP overdose results in a potentially fatal condition involving acute oxidative stress in the liver. APAP toxicity is one of the most common forms of DILI, resulting in over 30,000 hospitalizations and hundreds of deaths per year.24 APAP is metabolized in hepatocytes primarily via sulfation and glucuronidation, with a small fraction being metabolized into the reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI)25 primarily by cytochrome P450 (CYP) 2E1.26 At subtoxic doses, NAPQI is detoxified by glutathione (GSH), either by conjugation or reduction back to APAP.25 In overdose situations where GSH is depleted, NAPQI is thought to induce hepatocyte cell death via conjugation to intracellular proteins, particularly mitochondrial proteins, leading to reactive oxygen species (ROS) leakage from the electron transport chain, mitochondrial dysfunction, and acute oxidative stress.27
Here, we report the characterization of HMGB1 oxidation states from an in vitro hepatocarcinoma cell model of APAP overdose using stable isotope dilution two-dimensional nanoultra-high- performance liquid chromatography parallel reaction monitoring/high-resolution mass spectrometry (2D-nano-UHPLC-PRM/HRMS). HepaRG cells, which have similar morphology, CYP2E1 expression, and APAP sensitivity to primary human hepatocytes,28 were used as an in vitro hepatic model. Covalent labeling of free Cys thiols with acrylamide with or without preceding Cys reduction allowed for differentiation between free thiols and disulfide Cys residues in HMGB1. A [13C15N]-HMGB1 internal standard, prepared through stable isotope labeling by amino acids in cell culture (SILAC), was used for normalization and quantification. HMGB1 digestion with trypsin or Glu-C endoproteinases yielded peptides containing all three HMGB1 Cys-residues, allowing direct detection and quantification of carbamidoethyl (CAE)-Cys derivatives, Cys-disulfides, and terminally oxidized cysteic acid. Four distinct HMGB1 proteoforms that were secreted by high-dose APAP from the HepaRG hepatocytes were characterized, and their relative amounts established.
Materials and Methods
Chemicals and Reagents
Reagents and solvents were LC/MS-grade quality unless otherwise noted. Biosynth Carbosynth (previously Vivitide) (Gardner, MA) provided peptides with the sequences M[O]SSYAFFVQTC[O3H]RE, M[O]SSYAFFVQTCRE, RPPSAFFLFCSEYRPK, and RPPSAFFLFC[O3H]SEYRPK (>95%). Burdick and Jackson (Muskegon, MI) supplied LC/MS grade water and acetonitrile. Cambridge Isotope Laboratories (Andover, MA) supplied [13C615N2] lysine, [13C915N1] tyrosine, and [2H6] acetic anhydride. EMSCO/FISHER (Philadelphia, PA) supplied Pierce Protein A/G magnetic agarose beads, formic acid, Dulbecco’s phosphate buffered saline (DPBS), Gibco HepaRG cryopreserved cells, Gibco HepaRG thaw, plate, and general-purpose medium supplement, and Gibco Williams E medium with glutaMAX. Fisher Scientific (Pittsburgh, PA) supplied ammonium hydroxide (Optima). MilliporeSigma (Billerica, MA) supplied the anti-HMGB1 rabbit polyclonal antibody (pAb) against the C-terminal acidic tail of HMGB1 (H9539), APAP, GSH, acrylamide, ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail, l-cysteine, chloramine-T hydrate, triethanolamine, and ethanolamine. Novus Biologicals (Centennial, CO) supplied polarity sensitive indicator of viability (pSIVA) and propidium iodide (PI). Promega (Madison, WI) supplied mass spectrometry grade trypsin endoproteinase. Thermo Fisher Scientific (Waltham, MA) supplied tris(2-carboxyethyl)-phosphine (TCEP), endoproteinase Glu-C from Staphylococcus aureus V8, Tris HCl pH 8 solution, and dimethyl pimelimidate (DMP). Horseradish peroxidase (HRP) was supplied by Santa Cruz Biotechnology, Dallas, TX). PerkinElmer (Waltham, MA) supplied the Western Lightning Plus electrochemical luminescence (ECL) reagent.
APAP Hepatocyte Overdose Cell Model
Differentiated HepaRG cells were seeded at a density of 0.5 × 106 cells per well in 24-well plates in Williams E medium with glutaMAX and 1× Gibco HepaRG thaw, plate, and general-purpose medium supplement added. Cells were cultured at 37 °C with 21% O2 and 5% CO2 for 8 days to allow full expression of cytochrome P450 enzymes, according to manufacturer instructions (Biopredic International, Rennes, France). Williams E medium containing 10 mM APAP was added to cells for 24 h to model the toxic overdose of APAP. A 10 mM APAP concentration was chosen to model severe APAP toxicity as, according to the Rumack–Matthew nomogram, hepatotoxicity becomes likely in vivo starting at serum APAP concentrations of approximately 3 mM at 4 h postoral ingestion.29 Given maximal APAP serum concentrations occurring 0.5–1.5 h after oral administration and an elimination half-life of 2.6 h, maximal serum concentrations of 10 mM APAP are biologically possible and relevant.30 Media and released cell proteins were collected following APAP exposure to high concentrations of APAP.
Expression and Purification of SILAC-Labeled HMGB1
SILAC HMGB1 internal standard was prepared and purified as described previously.31 Briefly, the coding sequence of human HMGB1 was cloned into a pRK5 plasmid and linked to the glutathione S-transferase (GST) tag, and the plasmid was expanded in Escherichia coli. Human kidney HEK293 cells (ATCC CRL-1573) were transfected with GST-HMGB1 pRK5 plasmid using lipofectamine 2000 transfection reagent (Invitrogen). Transfected HEK293 cells were cultured in DMEM/F12/SILAC medium containing 0.2 mM [13C915N1] tyrosine and 0.5 mM [13C615N2] lysine for at least three passages. Transfected cells were harvested in NP-40 lysis buffer containing protease inhibitor cocktail after 24 h transfection. Lysis via sonication followed by centrifugation to remove cellular debris yielded SILAC-HMGB1-containing supernatant that was incubated with GSH Sepharose beads at 4 °C overnight. Beads were washed, and SILAC-HMGB1 was released via TEV enzymatic cleavage overnight at 4 °C.
Western Blots and Cell Staining
HMGB1 was detected via an HMGB1 rabbit pAb (H9539) and antirabbit HRP. Western blots were developed by use of ECL reagents. Necrosis-specific and apoptosis-specific staining were done with pSIVA and PI dyes, respectively, following washing of live cells with PBS and fixation of cells with 5% formaldehyde.
HMGB1 Cys-CAE Derivatization
Media from APAP (10 mM, 24-h exposure) or control HepaRG cell samples was split into two aliquots, and an identical amount of reduced SILAC HMGB1 internal standard (100 ng) was spiked into each. One aliquot for each sample was reduced via addition of TCEP to a final concentration of 50 mM and 30 min incubation at 37 °C. A concentrated solution of acrylamide in Tris HCl, pH 8.0 was added to reduced and nonreduced aliquots to yield a final concentration of 0.5 M acrylamide and 30 mM Tris. After acrylamide addition, samples were incubated at room temperature in the dark for 1 h. An equimolar Cys quench to remove residual acrylamide was done following acrylamide derivatization by incubation for 30 min at room temperature.
HMGB1 Immunoaffinity Precipitation
Protein A/G magnetic beads were washed twice with DPBS and twice with buffer A (0.1 M sodium phosphate, pH 7.4) before use. A total of 40 μL dispersed, prewashed beads was incubated with 100 μL buffer A and 100 μL Anti-HMGB1 rabbit pAb for 2-h at 4 °C, followed by the addition of 400 μL buffer A and overnight incubation at 4 °C. Beads were then washed twice with buffer A and twice with 1 mL of cross-linking buffer (0.2 M triethanolamine, pH 8). Beads were suspended in 1 mL of 25 mM DMP prepared in cross-linking buffer and incubated at room temperature for 1 h. Beads were washed once with 1 mL of blocking buffer (0.1 M ethanolamine, pH 8.2) and incubated at room temperature for 30 min in 1 mL of blocking buffer. Blocking buffer was then removed, and the beads were washed twice with PBS, followed by a 10 min incubation in elution buffer (0.1 M glycine-HCl, pH 2.5) at room temperature and an additional two washes with PBS. Beads were aliquoted into 10 clean protein LoBind tubes, and control or APAP-overdosed cell media acrylamide labeled, as described previously, was added for overnight incubation. HMGB1 was eluted with 15 min vigorous shaking of beads in 100 μL elution buffer, which was subsequently neutralized via addition of 50 μL 250 mM NH4HCO3. Postelution, the beads were washed once with 10 mM NH4HCO3, pH 6.0 with wash solution added to neutralization buffer to collect any residual HMGB1 still bound. The combined elute was dried under N2 for subsequent resuspension and digestion.
In Solution Lysine Acetylation and Enzymatic Digest
Samples digested with Glu-C were first acetylated with [2H6] acetic anhydride (AA) as follows. Dried samples were resuspended in a solution containing 50 μL of acetonitrile, 100 μL of 100 mM NH4HCO3, and 5 μL of AA pH adjusted to pH 8.0 via dropwise addition of ammonium hydroxide. Samples were incubated at 37 °C for 1 h. Glu-C samples were then adjusted to pH 4 via dropwise addition of acetic acid, and 500 ng of Glu-C was added per sample. Samples digested with trypsin were not acetylated and were directly resuspended in 100 μL of 25 mM NH4HCO3 containing 40 ng of trypsin. Both digests were incubated overnight at 37 °C and dried under N2 the following day. Samples were stored dry at −80 °C until LC/MS analysis, at which point they were resuspended in 30 μL water/acetonitrile (99.5:0.5 v/v) containing 0.1% formic acid.
2D-Nano-UHPLC-PRM/HRMS
UHPLC-HRMS was conducted on a Q Exactive HF hybrid quadrupole-Orbitrap mass spectrometer coupled to a Dionex Ultimate 3000 RSLC nano with capillary flowmeter chromatographic system (Thermo Fisher Scientific, San Jose, CA). A trapping column (Acclaim PepMap C18 cartridge 1 mm × 5 mm, 100 Å, Thermo Scientific) and analytical column (C18 AQ nano-LC column with a 10 μm pulled tip 75 μm × 25 cm, 3 μm particle size; Columntip, New Haven, CT) were used in the 2D system for preconcentration and peptide separation, respectively. The 2D-nano-LC system was controlled by Xcalibur software from the Q-Exactive mass spectrometer. Samples were loaded onto the trapping column with an 8 μL injection and loading solvent of water/acetonitrile (99.7:0.3 v/v) containing 0.2% formic acid. Peptides were separated on the analytical column with a linear gradient at a flow rate of 0.4 μL/min: 2% B at 2 min, 5% B at 15 min, 35% B at 40 min, 99% B at 50–60 min, and 2% B at 58–75 min. Solvent A was water/acetonitrile (99.5:0.5 v/v) containing 0.1% formic acid, and solvent B was acetonitrile/water (98:2 v/v) containing 0.1% formic acid. Nanospray Flex ion source (Thermo Scientific) was used for ionization. Spectrometer analysis conditions were: spray voltage 2500 V, ion transfer capillary temperature 250 °C, ion polarity positive, S-lens RF level 53, in-source collision-induced dissociation (CID) 1.0 eV. The full-scan parameters were resolution 60,000, automatic gain control (AGC) target 1 × 106, maximum IT 100 ms, scan range m/z 300–1600. The PRM parameters were resolution 30,000, AGC target 5 × 105, maximum IT 80 ms, loop count 15, isolation window 1.5 Da, NCE 25. PRM transitions for peptides of interest were scheduled as described in Tables S1–S3.
HMGB1 Cys Oxidation State Quantification
Relative percent disulfide and thiol oxidation state at each HMGB1 Cys residue was calculated using the acrylamide labeled signal in TCEP reduced and nonreduced samples. The four most intense transitions for each acrylamide peptide were selected, and the peak area ratio of unlabeled to SILAC- labeled peptide was calculated for each in Skyline (MacCoss Laboratory, University of Washington, Seattle, WA). The average area of the four transitions for each peptide in nonreduced samples was calculated as the signal representing free thiol Cys residues. The same calculation for TCEP-reduced samples was used as the signal for free thiol and disulfide Cys. Subtraction of the two yielded the signal for disulfide Cys residues, allowing relative quantitation. Sulfonic acid oxidized Cys peptides were also quantified with the peak area ratio of the four most intense transitions. Sulfonic acid peptide signal intensity was adjusted for ionization efficiency differences compared to acrylamide peptides by multiplying sulfonic acid area ratios by a factor of 5.1. This was based on analysis of the difference in peak area per mol synthetic acrylamide or sulfonic acid peptide analyzed via 2D-Nano-UHPLC-PRM/HRMS (Table S4). Relative quantitation of the three oxidation states was then performed directly. Statistical analysis was performed with GraphPad Prism (v9.3, GraphPad Software Inc., La Jolla, CA).
Direct Disulfide Connectivity Characterization
Recombinant HMGB1 was oxidized with 0.5 mM chloramine-T in the presence of 0.5 mM reduced GSH in 25 mM NH4HCO3, pH 7.0 via a 1 h incubation at 37 °C. Immunopurification as described previously followed by elution, and overnight trypsin digestion with 60 ng of trypsin at pH 7.0 and 37 °C was done to generate disulfide-linked peptides for optimization of 2D-Nano-UHPLC-PRM/HRMS and demonstration of peptide detectability. Disulfide-linked HMGB1 peptides were detected in biological samples via immunoaffinity precipitation (IP), as described previously, of 600 μL APAP-overdosed (10 mM APAP 24-h exposure) HepaRG media followed by digestion with 60 ng of trypsin for 18 h at pH 7.0 and 37 37 °C.
Total HMGB1 Quantification
Total HMGB1 was quantified as described previously.31 Briefly, HMGB1 was immunopurified from media with a constant amount of SILAC-HMGB1 added prior to purification. Eluent from immunopurification steps was further purified via gel electrophoresis under reducing conditions and Coomassie blue stained to allow for HMGB1 bands to be cut out of the gel. Following destaining, samples were acetylated with [2H6] acetic anhydride in-gel and digested with Glu-C overnight in-gel. Peptides freed from the gel pieces with sonication following digestion were analyzed via 2D-Nano-UHPLC-PRM/HRMS, as described previously, but with PRM for peptides E26HKKKHPDASVNFSE40, K57GKFE61, K146KAAKLKE153, and K180SKKKKE186 instead of the Cys-containing peptides used in oxidation state quantitation. The three most intense transitions were selected for each peptide, and the peak area ratio of each unlabeled peptide to SILAC- labeled peptide pair was calculated. Calibration curves (Supplementary Figure 1) were used to calculate absolute amount of each peptide present, and the average of the four was taken as the total amount of HMGB1 present.
Results
HMGB1 Release Induced by High Concentrations of APAP
HMGB1 release had a dose-dependent relationship with APAP concentration above the threshold of APAP toxicity of 5–10 mM in a hepatocarcinoma cell model (Figure 1A). High concentrations of APAP led to cell death primarily through oncotic necrosis rather than by apoptosis followed by secondary necrosis. This indicated that HMGB1 release at high concentrations of APAP was through passive diffusion following loss of membrane integrity, not by regulated secretion (Figure 1b). After a 24 h exposure to 20 mM APAP, most of the HMGB1 was secreted from hepatic cells and released into the media at levels that were much greater than those found with control cells or activated immune cells (Figure 2A,B).
Figure 1.

APAP induces dose-dependent HMGB1 release via necrosis, indicating passive HMGB1 release. (A) HMGB1 Western blot of HepaRG media following 24 h incubation with various concentrations of APAP. (B) HepaRG cells exposed to 10 mM APAP for 24 h stained with apoptosis-specific stain pSIVA and (C) necrosis specific-stain PI. (D) HepaRG cells exposed to 100 μM of known apoptosis-inducing agent doxorubicin stained with apoptosis-specific stain pSIVA and (E) necrosis specific-stain PI.
Figure 2.

Comparison and absolute quantification of HMGB1 via stable isotope dilution IP 2D-nano-UHPLC-PRM/MS in (A) the cell lysate and media of HepaRG cells following 24 h exposure to 10 mM APAP and (B) the media of various cell contexts (n = 3).
Cys-Redox State Quantification
Unbiased quantification of thiol, disulfide, and terminally oxidized sulfonic acid HMGB-Cys residues was done through differential reduction followed by CAE derivatization of free thiols with acrylamide (Figure 3). Covalent modification of free thiols with acrylamide prevented artifactual oxidation during analysis. Apart from a small amount of terminal oxidation of Cys-106 to cysteic acid, no other HMGB1 Cys-oxidation states were detected, thus quantification was mainly limited to these three Cys-oxidation states. TCEP reduction followed by CAE derivatization made it possible to calculate the amount total-Cys. Cys-disulfide was then determined from the difference between free-Cys and total-Cys. The ionization efficiency of terminally oxidized Cys-containing HMGB1 peptides was approximately five times lower than the corresponding CAE-modified peptides, allowing adjustment for those differences when making quantitative comparisons (Supplementary Table 1). Peptides containing Cys-23 and Cys-106 were obtained via trypsin digest, while a peptide containing Cys-45 was obtained via Glu-C digest. The methionine residue within the Cys-23-containing tryptic peptide was entirely oxidized to sulfoxide with no evidence of sulfone formation. Oxidation of other methionine residues was not investigated. Lysine residues in Glu-C peptides were acetylated with [2H6]-acetic anhydride to improve chromatographic behavior. Typical chromatograms for endogenous HMGB1 and SILAC standard Cys-containing tryptic peptides are shown in Figure 4; typical mass spectra are shown in Supplementary Figures 2–6. Relative HMGB1 redox state quantitation showed that Cys-23 and Cys-45 had matching oxidation states, Cys-23 was present as 82% Cys-disulfide and 18% as free-Cys, while Cys-45 was 87% Cys-disulfide and 13% free-Cys (Figure 5A,B). Cys-106 was 46% Cys-disulfide, 42% free-Cys, and 12% terminally oxidized to cysteic acid (Figure 5C).
Figure 3.
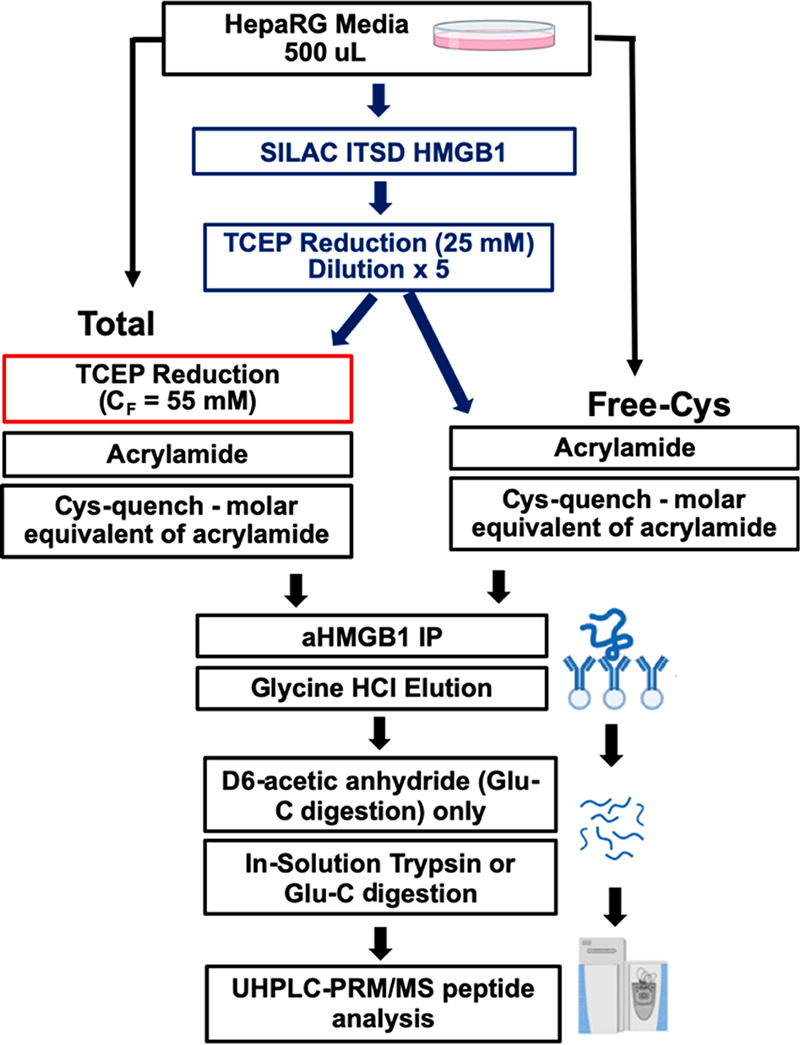
HMGB1 purification and derivatization protocol. APAP-exposed HepaRG cell media was split into two samples, each spiked with TCEP reduced stable isotope labeling by amino acids in cell culture (SILAC) HMGB1 internal standard. Differential TCEP reduction was followed by acrylamide derivatization, HMGB1 IP, and HR-LC/MS analysis of trypsin and Glu-C peptides.
Figure 4.

2D-Nano-UHPLC-PRM/MS chromatograms CAE-Cys derivatives or sulfonic acid (O3H) tryptic HMGB1 peptides. K* = [13C615N2] lysine, Y* = [13C915N1].
Figure 5.

Relative abundance of free-Cys, Cys-disulfide, and terminally oxidized Cys-residues at Cys-23 (A), Cys-45 (B), and Cys-106 (C). Terminal oxidation was not detected on Cys-23 or Cys-45 but accounted for 12% of Cys-106 residues.
Characterization of HMGB1 Cys-disulfides
Cys-disulfide HMGB1 peptides were characterized by conducting protease digestion without TCEP reduction in order to establish the disulfide connectivity. Recombinant HMGB1 was oxidized with chloramine-T in the presence of 0.5 mM GSH. Chloramine-T is known to induce protein disulfide bonds via generation of hydroxyl radicals.32,33 The solution was kept below pH 7.0 to avoid disulfide scrambling and thiol–disulfide exchange commonly observed at higher pH ranges.34 Chloramine T oxidation yielded the cross-linked Cys-23 to Cys-45 disulfide tryptic peptide M(O)13SSYAFFVQTC23(C45SER46)R24, the Cys-23 to GSH disulfide peptide [M(O)13SSYAFFVQTC23(CEG)R24], and the cross-linked Cys-106 to GSH disulfide tryptic peptide [R97PPSAFFLFC106(CEG)SEYRPK112] (Figure 6), representative mass spectra for which are shown in Supplementary Figures 7 and 8, respectively. Cross-linked disulfide peptides between Cys-23 and Cys-106, Cys-45 and Cys-106, and Cys-45 and GSH were not detected. The cross-linked Cys-23 to Cys-45 disulfide tryptic peptide M(O)13SSYAFFVQTC23(C45SER46)R24 was also observed in the HMGB1 secreted from hepatocytes by high concentrations of APAP (Figure 7). However, C-23 and C-106 glutathionylated tryptic peptides characterized in the chloramine T reaction with HMGB1 and GSH were not detected. HMGB1 tryptic peptides with metabolic modification of the C-106 glutathionylation or C-106 cystine were also not detected in hepatocyte secretion samples. Dimerization of HMGB1 via Cys-106 intermolecular disulfides cannot be ruled out due to difficulty in generating a detectable Cys-106 disulfide peptide, but no evidence of dimerization was observed in nonreducing Western blots of biological samples.
Figure 6.

2D-Nano-UHPLC-PRM/MS chromatograms of disulfide-linked peptides from recombinant HMGB1 oxidized with 0.5 mM chloramine-T in the presence of 0.5 mM GSH. (A) Cys-23 to Cys-45 disulfide peptide and (B) Cys-106 to GSH disulfide peptide.
Figure 7.

2D-nano-UHPLC-PRM/MS chromatogram of Cys-23 to Cys-45 disulfide-linked HMGB1 peptide from APAP overdose hepatocyte cell model. Low disulfide peptide ionization efficiency relative to sample matrix components results in some unassigned ions. The unassigned ions eluted at slightly different retention times to the Cys-23 to Cys-45 disulfide-linked HMGB1 peptide.
Discussion
HMGB1 is passively released during acute pathological conditions such as APAP overdose and has an important role in the subsequent immune response.12 Despite the potential for specific HMGB1 proteoforms to serve as pathology-specific biomarkers and their relevance for the biological response to pathology, there is no consensus on what HMGB1 proteoforms are secreted under different conditions. The field is marred by contradictory and ill-designed studies that have served to obfuscate the role of HMGB1 in many pathological conditions.5−23 There are three main reasons for this untenable situation. First, most of the >1600 studies of circulating HMGB1 that were designed to assess the role of HMGB1 in various clinical conditions have analyzed it in serum rather than plasma.35 HMGB1, which is present in platelets35−37 and other white blood cells,38−40 is secreted during the clotting process when serum is prepared. Therefore, studies of serum HMGB1 provide little insight into whether HMGB1 is involved in the particular disease state being studied let alone what proteoforms are involved in that disease. Second, it is often suggested that acetylation of lysine residues on nuclear localization signal (NLS)1 and/or NLS2 (Scheme 1) on HMGB1 is required before HMGB1 can be secreted.41−44 This suggestion is based on an incorrect interpretation of matrix-assisted laser desorption mass spectra in an early publication45 and on LC-MS data that were later retracted.46 Using a novel technique involving deuteroacetylation of HMGB1 lysine residues coupled with IP and UHPLC-HRMS analysis, we definitively showed that lysine residues on NLS1 or NLS2 are not acetylated when HMGB1 is secreted into the serum.31 Third, none of the studies on the oxidation state of the thee Cys-HMGB1 residues have used rigorously validated methodology.23 In addition, it has been assumed that Cys-derivatization with reagents such as iodoacetamide always go to 100% completion. This means that when reduction of disulfide residues and a second derivatization step is used (such as with N-ethyl maleimide) to determine total Cys-levels, overestimates of disulfide content are possible. In order to overcome these problems, we have employed efficient HMGB1 Cys-derivation with acrylamide to a CAE derivative in the presence of a stable isotope labeled HMGB1 internal standard followed by IP and UHPLC-HRMS analysis. This provides the free Cys-concentrations. A second experiment is then conducted using TCEP reduction of Cys-disulfides in the presence of the same labeled internal standard to give the total (free + disulfide) Cys-concentration. This has made it possible to rigorously quantify at least four of the discrete reduced and oxidized proteoforms that are secreted following incubation of hepatocytes with concentrations of APAP typical of those found in overdose situations (Figure 8).
Figure 8.

Oxidative proteoforms detected following treatment of hepatocytes with high concentrations of APAP and their relative proportions.
In the first of four detected proteoforms, HMGB1 was found to be fully reduced, with all three Cys-residues in the free thiol form. In the three other proteoforms, HMGB1 was oxidized to a disulfide between Cys-23 and Cys-45, with variation in oxidation state at Cys-106. The preferential disulfide formation between Cys-23 and Cys-45 was inferred based on identical amounts of disulfide formation and structural proximity. This was confirmed via direct characterization of a disulfide linked Cys-23/Cys-45 peptide ([M(O)13SSYAFFVQTC23(C45SER47)R24] isolated by trypsin digestion of secreted HMGB1 (Figure 7). The Cys-23/Cys-45 disulfide bond potentially alters the structure of the A box region of HMGB1 (amino acids 1–79; Figure 1), which contains both Cys-23 and Cys-45. The A box of HMGB1 has previously been implicated as the driver of HMGB1’s chemokine effects, as it has been shown to induce migration of immune cells at ten times lower concentrations than that of the B box of HMGB1 (amino acids 89–63; Scheme 1).47 Oxidation and Cys-23 to Cys-45 disulfide distortion of the A box region therefore may disrupt HMGB1’s chemokine signaling effects, resulting in a different immune response to pathology involving acute oxidative stress. Terminal oxidation to cysteic acid was previously proposed to occur at all three Cys-residues in HMGB1 released following APAP-overdose.23 Following acute oxidative stress, terminal oxidation was thought to inactivate HMGB1 and help resolve the inflammatory response. Terminal oxidation of HMGB1 was observed to occur only at Cys-106 and in only 12% of HMGB1, providing further evidence that previously proposed models of HMGB1 oxidation are incorrect.
A Cys-106 disulfide was unequivocally characterized; however, no evidence of disulfide formation with Cys-23, Cys-45, or GSH was obtained. It was very surprising given the relatively high concentrations of GSH in the hepatocytes. We cannot discount the possibility that one or more of the lysine residues close to Cys-106 could have been acetylated, which would have prevented the generation of a tryptic peptide containing Cys-106. However, this seems very unlikely as we were unable to detect any acetylated lysine residues when the Cys-106-containing peptide was analyzed as a CAE derivative after Glu-C digestion. Some 45% of the Cys-106 was present as a disulfide in the HMGB1 secreted from hepatocytes by high doses of APAP. Therefore, Cys-106 must have formed an intermolecular disulfide bond with Cys-residues on other intracellular proteins. This suggests that HMGB1 may form an intermolecular disulfide through specific binding to an unexpected partner or indiscriminately with a range of proteins, challenging our ability to identify them. Metabolic processing of HMGB1 intermolecular disulfides may additionally produce unexpected modifications. The existence of other proteoforms of HMGB1 (such as NAPQI-adducts) that were either not recovered by the immunopurification step and/or not detected by LC-MS analysis cannot be ruled out. Therefore, other proteoforms of HMGB1 could have been present. The formation of intermolecular disulfides of HMGB1 by cross-linking Cys-106 to other intracellular proteins offers a potential explanation for the functional diversity of HMGB1: Different protein disulfides could conceivably have different activities on the relevant receptors activated by HMGB1. Characterizing these cross-linked HMGB1 disulfides poses a formidable challenge. Fortunately, the stable isotope dilution UHPLC-HRMS method that has been developed for HMGB1 Cys-106 disulfides can be readily adapted for plasma samples. This will allow the importance of these disulfides to be assessed in APAP overdose as well as in different inflammatory diseases, while structural studies are conducted to fully characterize the relevant cross-linked proteins. Additionally, the methods developed for HMGB1 cysteine oxidation characterization can be extended to other proteins involved in oxidative pathology. Finally, we anticipate that quantifying Cys-23/Cys-34 cross-linked HMGB1 in plasma could provide a more specific biomarker of APAP overdose than the current method of analyzing all HMGB1 proteoforms. It is conceivable that similar HMGB1 proteoforms will arise as a result of injury induced by other liver toxicants. Previous studies have reported the presence of human serum albumin Cys-34 NAPQI-adducts after APAP overdose.48,49 Therefore, quantifying HMGB1 proteoforms together with albumin Cys-34 NAPQI-adducts in plasma49,50 offers a way to distinguish APAP-mediated liver injury from DILI as well as more general toxicant-mediated liver injury.
Glossary
Abbreviations
- 2D
two-dimensional
- APAP
acetaminophen
- DILI
drug-induced liver injury
- DMP
dimethyl pimelimidate
- ECL
electrochemical luminescence
- HMGB1
high mobility group box 1
- NAPQI
N-acetyl-p-benzoquinoneimine
- GSH
glutathione
- GST
glutathione S-transferase
- HRMS
high-resolution mass spectrometry
- HRP
horseradish peroxidase
- pAb
polyclonal antibody
- PI
propidium iodide
- PRM
parallel reaction monitoring
- pSIVA
polarity-sensitive indicator of viability
- SILAC
stable isotope labeling by amino acids in cell culture
- TCEP
tris(2-carboxyethyl)-phosphine
- UHPLC
ultrahigh-performance liquid chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.2c00161.
Ionization efficiency measurement data; calibration curves for absolute quantitation of HMGB1; Q-Exactive Orbitrap high-resolution product ion mass spectra for carbamidoethyl and cysteic acid HMGB1 peptides and respective SILAC internal standards; Q-Exactive Orbitrap high-resolution product ion mass spectra for disulfide linked HMGB1 peptides (PDF)
This publication was made possible by funding from P30 ES013508 (I.A.B.) and T32ES019851 (R.P.) from the National Institute of Environmental Health Sciences (NIEHS), NIH, and DHHS.
The authors declare no competing financial interest.
Dedication
This manuscript is dedicated to Dr. Larry Marnett to honor his contribution to 35-years of Chemical Research in Toxicology and our wonderful years working together.
Supplementary Material
References
- Rauvala H.; Pihlaskari R. Isolation and some characteristics of an adhesive factor of brain that enhances neurite outgrowth in central neurons. J. Biol. Chem. 1987, 262, 16625–16635. 10.1016/S0021-9258(18)49302-7. [DOI] [PubMed] [Google Scholar]
- Tsuda K.; Kikuchi M.; Mori K.; Waga S.; Yoshida M. Primary structure of non-histone protein HMG1 revealed by the nucleotide sequence. Biochemistry 1988, 27, 6159–6163. 10.1021/bi00416a050. [DOI] [PubMed] [Google Scholar]
- Scaffidi P.; Misteli T.; Bianchi M. E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- Bell C. W.; Jiang W.; Reich C. F. III; Pisetsky D. S. The extracellular release of HMGB1 during apoptotic cell death. American Journal of Physiology-Cell Physiology 2006, 291, C1318–C1325. 10.1152/ajpcell.00616.2005. [DOI] [PubMed] [Google Scholar]
- Chen G.; Li J.; Ochani M.; Rendon-Mitchell B.; Qiang X.; Susarla S.; Ulloa L.; Yang H.; Fan S.; Goyert S. M.; et al. Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14-and TNF-dependent mechanisms. Journal of leukocyte biology 2004, 76, 994–1001. 10.1189/jlb.0404242. [DOI] [PubMed] [Google Scholar]
- Hori O.; Brett J.; Slattery T.; Cao R.; Zhang J.; Chen J. X.; Nagashima M.; Lundh E. R.; Vijay S.; Nitecki D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- Park J. S.; Svetkauskaite D.; He Q.; Kim J.-Y.; Strassheim D.; Ishizaka A.; Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 2004, 279, 7370–7377. 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Yu M.; Wang H.; Ding A.; Golenbock D. T.; Latz E.; Czura C. J.; Fenton M. J.; Tracey K. J.; Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- Schiraldi M.; Raucci A.; Muñoz L. M.; Livoti E.; Celona B.; Venereau E.; Apuzzo T.; De Marchis F.; Pedotti M.; Bachi A.; et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. Journal of experimental medicine 2012, 209, 551–563. 10.1084/jem.20111739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu I. E.; Bianchi M. E.; Bacci M.; Manfredi A. A.; Rovere-Querini P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. Journal of leukocyte biology 2007, 81, 84–91. 10.1189/jlb.0306171. [DOI] [PubMed] [Google Scholar]
- Bartholdi D.; Schwab M. E. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: An in situ hybridization study. European Journal of Neuroscience 1997, 9, 1422–1438. 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Xu Y.; Sparkenbaugh E. M.; Li M.; Han X.; Zhang X.; Xia K.; Piegore M.; Zhang F.; Zhang X.; Henderson M.; Pagadala V.; Su G.; Tan L.; Park P. W.; Stravitz R. T.; Key N. S.; Linhardt R. J.; Pawlinski R.; Xu D.; Liu J. Design of anti-inflammatory heparan sulfate to protect against acetaminophen-induced acute liver failure. Sci. Transl Med. 2020, 12, aav8075. 10.1126/scitranslmed.aav8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine D. J.; Harris H. E.; Andersson U.; Tracey K. J.; Bianchi M. E. A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Molecular medicine 2014, 20, 135–137. 10.2119/molmed.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B.; Wang C.; Wang M.; Li W.; Chen F.; Tracey K. J.; Wang H. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev. Clin Immunol 2014, 10, 713–727. 10.1586/1744666X.2014.909730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. E.; Frigerio F.; Ravizza T.; Ricci E.; Tse K.; Jenkins R. E.; Sills G. J.; Jorgensen A.; Porcu L.; Thippeswamy T.; et al. RETRACTED: Molecular isoforms of high-mobility group box 1 are mechanistic biomarkers for epilepsy. J. Clin. Invest. 2019, 129, 2166–2166. 10.1172/JCI129285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez C.; Huebener P.; Pradere J.-P.; Antoine D. J.; Friedman R. A.; Schwabe R. F. EXPRESSION OF CONCERN: HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J. Clin. Invest. 2018, 128, 2436–2451. 10.1172/JCI91786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine D. J.; Williams D. P.; Kipar A.; Laverty H.; Park B. K. EXPRESSION OF CONCERN: Diet restriction inhibits apoptosis and HMGB1 oxidation and promotes inflammatory cell recruitment during acetaminophen hepatoxicity. Mol. Med. 2010, 16, 479–490. 10.2119/molmed.2010.00126. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Huebener P.; Pradere J.-P.; Hernandez C.; Gwak G.-Y.; Caviglia J. M.; Mu X.; Loike J. D.; Jenkins R. E.; Antoine D. J.; Schwabe R. F. EXPRESSION OF CONCERN: The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin Invest 2015, 125, 539–550. 10.1172/JCI76887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano A.; Antoine D. J.; Pellegrini L.; Baumann F.; Pagano I.; Pastorino S.; Goparaju C. M.; Prokrym K.; Canino C.; Pass H. I.; Carbone M.; Yang H. EXPRESSION OF CONCERN: HMGB1 and Its Hyperacetylated Isoform are Sensitive and Specific Serum Biomarkers to Detect Asbestos Exposure and to Identify Mesothelioma Patients. Clin. Cancer Res. 2016, 22, 3087–3096. 10.1158/1078-0432.CCR-15-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khambu B.; Huda N.; Chen X.; Antoine D. J.; Li Y.; Dai G.; Kohler U. A.; Zong W. X.; Waguri S.; Werner S.; Oury T. D.; Dong Z.; Yin X. M. EXPRESSION OF CONCERN: HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J. Clin Invest 2018, 128, 2419–2435. 10.1172/JCI91814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmblad K.; Schierbeck H.; Sundberg E.; Horne A. C.; Harris H. E.; Henter J. I.; Antoine D. J.; Andersson U. EXPRESSION OF CONCERN: High systemic levels of the cytokine-inducing HMGB1 isoform secreted in severe macrophage activation syndrome. Mol. Med. 2014, 20, 538–547. 10.2119/molmed.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sun Q.; Loughran P.; Shapiro R.; Shrivastava I. H.; Antoine D. J.; Li T.; Yan Z.; Fan J.; Billiar T. R.; Scott M. J. Retracted: Redox-dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology 2017, 65, 253–268. 10.1002/hep.28893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Lundbäck P.; Ottosson L.; Erlandsson-Harris H.; Venereau E.; Bianchi M. E.; Al-Abed Y.; Andersson U.; Tracey K. J.; Antoine D. J. RETRACTED: Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Molecular medicine 2012, 18, 250–259. 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Blieden M.; Paramore L. C.; Shah D.; Ben-Joseph R. A perspective on the epidemiology of acetaminophen exposure and toxicity in the United States. Expert review of clinical pharmacology 2014, 7, 341–348. 10.1586/17512433.2014.904744. [DOI] [PubMed] [Google Scholar]
- Blair I. A.; Boobis A. R.; Davies D. S.; Cresp T. M. Paracetamol oxidation: synthesis and reactivity of N-acetyl-p-benzoquinoneimine. Tet.Lett. 1980, 21, 4947–4950. 10.1016/S0040-4039(00)71162-2. [DOI] [Google Scholar]
- Patten C. J.; Thomas P. E.; Guy R. L.; Lee M.; Gonzalez F. J.; Guengerich F. P.; Yang C. S. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem. Res. Toxicol. 1993, 6, 511–518. 10.1021/tx00034a019. [DOI] [PubMed] [Google Scholar]
- Fernando C. R.; Calder I. C.; Ham K. N. Studies on the mechanism of toxicity of acetaminophen. Synthesis and reactions of N-acetyl-2, 6-dimethyl-and N-acetyl-3, 5-dimethyl-p-benzoquinone imines. Journal of medicinal chemistry 1980, 23, 1153–1158. 10.1021/jm00185a001. [DOI] [PubMed] [Google Scholar]
- Jaeschke H.; Xie Y.; McGill M. R. Acetaminophen-induced liver injury: from animal models to humans. Journal of clinical and translational hepatology 2014, 2, 153. 10.14218/JCTH.2014.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumack B. H.; Peterson R. C.; Koch G. G.; Amara I. A. Acetaminophen overdose: 662 cases with evaluation of oral acetylcysteine treatment. Archives of internal medicine 1981, 141, 380–385. 10.1001/archinte.141.3.380. [DOI] [PubMed] [Google Scholar]
- Forrest J. A.; Clements J.; Prescott L. Clinical pharmacokinetics of paracetamol. Clinical pharmacokinetics 1982, 7, 93–107. 10.2165/00003088-198207020-00001. [DOI] [PubMed] [Google Scholar]
- Weng L.; Guo L.; Vachani A.; Mesaros C.; Blair I. A. Quantification of serum high mobility group box 1 by liquid chromatography/high-resolution mass spectrometry: implications for its role in immunity, inflammation, and cancer. Analytical chemistry 2018, 90, 7552–7560. 10.1021/acs.analchem.8b01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anraku M.; Kragh-Hansen U.; Kawai K.; Maruyama T.; Yamasaki Y.; Takakura Y.; Otagiri M. Validation of the chloramine-T induced oxidation of human serum albumin as a model for oxidative damage in vivo. Pharm. Res. 2003, 20, 684–692. 10.1023/A:1023219420935. [DOI] [PubMed] [Google Scholar]
- Shechter Y.; Burstein Y.; Patchornik A. Selective oxidation of methionine residues in proteins. Biochemistry 1975, 14, 4497–4503. 10.1021/bi00691a025. [DOI] [PubMed] [Google Scholar]
- Sung W.-C.; Chang C.-W.; Huang S.-Y.; Wei T.-Y.; Huang Y.-L.; Lin Y.-H.; Chen H.-M.; Chen S.-F. Evaluation of disulfide scrambling during the enzymatic digestion of bevacizumab at various pH values using mass spectrometry. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 2016, 1864, 1188–1194. 10.1016/j.bbapap.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Pawlinski R. Platelet HMGB1: the venous clot coordinator. Blood 2016, 128, 2376–2378. 10.1182/blood-2016-09-738740. [DOI] [PubMed] [Google Scholar]
- Vogel S.; Rath D.; Borst O.; Mack A.; Loughran P.; Lotze M. T.; Neal M. D.; Billiar T. R.; Gawaz M. Platelet-derived high-mobility group box 1 promotes recruitment and suppresses apoptosis of monocytes. Biochem. Biophys. Res. Commun. 2016, 478, 143–148. 10.1016/j.bbrc.2016.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Li F.; Augi T.; Williamson K. M.; Onishi K.; Hogan M. V.; Neal M. D.; Wang J. H. Platelet HMGB1 in Platelet-Rich Plasma (PRP) promotes tendon wound healing. PLoS One 2021, 16, e0251166. 10.1371/journal.pone.0251166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabart P.; Kalousek I.; Jandova D.; Hrkal Z. Differential expression of nuclear HMG1, HMG2 proteins and H1(zero) histone in various blood cells. Cell Biochem Funct 1995, 13, 125–133. 10.1002/cbf.290130209. [DOI] [PubMed] [Google Scholar]
- Tang D.; Shi Y.; Kang R.; Li T.; Xiao W.; Wang H.; Xiao X. Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc Biol. 2007, 81, 741–747. 10.1189/jlb.0806540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schierbeck H.; Wahamaa H.; Andersson U.; Harris H. E. Immunomodulatory drugs regulate HMGB1 release from activated human monocytes. Mol. Med. 2010, 16, 343–351. 10.2119/molmed.2010.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotze M. T.; Tracey K. J. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol 2005, 5, 331–342. 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- Venereau E.; De Leo F.; Mezzapelle R.; Careccia G.; Musco G.; Bianchi M. E. HMGB1 as biomarker and drug target. Pharmacol. Res. 2016, 111, 534–544. 10.1016/j.phrs.2016.06.031. [DOI] [PubMed] [Google Scholar]
- Tripathi A.; Shrinet K.; Kumar A. HMGB1 protein as a novel target for cancer. Toxicol Rep 2019, 6, 253–261. 10.1016/j.toxrep.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.; Kang R.; Tang D. The mechanism of HMGB1 secretion and release. Exp Mol. Med. 2022, 54, 91–102. 10.1038/s12276-022-00736-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldi T.; Talamo F.; Scaffidi P.; Ferrera D.; Porto A.; Bachi A.; Rubartelli A.; Agresti A.; Bianchi M. E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoine D. J.; Jenkins R. E.; Dear J. W.; Williams D. P.; McGill M. R.; Sharpe M. R.; Craig D. G.; Simpson K. J.; Jaeschke H.; Park B. K. RETRACTED: Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol 2012, 56, 1070–1079. 10.1016/j.jhep.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Degryse B.; Bonaldi T.; Scaffidi P.; Müller S.; Resnati M.; Sanvito F.; Arrigoni G.; Bianchi M. E. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell Biol. 2001, 152, 1197–1206. 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsten M. C.; Commandeur J. N.; Fidder A.; Hulst A. G.; Touw D.; Noort D.; Vermeulen N. P. Liquid chromatography/tandem mass spectrometry detection of covalent binding of acetaminophen to human serum albumin. Drug Metab. Dispos. 2007, 35, 1408–1417. 10.1124/dmd.106.014233. [DOI] [PubMed] [Google Scholar]
- Geib T.; LeBlanc A.; Shiao T. C.; Roy R.; Leslie E. M.; Karvellas C. J.; Sleno L. Absolute quantitation of acetaminophen-modified human serum albumin in acute liver failure patients by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 1573–1582. 10.1002/rcm.8206. [DOI] [PubMed] [Google Scholar]
- Ozawa M.; Kubo T.; Lee S. H.; Oe T. LC-MS analyses of N-acetyl-p-benzoquinone imine-adducts of glutathione, cysteine, N-acetylcysteine, and albumin in a plasma sample: A case study from a patient with a rare acetaminophen-induced acute swelling rash. J. Toxicol Sci. 2019, 44, 559–563. 10.2131/jts.44.559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.